Overview of pMuTT's Core Functionality¶
Originally written for Version 1.2.1
Last Updated for Version 1.2.13
Topics Covered¶
- Using constants and converting units using the
constantsmodule - Initializing
StatMechobjects by specifying all modes and by usingpresetsdictionary - Initializing empirical objects such as
Nasaobjects using aStatMechobject or from a previously generated Nasa polynomial - Initializing
ReferenceandReferencesobjects to adjust DFT's reference to more traditional references - Input (via Excel) and output
Nasapolynomials to thermdat format - Initializing
Reactionobjects from strings
Useful Links:¶
- Github: https://github.com/VlachosGroup/pMuTT
- Documentation: https://vlachosgroup.github.io/pMuTT/index.html
- Examples: https://vlachosgroup.github.io/pMuTT/examples.html
Constants¶
pMuTT has a wide variety of constants to increase readability of the code. See Constants page in the documentation for supported units.
from pmutt import constants as c
1.987
print(c.R('kJ/mol/K'))
print('Some constants')
print('R (J/mol/K) = {}'.format(c.R('J/mol/K')))
print("Avogadro's number = {}\n".format(c.Na))
print('Unit conversions')
print('5 kJ/mol --> {} eV/molecule'.format(c.convert_unit(num=5., initial='kJ/mol', final='eV/molecule')))
print('Frequency of 1000 Hz --> Wavenumber of {} 1/cm\n'.format(c.freq_to_wavenumber(1000.)))
print('See expected inputs, supported units of different constants')
help(c.R)
help(c.convert_unit)
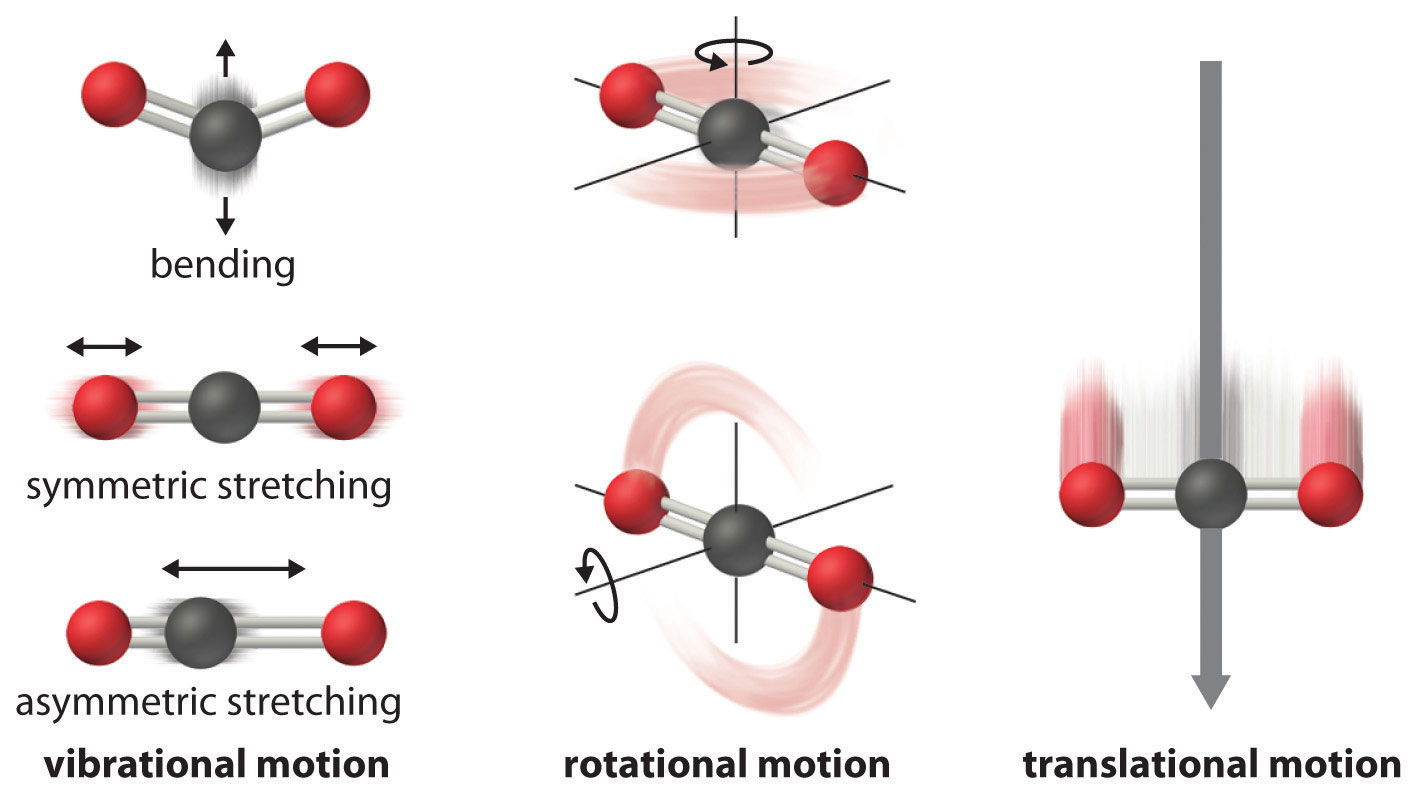
For this example, we will use a butane molecule as an ideal gas:
- translations with no interaction between molecules
- harmonic vibrations
- rigid rotor rotations
- ground state electronic structure -
- ground state nuclear structure).
from ase.build import molecule
from pmutt.statmech import StatMech, trans, vib, rot, elec
butane_atoms = molecule('trans-butane')
'''Translational'''
butane_trans = trans.FreeTrans(n_degrees=3, atoms=butane_atoms)
'''Vibrational'''
butane_vib = vib.HarmonicVib(vib_wavenumbers=[3054.622862, 3047.573455, 3037.53448,
3030.21322, 3029.947329, 2995.758708,
2970.12166, 2968.142985, 2951.122942,
2871.560685, 1491.354921, 1456.480829,
1455.224163, 1429.084081, 1423.153673,
1364.456094, 1349.778994, 1321.137752,
1297.412109, 1276.969173, 1267.783512,
