---
title: "Why data visualization?"
author: Abhijit Dasgupta
date: BIOF 339
---
```{r setup, include=F, child = here::here('slides/templates/setup.Rmd')}
```
```{r, echo=FALSE, results='asis'}
update_header('## The failed promise of data summaries')
```
---
It is tempting to try and summarize data just by some data summaries, like means, medians or standard deviations.
Summaries, by their very nature, compress information
Which means .heatinline[some information gets thrown out]
--
Visualization gives us a way to see patterns in the data that would not be obvious from data summaries
It also allows us to use our natural visual ability of pattern recognition to understand our data
---
### Anscombe's data
.pull-left[
```{r 05-Plotting-5, echo=F}
pacman::p_load(datasauRus)
d <- anscombe %>%
mutate(ID = 1:n()) %>%
tidyr::gather(variable, value, -ID) %>%
separate(variable, c('dim','data'), sep=1) %>%
mutate(data = paste0('dataset', data)) %>%
spread(dim, value) %>%
arrange(data)
ggplot(d, aes(x, y))+
geom_point() +
facet_wrap(~data) +
theme_bw() +
labs(caption = "Anscombe, 1973")
```
]
.pull-right[
| Statistic | Value |
|:----------|------:|
| `mean(x)` | 9 |
| `mean(y)` | 7.5 |
| `var(x)` | 11 |
| `var(y)` | 4.13 |
| `cor(x,y)` | 0.82 |
The variables for each data set have the same values of data summaries
]
---
### The [DataSaurus](https://www.autodeskresearch.com/publications/samestats) dozen
.pull-left[
```{r 05-Plotting-6, echo=F}
ggplot(datasaurus_dozen, aes(x=x, y=y, colour=dataset))+
geom_point()+
theme_void()+
theme(legend.position = "none")+
facet_wrap(~dataset, ncol=3)+
labs(caption="Matejka & Fitzmaurice, 2017")
```
]
.pull-right[
| Statistic | Value |
|:----------|------:|
| `mean(x)` | 54.3 |
| `mean(y)` | 47.8 |
| `var(x)` | 281 |
| `var(y)` | 725 |
| `cor(x,y)` | -0.07 |
The variables for each data set have the same values of data summaries
]
---
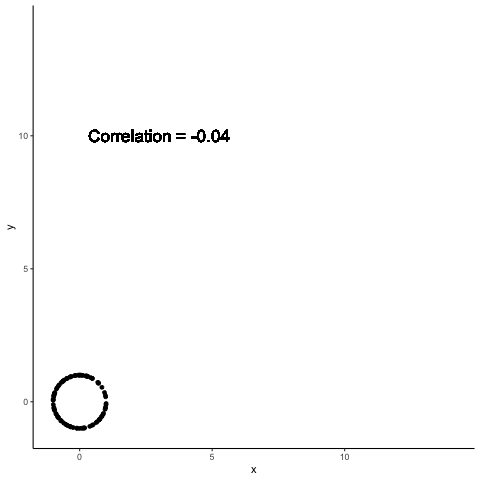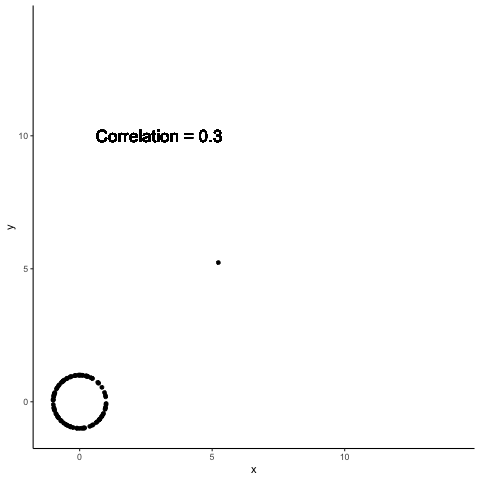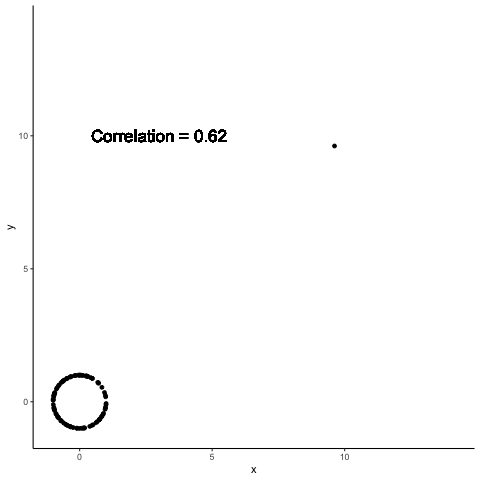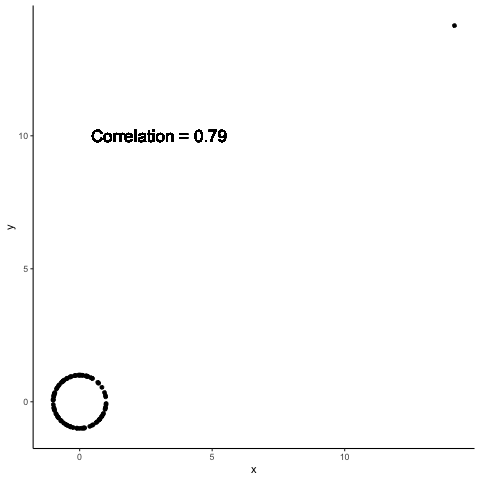
.left-column30[
A single point can completely change the computed correlation
]
.right-column70[
]
---
Data summaries are meant to help distinguish between different data sets
Both Anscombe and Datasaurus show that this promise is not met by standard data summaries
