{
"cells": [
{
"cell_type": "raw",
"metadata": {},
"source": [
"---\n",
"description: Reconstructing developmental or differentiation pathways\n",
" from individual cell gene expression profiles to understand cellular\n",
" transitions and relationships.\n",
"subtitle: Scanpy Toolkit\n",
"title: Trajectory inference using PAGA\n",
"---"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"\n",
"\n",
"> **Note**\n",
">\n",
"> Code chunks run Python commands unless it starts with `%%bash`, in\n",
"> which case, those chunks run shell commands.\n",
"\n",
"
\n",
"\n",
"Partly following this PAGA\n",
"[tutorial](https://scanpy-tutorials.readthedocs.io/en/latest/paga-paul15.html)\n",
"with some modifications.\n",
"\n",
"## Loading libraries"
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: libraries\n",
"import numpy as np\n",
"import pandas as pd\n",
"import matplotlib.pyplot as pl\n",
"from matplotlib import rcParams\n",
"import scanpy as sc\n",
"\n",
"import scipy\n",
"import numpy as np\n",
"import matplotlib.pyplot as plt\n",
"import warnings\n",
"\n",
"warnings.simplefilter(action=\"ignore\", category=Warning)\n",
"\n",
"# verbosity: errors (0), warnings (1), info (2), hints (3)\n",
"sc.settings.verbosity = 3\n",
"sc.settings.set_figure_params(dpi=100, frameon=False, figsize=(5, 5), facecolor='white', color_map = 'viridis_r') "
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Preparing data\n",
"\n",
"In order to speed up the computations during the exercises, we will be\n",
"using a subset of a bone marrow dataset (originally containing about\n",
"100K cells). The bone marrow is the source of adult immune cells, and\n",
"contains virtually all differentiation stages of cell from the immune\n",
"system which later circulate in the blood to all other organs.\n",
"\n",
"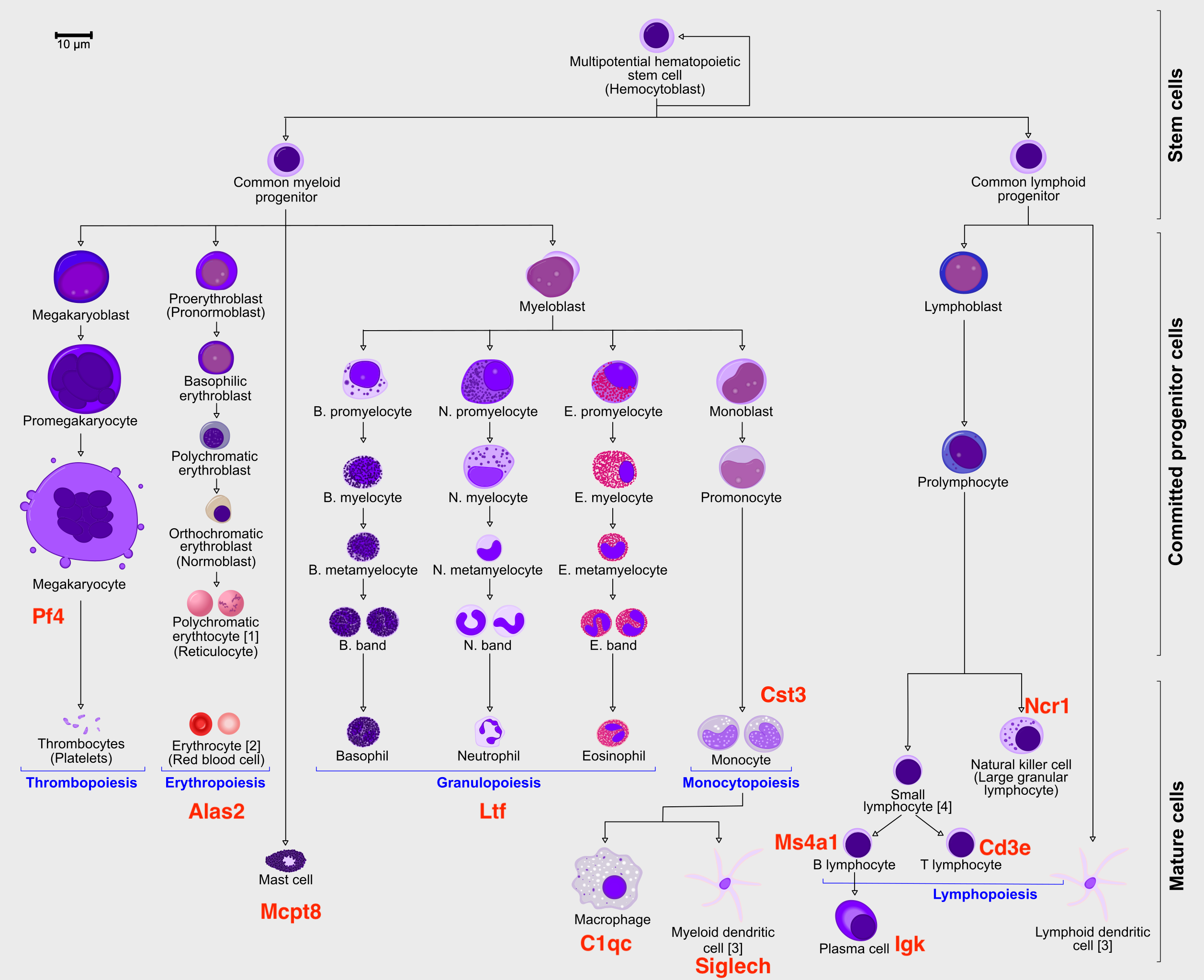\n",
"\n",
"If you have been using the **Seurat**, **Bioconductor** or **Scanpy**\n",
"toolkits with your own data, you need to reach to the point where can\n",
"find get:\n",
"\n",
"- A dimensionality reduction where to perform the trajectory (for\n",
" example: PCA, ICA, MNN, harmony, Diffusion Maps, UMAP)\n",
"- The cell clustering information (for example: from Louvain, k-means)\n",
"- A KNN/SNN graph (this is useful to inspect and sanity-check your\n",
" trajectories)\n",
"\n",
"In this case, all the data has been preprocessed with Seurat with\n",
"standard pipelines. In addition there was some manual filtering done to\n",
"remove clusters that are disconnected and cells that are hard to\n",
"cluster, which can be seen in this\n",
"[script](https://github.com/NBISweden/workshop-scRNAseq/blob/master/scripts/data_processing/slingshot_preprocessing.Rmd)\n",
"\n",
"The file trajectory_scanpy_filtered.h5ad was converted from the Seurat\n",
"object using the SeuratDisk package. For more information on how it was\n",
"done, have a look at the script:\n",
"[convert_to_h5ad.R](https://github.com/NBISweden/workshop-scRNAseq/blob/master/scripts/data_processing/convert_to_h5ad.R)\n",
"in the github repo.\n",
"\n",
"You can download the data with the commands:"
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: fetch-data\n",
"import os\n",
"import subprocess\n",