1150.401492, 1027.841298, 1018.203753,
945.310074, 929.15992, 911.661049,
808.685354, 730.986587, 475.287654,
339.164649, 264.682213, 244.584138,
219.956713, 115.923768, 35.56194])
'''Rotational'''
butane_rot = rot.RigidRotor(symmetrynumber=2, atoms=butane_atoms)
'''Electronic'''
butane_elec = elec.GroundStateElec(potentialenergy=-73.7051, spin=0)
'''StatMech Initialization'''
butane_statmech = StatMech(name='butane',
trans_model=butane_trans,
vib_model=butane_vib,
rot_model=butane_rot,
elec_model=butane_elec)
H_statmech = butane_statmech.get_H(T=298., units='kJ/mol')
S_statmech = butane_statmech.get_S(T=298., units='J/mol/K')
print('H_butane(T=298) = {:.1f} kJ/mol'.format(H_statmech))
print('S_butane(T=298) = {:.2f} J/mol/K'.format(S_statmech))
from pprint import pprint
from ase.build import molecule
from pmutt.statmech import StatMech, presets
idealgas_defaults = presets['idealgas']
pprint(idealgas_defaults)
butane_preset = StatMech(name='butane',
atoms=molecule('trans-butane'),
vib_wavenumbers=[3054.622862, 3047.573455, 3037.53448,
3030.21322, 3029.947329, 2995.758708,
2970.12166, 2968.142985, 2951.122942,
2871.560685, 1491.354921, 1456.480829,
1455.224163, 1429.084081, 1423.153673,
1364.456094, 1349.778994, 1321.137752,
1297.412109, 1276.969173, 1267.783512,
1150.401492, 1027.841298, 1018.203753,
945.310074, 929.15992, 911.661049,
808.685354, 730.986587, 475.287654,
339.164649, 264.682213, 244.584138,
219.956713, 115.923768, 35.56194],
symmetrynumber=2,
potentialenergy=-73.7051,
spin=0,
**idealgas_defaults)
H_preset = butane_preset.get_H(T=298., units='kJ/mol')
S_preset = butane_preset.get_S(T=298., units='J/mol/K')
print('H_butane(T=298) = {:.1f} kJ/mol'.format(H_preset))
print('S_butane(T=298) = {:.2f} J/mol/K'.format(S_preset))
from pmutt.statmech import EmptyMode
empty = EmptyMode()
print('Some EmptyMode properties:')
print('q = {}'.format(empty.get_q()))
print('H/RT = {}'.format(empty.get_HoRT()))
print('S/R = {}'.format(empty.get_SoR()))
print('G/RT = {}'.format(empty.get_GoRT()))
Empirical Objects¶
Empirical forms are polynomials that are fit to experimental or ab-initio data. These forms are useful because they can be evaluated relatively quickly, so that downstream software is not hindered by evaluation of thermochemical properties.
