
" ] }, { "cell_type": "markdown", "metadata": {}, "source": [ "

" ] }, { "cell_type": "markdown", "metadata": {}, "source": [ "

\n", "


 "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
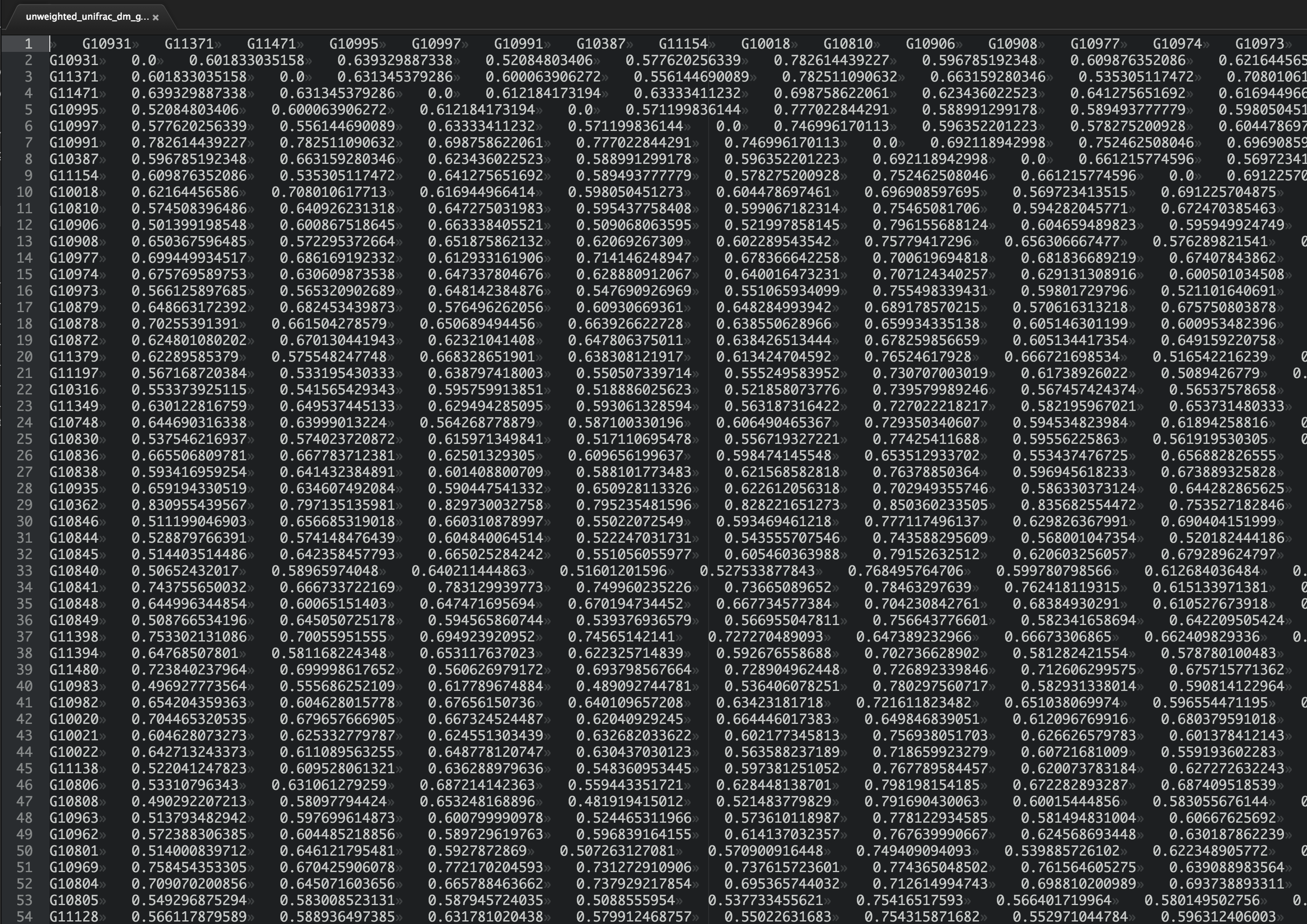
"Do you have a good feeling for the patterns here? What are the most similar samples? What are the most dissimilar samples?"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Chances are, you can't just squint at that table and understand what's going on (but if you can, I'm hiring!). The problem is exacerbated by the fact that in modern microbial ecology studies we may have thousands or tens of thousands of samples, not \"just\" hundreds as in the table above. We need tools to help us take these raw distances and convert them into something that we can interpret. In this section we'll look at some techniques, one of which we've covered previously, that will help us interpret large distance matrices."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Do you have a good feeling for the patterns here? What are the most similar samples? What are the most dissimilar samples?"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Chances are, you can't just squint at that table and understand what's going on (but if you can, I'm hiring!). The problem is exacerbated by the fact that in modern microbial ecology studies we may have thousands or tens of thousands of samples, not \"just\" hundreds as in the table above. We need tools to help us take these raw distances and convert them into something that we can interpret. In this section we'll look at some techniques, one of which we've covered previously, that will help us interpret large distance matrices."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"" ] }, { "cell_type": "markdown", "metadata": {}, "source": [ "One excellent paper that includes a comparison of several different strategies for interpreting beta diversity results is [Costello *et al.* Science (2009) Bacterial Community Variation in Human Body Habitats Across Space and Time](https://www.sciencemag.org/content/326/5960/1694.full). In this study, the authors collected microbiome samples from 7 human subjects at about 25 sites on their bodies, at four different points in time." ] }, { "cell_type": "markdown", "metadata": {}, "source": [ "Figure 1 shows several different approaches for comparing the resulting UniFrac distance matrix (this image is linked from the *Science* journal website - copyright belongs to *Science*):" ] }, { "cell_type": "markdown", "metadata": {}, "source": [ "
 "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Let's generate a small distance matrix representing just a few of these body sites, and figure out how we'd generate and interpret each of these visualizations. The values in the distance matrix below are a subset of the unweighted UniFrac distance matrix representing two samples each from three body sites from the Costello *et al.* (2009) study."
