---
title: "A Complete Example"
output:
rmarkdown::html_vignette:
toc: true
description: >
Step through a complex subtyping workflow.
vignette: >
%\VignetteIndexEntry{A Complete Example}
%\VignetteEngine{knitr::rmarkdown}
%\VignetteEncoding{UTF-8}
---
```{r, include = FALSE}
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>"
)
```
```{r echo = FALSE}
options(crayon.enabled = FALSE, cli.num_colors = 0)
```
Download a copy of the vignette to follow along here: [a_complete_example.Rmd](https://raw.githubusercontent.com/BRANCHlab/metasnf/main/vignettes/a_complete_example.Rmd)
We recommend you go through the [simple example](https://branchlab.github.io/metasnf/articles/a_simple_example.html) before working through this one.
This vignette walks through how the core functionality of this package, including:
1. Setting up the data
2. Building a space of settings to cluster over
3. Running SNF
4. Identifying and visualizing meta clusters
5. Characterizing and selecting top meta clusters
6. Selecting a representative cluster solution from a meta cluster
7. Cluster solution characterization
9. Cluster solution validation
## Data Set-up
### Pre-processing
Your data should be loaded into the R environment in the following format:
1. The data is in one or more `data.frame` class objects
2. The data is in wide form (one row per observation to cluster)
3. All data frames should have exactly one column that uniquely identifies each observation
4. All data should be complete (no missing values)
If you wish to use imputation to handle missingness in your data, you can take a look at the [imputation vignette](https://branchlab.github.io/metasnf/articles/imputations.html) which outlines a basic workflow for meta clustering across multiple imputations of the same dataset.
The package comes with a few mock data frames based on real data from the Adolescent Brain Cognitive Development study:
- `anxiety` (anxiety scores from the CBCL)
- `depress` (depression scores from the CBCL)
- `cort_t` (cortical thicknesses)
- `cort_sa` (cortical surface areas in mm^2)
- `subc_v` (subcortical volumes in mm^3)
- `income` (household income on a 1-3 scale)
- `pubertal` (pubertal status on a 1-5 scale)
Here's what the cortical thickness data looks like:
```{r}
library(metasnf)
class(cort_t)
dim(cort_t)
str(cort_t[1:5, 1:5])
cort_t[1:5, 1:5]
```
The first column `unique_id` is the unique identifier (UID) for all observations in the data.
Here's the household income data:
```{r}
dim(income)
str(income[1:5, ])
income[1:5, ]
```
Putting everything in a list will help us get quicker summaries of all the data.
```{r}
df_list <- list(
anxiety,
depress,
cort_t,
cort_sa,
subc_v,
income,
pubertal
)
# The number of rows in each data frame:
lapply(df_list, dim)
# Whether or not each data frame has missing values:
lapply(df_list,
function(x) {
any(is.na(x))
}
)
```
Some of the data has missing values and not all of the data frames have the same number of participants.
SNF can only be run with complete data, so you'll need to either use complete case analysis (removal of observations with any missing values) or impute the missing values to proceed with the clustering.
As mentioned above, `metasnf` can be used to visualize changes in clustering results across different imputations of the data.
For now, we'll just examine the simpler complete-case analysis approach by reducing our data frames to only common and complete observations.
This can be made easier using the `get_complete_uids` function.
```{r}
complete_uids <- get_complete_uids(df_list, uid = "unique_id")
print(length(complete_uids))
# Reducing data frames to only common observations with no missing data
anxiety <- anxiety[anxiety$"unique_id" %in% complete_uids, ]
depress <- depress[depress$"unique_id" %in% complete_uids, ]
cort_t <- cort_t[cort_t$"unique_id" %in% complete_uids, ]
cort_sa <- cort_sa[cort_sa$"unique_id" %in% complete_uids, ]
subc_v <- subc_v[subc_v$"unique_id" %in% complete_uids, ]
income <- income[income$"unique_id" %in% complete_uids, ]
pubertal <- pubertal[pubertal$"unique_id" %in% complete_uids, ]
```
### Generating the data list
The `data_list` class is a structured list of data frames (like the one already created), but with some additional metadata about each data frame.
It should only contain the data frames we want to directly use as inputs for the clustering.
Let's say we are working in a context where the anxiety and depression data are especially important outcomes and we want to know if we can find subtypes using the other data which still do a good job of separating out observations by their anxiety and depression scores.
We'll start by initializing a data list that stores our input features.
```{r}
# Note that you do not need to explicitly name every single named element
# (data = ..., name = ..., etc.)
input_dl <- data_list(
list(
data = cort_t,
name = "cortical_thickness",
domain = "neuroimaging",
type = "continuous"
),
list(
data = cort_sa,
name = "cortical_surface_area",
domain = "neuroimaging",
type = "continuous"
),
list(
data = subc_v,
name = "subcortical_volume",
domain = "neuroimaging",
type = "continuous"
),
list(
data = income,
name = "household_income",
domain = "demographics",
type = "continuous"
),
list(
data = pubertal,
name = "pubertal_status",
domain = "demographics",
type = "continuous"
),
uid = "unique_id"
)
```
This process removes any observations that did not have complete data across all provided input data frames.
