# mLLMCelltype: Multi-LLM Consensus Framework for Cell Type Annotation
mLLMCelltype is a multi-LLM consensus framework for automated cell type annotation in single-cell RNA sequencing (scRNA-seq) data. The framework integrates multiple large language models including OpenAI GPT-5.5, Anthropic Claude Opus 4.7 and Sonnet 4.6, Google Gemini 3, X.AI Grok 4.3, DeepSeek V4, Alibaba Qwen 3.6, Z.AI GLM 5.1, MiniMax M2.7, Stepfun 3.5, and OpenRouter to improve annotation accuracy through consensus-based predictions.
## Abstract
mLLMCelltype is an open-source tool for single-cell transcriptomics analysis that uses multiple large language models to identify cell types from gene expression data. The software implements a consensus approach where multiple models analyze the same data and their predictions are combined, which helps reduce errors and provides uncertainty metrics. This methodology offers advantages over single-model approaches through integration of multiple model predictions. mLLMCelltype integrates with single-cell analysis platforms such as Scanpy and Seurat, allowing researchers to incorporate it into existing workflows. The method does not require reference datasets for annotation.
The mLLMCelltype paper was published online in *Communications Biology* on June 8, 2026.
Across 49 benchmark datasets ([Yang et al., 2026](https://doi.org/10.1038/s42003-026-10420-8)), mLLMCelltype achieved 77.2% mean accuracy, improving on the best single-LLM baseline (61.5%) by 15.7 percentage points.
## Table of Contents
- [Key Features](#key-features)
- [Installation](#installation)
- [Usage Examples](#usage-examples)
- [Visualization Example](#visualization-example)
- [Citation](#citation)
- [Contributing](#contributing)
**Web Application**: A browser-based interface is available at [mllmcelltype.com](https://mllmcelltype.com) (no installation required).
**See also**: [FlashDeconv](https://github.com/cafferychen777/FlashDeconv) — cell type deconvolution for spatial transcriptomics (Visium, Visium HD, Stereo-seq).
## Key Features
- **Multi-LLM Consensus**: Integrates predictions from multiple LLMs to reduce single-model limitations and biases
- **Model Support**: Compatible with 10+ LLM providers including OpenAI, Anthropic, Google, and others
- **Iterative Discussion**: LLMs evaluate evidence and refine annotations through multiple rounds of discussion
- **Uncertainty Quantification**: Provides Consensus Proportion and Shannon Entropy metrics to identify uncertain annotations
- **Cross-Model Validation**: Reduces incorrect predictions through multi-model comparison
- **Noise Tolerance**: Maintains accuracy with imperfect marker gene lists
- **Hierarchical Annotation**: Supports multi-resolution analysis with consistency checks
- **Reference-Free**: Performs annotation without pre-training or reference datasets
- **Documentation**: Records complete reasoning process for transparency
- **Integration**: Compatible with Scanpy/Seurat workflows and marker gene outputs
- **Extensibility**: Supports addition of new LLMs as they become available
For changelog and updates, see [NEWS.md](R/NEWS.md).
## Installation
### R Version
```r
# Install from CRAN (recommended)
install.packages("mLLMCelltype")
# Or install development version from GitHub
devtools::install_github("cafferychen777/mLLMCelltype", subdir = "R")
```
### Python Version
[](https://colab.research.google.com/drive/1ZgmtlaORogSy0-QsaF0CHwFWOyOD26d2?usp=sharing)
**Quick Start**: Try mLLMCelltype in Google Colab without any installation. Click the badge above to open an interactive notebook with examples and step-by-step guidance.
```bash
# Install from PyPI
pip install mllmcelltype
# Or install from GitHub (note the subdirectory parameter)
pip install git+https://github.com/cafferychen777/mLLMCelltype.git#subdirectory=python
```
#### Important Note on Dependencies
mLLMCelltype uses a modular design where different LLM provider libraries are optional dependencies. Depending on which models you plan to use, you'll need to install the corresponding packages:
```bash
# For using OpenAI models (GPT-5.5, etc.)
pip install "mllmcelltype[openai]"
# For using Anthropic models (Claude)
pip install "mllmcelltype[anthropic]"
# For using Google models (Gemini)
pip install "mllmcelltype[gemini]"
# To install all optional dependencies at once
pip install "mllmcelltype[all]"
```
If you encounter errors like `ImportError: cannot import name 'genai' from 'google'`, it means you need to install the corresponding provider package. For example:
```bash
# For Google Gemini models
pip install google-genai
```
### Supported Models
- **OpenAI**: GPT-5.5/GPT-5.4/GPT-5.4-mini ([API Key](https://platform.openai.com/settings/organization/billing/overview))
- **Anthropic**: Claude-Opus-4.7/Claude-Sonnet-4.6/Claude-Haiku-4.5 ([API Key](https://console.anthropic.com/))
- **Google**: Gemini-3.1-Pro-Preview/Gemini-3-Flash-Preview/Gemini-3.1-Flash-Lite ([API Key](https://ai.google.dev/?authuser=2))
- **Alibaba**: Qwen3.6-Plus/Qwen3.6-Flash/Qwen3.6-Max-Preview ([API Key](https://www.alibabacloud.com/en/product/modelstudio))
- **DeepSeek**: DeepSeek-V4-Flash/DeepSeek-V4-Pro ([API Key](https://platform.deepseek.com/usage))
- **Minimax**: MiniMax-M2.7/MiniMax-M2.7-highspeed ([API Key](https://intl.minimaxi.com/user-center/basic-information/interface-key))
- **Stepfun**: Step-3.5-Flash/Step-3 ([API Key](https://platform.stepfun.com/account-info))
- **Zhipu/Z.AI**: GLM-5.1/GLM-5/GLM-5-Turbo ([API Key](https://bigmodel.cn/))
- **X.AI**: Grok-4.3 ([API Key](https://accounts.x.ai/))
- **OpenRouter**: Access to multiple models through a single API ([API Key](https://openrouter.ai/keys))
- Supports models from OpenAI, Anthropic, Meta, Google, Mistral, and more
- Format: 'provider/model-name' (e.g., 'openai/gpt-5.5', 'anthropic/claude-opus-4.7')
- Free models available with `:free` suffix (e.g., 'deepseek/deepseek-v4-pro:free', 'meta-llama/llama-4-maverick:free')
- **Note**: Free tier limits: 50 requests/day (1000/day with $10+ credits), 20 requests/minute. Some models may be unavailable.