]
},
{
"cell_type": "code",
"execution_count": 33,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
" body site individual\n",
"A gut subject 1\n",
"B gut subject 2\n",
"C tongue subject 1\n",
"D tongue subject 2\n",
"E skin subject 1\n",
"F skin subject 2"
]
},
"execution_count": 33,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"sample_ids = ['A', 'B', 'C', 'D', 'E', 'F']\n",
"_columns = ['body site', 'individual']\n",
"_md = [['gut', 'subject 1'],\n",
" ['gut', 'subject 2'],\n",
" ['tongue', 'subject 1'],\n",
" ['tongue', 'subject 2'],\n",
" ['skin', 'subject 1'],\n",
" ['skin', 'subject 2']]\n",
"\n",
"human_microbiome_sample_md = pd.DataFrame(_md, index=sample_ids, columns=_columns)\n",
"human_microbiome_sample_md"
]
},
{
"cell_type": "code",
"execution_count": 34,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"6x6 distance matrix\n",
"IDs:\n",
"'A', 'B', 'C', 'D', 'E', 'F'\n",
"Data:\n",
"[[ 0. 0.35 0.83 0.83 0.9 0.9 ]\n",
" [ 0.35 0. 0.86 0.85 0.92 0.91]\n",
" [ 0.83 0.86 0. 0.25 0.88 0.87]\n",
" [ 0.83 0.85 0.25 0. 0.88 0.88]\n",
" [ 0.9 0.92 0.88 0.88 0. 0.5 ]\n",
" [ 0.9 0.91 0.87 0.88 0.5 0. ]]"
]
},
"execution_count": 34,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"dm_data = np.array([[0.00, 0.35, 0.83, 0.83, 0.90, 0.90],\n",
" [0.35, 0.00, 0.86, 0.85, 0.92, 0.91],\n",
" [0.83, 0.86, 0.00, 0.25, 0.88, 0.87],\n",
" [0.83, 0.85, 0.25, 0.00, 0.88, 0.88],\n",
" [0.90, 0.92, 0.88, 0.88, 0.00, 0.50],\n",
" [0.90, 0.91, 0.87, 0.88, 0.50, 0.00]])\n",
"\n",
"human_microbiome_dm = DistanceMatrix(dm_data, sample_ids)\n",
"print(human_microbiome_dm)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"#### [3.1.4.2.1](#4.2.1) Distribution plots and comparisons [edit]"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"\n",
"First, let's look at the analysis presented in panels E and F. Instead of generating bar plots here, we'll generate box plots as these are more informative (i.e., they provide a more detailed summary of the distribution being investigated). One important thing to notice here is the central role that the sample metadata plays in the visualization. If we just had our sample ids (i.e., letters ``A`` through ``F``) we wouldn't be able to group distances into *within* and *between* sample type categories, and we therefore couldn't perform the comparisons we're interested in."
]
},
{
"cell_type": "code",
"execution_count": 35,
"metadata": {},
"outputs": [],
"source": [
"def within_between_category_distributions(dm, md, md_category):\n",
" within_category_distances = []\n",
" between_category_distances = []\n",
" for i, sample_id1 in enumerate(dm.ids):\n",
" sample_md1 = md[md_category][sample_id1]\n",
" for sample_id2 in dm.ids[:i]:\n",
" sample_md2 = md[md_category][sample_id2]\n",
" if sample_md1 == sample_md2:\n",
" within_category_distances.append(dm[sample_id1, sample_id2])\n",
" else:\n",
" between_category_distances.append(dm[sample_id1, sample_id2])\n",
" return within_category_distances, between_category_distances"
]
},
{
"cell_type": "code",
"execution_count": 36,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"[0.34999999999999998, 0.25, 0.5]\n",
"[0.82999999999999996, 0.85999999999999999, 0.82999999999999996, 0.84999999999999998, 0.90000000000000002, 0.92000000000000004, 0.88, 0.88, 0.90000000000000002, 0.91000000000000003, 0.87, 0.88]"
]
},
"execution_count": 36,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"within_category_distances, between_category_distances = within_between_category_distributions(human_microbiome_dm, human_microbiome_sample_md, \"body site\")\n",
"print(within_category_distances)\n",
"print(between_category_distances)"
]
},
{
"cell_type": "code",
"execution_count": 37,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"
"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Let's generate a small distance matrix representing just a few of these body sites, and figure out how we'd generate and interpret each of these visualizations. The values in the distance matrix below are a subset of the unweighted UniFrac distance matrix representing two samples each from three body sites from the Costello *et al.* (2009) study."