The structure of the data list is a nested list tracking the data, the name of the data frame, what domain (broader source of information) the data frame belongs to, and the type of feature stored in that data frame.
Options for feature type include "continuous", "discrete", "ordinal", "categorical", and "mixed".
The `uid` parameter is the name of the column in the data frames that uniquely identifies each observation.
Upon data list creation, the UID will be converted to `"uid"` and the UIDs themselves will be prefixed with `"_uid"` for ease of management across other functions in the package.
We can get a summary of our constructed data list with the `summary` function:
```{r}
summary(input_dl)
```
Each input data frame now has the same 87 observations with complete data.
The width refers to the number of features in each data frame (not including the UID column).
`data_list` now stores all the features we intend on using for the clustering.
We're interested in knowing if any of the clustering solutions we generate can distinguish children apart based on their anxiety and depression scores.
To do this, we'll also create a data list storing only features that we'll use for evaluating our cluster solutions and not for the clustering itself.
We'll refer to this as the target data list.
```{r}
target_dl <- data_list(
list(anxiety, "anxiety", "behaviour", "ordinal"),
list(depress, "depressed", "behaviour", "ordinal"),
uid = "unique_id"
)
summary(target_dl)
```
Note that it is not necessary to make use of a partition of input and out-of-model measures in this way.
If you'd like to have no target data list and instead use every single feature of interest for the clustering, you can stick to just using one data list.
## Defining sets of hyperparameters to use for SNF and clustering
The SNF config stores all the information about the settings and functions to be used for each SNF run to be completed.
Calling the `snf_config` function with a specified number of rows will automatically build a randomly populated `snf_config` class object.
```{r}
set.seed(42)
my_sc <- snf_config(
dl = input_dl,
n_solutions = 20,
min_k = 20,
max_k = 50
)
my_sc
```
The SNF config contains multiple parts that all play a role in determining how the data in a data list will be converted into a cluster solution.
### The settings data frame
The first part is the settings data frame:
```{r}
my_sc$"settings_df"
```
The settings data frame stores information about:
- **SNF hyperparameters**: `alpha`, `k`, and `t` hyperparameters that are directly used by the SNF algorithm.
- **SNF scheme**: the specific way in which input data gets collapsed into a final fused network (discussed further in the [SNF schemes vignette](https://branchlab.github.io/metasnf/articles/snf_schemes.html)).
- **Clustering functions**: Which clustering algorithm will be applied to the final fused network produced by SNF.
- **Distance function**: Which distance metric will be used when converting continuous, discrete, ordinal, categorical, or mixed type data into intermediate distance matrices (discussed further in the [distance metrics vignette](https://branchlab.github.io/metasnf/articles/distance_metrics.html)).
- **Component dropout** - Information on whether or not a particular input data frame in the data list will be in included or excluded from the corresponding SNF run (discussed further in the [SNF config vignette](https://branchlab.github.io/metasnf/articles/snf_config.html)).
The underlying structure of a settings data frame can be viewed by converting it from a `settings_df` class object into a regular `data.frame` class object:
```{r}
head(as.data.frame(my_sc$"settings_df"))
```
Each row of the settings data frame corresponds to a set of hyperparameters that can be used for generating a cluster solution.
Without specifying any additional parameters, the `snf_config` function randomly populates these columns and ensures that no generated rows are identical.
Further customization of the `settings_df` and the other parts of the SNF config will enable you to generate a broader space of cluster solutions from your data and ideally get you closer to finding a more useful solution for your context.
More on `settings_df` customization can be found in the [SNF config vignette](https://branchlab.github.io/metasnf/articles/snf_config.html).
Setting a seed prior to calling `snf_config()` ensures that the same random settings are generated each time we run the code.
### Other parts of the SNF config
There are three other parts to an SNF config object: a distance functions list (`dist_fns_list` class object) that stores distance metric functions, a clustering functions list (`clust_fns_list` class object) that stores clustering algorithm functions, and a weights matrix (`weights_matrix`) class object that stores feature weights.
```{r}
names(my_sc)
```
Customizing each of these parts is explained in more detail in separate vignettes:
- [distance metrics](https://branchlab.github.io/metasnf/articles/distance_metrics.html)
- [clustering functions list](https://branchlab.github.io/metasnf/articles/clustering_algorithms.html)
- [weights matrix](https://branchlab.github.io/metasnf/articles/feature_weights.html)
For now, we'll leave them as the default values generated during our `snf_config` call.
## Running SNF and clustering
The `batch_snf()` function integrates the data in the data list using all the hyperparameters, and functions contained in the SNF config.