## Usage Examples
### Python
```python
# Example of using mLLMCelltype for single-cell RNA-seq cell type annotation with Scanpy
import scanpy as sc
import pandas as pd
from mllmcelltype import annotate_clusters, interactive_consensus_annotation
import os
# Logging is initialized when annotation functions run.
# Call setup_logging() first only if you want a custom log directory or level.
# Load your single-cell RNA-seq dataset in AnnData format
adata = sc.read_h5ad('your_data.h5ad') # Replace with your scRNA-seq dataset path
# Perform Leiden clustering for cell population identification if not already done
if 'leiden' not in adata.obs.columns:
print("Computing leiden clustering for cell population identification...")
# Preprocess single-cell data: normalize counts and log-transform for gene expression analysis
if 'log1p' not in adata.uns:
sc.pp.normalize_total(adata, target_sum=1e4) # Normalize to 10,000 counts per cell
sc.pp.log1p(adata) # Log-transform normalized counts
# Dimensionality reduction: calculate PCA for scRNA-seq data
if 'X_pca' not in adata.obsm:
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5) # Select informative genes
sc.pp.pca(adata, use_highly_variable=True) # Compute principal components
# Cell clustering: compute neighborhood graph and perform Leiden community detection
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=30) # Build KNN graph for clustering
sc.tl.leiden(adata, resolution=0.8) # Identify cell populations using Leiden algorithm
print(f"Leiden clustering completed, identified {len(adata.obs['leiden'].cat.categories)} distinct cell populations")
# Identify marker genes for each cell cluster using differential expression analysis
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon') # Wilcoxon rank-sum test for marker detection
# Extract top marker genes for each cell cluster to use in cell type annotation
marker_genes = {}
for i in range(len(adata.obs['leiden'].cat.categories)):
# Select top 10 differentially expressed genes as markers for each cluster
genes = [adata.uns['rank_genes_groups']['names'][str(i)][j] for j in range(10)]
marker_genes[str(i)] = genes
# IMPORTANT: mLLMCelltype requires gene symbols (e.g., KCNJ8, PDGFRA) not Ensembl IDs (e.g., ENSG00000176771)
# If your AnnData object uses Ensembl IDs, convert them to gene symbols for accurate annotation:
# Example conversion code:
# if 'Gene' in adata.var.columns: # Check if gene symbols are available in the metadata
# gene_name_dict = dict(zip(adata.var_names, adata.var['Gene']))
# marker_genes = {cluster: [gene_name_dict.get(gene_id, gene_id) for gene_id in genes]
# for cluster, genes in marker_genes.items()}
# IMPORTANT: mLLMCelltype preserves cluster IDs as provided.
# Cluster IDs may be numeric or character values (e.g., "0", "T_cells", "7_0").
# Use stable, unique IDs so outputs can be mapped back to the original clusters.
# Configure API keys for the large language models used in consensus annotation
# At least one API key is required for multi-LLM consensus annotation
os.environ["OPENAI_API_KEY"] = "your-openai-api-key" # For GPT-5.5/5.4 models
os.environ["ANTHROPIC_API_KEY"] = "your-anthropic-api-key" # For Claude Opus 4.7/Sonnet 4.6 models
os.environ["GEMINI_API_KEY"] = "your-gemini-api-key" # For Google Gemini 3 models
os.environ["QWEN_API_KEY"] = "your-qwen-api-key" # For Alibaba Qwen3 models
# Additional optional LLM providers for enhanced consensus diversity:
# os.environ["DEEPSEEK_API_KEY"] = "your-deepseek-api-key" # For DeepSeek-V3 models
# os.environ["ZHIPU_API_KEY"] = "your-zhipu-api-key" # For Zhipu GLM-4 models
# os.environ["STEPFUN_API_KEY"] = "your-stepfun-api-key" # For Stepfun models
# os.environ["MINIMAX_API_KEY"] = "your-minimax-api-key" # For MiniMax models
# os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key" # For accessing multiple models via OpenRouter
# Execute multi-LLM consensus cell type annotation with iterative deliberation
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes, # Dictionary of marker genes for each cluster
species="human", # Specify organism for appropriate cell type annotation
tissue="blood", # Specify tissue context for more accurate annotation
models=["gpt-5.5", "claude-sonnet-4-6", "gemini-3.1-pro-preview", "qwen3.6-plus"], # Multiple LLMs for consensus
consensus_threshold=1, # Minimum proportion required for consensus agreement
max_discussion_rounds=3 # Number of deliberation rounds between models for refinement
)
# Alternatively, use OpenRouter for accessing multiple models through a single API
# This is especially useful for accessing free models with the :free suffix
os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key"
# Example using free OpenRouter models (no credits required)
free_models_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="human",
tissue="blood",
models=[
{"provider": "openrouter", "model": "meta-llama/llama-4-maverick:free"}, # Meta Llama 4 Maverick (free)
{"provider": "openrouter", "model": "venice/uncensored:free"}, # Venice Uncensored (free)
{"provider": "openrouter", "model": "deepseek/deepseek-v4-pro:free"}, # DeepSeek V4 Pro (free)
{"provider": "openrouter", "model": "meta-llama/llama-3.3-70b-instruct:free"} # Meta Llama 3.3 70B (free)
],
consensus_threshold=0.7,
max_discussion_rounds=2
)
# Retrieve final consensus cell type annotations from the multi-LLM deliberation
final_annotations = consensus_results["consensus"]
# Integrate consensus cell type annotations into the original AnnData object
adata.obs['consensus_cell_type'] = adata.obs['leiden'].astype(str).map(final_annotations)
# Add uncertainty quantification metrics to evaluate annotation confidence
adata.obs['consensus_proportion'] = adata.obs['leiden'].astype(str).map(consensus_results["consensus_proportion"]) # Agreement level
adata.obs['entropy'] = adata.obs['leiden'].astype(str).map(consensus_results["entropy"]) # Annotation uncertainty
# Prepare for visualization: compute UMAP embeddings if not already available
# UMAP provides a 2D representation of cell populations for visualization
if 'X_umap' not in adata.obsm:
print("Computing UMAP coordinates...")