]
},
{
"cell_type": "code",
"execution_count": 33,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
" body site individual\n",
"A gut subject 1\n",
"B gut subject 2\n",
"C tongue subject 1\n",
"D tongue subject 2\n",
"E skin subject 1\n",
"F skin subject 2"
]
},
"execution_count": 33,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"sample_ids = ['A', 'B', 'C', 'D', 'E', 'F']\n",
"_columns = ['body site', 'individual']\n",
"_md = [['gut', 'subject 1'],\n",
" ['gut', 'subject 2'],\n",
" ['tongue', 'subject 1'],\n",
" ['tongue', 'subject 2'],\n",
" ['skin', 'subject 1'],\n",
" ['skin', 'subject 2']]\n",
"\n",
"human_microbiome_sample_md = pd.DataFrame(_md, index=sample_ids, columns=_columns)\n",
"human_microbiome_sample_md"
]
},
{
"cell_type": "code",
"execution_count": 34,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"6x6 distance matrix\n",
"IDs:\n",
"'A', 'B', 'C', 'D', 'E', 'F'\n",
"Data:\n",
"[[ 0. 0.35 0.83 0.83 0.9 0.9 ]\n",
" [ 0.35 0. 0.86 0.85 0.92 0.91]\n",
" [ 0.83 0.86 0. 0.25 0.88 0.87]\n",
" [ 0.83 0.85 0.25 0. 0.88 0.88]\n",
" [ 0.9 0.92 0.88 0.88 0. 0.5 ]\n",
" [ 0.9 0.91 0.87 0.88 0.5 0. ]]"
]
},
"execution_count": 34,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"dm_data = np.array([[0.00, 0.35, 0.83, 0.83, 0.90, 0.90],\n",
" [0.35, 0.00, 0.86, 0.85, 0.92, 0.91],\n",
" [0.83, 0.86, 0.00, 0.25, 0.88, 0.87],\n",
" [0.83, 0.85, 0.25, 0.00, 0.88, 0.88],\n",
" [0.90, 0.92, 0.88, 0.88, 0.00, 0.50],\n",
" [0.90, 0.91, 0.87, 0.88, 0.50, 0.00]])\n",
"\n",
"human_microbiome_dm = DistanceMatrix(dm_data, sample_ids)\n",
"print(human_microbiome_dm)"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"#### [3.1.4.2.1](#4.2.1) Distribution plots and comparisons [edit]"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"\n",
"First, let's look at the analysis presented in panels E and F. Instead of generating bar plots here, we'll generate box plots as these are more informative (i.e., they provide a more detailed summary of the distribution being investigated). One important thing to notice here is the central role that the sample metadata plays in the visualization. If we just had our sample ids (i.e., letters ``A`` through ``F``) we wouldn't be able to group distances into *within* and *between* sample type categories, and we therefore couldn't perform the comparisons we're interested in."
]
},
{
"cell_type": "code",
"execution_count": 35,
"metadata": {},
"outputs": [],
"source": [
"def within_between_category_distributions(dm, md, md_category):\n",
" within_category_distances = []\n",
" between_category_distances = []\n",
" for i, sample_id1 in enumerate(dm.ids):\n",
" sample_md1 = md[md_category][sample_id1]\n",
" for sample_id2 in dm.ids[:i]:\n",
" sample_md2 = md[md_category][sample_id2]\n",
" if sample_md1 == sample_md2:\n",
" within_category_distances.append(dm[sample_id1, sample_id2])\n",
" else:\n",
" between_category_distances.append(dm[sample_id1, sample_id2])\n",
" return within_category_distances, between_category_distances"
]
},
{
"cell_type": "code",
"execution_count": 36,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"[0.34999999999999998, 0.25, 0.5]\n",
"[0.82999999999999996, 0.85999999999999999, 0.82999999999999996, 0.84999999999999998, 0.90000000000000002, 0.92000000000000004, 0.88, 0.88, 0.90000000000000002, 0.91000000000000003, 0.87, 0.88]"
]
},
"execution_count": 36,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"within_category_distances, between_category_distances = within_between_category_distributions(human_microbiome_dm, human_microbiome_sample_md, \"body site\")\n",
"print(within_category_distances)\n",
"print(between_category_distances)"
]
},
{
"cell_type": "code",
"execution_count": 37,
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"