"\n",
"# download pre-computed data if missing or long compute\n",
"fetch_data = True\n",
"\n",
"# url for source and intermediate data\n",
"path_data = \"https://nextcloud.dc.scilifelab.se/public.php/webdav\"\n",
"curl_upass = \"zbC5fr2LbEZ9rSE:scRNAseq2025\"\n",
"\n",
"path_results = \"data/trajectory\"\n",
"if not os.path.exists(path_results):\n",
" os.makedirs(path_results, exist_ok=True)\n",
"\n",
"path_file = \"data/trajectory/trajectory_seurat_filtered.h5ad\"\n",
"if not os.path.exists(path_file):\n",
" file_url = os.path.join(path_data, \"trajectory/trajectory_seurat_filtered.h5ad\")\n",
" subprocess.call([\"curl\", \"-u\", curl_upass, \"-o\", path_file, file_url ]) "
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Reading data\n",
"\n",
"We already have pre-computed and subsetted the dataset (with 6688 cells\n",
"and 3585 genes) following the analysis steps in this course. We then\n",
"saved the objects, so you can use common tools to open and start to work\n",
"with them (either in R or Python)."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: read-data\n",
"adata = sc.read_h5ad(\"data/trajectory/trajectory_seurat_filtered.h5ad\")\n",
"adata.var"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: check-data\n",
"# check what you have in the X matrix, should be lognormalized counts.\n",
"print(adata.X[:10,:10])"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Explore the data\n",
"\n",
"There is a umap and clusters provided with the object, first plot some\n",
"information from the previous analysis onto the umap."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: plot-umap\n",
"sc.pl.umap(adata, color = ['clusters','dataset','batches','Phase'],legend_loc = 'on data', legend_fontsize = 'xx-small', ncols = 2)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"It is crucial that you performing analysis of a dataset understands what\n",
"is going on, what are the clusters you see in your data and most\n",
"importantly How are the clusters related to each other?. Well, let's\n",
"explore the data a bit. With the help of this table, write down which\n",
"cluster numbers in your dataset express these key markers.\n",
"\n",
" Marker Cell Type\n",
" --------- ----------------------------\n",
" Cd34 HSC progenitor\n",
" Ms4a1 B cell lineage\n",
" Cd3e T cell lineage\n",
" Ltf Granulocyte lineage\n",
" Cst3 Monocyte lineage\n",
" Mcpt8 Mast Cell lineage\n",
" Alas2 RBC lineage\n",
" Siglech Dendritic cell lineage\n",
" C1qc Macrophage cell lineage\n",
" Pf4 Megakaryocyte cell lineage"
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: plot-markers\n",
"markers = [\"Cd34\",\"Alas2\",\"Pf4\",\"Mcpt8\",\"Ltf\",\"Cst3\", \"Siglech\", \"C1qc\", \"Ms4a1\", \"Cd3e\", ]\n",
"sc.pl.umap(adata, color = markers, use_raw = False, ncols = 4)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Rerun analysis in Scanpy\n",
"\n",
"Redo clustering and umap using the basic Scanpy pipeline. Use the\n",
"provided \"X_harmony_Phase\" dimensionality reduction as the staring\n",
"point."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: process\n",
"# first, store the old umap with a new name so it is not overwritten\n",