# Make sure neighbors are computed first
if 'neighbors' not in adata.uns:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=30)
sc.tl.umap(adata)
print("UMAP coordinates computed")
# Visualize results with enhanced aesthetics
# Basic visualization
sc.pl.umap(adata, color='consensus_cell_type', legend_loc='right', frameon=True, title='mLLMCelltype Consensus Annotations')
# More customized visualization
import matplotlib.pyplot as plt
# Set figure size and style
plt.rcParams['figure.figsize'] = (10, 8)
plt.rcParams['font.size'] = 12
# Create a more publication-ready UMAP
fig, ax = plt.subplots(1, 1, figsize=(12, 10))
sc.pl.umap(adata, color='consensus_cell_type', legend_loc='on data',
frameon=True, title='mLLMCelltype Consensus Annotations',
palette='tab20', size=50, legend_fontsize=12,
legend_fontoutline=2, ax=ax)
# Visualize uncertainty metrics
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(16, 7))
sc.pl.umap(adata, color='consensus_proportion', ax=ax1, title='Consensus Proportion',
cmap='viridis', vmin=0, vmax=1, size=30)
sc.pl.umap(adata, color='entropy', ax=ax2, title='Annotation Uncertainty (Shannon Entropy)',
cmap='magma', vmin=0, size=30)
plt.tight_layout()
```
### Using a Single Free OpenRouter Model
For users who prefer a simpler approach with just one model, the DeepSeek V4 Pro free model via OpenRouter can be used without API credits:
```python
import os
from mllmcelltype import annotate_clusters
# Note: Logging is automatically configured
# Set your OpenRouter API key
os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key"
# Define marker genes for each cluster
marker_genes = {
"0": ["CD3D", "CD3E", "CD3G", "CD2", "IL7R", "TCF7"], # T cells
"1": ["CD19", "MS4A1", "CD79A", "CD79B", "HLA-DRA", "CD74"], # B cells
"2": ["CD14", "LYZ", "CSF1R", "ITGAM", "CD68", "FCGR3A"] # Monocytes
}
# Annotate using DeepSeek V4 Pro free model
annotations = annotate_clusters(
marker_genes=marker_genes,
species='human',
tissue='peripheral blood',
provider='openrouter',
model='deepseek/deepseek-v4-pro:free' # Free model with advanced reasoning
)
# Print annotations
for cluster, annotation in annotations.items():
print(f"Cluster {cluster}: {annotation}")
```
This approach uses a free model and does not require API credits.
#### Extracting Marker Genes from AnnData Objects
If you're using Scanpy with AnnData objects, you can easily extract marker genes directly from the `rank_genes_groups` results:
```python
import os
import scanpy as sc
from mllmcelltype import annotate_clusters
# Note: Logging is automatically configured
# Set your OpenRouter API key
os.environ["OPENROUTER_API_KEY"] = "your-openrouter-api-key"
# Load and preprocess your data
adata = sc.read_h5ad('your_data.h5ad')
# Perform preprocessing and clustering if not already done
# sc.pp.normalize_total(adata, target_sum=1e4)
# sc.pp.log1p(adata)
# sc.pp.highly_variable_genes(adata)
# sc.pp.pca(adata)
# sc.pp.neighbors(adata)
# sc.tl.leiden(adata)
# Find marker genes for each cluster
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
# Extract top marker genes for each cluster
marker_genes = {
cluster: adata.uns['rank_genes_groups']['names'][cluster][:10].tolist()
for cluster in adata.obs['leiden'].cat.categories
}
# Annotate using DeepSeek V4 Pro free model
annotations = annotate_clusters(
marker_genes=marker_genes,
species='human',
tissue='peripheral blood', # adjust based on your tissue type
provider='openrouter',
model='deepseek/deepseek-v4-pro:free' # Free model
)
# Add annotations to AnnData object
adata.obs['cell_type'] = adata.obs['leiden'].astype(str).map(annotations)
# Visualize results
sc.pl.umap(adata, color='cell_type', legend_loc='on data',
frameon=True, title='Cell Types Annotated by DeepSeek V4 Pro')
```
This method automatically extracts the top differentially expressed genes for each cluster from the `rank_genes_groups` results, making it easy to integrate mLLMCelltype into your Scanpy workflow.
### R
> **Note**: For more detailed R tutorials and documentation, please visit the [mLLMCelltype documentation website](https://cafferyang.com/mLLMCelltype/).
#### Using Seurat Object
```r
# Load required packages
library(mLLMCelltype)
library(Seurat)
library(dplyr)
library(ggplot2)
library(cowplot) # Added for plot_grid
# Load your preprocessed Seurat object
pbmc <- readRDS("your_seurat_object.rds")
# If starting with raw data, perform preprocessing steps
# pbmc <- NormalizeData(pbmc)
# pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# pbmc <- ScaleData(pbmc)
# pbmc <- RunPCA(pbmc)
# pbmc <- FindNeighbors(pbmc, dims = 1:10)
# pbmc <- FindClusters(pbmc, resolution = 0.5)
# pbmc <- RunUMAP(pbmc, dims = 1:10)
# Find marker genes for each cluster
pbmc_markers <- FindAllMarkers(pbmc,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
# Set up cache directory to speed up processing
cache_dir <- "./mllmcelltype_cache"
dir.create(cache_dir, showWarnings = FALSE, recursive = TRUE)
