fix ti/spring command¶
Syntax¶
fix ID group-ID ti/spring K t_switch t_equil keyword value ...
- ID, group-ID are documented in fix command
- ti/spring = style name of this fix command
- K = spring constant (force/distance units)
- t_switch/t_equil = number of steps of the switching/equilibration procedure
- zero or more keyword/value pairs may be appended to args
- keyword = function
function value = function-ID function-ID = ID of the switching function (1 or 2)
Example:
fix ref all ti/spring 50.0 2000 1000 function 2
Description¶
This fix allows you to compute the free energy of solids by performing a thermodynamic integration between the solid of interest and an Einstein crystal (Frenkel). The thermodynamic integration is performed using the nonequilibrium method of Adiabatic Switching (Watanabe, de Koning96).
A spring force is applied independently to each atom in the group to tether it to its initial position. The initial position for each atom is its location at the time the fix command was issued. More details about the springs are available in fix spring/self. The forces on the atoms are dynamically scaled during the simulation, the rescaling is done in the following manner:
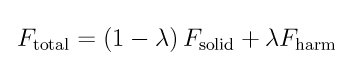
where F_harm is the force due to the springs, F_solid is the total force on the atoms due to the interatomic potential and lambda is the coupling parameter of the thermodynamic integration.
The fix acts as follows: during the first t_equil steps after the fix is defined the value of lambda is zero, this is the period to equilibrate the system in the lambda = 0 state. After this the value of lambda changes continuously from 0 to 1 according to the function defined using the keyword function (described below), this is done in t_switch steps. Then comes the second equilibration period of t_equil to equilibrate the system in the lambda = 1 state. After that the switching back to the lambda = 0 state is made using t_switch timesteps and following the same switching function. After this period the value of lambda is kept equal to zero and the fix has no action in the dynamics of the system anymore.
The description of thermodynamic integration in both directions is done in de Koning97, the main reason is to try to eliminate the dissipated heat due to the nonequilibrium process.
The function keyword allows the use of two different switching rates, the option 1 results in a constant rescaling where the lambda parameter changes at a constant rate during the switching time according to the switching function
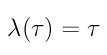
where tau is the scaled time variable t/t_switch. The option number 2 performs the switching at a rate defined by the following switching function
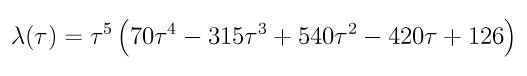
This function has zero slope as lambda approaches its extreme values (0 and 1), according to (de Koning96) this results in smaller fluctuations on the integral to be computed on the thermodynamic integration.
Note
It is importante to keep the center of mass fixed during the
thermodynamic integration, a non-zero total velocity will result in
divergencies during the integration due to the fact that the atoms are
‘attatched’ to its equilibrium positions by the Einstein
crystal. Check the option zero of fix langevin
and velocity. The use of the Nose-Hoover thermostat
(fix nvt) is NOT recommended due to its well documented
issues with the canonical sampling of harmonic degrees of freedom
(notice that the chain option will NOT solve this problem). The
Langevin thermostat (fix langevin) works fine.
Restart, fix_modify, output, run start/stop, minimize info¶
This fix writes the original coordinates of tethered atoms to binary restart files, so that the spring effect will be the same in a restarted simulation. See the read restart command for info on how to re-specify a fix in an input script that reads a restart file, so that the operation of the fix continues in an uninterrupted fashion.
The fix modify energy option is supported by this fix to add the energy stored in the per-atom springs to the system’s potential energy as part of thermodynamic output.
This fix computes a global scalar and a global vector quantities which can be accessed by various output commands. The scalar is an energy which is the sum of the spring energy for each atom, where the per-atom energy is 0.5 * K * r^2. The vector has 2 positions, the first one is the coupling parameter lambda and the second one is the time derivative of lambda. The scalar and vector values calculated by this fix are “extensive”.
No parameter of this fix can be used with the start/stop keywords of the run command.
The forces due to this fix are imposed during an energy minimization, invoked by the minimize command.
Note
If you want the per-atom spring energy to be included in the total potential energy of the system (the quantity being minimized), you MUST enable the fix modify energy option for this fix.
An example script using this command is provided in the examples/USER/misc/ti directory.
Restrictions¶
This command is part of the USER-MISC package. It is only enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more info.
Default¶
The keyword default is function = 1.
(Frenkel) Daan Frenkel and Anthony J. C. Ladd, J. Chem. Phys. 81, 3188 (1984).
(Watanabe) M. Watanabe and W. P. Reinhardt, Phys Rev Lett, 65, 3301 (1990).
(de Koning 96) M. de Koning and A. Antonelli, Phys Rev E, 53, 465 (1996).
(de Koning 97) M. de Koning and A. Antonelli, Phys Rev B, 55, 735 (1997).