{
"metadata": {
"name": "",
"signature": "sha256:e05e3dfc60d83e593fee372dc9629cd81d5978c004d6a6662e5e30c865ad41ca"
},
"nbformat": 3,
"nbformat_minor": 0,
"worksheets": [
{
"cells": [
{
"cell_type": "markdown",
"metadata": {},
"source": [
"This example is taken from the YAHMM wiki on silent states and edited."
]
},
{
"cell_type": "heading",
"level": 1,
"metadata": {},
"source": [
"Global Sequence Alignment using YAHMM"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Global sequence alignment is a problem in bioinformatics which involves aligning two sequences against each other in the maximally likely way. Lets say that you have the two sequences, ACT and ACG, and you wish to align them. The most obvious alignment is:\n",
"\n",
"```\n",
"ACT\n",
"||\n",
"ACG\n",
"```\n",
"It's a close match, but one sequence has a G and one has a T at the end. You know from biology that nucleotides can mutate over time, be removed, or be added. In this case, you have to weigh the two hypotheses of 'did this nucleotide mutate' vs 'was a nucleotide removed and then another one added later'. Almost always the nucleotide mutation hypothesis wins out.\n",
"\n",
"\n",
"Picture from Biological Sequence Analysis: Probabilistic Methods of Proteins and Nucleic Acids by Durbin, Eddy, Krogh, and Mitchison
\n",
"\n",
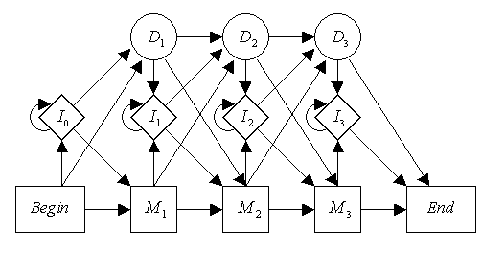
"This alignment can be done through the use of a profile HMM, pictured above. It contains matches along the bottom, inserts in the middle, and deletes along the top. The deletes are silent states, which are almost always pictured as circles in cartoons. In this example, the profile has M1='A', M2='C', and M3='T', with some pseudocounts for the other possibilities, and the sequence we wish to align is 'ACG'. The most likely path would be to follow the match states through, even though the 'G' does not align to the 'T' at M3.\n",
"\n",
"Lets look at another sequence alignment.\n",
"```\n",
"ACGTTGCGACGACAAAACT\n",
"||||| | ||\n",
"ACGTTCC----------CT\n",
"```\n",
"The bottom sequence seems to be missing a chunk. This is a common phenomena, and can be related to a protein losing an entire domain over a long stretch of time for some evolutionary reason. If the top sequence was the profile, and the bottom sequence was being aligned to the model, we couldn't take the match states through. After reaching the aligning the seventh nucleotide ('C') to a match state, there's a long stretch to get to the next match state. \n",
"\n",
"This is where silent states play a role. You don't need to emit a character to travel along them, though you do have to suffer the transition probability cost. We can skip from what would be M7 to D8, all the way down to D17, then transition back to M18 to align to the 'C' and 'T' at the end of the model, all without emitting a character while going along delete states. This is an extremely useful part of hidden Markov models, and not all packages contain them. \n",
"\n",
"We can see that the deletes allow for transitions from the beginning to the very end, without having to align to more than one character generating state, the first insert state. This allows the profile in the HMM to have sequences of variable length align to it, without cluttering the model with edges from every insert and match to every future insert and match. Lets create this very example.\n",
"\n",
"Insertions will be modelled as symbol-emitting states with uniform probabilities for each nucleotide. We believe that the probability of an insertion into the actual sequence does not depend on the position, and so the underlying distribution behind each of these states should be tied. This means that when these states are trained, they are all considered a single distribution, and will all have the same distribution post-training.\n",
"\n",
"Doing this by hand is a lot of edges and transitions. Typically, you can use a loop to build a model like this. An example is at https://github.com/jmschrei/PyPore/blob/master/alignment.py, under ProfileAligner._build_global(), which builds a global alignment HMM for Gaussian emissions, but can easily be changed to represent discrete characters by changing the GaussianKernelDensity distribution to DiscreteDistribution."
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Now, lets build this model using YAHMM. We start off by creating the Model instance. "
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"from yahmm import *\n",
"model = Model( name=\"Global Sequence Aligner\" )"
],
"language": "python",
"metadata": {},
"outputs": [],
"prompt_number": 1
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"The graphical structure of the model is now defined node by node, and edge by edge. This allows for complicated structures to be made easily, even if they do require a few more lines of code. "
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"# Define the distribution for insertions\n",
"i_d = DiscreteDistribution( { 'A': 0.25, 'C': 0.25, 'G': 0.25, 'T': 0.25 } )\n",
"\n",
"# Create the insert states\n",
"i0 = State( i_d, name=\"I0\" )\n",
"i1 = State( i_d, name=\"I1\" )\n",
"i2 = State( i_d, name=\"I2\" )\n",
"i3 = State( i_d, name=\"I3\" )\n",
"\n",
"# Create the match states\n",
"m1 = State( DiscreteDistribution({ \"A\": 0.95, 'C': 0.01, 'G': 0.01, 'T': 0.02 }) , name=\"M1\" )\n",
"m2 = State( DiscreteDistribution({ \"A\": 0.003, 'C': 0.99, 'G': 0.003, 'T': 0.004 }) , name=\"M2\" )\n",
"m3 = State( DiscreteDistribution({ \"A\": 0.01, 'C': 0.01, 'G': 0.01, 'T': 0.97 }) , name=\"M3\" )\n",
"\n",
"# Create the delete states\n",
"d1 = State( None, name=\"D1\" )\n",
"d2 = State( None, name=\"D2\" )\n",
"d3 = State( None, name=\"D3\" )\n",
"\n",
"# Add all the states to the model\n",
"model.add_states( [i0, i1, i2, i3, m1, m2, m3, d1, d2, d3 ] )\n",
"\n",
"# Create transitions from match states\n",
"model.add_transition( model.start, m1, 0.9 )\n",
"model.add_transition( model.start, i0, 0.1 )\n",
"model.add_transition( m1, m2, 0.9 )\n",
"model.add_transition( m1, i1, 0.05 )\n",
"model.add_transition( m1, d2, 0.05 )\n",
"model.add_transition( m2, m3, 0.9 )\n",
"model.add_transition( m2, i2, 0.05 )\n",
"model.add_transition( m2, d3, 0.05 )\n",
"model.add_transition( m3, model.end, 0.9 )\n",
"model.add_transition( m3, i3, 0.1 )\n",
"\n",
"# Create transitions from insert states\n",
"model.add_transition( i0, i0, 0.70 )\n",
"model.add_transition( i0, d1, 0.15 )\n",
"model.add_transition( i0, m1, 0.15 )\n",
"\n",
"model.add_transition( i1, i1, 0.70 )\n",
"model.add_transition( i1, d2, 0.15 )\n",
"model.add_transition( i1, m2, 0.15 )\n",
"\n",
"model.add_transition( i2, i2, 0.70 )\n",
"model.add_transition( i2, d3, 0.15 )\n",
"model.add_transition( i2, m3, 0.15 )\n",
"\n",
"model.add_transition( i3, i3, 0.85 )\n",
"model.add_transition( i3, model.end, 0.15 )\n",
"\n",
"# Create transitions from delete states\n",
"model.add_transition( d1, d2, 0.15 )\n",
"model.add_transition( d1, i1, 0.15 )\n",
"model.add_transition( d1, m2, 0.70 ) \n",
"\n",
"model.add_transition( d2, d3, 0.15 )\n",
"model.add_transition( d2, i2, 0.15 )\n",
"model.add_transition( d2, m3, 0.70 )\n",
"\n",
"model.add_transition( d3, i3, 0.30 )\n",
"model.add_transition( d3, model.end, 0.70 )\n",
"\n",
"# Call bake to finalize the structure of the model.\n",
"model.bake()"
],
"language": "python",
"metadata": {},
"outputs": [],
"prompt_number": 2
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"The profile created will be of the 3 nucleotide sequence 'ACT', with pseudocounts at each position to allow for the possibility of small deviations. Lets calculate the viterbi path across a few small sequences. "
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"for sequence in map( list, ('ACT', 'GGC', 'GAT', 'ACC') ):\n",
" logp, path = model.viterbi( sequence )\n",
" print \"Sequence: '{}' -- Log Probability: {} -- Path: {}\".format(\n",
" ''.join( sequence ), logp, \" \".join( state.name for idx, state in path[1:-1] ) )"
],
"language": "python",
"metadata": {},
"outputs": [
{
"output_type": "stream",
"stream": "stdout",
"text": [
"Sequence: 'ACT' -- Log Probability: -0.513244900357 -- Path: M1 M2 M3\n",
"Sequence: 'GGC' -- Log Probability: -11.0481012413 -- Path: I0 I0 D1 M2 D3\n",
"Sequence: 'GAT' -- Log Probability: -9.12551967402 -- Path: I0 M1 D2 M3\n",
"Sequence: 'ACC' -- Log Probability: -5.08795587886 -- Path: M1 M2 M3\n"
]
}
],
"prompt_number": 3
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"The first and last sequence are entirely matches, meaning that it thinks the most likely alignment between the profile ACT and ACT is A-A, C-C, and T-T, which makes sense, and the most likely alignment between ACT and ACC is A-A, C-C, and T-C, which includes a mismatch. Essentially, it's more likely that there's a T-C mismatch at the end then that there was a deletion of a T at the end of the sequence, and a separate insertion of a C.\n",
"\n",
"The two middle sequences don't match very well, as expected! G's are not very likely in this profile at all. It predicts that the two G's are inserts, and that the C matches the C in the profile, before hitting the delete state because it can't emit a T. The third sequence thinks that the G is an insert, as expected, and then aligns the A and T in the sequence to the A and T in the master sequence, missing the middle C in the profile.\n",
"\n",
"By using deletes, we can handle other sequences which are shorter than three characters. Lets look at some more sequences of different lengths. "
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"for sequence in map( list, ('A', 'GA', 'AC', 'AT', 'ATCC', 'ACGTG', 'ATTT', 'TACCCTC', 'TGTCAACACT') ):\n",
" logp, path = model.viterbi( sequence )\n",
" print \"Sequence: '{}' -- Log Probability: {} -- Path: {}\".format(\n",
" ''.join( sequence ), logp, \" \".join( state.name for idx, state in path[1:-1] ) )"
],
"language": "python",
"metadata": {},
"outputs": [
{
"output_type": "stream",
"stream": "stdout",
"text": [
"Sequence: 'A' -- Log Probability: -5.40618101242 -- Path: M1 D2 D3\n",
"Sequence: 'GA' -- Log Probability: -10.8868199358 -- Path: I0 M1 D2 D3\n",
"Sequence: 'AC' -- Log Probability: -3.62447187905 -- Path: M1 M2 D3\n",
"Sequence: 'AT' -- Log Probability: -3.64488075068 -- Path: M1 D2 M3\n",
"Sequence: 'ATCC' -- Log Probability: -10.6743329646 -- Path: M1 D2 M3 I3 I3\n",
"Sequence: 'ACGTG' -- Log Probability: -10.3938248352 -- Path: M1 M2 I2 I2 I2 D3\n",
"Sequence: 'ATTT' -- Log Probability: -8.67126440175 -- Path: M1 I1 I1 D2 M3\n",
"Sequence: 'TACCCTC' -- Log Probability: -16.9034517961 -- Path: I0 I0 I0 I0 D1 M2 M3 I3\n",
"Sequence: 'TGTCAACACT' -- Log Probability: -16.4516996541 -- Path: I0 I0 I0 I0 I0 I0 I0 M1 M2 M3\n"
]
}
],
"prompt_number": 4
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Again, more of the same expected. You'll notice most of the use of insertion states are at I0, because most of the insertions are at the beginning of the sequence. It's more probable to simply stay in I0 at the beginning instead of go from I0 to D1 to I1, or going to another insert state along there. You'll see other insert states used when insertions occur in other places in the sequence, like 'ATTT' and 'ACGTG'. "
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Now that we have the path, we need to convert it into an alignment, which is significantly more informative to look at."
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"def path_to_alignment( x, y, path ):\n",
" \"\"\"\n",
" This function will take in two sequences, and the ML path which is their alignment,\n",
" and insert dashes appropriately to make them appear aligned. This consists only of\n",
" adding a dash to the model sequence for every insert in the path appropriately, and\n",
" a dash in the observed sequence for every delete in the path appropriately.\n",
" \"\"\"\n",
" \n",
" for i, (index, state) in enumerate( path[1:-1] ):\n",
" name = state.name\n",
" \n",
" if name.startswith( 'D' ):\n",
" y = y[:i] + '-' + y[i:]\n",
" elif name.startswith( 'I' ):\n",
" x = x[:i] + '-' + x[i:]\n",
"\n",
" return x, y\n",
"\n",
"for sequence in map( list, ('A', 'GA', 'AC', 'AT', 'ATCC' ) ):\n",
" logp, path = model.viterbi( sequence )\n",
" x, y = path_to_alignment( 'ACT', ''.join(sequence), path )\n",
" \n",
" print \"Sequence: {}, Log Probability: {}\".format( ''.join(sequence), logp )\n",
" print \"{}\\n{}\".format( x, y )\n",
" print"
],
"language": "python",
"metadata": {},
"outputs": [
{
"output_type": "stream",
"stream": "stdout",
"text": [
"Sequence: A, Log Probability: -5.40618101242\n",
"ACT\n",
"A--\n",
"\n",
"Sequence: GA, Log Probability: -10.8868199358\n",
"-ACT\n",
"GA--\n",
"\n",
"Sequence: AC, Log Probability: -3.62447187905\n",
"ACT\n",
"AC-\n",
"\n",
"Sequence: AT, Log Probability: -3.64488075068\n",
"ACT\n",
"A-T\n",
"\n",
"Sequence: ATCC, Log Probability: -10.6743329646\n",
"ACT--\n",
"A-TCC\n",
"\n"
]
}
],
"prompt_number": 5
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"In addition to getting this alignment, we can do some interesting things with this model! Lets score every sequence of length 5 of less and see what the distribution looks like."
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"import itertools\n",
"\n",
"sequences = reduce( lambda x, y: x+y, [[ seq for seq in it.product( 'ACGT', repeat=i ) ] for i in xrange( 1,6 )] )\n",
"scores = map( model.log_probability, sequences )\n"
],
"language": "python",
"metadata": {},
"outputs": [],
"prompt_number": 6
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Now lets plot a kernel density of that distribution."
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"import seaborn as sns\n",
"%pylab inline\n",
"\n",
"plt.figure( figsize=(10,5) )\n",
"sns.kdeplot( numpy.array( scores ), shade=True )\n",
"plt.ylabel('Density')\n",
"plt.xlabel('Log Probability')\n",
"plt.show()"
],
"language": "python",
"metadata": {},
"outputs": [
{
"output_type": "stream",
"stream": "stdout",
"text": [
"Populating the interactive namespace from numpy and matplotlib\n"
]
},
{
"output_type": "stream",
"stream": "stderr",
"text": [
"WARNING: pylab import has clobbered these variables: ['log', 'i0', 'random', 'exp']\n",
"`%matplotlib` prevents importing * from pylab and numpy\n"
]
},
{
"metadata": {},
"output_type": "display_data",
"png": "iVBORw0KGgoAAAANSUhEUgAAAmcAAAFICAYAAAAVoFlvAAAABHNCSVQICAgIfAhkiAAAAAlwSFlz\nAAALEgAACxIB0t1+/AAAIABJREFUeJzt3Xl41PW99//n7Flmsm+EbOygKLJ46kbdatVaj1KW4vFY\ne8pRzzlXa69WbfV3zq3YlqXH9nSVq/fxvnus9G7xqHjaUhVEKbRUQEB2CGs2CEkI2SaTZLbv74+Q\nAEoWIDPfmcnrcV1cSeY735l3+GYyr3xWi2EYBiIiIiISE6xmFyAiIiIiZymciYiIiMQQhTMRERGR\nGKJwJiIiIhJDFM5EREREYojCmYiIiEgMsUfqgcPhMAsXLuTgwYM4HA4WLVpESUlJ7/FVq1bxyiuv\nYLPZGD9+PAsXLsRisTBr1izcbjcAxcXFLF68OFIlioiIiMSciIWztWvXEggEWLFiBTt37mTp0qUs\nW7YMgM7OTn7yk5+watUqXC4XTzzxBOvWrePGG28EYPny5ZEqS0RERCSmRaxbc/v27cycOROAKVOm\nsGfPnt5jLpeLV199FZfLBUAwGCQpKYkDBw7Q0dHBggULePjhh9m5c2ekyhMRERGJSRFrOfN6vb3d\nkwA2m41wOIzVasVisZCVlQV0t5J1dHRwww03cPDgQRYsWMDcuXOpqKjgkUceYfXq1VitGhonIiIi\nw0PEwpnb7aa9vb33655gdu7XL7zwApWVlfzsZz8DoKysjNLS0t7PMzIyaGhoID8/v8/nMQwDi8US\noe9CREREJLoiFs6mTZvGunXruPvuu9mxYwcTJkw47/izzz6Ly+XixRdf7A1XK1eupLy8nOeee466\nujq8Xi+5ubn9Po/FYqGhoS1S34ZEWG6uR9cvTunaxTddv/ilaxffcnM9A97HEqmNzw3DYOHChZSX\nlwOwZMkS9u7di8/nY/LkycyePZsZM2b03v/hhx/mlltu4ZlnnuHEiRMAPPXUU1xzzTUDPpd+SOOX\nfsnEL127+KbrF7907eKbqeEsmvRDGr/0SyZ+6drFN12/+KVrF98GE8400l5EREQkhiiciYiIiMQQ\nhTMRERGRGKJwJiIiIhJDFM5EREREYojCmYiIiEgMUTgTERERiSEKZyIiIiIxROFMREREJIYonImI\niIjEEIUzERERkRiicCYiIiISQxTORERERGKIwpmIiIhIDFE4ExEREYkhCmciIiIiMUThTCQGhcJh\ngqGw2WWIiIgJ7GYXICLnC4cNfrhiB1V1XubfPo4bryrAYrGYXZaIiESJWs5EYszqD6s4UNWMryvI\nL9/azw9f3cGp5g6zyxIRkShROBOJIcdPtfPmhqOkJNl5+M4JjBrhYV9FE9/51Va6/CGzyxMRkShQ\nOBOJEaFwmP+7ah/BkMGd1xaTn5XCnJvHMH18Lt6OADuPnDK7RBERiQKFM5EY8famKipOtnFlWSbj\nijIAsFgsTBmbDcDmfXVmliciIlGicCYSA4KhMKu3VJHssnP79KLzjuWkJ5OdnsSuI434OoMmVSgi\nItGicCYSA8qrm2nvDDKpJIMk5ycnUU8qySQUNvjoUIMJ1YmISDQpnInEgO3l3aFrXHHGBY9PLO2+\nfct+dW2KiCQ6hTMRk4UNg20HG0hy2ijOdV/wPlmeJPIzk9lb0YS3IxDlCkVEJJoUzkRMdvR4K63t\nfsYVZWC19r3Y7MTSTMJhg23l9VGsTkREok3hTMRk2w52h63xxen93m9iSSYAm9W1KSKS0BTORExk\nGAZbDzTgtFspzff0e9/0VCeF2SmUVzara1NEJIEpnImYqKrOS2NrJ2NGpmO3DfxyLC3wYAAVJ1sj\nX5yIiJhC4UzERNsOds/SHN/HLM2PK8hKAaCiti1iNYmIiLkUzkRMtP1gPTabhVEj+u/S7JGf2R3O\nKk8qnImIJCqFMxGTtLT7OXHKR3GuG6fdNqhzPCkOUlx2jtWqW1NEJFEpnImYpLyqCYCS/AuvbXYh\nFouFgqwUTrd10erzR6o0ERExkcKZiEnKq5sBKM4bXJdmj/wsdW2KiCQyhTMRk5RXNWG3WXsH+Q/W\n2UkB6toUEUlECmciJmj1dY83G5mTiq2fXQEupDecqeVMRCQhKZyJmOBgVXeX5sWMN+vhSXGQmmRX\ny5mISIJSOBMxwdnxZhcfzqC79azJ66elXZMCREQSjcKZiAkOVDZht1kYcZHjzXoU9E4KUOuZiEii\nUTgTiTJvR4Djp9opzE7FNogtmy4kXzsFiIgkLIUzkSg7WN0z3uziltA4lyYFiIgkLoUzkSg7cGbx\n2UsdbwbgTnaQmqydAkREEpHCmUiUlVc1Y7NZGJF9aePNeozISqWl3U+Lt2uIKhMRkVigcCYSRe2d\nAWrqvRRmp2K/xPFmPXIzkgA4cap9KEoTEZEYoXAmEkUHq5sxuLwuzR5Znu5wdvK077IfS0REYofC\nmUgUlfcsPjsU4SzNBUBto8KZiEgiUTgTiaIDVU3YrBZGZKde9mNlpXW3nNU2qltTRCSR2CP1wOFw\nmIULF3Lw4EEcDgeLFi2ipKSk9/iqVat45ZVXsNlsjB8/noULF2IYRr/niMQzX2eQ6novI3NScdgv\n/+8il8NGarKdWnVriogklIi1nK1du5ZAIMCKFSt48sknWbp0ae+xzs5OfvKTn7B8+XJ++9vf4vV6\nWbduXb/niMS7QzXNGAYU5136+mYfl+1J4nRrF12B0JA9poiImCti4Wz79u3MnDkTgClTprBnz57e\nYy6Xi1dffRWXq3vMTDAYxOVy9XuOSLwbyvFmPXq6NuvUeiYikjAiFs68Xi9u99k3IZvNRjgcBsBi\nsZCVlQXA8uXL6ejo4MYbb+z3HJF4d6CqCavVQmHO5Y8369EzKUAzNkVEEkfExpy53W7a288OVA6H\nw1it1vO+fuGFF6isrORnP/vZoM7pS27u0HUTSfQNh+vn6wxQVeelKN9Nbs7QtZyVjEgHjtPaGTLl\n/3E4XLtEpusXv3TtElvEwtm0adNYt24dd999Nzt27GDChAnnHX/22WdxuVy8+OKLWCyWQZ3Tl4YG\n7S8Yr3JzPcPi+u060kjYMCjMSqG5eehauVxn/nY5Ut0U9f/H4XLtEpWuX/zStYtvgwnWEQtnd9xx\nBxs3bmT+/PkALFmyhFWrVuHz+Zg8eTJvvPEGM2bM4Etf+hIADz/88AXPEUkE5dXd+2kO5XgzgLRU\nJzabRbsEiIgkkIiFM4vFwvPPP3/ebaNGjer9fP/+/Rc87+PniCSC8spmrBYozB268WZwZvymx0Xd\naR9hw8B6phVaRETilxahFYkwX2eQipOtFGSl4LTbhvzxs9KS8AfDNLdpA3QRkUSgcCYSYfsqThM2\nYFRhWkQeP7tnpwDN2BQRSQgKZyIRtvPIKQBGj0iPyONnec4sp6E9NkVEEoLCmUgEhQ2DXUcaSUmy\nU5CVHJHn6FmIVuFMRCQxKJyJRFDlyTbafAFGj0jrXTJmqPW0nGkDdBGRxKBwJhJBu440AjAmQuPN\nAJwOG+5kBycUzkREEoLCmUgE7TpyCqsFygoiF84AstNcNHv9dPqDEX0eERGJPIUzkQhpbfdTUdvG\nyFw3LufQL6FxrrMboHdE9HlERCTyFM5EImT30UYMYHQEuzR7ZLq7x53VNyuciYjEO4UzkQiJxniz\nHhlnJgXUN2nGpohIvFM4E4mAYCjMnqONpKU4eheJjaTelrMmtZyJiMQ7hTORCNhW3kCHP8T44oyI\nLaFxrnS3E1C3pohIIlA4E4mA97bVADB1XG5Uns9us+JJcajlTEQkASiciQyxypNtHD7ewqgRHjLP\njAWLhgy3i+a2LgLBUNSeU0REhp7CmcgQe397d6vZtPHRaTXrkeF2YQANzZ1RfV4RERlaCmciQ8jb\nEWDT3jrSU52MHhH5WZrnyvRo3JmISCJQOBMZQn/ZVUsgFGbquJyoTAQ4V4ZmbIqIJASFM5EhEgqH\neX97DXabhatGZ0f9+XvCWYPCmYhIXFM4Exkir607wqmWTiaPyibZZY/685/dJUAL0YqIxDOFM5Eh\nsGnvSdZ8WE1Wmoubryk0pQaX00ayy0adWs5EROKawpnIZaqqa+O/3j6A025l1szRuByR3eS8Pxmp\nLhpbOgmHDdNqEBGRy6NwJnKJgqEwm/fV8dPXdxEIhrnn+rKobNXUn0yPi1DY4HSrltMQEYlX0R8Y\nIxKHWrxd1DS00+bz094Z5HRrJxv3nKS13Q/AzKtHMK4o3eQqz26AXtfcQU5GssnViIjIpVA4E+lD\n5ck2fr/xGEdPtNJyJoSdy+WwMmNCLlPH5UZ1J4D+ZJzZY7OhqQPKzK1FREQujcKZyMcEgmF+v/EY\nb2+qJGyAO9nB2JHp5GUmk5rkIMlpI9lpozAnFaeJ48su5OyMTU0KEBGJVwpnIueob/Lxk9d3Udvo\nIy3FwV2fKqGsILor/V8OLUQrIhL/FM5EzvB1BvnJa7uoPe3jmrE53HxNoakzLy9FSpIdh91KfZPW\nOhMRiVcKZyJAOGzwn3/YS+1pHzMm5HLbtCKzS7okFouFDLeT+uYODMOI+hZSIiJy+bSUhgjwxvoj\n7DrSSFmBh1uuGWl2OZcl0+3CHwj3ziQVEZH4onAmw9628nre3lxFpsfF395YhtUa361NGZoUICIS\n1xTOZFjr6Ary63cPYrNa+MLM0SQ547+nv2etM00KEBGJTwpnMqz9fuMxWrx+PnVFPtnp5q7uP1Qy\nNWNTRCSuKZzJsFXT4OXdD6tJT3XyqUn5ZpczZHoXolW3pohIXFI4k2HJMAx+vaacsAGfmV6Ew544\nLwVPihOr1aKWMxGROJU470giF2HzvjoOVrcwdmQaY0aavyfmULJaLaSnOqnTWmciInFJ4UyGnWAo\nzBvrj2KzWuJ2PbOBZLpdtHcG8XUGzS5FREQuksKZDDvrd5ygsbWTa8bl9C47kWh6Zmxq3JmISPxR\nOJNhpdMf5Pcbj+GwW7n+isSZBPBxPZMC1LUpIhJ/FM5kWHl3aw1tvgDXTswjJclhdjkR09MiqJYz\nEZH4o3Amw4a3I8DbmypJdtm4dmKe2eVEVKYWohURiVsKZzJsvL2pkk5/iOuuKMDlsJldTkSlp/Z0\nayqciYjEG4UzGRZa2v28t60Gd7KDqeNyzC4n4uw2K54UB/UacyYiEncUzmRYeHtTJf5gmOuvzMdu\nGx4/9pluF81eP/5AyOxSRETkIgyPdykZ1pq9XazbfhxPioOrRmebXU7U9C6n0dJpciUiInIxFM4k\n4b21qZJAKMz1VxYMm1YzOGePTY07ExGJK8PnnUqGpaa2Lv700XHSUhxcNSrL7HKiKvPMchr1Wk5D\nRCSuKJxJQnt7UyXBkMH1VxZgG0atZnB2rTNNChARiS/D691KhpVWn5/1O0/gSXEweZi1msHZMWda\n60xEJL7YI/XA4XCYhQsXcvDgQRwOB4sWLaKkpOS8+3R0dPAP//APLF68mNGjRwMwa9Ys3G43AMXF\nxSxevDhSJUqCe29rDYFgmE9fPWLYtZoBuBw2kl12dWuKiMSZiIWztWvXEggEWLFiBTt37mTp0qUs\nW7as9/ju3bt57rnnqK+vx2KxANDV1QXA8uXLI1WWDBMdXUHe21ZDktPGVWOGzwzNj8twO6lr6iAU\nDmOzDr+AKiISjyL223r79u3MnDkTgClTprBnz57zjgcCAZYtW8aoUaN6bztw4AAdHR0sWLCAhx9+\nmJ07d0aqPElw63ecwNcVZMaEPJz2xN4NoD+ZbhfhsMHp1i6zSxERkUGKWMuZ1+vt7Z4EsNlshMNh\nrGf+ep82bdonzklOTmbBggXMnTuXiooKHnnkEVavXt17Tl9ycz1DW7xE1VBfv0AwxLvbqnHardwy\nozihNzgfSEGum32VTXSFI/M60Wsvvun6xS9du8QWsXDmdrtpb2/v/frcYNaXsrIySktLez/PyMig\noaGB/Pz8fs9raGi7/ILFFLm5niG/fht2nqCptYsZE3LxdwbwdwaG9PHjSbKj+zVXfqyRoqzkIX3s\nSFw7iR5dv/ilaxffBhOsI9atOW3aNDZs2ADAjh07mDBhwoDnrFy5kqVLlwJQV1eH1+slNzc3UiVK\nAjIMg7Vbq7Fa4NqJeWaXY7osTxIAJ09rOQ0RkXgRsZazO+64g40bNzJ//nwAlixZwqpVq/D5fMyb\nN++C58yZM4dnnnmGBx98sPecgVrbRM51rLaNmoZ2xhWl40lxml2O6bLSupfTONnYPsA9RUQkVkQs\nnFksFp5//vnzbjt38H+Pc2dm2u12XnjhhUiVJMPAhp3HAZgyJsfkSmKDy2EjNclOrVrORETihpql\nJGF0dAXZtK8OT4qDsgINlu2RlZZEU2sX/kDI7FJERGQQFM4kYWzeX4c/EObqMdlYrRazy4kZWWku\nDKBOOwWIiMQFhTNJGBt2nMBigatHD99FZy9EkwJEROKLwpkkhKq6NipOtjF6RJomAnyMJgWIiMQX\nhTNJCBt2ngDgak0E+AS1nImIxBeFM4l7YcNga3kDyS4bYwrTzC4n5qSnOrFZLdQ2KpyJiMQDhTOJ\nexW1bbS2+xlTmK6JABdgtVrIcDs5edqHYRhmlyMiIgMYMJy99NJLNDQ0RKMWkUuy43D3z+fYkekm\nVxK7stKS6PSHaG33m12KiIgMYMBw1tXVxd///d/zyCOP8PbbbxMIDN99CiU2fXTwFDarRWub9UPj\nzkRE4seA4eyrX/0q77zzDo899hibN2/mvvvu4zvf+Q779++PRn0i/TrV3MHxU+2U5ntwOmxmlxOz\nemZsaqcAEZHYN6gxZ52dndTU1FBdXY3VaiU9PZ1Fixbxgx/8INL1ifRrx+FTAIwtUpdmf7LSzrSc\naVKAiEjMG3BvzSeeeIJNmzbx6U9/mn/+539mxowZAPj9fm666SaefPLJiBcp0pePDnWHM83S7F+W\n58xaZ2o5ExGJeQOGs+uvv57vfOc7pKam9t7m9/txOp2sWrUqosWJ9MfXGaS8upn8zGQtPDuAZJed\nZJeNWi1EKyIS8wbs1nzttdfOC2ahUIjZs2cDkJeXF7nKRAaw51gj4bChLs1ByvIkcaqlk0AwbHYp\nIiLSjz5bzh566CE+/PBDACZOnNh7u81m4/bbb498ZSID2HGmS1NLaAxOVpqL46faqW/uYGRO6sAn\niIiIKfoMZ8uXLwfge9/7Hv/2b/8WtYJEBsMwDPZVNpGaZCcvI9nscuLCuZMCFM5ERGJXn+Fs3bp1\n3HrrrVx55ZX8z//8zyeO33///REtTKQ/DS2dtLb7mVCcgcWiXQEGIye9O5wdb/AyfUKuydWIiEhf\n+gxnu3fv5tZbb2Xz5s0XfPNTOBMzHapuBmBkrlqABisvIwWAqnqvyZWIiEh/+gxnjz/+OABLly7t\nva2trY3a2lrGjx8f+cpE+nH4eAsAI3PcJlcSP9zJdpJddqrq2swuRURE+jGo2ZrPPPMMjY2N3HPP\nPTz++OP86Ec/ikZtIn06VNOMw24lP1PjzQbLYrGQl5HMqZZOfJ3ahk1EJFYNGM5+85vf8O1vf5s/\n/vGP3H777axatYo///nP0ahN5IK8HQFOnPIxIisFq1XjzS5GT5itVtemiEjMGtT2TRkZGaxfv56b\nb74Zu91OV1dXpOsS6dORni7NXHVpXqy8M+FM485ERGLXgOFs7NixPPbYY1RXV3PDDTfw9a9/nauu\nuioatYlcUM94syJNBrhoPeGsuk7hTEQkVg24fdPixYvZsWMH48aNw+l0MmvWLG666aZo1CZyQYdq\nmrFYoFBrdV20LE8SNqtFkwJERGLYgOHM5/NRXl7O5s2be2/bs2cPX/3qVyNamMiFBENhjp1oIyc9\nCZfDZnY5ccdqtZCTnsTxU+0EQ2HstkGNbBARkSga8Dfz17/+dbZs2YJhGNGoR6RflSfbCITCFGm8\n2SXLz0ohFDaobfSZXYqIiFzAgC1njY2NvPzyy1EoRWRgh2p61jdTl+al6tnuqqqujeI8hVwRkVgz\nYMvZpEmTOHDgQDRqERnQoZqenQEUKi5VnpbTEBGJaQO2nB08eJBZs2aRnZ2N0+kEuhezfO+99yJe\nnMjHHT3RSmqynbQUh9mlxK3cnpazek0KEBGJRQOGs5///OdAdyDTuDMxU6vPT0u7n9GFadrs/DK4\nHDYy3E6q6rwYhqH/SxGRGDNgt2ZRURHbt2/nv//7v8nMzGTr1q0UFRVFozaR8/R0w2nLpsuXn5mC\nrzNIU5sWlBYRiTUDhrMXXniB9evXs2bNGoLBIG+88QZLliyJRm0i5+lZOLWnW04uXe9OAVqMVkQk\n5gwYzv7yl7/wwgsv4HK5SE9P57/+67/YsGFDNGoTOU/1mTFSeWo5u2z5mSnA2d0WREQkdgwYzmy2\n8xf69Pv9n7hNJBoq67w47FYy3S6zS4l7RbmpWC2wr+K02aWIiMjHDDgh4K677uIb3/gGLS0tvPzy\ny/zud7/jnnvuiUZtIr0CwRAnT/soyEzWAPYh4HTYGJGdSmVdG77OAClJmv0qIhIrBgxnN998M3l5\neVRXV7Nt2zYef/xxbr311mjUJtLrxCkf4bBB3pnuOLl8pQUejp9q50BVM9PG55pdjoiInNFnOGts\nbOTxxx/n0KFDlJaWYrPZ2LRpE52dnUyfPp20tLRo1inDXJXGmw250nwPf91zkv0VTQpnIiIxpM8x\nZ9/5zneYPn06Gzdu5LXXXuO1115j48aNTJw4kcWLF0ezRpHemZp5mqk5ZAqzU7DbrOyr1LgzEZFY\n0mc4Ky8v55vf/CYOx9mxKE6nk2984xvs3bs3KsWJ9Kg6s8ZZTkaSyZUkDpvNSnFeKrWNPq13JiIS\nQ/oMZ0lJF34TtFqtmq0pUWUYBtV1bWR6XDjt+tkbSqX5HgAOVDaZXImIiPQYcCkNEbM1tnbS4Q9p\nvFkE9IQzdW2KiMSOPicEHD58mNtuu+2Cx+rr6yNWkMjHabxZ5ORlJpPktLG/okn7bIqIxIg+w9k7\n77wTzTpE+tSzp6ZazoaexWKhNN9DeXUz9c0dvTsHiIiIefoMZ9rcXGJFz2QAtZxFRsmZcLbn6Gny\npyuciYiYTWPOJOZV1bWR5LThTtYq9pEwdmQaVgu8v70GwzDMLkdEZNhTOJOY5g+EaGzpJDc9SeOh\nIsST4mRiSSa1jT72HLu0iQGBYIhdRxr5zbsHWbOlimAoPMRViogMHwNu3yRippOnfRhAVrrWN4uk\nayfmsa+yidVbqrhqdPagz+vyh/jN2oNs2V9PVyDUe/tf95xkweevoDjPHYlyRUQSmlrOJKadPO0D\nIDtN4SyS8rNSKMlzs6+iqXcCxkAaWzpY8v+28eddtSQ5bcyYkMu8W8dw1egsquq9fOflD3l3a3WE\nKxcRSTwRC2fhcJhnn32W+fPn89BDD1FVVfWJ+3R0dDB//nyOHj066HNkeKltVDiLlhkT8wBYs2Xg\n111VXRvf/PEGquq8XDU6m3/8/BXcNq2IsoI07v5UKXNuHkOS08aK9w5x4lR7pEsXEUkoEQtna9eu\nJRAIsGLFCp588kmWLl163vHdu3fz4IMPUlNT0zuWaKBzZPipbex+Y89SOIu4MYVpZHlcbNpXR7O3\n7+2cdhw+xeJfb+N0ayc3X1PIXX9TjM16/njA0YVpfPbaYgwDXl9/JNKli4gklIiFs+3btzNz5kwA\npkyZwp49e847HggEWLZsGaNGjRr0OTL8nDjVjt1mJS1FMzUjzWKxMGNiHqGwwUt/2Ie3I3DeccMw\nePfDan72xi7CYYO/u3Min5qU3+dEjbEj0xmZk8qOQ6c4WN0cjW9BRCQhRGxCgNfrxe0+OxjYZrMR\nDoexWrvz4LRp0y76nL7k5nqGqGoxQ1/XLxw2qGvqIDczmczM1ChXNTzdNLWIyjov+yubWPzrbfzb\nP3yKojw3h2qaWbOpkne3VOFOcfCluydRlDfw6+7zM0fzv9/czZt/PsYLj8/UjNsYo9+d8UvXLrFF\nLJy53W7a28+ONRlMyLqUcwAaGtouvVAxVW6up8/rd6q5g0AwTEaqk+ZmX5QrG77uvb6ULI+TD/bW\n8c0fr8dms9LRFQQgJz2JOTePwe3s3oB+oOuSnmRnXFE65VVNrN54lOkT8iJevwxOf689iW26dvFt\nMME6Yt2a06ZNY8OGDQDs2LGDCRMmROQcSVy1Z2ZqZqW5TK5keLFaLcy8upD7bizDZrPgsFuZMjab\n+24axUOfnUBaqvOiHu/TUwqxWGDl+qNa5FZEZBAi1nJ2xx13sHHjRubPnw/AkiVLWLVqFT6fj3nz\n5g36HBm+as/M8tNMTXNMKMlkfHEGwGV1R2anJTGhOIMDVc1U1rVRVpA2VCWKiCSkiIUzi8XC888/\nf95t5w7+77F8+fJ+z5Hhq1ZrnJluqMaITSrN5EBVM1v21yuciYgMQIvQSszqaTnL9KhbM96NGpGG\n02Fly746dW2KiAxA4UxiVm2jj/RUJ3abfkzjnd1mZdzIdE63dXHkRKvZ5YiIxDS960lM8nYEaOsI\nkK09NRPGxJJMALbsrzO5EhGR2KZwJjHppLZtSjhlBR5cThsf7q8nrK5NEZE+KZxJTDq7bZPGmyUK\nm83K+KIMWtr9HNKOASIifVI4k5ikmZqJaVJJ99IcWw7Um1yJiEjsUjiTmHRCa5wlpJJ8D8kuO1sP\nqGtTRKQvCmcSk2ob20l22Uh2RWwpPjGB1WphTGEabb4A1XVes8sREYlJCmcScwLBMKdaOtVqlqDK\nCrr3ldtbcdrkSkREYpPCmcSc+iYfhgFZCmcJqbQnnB1TOBMRuRCFM4k5tVpGI6GlJjnIzUjiUE0z\n/kDI7HJERGKOwpnEHC2jkfjKCtIIhgwO1mhJDRGRj1M4k5ijZTQSX8+4s30VTSZXIiISexTOJOac\nONWOzWohLcVpdikSIUW5bmxWi8adiYhcgMKZxJSwYXDytI8sjwur1WJ2ORIhDruVkTmpVNd7aW33\nm12OiEhMUTiTmNLc1oU/ECZLG54nvLIRZ7o2K9V6JiJyLoUziSmaqTl8lBWkAbDvmMadiYicS+FM\nYkrvTE1qoxe8AAAc7klEQVSPZmomuvzMZJKcNvYea8TQVk4iIr0UziSm9LacqVsz4VksFkrzPTR5\n/dQ3d5hdjohIzFA4k5hytuVM4Ww4KMl3A1BepfXORER6KJxJTKlt9JGW4sBh14/mcFCc1x3ODlRp\n3JmISA+9A0rM8HUGaWn3azLAMJKdlkSyy86ByiaNOxMROUPhTGJG7ekzXZoabzZsWCwWSvLcNGvc\nmYhIL4UziRkntYzGsKRxZyIi51M4k5jRM1NTG54PLz3jzso17kxEBFA4kxjSM1MzWzM1h5XucWc2\njTsTETlD4UxixonGdlxOGylJdrNLkSjqHnfWvd5Zg8adiYgonElsCIbCNDR3ku1xYbFow/Ph5uyS\nGhp3JiKicCYxoaG5g3DY0GSAYerspACNOxMRUTiTmHB2MoDC2XDUO+6sqlnjzkRk2FM4k5jQOxlA\na5wNS73jztq6tN6ZiAx7CmcSE7SMhpTkewDYX6GuTREZ3hTOJCbUNrZjs1rISFU4G67KCrrD2b6K\n0yZXIiJiLoUzMZ1hGNQ2+shwu7BaNVNzuMpwO0lLcbCvsolwWOPORGT4UjgT0zV7/XT6QxpvNsxZ\nLBZKC9LwdQapqm8zuxwREdMonInpTvZMBtB4s2HvbNemxp2JyPClcCamqz19ZjKAtm0a9nrWO9t7\nTOPORGT4UjgT09We6g5n6taU1CQHuRlJHKppxh8ImV2OiIgpFM7EdD1rnGV51K0pUFaQRjBkcPh4\ni9mliIiYQuFMTFfb2I472YHTYTO7FIkBpfkadyYiw5vCmZjK1xmgyevXZADpVZSXitVqYa/WOxOR\nYUrhTEx1vMELQHZ6ssmVSKxw2m2MzE6h6mQb3o6A2eWIiESdwpmYqqb+TDhTy5mcY3RhOgaw49Ap\ns0sREYk6hTMxVU840zIacq7xxRkAbCuvN7kSEZHoUzgTU9WcWQk+K03hTM7K9LjIzUhiz7HTdHQF\nzS5HRCSqFM7EVNUn23A6rLiT7WaXIjFmQnEmobDBzsPq2hSR4UXhTEwTCoc5caqdLE8SFos2PJfz\njS9OB2BreYPJlYiIRFfEmivC4TALFy7k4MGDOBwOFi1aRElJSe/x999/n2XLlmG325k9ezZz584F\nYNasWbjd3Vu4FBcXs3jx4kiVKCarb+ogFDbI0c4AcgE56clkpbnYfbSRLn8Il1Pr4InI8BCxcLZ2\n7VoCgQArVqxg586dLF26lGXLlgEQCARYunQpb7zxBklJSTzwwAPcfvvtpKamArB8+fJIlSUxpPrM\nZIDcDC2jIRc2oTiDD/bWsftoIzMm5pldjohIVESsW3P79u3MnDkTgClTprBnz57eY0eOHKGkpASP\nx4PD4WD69Ols2bKFAwcO0NHRwYIFC3j44YfZuXNnpMqTGFDT0BPO1HImFzbhzKzNrZq1KSLDSMRa\nzrxeb2/3JIDNZiMcDmO1WvF6vXg8nt5jqamptLW1MXr0aBYsWMDcuXOpqKjgkUceYfXq1VitGhqX\niGrqu/fUVMuZ9CU3I5kMt5OdhxvxB0La4ktEhoWIhTO32017e3vv1z3BDMDj8Zx3rL29nfT0dMrK\nyigtLQWgrKyMjIwMGhoayM/P7/e5cnM9/R6X2HT8zJ6aIwvSzS5FLlFGRkrEn2PKuDzWf1TD3uoW\nPvup0og/33Ci353xS9cusUUsnE2bNo1169Zx9913s2PHDiZMmNB7bPTo0VRWVtLS0kJycjIffvgh\nCxYsYOXKlZSXl/Pcc89RV1eH1+slNzd3wOdqaGiL1LchEeLrDNLQ1MHYonSam31mlyOXICMjJSrX\n7oqSdP6y8zivrilnSlkmVqtm9g6F3FyPfnfGKV27+DaYYB2xcHbHHXewceNG5s+fD8CSJUtYtWoV\nPp+PefPm8fTTT7NgwQLC4TBz5swhLy+POXPm8Mwzz/Dggw/2nqMuzcR0/FT3eLOC7FSTK5FY50lx\nMnlUFjuPNPLhgXo+dUX/LemDZRiGlnARkZhkMQzDMLuIy6W/IOLPuo+Os3x1OXNuHcfoAvfAJ0jM\niVbLGUCzt4uXVu2jMCeV57/yN1gvMVQdPt7CnqONHKhq4sjxVnIzkrlmbA5TxmYzrihjWLXKqfUl\nfunaxTdTW85E+tOzp2ZBduTHLEn8y3C7mFSSyb7KJnYdbuSacTkXdX5TWxe/XlPOR+dspJ6dlsSp\nlk7e2VLFO1uqmFiawVdnXU1Kkn4tioi59FtITFFd78VigdzMFNq9nWaXI3Hguivz2VfZxKq/VjBl\nbPaguiTDhsH6HSd4bd1hOv0hinJTuXZiHsV5bpKcdgLBMFX1bWw/2MCBymaW/r9tfGPeNWR6XFH4\njkRELkwDuiTqDMOgpsFLlseFw64fQRmcnPRkxo5M52htK+s+Oj7g/dt8fn76+i6Wry7HMAzuvLaY\nB24fx7iiDJKc3X+XOuxWxhSmM/vTY5g6LoeahnYWLd9KbWP7AI8uIhI5emeUqGts7aTTH9L6ZnLR\nbp06khSXnV+vOcjG3bV93q+8qonnfrmFXUcaKc33sOCeK5gyNqfP1jar1cJnphcx8+oRnG7t4sev\n7aTTH4zUtyEi0i+FM4k6LT4rlyrT42LerWNJctr45Vv72bK/7rzjxxu8/Ocf9vLvv/2IlnY/n54y\ngnm3jsGd7BjwsS0WC9dfWcDfTMqjobmTFe8ditS3ISLSL405k6irbtCemnLp8jKTmXvLWF5dd4j/\n/bu9rFx/lLzM7p+lPcdOA91bgn12RjEjcy9+JvBNV42goraNDTtruXpMDtPGD7zWoojIUFI4k6jr\nmamZp3Aml2hEdgrzbhnLn3Ycp7G1i/rmDgAKslK4YXIBYwrTLnkNM7vNyudvKOWV1eW8/PZ+xhSm\nke7WBAERiR6FM4m6mgYvLocVT8rAXU0ifSnMSeXvPjMegK5AiM6uIGmpziFZWDYnPZmbrxnJe9tq\n+NXqch6fffVlP6aIyGBpzJlEVSAY4uRpHznpyVqdXYaMy2Ej3e0a0p+paeNyKMpNZcehU+yvbBqy\nxxURGYjCmURVVb0Xw4D8THVpSmyzWCzcOrUIgFffP0Q4/jdTEZE4oXAmUXX0eCsAI3K0p6bEvhHZ\nKVxRmklVnZfNe+sGPkFEZAgonElUHTnRAkChNjyXODFzSiE2q4XX1x/BHwiZXY6IDAMKZxJVR463\nkOyykeF2ml2KyKCkpzqZMSGXprYu3t1abXY5IjIMKJxJ1LR4u2hs7aIwO1WTASSuXHdFAckuG299\nUEl7Z8DsckQkwSmcSdQcOdE93qxQ480kzricNj41KZ8Of4g1W9R6JiKRpXAmUaPxZhLPpo7LJSXJ\nzpqt1Xg71HomIpGjcCZRc+TMTM2C7BSTKxG5eA67lesm5dPlD7F6S5XZ5YhIAlM4k6gIhcNU1LaS\nk56Ey2EzuxyRSzJlbA6pSXbe3VpNq89vdjkikqAUziQqjje04w+GNd5M4prDbuX6KwvwB8K8s1mt\nZyISGQpnEhW9kwE03kzi3NVjsnEnO3hvWw0t7Wo9E5Ghp3AmUXHk+JnJADkabybxzW7rbj0LBMO8\nvanS7HJEJAEpnElUHDnegtNhJTstyexSRC7b1aOz8KQ4WLf9OE1tXWaXIyIJRuFMIs7bEaCuqYMR\nWVp8VhKDzWblhskFBEJh3lLrmYgMMYUzibj9lU0AFOVpvJkkjsmjsklPdfKnj45zurXT7HJEJIEo\nnEnE7TpyCoDRI9JNrkRk6NisFm6YXEAobLDqA7WeicjQUTiTiAobBruONJKSZKcgK9nsckSG1JVl\nWWS6nWzYeYLaxnazyxGRBKFwJhFVebKNNl+A0SPSNN5MEo7VauGWqSMJhw1eff+w2eWISIJQOJOI\n2nWkEYAxhWkmVyISGWNHplOS52bXkUb2HG00uxwRSQAKZxJRu46cwmKBsgKFM0lMFouF26aNBOC3\n7x0iFA6bXJGIxDuFM4mY1nY/FbVtFOWk4nJqP01JXHmZKVw9JpvaRh9/+uiE2eWISJxTOJOI2X20\nEQMYPVKzNCXxzbx6BE67lTc3HNXCtCJyWRTOJGI03kyGk9QkB5+eUoivK8gv/7iPsGGYXZKIxCmF\nM4mIUDjMnqONpKU4tGWTDBtTx+UwaoSHvRVNvLetxuxyRCROKZxJROyvbKLDH2J0YbqW0JBhw2Kx\ncPenSkl22Xht3WGON3jNLklE4pDCmUTEuu3HAZg8KsvkSkSiy53s4K6/KSUYMvjF7/fS0RU0uyQR\niTMKZzLkGls62XH4FPmZyYzITjG7HJGoG1eUztRxORxvaOdH/72TTr8CmogMnsKZDLk/7TiOYcDU\ncbnq0pRh6/ZpRUwsyeDw8RZ+8vouugIhs0sSkTihcCZDKhAMs2HnCVxOG5NKM80uR8Q0VquFe64v\nY1xROuVVzfz09V20tvvNLktE4oDCmQypbeX1tPkCXD06G4ddP14yvNmsFv72hjLGFKaxv7KJ/++l\nTWzcXYuhZTZEpB9695Qh9d727uUDrhmbY3IlIrHBZrMya+Zobp82kkAwzP/9435e+O1HfLD3JL7O\ngNnliUgMsptdgCSOIydaOHK8lVEjPGR6XGaXIxIzrFYL0yfkMbYogzUfVnGgqpkDVc1YrRbGjUwn\nOz2J9FQnqckOwmGDYChMMNT9MRQyCITChEJhgmGDUNjAnewgPdVJeqqTEdkpFOW5SU1ymP1tisgQ\nUTiTIREMhXnlnXIAPjUp3+RqRGJTeqqTOTePobG1k4PVLRyqaaa8uhmqL/+xMz0uxhdnMKk0k0ml\nmeRmJF/+g4qIKRTOZEis+bCa6novV43KoiTfY3Y5IjHLYrGQk55MTnoyN0wuwB8M4esM0t4ZpLMr\niNVqwdbzz2bFZrWcd5vFYqGjK4ivM0hbh59TLZ3UN3VQ39TB5n11bN5XB0B2ehJXlmUyqTSLSWWZ\npKU4Tf7ORWSwFM7kstWd9vG7Px8jJcnOLVNHml2OSFxx2m043TYy3IMfCuBO/mQXpmEYNLZ2Ulnn\npfJkG9X1bWzYWcuGnbUAlOS7mTwqmytHZTF2ZPqQ1S8iQ0/hTC5L2DD4r7cPEAiFufu6EpJd+pES\nMcO5LXLTx+cSDhvUNfmoPNlGxck2ahraqarz8tamSpx2K1ePzWFMYRoTSjIoyfNgtWpNQpFYoXdS\nuWSGYfDf7x/mYHUzY0emMaE4w+ySROQMq9XCiOxURmSnct2V3d2n1fVeKmrbOHayla0H6tl6oB6A\nJKeN8cUZTCjOYHxJBqX5Huw2TeYXMYvCmVyScNjgV+8c4M+7aslOS+LOa0u0G4BIDHPabYwpTGdM\n4ZkuTbuNvYcaqG7wUl3Xxq4jjew60giAw26lNN/D6MI0Ro1IY3RhGjnpSXqNi0SJwplcNH8gxP9Z\ntY+t5Q0UZCUz55axpKg7UySuZLhdXDkqiytHZQHQ5gtQ0+Clut7L8VNejpxo4fDxlt77u5MdlOa7\nGZGdSkF2CiOyUijITiXD7VRoExliEXtHDYfDLFy4kIMHD+JwOFi0aBElJSW9x99//32WLVuG3W5n\n9uzZzJ07d8BzxFzBUJi/7KrldxuP0eL1U5Sbyuybx+By2MwuTUQukyfF0bsMB4A/GKLudAe1je3U\nnvZRe6qdvRVN7K1oOu88l8NGbkYS6W4XaSlO0lIdZz468aQ4SE12kJrkIDXJTkqSHZtV3aUiA4lY\nOFu7di2BQIAVK1awc+dOli5dyrJlywAIBAIsXbqUN954g6SkJB544AFuu+02tm3b1uc5Yo5w2ODY\nyVb2HD3NB3tOUt/cgd1m5bor8rn+ygJt0SSSoJx2G8V5borz3L23dflDnG7r5HRrV+/HxtZO6po6\nqGloH9TjJjltpCbZz4a2ZAeeZAeeFAfuZAeeFOcnPtf4NxluIhbOtm/fzsyZMwGYMmUKe/bs6T12\n5MgRSkpK8Hi618OaPn06H374ITt27OjzHBlaoXCY4w3tdPpDdAVCdJ352OkP0d4ZoKG5e92k4w3t\n+LqCQPcA46njcrj+yoILTuUXkcTmctp6Jxl8XCAYxtcZwNfVvWabrzOIrzNApz9Ehz9Epz9Ipz9E\nZ1eQDn+IVp+PQDA8qOdNctrwpDhJOye0uVO6A12S047TbsVx5p/TbsNht/auCWexgPXMRzj7ueVj\nHwEMo/sPUsMwuj8/52PYMDDCPbcZ3Ts5hA2CwfDZj6FzPg+Hz9x2/o4PPf8CIYNQKNy7+0PP11ar\nBYfNit3W/f3Yz/ncYbNit1tI8yQR6Apit597v7Pn2Xvua7N038dq7f0eLRYLFuj++pzPPSlO/V6P\nIRELZ16vF7f77F9cNpuNcDiM1WrF6/X2BjOA1NRU2tra+j1HhtZr646w5sP+lyW3WCAtxcnVRdmM\nGpFGab6bJKfGlonIJznsVtLdLtIvYr22YCjcHd66gvi6gnSc+efrPPt1z+e+zgCNLR2EE3jPeJvV\n0hsIo81us/AfX71JAS1GROyd1u12095+tpn73JDl8XjOO9be3k5aWlq/5/QnN1cr0l+sr82fxtfm\nTzO7DBEREfmYiDVJTZs2jQ0bNgCwY8cOJkyY0Hts9OjRVFZW0tLSgt/v58MPP2Tq1Kn9niMiIiIy\nHFgMIzINqIZhsHDhQsrLuzfDXrJkCXv37sXn8zFv3jzWrVvHiy++SDgcZs6cOfzd3/3dBc8ZNWpU\nJMoTERERiUkRC2ciIiIicvE00l5EREQkhiiciYiIiMQQhTMRERGRGKJwJiIiIhJD4nJF0ba2Np56\n6ina29sJBAI8/fTTXHPNNezYsYPFixdjs9m48cYb+epXv2p2qdKPd999l3feeYcf/vCHvV//+7//\nOwUFBQA8/vjjXHvttWaWKH34+LXTay/+GIbBpz/9acrKygCYOnUq3/zmN80tSvql/afj36xZs3oX\n2y8uLmbx4sUXvF9chrOXX36ZG264gS996UscO3aMJ554gpUrV/Lcc8/x85//nOLiYh599FH279/P\npEmTzC5XLuB73/seGzdu5Iorrui9be/evTz11FN89rOfNbEyGciFrt3ChQv52c9+ptdeHKmqquLK\nK6/kF7/4hdmlyCD1t2e1xL6uri4Ali9fPuB947Jb88tf/jJf/OIXAQgGg7hcLrxeL4FAgOLiYgBu\nuukm/vrXv5pZpvRj2rRpLFy4kHNXctm7dy9vvPEGDz74IN///vcJhUImVih9+fi183q9+P1+vfbi\nzN69e6mvr+dLX/oSjz76KMeOHTO7JBlAf3tWS+w7cOAAHR0dLFiwgIcffpidO3f2ed+Ybzl77bXX\neOWVV867bcmSJUyePJmGhga+9a1v8a//+q+f2JczNTWV6ur+946UyOvr+n3uc59j8+bN591+4403\n8pnPfIaioiKeffZZVqxYwYMPPhjNcuUcg712eu3Fvgtdy+eee47HHnuMO++8k23btvHUU0/x+uuv\nm1ShDIb2n45vycnJLFiwgLlz51JRUcEjjzzC6tWrL3j9Yj6czZ07l7lz537i9vLycp544gm+/e1v\nM2PGDLxe73n7cnq9XtLS0qJZqlxAX9fvQmbPno3H071P6u23386aNWsiWZoMYLDX7uN74uq1F3su\ndC07Ozux2WwATJ8+nfr6ejNKk4twqftPS2woKyujtLS09/OMjAwaGhrIz8//xH3j8qoePnyYr3/9\n6/zwhz/sbeJ1u904HA6qq6sxDIONGzcyY8YMkyuVwTIMg/vuu4+6ujoAPvjgAyZPnmxyVTIYeu3F\npxdffJFf/epXQHd3S2FhockVyUC0/3R8W7lyJUuXLgWgrq4Or9dLbm7uBe8b8y1nF/If//EfBAIB\nvve97wGQlpbGiy++yPPPP8+TTz5JKBTipptu4uqrrza5UumPxWLBYrH0fr5o0SK+9rWv4XK5GDdu\nHPPmzTO5QunLudcO0GsvDj366KM89dRTrF+/HrvdzpIlS8wuSQZwxx13sHHjRubPnw+gaxZn5syZ\nwzPPPNM7XGfJkiV9tnxqb00RERGRGBKX3ZoiIiIiiUrhTERERCSGKJyJiIiIxBCFMxEREZEYonAm\nIiIiEkMUzkRERERiiMKZiJhu8+bNPPTQQ0P+uLfddhv33HMP999/P/fddx9f+MIXPrFt2EAeeugh\n9u7dO+j7r1y5ku9+97ufuL2uro5HH30UgKeffpo333yT+vr63tvef/99Xn755YuqTUQSU1wuQisi\nMlgvvfRS7+r3f/nLX/jGN77Bn//8596tiwbjYpaDPHdx3nPl5+fzn//5n733sVgs5OXl9d62d+/e\nPs8VkeFFLWciEtN+8YtfcM8993Dvvffy/e9/n3A4DMArr7zCnXfeyZw5c/jWt77Fz3/+8wEfa8aM\nGZw+fZqWlhaefvpp/umf/onPfe5z/OlPf2LHjh3MnTuX++67jy9/+ctUVVX1nvfKK6/whS98gS98\n4Qts3boV6G4JW7BgAV/84he57bbb+OEPfwh0B7nDhw/zwAMPcO+99/KjH/0IgJqaGm677bbz6um5\n7ciRI6xYsYIVK1bw+uuvc9ttt1FRUQGAz+fjlltuwe/3X/b/pYjEB7WciUjMWr9+PevWrePNN9/E\nZrPxta99jd/+9rdMnz6d3/zmN6xcuRKHw8FDDz1ESUnJBR/j3Fav3/3ud5SVlZGVlQVAZmYmv/jF\nL/D7/dx111389Kc/ZfLkybzzzjt885vf5PXXXwe6t4hbuXIlBw4c4F/+5V9YvXo1f/zjH7n33nu5\n//77aWtr45ZbbuErX/kKANXV1bz55pukpKTw8MMPs379esaMGdPn9zlmzBgeeOABoHuLl9raWn7/\n+9/z+OOPs2bNGm699VacTueQ/J+KSOxTy5mIxKxNmzbx+c9/HqfTic1mY/bs2WzatIlNmzZx6623\nkpqaitPp5J577unzMR599FHuv/9+7rnnHtauXcuPf/xjoLtrccqUKQBUVFSQnp7O5MmTAbjrrruo\nqqrC6/UCMHfuXAAmTpxIRkYGx44d4ytf+QoFBQX88pe/ZNGiRQSDQTo6OrBYLHz2s58lPT0dh8PB\n3XffzQcffDBgl+W5IXLWrFmsWrUKgDfffJNZs2Zd4v+giMQjtZyJSMwyDOO80GIYBsFgEKvV2tu9\n2XN7X84dc/ZxLpcL4LzHOvcxQ6EQwHnj0wzDwGazsXTpUmpqarj33nv5zGc+wwcffNBbx7mbGYfD\nYez2i/tVW1RURGFhIWvWrOH06dPaSF5kmFHLmYjErOuuu44//vGPdHV1EQwGeeONN7juuuu4/vrr\nWb9+PV6vF7/fz5o1ay5rMP3o0aNpbm5m9+7dALz11luMHDmS9PR0AP7whz8AsHv3btrb2yktLeWv\nf/0rCxYs4M477+TEiRPU1dURDocxDIN169bh9Xrp6urirbfe4oYbbvhEyPx4oLTb7QSDwd6vZ8+e\nzaJFi7jvvvsu+fsSkfikljMRMZ3FYmHbtm1MnTq197b77ruPhQsXsn//fmbPnk0wGGTmzJk89NBD\nWK1WHnroIebPn09KSgqZmZkkJSVd0vMCOJ1OfvSjH/Hd736Xjo4OMjIyegfyAzQ3N3P//fdjt9v5\nwQ9+gN1u57HHHuNb3/oW2dnZjB07luuuu46amhosFgujRo3iH//xH2lra+Nv//ZvueGGG3qP9Tzv\nuf8Arr32Wr797W+Tm5vLgw8+yB133MH/+l//S+FMZBiyGBczR1xEJAZUVFTwpz/9iS9/+csA/Mu/\n/Avz5s3jlltuMbWuoWIYBhs2bODVV19l2bJlZpcjIlGmljMRiTuFhYXs3r2be++9F4CZM2cmTDAD\nWLx4MevXr+ell14yuxQRMYFazkRERERiiCYEiIiIiMQQhTMRERGRGKJwJiIiIhJDFM5EREREYojC\nmYiIiEgM+f8BNNeg5dDcu/IAAAAASUVORK5CYII=\n",