However, note that StatMech objects can calculate more properties than the currently supported empirical objects.
from pmutt.empirical.nasa import Nasa
butane_nasa = Nasa.from_model(name='butane',
model=butane_statmech,
T_low=298.,
T_high=800.,
elements={'C': 4, 'H': 10},
phase='G')
H_nasa = butane_nasa.get_H(T=298., units='kJ/mol')
S_nasa = butane_nasa.get_S(T=298., units='J/mol/K')
print('H_butane(T=298) = {:.1f} kJ/mol'.format(H_nasa))
print('S_butane(T=298) = {:.2f} J/mol/K'.format(S_nasa))
Although it is not covered here, you can also generate empirical objects from experimental data using the .from_data method. See Experimental to Empirical example.
Initializing Nasa Directly¶
We can also initialize the NASA polynomial if we have the polynomials. Using an entry from the Reaction Mechanism Generator (RMG) database.
import numpy as np
butane_nasa_direct = Nasa(name='butane',
T_low=100.,
T_mid=1147.61,
T_high=5000.,
a_low=np.array([ 2.16917742E+00,
3.43905384E-02,
-1.61588593E-06,
-1.30723691E-08,
5.17339469E-12,
-1.72505133E+04,
1.46546944E+01]),
a_high=np.array([ 6.78908025E+00,
3.03597365E-02,
-1.21261608E-05,
2.19944009E-09,
-1.50288488E-13,
-1.91058191E+04,
-1.17331911E+01]),
elements={'C': 4, 'H': 10},
phase='G')
H_nasa_direct = butane_nasa_direct.get_H(T=298., units='kJ/mol')
S_nasa_direct = butane_nasa_direct.get_S(T=298., units='J/mol/K')
print('H_butane(T=298) = {:.1f} kJ/mol'.format(H_nasa_direct))
print('S_butane(T=298) = {:.2f} J/mol/K'.format(S_nasa_direct))
Compare the results between butane_nasa and butane_nasa_direct to the Wikipedia page for butane.
print('H_nasa = {:.1f} kJ/mol'.format(H_nasa))
print('H_nasa_direct = {:.1f} kJ/mol'.format(H_nasa_direct))
print('H_wiki = -125.6 kJ/mol\n')
print('S_nasa = {:.2f} J/mol/K'.format(S_nasa))
print('S_nasa_direct = {:.2f} J/mol/K'.format(S_nasa_direct))
print('S_wiki = 310.23 J/mol/K')
Notice the values are very different for H_nasa. This discrepancy is due to:
- different references
- error in DFT
We can account for this discrepancy by using the Reference and References objects.
Referencing¶
To define a reference, you must have:
- enthalpy at some reference temperature (
HoRT_refandT_ref) - a
StatMechobject
In general, use references that are similar to molecules in your mechanism. Also, the number of reference molecules must be equation to the number of elements (or other descriptor) in the mechanism. Full description of referencing scheme here.