The previous example shows how a single point can change data summaries
---
layout: true
---
## Why visualize data?
- Summary statistics cannot always distinguish datasets
- Take advantage of humans' ability to visually recognize and remember patterns
- Find discrepancies in the data more easily
---
class: middle, center
# Some examples
---
```{r, echo=FALSE, results='asis'}
update_header('## Gallery')
```
---
```{r 05-Plotting-7, echo = F, fig.align='center'}
# install.packages('ggpubr')
pacman::p_load(ggpubr)
data("ToothGrowth")
df <- ToothGrowth
p <- ggboxplot(df, x = "dose", y = "len",
color = "dose", palette =c("#00AFBB", "#E7B800", "#FC4E07"),
add = "jitter", shape = "dose")
my_comparisons <- list( c("0.5", "1"), c("1", "2"), c("0.5", "2") )
p + stat_compare_means(comparisons = my_comparisons)+ # Add pairwise comparisons p-value
stat_compare_means(label.y = 50)
```
???
This is a typical plot in scientific journals
---
### Kaplan-Meier plots
```{r km1, eval = T, echo = F}
pacman::p_load(survminer)
pacman::p_load(survival)
fit <- survfit(Surv(time, status) ~ sex, data = lung)
ggsurvplot(fit, pval = TRUE, conf.int = TRUE,
risk.table = TRUE, risk.table.y.text.col = TRUE)
```
---
```{r 05-Plotting-8, echo = F, fig.align = 'center'}
pacman::p_load('cowplot')
p1 <- ggplot(iris, aes(Sepal.Length, Sepal.Width, color = Species)) +
geom_point() + facet_grid(. ~ Species) + stat_smooth(method = "lm") +
background_grid(major = 'y', minor = "none") +
panel_border() + theme(legend.position = "none")
# plot B
p2 <- ggplot(iris, aes(Sepal.Length, fill = Species)) +
geom_density(alpha = .7) + theme(legend.justification = "top")
p2a <- p2 + theme(legend.position = "none")
# plot C
p3 <- ggplot(iris, aes(Sepal.Width, fill = Species)) +
geom_density(alpha = .7) + theme(legend.position = "none")
# legend
legend <- get_legend(p2)
# align all plots vertically
plots <- cowplot::align_plots(p1, p2a, p3, align = 'v', axis = 'l')
# put together bottom row and then everything
bottom_row <- cowplot::plot_grid(plots[[2]], plots[[3]], legend,
labels = c("B", "C"), rel_widths = c(1, 1, .3), nrow = 1)
cowplot::plot_grid(plots[[1]], bottom_row, labels = c("A"), ncol = 1)
detach('package:cowplot')
```
???
We can put ggplot figures together in a panel with some annotations very easily
using the cowplot package.
These graphs can be cleaned up some.
---
```{r 05-Plotting-9, echo = F, fig.align="center"}
# pacman::p_load
pacman::p_load(ggridges)
pacman::p_load(ggplot2)
# basic example
ggplot(diamonds, aes(x = price, y = cut, fill = cut)) +
geom_density_ridges() +
theme_ridges() +
scale_x_continuous(labels = scales::label_dollar())+
theme(legend.position = "none")
```
???
This is a plot of the diamonds dataset that comes with ggplot2
---
### Manhattan plot
```{r 05-Plotting-10, echo = F, fig.align="center", out.height = "500px", out.width="600px", message = F}
pacman::p_load(qqman)
manhattan(gwasResults, annotatePval = 0.01)
detach('package:qqman')
```
???