"adata.obsm['X_umap_old'] = adata.obsm['X_umap']\n",
"\n",
"sc.pp.neighbors(adata, n_pcs = 30, n_neighbors = 20, use_rep=\"X_harmony_Phase\")\n",
"sc.tl.umap(adata, min_dist=0.4, spread=3)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: cluster\n",
"sc.pl.umap(adata, color = ['clusters'],legend_loc = 'on data', legend_fontsize = 'xx-small', edges = True)\n",
"\n",
"sc.pl.umap(adata, color = markers, use_raw = False, ncols = 4)\n",
"\n",
"# Redo clustering as well\n",
"sc.tl.leiden(adata, key_added = \"leiden_1.0\", resolution = 1.0) # default resolution in 1.0\n",
"sc.tl.leiden(adata, key_added = \"leiden_1.2\", resolution = 1.2) # default resolution in 1.0\n",
"sc.tl.leiden(adata, key_added = \"leiden_1.4\", resolution = 1.4) # default resolution in 1.0\n",
"\n",
"#sc.tl.louvain(adata, key_added = \"leiden_1.0\") # default resolution in 1.0\n",
"sc.pl.umap(adata, color = ['leiden_1.0', 'leiden_1.2', 'leiden_1.4','clusters'],legend_loc = 'on data', legend_fontsize = 'xx-small', ncols =2)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: annotate\n",
"#Rename clusters with really clear markers, the rest are left unlabelled.\n",
"\n",
"annot = pd.DataFrame(adata.obs['leiden_1.4'].astype('string'))\n",
"annot[annot['leiden_1.4'] == '10'] = '10_megakaryo' #Pf4\n",
"annot[annot['leiden_1.4'] == '17'] = '17_macro' #C1qc\n",
"annot[annot['leiden_1.4'] == '11'] = '11_eryth' #Alas2\n",
"annot[annot['leiden_1.4'] == '18'] = '18_dend' #Siglech\n",
"annot[annot['leiden_1.4'] == '13'] = '13_mast' #Mcpt8\n",
"annot[annot['leiden_1.4'] == '0'] = '0_mono' #Cts3\n",
"annot[annot['leiden_1.4'] == '1'] = '1_gran' #Ltf\n",
"annot[annot['leiden_1.4'] == '9'] = '9_gran'\n",
"annot[annot['leiden_1.4'] == '14'] = '14_TC' #Cd3e\n",
"annot[annot['leiden_1.4'] == '16'] = '16_BC' #Ms4a1\n",
"annot[annot['leiden_1.4'] == '8'] = '8_progen' # Cd34\n",
"annot[annot['leiden_1.4'] == '4'] = '4_progen' \n",
"annot[annot['leiden_1.4'] == '5'] = '5_progen'\n",
"\n",
"adata.obs['annot']=annot['leiden_1.4'].astype('category')\n",
"\n",
"sc.pl.umap(adata, color = 'annot',legend_loc = 'on data', legend_fontsize = 'xx-small', ncols =2)\n",
"\n",
"annot.value_counts()\n",
"#type(annot)\n",
"\n",
"# astype('category')"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: plot-annot\n",
"# plot onto the Seurat embedding:\n",
"sc.pl.embedding(adata, basis='X_umap_old', color = 'annot',legend_loc = 'on data', legend_fontsize = 'xx-small', ncols =2)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Run PAGA\n",
"\n",
"Use the clusters from leiden clustering with leiden_1.4 and run PAGA.\n",
"First we create the graph and initialize the positions using the umap."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: paga\n",
"# use the umap to initialize the graph layout.\n",
"sc.tl.draw_graph(adata, init_pos='X_umap')\n",
"sc.pl.draw_graph(adata, color='annot', legend_loc='on data', legend_fontsize = 'xx-small')\n",
"sc.tl.paga(adata, groups='annot')\n",
"sc.pl.paga(adata, color='annot', edge_width_scale = 0.3)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"As you can see, we have edges between many clusters that we know are are\n",
"unrelated, so we may need to clean up the data a bit more.\n",
"\n",