The resulting structure is a `solutions_df` class object, which contains an index column (`solution`), a column tracking the number of clusters in the cluster solution (`nclust`), a column labeling which meta cluster a solution belongs to (`mc`; more on this later) and columns specifying which cluster each subject was assigned to for each corresponding solution.
```{r eval = FALSE}
sol_df <- batch_snf(input_dl, my_sc)
sol_df
```
```{r echo = FALSE}
sol_df <- cache_a_complete_example_sol_df
sol_df
```
Just like that, 20 different cluster solutions have been generated!
In practice, you may end up wanting to create hundreds or thousands of cluster solutions at a time.
If you have access to a multi-core system, `batch_snf` can be run with parallel processing enabled.
See `?batch_snf` or the [parallel processing vignette](https://branchlab.github.io/metasnf/articles/parallel_processing.html) for more information.
You can pull the clustering results out of each row in a more convenient, transposed format using the `t()` function:
```{r}
cluster_solutions <- t(sol_df)
cluster_solutions
```
When transposed, the `solutions_df` class object is converted to a `t_solutions_df` class object, which only contains columns indicating what cluster each observation was assigned to for each solution.
Calling `t()` on a `t_solutions_df` class object will return it back to its original `solutions_df` form.
## Identifying and visualizing meta clusters
Now that we have access to 20 different clustering solutions, we'll need to find some way to pick an optimal one to move forward with for additional characterization.
In this case, plotting or running stats manually on each of the solutions might be a reasonable way to determine which ones may be the most suitable for our purpose.
But when the number of solutions generated goes up into the hundreds (or thousands), these approaches can become impractical or inefficient.
The main approach we recommend using is the meta clustering approach described in Caruana et al., 2006.
Meta clustering is a useful approach when the criteria for cluster solution evaluation is difficult to formalize and fully automate.
Meta clustering consists of clustering the *clustering solutions themselves* to yield a manageable number of qualitatively similar meta cluster solutions that are representative of the full set of solutions.
Characterization of these representative solutions then enables efficient identification of a maximally useful cluster solution for the user's context.
The first step in meta clustering is to calculate the [adjusted Rand index](https://en.wikipedia.org/wiki/Rand_index) (ARI) between each pair of cluster solutions.
This metric tells us how similar the solutions are to each other, thereby allowing us to find clusters of cluster solutions.
```{r}
sol_aris <- calc_aris(sol_df)
head(sol_aris)
```
We can visualize the resulting inter-cluster similarities with a heatmap.
Heatmaps in the `metasnf` package are constructed using the [`ComplexHeatmap`](https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html) and [`InteractiveComplexHeatmap`](https://bioconductor.org/packages/release/bioc/html/InteractiveComplexHeatmap.html) packages from [Bioconductor](https://bioconductor.org/).
If you don't already have these packages, you can install them with the following code:
```{r eval = FALSE}
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# To make heatmaps with metasnf
BiocManager::install("ComplexHeatmap")
# To use heatmap shiny app (later in vignette) with metasnf
BiocManager::install("InteractiveComplexHeatmap")
```
First, call `get_matrix_order()` to get a hierarchical clustering-based ordering of the rows in the adjusted rand indices.
```{r}
meta_cluster_order <- get_matrix_order(sol_aris)
# Just a vector of numbers
meta_cluster_order
```
This order can be passed into the `meta_cluster_heatmap` function to get a clearer view of existing meta clusters.
```{r eval = FALSE}
ari_hm <- meta_cluster_heatmap(
sol_aris,
order = meta_cluster_order
)
ari_hm
```
```{r eval = FALSE, echo = FALSE}
save_heatmap(
heatmap = ari_hm,
path = "vignettes/meta_cluster_heatmap.png",
width = 330,
height = 300,
res = 100
)
```
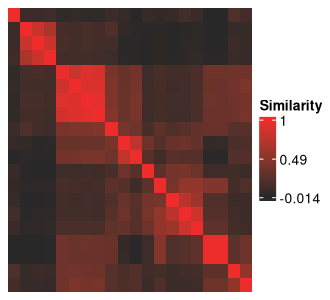
The clustering solutions are along the rows and columns of the above figure, and the cells at the intersection between two solutions show how similar they are to each other based on their ARI.
The diagonals should always be red, representing the maximum value of 1, as they show self-similarity.
Complete-linkage, Euclidean-distance based hierarchical clustering is being applied to these solutions to obtain the row ordering.
This is also the default approach used by the `ComplexHeatmap` package, the backbone of all heatmap functions in `metasnf`.
This heatmap is integral to identifying the meta clusters created from your data using the space of parameters defined in the SNF config.
We will later see how to further customize this heatmap to add in rich information about how each plotted cluster solution differs on various measures of quality, different clustering settings, and different levels of separation on input and out-of-model measures.
First, we'll divide the heatmap into meta clusters by visual inspection.