"text": [
""
]
}
],
"prompt_number": 7
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"There seem to be five local maxima. We can interrogate if these correspond to each of the sequence lengths by just plotting them out individually."
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"plt.figure( figsize=(10,5) )\n",
"for i in xrange( 1, 6 ):\n",
" subset = it.product( 'ACGT', repeat=i )\n",
" subset_scores = map( model.log_probability, subset )\n",
" sns.kdeplot( numpy.array( subset_scores ), color='rgbyc'[i-1], shade=True, label=\"Sequence Length \" + str(i) )\n",
"plt.ylabel('Density')\n",
"plt.xlabel('Log Probability')\n",
"plt.legend()"
],
"language": "python",
"metadata": {},
"outputs": [
{
"metadata": {},
"output_type": "pyout",
"prompt_number": 8,
"text": [
""
]
},
{
"metadata": {},
"output_type": "display_data",
"png": "iVBORw0KGgoAAAANSUhEUgAAAmcAAAFICAYAAAAVoFlvAAAABHNCSVQICAgIfAhkiAAAAAlwSFlz\nAAALEgAACxIB0t1+/AAAIABJREFUeJzs3XlgVOXZ+P3vObMlmWQySUjClgWBgAVFQcGl1vogWC0q\nVksRBPwhq/CAFRABxYhGQMBHajXVCEa24oKypKVSBEXxFZFNbEGWQICQxOx7Jpk55/0jmWBkSUJm\nMglen3+yzDnnvieHGa65l+tSdF3XEUIIIYQQLYLq6w4IIYQQQohzJDgTQgghhGhBJDgTQgghhGhB\nJDgTQgghhGhBJDgTQgghhGhBJDgTQgghhGhBjN66sKZpxMfHc+TIEUwmEwkJCURHR5933LPPPovd\nbmfatGkNPkcIIYQQ4krltZGzrVu3UlVVxdq1a5k+fToLFiw475i1a9dy9OhRFEVp8DlCCCGEEFcy\nrwVne/fu5bbbbgOgV69efP/99+c9/t133/GnP/0Jdx7c+s4RQgghhLjSeS04KykpITAwsPZng8GA\npmkA/Pjjj7z++uvMnTuXnxYouNQ5QgghhBC/BF5bcxYYGEhpaWntz5qmoarVseAnn3xCfn4+Y8eO\nJScnh4qKCq666qpLnnMxuq7XTosKIVqpgwfhllugpKT651tvhc2bISjIt/0SQggf8Fpw1rt3b7Zv\n387dd9/N/v376datW+1jI0aMYMSIEQB8/PHHnDhxggceeIAtW7Zc9JyLURSF7Oxibz0N4WXh4UFy\n/1opT9472/SZWEpKKH90DIYfDmHeuZPS+Yso+/MMj1xfnE9ee62X3LvWLTy8/g+dXgvOBgwYwM6d\nOxk6dCgA8+fPJyUlhbKyMoYMGdLgc4QQVzbj3m+xbNmM8+pf4bhvMJTfhWn/PvzefpOyx6eAxeLr\nLgohRLNS9J8u+mql5BNE6yWfAFsvT9072+hHsKRspOSF+Th7XguAX/Lb+G34mKLX/objT8Oa3IY4\nn7z2Wi+5d61bQ0bOJAmtEMJ3Kiowf/pvXO071AZmAI577kNXVPxXLPdh54QQwjckOBNC+Iz5y89R\nysupurFfnd/rERG4usZh3PMtSkG+j3onhBC+IcGZEMJnzJ/8C+C84AygqncfFE3D9MXnzd0tIYTw\nKQnOhBC+oeuYt2xGswbi6n71eQ87r+8NgHn7p83dMyGE8CkJzoQQPmE4egRDxlmcvfuAwXDe467O\nXdECAzFv2wqtf9+SEEI0mARnQgifMO7ZDYDz6l9d+ACDAWev6zGcTcdw/Fgz9kwIIXxLgjMhhE+Y\nvq0Ozlxx3S96jLN7deBm3LenWfokxC/JypXJPPHE40yePI4pUybwww+Hfd2lBlu27E3Wr1/n8etW\nVlaSkrK+UW2cOXOaUaOGerQfXktCK4QQl2LasxvdZMYVE3vRY1xdugJg3L8Xxx89++YnREthjX8G\ny6b1DT9BVQjVLj3V77h3MKXxL1708RMnUvnqqx0kJlanqzl69AgJCfEkJ69peD98yFtlG3Nzc9i0\naQODBg1uUBv/+tc/+PDD9ygoKPBoPyQ4E0I0v5ISDIf/i6tbdzBe/G3I1ekqdFXFJCNnQnhUYGAg\nWVlZpKRsoF+/m+naNY6kpHcBOH78GEuXLkbXdYKDg5k1ay5+fv68/HICqanHiYxsS3r6ad55Zw0J\nCfHceedd9Ot3M19//RXbtv2b2bOfY9u2rbz//hpUVeXaa69jwoTJLFv2JpmZGeTn55GZmcmUKU/S\nt+9N7Nz5BcnJSeg6xMV1Y8aM2ezfv5ekpERUVaVDh47MmDEb4yXeK9w+/HAtW7duQVGgf/+BPPTQ\nUBIS4jGbzWRkZJCbm8OcOc8RF9edlJT1fPTRBwQFBWMyGenffyDffXeAkydTSU5+G4Avv/yc7ds/\npaiogDFjJnLrrbfVac9mC+avf32LP/3pfo/eHwnOhBDNzvTdfhRNwxlXT/1ciwUtKgbjwe/A6bxk\nICdEa1Ua/+IlR7l+Ljw8iLwmVggID49gwYIlrFv3Pu+8k4Sfnx/jxj3O7bf/DwsXvsicOfHExMSS\nkrKB1atX0KVLHBUVFSQlvUtOTg7Dhj0IVI9guUeY3F+LiopYvvwtli1bicVi4YUX5rJ79y4URcFs\nNrN48V/YvXsXa9eupk+fG3n11UUkJa3AbrezZs1KsrKyePnlBBITl2O323n77b+xeXMK9947+JLP\n6cSJVLZt20pi4jI0TePJJyfTt+/NKIpC27btmTFjNps2rWfjxo8ZO3Yiq1evIDn575hMJqZMmQDA\nqFGjSU09xqOPjmHZsjcJD49k5sw57Nu3hzVrVpwXnN1yy6+bdB8uRt7phBDNztiA9WZuzq5xWNJO\nYDh8CFfPa7zdNSF+EdLTz2C1BjJr1lwADh8+xPTpU7j++htISzvB4sXVta2dTidRUdFYrVZ69Kh+\n/bVp04bY2E7nXVPTtJprn6agIJ/p06cAUFZWRnr6GQC6do0DICIikspKB4WFBQQFBWG32wEYNmwE\n+fl55Obm8uyzMwFwOBz07XtTvc8pNfU4mZkZtYFWSUkxZ86cBqpH5NztHjx4gDNnzhAbexWWmtq9\nPWsqlPy0oqWiKHTrVv0eFRoaRkVFRb198BQJzoQQzc7434MAuK7qXO+xrq5dYesnmA7sk+BMCA85\nduwoGzd+zMKFr2A0GomKiiIoKAiDQSU6OpZnn51HREQk+/fvpbCwED8/PzZtWs+QIQ9TVFRUG/SY\nzWZycrIBOHKkekNBu3YdiIiI5NVX38BgMJCSsoHu3X/Fjh3bgbrruEJCQikuLqGoqAibzcbSpUsY\nOPB3REREsHDhKwQEWNmx4zNsNlu9zykmJpZOnTqzZMlfAFi7dhWdO3fhs8/O5Up0B18dO3bk1KmT\nOBwOTCYThw79h5iYWFRVrQ0yfVl6XIIzIUSzMx76L7rFDy0ist5jXZ1rNgUc2AfDR3q7a0L8Itx+\n+x2kpZ1gzJiR+Pv7o+s6kyZNxWoNZPr0WbzwwlxcLheKojBr1lw6doxi3749TJgwmpCQUMxmMwCD\nBg1m/vx5bNmymaioGADsdjtDhw5n8uSxuFwa7dq1Z8CAu4C6C/ndU6LTps3kqaeeQFVV4uK6c/XV\nPZg6dRrTp09F1zWs1kCeeWbeec9h1apkUlI2AGC1Wlm6NJE+fW5k4sTHqKyspEePnoSHR9Rp1/01\nONjO8OGjmDRpLDabDYfDgdFoJCQkFKezisTE17BYLOf19+I8u0FB0X0ZGnpIdhPn3oXvhIcHyf1r\npS773lVV0SamLa7YTpQs+r/6j6+sJHjog1T1u4nCjf9qfHviguS113q1hHs3fPhDrF79oU/70BQu\nl4vVq99l5MjR6LrO5MnjGDduEr16Xef1tsPDg+o9RkbOhBDNypB6HMVZhSsmpmEnmM1obdti/OFQ\ndaUAL22hF0I0nLdSWTQXg8FAeXk5o0c/gslkokePns0SmDWUBGdCiGZlPPxfALRL5Df7OS0qGtM3\nX6Pk5KCHh3upZ0KIhlq16gNfd6HJxo+fxPjxk3zdjQuSCgFCiGZlOFQdnLmiGzhyBriiogEwHmk9\nGcyFEOJySXAmhGhWxsOHgMsLzgw15wohxJVMgjMhRLMyHPoPWlAQuj2kwedo0TJyJoT45ZDgTAjR\nfBwODGkn0TpGN2phv6t9R3RFkZEzIcQvgmwIEEI0G0PaSRRNw9WhQ+NOtFjQIiIx/iAjZ0J4ysqV\nyezZ8w1OpxNVVZk06YnajPgt3bJlbxIW1obBgx/06HUrKyvZsuWfDBo0uEFtvP76Ug4ePIDL5eS+\n+/5Qb4mphpLgTAjRbAypxwHQ2rVv9LlaVDSmb79Byc9DDwn1dNeE8Jn4r55h0/H1DT5eVRU07dIp\nSu/tPJj4Wy5er/PEiVS++moHiYnLATh69AgJCfEkJ69pcD98yVupPHJzc9i0aQODBg2ut429e7/l\n7Nkz/O1vy6mqqmLEiCHcccedBAYGNrkfEpwJIZpNU4IzV7v2mADDiVScEpwJ0SSBgYFkZWWRkrKB\nfv1upmvXOJKS3gXg+PFjLF26GF3XCQ4OZtasufj5+fPyywmkph4nMrIt6emneeedNSQkxHPnnXfR\nr9/NfP31V2zb9m9mz36Obdu28v77a1BVlWuvvY4JEyazbNmbZGZmkJ+fR2ZmJlOmPEnfvjexc+cX\nJCcnoevVNTBnzJjN/v17SUpKRFVVOnToyIwZszEa6w9ZPvxwLVu3bkFRoH//gTz00FASEuIxm81k\nZGSQm5vDnDnPERfXnZSU9Xz00QcEBQVjMhnp338g3313gJMnU0lOfhuAL7/8nO3bP6WoqIAxYybW\nKXzes+e1dO3arfZnl0trUB8bQoIzIUSzcQdnrvaNnNYEtHbtqq9xIhVn7xs82i8hfCn+lhcvOcr1\nc56oEBAeHsGCBUtYt+593nknCT8/P8aNe5zbb/8fFi58kTlz4omJiSUlZQOrV6+gS5c4KioqSEp6\nl5ycHIYNq57qc5dgcn8PUFRUxPLlb7Fs2UosFgsvvDCX3bt3oSgKZrOZxYv/wu7du1i7djV9+tzI\nq68uIilpBXa7nTVrVpKVlcXLLyeQmLgcu93O22//jc2bU+qdMjxxIpVt27aSmLgMTdN48snJ9O17\nM4qi0LZte2bMmM2mTevZuPFjxo6dyOrVK0hO/jsmk6m2WPqoUaNJTT3Go4+OYdmyNwkPj2TmzDns\n27eHNWtW1AnOzGYzZrMZp9PJiy8+x/33P4Cfn1+T7oubBGdCiGZjOH4UAK1t20af6x5tM5xI9Wif\nhPglSk8/g9UayKxZcwE4fPgQ06dP4frrbyAt7QSLF88HwOl0EhUVjdVqpUePawBo06YNsbGdzrum\nu2B4evppCgrymT59CgBlZWWkp58BoGvXOAAiIiKprHRQWFhAUFAQdrsdgGHDRpCfn0dubi7PPjsT\nAIfDQd++N9X7nFJTj5OZmVEbaJWUFNcWaI+L61bb7sGDBzhz5gyxsVdhsViA6lEwqFvsXFGU2jV4\noaFhVFRUnNdmUVERzz77NL179+GRRx6tt48NJcGZEKLZGFKPo4WFgaXxny61tudGzoQQTXPs2FE2\nbvyYhQtfwWg0EhUVRVBQEAaDSnR0LM8+O4+IiEj2799LYWEhfn5+bNq0niFDHqaoqKg26DGbzeTk\nZANwpCbVTbt2HYiIiOTVV9/AYDCQkrKB7t1/xY4d2/l5gfCQkFCKi0soKirCZrOxdOkSBg78HRER\nESxc+AoBAVZ27PgMm81W73OKiYmlU6fOLFnyFwDWrl1F585d+OyzT2uPcQdfHTt25NSpkzgcDkwm\nE4cO/YeYmFhUVa0NMusrPe5wVPDEExN5+OERDBjwuwb81RtOgjMhRPMoK8OQcZaqmk+ojaWFR6Ab\nDLVTo0KIy3f77XeQlnaCMWNG4u/vj67rTJo0Fas1kOnTZ/HCC3NxuVwoisKsWXPp2DGKffv2MGHC\naEJCQjGbzQAMGjSY+fPnsWXLZqKiqhNL2+12hg4dzuTJY3G5NNq1a8+AAXcBdRfyu6dEp02byVNP\nPYGqqsTFdefqq3swdeo0pk+fiq5rWK2BPPPMvPOew6pVyaSkbADAarWydGkiffrcyMSJj1FZWUmP\nHj0JD4+o0677a3CwneHDRzFp0lhsNhsOhwOj0UhISChOZxWJia9hsVjO6+9PrV+/jrNnz7Jx48ds\n3PgxALNnP0e7y1hT+3OKXl9oeJk0TSM+Pp4jR45gMplISEgguiaRJMAnn3xCUlISiqJw7733MnLk\nSAAeeOCB2p0OUVFRvPTSS/W21dS5d+E7nlg7IXyjsffO8N//EPrbm3EMvJvyiZMvq82gx8eiOCrI\nPXzyss4X58hrr/VqCfdu+PCHWL36Q5/2oSlcLherV7/LyJGj0XWdyZPHMW7cpGYpfh4eHlTvMV4b\nOdu6dStVVVWsXbuWAwcOsGDBAt544w2g+o/yyiuvsG7dOgICArjnnnu477778Pf3B2DlypXe6pYQ\nwkdqd2q2v/xPlVr79pj2fItSWIAebPdU14QQjeStVBbNxWAwUF5ezujRj2AymejRo2ezBGYN5bXg\nbO/evdx2W/Wuhl69evH999/XPmYwGNi8eTOqqpKTk4OmaZhMJg4fPkx5eTmPPfYYTqeTJ598kl69\nenmri0KIZmQ4eQI4t3bscmht29dey9nreo/0SwjReKtWfeDrLjTZ+PGTGD9+kq+7cUFeK99UUlJS\nJxGbwWCoXWQHoKoqW7ZsYfDgwfTr1w9/f3/8/f157LHHWLZsGc8//zzTp0+vc44QovUynE4DwBUR\nednXcLnTaci6MyHEFcxrI2eBgYGUlpbW/qxpGqpaNxYcOHAgAwYM4Omnn2b9+vUMGjSImJjqBYWx\nsbHY7Xays7OJjLz0m3lD5m9FyyX3r/Vq1L3LTAfA1jUWAgMur8HOsdXXyD4L8u+myeS113rJvbuy\neS046927N9u3b+fuu+9m//79dOt2LotuSUkJEyZMYPny5ZjNZvz9/VFVlY8++ogffviB5557jqys\nLEpKSggPD6+3LV8vjBSXryUsbBWXp7H3LuToMVRrIEVOFQrKLqtN1WrHBpT/cIwS+XfTJPLaa73k\n3rVuPt0QMGDAAHbu3MnQoUMBmD9/PikpKZSVlTFkyBDuu+8+HnnkEYxGI927d+f+++/H5XIxa9Ys\nhg8fXnvOz0fbhBCtkK5jOH0KV4eOTbqMFlG9Ld6QdtIDnRJCiJbJa6k0mpN8gmi95BNg69WYe6dk\nZdHmmq5U3nwrZU/NblK7tkeHoYWEkv/NgSZd55dOXnutl6fu3cqVyezZ8w1OpxNVVZk06YnajPgt\n3bJlbxIW1obBgx/06HUrKyvZsuWfDBo0uEFtvPnm6+zZsxtFUZgwYTLXX9+n3jZ8OnImhBBu7s0A\nWhM2A7hp4ZHVOz81DWRkXVwB4uMtbNrU8P+OVRU0zXrJY+6910l8vOOij584kcpXX+0gMXE5AEeP\nHiEhIZ7k5DUN7ocveSuVR25uDps2bWDQoMH1tnHkyGEOHfoPb72VTGZmBk8/Pc1jfz8JzoQQXmc4\n5cHgLDIS47EjqFmZtfU2hRCNExgYSFZWFikpG+jX72a6do0jKeldAI4fP8bSpYvRdZ3g4GBmzZqL\nn58/L7+cQGrqcSIj25Kefpp33llDQkI8d955F/363czXX3/Ftm3/Zvbs59i2bSvvv78GVVW59trr\nmDBhMsuWvUlmZgb5+XlkZmYyZcqT9O17Ezt3fkFychK6Xl0Dc8aM2ezfv5ekpERUVaVDh47MmDEb\no7H+kOXDD9eydesWFAX69x/IQw8NJSEhHrPZTEZGBrm5OcyZ8xxxcd1JSVnPRx99QFBQMCaTkf79\nB/Lddwc4eTKV5OS3Afjyy8/Zvv1TiooKGDNmYp3C53Fx3Vmy5DUAMjLOEhTkuU0aEpwJIbyuNjir\nZ+d1Q7gDPPXUKQnOxBUhPt5xyVGun6ue1iyt/8BLXiOCBQuWsG7d+7zzThJ+fn6MG/c4t9/+Pyxc\n+CJz5sQTExNLSsoGVq9eQZcucVRUVJCU9C45OTkMG1Y91ecuweT+HqqLgS9f/hbLlq3EYrHwwgtz\n2b17F4qiYDabWbz4L+zevYu1a1fTp8+NvPrqIpKSVmC321mzZiVZWVm8/HICiYnLsdvtvP3239i8\nOYV77x18yed04kQq27ZtJTFxGZqm8eSTk+nb92YURaFt2/bMmDGbTZvWs3Hjx4wdO5HVq1eQnPx3\nTCZTbbH0UaNGk5p6jEcfHcOyZW8SHh7JzJlz2LdvD2vWrKgTnEF1mrA333yddeve589/ntGke/JT\nEpwJIbxOPX0K8NDIWc01DKfTcPa7qcnXE+KXKD39DFZrILNmzQXg8OFDTJ8+heuvv4G0tBMsXjwf\nAKfTSVRUNFarlR49rgGgTZs2xMZ2Ou+a7ryk6emnKSjIZ/r0KQCUlZWRnn4GgK5d4wCIiIikstJB\nYWEBQUFB2O3VFT+GDRtBfn4eubm5PPvsTAAcDgd9+9b/Wk9NPU5mZkZtoFVSUlxboD0urlttuwcP\nHuDMmTPExl6FxWIBoGdNzd+fLsNXFKV2DV5oaBgVFRUXbHf8+EmMGPH/GD/+UXr1up727TvU29f6\nSHAmhPA69+5KzwRnNTs2a0bjhBCNd+zYUTZu/JiFC1/BaDQSFRVFUFAQBoNKdHQszz47j4iISPbv\n30thYSF+fn5s2rSeIUMepqioqDboMZvN5ORkA9VrsADatetAREQkr776BgaDgZSUDXTv/it27NgO\n1F3HFRISSnFxCUVFRdhsNpYuXcLAgb8jIiKChQtfISDAyo4dn2Gz2ep9TjExsXTq1JklS/4CwNq1\nq+jcuQufffZp7THu4Ktjx46cOnUSh8OByWTi0KH/EBMTi6qqtUFmffsl9+79ls8++5Qnn5yJ2WzG\naDR6LMOEBGdCCK9TT59CCw6Gmk+pTVE7rVkzGieEaLzbb7+DtLQTjBkzEn9/f3RdZ9KkqVitgUyf\nPosXXpiLy+VCURRmzZpLx45R7Nu3hwkTRhMSEorZbAZg0KDBzJ8/jy1bNhMVVZ1E3m63M3TocCZP\nHovLpdGuXXsGDLgLqLuQ3z0lOm3aTJ566glUVSUurjtXX92DqVOnMX36VHRdw2oN5Jln5p33HFat\nSiYlZQMAVquVpUsT6dPnRiZOfIzKykp69OhJeHhEnXbdX4OD7QwfPopJk8Zis9lwOBwYjUZCQkJx\nOqtITHwNi8VyXn9/6rrrerNt21YmTnwMTdN48MEhtG1CebqfklQawqdkO3/r1eB7p+u0iY7A1SGK\nkiVLm96ww4F96B+ovO12Ctdtavr1fqHktdd6tYR7N3z4Q6xe/aFP+9AULpeL1avfZeTI0ei6zuTJ\n4xg3blKzFD+XVBpCCJ9T8vNQHA60BlT7aBCLBS3YLtOaQviQt1JZNBeDwUB5eTmjRz+CyWSiR4+e\nzRKYNZQEZ0IIr1LTq2tq6mFhHrum1qYNhtOnQNehlf8nIURrtGrVB77uQpONHz+J8eMn+bobFyQZ\nHIUQXmU4Wx2caW08NHJWcy2lshIlN9dj1xRCiJZCgjMhhFeptcFZG49dU6+5liEj3WPXFEKIlkKC\nMyGEV9WOnIV5LjjTwqpH4dxTpkIIcSWR4EwI4VVqTfJJ3aPTmtXr19yjckIIcSWR4EwI4VXq2XR0\nRUELCfXYNd2jcIaMsx67phC/NCtXJvPEE48zefI4pkyZwA8/HPZ1lxps2bI3Wb9+ncevW1lZSUrK\n+ka1UVFRwaOPDmPXrv/PY/2Q3ZpCCK8ypJ9BD7aDyeSxa7pH4dyjckK0ZpmZz1BUtL7Bxx87pqBp\nl05RarMNpm3bFy/6+IkTqXz11Q4SE5cDcPToERIS4klOXtPgfviSt1J55ObmsGnTBgYNGtzgNl55\nZSGqqni0TxKcCSG8R9dRM87iio7x6GW1kFB0RZFpTSEuU2BgIFlZWaSkbKBfv5vp2jWOpKR3ATh+\n/BhLly5G13WCg4OZNWsufn7+vPxyAqmpx4mMbEt6+mneeWcNCQnx3HnnXfTrdzNff/0V27b9m9mz\nn2Pbtq28//4aVFXl2muvY8KEySxb9iaZmRnk5+eRmZnJlClP0rfvTezc+QXJyUnoenUNzBkzZrN/\n/16SkhJRVZUOHToyY8ZsjMb6Q5YPP1zL1q1bUBTo338gDz00lISEeMxmMxkZGeTm5jBnznPExXUn\nJWU9H330AUFBwZhMRvr3H8h33x3g5MlUkpPfBuDLLz9n+/ZPKSoqYMyYiecVPl+zZiXXXtvL4/dH\ngjMhhNcoOTkolZUe3QwAgMmEHmzHICNn4grQtu2Llxzl+jlPVAgID49gwYIlrFv3Pu+8k4Sfnx/j\nxj3O7bf/DwsXvsicOfHExMSSkrKB1atX0KVLHBUVFSQlvUtOTg7Dhj0InCvB5P4eoKioiOXL32LZ\nspVYLBZeeGEuu3fvQlEUzGYzixf/hd27d7F27Wr69LmRV19dRFLSCux2O2vWrCQrK4uXX04gMXE5\ndrudt9/+G5s3p3DvvYMv+ZxOnEhl27atJCYuQ9M0nnxyMn373oyiKLRt254ZM2azadN6Nm78mLFj\nJ7J69QqSk/+OyWSqLZY+atRoUlOP8eijY1i27E3CwyOZOXMO+/btYc2aFXWCs2+//Yb09NMMGzab\nAwf211uLszEkOBNCeI071YUnNwO4aWFhkohWiMuUnn4GqzWQWbPmAnD48CGmT5/C9dffQFraCRYv\nng+A0+kkKioaq9VKjx7XANCmTRtiYzudd013wfD09NMUFOQzffoUAMrKykiv+SDVtWscABERkVRW\nOigsLCAoKAi73Q7AsGEjyM/PIzc3l2efnQmAw+Ggb9+b6n1OqanHyczMqA20SkqKawu0x8V1q233\n4MEDnDlzhtjYq7DU1Pvt2fNaoG6xc0VR6NatOwChoWFUVFTUae8f/9hIZmYG//u/4zl16iRHjvxA\nWFgbunTpWm9f6yPBmRDCa9ypLjyZ48xNaxOO8fgxlNzc2rxnQoiGOXbsKBs3fszCha9gNBqJiooi\nKCgIg0ElOjqWZ5+dR0REJPv376WwsBA/Pz82bVrPkCEPU1RUVBv0mM1mcnKyAThypHpDQbt2HYiI\niOTVV9/AYDCQkrKB7t1/xY4d24G6H6RCQkIpLi6hqKgIm83G0qVLGDjwd0RERLBw4SsEBFjZseMz\nbDZbvc8pJiaWTp06s2TJXwBYu3YVnTt34bPPPq09xh18dezYkVOnTuJwODCZTBw69B9iYmJRVbU2\nyKxvJOy5586Ndr700vPceeddHgnMQIIzIYQXqWerPy17fFqTuolonRKcCdEot99+B2lpJxgzZiT+\n/v7ous6kSVOxWgOZPn0WL7wwF5fLhaIozJo1l44do9i3bw8TJowmJCQUs9kMwKBBg5k/fx5btmwm\nKqp6bandbmfo0OFMnjwWl0ujXbv2DBhwF1B3Ib97SnTatJk89dQTqKpKXFx3rr66B1OnTmP69Kno\nuobVGsgTcKTBAAAgAElEQVQzz8w77zmsWpVMSsoGAKxWK0uXJtKnz41MnPgYlZWV9OjRk/DwiDrt\nur8GB9sZPnwUkyaNxWaz4XA4MBqNhISE4nRWkZj4GhaL5bz+NhdF9+QkqY80de5d+I4n1k4I32jI\nvbO+8BwBr/0fxS8twnX1rzzavuXjdfivWE7hyveovOtuj177l0Bee61XS7h3w4c/xOrVH/q0D03h\ncrlYvfpdRo4cja7rTJ48jnHjJjVL8fPw8KB6j5GRMyGE17hTXXhnWjOsThtCiObTnKNI3mAwGCgv\nL2f06EcwmUz06NGzWQKzhpLgTAjhNWr6GXRFQfdgAlo3SUQrhO+sWvWBr7vQZOPHT2L8+Em+7sYF\nSYUAIYTXGM6mo9tDoAH5iRpLEtEKIa5UEpwJIbxD01AzMrwypQmSiFYIceWS4EwI4RVKdjaKswrN\nCznOAElEK4S4YklwJoTwCncCWm+NnEF1Ilo142x1IlohhLhCeG1DgKZpxMfHc+TIEUwmEwkJCURH\nR9c+/sknn5CUlISiKNx7772MHDmy3nOEEK2HOwGt7oUcZ261iWjz8tDDwrzWjhBXopUrk9mz5xuc\nTieqqjJp0hO1GfFbumXL3iQsrA2DBz/o0etWVlayZcs/GTRocIPaePrpJyksLMRoNOLn58eiRUs9\n0g+vBWdbt26lqqqKtWvXcuDAARYsWMAbb7wBVOcXeeWVV1i3bh0BAQHcc8893Hvvvezevfui5wgh\nWheDOwGtt6Y1+Uki2rNncEpwJlqp+MwzbCrKb/Dx6jEFTbv0aPG9thDi23a86OMnTqTy1Vc7SExc\nDsDRo0dISIgnOXlNg/vhS95K5ZGbm8OmTRsYNGhwg9o4c+YMq1a97/F+eC0427t3L7fdVl0gtFev\nXnz//fe1jxkMBjZv3oyqquTk5KBpGiaT6ZLnCCFal9rSTd4cOQur2bF59ixc08tr7QhxpQkMDCQr\nK4uUlA3063czXbvGkZT0LgDHjx9j6dLF6LpOcHAws2bNxc/Pn5dfTiA19TiRkW1JTz/NO++sISEh\nnjvvvIt+/W7m66+/Ytu2fzN79nNs27aV999fg6qqXHvtdUyYMJlly94kMzOD/Pw8MjMzmTLlSfr2\nvYmdO78gOTkJXa+ugTljxmz2799LUlIiqqrSoUNHZsyYjbEBu74//HAtW7duQVGgf/+BPPTQUBIS\n4jGbzWRkZJCbm8OcOc8RF9edlJT1fPTRBwQFBWMyGenffyDffXeAkydTSU5+G4Avv/yc7ds/paio\ngDFjJtYpfJ6Xl0tJSTFPPfVnSkqKeeSRR7nlll975P54LTgrKSkhMDCw9meDwYCmaahq9TI3VVXZ\nsmUL8+bN44477iAgIKDec4QQrYfaHGvOJBGtuALEt+14yVGun/NEhYDw8AgWLFjCunXv8847Sfj5\n+TFu3OPcfvv/sHDhi8yZE09MTCwpKRtYvXoFXbrEUVFRQVLSu+Tk5DBsWPVUn7sEk/t7gKKiIpYv\nf4tly1ZisVh44YW57N69C0VRMJvNLF78F3bv3sXatavp0+dGXn11EUlJK7Db7axZs5KsrCxefjmB\nxMTl2O123n77b2zenMK99w6+5HM6cSKVbdu2kpi4DE3TePLJyfTtezOKotC2bXtmzJjNpk3r2bjx\nY8aOncjq1StITv47JpOptlj6qFGjSU09xqOPjmHZsjcJD49k5sw57Nu3hzVrVtQJzpxOJw8//Ah/\n/OPDFBYWMnHiY1x9dQ9CQkKadG/Ai8FZYGAgpaWltT9fKMgaOHAgAwYM4Omnn2b9+vUNOudCGlIK\nQbRccv9ar0veu9xsUBSCYzt4Jc8ZADHV/6EFFeYQJP+OGk1ee61XU+/dqVOniI5uyyuvLALg+++/\nZ+zYsQwY8FtOnTrJ0qUvA9UBSGxsLEVFOfTrdwPh4UGEhwfRtWsXwsOD8PMzERzsT3h4EEFBFvz8\nTJSW5lJUVMCsWX8GoLS0lKKiHKxWC9HR1xIeHkS3bp3QdSdGo5OQEDtdu0YBMHXq4+Tm5pKXl8u8\nebMBcDgc3HrrrXWes9VqISjIr87vdu9OJzs7i2nTqhPLlpWVUFycg5+fiRtvvK6m37EcPfpfSkvz\n6NYtjo4dqz889u17A0FBfoSEBGAyGQgPDyIw0I/Y2OrzOneOwuWqqtNeSIg/V101Cn9/fyIjg7nm\nmh4UF2cTF9f0tfJeC8569+7N9u3bufvuu9m/fz/dunWrfaykpIQJEyawfPlyzGYz/v7+qKp6yXMu\nxdc1xsTlawk14sTlqe/ehZw+g2qzUVRSCVR6pQ+qXxA2oOJoKsXy76hR5LXXenni3n3zzX42bvyY\nhQtfwWg0EhgYhtUaSH5+GVFRMTz99HNERESyf/9eCgsL8fPzY9Om9dxzzx8oKirixImTZGcXo2kK\nx4+folu3Ynbv3kdFRRX+/iG0aRPBokWvYTAYSEnZQExMHGlp6VgsDrKzi8nPL6OqyoWmmcnPL+T4\n8XRsNhtLly5h4MDfER4ewYsvLiIgwMqOHZ9hs9nqPOfSUgcWS0Wd34WEtCU6uhNLlvwFgLVrV9Gm\nTQcqKqooLCwnO7uYgoIyKiqqsFpDOXr0GGfO5GAymdizZx/h4e0pKCjH4agiO7uYkpKK2jbc/f1p\ne19//RXr1r3HokVLKSsr49ChH7DbI+u9Nz6trTlgwAB27tzJ0KFDAZg/fz4pKSmUlZUxZMgQ7rvv\nPh555BGMRiPdu3fn/vvvBzjvHCFEK6TrGLIycUVEerUZraYslColnIRolNtvv4O0tBOMGTMSf39/\ndF1n0qSpWK2BTJ8+ixdemIvL5UJRFGbNmkvHjlHs27eHCRNGExISitlsBmDQoMHMnz+PLVs2ExUV\nA4Ddbmfo0OFMnjwWl0ujXbv2DBhwF1B3Ib97SnTatJk89dQTqKpKXFx3rr66B1OnTmP69KnouobV\nGsgzz8w77zmsWpVMSsoGAKxWK0uXJtKnz41MnPgYlZWV9OjRk/DwiDrtur8GB9sZPnwUkyaNxWaz\n4XA4MBqNhISE4nRWkZj4GhaL5bz+/tRNN93Ct99+w/jx/w9VVZkwYTI2W7BH7o+i660/QZB8+mu9\n5NN763Wpe6cUF9Gmc0eq+txA6TPPe7UftlEPo0VEkv/VHq+2c6WR117r1RLu3fDhD7F69Yc+7UNT\nuFwuVq9+l5EjR6PrOpMnj2PcuEnNUvzcpyNnQohfLjUzEwAtxPvpLfSQ0Nr2hBDNw1upLJqLwWCg\nvLyc0aMfwWQy0aNHz2YJzBpKgjMhhMepWdXBku6BXUv10ULDMKWdhJIS+MlubyGE96xa9YGvu9Bk\n48dPYvz4Sb7uxgVJjgohhMepmRlAdeDkbVpo9bozw48yeiaEuDJIcCaE8Ljaac2awMmb9JoAUKY2\nhRBXCgnOhBAeVzut2YwjZ+7ROiGEaO0kOBNCeJya5Z7WbIaRM3c6jawsr7clhBDNQYIzIYTHqRkZ\n6IqCHmz3elu1uc5k5EwIcYWQ4EwI4XGGzIzqwMxg8Hpb7k0H7tE6IYRo7SQ4E0J4lq6j/pjVLFOa\nALrdjq4oqBkSnAkhrgwSnAkhPEopLkIpL2+WzQAAGI3otmCZ1hRCXDEkOBNCeNS56gDeT0DrpoWG\nyoYAIcQVQ4IzIYRHuUewmm3kDNBDQ1HLSlFKpFakEKL1k+BMCOFR7hxnzbXmDM7V8JREtEKIK4EE\nZ0IIj2rOouduuiSiFUJcQSQ4E0J4lDulhR7anGvO3CNnEpwJIVo/Cc6EEB7li5GzcyWcZFpTCNH6\nSXAmhPAoQ2YGuqKiBwc3W5u105pZEpwJIVo/Cc6EEB6lZmag25unOoBbbQknqRIghLgCSHAmhPAc\nXUfNymzWnZoAerAdXVGlSoAQ4oogwZkQwmOUokIUh6N2mrHZGAzo9mAMsiFACHEFkOBMCOExvtgM\n4KaFhFWvOdP1Zm9bCCE8SYIzIYTHuFNZNPe0JoAeFopSUYFSVNjsbQshhCdJcCaE8BhflG5yq60S\nIDU2hRCtnARnQrRA2c4q1ubn8EVJEYUup6+702DuwKg5i567aVIlQAhxhTD6ugNCiLrOVlVy34kf\nOFVVCUCQqvLPTt3p5ufv457V71x1gOYfOdNDJDgTQlwZZORMiBYkq6qKwTWB2V1Bwfw+yE6xpjEh\n/QSVmubr7tXLkNn8Rc/dpEqAEOJKISNnQrQgCT+mc7KqkgeDQxluD0NRFCp0jU9Ling5O4NnIjv4\nuouXpGacRVdVdFvzVQdwc4/WSSJaIURr57XgTNM04uPjOXLkCCaTiYSEBKKjo2sfT0lJYcWKFRgM\nBuLi4oiPj0dRFB544AECAwMBiIqK4qWXXvJWF4VoUU5VOvigIJeOJjPDagIzgMdCIzhYUcZfczIZ\nExpOW5PZxz29ODUrE90eAmrzD8rXjpzJhgAhRCvnteBs69atVFVVsXbtWg4cOMCCBQt44403AKio\nqGDp0qWkpKRgsViYNm0a27dv59ZbbwVg5cqV3uqWEC3WX3IycQEPBYei1gRmAP6qygO2UN7M+5EP\nCvP43zZtfdfJS6mpDuCKjvFN87ZgdFXFkHHWJ+0LIYSneO3j7d69e7ntttsA6NWrF99//33tYxaL\nhffeew+LxQKA0+nEz8+Pw4cPU15ezmOPPcaoUaM4cOCAt7onRIuSUVXJ3wtyaWs08Wtr0HmP/9oa\nhBF4ryAXvYUmWVUK8lEqK32yGQAAVUW3h8iGACFEq+e1kbOSkpLa6UkAg8GApmmoqoqiKITWTEGs\nXLmS8vJybrnlFo4cOcJjjz3GH//4R06ePMnYsWP55JNPUOuZIgkPP/8/M9F6yP2Dv508SZWu80jb\nSMLsAec9bgduLQrm88JCTvnBDbaW8Terc++y0gAwtY3AfoHn0DwdagMnTxLeJhB+MvooLkxee62X\n3Lsrm9eCs8DAQEpLS2t/dgdmP/150aJFpKWl8dprrwEQGxtLTExM7fd2u53s7GwiIyMv2VZ2drEX\