In this example, we will use ethane and propane as references.
from pmutt.empirical.references import Reference, References
ethane_ref = Reference(name='ethane',
elements={'C': 2, 'H': 6},
atoms=molecule('C2H6'),
vib_wavenumbers=[3050.5296, 3049.8428, 3025.2714,
3024.4304, 2973.5455, 2971.9261,
1455.4203, 1454.9941, 1454.2055,
1453.7038, 1372.4786, 1358.3593,
1176.4512, 1175.507, 992.55,
803.082, 801.4536, 298.4712],
symmetrynumber=6,
potentialenergy=-40.5194,
spin=0,
T_ref=298.15,
HoRT_ref=-33.7596,
**idealgas_defaults)
propane_ref = Reference(name='propane',
elements={'C': 3, 'H': 8},
atoms=molecule('C3H8'),
vib_wavenumbers=[3040.9733, 3038.992, 3036.8071,
3027.6062, 2984.8436, 2966.1692,
2963.4684, 2959.7431, 1462.5683,
1457.4221, 1446.858, 1442.0357,
1438.7871, 1369.6901, 1352.6287,
1316.215, 1273.9426, 1170.4456,
1140.9699, 1049.3866, 902.8507,
885.3209, 865.5958, 735.1924,
362.7372, 266.3928, 221.4547],
symmetrynumber=2,
potentialenergy=-57.0864,
spin=0,
T_ref=298.15,
HoRT_ref=-42.2333,
**idealgas_defaults)
refs = References(references=[ethane_ref, propane_ref])
print(refs.offset)
Passing the References object when we make our Nasa object produces a value closer to the one listed above.
butane_nasa_ref = Nasa.from_model(name='butane',
model=butane_statmech,
T_low=298.,
T_high=800.,
elements={'C': 4, 'H': 10},
references=refs)
H_nasa_ref = butane_nasa_ref.get_H(T=298., units='kJ/mol')
S_nasa_ref = butane_nasa_ref.get_S(T=298., units='J/mol/K')
print('H_butane(T=298) = {:.1f} kJ/mol'.format(H_nasa_ref))
print('S_butane(T=298) = {:.2f} J/mol/K'.format(S_nasa_ref))
Input and Output¶
Excel¶
Encoding each object in Python can be tedious and so you can read from Excel spreadsheets using pmutt.io.excel.read_excel. Note that this function returns a list of dictionaries. This output allows you to initialize whichever object you want. There are also special rules that depend on the header name.
import os
from pathlib import Path
from pmutt.io.excel import read_excel
# Find the location of Jupyter notebook
# Note that normally Python scripts have a __file__ variable but Jupyter notebook doesn't.
# Using pathlib can overcome this limiation
try:
notebook_folder = os.path.dirname(__file__)
except NameError:
notebook_folder = Path().resolve()
os.chdir(notebook_folder)
# The Excel spreadsheet is located in the same folder as the Jupyter notebook
refs_path = os.path.join(notebook_folder, 'refs.xlsx')
refs_data = read_excel(refs_path)
pprint(refs_data)
Initialize using **kwargs syntax.
ref_list = []
for record in refs_data:
ref_list.append(Reference(**record))
refs_excel = References(references=ref_list)
print(refs_excel.offset)
Butane can be initialized in a similar way.
# The Excel spreadsheet is located in the same folder as the Jupyter notebook
butane_path = os.path.join(notebook_folder, 'butane.xlsx')
butane_data = read_excel(butane_path)[0] # [0] accesses the butane data
butane_excel = Nasa.from_model(T_low=298.,
T_high=800.,
references=refs_excel,
**butane_data)
H_excel = butane_excel.get_H(T=298., units='kJ/mol')
S_excel = butane_excel.get_S(T=298., units='J/mol/K')
print('H_butane(T=298) = {:.1f} kJ/mol'.format(H_excel))
print('S_butane(T=298) = {:.2f} J/mol/K'.format(S_excel))
Thermdat¶
The thermdat format uses NASA polynomials to represent several species. It has a very particular format so doing it manually is error-prone. You can write a list of Nasa objects to thermdat format using pmutt.io.thermdat.write_thermdat.