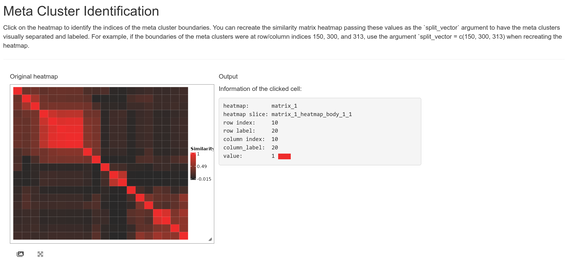
The indices of our meta cluster boundaries can be passed into the `meta_cluster_heatmap` function as the `split_vector` parameter.
You can determine this vector either by trial and error (repeated replotting with different `split_vector` values) or by using the `shiny_annotator` function, a wrapper around functionality from the `InteractiveComplexHeatmap` package.
While the shiny app is running, your R console will be unresponsive.
Clicking on meta cluster cell boundaries and tracking the row/column indices (printed to the R console as well as displayed in the app) can get us the following vector:
A demonstration of the shiny app can be seen here: [Meta Cluster Identification](https://pvelayudhan.shinyapps.io/shiny/)
```{r eval = FALSE}
shiny_annotator(ari_hm)
```
```{r}
split_vec <- c(2, 5, 12, 17)
```
This vector can be used to populate the meta cluster column in our solutions data frame as well as visualize our meta cluster boundaries more clearly.
```{r}
mc_sol_df <- label_meta_clusters(
sol_df,
order = meta_cluster_order,
split_vector = split_vec
)
mc_sol_df
```
```{r eval = FALSE}
ari_mc_hm <- meta_cluster_heatmap(
sol_aris,
order = meta_cluster_order,
split_vector = split_vec
)
ari_mc_hm
```
```{r eval = FALSE, echo = FALSE}
save_heatmap(
heatmap = ari_mc_hm,
path = "vignettes/ari_mc_hm.png",
width = 330,
height = 300,
res = 100
)
```
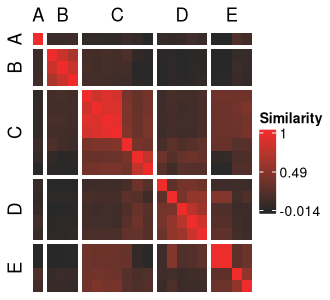
At this point, we have our meta clusters but are not yet sure of how they differ in terms of their structure across our input and out-of-model features.
## Characterizing cluster solutions
### Calculating associations between cluster solutions and initial data
We start by running the `extend_solutions` function, which will calculate p-values representing the strength of the association between cluster membership (treated as a categorical feature) and any feature present in a provided data list and/or target_list.
`extend_solutions` also adds summary p-value measures (min, mean, and max) of any features present in the target list.
This function can be quite slow depending on how large your data is; you can monitor its progress by setting `verbose = TRUE`.
```{r eval = FALSE}
ext_sol_df <- extend_solutions(
mc_sol_df,
dl = input_dl,
target_dl = target_dl
)
ext_sol_df
```
```{r echo = FALSE}
ext_sol_df <- cache_a_complete_example_ext_sol_df
ext_sol_df
```
The difference between the data list passed into the `dl` parameter and the one passed into the `target_dl` parameter is that the `target_dl` features are the only ones used for generating the p-value summary columns.
```{r eval = FALSE}
# No features are used to calculate summary p-values
ext_sol_df_no_summaries <- extend_solutions(
mc_sol_df,
dl = c(input_dl, target_dl)
)
# Every features are used to calculate summary p-values
ext_sol_df_all_summaries <- extend_solutions(
mc_sol_df,
target_dl = c(input_dl, target_dl)
)
```
### Visualizing feature associations with meta clustering results
This `ext_sol_df` we created can be converted into a standard data frame and passed into the `meta_cluster_heatmap` function to easily visualize the level of separation on each of our features for each of our cluster solutions.
```{r eval = FALSE}
annotated_ari_hm <- meta_cluster_heatmap(
sol_aris,
order = meta_cluster_order,
split_vector = split_vec,
data = as.data.frame(ext_sol_df),
top_hm = list(
"Depression p-value" = "cbcl_depress_r_pval",
"Anxiety p-value" = "cbcl_anxiety_r_pval",
"Overall outcomes p-value" = "mean_pval"
),
bottom_bar = list(
"Number of Clusters" = "nclust"
),
annotation_colours = list(
"Depression p-value" = colour_scale(
ext_sol_df$"cbcl_depress_r_pval",
min_colour = "purple",
max_colour = "black"
),
"Anxiety p-value" = colour_scale(
ext_sol_df$"cbcl_anxiety_r_pval",
min_colour = "green",
max_colour = "black"
),
"Overall outcomes p-value" = colour_scale(
ext_sol_df$"mean_pval",
min_colour = "lightblue",
max_colour = "black"
)
)
)
```
```{r eval = FALSE, echo = FALSE}
save_heatmap(
heatmap = annotated_ari_hm,
path = "vignettes/annotated_ari_hm.png",
width = 700,
height = 500,
res = 100
)
```
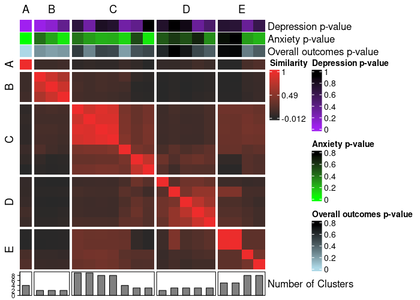
The `meta_cluster_heatmap` function wraps around a more generic `similarity_matrix_heatmap` function in this package, which itself is a wrapper around `ComplexHeatmap::Heatmap()`.