nnoFoDuHhQb/4+6frOivPZmJC4XrVQkFh+QWP+7XFyucUknjyNAvaRV/wmOb083tnOnQcO1ButeEo\nKPNJn6zBIZgcP5Bz9FRtag1xYfLaa73k3rVuDQmsvTat2bt3b3bs2AHA/v376datW53H586dS2Vl\nJa+//nrt9OZHH33EggULAMjKyqKkpITw8HBvdVGIFuGQo4KjlRX0CQggQDVc9Ljr/a0EqwbWF+ah\ntcCpzXPVAXwXFEk6DSHElcBrI2cDBgxg586dDB06FID58+eTkpJCWVkZPXv2ZN26ddxwww2MHDkS\ngFGjRvHQQw8xa9Yshg8fXntOfVOaQrR26wvzALjNarvkcQZFoU+AlW0lRfynopxr/H00dXgRapbv\nip67abXpNDJxXf0rn/VDCCGawmvBmaIoPP/883V+16lTp9rvDx06dMHzFi1a5K0uCdHi6LrOR4V5\n+CkKffyt9R5/jV8A20qK+LK0uOUGZz4cOdNrykbJpgAhRGsmw1JC+NCBijJOVVXSNyAQSwNGia+p\nKeH0RWmRt7vWaO7qAL5c63Wu+LlMawohWi8JzoTwoS3FhQDcFBBYz5HVwowm2htNfFVaQlULW3em\nZp5FNxjQbZeenvUm96idQUbOhBCtmARnQvjQp8WFGIBrGzFFea1/AGW6xoHy0voPbkZqpu+qA7jV\nlnCSDQFCiFZMgjMhfCTHWcX+ijK6W/yxXmKX5s9d41cdyH1Z2oK20us6alYWmo/TV+hBQegGo9TX\nFEK0ahKcCeEjn5UUoQO9A+rfCPBTPWuCsx0lLSc4U/LzUKoq0cJ8t1MTqK4SEGJHzZDgTAjReklw\nJoSPfFpSvai/dwN2af6UzWAgymRmb3lpi8l3praAzQBuWmhYdfHzFvK3EUKIxpLgTAgfcOk620sK\nCTUYiDGZG31+F7MfZbrG8UqHF3rXeO7UFb5Mo+GmhYahOKtQ8vJ83RUhhLgsEpwJ4QPfVZSR53Jx\nvb8V5TJqQHauqaqxv4VsCnCnrvBZ0fOfcFcoUDPO+rgnQghxeSQ4E8IHvqhZL9bL7/ISyXY2+wFw\noNw3NSx/ztDCRs4ADLIpQAjRSnmtQoAQ4uJ21CSRvdws/53MFlS8O3LmdOZQXLyZ0tLP8PO7ltDQ\nsajqhft7rjqA70fO3DtGJZ2GEKK1kuBMiGZWoWnsKish2mTGbri8l6BFVYkymTlYUY5T1zFextTo\npZSW7iAt7Y/oejkAhYUfkJPzGh06vEZQ0N3nHX9uQ0CIR/txOc7lOpORMyFE6yTTmkI0s2/LS3Ho\nOr2aWBuzs8WPcl3jqKPCQz2rVla2i7S0Ieh6FXb7KNq3f43g4KFoWgGnT4/G4Th+3jlqZga6wYge\n5LvqAG7uqVUZORNCtFYSnAnRzL6oSaFxzWWuN3Pr4l53VuG5dWeVlWmkpT2IrlcQEfE0dvsQzOar\nCAkZQVjYVHS9lPT0seh6VZ3z1MwM9NAQ8PAI3uXQw9wjZ7IhQAjROklwJkQz21FajAr0qClifrnc\nOzY9WcYpI2MmmlZEWNgkAgJurvNYYOBvsVp/S3n5t2RnLzn3gKah/phVW3Tc1/QAK7rZLIlohRCt\nVr3BWVJSEtnZ2c3RFyGueMUuF/vLS+li8SOgESWbLiTWVL0pwFM7NouLN1NS8k8slp4EBt51wWPC\nwh5HVe3k5v4Vp7N6x6mSm4vidPq+OoCboqCFhMqaMyFEq1VvcOZwOHjkkUcYO3Ysmzdvpqqqqr5T\nhBAX8VVZMS4uP4XGT5lVlXYmM4cd5ehNzIavaeVkZMwADISFTbpo7jVVtWKz/R5NKyIz893q32W1\nnM0AbnpoKGpONjidvu6KEEI0Wr3B2eTJk/nXv/7F+PHj2bVrF/fffz/z5s3j0KFDzdE/Ia4o7vxm\n13ogOAOIMZkp0TTOOpv2oSk//12qqk5hs92H2Rx9yWODgu4BjKSnL0XXtdp8Yi0hjYabFtYGRdOq\nAzQhhGhlGrTmrKKigjNnznD69GlUVSU4OJiEhAQWL17s7f4JcUX5orQIs6LQzc/PI9eLNlevOztc\nUX7Z19C0SnJyXkVRLAQH/7He4w0GO4GBd1BefoySki21uyK1FlBX0+1crjOZ2hRCtD71JlmaNm0a\nX3/9Nb/5zW+YOHEiN9xwAwCVlZX8+te/Zvr06V7vpBBXgmxnFYccFfTyC8CkeGYvTnRNXc5DjnL6\nBwVf1jUKC9/D6TyLzXY/BkPDrmGz3UdJyb/JzX2LyMx+QMso3eR2roRTBlzn484IIUQj1Ruc3Xzz\nzcybNw+r1Vr7u8rKSsxmMykpKV7tnBBXki9La6Y0m5jf7KeaOnKm6y5ycpYARmy2PzT4PLP5Kvz9\n4ygt/Qwtry3QMko3uWlhkohWCNF61fvx/YMPPqgTmLlcLh588EEAIiIivNczIa4wnl5vBtDWaMKE\nwuHLTERbXPwPKitTCQzsj9HYplHnBgf/BnBSaDsInButagncaT1Uqa8phGiFLjpyNmLECHbv3g1A\n9+7da39vMBjo37+/93smxBXm89IirIpKp5rRLk8wKAodTGaOOMpx6TqGRiaBzc19EwCbbXCj2w4O\nvo3MzLfJiz5JB5MJ3RrY6Gt4iy5VAoQQrdhFg7OVK1cC8OKLL/LMM880W4eEuBKlOio4XVXJTQGB\njQ6g6hNjNnOyykFapYOrLA3faFBRcZiysi/w87u23h2aF+LnF43JFEVB99NUtY1oEdUB3NwbAgwy\nrSmEaIUuGpxt376dO+64gx49erB+/frzHh88uPGftIX4pdpeU7Lpeg+uN3OLNlugtJjDjopGBWd5\neW8BEBQ06LLbDvC/mcKq0+T92oRn9p96iL8/un8AaoaUcBJCtD4XDc4OHjzIHXfcwa5duy6YkFKC\nMyEa7tOSQgCu97fWc2TjuXdsHnaUcw/2Bp3jchVRUPB3DIYwAgJuuuy2gyqvoZD3yelbScfLvop3\naCEhsiFACNEqXTQ4mzJlCgALFiyo/V1xcTEZGRnExcV5v2dCXCEqNI2dpSV0NJkJN5o8fn33js0f\nGrFjs6Dg7+h6KUFBf0BRLr+MlH9OEJYyKOhWQAeHhtKCyvVqYW0wHTwADgdYPLfOTwghvK1BuzVn\nzZpFbm4uv//975kyZQr/93//1xx9E+KKsKushHJd88qUJkAbgxGzonCksmE7NnVdr5nSNBIU9Lsm\ntW3Myse+F5z+VVSoJ5t0LU/T3ek0smRTgBCidak3OFuzZg0zZ87kH//4B/379yclJYUvvvii3gtr\nmsbcuXMZOnQoI0aM4NSpU3UeT0lJYciQITz88MM899xz6Lpe7zlCeMrpSgdv5GQxPO0otx37D79P\nPcyT6Wkc9FAR8Z/aVrPerLcXpjQBVEWhndFEqsOB1oAam6Wln1NZeRSr9TYMhoZNg16MISuXkL3V\n3xebDjTpWp52rkqABGdCiNal3iS0AHa7nc8//5wRI0ZgNBpxOBz1nrN161aqqqpYu3YtBw4cYMGC\nBbzxxhtAdTmopUuXkpKSgsViYdq0aWzfvh2n03nRc4TwhCOOcpb8mMHGonxcNb/zV1QqdI3d5aWs\nKsjhnqBgXuvQiSDD5U/3/dSnJYWYFYVfWfw9cr0LiTJZSKuqJMNZRYeaNWgX44mNAG6G7HwC91V/\nX2w8QITjgSZf01PctT4l15kQorWpNzjr0qUL48eP5/Tp09xyyy1MnTqVa665pt4L7927l9tuuw2A\nXr168f3339c+ZrFYeO+997DUrANxOp1YLBa++eabi54jRFMUu1wsyj7L27k/4qS6YPggWwjX+wcQ\nZjSh6Tr7ykv5oDCPfxYXcuLEYVZHd6Wj+dKBTn2OOSo44qjgBn8rZtV767Ham6rXsh11VFwyOKus\nPE1x8T8xm7tgsXRrcrvGrFz8csDksFNqPoiOE6Vhn/m8zp3rzCA7NoUQrUy976IvvfQS+/fvp2vX\nrpjNZh544AF+/etf13vhkpISAgPPJaU0GAxomoaqqiiKQmjNG+fKlSspLy/n1ltvZfPmzRc951LC\nw4Pq7Y9oubx5/3Rd593MTGampvJjVRXtzGYeb9+eW22283Yh97cH8Nu2bXgtPZ0Nubk8ePoou/v0\nIcx0+Yv430rLA2BAm1Dswd4bOeuqBUJhHlkm/ZJ/z9TUVYBGZOQfsNubPs1qyavehWpT48hVvkG1\nnyaYHk2+rkdEtwcgsCiPQHmPuCB572y95N5d2eoNzsrKyvjhhx/YtWtX7e++//57Jk+efMnzAgMD\nKS0trf3550GWpmksWrSItLQ0XnvttQadczHZ2cX1HiNapvDwIK/dv7NVlUxNP8nnpcWYFYVh9jDu\nt4VgRqWw6OKL50cFhmJ06qwrzOP+fQd4PzYO02UmWP17RiYGoAdmCgovr/5lQ4RUVX/dn1dItuXC\nxcs1rYL09DdR1SAU5SYKC5u2vi44OAD9zI9oZhNGZ2cwfUNG+S50R6cmXddTFHMgwUBFahrF8h5x\nHm++9oR3yb1r3RoSWNcb+UydOpVvvvkGvQELjX+qd+/e7NixA4D9+/fTrVvdKZS5c+dSWVnJ66+/\nXju9Wd85QjTUZyVF/ObYf/m8tJjefgH8tUMsf7SHNWhqUakJ5Pr5W9lZVsK8zDOX1YdURwX/qSjn\nOv8ArB5av3Yx7WumMo9eosZmUdHHuFx5BAYORFU9k1rC8GMurtAg/F2dASgxtpxNAXpICCC7NYUQ\nrU+9I2e5ubkkJyc3+sIDBgxg586dDB06FID58+eTkpJCWVkZPXv2ZN26ddxwww2MHDkSgFGjRl3w\nHCEaa0VeNjMzTqGiMCEsgoGBwRdMpHwpqqIwNbwdT2Wc4s28HxkQFMxvAm2NukZKUQEAtwR4f/rB\nX1UJNRg4dongLDf3LUAhKOgezzTqdGHIKcRxdQwGrJhckZQaf0DHhULTg9Eqp4LJ2LgPhXWYTGhB\nNqkSIIRodeoNzq6++moOHz5cp/h5QyiKwvPPP1/nd506nZvuOHTo0AXP+/k5QjTG6vwcpmecwqYa\nmBXRnu5+l7/Oy19VmdqmLTMzTjEl/SQ7uvTA1sARMF3XWVeYiwHoG9A8BcE7mMwcrCinVHNhVev2\ns7x8DxUVe/D374vJ1NYj7SnZ+Si6jhZWHbT6adEUG7IoN5wgwNXlsq556kc//rGrLSlfR/L1oVAi\nQhzcGFfADXH5DPufdNoEVzbqenpIqFQJEEK0OvUGZ0eOHOGBBx4gLCwMc83ONUVR+PTTT73eOSEa\n4/OSImacTSNQVXmpXVS9KSUaoovFjz8Gh/JeYR5zMk7xWseGraf6qqyEQ44Kbg0IJNDLU5pu7uAs\n1eHgmp8lvM3OfhUAm+0+j7WnZuYC4AqtHhn0c8VQbNpNqeFQo4MzlwsWvteVxR92QdcVFHQ6tSsj\nr8jEpq/bsunrtiSmdGLFU3u5sVtBg6+rhYVhOnUSpaQYPVAWUAshWod6g7O//vWvQHVA1th1Z0I0\nl6yqKkafPg4ozIro4JHAzO0hexi7y0t5rzCPe2wh3G2rP3HrW7lZAAyyhXisH/VxP+djlRV1gjOH\n4wjFxRsxm7vg53edx9pTM3MAcIXWjJy5ogEoMx6GynsbfJ2cQjNj/68Xnx0IJ8JewUO/OctN3fMJ\nCapC1yG70Mz2/eGs2daR38+5iYVj/8OjA0/TkJlqrWZXuJqViUuCMyFEK1Hv6uiOHTuyd+9e3n//\nfUJCQvj222/p2LGllTgWv3TxWacp1jRGh4bzqyZMZV6IUVF4ok1bTCj8+Wwa2c6qSx6fVungX8WF\ndDZb6Gbx82hfLqU2OPvZurOcnFcBneDgIY1ee3cpys9Gzkx6G1TdnxLDfxt8jcOnA/nNk7fy2YFw\nbozLZ+njB7n7xh8JCar+GysKRNgr+dNv03l+1CH8zC6e/Ns1LHq/YSNzujsRrVQJEEK0IvUGZ4sW\nLTs+tooAACAASURBVOLzzz9ny5YtOJ1O1q1bJwv1RYvyVWkx6wrz6Wy2MDDowmkkmirKbGF4SBvy\nXE4mnzmJ6xKjyG/l/ohO9aiZoigorjyMZVsxlXyAqWQdxvIvUFyFHu9jB2N1cHa88lwFj6qqMxQU\nrMVo7EBAwM0ebU/NqDtypqDg54qmyvAjVUpeveefzbXw4PM3kpHnzyP9T/PM8B8I9Hdd9PjrOhfx\n6uMHibBXsOC9rnxxMLTeNmqrBMi6M/H/s3feYVLV9/5/nXOmz+zObG/sskvvHWkiNuyxohIVvQlq\nbuI1UWP8Re+NRpMYE5Nr4s3VJBo1wYINRVBEEVRAEQTpnWXZ3uv0cs7vj9ldyvbdKeD9vp4nzxPm\nnPP5fJeRmfd+qkBwBtGjONuwYQNPPvkkRqMRu93Oiy++2D7uQiCIN6qm8fOKYiTgBykZKBGMDJ3K\ndxIdTDFbWOdq5g81nX/Zf+128o/6atIVHfOkLdjKr8BeMh5b9W1Ya+/BWvtjbFULsZeMwVZ+Mcam\n55BCdRE5X4pOh47wCI82qqt/CwSx269HkiK7oeB4zdnxLlZTaDAALt3+bp9tculY8Nh0yuvM3Hph\nMTeeW0ZvFiikO/z87IbDyJLGHU9Noq65+wHB7WnNCiHOBALBmUOPH4fKKcXMfr+/w2sCQbz41NnM\nfp+Xc6wJDI9yClGWJO5JzSJd0fHHmgpea6g96bpLDXFXWREa8ID8LEk1t6L4t+MzTMRpvZmmxHto\nSrgbp+UGfIaJKP59mBt+SWLpWZjqfz1gkaZIEhk6PYV+H5qm4fHsoLHxZfT6fGy28wdkuzPkqk7E\nmRquO3MpnXdjA/gCMrc8MYV9xYlcdlYlC87p26iLUblObrmglKoGEz96eiLdlcJqYr+mQCA4A+mx\nIeCSSy7h3nvvpampiZdeeonly5dz+eWXx+JsAkGPPF9fDcB3YlR4n6Ao/Dw9m19UlfKT8mMc8fu4\nIzmdJjXIz8qLOer3cb20iqmBV/EapuG0fZ+gvvMOT1ltxORZh9X9NqbmZzG0vIw36ef4ExaB1L9f\ngLL1BrZ4XNQFg7RUPghoJCffjtRPe90hVdSimgxoluMDbY2hQaBJuHRd15394qVRbNidyszR9dx5\neVGvCvtP5dqzy9lRmMhHW9N5ftVg7rjsWKf3tUfORM2ZQCA4g+gxcjZv3jzOO+88kpKS2Lp1Kz/+\n8Y/54Q9/GIuzCQTdUujzssbZzEijiaExLLwvMJr4XVYemTo9T9dWMv7gTs4+vJcv3U5m8DW3a0/h\ntH6XRsdjXQozAFV24LZeQ03qCzTb7gRULPX/ia3iO8j+A/06W1ZrU8DuhtW43Rswm6djNk/ul62e\nkCvrCCWd3AEpY8SgZuBRCtHoWD+2clMGz32QT16am58uOIzSz0yrLMN91x3BYgzy29eG0+zu/PdM\nze5Ak2QUUXMmEAjOILr8aKyrq+Pmm29m0aJFvPLKKyiKwqZNm3j11Vdpbm6O5RkFgk55ob4GiO24\nijZy9AZ+l5XHTY4UpputjDIaeVT3HL/lZ3gTvo/Tdiu9DglJBtzWa6hNeQ6P6Tx0/h0klF+MsfFp\n0IJ9OleWLlyDtb12KZJkJDn5jr7+aL0jEESubSSU0nFrglHNQZP8eOWSk14vrjZx1/9MwKAL8f8W\nHsJkUAd0hKSEANeeXUGD08BfV+R3fpOioDnsYkuAQCA4o+hSnD322GNMnTqVjRs38uabb/Lmm2+y\nceNGRo0axeOPPx7LMwoEHQhoGq831uGQFWbGaAL/qSQqCtc7UngoI4c/6Z/hnOCreE0X4jZf1S97\nqpJEk/0BGhy/RJUTMDf+DlvFFcj+ruu3TiVbFxaEJVoKKSk/Qq/P6ddZekKpaQBOrjdrw6iGfXqU\nw+2vBYISi/84mWa3nh9cUUReemSWwH9nVgWJlgD/8+4Q6rtoDlCTU8L7NcWcRoFAcIbQpTg7cOAA\n9913H3r98Q88g8HAvffey549e2JyOIGgK75wtdCkhphjTUAXxQ7N3qDzbMDofJmAbghNiXf1PmLW\nBT7jDGpT/orbdCE6/y4Syi/B2Pgn0Lqfr4bqYUjTIwBUKVOx2S4c0Dm6Q2ltBlCTOw52NYbC4syt\nOy7Ofv/GML4+mMQ542uZP6UmYuewGFVumFeG06vjT+8M6fQeLTkFyedDamyImF+BQCCIJl2KM5Op\n8xoeWZZFt6Yg7nzQulR8RpyiZu1oIUz1v0RDoinxHpAiU/umyQk0239Kg+NRVDkRc+OT2MovQ+f+\nqNMIkBSswFa1kGzfW+gJUCaPjMg5ukLXOuMsmNpxrpxBzQJNxq0cAmDLAQf//dYw0uw+7rry6EC1\nawcunV5FSqKPv7+fT0W9scN1VQyiFQgEZxiRHXwkEMQAVdP4oKWRBFmO+DaAvmJwLkUX2IfHdCFB\n/fCI2/cZz2qNos1HF9iLrfp7JJTNw1T/S/TOtzC0LMFcey+JpTPR+b7GZzyHdEXmWFCJahZP17a6\nKbXjKisZPQY1DY9yFKcXfvCniWjAvdcdxmLqeshsfzHoNb57Xim+gMJTbw/tcF1NaRVnFWUR9y0Q\nCATRoMtRGocPH+b88zufjVRdXR21AwkEPbHN46IqGOB8W2JUh872iOrG1PA7VMmE03Zb1NyEo2j3\n4bZeh9X1OibvekzNz510T1AZhMtyHR7zRWQ1QZlPok6F1CgFuZW27QCpHWvOAIzqIPxKFU+9r+do\npZWr55QzvqAlOocBLphcy2vrBvHq2kH8180HSbQcb6TQWsWZUl5OD4lhgUAgOC3oUpx9+OGHsTyH\nQNBr2lKa8WoEaMPgfANZrcNp/S6qkhJ1f0HdYJrsD9CU+BMMgf0owRI0yUpISSGgHwetGwAylXDI\nrCggk6oMrCOyK3Rt2wE6SWsCGNVsWtjK7uoK8tLdLLqgpNP7InYeRePS6dW8/Ekur3+ac9LcMzU1\nDQC5rDSqZxAIBIJI0aU4E8vNBacrHzubMEoSE0yW+B1CUzE2P4eGDrf5O7H1LRnxGyaCYWKnl9vE\n2bGgxLQoHUEpr0GTZUKOjg0BAP6WAjDCqJFfc/mwSRj00e+UvGhaNUvX5fD8B4O5/dJj7bVtakqr\nOCsXaU2BQHBmIGrOBGcUNcEAB3xeRhvNGHuzjDFK6DxrUIJFeEznoSqxn7PWHZm6sBA6Goje34+u\nshYtJYGupsj++tlbCYVkZk37kiFZ7qid40SSbAHmjKvnYJmNDbuPL0VXU1vTmmVCnAkEgjMDIc4E\nZxSbXE4Axsa5EcDYFK75cluuies5OiOzNZVZFIhSPV4ohFJdj5rWsRkA4L3PR/PO2qlUVQwnI2c3\ndLIpIFpcPiPckfn3D/KPv2g0oSYkIJdFN7UqEAgEkaLH3ZoCwenERne4qHxcHFOasv8get8X+AyT\nul3PFC+SZDCgcTQYnd+9lLompGCoU3FWWWfjoWcuxmQIkKCkISkHkMwlaJ78qJzlVEblOinIdLFq\nczpltSZyUr0AqCmpKOVloGl4Ql62Vm1hU/kXbKv6mkp3JXWeWoJqkHRLBpnWTKZkTGP+4IuZkDYJ\nWRK/wwoEgtgixJngjGKjqwWjJMV0l+apGFxvA+AxXxK3M3SHLEG6olEUkNC0Ac/E7YBSHh4ie6o4\nC4Yk7n7ySppdJu664TOMWmb4ftt+gjESZ5IUjp79ZflQXvool/+8KTxrTU1NZT1H+fvq21lRsgpX\nwNn+jFExkWhIRJZkjjQeYk/dLj4p/pgnt/yWbGsOd0+5h5tH34ZJF7//5gQCwf8thDgTnDG01ZtN\nNFnQx2uEhqaidy5Dlcx4jTPic4ZekKXTKPXJ1IYgLcL/yttmnKnpJ4uzP74yl6/25DF7whEum7MH\nzZMHgGzbDzWxE7LzJtTxwoeDeXXtIO6/cR/vNH3OX87fxx4LUPgmaeZ0Lsibz5iUsYxOHkOi8eSO\n0xZ/CztrtrO1agtflG/kwfU/48/b/ptfzHyUBSNuRIrzRgqBQPDtR4gzwRlDW73ZuDjWmym+zSih\nctym+RHbBhAN2jo2jwZl0nSRHaehVLSubjohcvbJlqH875uzyUpt4t6b1yFJoHoGoWkSim1/RP33\nhMmgMmtMHZ98k8H4D1+gZtByFLPETTthzpzbyDxvQbepygRDAnNy5jInZy63jV3M8sNv88HRldz1\nyZ2sOfYRT857qoOgEwgEgkgiiikEZwxftNabjY1jvZnBuQwAr+m8uJ2hNxyfdRb5KE975KxVnJVW\nJ3LPH69Arwvy0Pc/xGr2h2/UjGi+DGTbQSA689Y6Y5u2iV0T7gOgdtsVXJE4i+W1l/HKMphdZ+tT\nDZndaOfWsd/nz+c9w4ikUbxz+C3OfX02++t7v4xeIBAI+ooQZ4Izhs1uJzpgWLzqzTQfetcKQnIy\nfsOE+Jyhl2S2RsuORaEpQKloqzmz0+g08m+/vJ4ml5kfLljP0EF1J92revKQFA+SuTji5ziVcq2E\nX4V+xiPqvVQPXoo+oRrTvpv5nv0akuzZAJir+rd0PcOayeNn/57rRyyk1FnCle9czDdVWyN5fIFA\nIGhHiDPBGYFXVdnn9ZBvMMat3kzn2YCsNeM1nQtSlPYiRYj2tGY0ImcVtWiyhNuWzOLHFnCwJI0r\n5+3k4lkdo0mqJxcAxXYg4udow6O5+af6DHeFbmYzG8hXR3F36NfMGluLx2Nmy45heFPCUT5za9Sv\nPyiywk2jF3HXpJ/Q7GvmmuVXsLFsfaR+DIFAIGhHiDPBGcEer4cgMMIYv3ozvedjALzGWXE7Q29J\nksEoaRRFYRCtUlGLz+7gzscvZ8u+XM6ZfIg7r9nQaVfo8aaAyIszVVNZq67iB+qNvKUtwUoiC4N3\nc0fwv8jSBjNp3FEAPt04Fm9SeAeoubr/4qyNCwdfxP3Tf44/5OPm929gV+3OAdsUCASCExHiTHBG\nsN3jAuKZ0tTQuz8iJCUQ0I+Ozxn6gCRBhqJRFAyP04gYqkqw0smtwRd5f+MwJgwv5ae3fEJXyxpU\nb3SaAvZru7hfvYOntMdo0Zo4P3QN9waeZII6E4mwSsxMbyIjrYEt24fS6LXhTbRhruxfWvNUZmXP\n4d5pP8MTdHPTygWUtYi9nQKBIHJETZypqsrDDz/MwoULWbRoEcXFHWtOPB4PCxcupLCwsP21a665\nhkWLFrFo0SIeeuihaB1PcIbxTZs4Mxjj4l/x70IOVeE3nnXapzTbyFQ0PJpEdShyqc2GQx4uCH3M\nmy1XMXZoJQ/fvgq9vptif9WE5ktHth4gEk0BNVoVf1Af4WfqnRxiL+NDM7k38CQXhhZgoON/G5PH\nFxEKKWzcPApvsh1zVS2RUquzs8/mtrGLqXJXsnDldTT7miJiVyAQCKI2SmPNmjUEAgGWLl3Kjh07\neOKJJ3jmmWfar+/atYtHHnmE6urq9rlBPp8PgCVLlkTrWIIzlG88bsySTI7eEBf/endbSnNmXPz3\nh+PjNCQydAMXJHv2JPLv35tDKXauTF/DXT8tIRQM9Pic6s1F5/gayVSG5s3tl+8WrYl3taW8q76G\nX/KRrRZwRWgR+drIbp+bOKaI1Wsn8ekXY/Gm2HEUlWFoaMafHJlRGFcOvZpqdyUfHF3Jvev+g+cv\n/peYgyYQCAZM1CJn27ZtY+7cuQBMnDiR3bt3n3Q9EAjwzDPPUFBwfP3N/v378Xg8LF68mNtuu40d\nO3ZE63iCM4iWUIjDfi9DDEbkuDUDfISGDr9hSlz894e2BegDHadRV2fgP/9zHFdeOYfSGju/5BF+\nO+sZDPre7czUBtAU0KI18bL6Nxar1/KG9hIGTFwXvJMfBR/rUZgB2BM95OXWsO/QII5ZhwD979js\nDEmS+P64OxmVPIYVhcv5554XImZbIBD83yVqkTOn04nNZmv/s6IoqKqK3FqcMmVKxy85s9nM4sWL\nuf766ykqKuKOO+5g9erV7c90RVpaQmQPL4gpPb1/uxsa0IBxiTYc9jg0BPjLwL+boHk6lsTk2Pvv\nJwUK0AwVshF7HwNFqgrbtiWwcmUaL72UTXOzjrw8F49M/hffX/4YezLuBMBs7kUkUw2LIlPSEfBe\n0Sv/zWoTb/lf5i3fq3hwY8POVdJtzJbnY9D1LbU9ZXw5x0rS+dB7GTNYSWpLM5IjsrPyHjv/l9y+\n4nZ+sfHnzB99LpMyJ0XUfrQQn51nLuK9+3YTNXFms9lwuVztfz5RmHVFfn4+gwcPbv//DoeDmpoa\nMjIyun2upqZl4AcWxIW0tIQe3791teEOu1x0NDZ5YnGskzC0rMYCuHVTcTu9MfffF7xuhV1fpPDN\nZ2kU1xrgl/t4ZnkKz/9mNBZLEJsthN0eIDXVR2qqj6SkACZTCJMpRCAgU1VlorrayPbtDqqrw80X\nNpufO+7Yx2WXFTPu5bUANFvCv3h5PP6eD+XLxJwDQcNevD38/R3U9rJKW8bn6hr8kg+bZuey0C2c\npZ6PASMhwEMvfJ7AiCFHgUmsLD2HRwD1aDmNje4+2egJPVbunnQvv/nqURYsvZ61N27ErItfZ3Fv\n6M2/PcHpiXjvzmx6I6yjJs6mTJnCunXruPTSS9m+fTsjR/acgli2bBkHDhzgkUceoaqqCqfTSVpa\nWrSOKDhD2OUJf5EOM8SnU1PnDc+y8p3GKc2qEjNLnxrB12vTCfpbGxZkFTwHkHLdZGa68Hh0NDYq\nlJaaCfYwnNZm83PBBaXMnFnN5Mm1GI3hYn5zq1D2OpLodbJUtaD6k1vTmhqc8qRX8/K59hEfaO9w\nhHBXZxLpzA9exPRWUTYQ7IkecnNq2FY+mmrSItaxeSrTMs/i8iFX8n7hezy55bc8POuxqPgRCATf\nfqImzubPn8/GjRtZuHAhAL/97W9ZuXIlbrebG264odNnFixYwIMPPsjNN9/c/kxP0TbBt5/d3nAz\nQLouDqtgNRWdZz0hOYWQMij2/nvA61ZY/twQVr6YT9CvkJbrZOzsakbPqCFneBP/a9RRNyjEU3/+\nsr3AVNPA5dLR0GDE5dLj88n4/QqyrJGc7CUlxUdCQqDTuWWm2lo0ScJnt9MXqax58pDt25EMNWj+\ndIJakO1s4XPtIzZpn+PBjaRJjFanMEOdzzBtHHIES2LHjyqhpCyNd7maS6sORczuqdwy+ja2VH7F\nM9uf5jtDrmJyxtSo+RIIBN9eovZtJ0kSjz766EmvnVj838aJnZk6nY4nn3wyWkcSnIF4VJVCv4+R\nRlNcuuDkwD5ktR6P6UI6VStxpPyolSfunEpNmYXEVC+XLd7N+HOqTjpmqqpRoZOplyC1tWFTksBm\nC2KzBfvs01Rbiy8hAU3p2zgR1ZOLYt9OqXUVy71VbNDW0kJ49IRdS2GmehHTQ+fhILXPZ+oNY0cV\n88EnU3iLBVxbeV9UfACYdCbumvQTHvniIX689oesuWE9RiU+418EAsGZSxxCEQJB7zng86AC+XGa\nb6b3tKU0J8fFf1cc2Z3IE3dOw9loYO51RVxw8xEMpo7dkymtgqxclkkNDXDOmKZhqqulJTu7T48V\nBks50ljMlZnwpfWvrKoFq5bITPUiJqqzyNWGRTRK1hlJdjc5WXWsrTgfd3nfRWlfmJA2kYvyL+Wj\nolX85Zs/8dNp/y+q/gQCwbcPIc4EpzV7vOEGgHiJM53ncwD8htOn+27XFyn88ceT8XsVrvnxHqZf\nUtblvSmtA1fLJYmBrmo3NjagBIN4k3ruWG1R3Xzq38JHvi84EiolyQNXAtOsKciBOxiijUEhtsN8\nx48qpqxiMh/VnE2qqtLlWoMIcNuY77O5YhN/2voHrh+xkLzEwVHzJRAIvn2Igi7Bac0eb7gZIC7i\nTPOh831FQDcYVTk9Rmgc2ZXI7380haBf5rsP7uhWmAGktokzeeApWXNVFQDulJROr2uaxq7AIX7X\n8gK3ND7Is+43OBoqY7wynAXydQT8VoYnqAzXxsdcmAGMGxXeUrJMvRZTbUNUfVn0Fm4b+318IR8P\nb3wwqr4EAsG3DxE5E5zW7PF6kIA8fezFmc67FUnznjZRs6Y6A//948mEAjK3/vIbRk7veYl3iho5\ncWapDoszT/LJQlXTNDYFdvKG5yMOhIoASJeSmaWbxAzdeBLl8NgNr3sHCY7D6PTNBAOJAz5PX0lO\ncjHccoRP3Bdw95G/QXrnIjNSzBt0HquLVvHB0ZWsLV7D+XkXRtUfmoZhzWr0n61D8vnxn3Mu/ksv\nh3g00ggEggEh/tUKTls0TWOP10OmTo8pDl27im8TAH79xJj7PpVgQOLP902ivsrMRbcd6pUwA7AC\nRk2jIgLNDOaqagA8KceL9ncFDvG8exmHQuGo1HhlBBfoZzBUzu3QwOF1Z5DgOIw1oYim+oEmWfvH\njKxdHDoylG8+S2bMrOj6kiSJOyf8kPs//QkPrr+f9Qs3Y1Cis35Mam4i4Sc/wvj+ivbXzP/8B/45\nc2n+24to6elR8SsQCKKDEGeC05ayQIBmNcQ4U3yGeeq8reLMMC4u/k/klT+MZN+WZMbOqWLeDUd7\n/ZxEOHpWIUuoDKyO4cTIWZ3ayN/r3mK99xsAJiujucwwlyy567mEHncmANbE+Imz8cOOwhHYsHUo\nY4huahOgwD6EiwsuY9XRlfxzzz+4Y8IPI+/E58P+3evQb9lMcOw4PAtvAb0e47I3MWxcj+OqS2hc\nvQ4tMTL7RAUCQfQRNWeC05a9vnjWm/nR+bYS0OWjyfFdk7LrixQ+XJJPWq6TBffu7vNEj1RNIyBJ\n1A4weGaurkID3kk4xA8af8V67zcMkQdxv+nfWGy6tlthBuB1ZwFgTSga2EEGQOLgEAUU8nnRBAL+\n2Hz83TjyJsw6C3/Y8gRNvsaI27c99AD6LZvxz5mL89HHCY0bT2jkKNw//y98V1yF7shhEv7j38M7\nuQQCwRmBEGeC05a9cezUVHw7w/Vm+vEx930ifq/M84+OQZZVbnxgF0ZL75aNn0jbOI2KAaaG/fXl\nXH2zwlP+NwihcovlCu4x3Uq+ktOr5wP+REJBE9bEogGdYyA0O1K5hndwBa3s2hKbJg+70c6CETfQ\n4Gvgz9v+O6K29Z+uxbzkRYIFQ3DffQ+cOH9OkvD822IC4yZg/PB9jG+8FlHfAoEgeghxJjht2e8L\ni7P4NAOEU5qBOKc0lz07lOoSK7OvLiZ7aP926Z04TqO/HHEeYt5VFbw3PMRweTD/Zf4B80zTkPtk\nU8LjzsRsqULRRXa3ZW8J6QxcZP4EgK/WZcbM7+VDriTVnMbfdzxDcfOxyBgNBLD95wNosozn7nvA\n2MnOBkXB/eN70QwGrL96BMkp9jEKBGcCQpwJTlv2e70YJYm0OHSbtdeb6eMnzooP2ljxQgGOdA8X\n3nKk33YGOk5jU+OXPHDgpxQmwQ/3p3C36SaS5P51W3pb684stggJlH4wMukoaVTz1br0mGX6jIqR\nW0bfhl/188TmX0fEpulfL6I7dBD//EsIFQzt8j4tLR3vtdej1FRjfvqpiPgWCATRRYgzwWlJSNM4\n7PeSozf0MToTAbQQim8zQSU7bvPNVBWef2Qsakjmqrv2dTr9v7cMZJzGqpr3efzIr5A0jeWvwR0V\nw5Gl/n9snNgUEC+cySlcyXs01Zs4uNMRM79zB80jP7GAtw++wf76fQMzFghg+cuf0AwGvAtv7vF2\n39XXotodmJ//G1Jz08B8CwSCqCPEmeC05Jjfh1/T4pLSVPx7kDUXfkN8OgoBvlyVxaEdSYw7u7LX\nYzO6wgKYNK3P4uz1itd4pvgvWGUrv2u4hCsPQHPSwDr+vK6wOLPFsSmg0Z7O1bwLwFefZsTMryzJ\n3DR6ERoav9/8mwHZMi57E6WsFP/8i9EcvRCYRhO+71yF7GzB9NILA/ItEAiijxBngtOS/T4vAHmG\n6MyF6g7FtwUAv35MzH1DeKbZG08PQ9GpXPK9QwO2JxGuO6uUJHobf3uz8g1eLv8Xybpk7h30U6ZX\nhj8qWpIHJs78vmRCIUNcI2eNjjQuZA0mnY9NazNpzfrGhGkZZzE8aSQrC99jZ832/hnRNCz/+2c0\nRcF75bW9fsx/yWVoZjPmv/0v+Hz98y0QCGKCEGeC05IDrc0Ag/SxF2c679cABOIkzj5dNojqEivT\nLyklOcsTEZupqkZQkqjtRYr4naq3+VfZiyTpkrg75x7SDOkk1tQB0JI00DSghNedjtlahizHRyA0\nOdIw4WNm6k4qS6yUFNpi5luSJG4etQiAx7/6Vb9s6L7ahG7/PgKz5vRpuKxmteG78GKUmmqMH77f\nL98CgSA2CHEmOC050BY5i0da0/c1ISmRkJIdc99+r8zbzwxFbwxy3sLCiNltG6fRU2pzTe3HvFD6\nPHbFwX/k/IQUfXjFUUJtPQDNA4ycAXjd2UiSFrd5Z42O8Dy2SyytXZtrY5faBJiQNolxKeNZW/wx\nX1Vs6vPz5iUvAuC/+NI+P+uffzEApiUv9flZgUAQO4Q4E5yW7Pd64tKpKQXLUULlBAxj6PO01wiw\n+pU8GmtMzL6qmIRkf8Ts9macxo7mb/jLsT9jkS38R87dpOmPD5VNrKnDbbUQjECa2eNqHUZrj5z4\n7AstCcmokszlofeQZZVNMRypAa3RszG3AvD4pkfR+pBXlRrqMS5fRigrm+DYvs/gU3PzCI4eg+Hz\nT6EwPn//AoGgZ4Q4E5x2xLNTU+fbCkBAPzqmfgHcLTqWPzcEsy3AOQuKImo7tYeOzWOeIn5z5NeA\nxB1ZPyDDcIJgUVUSautpGWAzQBseVzgimZDY//EgA0GTFZoSUxjccICCUc0U7rNTU9HJjLAoMip5\nDFPSp/FlxUY+L/20188Z33kbye/HP/+Sfv/y4LuoNeL24ov9el4gEEQfIc4Epx3HOzXj0QwQFmf+\nOIizNa/n4mo2MPe6Isy2YERtp3Qz68wZbOFXhx/Fo7q5JWMRQ83DTrpuaWpBFwzSkhyZsRN+LyZW\nWAAAIABJREFUX1J4U0CcImcQrjtLaK5n3PjwMvfNMezabOOm0W21Z72Pnpnefh1NkvCfM6/ffgMz\nZ6MZTfDKK8S0G0IgEPQaIc4Epx1tnZq5cVjbpPN+jYZCQD88pn79PpkP/jUYoyXAzCtKIm7fApg7\nGaehaip/PPokVf5KLk66hKkJ0zo829YMMNAxGseR8LiyMFuq0OmcEbLZNxod4UL6Gbl7AdgU47oz\ngKGOYczMms031dtYXbSqx/vlY0Xh5ebjxqOlpPbfsclEYMZMOHoU3bav+29HIBBEDSHOBKcdB1s7\nNXNjHTlTPSj+XQR0Q0GKbZpr/fJsmmpNzLisFJM1slGzNlI0japTxmm8XvEaXzdvYZR5NJcmX97p\nc4m1rZ2aEWgGaKMttWlNjE/0rKm1KSDfX0hOvpO925JpbtTH/Bw3jVqEjMwTm3+FqnW/rsD0zlsA\nBM45b8B+/eecC4TnpQkEgtMPIc4Epx3tkbMYizPFvwuJIAFDbFOaaghWvFCAolOZfXX01hqlqhoh\nSaK6tVZpe/M3vFbxCsm6ZG7L/LcuJ/9HbozGcdrEmS1Oqc22jk1HbTljptSjqjJb1/d+LEWkyE3M\nY+6geeyt28P7he91e6/xnbfRdDoCs2YP2G9w4mRITMT47jJitsNKIBD0GiHOBKcdB7weDJJEui62\nkYx4NQNs/jiDqmIrUy4sJzGCHZqn0rZjs0yWaAo28d9Hn0RG5vuZt2NVup71FckxGm143OGOTVuc\nmgKa7GEh5qgtZ/Tk8M/31brYpzYBbhj5XWRJ5nebf0NI7XxMsFx4BN2+PQQnTUazRmAum04HZ5+N\nUlONbuuWgdsTCAQRRYgzwWlFW6fmoDh0aire2G8G0DR47/khSJLG3OuKouorvbVjs1iSeLroTzQE\nG7g85TvkmQZ3+9zxyFnkxFkwkEDAn4DNfgSIfVH68chZBWmZXlIzPXzzRRo+T+w/ErNtOZybewEH\nGw6w/MiyTu8xrgxH1QIz50TO8ZywLeMqMZBWIDjdEOJMcFpxzO/DF49OTU1D5/uakJyKqqT1fH+E\n2LclmaN77YydXUVqjjuqvtJbI2dfBkrZ3LSJ4eYRnO+4oMfnIjnj7EQ8rmwMxiYMprqI2u0NLqud\ngM6Ao6YcgDGT6/H7FLZvit17fyI3jFiIIin8fvPjBNWONYfG999Dk2UC02dEzunUqWhGI4ZVKyNn\nUyAQRAQhzgSnFW2bAQbFeDOAHCxGVutivk/zo9dyAZhzdXHUfaVoIGsaBzQvFtnCoozbuqwza0MK\nhUioqaM5JSni53E7BwGQ4DgYcds9Ikk02tNIqi0DTYt7ajPDmskFefMpbDrC2wffOOmaXF6G/put\nBMdNQEtMjJxTo5HApCnojhxGORSH90AgEHSJEGeC04q2nZqxXniu+Nr2aY6Kmc/6aiNb1mSQkd9C\n3pjGqPuTNBXFW4FmyWNB2o04dD0X+CfUNaCoKo2pyRE/j9sZFqaJjgMRt90bGpIzMHlcWJyNZA92\nkeDws+WzdELB2G+GAFgw4kZ0so4/bHmCQCjQ/rphzUcABM6KYNSslcCMWWEfIrUpEJxWRE2cqarK\nww8/zMKFC1m0aBHFxR0jAx6Ph4ULF1LYukakN88Ivt0cj5zFVpzp2sSZIXaRs3VvDUINycy8oiQm\nm6I2Va0g0HIQdFaG2nv3Re+oDA9pbUqNfOTM685EVXXxiZwB9cnhpoTkqhJkGUZPqsfZbGDvN5H/\nWXtDmiWd+YMv4VhLEa8feLX99TZxFpw6PeI+g1Ono8kyxlUrIm5bIBD0n6iJszVr1hAIBFi6dCn3\n338/TzzxxEnXd+3axc0330xpaSlS6zdTT88Ivv3s94U7NTNi3KmpeL9Gw0BANyQm/oIBiTWv52K0\nBJh0XkXU/dV7K1hd+g8Ub7jGqlLfuzlu9qoaIDriTNMUPK4sLLYSFCW69Xad0ZAcXlGVXBUe+jt6\ncgMAX62N7a7NE1kw/Ab0sp4/fv07/CE/+HwYPltHKGcQamZWxP1piYkEx4xFt20rclVlxO0LBIL+\nETVxtm3bNubOnQvAxIkT2b1790nXA4EAzzzzDAUFBb1+RvDtJqRpHPbFoVNTdaIE9uPXDwcpNqJw\n67p0GmtMTLmwHKO58/EJkULTNN4tepqA6mOcIdyZWanrXU3fcXEW+bQmhFObkqRhsx+Oiv3uqG8T\nZ9VhcVYwohmjOcimdRlx22qUbE7hkvzLKHOW8sq+f6H/ciOSx00gClGzNoJnzUTSNAyre95SIBAI\nYkPUxJnT6cRmOz6PR1EU1BOGHU6ZMoXMzMw+PSP4dtPWqRnr4bM63zdIqDGdb/bRa3kAzLisNOq+\ndtV/zuHmbeRaxzJGHxZnVb1suHC0ibMoNARAfJsC2sVZVbh8QtFpjJzQQF2VmcJ9ESy87yPXDr8e\ng2zgqa1Pon7yAQDBqR3XakWKwFkzATB8KOrOBILTBV20DNtsNlwuV/ufVVVFlrvXgv15BiAtLaH/\nBxXEnbb378taHwAjEqw47ObYHcC7AwAlcSJWa/TXNpUcsrD3qxSGTW5g8MgAED0x6g26eL/4ryiS\nnosGf48EvYSkaVQbTJjNPftNqqrBazFDUiKdyTmjcWCRxlAgHwBHyiHqKmIrykOmDAJ6I2k1Zdhs\n4fd9yiwnO79KY8eXg5g6uyim52nDgYVrRl/D63te5+XSZfzYbMY2cxpEoUnG4bCAYwjk52Pc8Dlp\nVgUsloj7EUQe8b337SZq4mzKlCmsW7eOSy+9lO3btzNy5MioPANQU9My0OMK4kRaWkL7+7e5JjzO\nIFWVaWzyxOwM1sZN6IGW0DBUpzfq/t57MR+A6ZcW4/FEbyMAwIpjz9MSqGdG2tVYSCHkC2C3BClT\nDLg9frpLHkuqSkJVLbXZGfh8gQ7XjUZ9p6/3DT1eTyoW2yE8Xg9oygDt9Y36pAySKotxtnhAkhg0\n1I+iG8y691O4dvHemJ7lRC7LvYr39r3L46NrubX+LHAHw/+LIA6HhcbGcK2fafI0TO+8RdO77+Of\nf0lE/Qgiz4mfm4Izj94I66ilNefPn4/BYGDhwoU88cQTPPjgg6xcuZI33nijT88I/u8Ql52amori\n20pQyUKVI7c7siu8LoXP3skhIdnLmJnVUfVV5jrEpqr3cBgymJpyWfvrySE/HlmhWe7+dzNbXQNK\nKBSVMRon4m7JRdH5sMVhCXp9chYGv5eExnD61mhSGT62kdKjCRQficCapH6SaLRzi28MVTZ4dnb0\n6yDbatraOkMFAkF8iVrkTJIkHn300ZNeO7H4v40lS5Z0+4zg/w4HfB70Md6pKQcOI2vN+PTRK7g+\nkY3vZ+F16Zlz9TEUXfSqzlUtxLtHn0ZD5dzMW1Hk43+nqcEAhUYo0xux+7qOxrSN0WhOia5odbUU\nkJz+Dfbk3TibhkfV16nUn9Cx2ZIU3rc5/qw69u9IZsPqLG760aGYnudE7lsf5PXZ8Af7DhaFPNiU\n6KX6Q6NGo1osGD5eDU9oxGS2i0Ag6BIxhFZwWhDSNA61dmoqMfxiaJtv5o9BM4CmwepX85BllemX\nlEXV1+bqDyhzH2RE4kxybSfPbksNhlOpZT2M00iqqAKgIT0lOodsxdWcj6aBI2VXVP10Rts4jZTK\nY+2vjZrYgN4QYsOH2XHr2lQ8XoZ/uZ8f7bVRrzr5a+3yKDtUCE6eilJagnIwPkOBBQLBcYQ4E5wW\nFAf8cenUPL4ZIPrDZw9td1ByMJExs6tJTPFFzU+Lv57VpS9ikM2cnbmww/W0VnFW2ltxlhZdcRYK\nmfG6M7HZDyMr0a/5O5Ha1BwAUk8QZwajysgJjVSUWDl6ID5dm6lbdqIEAtwgTSJRtvDn6reoCzZF\n1Wd7avPj1VH1IxAIekaIM8FpwQFv69qmGO/U1Hm/RpXMBHV5UffVPj7j8pKo+lld+gK+kItZ6Quw\n6uwdrieoQQxqqGdxVh4WZ01RFmcAzuYhyHKIxKT9Ufd1InUp2QCklh896fXx02sB2LA68oNfe0PG\nhi0AuMeP5cak83GqHv5Y9XpUfQYnT0WTJFF3JhCcBghxJjgtaNupmRvDnZpSqB4leCS8T1OKbpdg\nU52Br1ZnkpbrZMiEhqj5KXHuZ1vtx6Qa8xiXdG6n90iEo2e1OgO+blLIjvIqWuyJBIz9e0/8ssym\nlCy+Tsog2EOq2tUcrke1J8d28LTfaKYpIZnUiqKTXh8+vhGDKU6pTU0jc/0W/BYTjcNyudQ+g3Rd\nEs/XvU+xvyp6bh0OQkOHod/8JVJzdKN0AoGge4Q4E5wWHIhDp6bi2wbEJqW57u1BBAPR3aOpaior\njj0LwDmZNyFLXf/zTgv60SSpy7ozvcdLQkNjv+vN1qfl8F8TzublgrG8NHQ8j4yfwzeO9C7vdzsH\noao67MmxrzurS80hsbEGo9vZ/pperzF6Uj01lWYO7op+F++JJBQWY6msoWb8CDRFQS/pWJQ8n4AW\n5DeVS3o2MAAC085CCgbRf7o2qn4EAkH3CHEmOC3YH4dOzVg1A6ghWLM0F4MpyOTzo7dHc0fdWkpd\n+xmWOJ0ca/czAntqCmjr1Gzshzjb4Ujj9cGjCUky0+sqmFRfhUun559DxlFq7nw8habpcLfkYk0o\nxWCs77PPgdBWd3ZiUwDA+Ol1QOxTmxkbwv9dVk86/h6eY5tIgSGLNxrWsd0dvQ7StuXqRpHaFAji\nihBngrjT1qmZo4ttp2Z42blEQN+7Ycf95ZvP06irNDPpvApM1sgOEm3DF/KwquQfKJKeOek39Hh/\nWg/iLKk8vAS7r80AVUYL/yoYi05VubbkILNqyzmnppRLywsJyjL/GDoen9x5CrmldYxGUtrXffI5\nUOramgIqik56feiYJkyWIBtWZxGK7vrTk8hYvwVNkqgZP6L9NVmSuT31cgAeLPs7WpRyraEhQ1Ht\njnDdmVidJxDEDSHOBHGnrVMzL4b1ZmgBdP7tBHV5aHJ0h43GohHgs/KlOAP1TEm5lERDao/3J4UC\nyJpGSVfirKLvkTMN+FfBWHyKjgsqi0jzHd/yUOBqZnJ9FTUmK+/lDO30+eaGsEhOydjca5+RoL1j\ns+LkpgCdTmP89Doa60xs/zItJmfRtbhI2b6XxoIc/PaT/7ucYB7KTMsYNrn3srxpQ3QOIMsEpkxD\nrq1Bt3N7dHwIBIIeEeJMEHf2x6FTU/HvQ9I8Ua83qzxmYeeGNPLGNJA1xNnzA/2g3lvB+sq3semS\nmJp6Wc8PAAqQHPRToTfSWVCoPXLWB3G205HGMZudYS0NjGzp2PQwu6aMRL+PjWk5tHSSvg4GEnE7\ns0lM2o9O39xrvwOlvWOzoqjDtcmzw5sD1r43KCZnSf9qO3IoRPXEzqO530u9FB0Kvyj/B141Oqu/\ngtPESA2BIN4IcSaIO/taIyyxjJwpMao3W/N6LgAzoxg1W1XyHCEtwJyMG9DLvRe4mUE/QUnudKRG\ncmkFPqMRV2LvliurwIqcoUiaxqyazgfsKmhMbqgiKCt8lp7b6T3NDaORJI3k9K29/jkGiseSgNNq\nJ73sSIdrgwqcpGZ62PxpBi1N0a+HbBuhcWK92Ylk61P5jn02pYEa/lKzLCpnCEycjKYoYqSGQBBH\nhDgTxJ19rZGzwYbYRc507cNnoyfO/F6ZT5flYHX4GHd2dEYgHGnezp6GjWSZhzE8cUafns0KhDtk\nCw2Wk16XAwGSKqupz0zr9RqfLSlZVJptjG6qIynQ9YDdMU21mIIBPkvP7bT2rLlhFADJ6bFNbVan\n5+GoqzypYxPCP/7Us6sJBmTWf5gd3UOoKhkbtuBLtNKUn9PlbTcmnY9DsfGHqqUU+aLQYGK1Ehw1\nGt32bUg1NZG3LxAIekSIM0Hc2ev1YJZk0pSorXrtgOL9mpCUQEjp+ktwoHy5KgtXs4HpF5eh00e+\ngDukhVh57FlA4pzMm5H62EyR1SqijhpO3tmYVFGNrKphcdYLNOCjzHxkTWVGXXm39+o1jYmNNXh0\ner5M7Sh2An4HHlcm9uTd6HTRSQN3RnV6uC6ws+jZxJm1SLLGJ8ujm9q0HyjEVNdI9YSRIHf90WxV\nTCxOuRyfFuCBsr9GpTkgOPUsJE3DsPbjiNsWCAQ9I8SZIK54QyEK/V4GGwx9Fhf9RQpWoITKwvVm\nUfT50Wu5SLLG9EtKo2J/S/UHVHmKGOM4m3Rzfp+fT1SDWEJBCo0WTvx6TykNC6y6XoqzAwnJVJmt\nDG9pICEY6PH+CY3VyJrGppTOR1Q01Y9FllXScj7rlf9IUJ3RKs5KD3e4lmAPMHxcI4X77Bw71Ls0\nb3/obIRGV8yzTWSCeSgft3zN+81fRvwsgba6M5HaFAjighBngriyz+0mRKxTmuF6poAhes0AR3Yl\nUrjbwcjpNSRlRH5fpDvYzMel/0Qvm5iVfl2/bEhAVtBHs6KnXjleT5VSEhZnvY2cfdpaPzaxobpX\n95tDIQa7mii1JlJhsna43lg7EVXVkTnoY8LVbNGnLXKWUdr5DLEpc8I/29r3ohdpzdiwBVWWqR03\nvMd7JUnih6lXoUPhgdK/0hxyR/Qs6qBcQmlpGNatgWB0xr8IBIKuEeJMEFd2uVwADI5hp6bOG65n\nimYzwMet4zNmXhGdRoBPyl7BE2rhrNQrsXSyP7O3tKU2C09IbfYlclZrMLPHkUqGx0mmt/cCYWRz\neNDslpTMDtdCITNNdWMxWWpwpMZmnENdSjYhWSG9pGPkDGDkhEbM1gCfrswh4I/8x6ahoYnkXQdo\nGJZHwGru+QFgkCGN65POpSJYx8Plz0f2QJJEcOpZyM3N6Ld8FVnbAoGgR4Q4E8SVdnEWy05N7yY0\n9FEbPtvSqOeLVVkkZ7kYNrku4var3EVsqnoPuyGDiSnzB2SrrSng6AlNAckl5bhtVry2jlGtU/k8\nfRCaJDGpl1GzNoY4G9GHQmxJzuw0NlZfPS18vtxIptU0ZNkPdKzRUhUdtSnZ4ZqzToav6nQaU+bU\n0NxojMrGgPQvtiJpWq9SmidyfdK5FBgy+Wf9aj5pjmyHa2Bq+D0QqU2BIPYIcSaIK7uc4aLvWM04\nk0JNKIG9+PUjQYqOIPzsnRwCPoWZl5d2V9fdLzRNY0Xxs2iozM1YiCINrIkiLehH0VQKjWFxpvd4\nsdfW9ypq5pVlvkjNwRIMMKylsU9+dZrGMGcDDUYzhbaOuyu9nkxcLYNwpO7CYivqk+0TMVnKyRv2\nGpPP/gkzLriNGRd8j0lz7mPwiCWYLJUn3VuTkYfB7yWpi1EgM8+vRJI0VrySH/Fl6Jmfh6O5VZP7\nFs3VSzruSb8eBZm7S/5MU8gVsTMFx09AMxgwrBHzzgSCWCPEmSCu7HC5SFF02JTOV/pEGsW3BQmN\ngGF8VOyrKny8NBedIcSU+Z1/yQ+EPQ0bKWzeTp5tPPm2iQO2pxBObZbrTTTJOlJLwmeuy+p6SXkb\nmxwZeHU6xjfWoHQSjeqJttTmtuSMTq/XVJwNQMHol+hr7ZmseCgY9SKT5/yMnIKV6PUteN0ZOJsH\nYzA2kj34QybM/DmZuR+1267MyAcgq/hApzYdKX5GT67n6AE7+75J6tN5ukMKBMjY+DWutCScOT3/\nvZ/KEGM2NyadT0WwjgfKno1c96bRRHDseHT79iKXRm9On0Ag6IgQZ4K4UR8MUun3xzSlqfNuAsCv\nj44427kxleoSKxPnVWJJiGwhdUD18UHx35BROCfjpoh1txb4w7Vie0w20orCX8I1OR1rwU5EA9ak\nDELWVMY19m8WVo67BWMoyA5HWqfSy9U8lKb6kSQ6DpGWvb7Xdu0pO5k0+wEyc9fg9aRQcuRqDuy4\nh6P7v8exg7dwYPu9lBZehaYpFIz6J8PHPQOoVGQNASCraG+XtmddEI62rXg1vw8/afekbNuD3uWh\nevLofncPX590LiOMubzRsI5XG9ZE7GyBaWcBIrUpEMQaIc4EcWN3awF5fiw7Nb2b0FAIGKLTDPDh\ny4MBmHlFccRtf17xJo3+aialXESSsXvx1BcKWjc07DIltIuz2h7E2f7EZKpMFoa3NGAN9U+EKkCB\ns4kmg4lia2Kn91SWXEQopGfw8FcxGOu7tafTORk69q+MmfI7DMYGasrnULj3dpobxqJpx9O/mqaj\nqX4cR3b/ALczh9SsLxky5h9UZeahShLZRfu79DF4eAtZeU6+WpdJdXnne0n7SlY/U5onopMUHsj4\nLlbZxP2lz7DPeywiZwu21Z19IsSZQBBLhDgTxI2dreJsqCEyX3I9orpQ/LsI6IejSZH3WX7Uyo71\naeSNbiBneEtEbTf4qvis/HUsip3pqd+JqG27GiQ56OeAyYqlvJqgTkdDWvc7NT9pnQvW2/EZXTHU\nGd7BucPReY1bMJBIddm56A1Oxk57DIOpsyidRnLaFibOfoD07PV4XBkU7v0+1eXnniTKOtgO2jh2\naCEeVyYZOZ+SOWYFdSnZZB7bj6R2tnE0HNiadUElmirxwev5ff1xOzm6RuanmwiYjdSNGpi9DH0S\nP0lfgFfz829Fj9MSgfEaakYmoZxBGD7/DLyRHwkjEAg6R4gzQdzY5Ql/eQyJUeRM59uKRBC/flxU\n7K9+JSxYZl8V+ajZquLnCGp+5mTcgEHp3aiFvjDE5yYoyWzLyqE2Kx1N6fqjodRsY789lUF9HJ/R\nGXmuZnSqyk5H17VW9dXTqS4/G5OlhnHTHyM95xN0+hYMplqS079i3PRfMnLSn9DpnVSVnkfh/u/h\n9fQusqiGTBw7+F183iQGFbzHsfMdGH0ekqu6rrEaP70Oa6Kfj5fl4moZWENGQmEx1vIqasYPR9MN\nfEPGLOtYrrTP5qCvlNuP/Z6Q1rnI7AuBqdORPG70X2wYsC2BQNA7hDgTxI2dXjdWWSZDF/2F0gA6\nb3iSuj8KzQDuFh2fvZtDYqqXsbMHFk06lSPN29ndsJ5M81BG2mdG1HYbbXVnK2fN6jGl+UlmOHV7\nVuPA94XqNY08VxNVZiuVJksXd0nUlM+jqvRcDMYGho55genn/jtT5/6EkROfJsFxmObG4RzZezu1\nlbNB61tzSShkobTwGlRVpuXWI/iSIPto13VnOr3G7AsrcTv1LF9S0Cdfp5L5WXiG2EBSmqfy/ZTL\nmGQexkctW/hF+T8GbC/Yvi1AdG0KBLFCiDNBXHCGQhT6fQw3m2O2tknnWR+uN4tC5OzTZTn43Dpm\nXVGMoovcnIWQFmLFSfszo/NPNiPoJ8nt5q1zzuHokLwu76s3GNmanEmyz0OBJzKp26HO8BiOHd1E\nzwBqK+dwcOd/UFV6Hs6mITTWjaWmYjaHdv+AksM34Pem9vsMXncWVaUXIFm9HPgZZBXt6fb+medX\nYk30896SAhrr+9/Qkvn5ZjRJonpi5GbuKZLCzzNuJlefzrO1y3muduWA7AVHjUEzmzF+vJqIzxAR\nCASdIsSZIC7s9nrQgBGWrqIlkUUKNaH4dxDQj0STI+tTDcHqVwajM4SYfklkx2d8Wfku1a37MzPM\nA4vSdIcE3PDVl7RYrSyddXaX963IGYYqSUytryRSkrrA2YSkaexI6nm2WjCQSG3lbI4d+i5lR6+m\nuuy8AYmyE6mvno6raTD1s8CR1P2+SoNR5dzLy/B5dSx7YWi//BnqG0neuZ/64XkEEnoe+NsXrIqJ\nh7Nuwy5beaDsWV6uG0BBv15PYOJklGNFKEc636AgEAgiixBngriwq7VWabg58vVTnaHzfomEit8w\nOeK2t32WRnWphUnnVWBJ7Hnxd29p8FXxcdk/MSk2ZqdfHzG7XXH3m2+R4HKxYsQ4Ap1E6I7Y7GxJ\nySLd42qfURYJTGqIHHcLxVY7DTFc49URifLiy5ACUPvdCsy+7tO20+ZWY0/2seqNPGqr+t5gkv3J\nF0iaRuW0sf09cLdk6pP5dfbtJMgWflz6NEvrP+m3reBUkdoUCGKJEGeCuNDWqTkiZuIsPCfLZ5gU\ncdsr/hGejxXJRgBN03iv6H8JqD7OzrgRsy4hYrY7w+D1MXr/ARau/4wWg5HNp+y8VIE38kYBMK+6\nJOIfHG2pzZ1ddG3GCr8vGf3GXPypMNL8h27v1ek1zr+ylGBA4c3nhvXZV85HnwNQcVZ0Zu4B5Bsz\n+XX2YqyyiR+VPNXvFGdgSutIjY/FSA2BIBZETZypqsrDDz/MwoULWbRoEcXFJ39xrV27lgULFrBw\n4ULefPPN9tevueYaFi1axKJFi3jooYeidTxBnNnpcWGUJHKMMerU9HyOKpkJ6EdF1O6BbQ4OfpPE\nyOk1ZOY7I2Z3T8NGDjR9RY5lFKPscyJmtyvyCo8iaxoXHtiHoqq8kzuCY5awIAwhsaRgLGWWBEY3\n1ZLljdyKoDba686S+j4hP9LUlc/AXAy2/I1YDIXd3jtxZg0pGR7WvDOIwv2dz2rrDGNNPalbd1M/\nfDDelI7rqyLJEGM2v8peTKJs4YGyZ3mo7O997uLUkpMJFgxFv2kjkjOyY2IEAkFHoibO1qxZQyAQ\nYOnSpdx///088cQT7dcCgQBPPPEEL774IkuWLOH111+nvr4en88HwJIlS1iyZAmPP/54tI4niCMe\nVeWgz0u+wYgSg2YAKViOEiwMj9AY4C7KU3mvNWp2zvVHI2bTG3Kx4tgzyJKO87JujUnDRP7BIwA4\n05KYX1mET1b4y4gpvJ89hL8On8iWlCwyPU7OqS6Nin9bMECGx8XhhCRcSmTfo75SmTuYoX8FZI2C\n1D/R2aL0NhQFrvhuEaoq8z8PTyAQ6N17lbNmA5KmUT4jelGzExlmzOGPg+5qbxJYePRRqv0NfbIR\nnDYdKRBA/9mn0TmkQCBoJ2qfgtu2bWPu3LkATJw4kd27d7dfO3LkCHl5eSQkhH8znzp1Kps3byYr\nKwuPx8PixYsJBoPcd999TJw48P2BgtOLHR4XIWCEMTbDZ3Xe8HymSNeblRyysW1dOrmG1hgmAAAg\nAElEQVSjGskf27fF393xUclLtATqmJF2NUnGrIjZ7Y7Bh8IRoqIhgxnR0kBIkvg4M59V2WHxmeNu\n4TulhzFofdtx2ReGOhuoMlvZ7UhjRl1F1Pz0hN9sQitKw7G1FqZuIcnyBQ3urqOXw8Y2MWVONds2\nprPshaHc+IOei+azP1qPJklRTWn6/Qr7Dg1i5948yiqTqapx0FjzMIrqZ43iJkvnZ0yWxnkjNCYO\naWbu+DrSHf4u7QWmTsf05lIMa1bjvzyyg5AFAsHJRE2cOZ1ObDZb+58VRUFVVWRZxul0tgszAKvV\nSktLC0OGDGHx4sVcf/31FBUVcccdd7B69WpkWZTGfZvY4gmnxUYaY1NvpnevA8BvjKw4W/liuHty\n3vVH+7sSsQMlzv1sql6Bw5DJ1JTLImO0JzSN/ENHaHTYaUoKp9hGN9eT6XHh1BvQqSoZXlfUC1SH\ntjTyRdogdsRZnAGUDStg+F9q2PyCxJDUP7Ot+Cw0up7Hd8kNxzi028Gbzw9j5vlVDO5mQ4SpqpbU\n7XupG1mAL6n3qdDeEAgobNo6nLUbxrFz72D8geNnVpQQjkQXJkXCGdLhCUjs3pvN7taRbrKkce7E\nWm6YV8YVM6uwmk5OfYaGDUd1JGH8YAXO3z8F+tjMJxQI/i8SNXFms9lwuY7XprQJM4CEhISTrrlc\nLux2O/n5+QweHB5wmZ+fj8PhoKamhoyMjG59paVFt1haEFl2VYX3/p2VGhYCDnsURZoWQCv+FFWX\ngdE+EmOEVFRNuZGNK7NIz3Mx+dwmZHngy9tDWojle54GNC7OW4zFHJsxI6nlldgbm9gxbSIG/fEB\nrhkEyQi07s3Udz7Y1dDF6/0hgyDJfi/77ClgMmKMYpSuJyrHDmfS+s2YduYiTSwmP/09atw3d3m/\nzQY33lnK808O4ZnHJvE/b23DYOw8HTro7fDg2bp5U7DZIhM9rqxO4O2Vk1m9bgwtzrDNjLQmRg6r\nZuSwKrIzG0m0eTnx99xy7Rivul+mrDIRuWz2/2/vvuOrLu/+j7/OnjnnZJzszd5bkKFsFESUoVKK\nUnHdtra3tSr+OsRahWqtrRZvrdZBa4sLFBwMZcqQvcJMQsgiO+fkjJz9/f0RQFBC1smy1/PxOI8k\nh3Ouc4WTnPPOtT6Yj9/DxoNd2HjQSvRbPhb9qIAHZxSh117yPIwfBytXYj30Ddx4Y1j6LjSPeN/7\nYWu1cDZ48GA2bdrEjTfeyMGDB+nR49tDFjMzMzl79ix2ux2dTseePXtYuHAhK1eu5OTJkzz55JOU\nlpbidDqxWhvevVVeLhaodhaSJLGt2ka0QomqNghqsNlrW+3xlLU7MIZqcGuux+Xyhq3d917KIBiQ\nM2bmGbze+qeCmmJL8XsUu3LoZRlNrLorXm/4juW4mtQDdUsOTnbrgs/f+IXiapWiSbdvjExHNXuj\nEzikNTHAdqU6mm3jbEoSQYWC9L/7OfayjjjDqxRUTCAQqn/xflp3D4NGGjmwI5anHurGI0sPcqVB\n//6ffIUkk5HXvwc+Z8vqVZaUmflgzbV8tbUfwZACg97DmBHHGNI/l9iYmstu6/3Oj3+iLo3/kT/C\n4eSdrEv7O9WjliKr6E7Sod9Rvns2j77ahef+k8yiO06zYHI+cjkoho0kYuVKPG8txzG0/vPwhNZl\ntUaI971OrDHButXC2aRJk9i+fTt33HEHAEuWLOHTTz/F7XZz2223sWjRIhYuXEgoFGL27NnExsYy\ne/ZsnnjiCebNm3fxPmJK84flrN9HZTDAKL2x4RuHgbL2SwC8mmvC1mZFsZaNH6QQGe9m4PjwTL+V\nuPP4smg5eoWZUbG3h6XNxup27AQA2d2bd5hqOHVx2OrCmSW2XcNZQK2iOCOFlGN5fF02DWv8Z6RG\nvU5uxaNXvd/0H5+hqkzLjg2JRMd6+ckjxy+b8jacLSLqyEnK+3TFZ27+yEfRuUjeXz2SzTv6EArJ\niYmyM25UFv17n0WhaPwp/nLkDAyNok9oGIflO9ke9QWFE34MI3+GauciqnY9xCOv9eWdTdG8+rOT\n9OrRk6A1FvXna6C2FtroKBxB+G/TauFMJpPx1FNPXXZdRsa3J5yPGzeOcePGXd4ZpZLnn3++tbok\ndAB73XXHTbTderMNhNDgU/cPW5sfv9aFgF/OhB/lhqVUUzAU4IPc5whKAcYnLkCnbJvgCoAk0TXr\nBI4II6Xx7X+MRazXjdHv44glhqBMhqIdywUVdM8kJTsP8zYL3ltiSTB/TEnNTNy++kOsSiUx72cn\nef2PfVjzbgZRsR5uufPbnbxpn2yoa/v6Ic3rU1E0762+lq07eyNJcmJj7IwbdZR+vfKRy5v/f6VC\nzZDQ9QwOXUe+7BRH1Ls5Ou6P1Ax7Eb54icPHbmPkw1EkTPobY283kH68jMhVvyJm+BTiDfHEGeKJ\n0VnRKUVYE4RwaN8968J/nT3u85sBtK2/U1Puz0URyMWjGQGylq8JAygt0LFpZRLRSS4GjAvPqNnm\ncys4586hl3k0GRHhPyT3aqwlpViqbewfMoCw7WpoARmQ6bRxODKW08ZIejrCV4mgqc726srIzzeS\nuSeLr8fPJD36VTJi/kpW8V/P9/TKdIYgd/3vCV5b0od3XuxFxTkddz18ArXcT8qaL/HptZQMaVpV\ngLwCK+99PJLte3oiSTLirdWMG32UPj0LkIfxaZMhI03qQVqwB1OD8yjTFpE3cy2H++4j/4uHObf2\nEf6T2R9m/hhs/4R1/7zs/nqlgRhdDNG6GGJ0Vqw6K9G6GBIMCXSxdKOLpSvJESnIW6lGrCD8UIhw\nJrSpPbVOVMjIULd+OFO5z09pqoeHrc1V/9eFUFDOxHk5TZo+qk++8zgbi97FqIxkTPzcMPSwabod\nPQ5ATrfMNn/s+nQ5H84OR1rbNZxVxcVQZY0m7VAWG2zzcRh6EqnfQ6R+O9Xuq6+3Mkf5+Mkjx/nP\nK935bEU6xw9GsnTmW+gqqsmbOIKQuuGdjqEQHDiawep1Q9l/uG60LjGuivFjjtCzW1FYQ9mVyJET\nL6UQL6Uwoiu4F37NB58O52T2JCL/cogH035Mye+TqZAcVHuqsHvt2L027F47Rc4iglLgiu1qFBoy\nzF3oGdWTwXFDGRI3jAHWQagV4fkDShB+CEQ4E9qMPRjgmKeW7hotqjYYpVG5P0VCFrb1Zufy9Gxd\nnURsqpN+15W0uD1PwMV72UuQkJiUdC8aRdvszrxU7wNHADjep0cDt2w7SW4HmmCAQxYrs/NPtl+N\nOZmMnP49GfbVdtIOH6PQOAuj5lkyY/7CgYKhhKSr/4FhjffwwK+P8tl/0tm/PZa7lt7Pl+gY1sNO\nPFfeiSpJkHM2jm/2dWPrzt4Ul0YBkJZcxvUjj9GjS3G7DXDq9T7unLON7bt7sm5jf5bmrOWRf69m\n2tPa7/VJkiTcAffFsFZRW0axs5giZyFFzkLy7LmcqDrGx9kr69pW6hmVdB3jUydwY8ZNJBqT2uE7\nFISOQ4Qzoc1sdzkJAQO0rR9CZIEilN59eFX9CSmiwtLmf17sjhSSMfHH2VfchdcUkiTxSd7LVPtK\nGRpzE8mGXmHpY1Mo/X56HDlGaZyVSmtMmz9+feRAhtPGCXMM+QYT6a6aBu/TWnL692LYV9vpuvsg\nOdfcTaVrHDHGjaREvsXZqv9p8P5qTYhbF+QyID6brz5K4u/cz9+XQfrqUlISK4mJdmA0eKioiqC0\n3ExeQSxV1XUbBRSKIIP75XLtsJMkxTftNP/WIpPB6OEn6G08xfJPxvHcZzOZrM3n3sezUKqkS24n\nw6AyYFAZzget3pe1I0kSJe4STlef5ETVcQ6VHWDD2bVsOLuWJ7Y9ytC4YdzabTYzu91GtC66jb9L\nQWh/IpwJbWaLs+5NdoDO0OqPpXatAcCjvT4s7R3dFcWeDfGk9qqmz6iyFre3v2IDh6o2EafL5Brr\njDD0sOkyj59C4/VyrG/bB8OGdDkfzg5ZrG0SzvwyORUaHVVqLdpQgPhaF4ZggMqEWGwxkWTsP4za\n5aZUNg2T9iBJke9S4ZyIy9etUe3/tPx5Xmc1vxn5KqvKpnI6N4G8gu+f32jQexjY5wy9uhfSPfMc\nGs2VpwbbW1SfEKt2z+XBc39m/UcDKcozsOjP+zCaGtdfmUxGgiGBBEMC1yWPBaDcXcbe0t3sKN7O\nvtJ97C3dw5M7fsON6VO5t///MDzh2jYpZSYIHYEIZ0Kb2eKqQSuT0a0NyjapXKuRkOPRtrxoeDAg\nY/mSXshkEtMfONHiaaViVw6f5L2MWq5jStL9KMJc77Ox+hw4DMCxvuEtBh8Oaa4alKEghyJjmVGU\n02qPU6LVsyU2hT3RCXi+U9Mz02FjXGk+/YcPYsxnG+m5fTeHJ4+lyHYHGTGv0C32DxwqfOOqlQMA\njLZyBuz4jBpzNObrdNwl30IwJMPp1GKvMVDrUWMyuYk0O9F20DB2JRXDerJ99SimxW1my75hLLpr\nJL9btofYxOadW2jVx3Jjxk3cmHETNk81Wwo3szF/A2tyP2FN7icMjh3CTwf9gqkZ01HIw3f4sSB0\nRGLLjNAmCn0+cn1e+mr1KFv5r1+5/yxK3yF86oFIcnOL29v4YTIFpyMYPKmIpKuU5WmM2oCDd0//\nnoDkY3LSfZjV7XR8hSTRZ99BvGo1OV07zmaAC5SSRJqrhjKtgXx9+E9C98tkfJaYybN9RrAtNgVF\nKEQfWznXlhcxuKqEJLeD3AgL/+jan7vvfZDspET6fvU1SBJObx+qXCMwak+RHvNKg481cu2/UAb8\n7Bw1A+n8fLhCLmE21ZKaXEGPrsUkxNo6VTADONnjGhQa+MR9I6MmFFGUZ+Sx+SPJzmr575xFG8mM\nrrfyl3HLeGb0c1wTP4IDZftZuO5Ornl3AG8cfhW33x2G70IQOiYRzoQ2sdV1YUqz9debqVyrAfBo\nr2txW06bivdf6oZGF2DynQ0XtL6akBTk/ZznqPaVMDTmpjY/NuNSSWcLiC0pI6tfL4KqjjmA3sde\nAcDX1uSwtmtTqflzz2F8kZiJPhDgxqIcfpJ7hAml+QyrKmF0eRGzCk7x4zNZdHFUk22OZsAb/2B7\nWgaJJ+p+Bs7Z5+Dxx5FkWUGUflu9j2W0lTNo68fYTDEc6TcmrN9Hewuo1BzpNwazo5JfZr7JtDvO\nUFOt5tf3jGDP1vD80SGTyegd3Ycnhv+Wlye8xpT0qZS4Svh/Xz/G8HcH8k7Wm/iDbVNJQxDakghn\nQpu4GM5aezOAFELtXIGEGo9mZIubW/GXbjhtasbNzSUiqmVlmtYVvMlJ+25SDH0Ybr21xX1riUE7\ndgNwcMiAdu3H1aS5aojwedkTFU+tIjzTWIU6I8/3uoYCg4le9grm5WXRzWm74gthlM/D1OJcJp87\nQ0Cp5Nbf/54zJTYAQpKWgqqFhCQV3eMWY1CfvOLjTfhwGSq/j+2jbyWkaFwIVqrsRMfvIKPH2/Qc\n9Bz9RzxB32t+S6/BS0jr9m+iYvcgl4evFFlL7Bs6GYBhX73PiAmlzH3wFKEgLHl4CGvfTw3rYyUZ\nk3hgwE95ffLbzOp2GzZvNY9u+V9G/WcoK09/QKgda7EKQrgpFi9evLi9O9FSbnd4ahsKrSMgSTx6\nLh+dXM78yJjLFvVqtSo83vBN5yg9X6N1vIFHOxaPblzDd7iKozujeGdJb2LTHMx6OIuWLHPZU/Y5\n6wrfwqKOZ0baI6jkmhb1rUUkidtfX47S7+e9H88m1Mzgo1DICYZa7wR/GRCUyzhrtGDxeUl3t2xj\nQKHOyF97DMGpVDOqoohR5UUoG6hAIANivLUk1zooVOlZM/QakouKiNMoCIRM+AJWLPo9xERspso1\n5rLamymnDzH5/b9yLj6D9VMWXP2QX1mQSOt+Mnosp0vvt4iO24PRnItOX4pS6UatsaEzlBJhOU1M\n/C7iU9eh0VZR60wiGGjeBhuVSkEg0LLaqB6dkYTiHDJyj3C630i0vSLo2tvO8QNR7PwqAa9HTv9r\nKsN6/IdWqaW/dSATUifhD/k5ULaf1Tkf81nuGtJMaWSY278MWWszGDTifa8TMxgafv0X4Uxoddtc\nDpZXVzDOaGLod2pqhjucaauXoPCfoibiZ4QU1ma3U+tSsPS+YXhcCu566gAWa/NHKrLt+3kvZyka\nhYGZ6Y9jVEU2u61wSMnNY9Inn3NoUD8ODBvU7HZaO5wBWHxeDkbGUqnRMbq88Crn8l9dkc7ASz2G\nUKtQMakkj/62iia1ZQz4uSYnm+wIC18npxHn85AQ9OENJBIIRmDR7yXGuAmHpw/eQDwqj5s7Xvol\nelcNq2b+LzXmKx9VolLbSUhdS7e+/0d88ia0+jLcziSqyoZSWjiO0sIJlBdfT8W50VSWjMBZk0nA\nF4FWV4E56gRxKV+hVLpx1nRFCjV8sO1ljx2GcAZQq4+g79HtaN1Ojg+dgCnST+/BVZw+auHA9liK\n8gwMva4sLKXOLqVT6hgSN4zrk8fh9LvYV7qbD0+9x56Sb+gT0w+rvv3LkbUWEc46NxHOhA7h5YoS\nDnvc3BVpJVZ1+RtIOMOZLFiBvvJRAsoUnMYFLSpH9M6zvcjaFcP1t51h0PjmHzhb4DzB26d+g4TE\n9JSHserCO9XTHDd8tIbU3DzW3DqV8rjmB9i2CGcqKYRNpSXPaCGh1kmCx9XkNs5pDfy1xxDcKjUT\nSs7Su6Z5VQeCBh0LVq1mY9/+7DVFke73EBP0