# Choose a model from any supported provider
# Supported models include:
# - OpenAI: 'gpt-5.5', 'gpt-5.4', 'gpt-5.4-mini'
# - Anthropic: 'claude-opus-4-7', 'claude-sonnet-4-6', 'claude-haiku-4-5-20251001'
# - DeepSeek: 'deepseek-v4-flash', 'deepseek-v4-pro'
# - Google: 'gemini-3.1-pro-preview', 'gemini-3-flash-preview', 'gemini-3.1-flash-lite'
# - Qwen: 'qwen3.6-plus', 'qwen3.6-flash', 'qwen3.6-max-preview'
# - Stepfun: 'step-3.5-flash', 'step-3.5-flash-2603', 'step-3'
# - Zhipu/Z.AI: 'glm-5.1', 'glm-5', 'glm-5-turbo'
# - MiniMax: 'MiniMax-M2.7', 'MiniMax-M2.7-highspeed', 'MiniMax-M2.5'
# - Grok: 'grok-4.3', 'grok-4.3-latest', 'grok-latest'
# - OpenRouter: Access to models from multiple providers through a single API. Format: 'provider/model-name'
# - OpenAI models: 'openai/gpt-5.5', 'openai/gpt-5.4-mini'
# - Anthropic models: 'anthropic/claude-opus-4.7', 'anthropic/claude-sonnet-4.6'
# - Google models: 'google/gemini-3.1-pro-preview', 'google/gemini-3-flash-preview'
# - X.AI models: 'x-ai/grok-4.3'
# - Stepfun models: 'stepfun/step-3.5-flash'
# Run LLMCelltype annotation with multiple LLM models
consensus_results <- interactive_consensus_annotation(
input = pbmc_markers,
tissue_name = "human PBMC", # provide tissue context
models = c(
"claude-sonnet-4-6", # Anthropic
"gpt-5.5", # OpenAI
"gemini-3.1-pro-preview", # Google
"qwen3.6-plus" # Alibaba
),
api_keys = list(
anthropic = "your-anthropic-key",
openai = "your-openai-key",
gemini = "your-google-key",
qwen = "your-qwen-key"
),
top_gene_count = 10,
controversy_threshold = 1.0,
entropy_threshold = 1.0,
cache_dir = cache_dir
)
# Print structure of results to understand the data
print("Available fields in consensus_results:")
print(names(consensus_results))
# Add annotations to Seurat object
# Get cell type annotations from consensus_results$final_annotations
cluster_to_celltype_map <- consensus_results$final_annotations
# Create new cell type identifier column
cell_types <- as.character(Idents(pbmc))
for (cluster_id in names(cluster_to_celltype_map)) {
cell_types[cell_types == cluster_id] <- cluster_to_celltype_map[[cluster_id]]
}
# Add cell type annotations to Seurat object
pbmc$cell_type <- cell_types
# Add uncertainty metrics
# Extract detailed consensus results containing metrics
consensus_details <- consensus_results$initial_results$consensus_results
# Create a data frame with metrics for each cluster
uncertainty_metrics <- data.frame(
cluster_id = names(consensus_details),
consensus_proportion = sapply(consensus_details, function(res) res$consensus_proportion),
entropy = sapply(consensus_details, function(res) res$entropy)
)
# Add uncertainty metrics for each cell
# Note: seurat_clusters is a metadata column automatically created by FindClusters() function
# It contains the cluster ID assigned to each cell during clustering
# Here we use it to map cluster-level metrics (consensus_proportion and entropy) to individual cells
# If you don't have seurat_clusters column (e.g., if you used a different clustering method),
# you can use the active identity (Idents) or any other cluster assignment in your metadata:
# Option 1: Use active identity
# current_clusters <- as.character(Idents(pbmc))
# Option 2: Use another metadata column that contains cluster IDs
# current_clusters <- pbmc$your_cluster_column
# For this example, we use the standard seurat_clusters column:
current_clusters <- pbmc$seurat_clusters # Get cluster ID for each cell
# Match each cell's cluster ID with the corresponding metrics in uncertainty_metrics
pbmc$consensus_proportion <- uncertainty_metrics$consensus_proportion[match(current_clusters, uncertainty_metrics$cluster_id)]
pbmc$entropy <- uncertainty_metrics$entropy[match(current_clusters, uncertainty_metrics$cluster_id)]
# Save results for future use
saveRDS(consensus_results, "pbmc_mLLMCelltype_results.rds")
saveRDS(pbmc, "pbmc_annotated.rds")
# Visualize results with SCpubr for publication-ready plots
if (!requireNamespace("SCpubr", quietly = TRUE)) {
remotes::install_github("enblacar/SCpubr")
}
library(SCpubr)
library(viridis) # For color palettes
# Basic UMAP visualization with default settings
pdf("pbmc_basic_annotations.pdf", width=8, height=6)
SCpubr::do_DimPlot(sample = pbmc,
group.by = "cell_type",
label = TRUE,
legend.position = "right") +
ggtitle("mLLMCelltype Consensus Annotations")
dev.off()
# More customized visualization with enhanced styling
pdf("pbmc_custom_annotations.pdf", width=8, height=6)
SCpubr::do_DimPlot(sample = pbmc,
group.by = "cell_type",
label = TRUE,
label.box = TRUE,
legend.position = "right",
pt.size = 1.0,
border.size = 1,
font.size = 12) +
ggtitle("mLLMCelltype Consensus Annotations") +
theme(plot.title = element_text(hjust = 0.5))
dev.off()
# Visualize uncertainty metrics with enhanced SCpubr plots
# Get cell types and create a named color palette
cell_types <- unique(pbmc$cell_type)
color_palette <- viridis::viridis(length(cell_types))
names(color_palette) <- cell_types
# Cell type annotations with SCpubr
p1 <- SCpubr::do_DimPlot(sample = pbmc,
group.by = "cell_type",
label = TRUE,
legend.position = "bottom", # Place legend at the bottom
pt.size = 1.0,
label.size = 4, # Smaller label font size
label.box = TRUE, # Add background box to labels for better readability
repel = TRUE, # Make labels repel each other to avoid overlap
colors.use = color_palette,
plot.title = "Cell Type") +
theme(plot.title = element_text(hjust = 0.5, margin = margin(b = 15, t = 10)),
legend.text = element_text(size = 8),