Consequently, any parameter that can be used with `ComplexHeatmap::Heatmap()` package can be used here.
This also makes the documentation for the `ComplexHeatmap` package one of the best places to learn more about what can be done for further customizing these heatmaps.
The data for the annotations don't necessarily need to come from functions in `metasnf`.
For example, to highlight the solutions where two very specific observations happened to cluster together, we can easily add that information in as another annotation.
By converting an `ext_solutions_df` class object into a data frame with the parameter `keep_attributes = TRUE`, the resulting data frame will retain the p-value information from the `ext_solutions_df` side of things, cluster assignment columns from the `solutions_df` side of things, and `settings_df` and `weights_matrix` columns from the embedded original SNF config.
Then, we can use `dplyr::mutate()` to create a new column flagging when two observations co-cluster.
```{r eval = FALSE}
annotation_data <- ext_sol_df |>
as.data.frame(keep_attributes = TRUE) |>
dplyr::mutate(
key_subjects_cluster_together = dplyr::case_when(
uid_NDAR_INVLF3TNDUZ == uid_NDAR_INVLDQH8ATK ~ TRUE,
TRUE ~ FALSE
)
)
annotated_ari_hm2 <- meta_cluster_heatmap(
sol_aris,
order = meta_cluster_order,
split_vector = split_vec,
data = annotation_data,
top_hm = list(
"Depression p-value" = "cbcl_depress_r_pval",
"Anxiety p-value" = "cbcl_anxiety_r_pval",
"Key Subjects Clustered Together" = "key_subjects_cluster_together"
),
bottom_bar = list(
"Number of Clusters" = "nclust"
),
annotation_colours = list(
"Depression p-value" = colour_scale(
ext_sol_df$"cbcl_depress_r_pval",
min_colour = "purple",
max_colour = "black"
),
"Anxiety p-value" = colour_scale(
ext_sol_df$"cbcl_anxiety_r_pval",
min_colour = "purple",
max_colour = "black"
),
"Key Subjects Clustered Together" = c(
"TRUE" = "blue",
"FALSE" = "black"
)
)
)
```
```{r eval = FALSE, echo = FALSE}
save_heatmap(
heatmap = annotated_ari_hm2,
path = "vignettes/annotated_ari_hm2.png",
width = 700,
height = 500,
res = 100
)
```
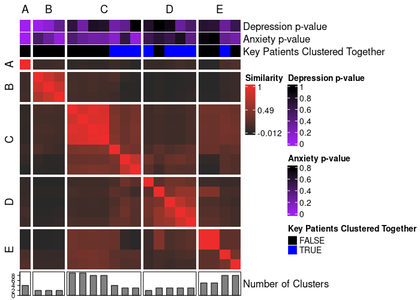
### Characterizing individual solutions representative of each meta cluster
Now that we've visually delineated our meta clusters, we can get a quick summary of what sort of separation exists across our input and held out features by taking a closer look at one representative cluster solution from each meta cluster.
This can be achieved by the `get_representative_solutions` function, which extracts one cluster solution per meta cluster based on having the highest average ARI with the other solutions in that meta cluster.
```{r eval = FALSE}
rep_solutions <- get_representative_solutions(sol_aris, ext_sol_df)
mc_manhattan <- mc_manhattan_plot(
rep_solutions,
dl = input_dl,
target_dl = target_dl,
hide_x_labels = TRUE,
point_size = 2,
text_size = 12,
threshold = 0.05,
neg_log_pval_thresh = 5
)
```
```{r eval = FALSE, echo = FALSE}
ggplot2::ggsave(
"vignettes/mc_manhattan.png",
mc_manhattan,
height = 4,
width = 8,
dpi = 100
)
```
`neg_log_pval_thresh` sets a threshold for the negative log of the p-values to be displayed.
At a value of 5, any p-value that is smaller than 1e-5 will be truncated to 1e-5.
The plot as it is is a bit unwieldy plot given how many neuroimaging ROIs are present.
Let's take out the cortical thickness and surface area measures to make the plot a little clearer.
We'll also be able to read some feature measures more clearly when we dial the number of features plotted back a bit, as well.