"## Filtering graph edges\n",
"\n",
"First, lets explore the graph a bit. So we plot the umap with the graph\n",
"connections on top."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: plot-graph\n",
"sc.pl.umap(adata, edges=True, color = 'annot', legend_loc= 'on data', legend_fontsize= 'xx-small')"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"We have many edges in the graph between unrelated clusters, so lets try\n",
"with fewer neighbors."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: redo-graph\n",
"sc.pp.neighbors(adata, n_neighbors=5, use_rep = 'X_harmony_Phase', n_pcs = 30)\n",
"sc.pl.umap(adata, edges=True, color = 'annot', legend_loc= 'on data', legend_fontsize= 'xx-small')"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"### Rerun PAGA again on the data"
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: draw-graph\n",
"sc.tl.draw_graph(adata, init_pos='X_umap')\n",
"sc.pl.draw_graph(adata, color='annot', legend_loc='on data', legend_fontsize = 'xx-small')"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: paga2\n",
"sc.tl.paga(adata, groups='annot')\n",
"sc.pl.paga(adata, color='annot', edge_width_scale = 0.3)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"## Embedding using PAGA-initialization\n",
"\n",
"We can now redraw the graph using another starting position from the\n",
"paga layout. The following is just as well possible for a UMAP."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: draw-graph-paga\n",
"sc.tl.draw_graph(adata, init_pos='paga')"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Now we can see all marker genes also at single-cell resolution in a\n",
"meaningful layout."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: plot-graph-paga\n",
"sc.pl.draw_graph(adata, color=['annot'], legend_loc='on data', legend_fontsize= 'xx-small')"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Compare the 2 graphs"
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: paga-compare\n",
"sc.pl.paga_compare(\n",
" adata, threshold=0.03, title='', right_margin=0.2, size=10, edge_width_scale=0.5,\n",
" legend_fontsize=12, fontsize=12, frameon=False, edges=True)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"\n",
"\n",
"> **Discuss**\n",
">\n",
"> Does this graph fit the biological expectations given what you know of\n",
"> hematopoesis. Please have a look at the figure in Section 2 and\n",
"> compare to the paths you now have.\n",
"\n",
"
\n",
"\n",
"## Gene changes\n",
"\n",
"We can reconstruct gene changes along PAGA paths for a given set of\n",
"genes\n",
"\n",
"Choose a root cell for diffusion pseudotime. We have 3 progenitor\n",
"clusters, but cluster 5 seems the most clear."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: pseudotime\n",
"adata.uns['iroot'] = np.flatnonzero(adata.obs['annot'] == '5_progen')[0]\n",
"\n",
"sc.tl.dpt(adata)"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Use the full raw data for visualization."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: plot-pt\n",
"sc.pl.draw_graph(adata, color=['annot', 'dpt_pseudotime'], legend_loc='on data', legend_fontsize= 'x-small')"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"\n",
"\n",
"> **Discuss**\n",
">\n",