legend.key.size = unit(0.3, "cm"),
plot.margin = unit(c(0.8, 0.8, 0.8, 0.8), "cm"))
# Consensus proportion feature plot with SCpubr
p2 <- SCpubr::do_FeaturePlot(sample = pbmc,
features = "consensus_proportion",
order = TRUE,
pt.size = 1.0,
enforce_symmetry = FALSE,
legend.title = "Consensus",
plot.title = "Consensus Proportion",
sequential.palette = "YlGnBu", # Yellow-Green-Blue gradient, following Nature Methods standards
sequential.direction = 1, # Light to dark direction
min.cutoff = min(pbmc$consensus_proportion), # Set minimum value
max.cutoff = max(pbmc$consensus_proportion), # Set maximum value
na.value = "lightgrey") + # Color for missing values
theme(plot.title = element_text(hjust = 0.5, margin = margin(b = 15, t = 10)),
plot.margin = unit(c(0.8, 0.8, 0.8, 0.8), "cm"))
# Shannon entropy feature plot with SCpubr
p3 <- SCpubr::do_FeaturePlot(sample = pbmc,
features = "entropy",
order = TRUE,
pt.size = 1.0,
enforce_symmetry = FALSE,
legend.title = "Entropy",
plot.title = "Shannon Entropy",
sequential.palette = "OrRd", # Orange-Red gradient, following Nature Methods standards
sequential.direction = -1, # Dark to light direction (reversed)
min.cutoff = min(pbmc$entropy), # Set minimum value
max.cutoff = max(pbmc$entropy), # Set maximum value
na.value = "lightgrey") + # Color for missing values
theme(plot.title = element_text(hjust = 0.5, margin = margin(b = 15, t = 10)),
plot.margin = unit(c(0.8, 0.8, 0.8, 0.8), "cm"))
# Combine plots with equal widths
pdf("pbmc_uncertainty_metrics.pdf", width=18, height=7)
combined_plot <- cowplot::plot_grid(p1, p2, p3, ncol = 3, rel_widths = c(1.2, 1.2, 1.2))
print(combined_plot)
dev.off()
```
#### Using CSV Input
You can also use mLLMCelltype with CSV files directly without Seurat, which is useful for cases where you already have marker genes available in CSV format:
```r
# Install the latest version of mLLMCelltype
devtools::install_github("cafferychen777/mLLMCelltype", subdir = "R", force = TRUE)
# Load necessary packages
library(mLLMCelltype)
# Configure unified logging (optional - uses defaults if not specified)
configure_logger(level = "INFO", console_output = TRUE, json_format = TRUE)
# Create cache directory
cache_dir <- "path/to/your/cache"
dir.create(cache_dir, showWarnings = FALSE, recursive = TRUE)
# Read CSV file content
markers_file <- "path/to/your/markers.csv"
file_content <- readLines(markers_file)
# Skip header row
data_lines <- file_content[-1]
# Convert data to list format, using numeric indices as keys
marker_genes_list <- list()
cluster_names <- c()
# First collect all cluster names
for(line in data_lines) {
parts <- strsplit(line, ",", fixed = TRUE)[[1]]
cluster_names <- c(cluster_names, parts[1])
}
# Then create marker_genes_list with numeric indices
for(i in 1:length(data_lines)) {
line <- data_lines[i]
parts <- strsplit(line, ",", fixed = TRUE)[[1]]
# First part is the cluster name
cluster_name <- parts[1]
# Use the original cluster ID as key (preserve input IDs as-is)
cluster_id <- as.character(cluster_name)
# Remaining parts are genes
genes <- parts[-1]
# Filter out NA and empty strings
genes <- genes[!is.na(genes) & genes != ""]
# Add to marker_genes_list
marker_genes_list[[cluster_id]] <- list(genes = genes)
}
# Set API keys
api_keys <- list(
gemini = "YOUR_GEMINI_API_KEY",
qwen = "YOUR_QWEN_API_KEY",
grok = "YOUR_GROK_API_KEY",
openai = "YOUR_OPENAI_API_KEY",
anthropic = "YOUR_ANTHROPIC_API_KEY"
)
# Run consensus annotation with paid models
consensus_results <-
interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "your tissue type", # e.g., "human heart"
models = c("gemini-3.1-pro-preview",
"gemini-3-flash-preview",
"qwen3.6-plus",
"grok-4.3",
"claude-sonnet-4-6",
"gpt-5.5"),
api_keys = api_keys,
controversy_threshold = 0.6,
entropy_threshold = 1.0,
max_discussion_rounds = 3,
cache_dir = cache_dir
)
# Alternatively, use free OpenRouter models (no credits required)
# Add OpenRouter API key to the api_keys list
api_keys$openrouter <- "your-openrouter-api-key"
# Run consensus annotation with free models
free_consensus_results <-
interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "your tissue type", # e.g., "human heart"
models = c(
"meta-llama/llama-4-maverick:free", # Meta Llama 4 Maverick (free)
"venice/uncensored:free", # Venice Uncensored (free)
"deepseek/deepseek-v4-pro:free", # DeepSeek V4 Pro (free)
"meta-llama/llama-3.3-70b-instruct:free" # Meta Llama 3.3 70B (free)
),
api_keys = api_keys,
consensus_check_model = "deepseek/deepseek-v4-pro:free", # Free model for consensus checking
controversy_threshold = 0.6,
entropy_threshold = 1.0,
max_discussion_rounds = 2,
cache_dir = cache_dir
)
# Save results
saveRDS(consensus_results, "your_results.rds")
# Print results summary
cat("\nResults summary:\n")
cat("Available fields:", paste(names(consensus_results), collapse=", "), "\n\n")
# Print final annotations
cat("Final cell type annotations:\n")
for(cluster in names(consensus_results$final_annotations)) {
cat(sprintf("%s: %s\n", cluster, consensus_results$final_annotations[[cluster]]))
}
```
**Notes on CSV format**:
- The CSV file should have values in the first column that will be used as cluster IDs (these can be cluster names, numbers like 0,1,2,3 or 1,2,3,4, etc.)