Manhattan plots are often used in GWAS studies. You can customize the annotations and
the line for the significance levels
---
### Circular Manhattan plot
```{r cmplot, include=F}
#install.packages('CMplot')
pacman::p_load(CMplot)
data(pig60K)
CMplot(pig60K, plot.type="c", chr.labels=paste("Chr",c(1:18,"X", 'Y'),sep=""), r=0.4, cir.legend=TRUE,
outward=FALSE, cir.legend.col="black", cir.chr.h=1.3 ,chr.den.col="black", file="jpg",
memo="", dpi=300)
detach('package:CMplot')
```
```{r 05-Plotting-11, echo=F, out.height="500", out.width="500"}
knitr::include_graphics(here::here('slides/lectures/week3','img/Circular-Manhattan.trait1.trait2.trait3.jpg'))
```
???
This gives a different representation of the manhattan plot. This example looks at three traits simultaneously
---
### Maps
.pull-left[
```{r map2, echo=F, eval=FALSE}
pacman::p_load(tmap)
tmap_mode('view') #<<
mp2 <- tm_shape(spData::world) + tm_polygons(col = 'pop', style='jenks',
palette='Reds', title='Population',
popup.vars = c('pop'), id='name_long')
tmap_save(mp2, file = here::here('docs/slides/lectures/week3/img/map2.html'))
```
]
.pull-right[
```{r map3, echo=F, eval=F, cache=F}
pacman::p_load(leaflet)
pacman::p_load(leaflet.providers)
pacman::p_load(htmlwidgets)
pacman::p_load(htmltools)
load(file.path(here::here('slides','lectures','data', 'exdata.rda')))
m <- leaflet(gpx) %>% addTiles() %>% addCircleMarkers(~Longitude, ~Latitude, radius=1)
withr::with_dir(here::here('docs/slides/lectures/week3/img'), saveWidget(m, file='map1.html'))
```
]
---
### Heatmap
.pull-left[
```{r heatmap, echo=FALSE, fig.height=5.5}
pacman::p_load(ComplexHeatmap)
pacman::p_load(circlize)
mat = readRDS(system.file("extdata", "measles.rds", package = "ComplexHeatmap"))
ha1 = HeatmapAnnotation(
dist1 = anno_barplot(
colSums(mat),
bar_width = 1,
gp = gpar(col = "white", fill = "#FFE200"),
border = FALSE,
axis_param = list(at = c(0, 2e5, 4e5, 6e5, 8e5),
labels = c("0", "200k", "400k", "600k", "800k")),
height = unit(2, "cm")
), show_annotation_name = FALSE)
ha2 = rowAnnotation(
dist2 = anno_barplot(
rowSums(mat),
bar_width = 1,
gp = gpar(col = "white", fill = "#FFE200"),
border = FALSE,
axis_param = list(at = c(0, 5e5, 1e6, 1.5e6),
labels = c("0", "500k", "1m", "1.5m")),
width = unit(2, "cm")
), show_annotation_name = FALSE)
year_text = as.numeric(colnames(mat))
year_text[year_text %% 10 != 0] = ""
ha_column = HeatmapAnnotation(
year = anno_text(year_text, rot = 0, location = unit(1, "npc"), just = "top")
)
col_fun = colorRamp2(c(0, 800, 1000, 127000), c("white", "cornflowerblue", "yellow", "red"))
ht_list = Heatmap(mat, name = "cases", col = col_fun,
cluster_columns = FALSE, show_row_dend = FALSE, rect_gp = gpar(col= "white"),
show_column_names = FALSE,
row_names_side = "left", row_names_gp = gpar(fontsize = 6),
column_title = 'Measles cases in US states 1930-2001\nVaccine introduced 1961',
top_annotation = ha1, bottom_annotation = ha_column,
heatmap_legend_param = list(at = c(0, 5e4, 1e5, 1.5e5),
labels = c("0", "50k", "100k", "150k"))) + ha2
draw(ht_list, ht_gap = unit(3, "mm"))
decorate_heatmap_body("cases", {
i = which(colnames(mat) == "1961")
x = i/ncol(mat)
grid.lines(c(x, x), c(0, 1), gp = gpar(lwd = 2, lty = 2))
grid.text("Vaccine introduced", x, unit(1, "npc") + unit(5, "mm"))
})
```
]
.pull-right[
```{r hm2, echo=FALSE, fig.height=5.5}
pacman::p_load(ComplexHeatmap)
