# What is Protein Folding?
Proteins are the biological molecules that "do" things, they are the molecular machines of biochemistry. Enzymes that break down food, hemoglobin that carries oxygen in blood, and actin filaments that make muscles contract are all proteins. They are made from long chains of amino acids, and the sequence of these chains is the information that is stored in DNA. However, its a large step to go from a 2D chain of amino acids to a 3D structure capable of working.
The process of this 2D structure folding on itself into a stable, 3D shape is called **protein folding**. For the most part, this process happens naturally and the end structure is in a much lower free energy state than the string. Like a bag of legos though, it is not enough to just know the building blocks being used, its the way they're supposed to be put together that matters. *"Form defines function"* is a common phrase in biochemsitry, and it is the quest to determine form, and thus function of proteins, that makes this process so important to understand and simulate.
# Why is Folding a Good Subnet Idea?
An ideal incentive mechanism defines an asymmetric workload between the validators and miners. The necessary proof of work (PoW) for the miners must require substantial effort and should be impossible to circumvent. On the other hand, the validation and rewarding process should benefit from some kind of privileged position or vantage point so that an objective score can be assigned without excess work. Put simply, **rewarding should be objective and adversarially robust**.
Protein folding is also a research topic that is of incredibly high value. Research groups all over the world dedicate their time to solving particular niches within this space. Providing a solution to attack this problem at scale is what Bittensor is meant to provide to the global community.
# Simulation Backend and Reproducability
Molecular dynamics (MD) simulations require a physics-based engine to run them, and SN25 utilizes the open-source project [OpenMM](https://openmm.org). As their tagline suggests, they are a "high performance, customizable molecular simulation" package.
One of the key advantages of using OpenMM for MD-simulations is the built-in capabilities for *reproducability*. This is a key component in the reward stack and all miners should be intimately familiar with this. For more information, please read this [document](./reproducibility.md).
# Reward Mechanism
Protein folding is a textbook example of this kind of asymmetry; the molecular dynamics simulation involves long and arduous calculations which apply the laws of physics to the system over and over again until an optimized configuration is obtained. There are no reasonable shortcuts.
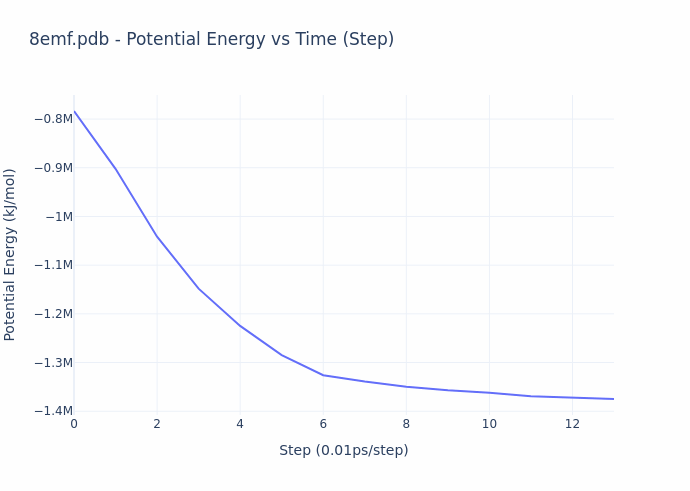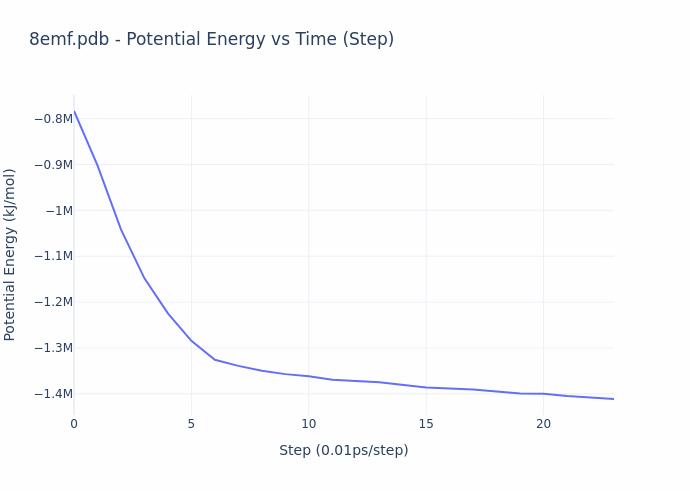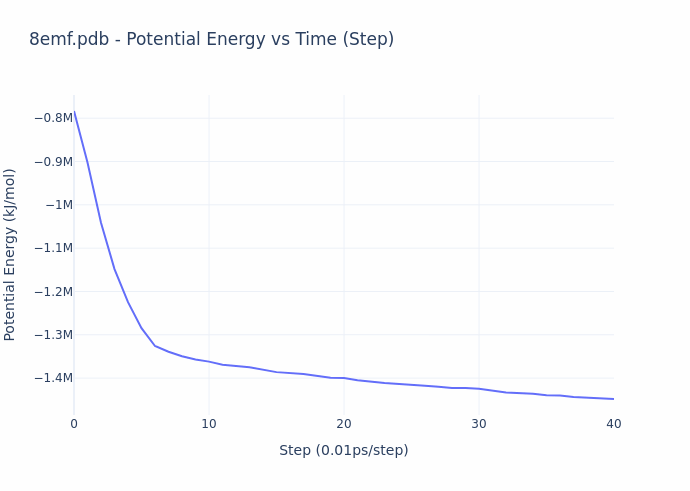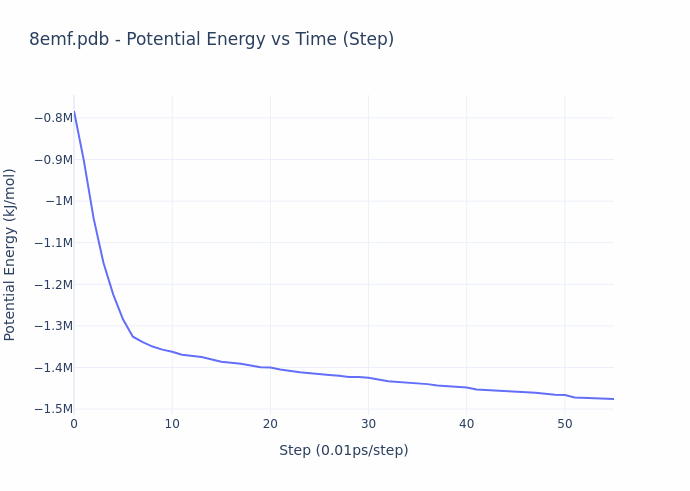
While the process of simulation is exceedingly compute-intensive, the evaluation process is actually straightforward. **The reward given to the miners is based on the ‘energy’ of their protein configuration (or shape)**. The energy value compactly represents the overall quality of their result, and this value is precisely what is decreased over the course of a molecular dynamics simulation. The energy directly corresponds to the configuration of the structure, and can be computed in closed-form. The gif below illustrates the energy minimization over a short simulation procedure.
When simulation reach convergence (ΔE/t < threshold), they obtain the structural form of proteins as they are observed in real physical environments, and this form gives rise to their biological function. Thus, the miners provide utility by preparing ready-for-study proteins on demand. An example of such a protein is shown below.