```{r eval = FALSE}
rep_solutions_no_cort <- dplyr::select(rep_solutions, -dplyr::contains("mrisdp"))
mc_manhattan2 <- mc_manhattan_plot(
ext_sol_df = rep_solutions_no_cort,
dl = input_dl,
target_dl = target_dl,
point_size = 4,
threshold = 0.01,
text_size = 12,
domain_colours = c(
"neuroimaging" = "cadetblue",
"demographics" = "purple",
"behaviour" = "firebrick"
)
)
```
```{r eval = FALSE, echo = FALSE}
ggplot2::ggsave(
"vignettes/mc_manhattan2.png",
mc_manhattan2,
height = 8,
width = 12,
dpi = 100
)
```
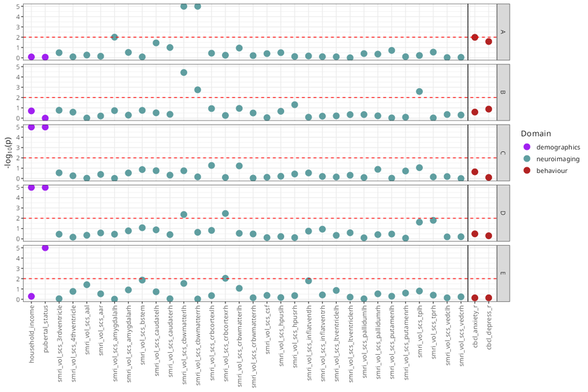
Note that the Manhattan plot automatically uses a vertical line to separate features in the data_list argument from those in the target_list.
Vertical boundaries can be controlled with the `xints` parameter.
### Relating results to `metasnf` hyperparameters
If you see something interesting in your heatmap, you may be curious to know how that corresponds to the settings that were in your settings data frame.
You could certainly stack on each setting to the ARI heatmap as annotations, but that may be a bit cumbersome given how many settings there are.
Another option is to start by taking a first look at entire `settings_df`, sorted by the meta cluster results, through the `settings_df_heatmap` function.
The settings can be passed in through either an `snf_config` or `settings_df` class object.
```{r eval = FALSE}
config_hm <- config_heatmap(
my_sc,
order = meta_cluster_order,
hide_fixed = TRUE
)
```
```{r eval = FALSE, echo = FALSE}
save_heatmap(
heatmap = config_hm,
path = "vignettes/config_hm.png",
width = 400,
height = 500,
res = 75
)
```
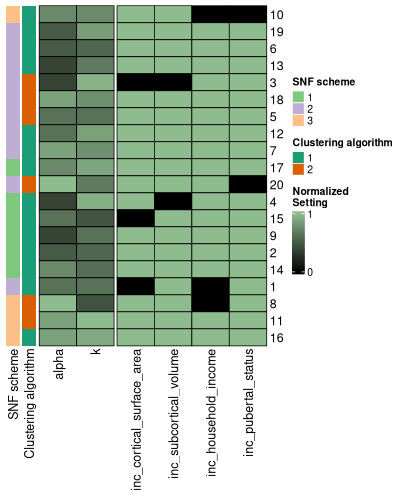
This heatmap rescales all the columns in the settings_df to have a maximum value of 1.
The purpose of re-ordering the settings data frame in this way is to see if any associations exist between certain settings values and pairwise cluster solution similarities.
If there are any particular important settings, you can simply add them into your adjusted rand index heatmap annotations.
Recall that the `solutions_df` class (and, by extension, the `ext_solutions_df` class that contains a `solutions_df` object as an attribute) contain the SNF config as an attribute, so no further data manipulation is needed to add a setting as a heatmap annotation.
Give it a try with the code below:
```{r eval = FALSE}
annotation_data <- ext_sol_df |>
as.data.frame(keep_attributes = TRUE) |>
dplyr::mutate(
key_subjects_cluster_together = dplyr::case_when(
uid_NDAR_INVLF3TNDUZ == uid_NDAR_INVLDQH8ATK ~ TRUE,
TRUE ~ FALSE
)
)
annotation_data$"clust_alg" <- as.factor(annotation_data$"clust_alg")
annotated_ari_hm3 <- meta_cluster_heatmap(
sol_aris,
order = meta_cluster_order,
split_vector = split_vec,
data = annotation_data,
top_hm = list(
"Depression p-value" = "cbcl_depress_r_pval",
"Anxiety p-value" = "cbcl_anxiety_r_pval",
"Key Subjects Clustered Together" = "key_subjects_cluster_together"
),
left_hm = list(
"Clustering Algorithm" = "clust_alg" # from the original settings
),
bottom_bar = list(
"Number of Clusters" = "nclust" # also from the original settings
),
annotation_colours = list(
"Depression p-value" = colour_scale(
ext_sol_df$"cbcl_depress_r_pval",
min_colour = "purple",
max_colour = "black"
),
"Anxiety p-value" = colour_scale(
ext_sol_df$"cbcl_anxiety_r_pval",
min_colour = "purple",
max_colour = "black"
),
"Key Subjects Clustered Together" = c(
"TRUE" = "blue",
"FALSE" = "black"
)
)
)
annotated_ari_hm3
```
### Quality measures
Quality metrics are another useful heuristic for the goodness of a cluster that don't require any contextualization of results in the domain they may be used in.
metasnf enables measures of silhouette scores, Dunn indices, and Davies-Bouldin indices.