from pmutt.io.thermdat import write_thermdat
# Make Nasa objects from previously defined ethane and propane
ethane_nasa = Nasa.from_model(name='ethane',
phase='G',
T_low=298.,
T_high=800.,
model=ethane_ref.model,
elements=ethane_ref.elements,
references=refs)
propane_nasa = Nasa.from_model(name='propane',
phase='G',
T_low=298.,
T_high=800.,
model=propane_ref.model,
elements=propane_ref.elements,
references=refs)
nasa_species = [ethane_nasa, propane_nasa, butane_nasa]
# Determine the output path and write the thermdat file
thermdat_path = os.path.join(notebook_folder, 'thermdat')
write_thermdat(filename=thermdat_path, nasa_species=nasa_species)
Similarly, pmutt.io.thermdat.read_thermdat reads thermdat files.
Reactions¶
You can also evaluate reactions properties. The most straightforward way to do this is to initialize using strings.
from pmutt.io.thermdat import read_thermdat
from pmutt import pmutt_list_to_dict
from pmutt.reaction import Reaction
# Get a dictionary of species
thermdat_H2O_path = os.path.join(notebook_folder, 'thermdat_H2O')
species_list = read_thermdat(thermdat_H2O_path)
species_dict = pmutt_list_to_dict(species_list)
# Initialize the reaction
rxn_H2O = Reaction.from_string('H2 + 0.5O2 = H2O', species=species_dict)
# Calculate reaction properties
H_rxn = rxn_H2O.get_delta_H(T=298., units='kJ/mol')
S_rxn = rxn_H2O.get_delta_S(T=298., units='J/mol/K')
print('H_rxn(T=298) = {:.1f} kJ/mol'.format(H_rxn))
print('S_rxn(T=298) = {:.2f} J/mol/K'.format(S_rxn))
Exercise¶
Write a script to calculate the Enthalpy of adsorption (in kcal/mol) of H2O on Cu(111) at T = 298 K. Some important details are given below.
Information Required¶
H2O:¶
- ideal gas
- atoms: You can use "ase.build.molecule" to generate a water molecule like we did with ethane, propane, and butane.
- vibrational wavenumbers (1/cm): 3825.434, 3710.2642, 1582.432
- potential energy (eV): -14.22393533
- spin: 0
- symmetry number: 2
Cu(111):¶
- only electronic modes
- potential energy (eV): -224.13045381
H2O+Cu(111):¶
- electronic and harmonic vibration modes
- potential energy (eV): -238.4713854
- vibrational wavenumbers (1/cm): 3797.255519, 3658.895695, 1530.600295, 266.366130, 138.907356, 63.899768, 59.150454, 51.256019, -327.384554 (negative numbers represent imaginary frequencies. The default behavior of pMuTT is to ignore these frequencies when calculating any thermodynamic property)
Reaction:¶
H2O + Cu(111) --> H2O+Cu(111)
Solution:¶
from ase.build import molecule
from pmutt.statmech import StatMech, presets
from pmutt.reaction import Reaction
# Using dictionary since later I will initialize the reaction with a string
species = {
'H2O(g)': StatMech(atoms=molecule('H2O'),
vib_wavenumbers=[3825.434, 3710.2642, 1582.432],
potentialenergy=-14.22393533,
spin=0,
symmetrynumber=2,
**presets['idealgas']),
'*': StatMech(potentialenergy=-224.13045381,
**presets['electronic']),
'H2O*': StatMech(potentialenergy=-238.4713854,
vib_wavenumbers=[3797.255519,
3658.895695,
1530.600295,
266.366130,
138.907356,
63.899768,
59.150454,
51.256019,
-327.384554], #Imaginary frequency!
**presets['harmonic']),
}
rxn = Reaction.from_string('H2O(g) + * = H2O*', species)
del_H = rxn.get_delta_H(T=298., units='kcal/mol')
print('del_H = {:.2f} kcal/mol'.format(del_H))