"> The pseudotime represents the distance of every cell to the starting\n",
"> cluster. Have a look at the pseudotime plot, how well do you think it\n",
"> represents actual developmental time? What does it represent?\n",
"\n",
"
\n",
"\n",
"By looking at the different know lineages and the layout of the graph we\n",
"define manually some paths to the graph that corresponds to specific\n",
"lineages."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: paths\n",
"# Define paths\n",
"\n",
"paths = [('erythrocytes', ['5_progen', '8_progen', '6', '3', '7', '11_eryth']),\n",
" ('lympoid', ['5_progen', '12', '16_BC', '14_TC']),\n",
" ('granulo', ['5_progen', '4_progen', '2', '9_gran', '1_gran']),\n",
" ('mono', ['5_progen', '4_progen', '0_mono', '18_dend', '17_macro'])\n",
" ]\n",
"\n",
"adata.obs['distance'] = adata.obs['dpt_pseudotime']"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Then we select some genes that can vary in the lineages and plot onto\n",
"the paths."
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: select-genes\n",
"gene_names = ['Gata2', 'Gata1', 'Klf1', 'Epor', 'Hba-a2', # erythroid\n",
" 'Elane', 'Cebpe', 'Gfi1', # neutrophil\n",
" 'Irf8', 'Csf1r', 'Ctsg', # monocyte\n",
" 'Itga2b','Prss34','Cma1','Procr', # Megakaryo,Basophil,Mast,HPC\n",
" 'C1qc','Siglech','Ms4a1','Cd3e','Cd34']"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: plot-genes\n",
"_, axs = pl.subplots(ncols=4, figsize=(10, 4), gridspec_kw={\n",
" 'wspace': 0.05, 'left': 0.12})\n",
"pl.subplots_adjust(left=0.05, right=0.98, top=0.82, bottom=0.2)\n",
"for ipath, (descr, path) in enumerate(paths):\n",
" sc.pl.paga_path(\n",
" adata=adata, \n",
" nodes=path, \n",
" keys=gene_names,\n",
" show_node_names=False,\n",
" ax=axs[ipath],\n",
" ytick_fontsize=12,\n",
" left_margin=0.15,\n",
" n_avg=50,\n",
" annotations=['distance'],\n",
" show_yticks=True if ipath == 0 else False,\n",
" show_colorbar=False,\n",
" color_map='Greys',\n",
" groups_key='annot',\n",
" color_maps_annotations={'distance': 'viridis'},\n",
" title='{} path'.format(descr),\n",
" return_data=True,\n",
" use_raw=False,\n",
" show=False)\n",
"\n",
"pl.show()"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"\n",
"\n",
"> **Discuss**\n",
">\n",
"> As you can see, we can manipulate the trajectory quite a bit by\n",
"> selecting different number of neighbors, components etc. to fit with\n",
"> our assumptions on the development of these celltypes.\n",
">\n",
"> Please explore further how you can tweak the trajectory. For instance,\n",
"> can you create a PAGA trajectory using the orignial umap from Seurat\n",
"> instead? Hint, you first need to compute the neighbors on the umap.\n",
"\n",
"
\n",
"\n",
"## Session info\n",
"\n",
"```{=html}\n",
"\n",
"```\n",
"```{=html}\n",
"\n",
"```\n",
"Click here\n",
"```{=html}\n",
"
\n",
"```"
]
},
{
"cell_type": "code",
"metadata": {},
"source": [
"#| label: session\n",
"sc.logging.print_versions()"
],
"execution_count": null,
"outputs": []
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"```{=html}\n",
" \n",
"```"
]
}
],
"metadata": {
"kernelspec": {
"display_name": "Python 3",
"language": "python",
"name": "python3"
}
},
"nbformat": 4,
"nbformat_minor": 4
}