To calculate these values, we'll need not only the cluster results but also the final fused network (the similarity matrices produced by SNF) that the clusters came from.
These similarity matrices can be collected from the `batch_snf` using the `return_sim_mats` parameter:
```{r eval = FALSE}
sol_df <- batch_snf(dl = input_dl, sc = my_sc, return_sim_mats = TRUE)
```
Now, the solutions data frame contains a non-empty list of similarity matrices as one of its attributes.
These similarity matrices are the final SNF-derived networks from each SNF run.
Using that attribute, we can calculate the above mentioned quality metrics:
```{r eval = FALSE}
silhouette_scores <- calculate_silhouettes(sol_df)
dunn_indices <- calculate_dunn_indices(sol_df)
db_indices <- calculate_db_indices(sol_df)
```
The first function is a wrapper around `cluster::silhouette` while the second and third come from the *clv* package.
*clv* isn't set as a mandatory part of the installation, so you'll ned to install it yourself to calculate these two metrics.
The original documentation on these functions can be helpful for interpreting and working with them:
1. [`cluster::silhouette` documentation](https://www.rdocumentation.org/packages/cluster/versions/2.1.4/topics/silhouette)
2. [`clv::clv.Dunn` documentation](https://www.rdocumentation.org/packages/clv/versions/0.3-2.1/topics/clv.Dunn)
3. [`clv::clv.Davies.Bouldin` documentation](https://www.rdocumentation.org/packages/clv/versions/0.3-2.1/topics/clv.Davies.Bouldin)
### Stability measures
metasnf offers tools to evaluate two different measures of stability:
1. Pairwise adjusted Rand indices (across resamplings of the clustering, on average, how similar was every pair of solutions according to the adjusted Rand index?)
2. Fraction clustered together (what is the average fraction of times that observations who clustered together in the full results clustered together in resampled results?)
You can learn more about running stability calculations in the [stability and coclustering vignette](https://branchlab.github.io/metasnf/articles/stability_measures.html).
### Evaluating separation across "target features" of importance
If you can specify a metric or objective function that may tell you how useful a clustering solution will be for your purposes in advance, that makes the cluster selection process much less arbitrary.
There are many ways to go about doing this, but this package offers one way through a `target_list`.
The `target_list` contains data frames what we can examine our clustering results over through linear regression (continuous data), ordinal regression (ordinal data), or the Chi-squared test (categorical data).
Just like when generating the initial `data_list`, we need to specify the name of the column in the provided data frames that is originally being used to uniquely identify the different observations from each other with the `uid` parameter.
We will next extend our `sol_df` with p-values from regressing the `target_list` features onto our generated clusters.
```{r}
ext_sol_df <- extend_solutions(sol_df, target_dl)
```
There is a heatmap for visualizing this too:
```{r eval = FALSE}
pval_hm <- pval_heatmap(ext_sol_df, order = meta_cluster_order)
pval_hm
```
```{r eval = FALSE, echo = FALSE}
save_heatmap(
heatmap = pval_hm,
path = "vignettes/pval_heatmap_ordered.png",
width = 400,
height = 500,
res = 100
)
```
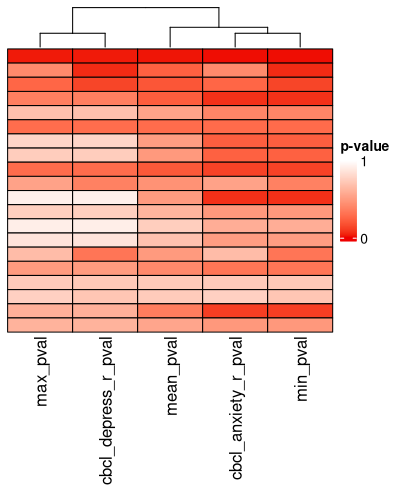
These p-values hold no real meaning for the traditional hypothesis-testing context, but they are reasonable proxies of the magnitude of the effect size / separation of the clusters across the features in question.
Here, they are just a tool to find clustering solutions that are well-separated according to the outcome measures you've specified.
Finding a cluster solution like this is similar to a supervised learning approach, but where the optimization method is just random sampling.
The risk for overfitting your data with this approach is considerable, so make sure you have some rigorous external validation before reporting your findings.
We recommend using *label propagation* (provided by the SNFtool package in the `groupPredict` function) for validation: take the top clustering solutions found in some training data, assign predicted clusters to some held out test observations, and then characterize those test observations to see how well the clustering solution seemed to have worked.
## Validating results with label propagation
Here's a quick step through of the complete procedure, from the beginning, with label propagation to validate our findings.
The `metasnf` package comes equipped with the function `train_test_assign` to provide random splitting for you.