- Cluster IDs are included in prompts so returned annotations can be mapped back to the original clusters
- Subsequent columns should contain marker genes for each cluster
- An example CSV file for cat heart tissue is included in the package at `inst/extdata/Cat_Heart_markers.csv`
Example CSV structure:
```
cluster,gene
0,Negr1,Cask,Tshz2,Ston2,Fstl1,Dse,Celf2,Hmcn2,Setbp1,Cblb
1,Palld,Grb14,Mybpc3,Ensfcag00000044939,Dcun1d2,Acacb,Slco1c1,Ppp1r3c,Sema3c,Ppp1r14c
2,Adgrf5,Tbx1,Slco2b1,Pi15,Adam23,Bmx,Pde8b,Pkhd1l1,Dtx1,Ensfcag00000051556
3,Clec2d,Trat1,Rasgrp1,Card11,Cytip,Sytl3,Tmem156,Bcl11b,Lcp1,Lcp2
```
You can access the example data in your R script using:
```r
system.file("extdata", "Cat_Heart_markers.csv", package = "mLLMCelltype")
```
### Using a Single LLM Model
If you only want to use a single LLM model instead of the consensus approach, use the `annotate_cell_types()` function. This is useful when you have access to only one API key or prefer a specific model:
```r
# Load required packages
library(mLLMCelltype)
library(Seurat)
# Load your preprocessed Seurat object
pbmc <- readRDS("your_seurat_object.rds")
# Find marker genes for each cluster
pbmc_markers <- FindAllMarkers(pbmc,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
# Choose a model from any supported provider
# Supported models include:
# - OpenAI: 'gpt-5.5', 'gpt-5.4', 'gpt-5.4-mini'
# - Anthropic: 'claude-opus-4-7', 'claude-sonnet-4-6', 'claude-haiku-4-5-20251001'
# - DeepSeek: 'deepseek-v4-flash', 'deepseek-v4-pro'
# - Google: 'gemini-3.1-pro-preview', 'gemini-3-flash-preview', 'gemini-3.1-flash-lite'
# - Qwen: 'qwen3.6-plus', 'qwen3.6-flash', 'qwen3.6-max-preview'
# - Stepfun: 'step-3.5-flash', 'step-3.5-flash-2603', 'step-3'
# - Zhipu/Z.AI: 'glm-5.1', 'glm-5', 'glm-5-turbo'
# - MiniMax: 'MiniMax-M2.7', 'MiniMax-M2.7-highspeed', 'MiniMax-M2.5'
# - Grok: 'grok-4.3', 'grok-4.3-latest', 'grok-latest'
# - OpenRouter: Access to models from multiple providers through a single API. Format: 'provider/model-name'
# - OpenAI models: 'openai/gpt-5.5', 'openai/gpt-5.4-mini'
# - Anthropic models: 'anthropic/claude-opus-4.7', 'anthropic/claude-sonnet-4.6'
# - Google models: 'google/gemini-3.1-pro-preview', 'google/gemini-3-flash-preview'
# - X.AI models: 'x-ai/grok-4.3'
# - Stepfun models: 'stepfun/step-3.5-flash'
# Run cell type annotation with a single LLM model
single_model_results <- annotate_cell_types(
input = pbmc_markers,
tissue_name = "human PBMC", # provide tissue context
model = "claude-sonnet-4-6", # specify a single model
api_key = "your-anthropic-key", # provide the API key directly
top_gene_count = 10
)
# Using a free OpenRouter model
free_model_results <- annotate_cell_types(
input = pbmc_markers,
tissue_name = "human PBMC",
model = "meta-llama/llama-4-maverick:free", # free model with :free suffix
api_key = "your-openrouter-key",
top_gene_count = 10
)
# Inspect the provider response lines. Parse or review this text before
# mapping annotations back onto a Seurat object.
cat(single_model_results)
```
#### Comparing Different Models
You can also compare annotations from different models by running `annotate_cell_types()` multiple times with different models:
```r
# Define models to test
models_to_test <- c(
"claude-sonnet-4-6", # Anthropic
"gpt-5.5", # OpenAI
"gemini-3.1-pro-preview", # Google
"qwen3.6-plus" # Alibaba
)
# API keys for different providers
api_keys <- list(
anthropic = "your-anthropic-key",
openai = "your-openai-key",
gemini = "your-gemini-key",
qwen = "your-qwen-key"
)
# Test each model and store results
results <- list()
for (model in models_to_test) {
provider <- get_provider(model)
api_key <- api_keys[[provider]]
# Run annotation
results[[model]] <- annotate_cell_types(
input = pbmc_markers,
tissue_name = "human PBMC",
model = model,
api_key = api_key,
top_gene_count = 10
)
# Store the provider response lines for review or downstream parsing.
message(sprintf("%s response:\n%s", model, results[[model]]))
}
```
### Advanced Consensus Configuration: Specifying the Consensus Check Model
The `consensus_check_model` parameter (R) / `consensus_model` parameter (Python) allows you to specify which LLM model to use for consensus checking and discussion moderation. This parameter is important for the accuracy of consensus annotation because the consensus check model:
1. Evaluates semantic similarity between different cell type annotations
2. Calculates consensus metrics (proportion and entropy)
3. Moderates and synthesizes discussions between models for controversial clusters
4. Makes final decisions when models disagree
We recommend using a capable model for consensus checking, as this directly impacts annotation quality.
#### Recommended Models for Consensus Checking
- **Anthropic**: `claude-opus-4-7`, `claude-sonnet-4-6`
- **OpenAI**: `o1`, `o1-pro`, `gpt-5.5`, `gpt-4.1`
- **Google**: `gemini-3.1-pro-preview`, `gemini-3-flash-preview`
- **Other**: `deepseek-v4-pro` / `deepseek-v4-pro`, `qwen3.6-plus`, `grok-4.3`
#### R Package Usage
```r
# Example 1: Specifying a consensus check model
consensus_results <- interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "human brain",
models = c("gpt-5.5", "claude-sonnet-4-6", "gemini-3.1-pro-preview", "qwen3.6-plus"),
api_keys = api_keys,
consensus_check_model = "claude-sonnet-4-6",
controversy_threshold = 0.7,
entropy_threshold = 1.0
)
# Example 2: Using an alternative consensus check model
consensus_results <- interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "mouse liver",
models = c("gpt-5.5", "gemini-3.1-pro-preview", "qwen3.6-plus"),
api_keys = api_keys,
consensus_check_model = "claude-sonnet-4-6",
controversy_threshold = 0.7,
entropy_threshold = 1.0
)
# Example 3: Using OpenAI's reasoning model
consensus_results <- interactive_consensus_annotation(
input = marker_genes_list,
tissue_name = "human immune cells",
models = c("gpt-5.5", "claude-sonnet-4-6", "gemini-3.1-pro-preview"),
api_keys = api_keys,
consensus_check_model = "o1",
controversy_threshold = 0.7,
entropy_threshold = 1.0
)
```
#### Python Package Usage
```python
# Example 1: Specifying a consensus model
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="human",
tissue="brain",
models=["gpt-5.5", "claude-sonnet-4-6", "gemini-3.1-pro-preview", "qwen3.6-plus"],
consensus_model="claude-sonnet-4-6",
consensus_threshold=0.7,
entropy_threshold=1.0
)
# Example 2: Using dictionary format
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="mouse",
tissue="liver",
models=["gpt-5.5", "gemini-3.1-pro-preview", "qwen3.6-plus"],
consensus_model={"provider": "anthropic", "model": "claude-sonnet-4-6"},
consensus_threshold=0.7,
entropy_threshold=1.0
)
# Example 3: Using Google's model for consensus
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="human",
tissue="heart",
models=["gpt-5.5", "claude-sonnet-4-6", "qwen3.6-plus"],
consensus_model={"provider": "google", "model": "gemini-3.1-pro-preview"},
consensus_threshold=0.7,
entropy_threshold=1.0
)
# Example 4: Default behavior (auto-selects from available API keys)
consensus_results = interactive_consensus_annotation(
marker_genes=marker_genes,
species="human",
tissue="blood",
models=["gpt-5.5", "claude-sonnet-4-6", "gemini-3.1-pro-preview"],
# If not specified, consensus_model is selected from available api_keys
consensus_threshold=0.7,
entropy_threshold=1.0
)
```
#### Notes on Consensus Model Selection
1. **Model Availability**: Ensure you have API access to your chosen consensus model. The system will use fallback models if the primary choice is unavailable.