pacman::p_load(circlize)
expr = readRDS(system.file(package = "ComplexHeatmap", "extdata", "gene_expression.rds"))
mat = as.matrix(expr[, grep("cell", colnames(expr))])
base_mean = rowMeans(mat)
mat_scaled = t(apply(mat, 1, scale))
type = gsub("s\\d+_", "", colnames(mat))
ha = HeatmapAnnotation(type = type, annotation_name_side = "left")
ht_list = Heatmap(mat_scaled, name = "expression", row_km = 5,
col = colorRamp2(c(-2, 0, 2), c("blue", "white", "orange")),
top_annotation = ha,
show_column_names = FALSE, row_title = NULL, show_row_dend = FALSE) +
Heatmap(base_mean, name = "base mean",
top_annotation = HeatmapAnnotation(summary = anno_summary(gp = gpar(fill = 2:6),
height = unit(2, "cm"))),
width = unit(15, "mm")) +
rowAnnotation(length = anno_points(expr$length, pch = 16, size = unit(1, "mm"),
axis_param = list(at = c(0, 2e5, 4e5, 6e5),
labels = c("0kb", "200kb", "400kb", "600kb")),
width = unit(2, "cm"))) +
Heatmap(expr$type, name = "gene type",
top_annotation = HeatmapAnnotation(summary = anno_summary(height = unit(2, "cm"))),
width = unit(15, "mm"))
ht_list = rowAnnotation(block = anno_block(gp = gpar(fill = 2:6, col = NA)),
width = unit(2, "mm")) + ht_list
draw(ht_list, ht_gap = unit(5, "mm"))
```
]
---
### OncoPrint
```{r, echo=F, fig.height=5}
pacman::p_load(ComplexHeatmap)
mat = read.table(system.file("extdata", package = "ComplexHeatmap",
"tcga_lung_adenocarcinoma_provisional_ras_raf_mek_jnk_signalling.txt"),
header = TRUE, stringsAsFactors = FALSE, sep = "\t")
mat[is.na(mat)] = ""
rownames(mat) = mat[, 1]
mat = mat[, -1]
mat= mat[, -ncol(mat)]
mat = t(as.matrix(mat))
col = c("HOMDEL" = "blue", "AMP" = "red", "MUT" = "#008000")
alter_fun = list(
background = function(x, y, w, h) {
grid.rect(x, y, w-unit(2, "pt"), h-unit(2, "pt"),
gp = gpar(fill = "#CCCCCC", col = NA))
},
# big blue
HOMDEL = function(x, y, w, h) {
grid.rect(x, y, w-unit(2, "pt"), h-unit(2, "pt"),
gp = gpar(fill = col["HOMDEL"], col = NA))
},
# big red
AMP = function(x, y, w, h) {
grid.rect(x, y, w-unit(2, "pt"), h-unit(2, "pt"),
gp = gpar(fill = col["AMP"], col = NA))
},
# small green
MUT = function(x, y, w, h) {
grid.rect(x, y, w-unit(2, "pt"), h*0.33,
gp = gpar(fill = col["MUT"], col = NA))
}
)
column_title = "OncoPrint for TCGA Lung Adenocarcinoma, genes in Ras Raf MEK JNK signalling"
heatmap_legend_param = list(title = "Alternations", at = c("HOMDEL", "AMP", "MUT"),
labels = c("Deep deletion", "Amplification", "Mutation"))
oncoPrint(mat,
alter_fun = alter_fun, col = col,
remove_empty_columns = TRUE, remove_empty_rows = TRUE,
column_title = column_title, heatmap_legend_param = heatmap_legend_param)
```
---
### Interactive graphs
```{r 05-Plotting-13, echo=F, eval=T}
pacman::p_load(plotly)
pacman::p_load(fs)
pacman::p_load(here)
p=ggplot(iris,
aes(x=Sepal.Length, y=Sepal.Width, color=Species, shape=Species)) +
geom_point(size=2, alpha=0.6)+
theme_bw()
mytext=paste("Sepal Length = ", iris$Sepal.Length, "\n" , "Sepal Width = ", iris$Sepal.Width, "\n", "Row Number: ",rownames(iris), sep="")
pp=plotly_build(p)
style( pp, text=mytext, hoverinfo = "text", traces = c(1, 2, 3) )
pg <- ggplotly(p)
# htmltools::save_html(pg, file = here::here('slides','lectures','week3','why_plot_files','pl.html'))
```
???
These graphs are clickable. For example, click on a symbol on the legend, or drag your mouse over a region with left button held down.