```{r eval}
# All the observations present in all data frames with no NAs
all_observations <- uids(input_dl)
all_observations
# Remove the "uid_" prefix to allow merges with the original data
all_observations <- gsub("uid_", "", all_observations)
# data frame assigning 80% of observations to train and 20% to test
assigned_splits <- train_test_assign(train_frac = 0.8, uids = all_observations)
# Pulling the training and testing observations specifically
train_obs <- assigned_splits$"train"
test_obs <- assigned_splits$"test"
# Partition a training set
train_cort_t <- cort_t[cort_t$"unique_id" %in% train_obs, ]
train_cort_sa <- cort_sa[cort_sa$"unique_id" %in% train_obs, ]
train_subc_v <- subc_v[subc_v$"unique_id" %in% train_obs, ]
train_income <- income[income$"unique_id" %in% train_obs, ]
train_pubertal <- pubertal[pubertal$"unique_id" %in% train_obs, ]
train_anxiety <- anxiety[anxiety$"unique_id" %in% train_obs, ]
train_depress <- depress[depress$"unique_id" %in% train_obs, ]
# Partition a test set
test_cort_t <- cort_t[cort_t$"unique_id" %in% test_obs, ]
test_cort_sa <- cort_sa[cort_sa$"unique_id" %in% test_obs, ]
test_subc_v <- subc_v[subc_v$"unique_id" %in% test_obs, ]
test_income <- income[income$"unique_id" %in% test_obs, ]
test_pubertal <- pubertal[pubertal$"unique_id" %in% test_obs, ]
test_anxiety <- anxiety[anxiety$"unique_id" %in% test_obs, ]
test_depress <- depress[depress$"unique_id" %in% test_obs, ]
# A data list with just training observations
train_dl <- data_list(
list(train_cort_t, "cortical_thickness", "neuroimaging", "continuous"),
list(train_cort_sa, "cortical_sa", "neuroimaging", "continuous"),
list(train_subc_v, "subcortical_volume", "neuroimaging", "continuous"),
list(train_income, "household_income", "demographics", "continuous"),
list(train_pubertal, "pubertal_status", "demographics", "continuous"),
uid = "unique_id"
)
# A data list with training and testing observations
full_dl <- data_list(
list(cort_t, "cortical_thickness", "neuroimaging", "continuous"),
list(cort_sa, "cortical_surface_area", "neuroimaging", "continuous"),
list(subc_v, "subcortical_volume", "neuroimaging", "continuous"),
list(income, "household_income", "demographics", "continuous"),
list(pubertal, "pubertal_status", "demographics", "continuous"),
uid = "unique_id"
)
# Construct the target lists
train_target_dl <- data_list(
list(train_anxiety, "anxiety", "behaviour", "ordinal"),
list(train_depress, "depressed", "behaviour", "ordinal"),
uid = "unique_id"
)
```
```{r eval = FALSE}
# Find a clustering solution in your training data
set.seed(42)
my_sc <- snf_config(
train_dl,
n_solutions = 5,
min_k = 10,
max_k = 30
)
train_sol_df <- batch_snf(
train_dl,
my_sc
)
ext_sol_df <- extend_solutions(
train_sol_df,
train_target_dl
)
```
```{r echo = FALSE}
ext_sol_df <- cache_a_complete_example_lp_ext_sol_df
```
```{r}
# The first row had the lowest minimum p-value across our outcomes
lowest_min_pval <- min(ext_sol_df$"min_pval")
which(ext_sol_df$"min_pval" == lowest_min_pval)
# Keep track of your top solution
top_row <- ext_sol_df[1, ]
# Use the solutions data frame from the training observations and the data list from
# the training and testing observations to propagate labels to the test observations
propagated_labels <- label_propagate(top_row, full_dl)
head(propagated_labels)
tail(propagated_labels)
```
You could, if you wanted, see how *all* of your clustering solutions propagate to the test set, but that would mean reusing your test set and removing the protection against overfitting conferred by this procedure.
```{r eval = FALSE}
propagated_labels_all <- label_propagate(
ext_sol_df,
full_dl
)
```
That's all!
If you have any questions, comments, suggestions, bugs, etc. feel free to post an issue at [https://github.com/BRANCHlab/metasnf](https://github.com/BRANCHlab/metasnf).
## References
Caruana, Rich, Mohamed Elhawary, Nam Nguyen, and Casey Smith. 2006. “Meta Clustering.” In Sixth International Conference on Data Mining (ICDM’06), 107–18. https://doi.org/10.1109/ICDM.2006.103.
Wang, Bo, Aziz M. Mezlini, Feyyaz Demir, Marc Fiume, Zhuowen Tu, Michael Brudno, Benjamin Haibe-Kains, and Anna Goldenberg. 2014. “Similarity Network Fusion for Aggregating Data Types on a Genomic Scale.” Nature Methods 11 (3): 333–37. https://doi.org/10.1038/nmeth.2810.