2. **Consistency**: Use the same model for all consensus checks within a project to ensure consistent evaluation criteria.
3. **Default Behavior**:
- R: Uses the first model that succeeds during initial annotation if not specified
- Python: Selects the consensus checker from providers available in `api_keys`; pass `consensus_model` explicitly for reproducible runs
The consensus check model must accurately assess semantic similarity between different cell type names (e.g., recognizing that "T lymphocyte" and "T cell" refer to the same cell type), understand biological context, and synthesize discussions from multiple models.
### Advanced Features: Cluster Selection and Cache Control (v1.3.1)
mLLMCelltype v1.3.1 introduces two parameters that give you fine-grained control over the annotation process:
#### 1. **clusters_to_analyze** - Selective Cluster Analysis
This parameter allows you to specify exactly which clusters to analyze without manually filtering your input data:
```r
# Example: Focus on specific clusters for T cell subtyping
consensus_results <- interactive_consensus_annotation(
input = pbmc_markers,
tissue_name = "human PBMC - T cell subtypes",
models = c("gpt-5.5", "claude-sonnet-4-6"),
api_keys = api_keys,
clusters_to_analyze = c(0, 1, 7), # Only analyze T cell clusters
controversy_threshold = 0.7
)
# Example: Re-analyze controversial clusters with different context
consensus_results <- interactive_consensus_annotation(
input = pbmc_markers,
tissue_name = "activated immune cells",
models = c("gpt-5.5", "claude-sonnet-4-6", "gemini-3.1-pro-preview"),
api_keys = api_keys,
clusters_to_analyze = c("3", "5"), # Focus on specific clusters
cache_dir = "consensus_cache"
)
```
**Benefits:**
- No need to subset your data manually
- Maintains original cluster numbering
- Reduces API calls and costs by only analyzing relevant clusters
- Useful for iterative refinement of specific cell populations
#### 2. **force_rerun** - Bypass Cache for Fresh Analysis
This parameter forces re-analysis of controversial clusters, bypassing cached results:
```r
# Example: Initial broad analysis
initial_results <- interactive_consensus_annotation(
input = markers,
tissue_name = "human brain",
models = c("gpt-5.5", "claude-sonnet-4-6"),
api_keys = api_keys,
use_cache = TRUE
)
# Example: Re-analyze with specific subtype context
subtype_results <- interactive_consensus_annotation(
input = markers,
tissue_name = "human brain - neuronal subtypes",
models = c("gpt-5.5", "claude-sonnet-4-6"),
api_keys = api_keys,
clusters_to_analyze = c(2, 3, 5), # Neuronal clusters
force_rerun = TRUE, # Force fresh analysis despite cache
use_cache = TRUE # Still benefit from cache for non-controversial clusters
)
```
**Important Notes:**
- `force_rerun` only affects controversial clusters requiring LLM discussion
- Non-controversial clusters still use cache for performance
- Useful when changing tissue context or focusing on subtypes
- Combines well with `clusters_to_analyze` for targeted re-analysis
#### Common Use Cases
1. **Iterative Subtyping Workflow:**
```r
# Step 1: General cell type annotation
general_types <- interactive_consensus_annotation(
input = data,
tissue_name = "human PBMC",
models = models,
api_keys = api_keys
)
# Step 2: Focus on T cells with subtype context
t_cell_subtypes <- interactive_consensus_annotation(
input = data,
tissue_name = "human T lymphocytes",
models = models,
api_keys = api_keys,
clusters_to_analyze = c(0, 1, 4, 7), # T cell clusters from step 1
force_rerun = TRUE # Fresh analysis with T cell context
)
# Step 3: Further refine CD8+ T cells
cd8_subtypes <- interactive_consensus_annotation(
input = data,
tissue_name = "human CD8+ T cells - activation states",
models = models,
api_keys = api_keys,
clusters_to_analyze = c(1, 4), # CD8+ clusters
force_rerun = TRUE
)
```
2. **Cost-Effective Re-analysis:**
```r
# Only re-analyze clusters that were controversial
controversial <- initial_results$controversial_clusters
refined_results <- interactive_consensus_annotation(
input = data,
tissue_name = "human PBMC - refined",
models = c("gpt-5.5", "claude-sonnet-4-6", "gemini-3.1-pro-preview"),
api_keys = api_keys,
clusters_to_analyze = controversial, # Only controversial ones
force_rerun = TRUE,
consensus_check_model = "claude-sonnet-4-6"
)
```
## Visualization Examples
### Cell Type Annotation Visualization
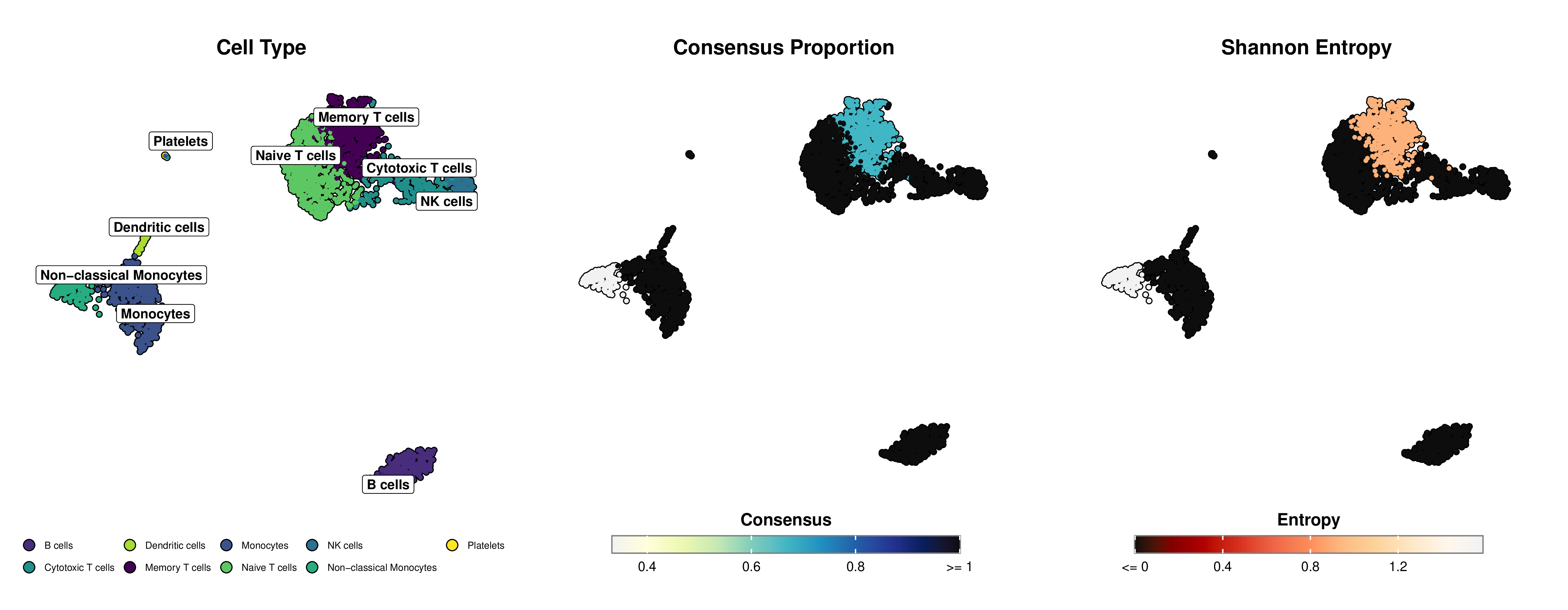
Below is an example of publication-ready visualization created with mLLMCelltype and SCpubr, showing cell type annotations alongside uncertainty metrics (Consensus Proportion and Shannon Entropy):