---
## Network graphs
```{r network, eval=!fs::file_exists(here::here('docs','slides','lectures','week3','anim/fn.html')), echo=FALSE}
pacman::p_load(htmlwidgets)
pacman::p_load(networkD3)
data(MisLinks, MisNodes)
fn <- forceNetwork(Links = MisLinks, Nodes = MisNodes, Source = "source",
Target = "target", Value = "value", NodeID = "name",
Group = "group", opacity = 0.4)
htmltools::save_html(fn, file = here::here('docs','slides','lectures','week3','anim','fn.html'))
```
---
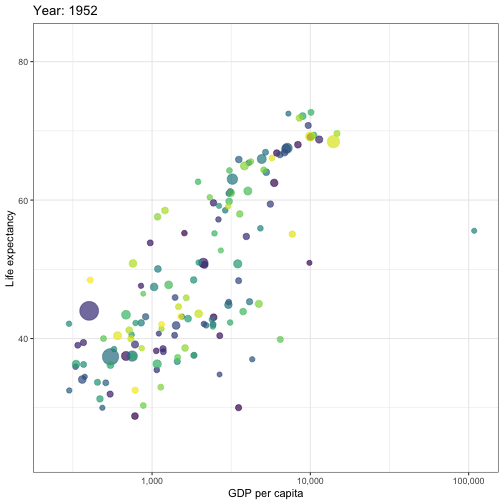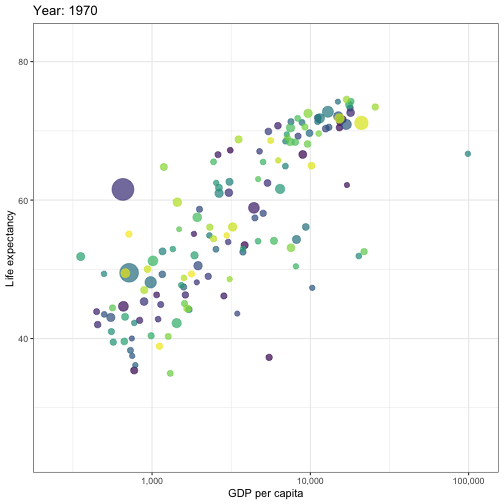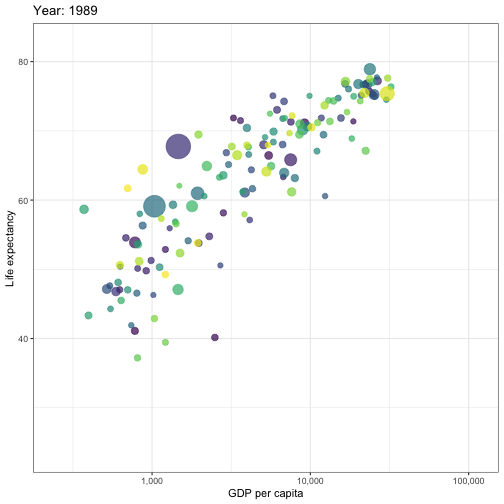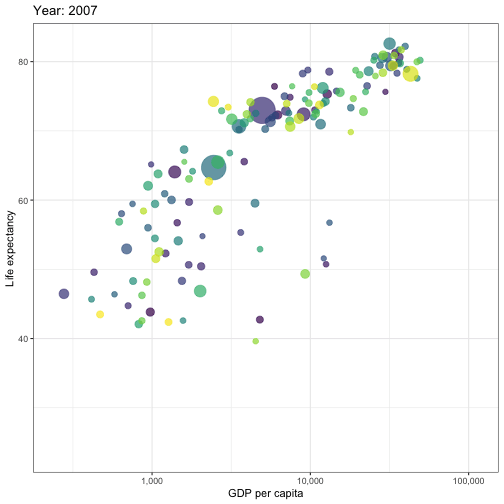
## Animated graphs
```{r aniplot, eval=!fs::file_exists('anim/gapminder.gif'), echo=F, results='hide', cache=FALSE}
pacman::p_load(ggplot2)
pacman::p_load(gganimate)
theme_set(theme_bw())
pacman::p_load(gapminder)
p <- ggplot(
gapminder,
aes(x = gdpPercap, y=lifeExp, size = pop, colour = country)
) +
geom_point(show.legend = FALSE, alpha = 0.7) +
scale_color_viridis_d() +
scale_size(range = c(2, 12)) +
scale_x_log10(labels = scales::label_comma()) +
labs(x = "GDP per capita", y = "Life expectancy")
p <- p + transition_time(year) +
labs(title = "Year: {frame_time}")
animate(p, renderer = gifski_renderer(), device='png')
anim_save('anim/gapminder.gif')
```
---
```{r, echo=F, results='asis'}
update_header()
```
---
class: middle, center, inverse
# For more in-depth looks at data viz, consider BIOF 439 in the Spring