U+tPIxgyYNbtJ870GZKkZMxbn5Jx7AC7hk/j8MCx\n32kphCkqi9Su75HZ+w0s0UeRyYLYKgZQlDeNipLRuJ0pBPwRSNK3U6GSpMDvs+BypFNZOgyfJxqd\noQhLzFFi4nfgcqTi9TQ+kIQrnNkssXQ9vZ+M7INkDZtIrdGCzhCk//AK8rMjOLonhqy9UQwbW4pG\nG/6pR6PayIiEaxmecC0lrmK+ObeT5cfe4pyrmIGxgzGq2rBWbRsR4axzE+FMaHcBSeLh4rOoZTIW\nRsci/05gCmc409S8icqzFZdhLn5184+HOLwjmuVLehOX5uD2x440ezqzxJ3HP048jj9Uy43JD5IW\n0a/ZfQoXtcfDj5e9gcNo4MO5M1sUYNsinAFEe90cscRSoDcxprywSQtlS7R6/tpjKC6VmvElZ+lb\nU9mivlQnxvLIa//gg+uv57A2gj5eJxGhILX+dFzebkRojxJt3EFwYA62BAv7Ro5Hpgig0ZVjijxB\nXPKXZPZ6k4TUDeiNhXg90ZQVj6H4zM04bD0JBhobJGR4a2OpLh+ChAxT5Clik7Yhk4Woqe7F1Wp/\nXhCucIZMRq0+gt7Hd6Hy1XJqYN1GHJVaov/wCirLtBzbH83uTXEMHl1OhLl1FvBbtJFcnzyebpE9\nyLad5uuirSzPehNJkhhgHYRK0bSRxY5MhLPOTYQzod1tdzl4p7qCsUYT1+i//8YTtnAW8mAofxAJ\nCbvpkWYXOq8s0bDknqEE/HLuWtz86czy2gL+cXIR7oCdiYkL6W4e0ax2wm3Y1h0M2rWXLROu43TP\nxh2gWp+2CmfaUBCXQsmZiLq1Z6nuxh1nUqrR89eeQ3CqNIwtPUu/87s/WyKgUhE06pjzyad8cP1Y\nsiUlgzw1qGWgsGno83oR0WdLsPeV4e7nITrhG+JTviIuafP5dWRnkMkk7JX9KCmYRFnReDzupMtG\nyJpGjtuRjsPeBWNEHpHWgxgi8qmuGNRgm2ELZ0BldAI9j+8m4/SBi6NnAAoF9B5URTAgI2tfDNu+\nSKDPkCqi41pnQ4NMJiPRmMiU9KlEaqPIqjzKV/nree/kv4nWRdMrus8P4iBbEc46NxHOhHa3rKKU\ngx43d0ZaiVN9/y/XcIUzteNfqN1rcOlvxadtXqFzv0/G0vuGUnLWyE0PnKD3teXNauecO5c3TjyO\nK1DNdfHz6Bs5tlnthJ0k8aNX38Jor+Hdu27Ho9e1qLm2CmcAsR43RyxWcowWhlWVoA1dPVSUavT8\ntccQHGoN15fmM8DW8mB2gS0qEr0Shn6zn8+HXYOzuILfPfs81727kricc/jKE9gb9yPsvq54PTF4\nPTG4atJx2HpSVnwdJQWTcNh64PeZacwIV2ME/BHYK/uiNZzDHHWcyJj92CoGXnWzQDjDGTIZzggL\nfY7txGiv4PiwiZf+E11612A0+Ti6N4YtnyeRkukkOaPpU9SNJZfJ6RrZjSnpNyJDxoGyfXya+wkb\nzq6lq6UbKab2X17QEiKcdW4inAntyh0K8fOiPLQyOfdcYUoTwhTOJD/68geQSV7slv+HJGteBYK3\nft+HfZviGDS+mMkLsps141foPMWbJxbhDtYwNv5O+kdNaFZfWkPPQ0eZsGYtB4YMYNfolo/ktWU4\nU0sh1KEgp01R5BrNXFN5rt7pzVyDmZd7DMGpUjOmrICBtuaF7Kupjo5CZdSgrbCztW8/io0mrss9\nzYFx17J51lR8WgM+bxRuZypOe1dcjgxqXckE/CZa6wQjSVJhr+qDQlmLOeokMQlf47B3x+epZzNC\nOMMZUBmdSGbuYTJP7Senz3AcUZeXp0pKd5GY5uLovmi2fZEIQO/BVa1ayF2lUNHfOpDrU8Zj99rY\nXfINK06+y9GKIwywDiBSG566u21NhLPOTYQzoV29Z6tkdU01N5si662nGY5wpnZ+iMb1IW79TXi1\no5vVxoYVKaz8v64kZNYw7zcHLyvi3FjHq3ey/PTv8IVqmZi4kD6R4anrGS5zX32b6PIK/vmTH+Ew\nm1rcXluGM4A4jxubWktORCSVGh29aypRXHIURgjYHJvC8sw++OQKJpacpX8YpjLr49NqiFTVHdGx\nq3tPjg0fhDlSD/L2nDaT47R3JeAzYo48gTXxazzueGpdKd+7ZbjDGTIZFdGJDDi8hYSzJzgwejrI\nLw+iMXEeuvW1cepIJPu2xZF73MSQ0eWoNa17RplBZeDaxFEMjhtKYU0+u87t4O2j/6DaW8Wg2CHo\nlC0bRW5rIpx1biKcCe1GkiR+UXSWymCAh63x6OtZVd/icBZyYSi/ByQvNvMiJHnTz3za/lkCf/9t\nX/QRfhYu2YfR0rQFy5IksaN0FR+e+TNy5NyQ/CDdzc2bWm0t3Y8c48aPVnO8d3c2TW7Z+W8XtHU4\nk1F3MO1Zg4nTpij2R8aiCQbxKpQctlh5P7Unu6xJqEIhphbl0s1pa/U+yYEMl51sYyRZFisxHjdJ\ntc5Wf9yGeNwJuJ3JmCJPYE3YSSikwmHrzqXTqGEPZ4DDHIPZXkGX0wfwGEwUZfb93m0izH4GjKig\nOF9P1r4Ytm+Ip8cAG9GxrX+wbrQuhgmpk0gzpXOq+iRbCzfzz6y3UMnV9LcOQCnvmNUyvkuEs85N\nhDOh3XzjdvFSZQnX6o1MjLDUe7uWhjNt9fOoPJtwGe7Aq722yfff+1UsL/9qABpdkLuf3UdcatPW\nwXiDtaw88yLbSj5ArzBzS9qjpBh7NbkfrUkWCrHwhb9hrHHwzr3zqQnDqBm0fTgDUCDRq6aSgExO\njtHCkchYdsUkcswcQ41aQ/eaKm4uysbqa17x7eZQSSFS3XZOmKI4FBlLN0c1UT5Pmz1+ffy+SBz2\nrhjN2UTH7ketsVFdOYAL06qtEc4ACpO60//QZroc283xIeMubg64lFoTYsDwCkJBGcf2R7Pxk2SU\nqhA9BlS36jQn1G0aSIlIZUr6VAwqI0crD7P+7FpWnv6ABEMi3SN7dPhNAyKcdW4inAnt5tcl+WT7\nvNwfHUussv4t7C0JZ3JfNvqKXxCUW7FZFoGsaX/1Htwaw4v/OwiFMsSCp/eT0qNpJ9CXuPN48+QT\n5DoOEqvN4Jb0XxGlSWxSG21h5JebGblxG7uvHcr261te0uqC9ghnUBct0tw1dHHaiPG4ifJ56Gmv\nZFxpPn1qKlG1QxkfXTBInMfNCXMUhyKtDKwuwxBs/0LmwYCBmureGEx5WKKPYjTlUFU+BElStVo4\nC6g12Cyx9M3aQVLuUQ6NnIZ0hZFzmQy69KohrWsNp7Ms7N0ax7H9kfQeXIXR1Pr/dwq5gp5RvZiY\nNgV/yM/+0r18nP0R2wq3kGpKIyUitcOGNBHOOjcRzoR2scvl5OmyIrprtMy1RF/1Ba7Z4UzyYyi/\nB0WwELvplwRVGU26+5fvpbBsUX9kMok7Fx8ko1/jp8CCUpCt597nvZwluAI2BkZNZkryA2gVHe+w\ny6iycu7508v4VCrevP8uvNrmbZa4kvYKZxfogwHivG5S3Q7ivG40ofatrWj2+9AHApwyRZNljmFQ\ndWmDu0rbQiikqdvJqSvFHHWCyJiDVJcPRi4ztko4A6i0JmO2ldP11H70jmqyB9S/FjTK6mXQteVU\nlGg5diCaDStTUapCdO9r/+6StVahUWgYHDeUUUnXUVFbzu6SXbx38t9szP+SaF0MXSxdO1xIE+Gs\ncxPhTGhzIUninoJcSgJ+fmVNwHqF4zMu1dxwpq1eitr9CbWaMbgMcxt9mGooCP96vgfv/7U7ugg/\nC35/gIz+1Y1+3ELnKf51+ikOVn6FThHBlOQHGBA9CbmsDd5FmkgRCHDv8y8TW1LGez+ezZmumeFt\nv53DWUcU63UTksnINkVx1BzDoOoyNB0goEmSAntVb5RKV91OzvgduB3dcbtar5TYmcx+dMk+SPdj\n31BrMFGc2afe26o1IfoNqyQmrpacE2b2bo1j95Y4ktKdxCW1zRS1SW1iTPL1DI4dQo2vhr2lu/k4\n+yPW5HyMUW2ke2RPFC0psBtGIpx1biKcCW3uI3sV/6guZ6TeyAxzw9vUmxPOlO716Kt+Q0CRiM3y\nFMgbd+Bs5Tktf/nlQLZ/moQ1xcm9S/eSkNm4xdt2XwVr8paxJn8ZDn8VPc2juCn1F1i1HfS8JEni\njtffof+eAxwY3J/Pb76hRdUArkSEsytLdjvwy+XkmKI4Yo6hn70cXbD9AxrIcAWI9rcAABbTSURB\nVNq7EgxoMUcdJyp2M8GgDqe9C+E6b+1SIYWS3C796X1sJ70PbMYeFUdpao/6eyeDuORahowuw+1Q\ncfxgFJvWJJOdZSatew2WqLZ5nY/WxTAm+XpGJY6hNlDLwfL9fJa7muXH3sLmtZEakYZF2771cUU4\n69waE85kkiR1+lfX8vLGnRgutK4zPi8Tc47hlSReSky/4qGz32Ux67DZG/+XsbJ2B4ay+SCFqIx6\nkYCq4dEgSYItq5JYvqQntS4VPYaVc9ujR9AZGw6Fdl85W4rfZ0/5FwQlPzGaVMbEzyXZ0PzyUK1O\nkrj53Q+YuPoLClKSeOlXD+LTNPxi0FRqlQKfvyOEjo5HArZbk9gfFY/J5+Wnpw90iF2cF+iNZ0np\nsgqlyoWtsi/ZWQ/g97ZO4IgpK2Deu39AX+vkq1k/ZeeUeY36Q6HwjIF1H6aRd8qETCYxfFwpt9yV\nS4/+rb8L91Jl7jI+zf2Ejflf4vLXPYfXJ4/jzj4/YUr6VNSK5lUjaQmrNUK873ViVmtEg7cR4UwI\nC08oxNQzJzjqqeXnMfGMMzZuR2BTwpnCswtj6XyQfFRbfotPc02D98k+bGbFi93J+iYajS7AtPtO\nMmRy0VXfGyRJotB1kl2lazhctZmgFCBCFc2wmJvpZRndIacwL1B5vcz5x78YsflrSmOtLHv4AeyR\n5lZ5LBHOGrY/MpavY1NQBYPMPXuca6pK2rtLF+kNXmISVhFhySHg15GffQelheNpjUNyrWX53Pbe\n85gcVRwbMp7P5z+Ox9Dwa4QkwanDFjauSab4bN2azl4Dq5g8K58RE0rR6tru588X9LGj+GvW533B\n8apjAJjUZm7ImMpNmTMYmzIerTJ8azqvRoSzzk2EM6FN1IZC3FOQwwZnDRONJn4aE9/o+zYqnEkS\nasfb6KoWAxI282/waq9+wv2ZYxGs/L+u7P2q7pTy7kPKueWh41hi6z/iwO4r53DlFg5WbuScOwcA\nszqOoTHT6GG+FkUTd4O2KUmi5+EsZr79b+KLzlGQksSrD92D09Twi0BziXDWODlGCxvi0/EpFFxT\ncY6ZhacwBlqn+HdTaDQqvF4fkdb9xCVtQqH04qxJJ//0Hdir+oX98YyOKm5Z+TIpRadwRkSydca9\nHBo5jaCq4ZEnSYK8Uya2rU3g9NG6ET6tLsDISSVcM7aE/sMr0enb7mexoCafDfnr2FG0jUpPJVB3\n0O3ktBuYlH4Do5OuI06fgMMBpaVyyspkVFfLcLvB7b78o98vQy6vO69XoZAufq5Wg9ksYTJJ5z/W\nfR0ZKdGrl5HqavG+11mJcCa0uqpAgAUFOexyOxmg1fNEbCKaJmyxaiicyf35aKsWo65dR1Bmwm5e\nhE8z6Iq3dTsV7Pwiga/eT+FMVt1oUWqvaiYvyCaz3/cX/YekIMWuHE7Z93DStpsC10lAQoac9IgB\n9I+cQIqhF7IOPFIGMPjrXYz/dC2puWcJyWRsGzuK1TOnEWjEtHJLiHDWeDaVhrUJGZTpDOgDPqYV\n5TKyoghVO7781oWzupCoVDmIS/4KS3QWADXVPSg+O5Xq8sGEcyRNFgoyfNdnjNr+MWq/F2dEJMeG\nTeRM72GUJ3bBbTSjCPiRh4K4zNFXbKOqTMOBnVYO7LBir9Kc73+I3oOr6D2oim59bXTrayfC3DoB\nOBSCmmo11RUaqso1nMivJiuvkryiWmqrzeBMAGc8Mmcikr91Kg/I5RATEyI+XiIhQSIuru7zpKQQ\nSUl1HxMTJXSdq/DBf412DWehUIjFixdz6tQpVCoVzzzzDKmp3y6e3rhxI6+88gpKpZJZs2YxZ86c\nBu9THxHO2l5Iklhlr+bXJQVUBQOM1Bv5X2s8qiYGmfrCmdyfjaZmOWrHcmT48ar6YDc/Tkhh/bYP\nITiXZ+DI9hj2b7FybHcUwYAcmVyi+9AKrr0pn25DKpHJ6o6/qPQUUVqbR4n7DIXOk5x1HsMXqnts\nGXIS9N3obhpOV9NQdMrWG3EKt+fu/B9UPh9Z/XrxxfQpFCe3zVlrIpw1TQg4dP7QXL9cgdnn4fqy\nAkZUnMMUaPvF3ZeGswu0uhJikzcTYa4bOfbWRlNeMorKkhG4namEa+OA0VHNsD1fMODgZnSeKx/8\n/MZv3qYkrf4NBKEQFJ4xcuqIhZOHIykpuLw6iCXaS2Kak8RUN5FWD5ZoH5YoL1p9ALUmhFobJBSU\nEQzICPjldZeAHL9PjrNGhcOmwl6txmFTU2NTY6+qC2Q11WpCofpf52TyIHJjFSFDIZKxCCLOgfEc\nGoOXGGMk8cZYEo0JJEUkkWCMx6DWIUkyQiEuXiQJ/H5wuWS4XBc+1n3ucMioqVFSVhaiqkqG31//\ncxIVFSI5WSIx8cof4+IkFB1jA+p/lXYNZ+vXr2fTpk0sWbKEQ4cO8dprr/HKK68A4Pf7mTZtGh99\n9BFarZa5c+fy2muvsW/fvnrvczUinLWdMz4v62psvF1dTq7Pi1om4w5LNDebIlE0YzfgxXAW8qDw\nH0dZuw1V7ZcovfsACMitOIx3U147kdICA8VnDBTnGjlz3ETOYTNux7ejQ9aMClKHniRx9FYCxlxq\nfJXY/RVU1BZR7sknKF2+AcCijiNR351UQ19SjH3QKppe+qkjiMovJBAK4GjFKcwrEeGsedwKJfuj\n4jhssRKQK5BLEhlOG/1sFWQ6baS4HW1ykO6VwtnFf9OWExW3G3PUMRSKuuDo81iwV/XB5Ug/f0kj\nGGjZ74w8GCCl4CTJhaeIqjyHxusmJFdQZU1i2x0P4dM2vn2XQ0lhrpGCM0aK8oxUlGixVWlACk+g\nVKmDGE1+Isw+Iiz+yz6PMPvqvrb40Rv9yOUQlAKc858k17ufIt8xzvlPUR0s/l67JkUMcZp04tTp\nWFWpWFSxmJXWixeTMgat3IBWbri4tMJi0WOzuZEkcLmgqkpGVZWMigoZ5eXy8x/rvq6oqD/AKRTS\nxRG3C6EtKUnCapWIipKIjq77GBUl0coD8f9V2jWcLV26lP79+zN16lQArrvuOrZu3QrAiRMn+NOf\n/sQbb7wBwJIlSxg0aBAHDx6s9z5XI8JZywQliVNeD14phE+S8EkSjmCQymCAyoCfikCAXJ+XLI+b\n4vNrZVTIGGWI4HZLFPENrBmR+3OQ+3OQhWqRSbUguZEHK5AFz6GhFFdVETrlGeTyujf6UEhOdt5I\nNm9bwKaNs6goiSDgu8IJ49GnkZK+gdSt0P0zMH3/hQ9AKVMTpUkkWptMtKbuEqNNQa8MTxmjdldb\ni6zW3eYPK8JZy3jlCk6aojhuiqJUa7i4g1EmSVh8HqzeWmK8biJ9XnTBAPqAH20wgDoUQiGFUEjS\n+Uvd5zIk1KEgUb7G1ai8Wji7QCbzE2E5TYTlNEZTDkrV5aPcPk8kXm8kPk8Ufm8kPp+ZYFBLKKAj\nGNQQCmqQJHndBTl+r4VaV3KDfQvpdEjxCY36Pq7G75dRXaHBaVfjrFHhcqjweetGyfw+OXK5hEIp\noVBIKJShi5/r9AH0EQH0Rj8GY91Hlbrlb5WekJMS/2nO+U9T7j9DVbCY6kAxtmAJIRrePa6SadDI\nDegUepDkyJEjk8mRIUMt1/FA8kv0NFxe11eSwOHgfFiTXwxsF8JbeXndejipgRAbEVG3/s1gkDAa\nOf9RwmDgso9qNahUEkolKJWgUtWFQJXqwud1P+qS9O0o4YURw4EDg2RmdvqVVg1qTDhrtRXOTqcT\no/HbE9MVCgWhUAi5XI7T6SQi4tvOGQwGHA7HVe8jtJ6nS4t4pbK0wduZ5QpG6A0M0OoZoTdiVtT9\n+Fw130s+IoonIZPqf8MI1EZzrOAaTp0aQlbWSPbunUxNTTRyeQi92UdcmhNTtIeouFpiUlzEJNWw\n0f8balSnUMt1qOU6VIpk1PKu6BQRGJSRGFSRGJUWDMpI9ErTFdeNhTr/ckvg/Ht6O5w3JoUkJHHO\nWbOpQwH6VZXRr6oMt0JJgcFEic5AuVaPXaXhlCmKUzR8VuB3PXDyAL1rKhu8nSRJDf/YSCpsVb2x\nVfUGJNTaKnT6EnT6UrT6EjTaSgwReUSYcxvdv/3b/oSn9uqbhiSZHImW/2wpVRLWhFqsCS0/yDYc\nP+kauYE0zUDSNAMvuz4kBakJlmMPluIK2XCFqnEFq3GFqnGHbPgkD76QG59Ui0+qJRDyEgz5kQgB\nEhIhlDIXvlDtFV+PIyLqwlVm5pVHZINBqK6uC2qVlTLsdtn56dPLL2432O1yPB4IBMJ/Nt6gQUHW\nrWv7PzQ7olYLZ0ajEZfr27UEl4asiIiIy/7N5XJhMpmuep+raUwKFeq3zNqLZbRise7M5haBlgPa\n85fvHgfxVYu6JAg/WGPHtncPrqqDd09oVwpAvJ9Daxxoc97gwYMvTkkePHiQHj2+XdiZmZnJ2bNn\nsdvt+Hw+9uzZw6BBg656H0EQBEEQhP8GrbbmTJIkFi9ezMmTJ4G6dWVZWVm43W5uu+02Nm3axLJl\nywiFQsyePZsf/ehHV7xPRkbTCloLgiAIgiB0Zj+Ic84EQRAEQRB+KMRKe0EQBEEQhA5EhDNBEARB\nEIQORIQzQRAEQRCEDkSEM0EQBEEQhA6k1c45a00Oh4NHH30Ul8uF3+9n0aJFDBw4kIMHD/Lss8+i\nUCgYNWoUP/vZz9q7q8JVbNiwgbVr1/LCCy9c/Pq5554jPr7ugMqf//znDBs2rD27KNTju8+d+N3r\nfCRJ4rrrriM9PR2AQYMG8ctf/rJ9OyVcVXPrTwsdx6233nrxsP2UlBSeffbZK96uU4azt99+m5Ej\nR3LnnXdy5swZHnnkEVauXMmTTz7J3/72N1JSUrjvvvs4fvw4vXq14uGqQrP94Q9/YPv27fTu3fvi\ndVlZWTz66KNMnjy5HXsmNORKz93ixYt5+eWXxe9eJ5Kfn0+fPn149dVX27srQiN9+eWX+P1+VqxY\nwaFDh1i6dGmj6k8LHYPXW1cp55///GeDt+2U05oLFizg9ttvByAQCKDRaHA6nfj9flJSUgAYPXo0\nO3bsaM9uClcxePBgFi9efFmpkaysLD766CPmzZvHH//4R4JBUbexI/ruc+d0OvH5fOJ3r5PJysqi\nrKyMO++8k/vuu48zZ860d5eEBuzfv58xY8YAMGDAAI4ePdrOPRKa4sSJE9TW1rJw4ULuuusuDh06\nVO9tO/zI2QcffMDy5csvu27JkiX07duX8vJyHnvsMX79619/ry6nwWCgoKCgrbsrfEd9z9/UqVP5\n5ptvLrt+1KhRTJw4keTkZH73u9+xYsUK5s2b15bdFS7R2OdO/O51fFd6Lp988knuv/9+pkyZwr59\n+3j00Uf58MMP26mHQmOI+tOdm06nY+HChcyZM4e8vDzuvfde1q1bd8Xnr8OHszlz5jBnzpzvXX/y\n5EkeeeQRHn/8cYYOHYrT6bysLqfT6cRkMrVlV4UrqO/5u5JZs2YREVFXV23ChAmsX7++NbsmNKCx\nz913a+KK372O50rPpcfjQaFQADBkyBDKysrao2tCEzS3/rTQMaSnp5OWlnbxc4vFQnl5OXFxcd+7\nbad8VrOzs/nFL37BCy+8cHGI12g0olKpKCgoQJIktm/fztChQ9u5p0JjSZLEjBkzKC0tBWDnzp30\n7du3nXslNIb43eucli1bxjvvvAPUTbckJia2c4+Ehoj6053bypUrWbp0KQClpaU4nU6sVusVb9vh\nR86u5M9//jN+v58//OEPAJhMJpYtW8ZTTz3Fr371K4LBIKNHj6Z///7t3FPhamQyGTKZ7OLnzzzz\nDA899BAajYZu3bpx2223tXMPhfpc+twB4nevE7rvvvt49NFH2bJlC0qlkiVLlrR3l4QGTJo0ie3b\nt3PHHXcAiOesk5k9ezZPPPHExeU6S5YsqXfkU9TWFARBEARB6EA65bSmIAiCIAjCD5UIZ4IgCIIg\nCB2ICGeCIAiCIAgdiAhngiAIgiAIHYgIZ4IgCIIgCB2ICGeCIAiCIAgdiAhngiC0u2+++Yb58+eH\nvd3x48czbdo0brnlFmbMmMHMmTO/VzasIfPnzycrK6vRt1+5ciVPP/30964vLS3lvvvuA2DRokWs\nWrWKsrKyi9dt3LiRt99+u0l9EwThh6lTHkIrCILQWK+//vrF0++//vprHn74YbZt23axdFFjNOU4\nyEsP571UXFwcf//73y/eRiaTERsbe/G6rKyseu8rCMJ/FzFyJghCh/bqq68ybdo0pk+fzh//+EdC\noRAAy5cvZ8qUKcyePZvHHnuMv/3tbw22NXToUKqqqrDb7SxatIgHHniAqVOnsnnzZg4ePMicOXOY\nMWMGCxYsID8//+L9li9fzsyZM5k5cyZ79+4F6kbCFi5cyO2338748eN54YUXgLogl52dzdy5c5k+\nfTovvvgiAIWFhYwfP/6y/ly4LicnhxUrVrBixQo+/PBDxo8fT15eHgBut5uxY8fi8/la/H8pCELn\nIEbOBEHosLZs2cKmTZtYtWoVCoWChx56iP/85z8MGTKEf//736xcuRKVSsX8+fNJTU29YhuXjnp9\n8sknpKenExUVBUBkZCSvvvoqPp+PG264gZdeeom+ffuydu1afvnLX/Lhhx8CdSXiVq5cyYkTJ3jw\nwQdZt24dn332GdOnT+eWW27B4XAwduxY7r77bgAKCgpYtWoVer2eu+66iy1bttClS5d6v88uXbow\nd+5coK7Ey7lz51i9ejU///nPWb9+PePGjUOtVofl/1QQhI5PjJwJgtBh7dq1i5tuugm1Wo1CoWDW\nrFns2rWLXbt2MW7cOAwGA2q1mmnTptXbxn333cctt9zCtGnT+PLLL/nLX/4C1E0tDhgwAIC8vDzM\nZjN9+/YF4IYbbiA/Px+n0wnAnDlzAOjZsycWi4UzZ85w9913Ex8fz5tvvskzzzxDIBCgtrYWmUzG\n5MmTMZvNqFQqbrzxRnbu3NnglOWlIfLWW2/l008/BWDVqlXceuutzfwfFAShMxIjZ4IgdFiSJF0W\nWiRJIhAIIJfLL05vXri+PpeuOfsujUYDcFlbl7YZDAYBLlufJkkSCoWCpUuXUlhYyPTp05k4cSI7\nd+682I9LixmHQiGUyqa91CYnJ5OYmMj69eupqqoSheQF4b+MGDkTBKHDGjFiBJ999hler5dAIMBH\nH33EiBEjuPbaa9myZQtOpxOfz8f69etbtJg+MzMTm83GkSNHAPj8889JSkrCbDYDsGbNGgCOHDmC\ny+UiLS2NHTt2sHDhQqZMmUJxcTGlpaWEQiEkSWLTpk04nU68Xi+ff/45I0eO/F7I/G6gVCqVBAKB\ni1/PmjWLZ555hhkzZjT7+xIEoXMSI2eCILQ7mUzGvn37GDRo0MXrZsyYweLFizl+/DizZs0iEAgw\nZswY5s+fj1wuZ/78+dxxxx3o9XoiIyPRarXNelwAtVrNiy++yNNPP01tbS0Wi+XiQn4Am83GLbfc\nglKp5E9/+hNKpZL777+fxx57jOjoaLp27cqIESMoLCxEJpORkZHBPffcg8Ph4Oabb2bkyJEX/+3C\n4156ARg2bBiPP/44VquVefPmMWnSJH7729+KcCYI/4VkUlP2iAuCIHQAeXl5bN68mQULFgDw4IMP\ncttttzF27Nh27Ve4SJLE1q1bee+993jllVfauzuCILQxMXImCEKnk5iYyJEjR5g+fToAY8aM+cEE\nM4Bnn32WLVu28Prrr7d3VwRBaAdi5EwQBEEQBKEDERsCBEEQBEEQOhARzgRBEARBEDoQEc4EQRAE\nQRA6EBHOBEEQBEEQOhARzgRBEARBEDqQ/w/bSVTeN1+L1gAAAABJRU5ErkJggg==\n",
"text": [
""
]
}
],
"prompt_number": 8
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Well, that hypothesis was incorrect! We can say that the red peak just above a log score of -10 is likely associated with the same peak in the previous graph, but the others are likely more complicated. One last thing we can do is look at the distribution of sequences which do not contain 'G' compared to those which do, because 'G' does not appear in the profile at all."
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [
"plt.figure( figsize=(10, 5) )\n",
"\n",
"subsequences = it.product( 'ACT', repeat=3 )\n",
"scores = map( model.log_probability, subsequences )\n",
"\n",
"sns.kdeplot( numpy.array( scores ), color='r', shade=True, label='Without G' )\n",
"\n",
"subsequences = filter( lambda seq: 'G' in seq, it.product( 'ACGT', repeat=3 ) )\n",
"scores = map( model.log_probability, subsequences )\n",
"\n",
"sns.kdeplot( numpy.array( scores ), color='b', shade=True, label='Including G')\n",
"\n",
"plt.ylabel('Density')\n",
"plt.xlabel('Log Probability')\n",
"plt.legend()"
],
"language": "python",
"metadata": {},
"outputs": [
{
"metadata": {},
"output_type": "pyout",
"prompt_number": 9,
"text": [
""
]
},
{
"metadata": {},
"output_type": "display_data",
"png": "iVBORw0KGgoAAAANSUhEUgAAAmcAAAFICAYAAAAVoFlvAAAABHNCSVQICAgIfAhkiAAAAAlwSFlz\nAAALEgAACxIB0t1+/AAAIABJREFUeJzs3Xd4VHXa//H3mTMlk95DEUggDQihSxFEdN0FC4oVC7qu\nbd1FbLiK6yK6IlhWHn+suuKu68quoqxlFTtKJ9RASOi9l4SWPpmZc35/DIkgkEySmTkz5H5d13M9\nJjPnnBt2CB++5f4quq7rCCGEEEKIoGAyugAhhBBCCPETCWdCCCGEEEFEwpkQQgghRBCRcCaEEEII\nEUQknAkhhBBCBBEJZ0IIIYQQQcTsrxtrmsbEiRPZvHkzFouFSZMm0b59+7rXZ8+ezXvvvYeqqmRm\nZjJx4kQURWHkyJFERkYC0K5dO1544QV/lSiEEEIIEXT8Fs7mzJmD0+lk5syZFBQUMGXKFN544w0A\nqquree2115g9ezY2m43HHnuMuXPnctFFFwEwY8YMf5UlhBBCCBHU/DatmZ+fz+DBgwHo3r07RUVF\nda/ZbDY+/PBDbDYbAC6Xi7CwMDZu3EhVVRV33303d955JwUFBf4qTwghhBAiKPlt5Ky8vLxuehJA\nVVU0TcNkMqEoCvHx8YBnlKyqqoqBAweyefNm7r77bm688UZ27tzJvffey7fffovJJEvjhBBCCNEy\n+C2cRUZGUlFRUfd1bTA79euXX36ZXbt2MW3aNABSU1Pp0KFD3X/HxsZSXFxMSkrKOZ+j6zqKovjp\nVyGEEEIIEVh+C2e9evVi7ty5DB8+nDVr1pCVlXXa6xMmTMBms/H666/XhatPPvmETZs28cwzz3Do\n0CHKy8tJSkqq9zmKolBcXOavX4Y4zyQlRcnnRXhFPiuiMeTzIryVlBTV4HsUfx18rus6EydOZNOm\nTQBMnjyZdevWUVlZSU5ODtdffz19+vSpe/+dd97JJZdcwvjx49m/fz8Ajz/+OD169GjwWfIHQnhL\nfoAKb8lnRTSGfF6EtwwNZ4EkfyCEt+QHqPCWfFZEY8jnRXjLm3AmK+2FEEIIIYKIhDMhhBBCiCAi\n4UwIIYQQIohIOBNCCCGECCISzoQQQgghgoiEMyGEEEKIICLhTAghhBCGeuih37FhwzoAnE4nv/rV\nEN5/f0bd62PG3MeWLZt55pmncLlcHDp0kMWLF9a9tnv3rmY9f/78uZSUlJzx/X379vL003/g/vvv\n4qGHHuAPf3iYHTu2N+tZ3vDbCQFCCCGECD0RE5/G9sVnPr2n4+prqZj4/Dlf79v3QgoKVtO5c1cK\nClbTr99Ali5dzK23jsbhcHDo0CEyMjJ59tkXAFi1agW7d+/ioosGnzxlqHktW//735mkpaUBiXXf\nq66uZvz4x3jiiT/RtWsOABs2rOPVV19k2rS3mvW8hsjImRBCCCEM1bdvfwoK1gCwdOkSrr76GsrL\ny6ioKGfdukJ69uwFwA03XE11dTX//ve7zJnzLYsWLQDgnXfe5qGHHuC++37N/v37AJg2bSr33fdr\n7rvv18yaNROASZMmsmxZXt1zXnjhWfLyFrFly2aef34iLperrqbFixfQu/eFdcEMoHPnrn4PZiAj\nZ0IIIYQ4RcXE5+sd5fKHjIxMdu/eCUBBQT733/97+vS5kJUrl7N16xb69RsIeM7TNplMjB59F7t3\n72LQoIv58MP/MHDgYH75y2G888505s37gQ4d0jh4cD/Tp7+Ly+Xid7+7h969+6AoSt153rX/f8CA\nQWRkZPL4409hNv8Uiw4c2E/btm3rvh4//jHKy8s5cqSE1157k6SkZL/9fsjImRBCCCEMZTKZSE/P\nYOnSJcTHJ2CxWOjf/yLWrl3D2rUFXHhh/9Per+s6p54+mZ2dDUB8fALV1dXs2rWT7t17AmA2m+na\ntRs7duw47R6aptVbU3JyKw4c2F/39eTJf2HatLeIiorG7a7/2uaScCaEEEIIw/Xt24/33nuHAQMu\nAiA3twebNm0EdKKiTj+P0mQy/SxcKae9npqaxtq1nmlSl8tFUVEB7dq1w2q1UlJSDMDmzRvruR8M\nHjyElSuXs25dUd339u7dQ3HxYZTTH+dzMq0phBBCCMP16dOPl156gQkTPFOqZrOZqKhoMjOzTnmX\nZ1qyU6d03nvvHbKysuumJ+veoSgMHDiI1atX8dvf/gan08lll11OZmY2V111LZMnP8d3331Nu3Yd\n6q7Jycnl+eefYerU1+uCoN1u58UXp/Lmm9M4cqQEt9uNqqqMHfsoKSmt/Pp7oeinjguGqOLiMqNL\nECEiKSlKPi/CK/JZEY0hnxfhraSkqAbfI9OaQgghhBBBRMKZEEIIIUQQkXAmhBBCCBFEJJwJIYQQ\nQgQRCWdCCCGEEEFEwpkQQgghRBCRcCaEEEIIQx04sJ/777/Lr9fcccfNAPz73++yYcO6Rj3r5/bt\n28vTT/+B+++/i4ceeoA//OFhduzY3qx7nkqa0AphMOXEcfSISDDLH0chhPEmTrTxxRe+/Xl09dUu\nJk50+PSeTXX77b9u1vXV1dWMH/8YTzzxp7pD0TdsWMerr77os0PR5W8DIQJN17EsWYT1xzlY5v2I\nuWgtWkorSv/zEa5u3Y2uTgghDDVmzH1kZmaxffs2Kioq+POfX6RVq1a8++7fWbRoAW63i2uvvYF+\n/QbUXXPDDVfzwQefYLFYePPNaaSmpjFs2JW89NILbNu2heTkFCoqKgCYNGkiv/jFrzhypIS8vMU4\nHA7279/LbbfdyfDhV7F+fRFTp75EeHgEsbFx2Gw2nnrqmbpnLV68gN69L6wLZgCdO3f1WTADCWdC\nBFz4i5OIePUlAHTVjDsjE3XLZmKv+iWlb/2TmmFXGFyhEKIlmzjRYegol6IodOmSw9ixjzF9+hvM\nmfMNF17Yn2XL8nj77X/hdrt5663XTzsM/dQjnGr/e8GCuTgc1Uyf/i7Hjx9n1KhrT3tdURQqKip4\n9dVp7N27hyeeeIThw6/ilVcmM2HC86SmpjF9+ht1Z3HWOnBgP23btq37evz4xygvL+fIkRJee+1N\nkpKSm/17IGvOhAggy49zCJ/6Mu7kFMqffpYT//6Q8hdfpfKJP4KmEX3nLdjf/CuE/qlqQgjRZLXn\naSYnp1BTU8OePbvp0qUriqJgNpv5/e8fOue1tadS7t69i+zsLgDExsbSoUPaGe/NyMgEICkpmZqa\nGgCOHCkhNdXz3u7de55xTXJyKw4c2F/39eTJf2HatLeIiorG7dbOeH9TSDgTIkBMB/YT/cA9oKpU\nPj4eV+8+EBYGgLPfAMonvYQeF0fkM09hmzXT4GqFEMJIpx9m3r59Kps2bUTXdVwuF48++iBOp7Pu\ndavVSklJMbqus2XLZgBSU9MoKloLQGlpKXv27D7zKT87NB08gXDnzh0AddefavDgIaxcuZx164rq\nvrd37x6Kiw9zlts1iUxrChEILhdR992F6dhRKu/9Le70jDPe4u6UTvmkl4h68LdEvDgJx8gbwGIx\noFghhAi8swWl2u9nZGTSr99AHnjgbjRNY+TIG7BarXXX3HrrHTz++EO0atWa6OhoAAYPvoT8/FXc\ne++dJCYmER+f0MAzPf/92GNPMnnyc9jtdiwWC4mJp09T2u12XnxxKm++OY0jR0pwu92oqsrYsY+S\nktLKB78ToOh66M+fFBeXGV2CCBFJSVGGfF4iJj1L+Gt/oWbgICrHPUl9/7yyv/0mtq9mU/bqNKpv\nvzOAVYpTGfVZEaFJPi/nj08+mcWll15ObGwsb7/9JhaLhV//+h6f3T8pKarB98i0phB+ZjqwH/vr\nr+FOTqHyd2PrDWYA1dffhG6xEv6XF8ERHFvPhRCipYiPj+fRR3/P739/L1u3bua6624KeA0yrSmE\nn9n/MR3F5cJx480QEdHg+/X4BBzDriDsi88Ie38G1Xf57l9sQggh6nfJJZdxySWXGVqDjJwJ4U8V\nFYT96x9o0THUXDzU68sc192IbrMRPvVlqK72Y4FCCCGCjYQzIfwo7KMPMJ044eldZrV6fZ0eG4vj\niqtRDx7APuOffqxQCCFEsJFwJoS/aBr2t15HN5txDL+y0Zc7rr0OPSwM+19fA803vXOEEEIEPwln\nQviJdc63mLdvo+biS9Bj4xp9vR4dQ81Fg1EP7Me8YrkfKhRCCBGMJJwJ4Sf2v70OgOPqa5t8D+fA\nQQDYvvjUJzUJIYQIfhLOhPADtagQ66IFOLt1R0s988gQb7lye6BFRGL736cytSmEEC2EhDMh/CBs\n5r8BcFw1onk3Mptx9huAeuigTG0KIUQLIeFMCF/TNGyff4YWEYGrZ+9m3855kUxtCiFESyLhTAgf\nM69cgXrwAM5+A3xyNqZMbQohRMsi4UwIH7N98RkAzgEX+eaGMrUphBAtioQzIXxJ07B98RlaeDiu\n7j19dlvZtSmEEC2HhDMhfMicvxJ1/z5cF/pmSrOWK7e7TG0KIUQLIeFMCB+yffE/AGoG+mhKs5bF\nIlObQgjRQkg4E8JXdB3bF5+h28Nx9ejl89s7+w8EwPrjdz6/txBCiOAh4UwIHzGvXoW6dw/OC/v5\ndEqzlqtrDrrJhHXhfJ/fWwghRPCQcCaEj9RNafpql+bPhYfj7pSOec1qlPIy/zxDCCGE4SScCeEL\nuo7t80/R7XafNJ49F1duDxSXC8uyPL89QwghhLEknAnhA+rWLah7duPs0QusVr89x5WTC4Bl0UK/\nPUMIIYSxzP66saZpTJw4kc2bN2OxWJg0aRLt27eve3327Nm89957qKpKZmYmEydORNf1eq8RIlhZ\nf/weAFevPn59jqtzZ3TVjEXWnQkhxHnLbyNnc+bMwel0MnPmTMaNG8eUKVPqXquurua1115jxowZ\nfPDBB5SXlzN37tx6rxEimFl/nAOAs6fvd2mexhaGOysLc9FalOPH/PssIYQQhvBbOMvPz2fw4MEA\ndO/enaKiorrXbDYbH374ITabDQCXy4XNZqv3GiGCVlUVliWLcLfvgJ6Q6PfHubp1R9E0LHlL/P4s\nIYQQgee3cFZeXk5kZGTd16qqop3sbK4oCvHx8QDMmDGDqqoqLrroonqvESJYWfIWozgcOP24EeBU\nzm7dPc9dJFObQghxPvLbmrPIyEgqKirqvtY0DZPJdNrXL7/8Mrt27WLatGleXXMuSUlRPqxcnO98\n/nlZugCAsMEDCYsN9+29z6ZvD7BaCc9bRLh89v1KfraIxpDPi/AVv4WzXr16MXfuXIYPH86aNWvI\nyso67fUJEyZgs9l4/fXXURTFq2vOpbhYej4J7yQlRfn88xL35VeoNhsn2qfD8Uqf3vtcIrK7YFm7\nhpINO9AT/T+V2hL547Mizl/yeRHe8ibE+y2cXX755SxevJhRo0YBMHnyZGbPnk1lZSU5OTl8/PHH\n9OnThzvuuAOAO++886zXCBHMTHt2Y96yGWefvn45FeBcXN1ysaxdg2XJQmpGjAzYc4UQQvif38KZ\noig8++yzp30vLS2t7r83bNhw1ut+fo0Qwcw69weAgK03q+U6ue7MunCBhDMhhDjPSBNaIZqhNpz5\n81SAs3GnZ6Db7bIpQAghzkMSzoRoKqcTy/y5uFNaobVuE9hnqyqurGzM27aiHD0S2GcLIYTwKwln\nQjSRZdUKTOVlfj8V4FzcGZ4NM5bVqwx5vhBCCP+QcCZEE1nmnVxv1qOnIc93ZXrCmXnVSkOeL4QQ\nwj8knAnRRNaF89FNprrDyAOtbuQsX8KZEEKcTyScCdEU5eWYV+fj7pQO4QFoPHsWekwM7pRWmFet\nAF03pAYhhBC+J+FMiCawLM9Dcblw5fYwtA53VjamEydQt281tA4hhBC+I+FMiCawLvQc2eTqZsyU\nZi1Xhqw7E0KI843fmtCKlq28HLZvN2G3Q0SETkKCjs1mdFW+Y1k4H91sxpXdGV2HuQWJ/JCfxL6S\nMPaWhBET4WLEgINc1f8gCdFOv9XhPrkpwLJqBY6bbvHbc4QQQgSOhDPhU6tXm/j3vy18/LGFykql\n7vtRUTq33urkN7+pIS0ttNdHKcePYS4swNW5K3PWXcCUmRms2hJX97pq0nBrJn5ck8Rjb3Xltkv3\n8uK96wmzaj6vxZ3WEd1slpEzIYQ4j0g4Ez7hdMLjj9t4/30rAAkJGhdf7ELToKpKYe1albfesjJ9\nuoXrrnPx4ovVREcbXHQTWZYsplyPYHTpP/jfny8EoH/no4wYcIC2idXERjgpPmFj8bp45uQn8d6c\n9qzbFcV/xq8iJa7Gx8VYcKd1xLy+CKqqwG737f2FEEIEnIQz0WxlZfCb39iZP99Mx44at91WQ/fu\nGqr603ucTli6VOWzzzyjasuWqUyfXkWfPr4fTfK3nV9u4BaWsWFvF7p2KOX+q3aS1qrytPekxDm4\nbtABrup3kGmfdWTe2iSGjruIj/60kpzUMp/W487MxrxlM+a1Bbj69ffpvYUQQgSebAgQzVJcrHDV\nVeHMn2+mTx83kyZV06vX6cEMwGKBwYPdvPRSNTfd5GTfPoWrrw7nH/+wGFN4Ey1ZojLk43FsoAvX\n9N/LpLvWnxHMTmW16Dx6wzbuuHw3B4+FccukPhwv9+2/iVxZ0u9MCCHOJxLORJNpGvz+92Fs2KDy\nq185efJJB2Fh9V+jqnDLLU6ee85BVBSMHx/G1KnWkGjTlZencsuoMCq1MF5PfoZ7rtx7Rgg9G0WB\nGy/ez81D9rG3xM6Dr+f69Ndb24zWLOFMCCHOCxLORJO9+aaFefPM9Orl5v77nV4FlVo5ORovvFBN\nUpLG5Mk2nn8+uAPa0qUqt9xix1mj8zHXM7R7SaPvMWroXnJSS5m9tBV//7qDz2rTUlqhRUVjWbXC\nZ/cUQghhHAlnoknWrDExaZKNmBidBx90oCgNX/NzrVvrvPCCgzZtNKZNs/Hcc8HZa2PNGhOjRtlx\nOOD1bm9wNbMp6di70fdRTTDuxi1Ehzv54z87U7gjyjcFKgrurCzUvXtQDh3yzT2FEEIYRsKZaLSK\nCrjvPjsul8LDDzuIjW36vRITdSZNqqZNG43XX7fy178G1xq0XbsUbrnFTnU1PPpoDbcenIrTFklp\n68wm3S8h2snD123D6TLx1DudfTZa6MrMBmTdmRBCnA8knIlGmz7dys6dJq65xkmPHs3fbRkbCxMn\nOkhI0HjuuTA++CA4NhEfPQqjRtk5csTE3Xc7uSxjJ5GHdnAktQe62vQa+2Ydp3fGMRYVJTK3INEn\ntbozPGHRXJDvk/sJIYQwjoQz0ShHj8K0aVaionRuusl3ne+TknSeecZBZKTOI4+E8fXXxga06moY\nPdrOtm0q11zj5IorXCQWzgfgSBOmNH/uzsv3ADDxvSw0H3QTcad1AsC8tqD5NxNCCGEoCWeiUaZN\ns1FernDDDU7Cw31773btdP70JwcWC9x7bxh5eY3YYeBDmgZjxoSxYoWZQYNc3HGHJ4QmnQxnJR37\nNPsZaa0rGZJbQuGOGD5b0rrZ99NjYtASEjEXrGn2vYQQQhhLwpnw2oEDCn//u4WEBI1hw1x+eUZm\npsYTTzjQNLjtNjuFhYH/iE6caOPzzy106eLmwQdrMJkAXSexcD419hjKkjv65Dm3X7YH1aTx/H8y\ncbqasKPiZ9yd0lGLD2M6dNAH1QkhhDCKhDPhtVdeseJwKIwa5cRq9d9zevbUGDu2hooKz5qvHTua\nH1y89fbbFv72Nytt22o8+aSj7tcZcXA74Uf2UdKxF5601nyt4h0M63uYHQcj+O+CNs2+n6vjyanN\nQpnaFEKIUCbhTHhl3z6F99+30LatxtChbr8/b/BgN/fc46S42MRNN4Vz6JD/A9onn5h5+mkbsbE6\nEyZ4muTWSixcAMCRtOavNzvVdYP2Y1J0pn/V/L5n7o7pgKw7E0KIUCfhTHjlX/+y4HYrjBzZuGaz\nzXHFFS5uvtnJrl0mbr7ZzokT/nvWnDkqY8aEYbfDhAnVJCef3uMiqXAeQJP6m9UnObaGvlnHWLMt\nllWbY5p1L3dHz3SrhDMhhAhtEs5EgxwOeO89C5GROoMG+X/U7FQ33+xk2DAn69er3H67nfJy3z9j\n6VKVu+6yYzLBH//oIC3tZ83HdJ3EwgVUR8ZTkei7zv61ruznaRz7djNPDdDjE9BiYjAXrPZFWUII\nIQwi4Uw06PPPzRw9auIXv3BhC3ATf0WBe+5xMmiQi2XLzFx/fTjHjvnu/vPmqYwaZcflgj/8wUGX\nLmf2tYjau5GwE4c9U5pNOQqhAd07nqBtQhWfLGpDyYlmLOZTFNwd01H37UU5esR3BQohhAgoCWei\nQe+8Y0VRdL/t0GyIqsLDD9cwdKiL1atVRowI5+DB5oekL74wc+utdmpqYNy4Gnr3PnvDsdr1Zr5o\noXE2JhNc0e8QTpeJ9+a0a9a93LWbAooKfVGaEEIIA0g4E/UqKDCxapVK794aKSnGnUyuqjBmTA1X\nXulk0yaV4cPDWb68aR9fTYO//c3CPfeEYTbDhAkO+vc/93RtUl3z2V5Nep43LutZTJjFzTvftMfl\nbnrwdHeSTQFCCBHqJJyJer3zjmeabfhw350G0FQmE9x9t5Pbb6/hwAGFa64JZ+pUK+5GLIPbv9/T\nnmPChDCiouDPf3bQrVs9Lfo1jcSiBVTGpFAZ17b5v4hziAhzc0mPEvaV2JlXkNDk+9SNnBVKM1oh\nhAhVEs7EOZWXw6efmklJ0XxyhqYvKApcf72L555zEBOjM3myjWHDwvnqK3O9xyCVlcH06RZycmDe\nPDM9e7qZOrWK9PT6f13Ru4qwlh/zHNnkh/Vmp7q0RzEAHy9ses8zLTkFLSJCTgoQQogQFhwnTIug\n9NVXZqqrFa691umrvqs+07WrxtSp1bz9tpVFi8z8+td2MjLcjBjhoksXjYwMjePHFXbvVsjPV5k5\n00JlpYLNBvfdV8OwYS6vslbdkU0+7m92NtntykmKcfDF0la8+tsi7LYmBGJFwZ3WCUvRWpSyUvSo\naN8XKoQQwq8knIlz+uQTC+BpCBuMoqPhscdquOkmJ59+amHBApW//OXs20nj4jRGjnRyww1WwPuN\nDYlFJ5vP+ri/2dkoCgzJLeG/C9vy7cpkrr2oaccwuTt5wpl5XRHO/gN9XKUQQgh/k3Amzqq4WGH+\nfJX0dDdt2hi3EcAb7drpjB1bw513wo4dJnbtMrF3r4moKJ2UFJ3WrTW6dNEwmyE21srx497dV3G7\nSCxaSEX8BVTHpPj3F3FSbTj7eGGbpoez2pMCCgsknAkhRAiScCbO6vPPzbjdChdfHJyjZmcTEwM9\nevhufVzM9gIsVWXs73qpT+7njdRWVbRPquTbVcmcqDATE9H49iV1mwJkx6YQQoSkIFtJJILFJ59Y\nUBSdiy4yprdZMKg9ssnX52k2ZEj3EpwuE7OXtmrS9VrrNui2MMxrZVOAEEKEIgln4gy7dimsWKGS\nk6MRH290NcapW2+W5r/+Zmdzca6nu/+sBU3ctamquFNTUTdvgupqH1YmhBAiECSciTN8+qlnI8DF\nF7fcUTPFWUPC+iWUJaXiiGp637GmaBXnIOuCMhYWJjT5OCd3WkcUtxvz5o0+rk4IIYS/STgTZ/j0\nUzNms86AAaGz3szX4rauwuyoDMguzbMZ2PUomq7w3aqkJl3vTusIyDFOQggRiiScidPs2qWwYYNK\nbq5GRITR1Rjnp/5m/jlPsyEXZnlOd/96edN2ibpTPeFMLVrrs5qEEEIEhoQzcZpvv/Vs4L3wwpY7\nagaQWDgfHYUjaT0Nef4FSdW0Tajih9VJVNc0/o+pu0MHdMUkI2dCCBGCJJyJ03zzjSec9e3bcsOZ\nqaaa+E3LKE3phDM8xrA6+nU+RlWNyoLCJqx5s4WhtWmDeV0R6MHdp04IIcTpJJyJOsePQ16ep/Fs\nfHzL/Qs9ftMyVKeDI52MmdKsdWG2Z2rzq6ZObaZ1xFRWimnPbl+WJYQQws8knIk6c+Z4Gs+29CnN\nn9abBbaFxs9ltysjOtzJN8uT6z3U/VzcqWmAbAoQQohQI+FM1JH1Zh6JhfPRFRNHU41Zb1ZLNUHf\nrGMcOh7Gmm2Nn16t3RRgXifhTAghQomEMwGAw+EZOUtJ0WjfvuVOaZqryojbspITrbNwhUUaXQ79\nTk5tfr0iudHXSjsNIYQITRLOBACLF6tUVHimNBXF6GqMk7h2Pia3i8MZ/YwuBYAenU5gUTW+XNb4\ndWd6XBxadDRmaachhBAhRcKZAGRKs1bK6u8BKM7ob3AlHnabRk5aKRt2R7P/iK1xFysK7rROqHt2\no5w47p8ChRBC+JyEMwHADz+YCQ/Xyc5uwsrz84Wuk5z/HU5bJMcv6Gp0NXV6ZXiC1byCxEZfW7cp\nYP06n9YkhBDCf/wWzjRNY8KECYwaNYrRo0eze/eZ2/mrqqoYNWoU27dvr/veyJEjGT16NKNHj+ap\np57yV3niFNu3K+zebSI3143ZbHQ1xonct5mI4t0Up/dFV4PnN6JX+gkAfljd+KOcatedqbIpQAgh\nQobf/gaaM2cOTqeTmTNnUlBQwJQpU3jjjTfqXi8sLOSZZ57h8OHDKCcXOTkcDgBmzJjhr7LEWcyd\n6/kY9OwpU5oAxRkDDK7kdO2SqkiIdjB3TSJuN6iq99fKpgAhhAg9fhs5y8/PZ/DgwQB0796doqKi\n0153Op288cYbpKWl1X1v48aNVFVVcffdd3PnnXdSUFDgr/LEKX780RPOevRowVOaQHJ+bTgLjs0A\ntRTFM3p2rNxKwfbGtdTQ2l6AbrFIOBNCiBDit5Gz8vJyIiN/akWgqiqapmEyefJgr15nNvi02+3c\nfffd3HjjjezcuZN7772Xb7/9tu6ac0lKivJt8S1ITQ0sWQLt2kFmpt3ocgIiNjb8jO+ZHFUkrl9E\neet0zG3aY3wTjdMNyKnk+3xYsrENl/Z1Nu7i1FQsmzaQFGenRc9bN4H8bBGNIZ8X4St++0kdGRlJ\nRUVF3denBrNzSU1NpUOHDnX/HRsbS3FxMSkp9bcRKC4ua37BLdSiRSoVFeFccomT48cb+Zd+CIqN\nDef48crJDgG3AAAgAElEQVQzvp+c/x1qTTWHOvWjvLzagMrql922GJPSga/yYhhz9Zn118fePhXb\nli0czcvHnd3ZTxWef5KSouRni/CafF6Et7wJ8X6b1uzVqxcLFiwAYM2aNWRlZTV4zSeffMKUKVMA\nOHToEOXl5SQlNX4RtPDe3LmeBUwtfr1Z/ncAHA6y9Wa1osLdpLctZ8XmWEorG/dvqp/WnUm/MyGE\nCAV+C2eXX345VquVUaNGMWXKFMaPH8/s2bP56KOPznnNDTfcQHl5ObfddhuPPvookydPbnC0TTTP\njz+asVh0cnJkvZnLYudo+1yjSzmnXukncLlNLCxMaNR1Px3jVNTAO4UQQgQDv01rKorCs88+e9r3\nTl38X+vUnZlms5mXX37ZXyWJnzl8WGHdOpXcXDe2RvY3PZ+EH9xB1IGtHMwehG62GF3OOfVMP87M\neRfww+pErux3yOvrfjoAXUbOhBAiFMiwVAs2b55MaULwttD4uawLygm3uRrfjDYiAndSsiec6S33\n3FQhhAgVEs5asAULaltotOxw1mr5lwAczgzucKaqkJNayo6DEewtCWvUte60jpiOHMF02PsRNyGE\nEMaQcNZC6TosXKgSHa3Tvn3LHU2xVBwnqXAex1tnURXb2uhyGpTbsRSARY1cd6bJpgAhhAgZEs5a\nqB07FA4cMNGtm5uWvOei1YqvMLldHOx6idGleKU2nC1o7KaAumOcZFOAEEIEuxb813LLtnChZ0qz\nW7eWvUuzdd7/ADjQ5RJjC/FSh+RKouxO5q9NaNTyMWmnIYQQoUPCWQu1aJFnM0C3bi13vZlaXUHK\n6jmUJaZSkZRqdDleMZk8o2f7j9jZcfDMkw7ORUtKRgsPl2OchBAiBEg4a4E0zbPeLCFBo3Xrlrve\nLGX196jO6pCZ0qzVpKlNRUFLTUPdvg0qG3fCgBBCiMBqMJy9/fbbFBcXB6IWESAbNpg4etREt24a\nimJ0NcZpvTS0pjRr5aadAGh8M9q0jiiahnnjen+UJYQQwkcaDGcOh4Pbb7+de++9l6+//hqn8/w/\nf/F8J1OaYHI6aLXiaypjWlHaOtPochqlbWI18VE1LGjsurPakwJkalMIIYJag+FszJgxfPPNN9x/\n//0sW7aMa665hueee44NGzYEoj7hB7WbAXJzW+5mgMS187BUlXmmNENs+FBRILfjCUpKbWzcE+n1\nde40OSlACCFCgVdrzqqrq9m7dy979uzBZDIRExPDpEmTeOWVV/xdn/AxlwuWLFFp3VojMbHlrjdr\ns/RzIPSmNGt1P7nurDFTm+52HdBNJjljUwghglyDZ2s+9thjLF26lIsvvpgHHniAPn36AFBTU8Og\nQYMYN26c34sUvrN2rYnycoWBA11Gl2IYxe2i9fLZVEfEc6xdjtHlNEm3tJ/C2X1X7vLuIqsVrU1b\nzOsKPbtCWnKDOyGECGINhrMBAwbw3HPPERERUfe9mpoarFYrs2fP9mtxwvcWLZL+Zsn532MrLWFn\nv+vBpBpdTpOkxDlIjHGwZH08uu79zKy7YyfUvXsw7dyB1rGTf4sUQgjRJA3+03nWrFmnBTO32831\n118PQHJysv8qE36Rl+cJI127ttzNAB1++BcAu3tdbXAlzZOTWsrRMiub9jZi3VntpoB1silACCGC\n1TnD2ejRo8nOzqagoIDs7Oy6/8vNzSXt5MJiEVpcLli61LPeLC7O6GqMYTleTKuVX3OiVUbI7dL8\nuZzUMgCWrIv3+pq6kwIknAkhRNA657TmjBkzAHj++ed5+umnA1aQ8J+iIhMVFQoDB7bcUbOU7/+N\nye1iT6+rQm6X5s917eBZd7ZkfTy/Gbbbq2vcqbU7NiWcCSFEsDpnOJs7dy5Dhw6la9eufPbZZ2e8\nfu211/q1MOF7S5a08ClNXaf1V//ArVrY1/1XRlfTbG0Tq4mNqGFRkffrzvTYWLS4OAlnQggRxM4Z\nzgoLCxk6dCjLli1DOctPfQlnoWfJEs//3F27tszNALFb84nYuY79XYfiDI8xupxmUxTomlrG4nUJ\n7DwYTlpr745lcqd2xLJ6FcrRI+jxjTtlQAghhP+dM5yNHTsWgClTptR9r6ysjAMHDpCZGdprdVoi\nt9uz3iw5ueX2N+vww3sA7AnxjQCnykktZfG6BJasj/c+nKV5wpl5XRHOwUP8XKEQQojG8mq35vjx\n4zly5AhXXnklY8eOZerUqYGoTfjQ+vUmSksVcnJa5qiZ6qjkggUf4ohJpjj9QqPL8ZmutZsC1sum\nACGEOF80GM7ef/99nnjiCb788ksuu+wyZs+ezcKFCwNRm/Chlt5Co/XSzz3HNfW9OmR7m51Nh+RK\nIsNcLCpqRDiTTQFCCBHUvGoRHhsby/z58xkyZAhmsxmHw+HvuoSP/bQZoAWOnOk66Z9PQ0fh4IUj\njK7Gp0wm6Jpayu7D4ewtCfPqGq11G3SrVUbOhBAiSDUYztLT07n//vvZs2cPAwcO5KGHHqJbt26B\nqE34iKZ5Rs4SEjSSk1veerPk1d8Tu30NB3IupSqpvdHl+Fzt1Gaet1Obqoq7fQfUzZugpsaPlQkh\nhGiKBo9veuGFF1izZg0ZGRlYrVZGjhzJoEGDAlGb8JHNm00cO2ZiyBBXqLf2apLM/74MwNaL7zC4\nEv+o63e2Lp4bL97v1TXujp0wb92CunkT7hz5x5YQQgSTBsNZZWUlmzZtYtmyZXXfKyoqYsyYMX4t\nTPhO7XqzLl1a3nqzhPWLSdywhEOZAyltnYn3Bx2Fjk6tKwizupu47mythDMhhAgyDU5rPvTQQyxf\nvhxdb3nTYeeLZctqw1nLW29WN2o25E6DK/EfVYXO7cvYuj+S4uNWr675acdmkT9LE0II0QQNjpwd\nOXKEd999NwClCH/Qdc/IWXS0Ttu2LStgx2xbTcrq7ylJ7cWx9rlGl+NXOamlrN4aS96GeEYMONjg\n+90dUtEVBXPR2gBUJ4QQojEaHDnr3LkzGzduDEQtwg/27FE4cMBE587uFrferCWMmtWq3RSw2NtD\n0O3haCmtPCNnMiouhBBBpcGRs82bNzNy5EgSEhKwWj1TJoqi8MMPP/i9ONF8S5e2zCnNuE3LabPs\nc463yaakU1+jy/G7zLblWFSNxY1Zd5bWEWveYkz796G1vcCP1QkhhGiMBsPZX//6V8ATyGTdWehp\nievNFJeTHm8+iKLrrB/+kHcngoc4i1knq10563ZFcbzcTGykq8Fr3GkdIW8x5qJCaiScCSFE0Ghw\nWvOCCy4gPz+fjz76iLi4OFauXMkFF8gP8lCRl6cSFqaTltZywlmnL/5KzK4idvcewdHUHkaXEzA5\nqaXousLSDd6NnrlT5RgnIYQIRg2Gs5dffpn58+fz3Xff4XK5+Pjjj5k8eXIgahPNVFKisHWrSlaW\nhnr+nFhUr/DDu8ie+TyO8Fg2/PJ3RpcTUDmpnn5n3q47c6fJMU5CCBGMGgxnixYt4uWXX8ZmsxET\nE8M///lPFixYEIjaRDP9NKXZQvqb6Tq5bz2Cuaaa9Vc8jDM8xuiKAiqrXTmqSWOJl+FMT0hEi4xE\nlR2bQggRVBoMZ+rPhlxqamrO+J4ITi1tM8AFCz+iVf63FHfsy77cXxpdTsCFWTXS21RQsD2a8iov\n/owqCu7Ujph37kApL/N/gUIIIbzSYDgbNmwYjzzyCCdOnODdd9/ltttu48orrwxEbaKZli5VUVWd\njIzzP5zFbFtNj9d/h8sSRuGIx1vEJoCzyUkrxa2ZWL4pzqv31zajVdev92dZQgghGqHBcDZkyBCG\nDh1KXFwcq1atYuzYsTzwwAOBqE00Q3k5FBWZ6NRJw2Yzuhr/Cjuyn/6TbkB1Olh943NUJrQzuiTD\n5Jzsd+bt1GbdMU6FBX6rSQghROOcs5XGkSNHGDt2LFu2bKFDhw6oqsrSpUuprq6md+/eREdHB7JO\n0UgrV6q43Qpdu57fo2ZqdQX9J92A/dhB1g97kEOdBxtdkqE6ty9DQfc+nHVKB5CTAoQQIoicc+Ts\nueeeo3fv3ixevJhZs2Yxa9YsFi9eTHZ2Ni+88EIgaxRNULverHPn83czgOJy0mfqb4jdUcCuPtew\nfeAtRpdkuIgwN2mtKlm5JZbqmgYHxtEuaIdutWJZnR+A6oQQQnjjnD+9N23axKOPPorFYqn7ntVq\n5ZFHHmHdunUBKU403dKlKoqi07nz+TlyZq44wYA/j6T18tmUpPWm6KpxLXad2c/lpJXidJlYtcWL\n3aqqijutI+qmDVBV5f/ihBBCNOic4SwsLOzsF5hMslszyNXUQH6+Srt2OpGRRlfje/biPVw8/jKS\n187lYPYgVtz+Mrra4GEXLUbXDp5+Z3nrvZzaTM9EcbulGa0QQgSJhuc9RMhZu9ZEdbVyXvY3i9u8\ngiF/uJjoPRvY0f8mVt4yBbfVbnRZQaWxh6C70jMAMBes9ltNQgghvHfO4YatW7dy6aWXnvW1w4cP\n+60g0XznY38zk9NB1oeTyfj0VRRdY90VD7NjwM1GlxWUYiJcXJBYxfKNcThdChZz/Wfi1m4KsKxZ\nTXUgChRCCFGvc4azb775JpB1CB9autTzP+v5Es5idhTQ67X7iNlVRGVsawque5ojab2MLiuo5aSV\n8s2KFNZuj6Z35ol636u1aYseFoZ5jWwKEEKIYHDOcCaHm4cmTfMc25ScrJGQUP+ISbBTqyvI+mgy\n6Z9Pw+R2savvtaz/1RjctgijSwt6OamecLZkfXyD4QxVxd2xE+rGDZ4GeefjQkUhhAghsubsPLNp\nk4kTJ5SQHzVrteIrLnuwF5mfTqU6Kolld0ylcMQTEsy81LXDyWa0Xm4KcKVnomiaHIIuhBBBQLa4\nnWd+Wm8WmpsBrCeK6f7Ww7TN+wzNZGbLxXeyZciv0axn3z0szi4xpoaUuGqWrItH08DUwD/D3Okn\n150V5OPqPyAAFQohhDgXCWfnmWXLQnczQOu8/9Hjb2OxlZZwtH0ua695kvLkNKPLClk5qaX8sDqZ\n9buj6o51Ohd3p5M7NtfIjk0hhDCahLPziK5DXp5KdLROmzahs95MrSqnx9/G0m7Bh7jNVtYNH8uO\n/jeBSfrpNUdOahk/rE5myfr4BsOZ1qo1Wni4bAoQQogg4Lc1Z5qmMWHCBEaNGsXo0aPZvXv3Ge+p\nqqpi1KhRbN++3etrxLnt3q1w4ICJrl3dIdMs3168h4uf+gXtFnzIsbZdWPC7f7Fj4C0SzHyga6qn\nGe2SIi/WnZlMuDulY962FaWs1M+VCSGEqI/fwtmcOXNwOp3MnDmTcePGMWXKlNNeLyws5LbbbmPv\n3r0oJ5NEQ9eI+uXleQJNqBx2HrdpOUMev5iYnYXs6nMNS+59i4qkVKPLOm+0inMQH1XD4nXx6F4M\npLprm9GuLfBzZUIIIerjt3CWn5/P4MGDAejevTtFRUWnve50OnnjjTdIS0vz+hpRv7y82v5mwb8Z\noHXe/xj09DBspSUUXfEIhSOekCOYfExRPOvOSkptbN3f8C5XWXcmhBDBwW9/G5aXlxN5Sr8kVVXR\nNA3TyW1jvXqd2US0oWvOJSkpykdVh7ZlyzwtqnJz7QTz8afxy78h5y93oJmtFP7mFY52HkQgO2tF\nRracnZ+9sqpYUAhrdrSib9cDDbw5F4DIjYVEyp8pQH62iMaRz4vwFb+Fs8jISCoqKuq+9iZkNeUa\ngOLi+hc7twQHDihs3x5Jnz5uysocRpdzTvEb8ugycSS6YmL57a9wtF1PKA/coUGRkWGUB/B5Rktv\ndRRox5yVkdw4qLL+N4dFEx0Zib50GUflzxRJSVHys0V4TT4vwlvehHi/TWv26tWLBQsWALBmzRqy\nsrL8co3w+Gm9WfBOaUbvWMuA56/D5HKyatQLHE3taXRJ5732yVVEhzu9OwRdUXCnZ6Lu2olSUuL/\n4oQQQpyV38LZ5ZdfjtVqZdSoUUyZMoXx48cze/ZsPvroo0ZdI7xTG86Ctb+ZvWQvA5+9BnNVGWuu\nn8DhrIuMLqlFUBTo0qGMfSV2dh9ueDrXld0ZAMvK5f4uTQghxDn4bVpTURSeffbZ07536uL/WjNm\nzKj3GuGdvDwVm02nY8fgC2eK20XvV+8i7MRh1l3xMPtzf2l0SS1KTmopSzfEs2R9PO2T99f7Xndt\nOFuxjJphVwSiPCGEED8jZ2ueB0pKFDZvVsnO1jAH4YbHrI+mkLhhCfu7DvU0lxUBVdvvLM+LqU1X\nZha6YsK8Yqm/yxJCCHEOEs7OA7XnaQbjerPEwvlkzZpCZWxr1l4znpDpjnseSWtVid3q9m7dmT0c\nd4cOWFbnQ02N/4sTQghxBgln54GfwllwTWlaTxTT+9W70BUT+Tc9h8su28yNoJqgS4dSth2I5MBR\nW4Pvd2d3QXE4MBetDUB1Qgghfk7C2XlgyRIVi0UnPT24wlmPNx/EfvwQm37xW463yzG6nBYtt6Nn\nanNRUUKD763bFLBcpjaFEMIIEs5C3IkTsG6diYwMDavV6Gp+krLqG9os+4Ij7buz7aJbjS6nxeuW\ndjKcFTY8tfnTpgDZsSmEEEaQcBbili9X0XUlqKY0TTXV5L79GJqiUnT1OPCikbDwr46tKwi3uVhQ\n2PDImZacghYbh3n5Urw6lFMIIYRPyd+aIW7JEs/2zGDaDJDx6atEHNrJjgE3UdYq3ehyBJ51Zzmp\npew8FMHekgb6nSkKrs5dUA8dxLR3T2AKFEIIUUfCWYjLy1NRVZ2srOAYOQs/sJ3M/75CdWQCW4be\nbXQ54hTdatedeTF65s76qd+ZEEKIwArCrljCW+XlsHatiU6dNMKC4SxvXSf37+NQXQ4Khv8RV1iE\n0RWJU3Q/ue5sQWECo4buq/e9rlOa0Tquu9HvtQUFtxtzYQHq9m2oO7bDwb1ERMRQc/ElOPsPhPBw\noysUQrQQEs5C2MqVKi5X8Kw3S14zh1b531KS1pv93X5hdDniZzqkVBJld7KgMAFdr7/lnLtjJ3SL\nBfOyFrBjU9exfvs1EZMmYt608bSXwoHwN/4fusWCs/9AKv70LK4evYypUwjRYkg4C2G1/c26dAmC\n9Wa6Tuf/PAfA+uFjpdlsEDKZICetlLz1Cew6ZCe1VdW532yx4O6Ujnl9kWeINjIycIUGkHlpHpHP\nPY1l5Qp0k4maiy/BlZWN1qo1kZkdKd+6C/Pa1ZgLCrAunI9l+GVUjXmYinFPgq3hnnFCCNEUEs5C\n2JIlKoqi07mz8SNnrZfPJm5bPvtzLqO0dabR5YhzyD0ZzhYWJZDaam+973Vld8G8cQOW1atwDh4S\noAoDJ+ydt4l86nEUTaOm/0Cqbx2N1q79T2+IDccVGY+rR08AzIUF2P/6GuGv/QXr17Mpe306ru49\nDapeCHE+kw0BIaq6GvLzVVJTdSKMXtqlaWT/5zl0xcSmS+8xuBhRn9p+Zwu92BRQt+5sWZ5fawo4\nXSdi0rNEPfkYelQUZZNeovKJP54ezM7C1a07Zf/3Oo4rrsK8eRMx11yBedWKABUthGhJJJyFqNWr\nVWpqlKBoodF28cfE7FnP3h7DqEhKNbocUY/2yVXERDiZvzahwRZm7i456IqCZeH8wBQXCDU1RI25\nn/DX/oK7dRvKp/wFd5eu3l9vt1N17wNUPD4epbqKmJtHohbKMVdCCN+ScBai8vJq15sZO6WpuF1k\nf/A8mkllyyW/MbQW0TBFge4dT3D4eBgb99S/jkyPisLdsZPnpICKigBV6Ee6TtSY+wmbNRNXRhbl\nk19Ba9W6SbdyDhxE5YOPoJSVEXvDCNSfbSQQQojmkHAWohYvDo7NAO3mvU/Uga3s7j2Cyvi2htYi\nvNMz/QQA8woSG3yvq3tPFJcTy7Il/i7L78JfmULYZx/jyu5M+Z9fQI+Jadb9nJdcStVvx2A6dpSY\n667CtGe3jyoVQrR0Es5CUHW159imDh00mvn3S/O43WTOegm3amXrkF8bWIhojB6dPOFs7hpvwlkP\nAKzz5vqzJL+zffpfIl6ejDs5hYon/wQ23zQGrPnlMKruuge1+DDRv/0NuFw+ua8QomWTcBaCVq1S\ncTgUcnONHTVrs+xzIg/tYG/P4VTHJBtai/BeYkwNbROrWLwunhpn/S1PXNld0C1WrPNDN5yZV60g\n6sHfotvDqfjjM80eMfs5x9XXUjNwMJYVywl/ZYpP7y2EaJkknIWghQs9U5rduhm43kzXyfj0VXQU\ntl90q3F1iCbpmX6cSoeZFZvi6n+j1YqrcxfMG9ahHD4cmOJ8SCkuJvqOW8DlomLcE2jtO/jhIQpV\nvxuDlpRM+P+9gmXJIt8/QwjRokg4C0ELF3r6mxm53iyxaAFxW/M52GUIFYn1tyAQwadn7dSmV+vO\nTk5tLgqxXZu6TtQjY1CLD1M9+te4evXx36MiIql49HEAoh64B+XoEb89Swhx/pNwFmLKyz1tNDp1\n0gztb5bx6VQAtg263bgiRJPlpJWimrRGrTuzLJjn56p8K2zGu9i++xpnt+44Roz0+/Pc2V2oHnUb\n6oH9RD3+sN+fJ4Q4f0k4CzHLl3vO08zNNW5KM3pnISmrv+dIak+Ot2tEjygRNMJtGlkXlLNmWwzH\ny+s/KMSd1gktMgrrvB9psDlakFC3byXyT0+iRURQOfZRz9lVAeC47kZc2Z2xffE/rD98F5BnCiHO\nPxLOQszChZ6/SLt1M25KM+Oz/wNg22AZNQtlPdJPoOkKCwobGD0zmXDldkfdvw91x7bAFNccTidR\nD9yLUlVF1QMPoic2PDroM6pK5W/HoJtMRD45zrO1WgghGknCWYhZuFBFVY07T9NevIe2C2dRmtyR\nwxkDDKlB+EbPJrTUsMyf58+SfCL8/72KZfUqaoYMxXnR4IA/X+uQiuPKEai7dhL++msBf74QIvRJ\nOAshx49DYaGJrCwNm82YGtK+no5Jc3t2aCr1t2EQwS2jbTkRYS7m5Cc1OFvpyj25KSDI152p27YQ\n/upLaPHxVN77gGF1VI+6DS02jvD/ewXTrp2G1SGECE0SzkLIkiVmdN24/mYmRxWp3/8TR3gM+7v9\nwpAahO+oque0gL0ldjbtrf8oJ61Va9zJKZ5zNoO10aquE/n4IyhOJ1X3/BZDd8yEh1N11z0oDgeR\nf3zCuDqEECFJwlkIWbDA2P5m7RZ+hLX8GLv7jkSzGDR0J3yqb+YxAL5bmdTge129+mAqPYElb7G/\ny2oS26yZWBctwNmnL87+A40uB+fgITi7dsP23deyOUAI0SgSzkLI3LlmwsJ0MjMNCGe6TsfZb6Ap\nKrv6+r8tgQiMXhnHAfh+VcMnPDgHXgSA7YvP/FpTUyhHjxD5p/HoNhuV9/4uOKbcFYWqe3+LrihE\n/PkZ0AxsGi2ECCkSzkLErl0KO3aY6NbNjbn+zgd+kbBuETG7ijjY9RI5quk8EhvpIqNtOUs3xnGi\nov4PlqtLDlp0DLbZn4Pb2KPDfi7i2T9hOnaU6lG3oScHz+dT65CKc8hQzOvXYftkltHlCCFChISz\nEDF/vucvzh49jPnXd8cv3wRgR/8bDXm+8J++mcdwuU3Ma+i0AFXF2X8AppJiLMuXBqY4L1jyFmP/\n4N+4U9NwXHWN0eWcofqW29HNZiIm/xlqaowuRwgRAiSchYh58zzrzXr0CPyIhb14D22WfcGJ1pkc\na58b8OcL/+qdWTu12fC6M+cAz9SmNVimNh0OIh8bi64oVD4wBkOGlRugJadQ86srUPfsJmzGu0aX\nI4QIARLOQoDLBQsWmElK0mjdOvAd2tO+no6ia+zof1NwrOURPpXepoKYCCffrUpucFmUKycXLSIS\n2xf/C4o1VOGvv4Z56xZqfnUF7sxso8s5p+obbkYPCyPilSmeM9iEEKIeEs5CwJo1JkpLFXr2dAc8\nG5lqqunw/bvU2KV9xvnKZPKMnhWfsLF2R3T9bzabcfYbgHroIOaVKwJT4DmYtm/z9DSLjaPq9jsN\nraUhemwsjmuuw3SkhPC3Xje6HCFEkJNwFgLmzTNuvVmbJZ9iKz/K7t5XS/uM81ifjNqWGo3YtTn7\nf36tqV66TtQfHkGpqaHqnvuN7WnmpeoRI9GiorG/MQ3lxHGjyxFCBDEJZyFg7lwVRdENOU8z7Zu3\n0VHY3ffagD9bBE6vjBOYTRqzl6U0+F5Xbg90e7inpYZBB6HbPpmFdcE8nD174xw4yJAaGi08HMe1\n12EqK8X+9t+MrkYIEcQknAW50lLIz1fJyNCIrL+Ju89F71hLwqZlFKf3ozK+bWAfLgIqIsxNbsdS\nCnfEsPuwvf43Wyw4L+yHum8v5jX5gSnwFMqxo0Q+/SS61UrV/UHS08xLjuFXoUVFYf/b6yhlpUaX\nI4QIUhLOgtzChWbcboXu3QM/pZn2zd8B2NnvuoA/WwTegC5HAZi9tOHRs5qTuzZtH3/k15rOJuL5\niZiOlFB9861oKa0C/vxmsdtxjBiJqfQE9r+/ZXQ1QoggJeEsyM2Z42mh0bt3YKc0zZWltJv/AZUx\nKRzONP4oHOF//TofQ0Hni7yGA4+rVx+02DjC3v83SnlZAKrzMC9bin3Gu7jbd8AxIjRPqnBccTVa\nRCT2N6cF9PdOCBE6JJwFMV2H7783ExWlk54e2JGzdvNnYnZUsrvPtWBSA/psYYy4SCedO5SxbFMc\nh49b63+zxYJj2BWYysuwffhBYAqsqSFq3FiAoO1p5pXwcBwjrsV0/Dhh7/zd6GqEEEFIwlkQKyw0\ncfiwid693aiBzEe6TtrX09FMZvb0vjqADxZGG9jlKLqu8NVyL6Y2fzUc3WzG/vabAel5Zn9zGuZN\nG3H8cjju7C5+f54/1Vx5NVp4BOFvvCZ9z4QQZ5BwFsS++84zMtCnT2CnNBM2LCF6zwYOdLkER1RC\nQJ8tjNW/s6elhjdTm3psHM5BQzBv34Z17hy/1qVu3ULEK1PQYmKpHh3cPc28oUdEUnP1NZiOHsX+\n3nMlezUAACAASURBVD+NLkcIEWQknAWx7783o6p6wI9sSvt6OgC7LgzNNT2i6VLiHHRsXc7CwoQG\nD0IHcFw1AsC/rSFcLqJ+fx+Kw0HVfQ+gR0b571kB5LhyBHpYGPbXX4PqaqPLEUIEEQlnQerwYYU1\na0xkZ2sB7a9pO3aQNnn/ozQpjaOpPQP3YBE0BnY5itNt4psVDTekdXdKx5XdBeuPc1C3bPZLPeH/\n9wqW1auoufiS0Olp5gU9KgrH8CtRiw8T9sG/jS5HCBFEJJwFqR9/VNF1JeBTmh3mvIfJ7WRXv+tD\nqn+U8J1BOZ6WGv9d2Mar9zuuvgYA+999P3pmXpNP+F9eREtIpOq+B3x+f6M5RoxEt1gJnzYVnE6j\nyxFCBAkJZ0Hq++8Dv95McbtI/fbvuKx29nUfFrDniuDSNrGa9DblzF2TSHFDuzYBZ78BaAmJhM38\nD6YD+31XSFUVUb+7F8XtpnLsI+gRAe7CHAB6bBw1v/gl6t492D6ZZXQ5QoggIeEsCNXUwNy5ZlJS\nNNq2DdzxOCkrvyH8yD729hiOKyz4zyoU/jO0RwluzcSni1s3/GZVpfrmW1GqqogcP843Beg6kU8/\ngXnrFhxXXYMrt4dv7huEqkdej66qhL/2l4DsehVCBD8JZ0EoL0+lvNwzpRnImcW0b05uBOgrGwFa\nusHdjmBSdD6c592xXTWXXY6rS1dsX83G+uUXzX6+ffob2Ge8iyutI1W3h/7uzProScnUDLkU89Yt\nWL/83OhyhBBBwG/hTNM0JkyYwKhRoxg9ejS7d+8+7fUff/yRG264gVGjRjFr1k/D+SNHjmT06NGM\nHj2ap556yl/lBbUvv/RMafbvH7gpzYj9W0lZ8wNH2nenrFV6wJ4rglNcpJPunU6QvzWWbfvDG77A\nZKLyd2PRzWYin3gUpfREk59t/e5rIiY8hRYXT8VTz4DN1uR7hQrHdTeim0yET33FsMPkhRDBw2/h\nbM6cOTidTmbOnMm4ceOYMmVK3WtOp5MpU6bwz3/+kxkzZvDhhx9y9OhRHA4HADNmzGDGjBm88MIL\n/iovaGmaJ5xFRel07hy4KY60bz2dynf1uz5gzxTBbWj3EgBmLfBu9ExrewHVN45CPXyIiD9PbNIz\n1XVFRN33G7BYqHhqAnpiYpPuE2q0tm1xDrgIS9FarD98Z3Q5QgiD+S2c5efnM3jwYAC6d+9OUVFR\n3Wvbtm2jffv2REVFYbFY6N27N8uXL2fj/2/vvuOjqvL/j7/u9DTSSAKkkNA70nsVpEkPTQRdUFzZ\n1V1ddVn364oNxNW1rLAo6k9xRVCKgiBGpQkIBDCAkVBDCcQkEBIySabe+/sjawRFCJApST7Px4MH\nMJm55zN5nOS+59x7zsnIoLS0lOnTp3PXXXexb98+T5Xnt1JT9eTl6ejSxXu7AujtJSR8/T72oHCy\nW/T1TqPC73Vtno/Z6GbZpnoVHsyxj07GHZ9AwHtvY/xm83W1pz98iNDJ49CVFFPyp7/gbtT4Bqqu\numzJEwBk9EwI4blwZrVaCQ7+eXaVXq9H/d/NrlarlZCQnxeSDAoKoqioiICAAKZPn87bb7/NU089\nxSOPPFL+mpri50uaLq+1Gbt1OabiAk51HIlmMHqtXeHfAswq3ZrncyIniF2Hwir2IqOx7PKmTkfo\n5HEVvv/MlPI5YYP6oT97htKp06rVemYVpSYm4ezYGWPqTozbt/q6HCGED3ls5+Dg4GCKi4vL/6+q\nKjpdWRYMCQm57GvFxcWEhoaSmJhI/fr1AUhMTCQsLIy8vDxiYq6+z19UVPVYMVzTYN06CAyEXr0s\nmK69ikGlaJTyNpqi41yf8QQHW7zTqA/VhPdYWW7vXsim/VF8uKkBg7pmVOxFXTvAs8/C008TOu1O\neOkl+POfr7xunqbBvHnw+ONgNML//R8Bt95KQOW+jRsWFlaB++0q0+/ugt27CFvwCowa6t22xU2r\nLuci4XseC2ft27dn48aNDBkyhLS0NJo2bVr+tQYNGnDy5EkKCwsJCAggNTWV6dOns3LlSg4dOsST\nTz5JTk4OVquVqKioa7aVl1fkqbfhVfv36zh1KojevV2UlDgoKfF8m2FH9lDr8G5+bNaL88ZwsFbv\nbWSCgy1Yq/l7rEyN69qoGxHH0g1RPDn5AOEhFVwotXlb9M/OI+i5p9A9/DC2nbuxjxqDs31HtPAI\ndNlnMX+yEvOKjzDuT0ONiKT48SdwN2wMBV7o+BUQFhZIgbdrqZdIUKs2GL/8kgspm3C16+Dd9sUN\ni4oKqTbnIuFZFQnxHgtnAwcOZNu2bUycOBGAuXPn8tlnn1FSUsL48eOZNWsW06dPR1VVkpOTiY6O\nJjk5mb/97W9Mnjy5/DU/jbbVBL6YpZm0fhEAJ2QigLgCnQ6GdM7hnfX1WbIhjj+MzKzwa90NG1E0\n7yWCn52NZdkSLMuWlD0eF4/uTBaKpqHpdDg7daHk/j+ihUd46F1ULfZxEzF+v5/Al1/k4uIPfV2O\nEMIHFE2r+neeVpdPK927B3LqlI733ivF4oUrb8aifAZPb4wtpDYb/7Ss7ExczcnI2fW7WGLg7hfa\nEx9dSurrm6+/mzgcGPanYTh8CP2hDPSZx1DjEnD07oOzW0+00FCP1H2zfDJyBmUL8M76C4bDh8jf\n9C3uFi29X4O4bjJyJirKpyNn4vqkp+s4elRP164urwQzgIQN/0XvtHGi85gaEczEjakV6KJX6/Ns\nSItiy4FI+rY9f30HMJlwdeyMq2NnzxRY3SgKtuSJBM95isDXXqJo4Tu+rkgI4WVyRvYTK1aU5eTe\nvb10SVNVSVq/CLfBRFY7ufFYXN2QTjkAvL2+vo8rqRlcHTvhTkzC/MlKdMeP+bocIYSXSTjzA6oK\nK1caCQzU6NDBO+EsOu0rgn88zpk2A3EG+udlJeE/msZbSYop5vNd0WSdk9muHqco2JInoKgqga+/\n4utqhBBeJuHMD+zYoefsWR3durm9tnxGw9WvA3CiyzjvNCiqNEWBEd1/xK3qeP2TJF+XUyM4u3bH\nXa8elqVL0J3J8nU5QggvknDmB5Yv/+mSpncWng05mU7Mvq85n9iOi/WaXvsFQgB9254jKtTOe18m\nkFfgpU8RNZlej33MeBSXk4AFr/m6GiGEF0k48zG7HVavNhIRodKypXd2Q2j42XwAjnef6JX2RPVg\n0GuM7XUWm0PPgjUyeuYNjj79UGtHEfD+uyh5eb4uRwjhJRLOfOyrrwxcvKjQq5d39tI0FeQSv2kp\nxRFx5DTt4fkGRbUyoH0uYUEO3lpXnwKrTPb2OIMB2+hkFJuNwDfm+7oaIYSXSDjzsZ9mafbp451L\nmklfvIXeZSez23jQeWlndVFtmI0ao3tmY7UZWLQu0dfl1AiOWweihoZhefsNlPPXuYyJEKJKknDm\nQ+fOKXzxhYH4eJXERM+vBaxz2Eha9yYOSzCn2w3zeHuiehrcKYdgi4sFaxIpLJbRM48zm7GNHY+u\nuJjA+a/6uhohhBdIOPOhZcsMOJ0KAwe6rrgndGWL27IMy8U8TnUchdvs5Q2dRbURaFYZ2+ssBVYT\nLyxr7OtyagTHoCGoEZEEvLUQJSfH1+UIITxMwpmPaBosXmzCaNTo29cLlzRVlUafvoaq6DnRNdnz\n7YlqbWT3bOqE23hzXX0OZwX5upzqz2TCNn5S2b1nr73k62qEEB4m4cxHtm3Tk5mpo0cPNyHX3mbr\nptXd9Rm1sjI40/Y2bKExnm9QVGtGg8b0ISdxuXX8/f8193U5NYLj1oG4Y+oQ8O7bsu6ZENWchDMf\nef99IwADB3ph1EzTaPLxC2goHO091fPtiRqhS7MLtG1QwFd7o0nZE+Xrcqo/gwHbhDtQnE4C//WC\nr6sRQniQhDMfOHdOYc0aA3FxKs2be35ts+jvviT8+Hdkt+xLcVSix9sTNYOiwL1DT6LTaTy2qCVF\npTL719Ocvfvijo3DsuR92XNTiGpMwpkPLFtmwOVSuO02L0wE0DSafjwPgKN97vZwY6KmqR9Tyuge\nZzmZE8ist1r6upzqT6/HNulOFLeboDlP+7oaIYSHSDjzMrcb3nnHexMBItO3Epmxg5wm3blYt4nH\n2xM1z+T+WTSsa2XJhjhWfFPX1+VUe87uPXE1boJl9SoMqTt9XY4QwgMknHnZ2rUGTp/W0a+fyysT\nAZouL7s35YiMmgkPMRo0Hh1/FIvRzUMLW3Eq1+Lrkqo3RaF02r0ABP/j8bKp30KIakVWkPQiTYPX\nXzehKBojRnh+1Cz8cCrR+zZwLqkDBQmtPd6eqLlia9u47/YTvLqqIVPmdWDNMzupFeidXS8qSlXh\n4OkQDmSGcPBUCIdOB5N93kJeoYn8IhOaVnYfndGgEhPmICHGQb0IK20aXqRdw0JaJ13EYvLO/rfX\n4m7WAkf3npi2b8X86Urso8b6uiQhRCWScOZFO3boSUvT07mzi9hYD3/a1TRaLH4CgMP9pnm2LSGA\nW9vlkXE6mC92x3DHnA58/EQqAWbfhpnTeRbWp8aweX8k29IjKLCaLvu62egmNMhJfFQpOp2Gpio4\n3Ap5hSaOZQcB4Xywoey5FpObvm3OMbhTLkM65xAd5vD+G7qEbcrdGHftIOjpf2AfPAwsMmIpRHUh\n4cyL5s8vOzGMGuX5EYXo774kKv0bcht3Iz+pvcfbE0JR4P7hmRSVGtiWHsm0F9ux+K97MRq8e9nt\nZE4AH2+px+pv63AgM7T88ahQO/1vyaNJnJX6MSXUjy4hJND9m8cxWQI4egqOngniyJlg0o6Fsn53\nDOt3x/DImy0Z3DGXKQNPc+steeh9MFFVrVMX+7DhWD5dRcCihZQ+8GfvFyGE8AhF06r+DQt5eUW+\nLuGaDh/W0bNnEE2auJk3z+7ZxlSVfg93o9bJdLbMfI+iurLFzk+Cgy1YrTZfl1GtOV0KT/+3KWnH\nwhjYPpdFD6cRGuTZDyT5F42s2l6XjzbVY9ehCAAMOpXWDS7StfkFOjQuICb8+n7urtRXsvPN7MwI\nZ8PeKDJzynZGSKpTzJ/HHGNC37OYjd4dKVSKrYTcfw+oGhe270atW8+r7YufRUWFVIlzkfC9qKhr\n33Au4cxLHnzQwtKlRh57zE63br/9ab0yxG36kI6v3kNW28GkJT/p0baqGgln3lFq1zHnwyakHQsj\nMaaY92ftpVVi5f6cFtv0rE+NZvmWenz1XRQutw4FjTYNLtK37Tm6tcgnyHLjP2tX6yuaBkfPBvF5\nagwb02rjcuuoE27jkfFHmTrgtFdHC00p6wn8z7+xDx/Jxbff91q74nISzkRFSTjzE4cP6+jdO5DY\nWI2XX7Z59BKIzmnn1j/cgiU/m01/WkZpuCxtcCkJZ97jVuGDr+P5eEssFpObx8YfYcawkzcVmOxO\nHV9/V5sV39Tj810xlDrKfpiS6hTT75Zz9G59jshazkqpv6J95fxFI59sr8v6XTHYnHoa1rUye+oh\nhnXJ8fw6hgCqSvDfH8OQcZDCDz7CMXCwFxoVvyThTFSUhDM/MX26hTVrjMyaZadLF8+OmjVYM582\n7zzG8W4T+GGo3IPySxLOvO/bH8J57ZMGWEuNRIfZ+EvyMZJ7nSWigiEq/6KRzQci+XpvFGt21OFi\nSdnWZ3XCbfRpc45erc9TP6a00uu+3r5ywWpk6cZY1qfGoGoKvVqf48UZ6TSJK6702n5Jd+okIQ8/\ngFq3Hvnf7IIg2Yze2ySciYqScOYH9u3TMXBgEI0auXnhBbtHP0mbCvMY8IdbUJwONjy8AmdQmOca\nq6IknPlGsU3Pqq11+XR7XWxOPTpFo1PTC/S75RwJUaXUq20jyOLCWmqgqMTAydxA0k+EcCCzFj+c\nCkHTyn5wIkIc9Gp9nt6tz9E4ttijP0832ley8iy8vb4+uw+HY9Sr/GFkJo+NP+LxmauW/76HZcVH\nlPzhTxQ/+YxH2xK/JuFMVJSEMz8wYUIAGzcaeOopG23aePaXc/vXZpCw8QPSh/6ZzG4TPNpWVSXh\nzLcKrAa+3BvNroxwDp0ORuPq6cpkUGkca6Vdo0JuaVhIo1grei8tnX0zfUXTYGdGOG+uTSSv0EzD\nulYWPLifzs0KKrnKS9hthDw4E935c1xI2Yy7dRvPtSV+RcKZqCgJZz62bZue0aMDad3azdNPe3aG\nZu3vt9DziSEU1m3C1vveRtPLKilXIuHMfxQWGzhyJpjzF02cv2jC7tQRYHITYHZTu5aD+jEl1I20\neS2M/VJl9BWbQ8f7X8WzZkcdFGDmiEwen3TYY6Nohu/2EPz0P3A1bc6FlE0QEOCRdsSvSTgTFSXh\nzIecTrj11kAyMvTMm2ejSRPPjZopTgf9H+pC8NkjbJ3xFoVxLTzWVlUn4UxUVGX2lfQTIby6qiHZ\n+RYa1rOy4AHPjaIFvLEA8/q1lNz7e4qfe8EjbYhfk3AmKqoi4Uz21vSQRYuMZGToGTDA5dFgBtD4\n01cJOXOYk53HSDATwg+1TCzitT/sZ0S3bI5nBzHk8W78472m2ByV/yu49O5puOPiCVy0ENPXKZV+\nfCGE50k484AzZxReeMFMSIjGlCme3eIlKPsYTT96HltwJBkDfu/RtoQQN85iUrl36EnmTvuB6DA7\n//6kIf0f7cGBzGt/ir4uZgslDz+KZjAQ8sD9KHl5lXt8IYTHSTjzgCeeMFNSojB1qoNatTzXjuJ0\n0PFfv0PvtJE+9M+4LMGea0wIUSlaJhbx7z/uZ0jnHzl4KoRbH+3Byysa4K7EVXbcSQ2x3XkXunN5\nhPzp/rJd34UQVYaEs0r2+ecGPvvMSLNmbvr39+yaZs0/fIbwo3s4fcsQslsP8GhbQojKYzGpzBx+\ngtlTDxIc4OLp/zZj2P915cSPlXcDv334KJxt22H+KoWgubK0xlW53WC3l90s7HJJmBU+JxMCKlF2\ntkKfPkGUlMA//2kjIcFz39qotK/p8dQIiiPi2DLzXdxmWXSyImRCgKgob/WViyUGFqxOYlt6JIFm\nF3OmHWTqwNOVsoabUlRE8GMPof8xm4uvv4F9/KSbP2gVpuTmYkzbg2HvHgwZB9GdPYMu+yy6vFyU\nSwKZptOh1qmDGpeAOy4Od9PmODt0wtW+A1rwlS9Dy4QAUVEyW9OL3G4YOzaA7dsN3Hefg8GDPbfR\ns6kgl/4PdcFUlM+2e9+kMLa5x9qqbiSciYryZl/RNNi8P5L/rEmixG5gUMccXp15gJjwm79nVZd1\nmuC//gXF6aRg1VpcnbtUQsVVhMOB8dttmFI+x5yyHv3JE5d9WTMYUCMi0SIi0MwWQANVRXE40J0/\nj5J//lehzd2sBfbbBuMYMgxX23agK7sAJeFMVJSEMy965RUTc+aY6dzZxaxZDo+tXK64XXR9dgwx\naV/zw6A/crznZM80VE1JOBMV5Yu+kldo4pWVDdl/PJTwYAevzjzA8G45N31cQ9p3BD3zD7TwCC58\nsRE1oX4lVOunVBXjt9uwLFuC6bPV6Kxl5wctIABX85a4mjTF3agJ7oYN0ULDuOova7cb5fx59JnH\nMBzKwHDoIPojh1GcZVuPuevUxT5iFLZJU4jo280vzkXC/0k485LNm/VMnBhAaGjZxuYemwSgadzy\nnwdI/PL/kdO4G6l3vlj+qU1UjIQzUVG+6iuqCp/trMN7KQk4XDom9s3i+Xt+IDTo5kbjTes+I3DR\nf3DHxVPwybpqF9CU3FwCFr+D5cP/oj99CgC1dhTOLt1wduqMq0UrMBpvviGbDWPaXow7d2BI3Ymu\n2Fr2eIcOFE24E1vyBAiWyVnit0k484KDB3UMGxaIzQZPPWWnRQvP3UjaZPk/afHBbArrNmH79AVy\nn9kNkHAmKsrXfeV0roWXljfiWHYwsbVLeeX+Awxof+6mjmletoSApR9Uq4Cm/yGdgDfmY1m+DMXp\nRDNbcPToiaPfANwtWnr2A6zTiXFPKqavUzDu3QOqilqrFra776H0nvtQ69T1XNuiypJw5mE//qgw\neHAgZ8/qeOghO717e252ZtymD+n46j2UhMaw7b63sIfU9lhb1ZmvT7ii6vCHvuJyKyzbFMtHm2NR\nNYXRPc7y3LSD1I248e3gzB99SMCH/8UdG1cW0OonVl7B3qKqmL5OIWDhfEzfbAbAXbce9uEjcfS9\n1SfbVoW5Sihd8QnmdZ+hu1iIZjBiH5NMyf0P4G7Zyuv1CP8l4cyDLlyAsWMD+f57PZMnO0hO9twE\ngJjUz+kybxJug5ltM97EGp3ksbaqO3844YqqwZ/6SuaPgcz/NIlDWSEEW1w8cechpg8+iV5/Y8cz\nf7yUgCXv465Tl4vvLcHVrkPlFuwpJSVYPvqQgDfmYzh2FABn6zbYh4/G1aGjT2/zCAsLpKCgBOx2\nTJs3Yl69Cv2ZLAAcffpRcv8DOPvdevV73ESNIOHMQ3JzFcaNC+DgQT0DB7q4/37PTQCI37SEdv/+\nPZrOwK6p/+J8UnvPNFRD+NMJV/g3f+srqgope6J5NyWBYpuBtg0KeWXmAW5pePGGjmf+dCWW994B\no5GiF1/FPtF/Jxfpfswm4O03sbz3NrqCAjS9AWfvPtiHj8Sd1NDX5QGXhLOfqCqGvbsxf7ISY/oB\nAFwtWlHy4EPYR4wGg8FHlQpfk3DmAWfPKowZE8jx4zoGD3Zy771Oj31Ya7BmPm3eeQyHJZjUKS9x\nIaGNZxqqQfzthCv8l7/2lQtWI++sT2DTvigURWNSvzM8PukwsbWvv1bD3t0E/usFdMXFlE67F+vT\nc8Fk8kDVN8awP42AhfMxf7ISxeVEDQnBMXgY9iHD0MIjfF3eZX4Vzi6hP3YU8ycrMG7fiqKquBPq\nU3L/A9gm3QmBgV6uVPiahLNKtmePjmnTAsjO1jFqlJOpU50eGTFT3C6aL3maJitfwhYcyc67XqGo\nTqPKb6gG8tcTrvA//t5X9h2rxaJ19TmZG4TF5Gbm8Ez+ODKT8BDndR1Hl51N0PPPoD91EleTplj/\n+QrObj08VHUFuFyYUtYTsPB1TDu2A+COjcM+YjSOPv3AbPZdbVdxtXD2E92P2Zg/XYXp6y9RnA7U\nyEhK772f0mn3ooWFe6lS4WsSziqJpsG77xr5+9/NqCrceaeTUaNcHglmAeey6PCv31H74HaKw2PZ\neferlETEVn5DNZS/n3CF/6gKfcWtwobvovjv1/HkF5kIDnBy/+0nmDkik7Dg67gP1mYj4N23MaV8\njqJp2CbcgfXJZ9Fqe2/ike70KSwfLMbywWL0OT8C4GzbDvuIUWX3xPn5vVoVCWc/UQoKMK9djenz\nz9AVF6MFBmKbdCel99yHu2FjD1cqfE3CWSXIyVH4+9/NrF5tJCRE4y9/sdO2rWeWy6i7YzXtXr8f\nU3EBZ1v248DIWTgDPLhzeg1UFU64wj9Upb5ic+hYnxrN8m9iKSw2EmRxMXXgaX5/eyYJ0RV/D/rD\nGQQsnI8h8zhqYBC2O6dSeu/9HpvRqeSfx7x2Tdklv61bUDQNLSAQR9/+2AcNqVIzSa8nnJUrLcGc\nsh7zmk/QnT8PgH3AbZTecx/OPv254Rkfwq9JOLsJqgqLFxt55hkzRUUKTZq4efRRB7VrV/63KzD7\nOC0X/x+xOz7FbTCTPuwhTnUY4fefFKuiqnTCFb5VFfuKzaHj89QYPtlWh/wiM3qdypDOufxu0Cn6\ntjlXsftj3W5MX6zDsvwjdBfy0XQ6HEOGYUueiLNP39/cW7JCNA39wR8wbd6IaeNXZYHMVTbC52ra\nDMdtg3F07wUWy4234SM3FM5+4nJh3Pkt5jWfYDiUAZRdyrXdMQXbHVNQY+MqsVLhaxLOboCqwtq1\nBl580cTBg3oCAjSmTnUycKCr0j/EGK0XaPrxPBqsXYjO7SQ/rhX7Rz8uS2V4UFU84QrfqMp9xelS\n+Ob7SD7ZVpfMH8sWq06ILmFSvzOM7XWWxrHFFTiIE+P2rZhXr8Jw/BgAmsGIs1t3HL374m7cFHej\nxrgTk349iUDTUAoL0OXloT+UgSH9AIYf0jGm7kR3Lq/8aa4GjXD26o2jey+06OhKe/++cFPh7BL6\no0cwpXyO6ZvNKDYbmqLg7N4T+5hx2G8f4XcTIcT1k3B2HYqKYNUqI2+9ZSQjQ49Op9G7t5spUxxE\nVPLPQsipH0j6/E0SNi3BYCumOKwuGbfNJLuVrIHjaVX5hCu8qzr0FU2Dw1nBfLE7mi0HIrE7yz5h\ntmlQyO1dchjcKYdWiUVX/7WjaeiPHMa4exeGPanlQa38yzodWkAgmExoZjOoKrr8fBTXrycmqOHh\nuNrcgrNtO1xt2qJFVp/FtCsrnJUrLcG09RtMX6eUj6ZpBmPZEiK3DcExcBBqfELltSe8RsLZNZSU\nwJYtetauNbJ6tYHSUgWdTqNPHzfJyU7q1au8b405P5s6uz8nbstHRKV/A0BprWgyu0/gRJdkVIP/\nTF+vzqrDCVd4R3XrK6V2HTszwtm8rzbfHQvFrZZd46wbUUrftufp1eo8PVrlEx9VetWwpuSfR3/k\nMPqzZ9CdyUJ/9izYSss2A3c4QFHQQsPQQkNRQ0NR68XhTkrCXT+xWo/6VHo4u4SSm4tp62aMW7dg\nyDxe/rireQscvfvi7NYTZ9duaBGRHmlfVC6fhjNVVZk9ezaHDx/GaDTy3HPPkZDwc8rfsGEDCxYs\nwGAwMHbsWMaNG3fN1/yWioaz4mJIS9OTmqpnxw4927bpsdvLfgvFxKgMGOCiXz83kZE3/y0xFuUT\nfmQ3EYdTid6bQsSR3T/X26AjJ7okk9u0B5peFiL0pup2whWeU537irVUz94jYew6FM6eI6FYS3/e\nEDws2EHbBhdp06Dwf39fpEGdYrk3/Ro8Gc4upeTmYtyzq2wk88D+slD8P67GTXC1bYer7S242tyC\nq2kzCWx+yKfhLCUlhY0bNzJ37lz27dvHG2+8wYIFCwBwOp0MGzaMFStWYLFYmDRpEm+88QZ7Vot6\nGQAADF5JREFU9uz5zddczU/hrLgYzp1TyM9XyMlRyMrScfq0jmPHdBw8qCMrS0HTfv5IGB+v0qmT\nm06d3DRpol7fYrKahrEoH0tBDuaCHIJyThKUfZTgs0cJOXWQkOyj5U9VFT35iW3Jad6bH5v1ojS8\n3nU0JCpTdT7hispVU/qKqsLJnED2Z9bi4KkQjmUH8WP+5TfkG/QqsbVt1I8uISG6lIToUuKjSoms\n5SA8xEF4sJOwYCehQS4M+ip/MeaGeCucXcbhQH/kcNk9fd8fwHDkEIrt8j6rRkTgbtQEd4OGuGPj\nUGPjcNeLRY2KRouMRI2IrJITMKqyioQzjw3b7N27l169egHQtm1bvv/++/KvHTt2jISEBEJCygrs\n0KEDqamppKWl/eZrruWDD4w8/LD5svB1qVq1NFq0UGnYUKVZM5WmTd03fC9ZVNrXdHl+AgZ76RW/\n7jQHk9egExfiW1IQ34oLCa1lSQwhhF/S6SCpbglJdUsY2b1sfbFim57j2YEcyw7i+Nkgzp63kFNg\nZsuBa98jFmRxYTSoGPUaRoOKXqdhNGgogNOt4HIrzBh2kj+NPn7NY4lrMJlwt2yFu2Ur7OMnld3v\nl30W/bGj6I8fQ38mC13WaQypuzDu2vGbh9HMZjSLBc1sAYvl538HBKAZDChuN7hcoNNhffJZXJ27\nePFN1kweC2dWq5Xg4ODy/+v1elRVRafTYbVay4MZQFBQEEVFRVd9zbU0aeKmSxc3FotGSAiEhWlE\nRalERWnUqaMSGvrr19zomKE9JJIL8a1xGS3YgyKwB0dQGlaH4sh4rJHxOILCf31jf9W/ta+a0KgG\nt1kKr6i5fSXQ7KJV4kVaJV6+b6fdqSOv0ETuBTO5hWaKSgxYS41YS/UUlZb9u9imx/W/EOZyKzic\nOopKFTQNDDoNvV7DqFer5a9En78nRYe7XhzuenHQq+/Pjzud6M7llf3Jy0V37hzKxUJ0RRdRLl5E\nsVrB6UBxOFCsVpQLF1AcDnA4UDQVTdGBXodmNl8221Z4jsfCWXBwMMXFP0/XvjRkhYSEXPa14uJi\natWqddXXXE1UVAhDh8LQoZX4Bq6qO/xlp7caE0KIaqbF//5UL9V1Ayblkr+vMM4hPMBDW3ZD+/bt\n2bJlCwBpaWk0bdq0/GsNGjTg5MmTFBYW4nA4SE1NpV27dld9jRBCCCFETeCxCQGapjF79mwOHToE\nwNy5c0lPT6ekpITx48ezceNG5s+fj6qqJCcnc8cdd1zxNUlJsiCrEEIIIWqOarHOmRBCCCFEdeGx\ny5pCCCGEEOL6STgTQgghhPAjEs6EEEIIIfyIhDMhhBBCCD9SpTd2/PLLL1m/fj0vvfRS+f9feOEF\n6tSpA8CDDz5Ip06dfFmi8CO/7C9paWnMmTMHvV5Pjx49+OMf/+jjCoU/0TSN3r17k5iYCEC7du14\n+OGHfVuU8Cs3uh+0qLlGjx5dvth+fHw8c+bMueLzqmw4e/bZZ9m2bRstWvy8kGF6ejqPPvoot912\nmw8rE/7oSv1l9uzZ/Pvf/yY+Pp4ZM2Zw8OBBmjdv7sMqhT85deoULVu2ZOHChb4uRfipr776CqfT\nydKlS9m3bx/PP/98hfaDFjWT3W4H4P3337/mc6vsZc327dsze/bsy7ZXSU9PZ8WKFUyePJl58+bh\ndrt9WKHwJ7/sL1arFYfDQXx8PAA9e/Zk+/btvixR+Jn09HRyc3OZOnUqM2bMIDMz09clCT9ztT2k\nhfiljIwMSktLmT59OnfddRf79u37zef6/cjZxx9/zOLFiy97bO7cuQwdOpSdOy/fQqlHjx4MGDCA\nuLg4/vGPf7B06VImT57szXKFj1W0v/xyH9egoCBOnz7ttTqFf7lSv3nyySe57777GDRoEHv27OHR\nRx9l+fLlPqpQ+KOb2Q9a1DwBAQFMnz6dcePGceLECe69916++OKLK/YXvw9n48aNY9y4cRV67tix\nY8s3VL/11ltJSUnxZGnCD1W0v/xyH1er1UqtWrU8WZrwY1fqNzabDb1eD0CHDh3Izc31RWnCj93o\nftCiZkpMTKR+/frl/w4LCyMvL4+YmJhfPbfa9CJN0xg5ciQ5OTkAfPvtt7Rq1crHVQl/FRwcjNFo\n5PTp02iaxrZt2+jYsaOvyxJ+ZP78+bz33ntA2eWIevXq+bgi4W9kP2hxPVauXMnzzz8PQE5ODlar\nlaioqCs+1+9Hzq5GURQURSn/93PPPccDDzyA2WymcePGjB8/3scVCn9yaX8BeOqpp3jkkUdwu930\n7NmTNm3a+LA64W9mzJjBo48+yubNmzEYDMydO9fXJQk/M3DgQLZt28bEiRMBpI+Iq0pOTuZvf/tb\n+e1Wc+fO/c2RVtlbUwghhBDCj1Sby5pCCCGEENWBhDMhhBBCCD8i4UwIIYQQwo9IOBNCCCGE8CMS\nzoQQQggh/IiEMyGEEEIIPyLhTAjhczt37mTKlCmVftz+/fszbNgwRo0axciRIxkzZsyvtn27lilT\nppCenl7h569cuZJnnnnmV4/n5OQwY8YMAGbNmsWqVavIzc0tf2zDhg28++6711WbEKJ6qtKL0Aoh\nxLUsWrSofHX/rVu38tBDD/HNN9+Ub81UEdezHOSlCx1fKiYmhjfffLP8OYqiEB0dXf5Yenr6b75W\nCFGzyMiZEMKvLVy4kGHDhjF8+HDmzZuHqqoALF68mEGDBpGcnMxjjz3G66+/fs1jdezYkfz8fAoL\nC5k1axa///3vGTp0KJs2bSItLY1x48YxcuRI7r77bk6dOlX+usWLFzNmzBjGjBnD7t27gbKRsOnT\npzNhwgT69+/PSy+9BJQFuaNHjzJp0iSGDx/Oyy+/DEBWVhb9+/e/rJ6fHjt27BhLly5l6dKlLF++\nnP79+3PixAkASkpK6Nu3Lw6H46a/l0KIqkFGzoQQfmvz5s1s3LiRVatWodfreeCBB/jwww/p0KED\nS5YsYeXKlRiNRqZMmUJCQsIVj3HpqNenn35KYmIiERERAISHh7Nw4UIcDgeDBw/mtddeo1WrVqxf\nv56HH36Y5cuXA1CrVi1WrlxJRkYGM2fO5IsvvmDt2rUMHz6cUaNGUVRURN++fZk2bRoAp0+fZtWq\nVQQGBnLXXXexefNmGjZs+Jvvs2HDhkyaNAko2+IlOzub1atX8+CDD5KSkkK/fv0wmUyV8j0VQvg/\nGTkTQvitHTt2cPvtt2MymdDr9YwdO5YdO3awY8cO+vXrR1BQECaTiWHDhv3mMWbMmMGoUaMYNmwY\nX331Fa+88gpQdmmxbdu2AJw4cYLQ0FBatWoFwODBgzl16hRWqxWAcePGAdCsWTPCwsLIzMxk2rRp\n1KlTh3feeYfnnnsOl8tFaWkpiqJw2223ERoaitFoZMiQIXz77bfXvGR5aYgcPXo0n332GQCrVq1i\n9OjRN/gdFEJURTJyJoTwW5qmXRZaNE3D5XKh0+nKL2/+9PhvufSes18ym80Alx3r0mO63W6Ay+5P\n0zQNvV7P888/T1ZWFsOHD2fAgAF8++235XVcupmxqqoYDNf3qzYuLo569eqRkpJCfn4+bdq0ua7X\nCyGqNhk5E0L4ra5du7J27Vrsdjsul4sVK1bQtWtXunXrxubNm7FarTgcDlJSUm7qZvoGDRpQUFDA\ngQMHAFi3bh2xsbGEhoYCsGbNGgAOHDhAcXEx9evXZ/v27UyfPp1BgwZx9uxZcnJyUFUVTdPYuHEj\nVqsVu93OunXr6N69+69C5i8DpcFgwOVylf9/7NixPPfcc4wcOfKG35cQomqSkTMhhM8pisKePXto\n165d+WMjR45k9uzZHDx4kLFjx+JyuejVqxdTpkxBp9MxZcoUJk6cSGBgIOHh4VgslhtqF8BkMvHy\nyy/zzDPPUFpaSlhYWPmN/AAFBQWMGjUKg8HAiy++iMFg4L777uOxxx4jMjKSRo0a0bVrV7KyslAU\nhaSkJO655x6KiooYMWIE3bt3L//aT+1e+gegU6dO/PWvfyUqKorJkyczcOBAnnjiCQlnQtRAinY9\nc8SFEMIPnDhxgk2bNnH33XcDMHPmTMaPH0/fvn19Wldl0TSNLVu2sGzZMhYsWODrcoQQXiYjZ0KI\nKqdevXocOHCA4cOHA9CrV69qE8wA5syZw+bNm1m0aJGvSxFC+ICMnAkhhBBC+BGZECCEEEII4Uck\nnAkhhBBC+BEJZ0IIIYQQfkTCmRBCCCGEH5FwJoQQQgjhR/4/GUaQIkvtDP8AAAAASUVORK5CYII=\n",
"text": [
""
]
}
],
"prompt_number": 9
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"Well, that's mildly informative. We see that sequences which contain 'G' in them are on average less likely, but still pretty close to sequences which do not contain 'G'. You'll notice in the code we filtered out any sequence which did not contain 'G' from the second group, to ensure all sequences scored contained at least one 'G' in them. "
]
},
{
"cell_type": "heading",
"level": 2,
"metadata": {},
"source": [
"YAHMM Information"
]
},
{
"cell_type": "markdown",
"metadata": {},
"source": [
"YAHMM is freely available under the MIT license at https://github.com/jmschrei/yahmm. Feel free to contribute or comment on the library! \n",
" \n",
"Installing YAHMM is easy, but simply calling `pip install yahmm`. Dependencies are numpy, scipy, matplotlib, networkx, and Cython. If you do not have these, and pip fails when building the prerequisites, the Anaconda distribution is a good way to get many scientific packages for Python, and has been shown on multiple platforms to allow YAHMM to install.\n",
"\n",
"If you have questions or comments, my email is jmschreiber91@gmail.com. Please let me know how you use YAHMM to solve your problems!"
]
},
{
"cell_type": "code",
"collapsed": false,
"input": [],
"language": "python",
"metadata": {},
"outputs": [],
"prompt_number": 9
}
],
"metadata": {}
}
]
}