*Figure: Left panel shows cell type annotations on UMAP projection. Middle panel displays the consensus proportion using a yellow-green-blue gradient (deeper blue indicates stronger agreement among LLMs). Right panel shows Shannon entropy using an orange-red gradient (deeper red indicates lower uncertainty, lighter orange indicates higher uncertainty).*
### Marker Gene Visualization
mLLMCelltype includes marker gene visualization functions that integrate with the consensus annotation workflow:
```r
# Load required libraries
library(mLLMCelltype)
library(Seurat)
library(ggplot2)
# After running consensus annotation
consensus_results <- interactive_consensus_annotation(
input = markers_df,
tissue_name = "human PBMC",
models = c("anthropic/claude-sonnet-4.6", "openai/gpt-5.5"),
api_keys = list(openrouter = "your_api_key")
)
# Create marker gene visualizations using Seurat
# Add consensus annotations to Seurat object
cluster_ids <- as.character(Idents(pbmc_data))
cell_type_annotations <- consensus_results$final_annotations[cluster_ids]
# Handle any missing annotations
if (any(is.na(cell_type_annotations))) {
na_mask <- is.na(cell_type_annotations)
cell_type_annotations[na_mask] <- paste("Cluster", cluster_ids[na_mask])
}
# Add to Seurat object
pbmc_data@meta.data$cell_type_consensus <- cell_type_annotations
# Create a dotplot of marker genes
DotPlot(pbmc_data,
features = top_markers,
group.by = "cell_type_consensus") +
RotatedAxis()
# Create a heatmap of marker genes
DoHeatmap(pbmc_data,
features = top_markers,
group.by = "cell_type_consensus")
```
**Marker Gene Visualization Features:**
- **DotPlot**: Shows both percentage of cells expressing each gene (dot size) and average expression level (color intensity)
- **Heatmap**: Displays scaled expression values with clustering of genes and cell types
- **Integration**: Works directly with consensus annotation results added to Seurat objects
- **Standard Seurat Functions**: Uses familiar Seurat visualization functions for consistency
For detailed instructions and advanced customization options, see the [Visualization Guide](https://cafferyang.com/mLLMCelltype/articles/visualization-guide.html).
## Citation
If you use mLLMCelltype in your research, please cite:
```bibtex
@article{yang2026llmconsensus,
author = {Yang, Chen and Zhang, Xianyang and Chen, Jun},
title = {Large language model consensus substantially improves the cell type annotation accuracy for scRNA-seq data},
journal = {Communications Biology},
year = {2026},
doi = {10.1038/s42003-026-10420-8},
url = {https://doi.org/10.1038/s42003-026-10420-8},
publisher = {Springer Nature}
}
```
You can also cite this in plain text format:
Yang, C., Zhang, X., & Chen, J. (2026). Large language model consensus substantially improves the cell type annotation accuracy for scRNA-seq data. *Communications Biology*. [Read the paper](https://doi.org/10.1038/s42003-026-10420-8)
## Contributing
We welcome contributions from the community. There are many ways you can contribute to mLLMCelltype:
### Reporting Issues
If you encounter any bugs, have feature requests, or have questions about using mLLMCelltype, please [open an issue](https://github.com/cafferychen777/mLLMCelltype/issues) on our GitHub repository. When reporting bugs, please include:
- A clear description of the problem
- Steps to reproduce the issue
- Expected vs. actual behavior
- Your operating system and package version information
- Any relevant code snippets or error messages
### Pull Requests
We encourage you to contribute code improvements or new features through pull requests:
1. Fork the repository
2. Create a new branch for your feature (`git checkout -b feature/amazing-feature`)
3. Commit your changes (`git commit -m 'Add some amazing feature'`)
4. Push to the branch (`git push origin feature/amazing-feature`)
5. Open a Pull Request
### Areas for Contribution
Here are some areas where contributions would be particularly valuable:
- Adding support for new LLM models
- Improving documentation and examples
- Optimizing performance
- Adding new visualization options
- Extending functionality for specialized cell types or tissues
- Translations of documentation into different languages
### Code Style
Please follow the existing code style in the repository. For R code, we generally follow the [tidyverse style guide](https://style.tidyverse.org/). For Python code, we follow [PEP 8](https://www.python.org/dev/peps/pep-0008/).
### Community
Join our [Discord community](https://discord.gg/pb2aZdG4) for discussion and questions about mLLMCelltype and single-cell RNA-seq analysis.
Thank you for helping improve mLLMCelltype!