{"cells":[{"metadata":{},"cell_type":"markdown","source":"# ** DIscBIO: a user-friendly pipeline for biomarker discovery in single-cell transcriptomics**"},{"metadata":{},"cell_type":"markdown","source":"The pipeline consists of four successive steps: data pre-processing, cellular clustering and pseudo-temporal ordering, determining differential expressed genes and identifying biomarkers."},{"metadata":{},"cell_type":"markdown","source":"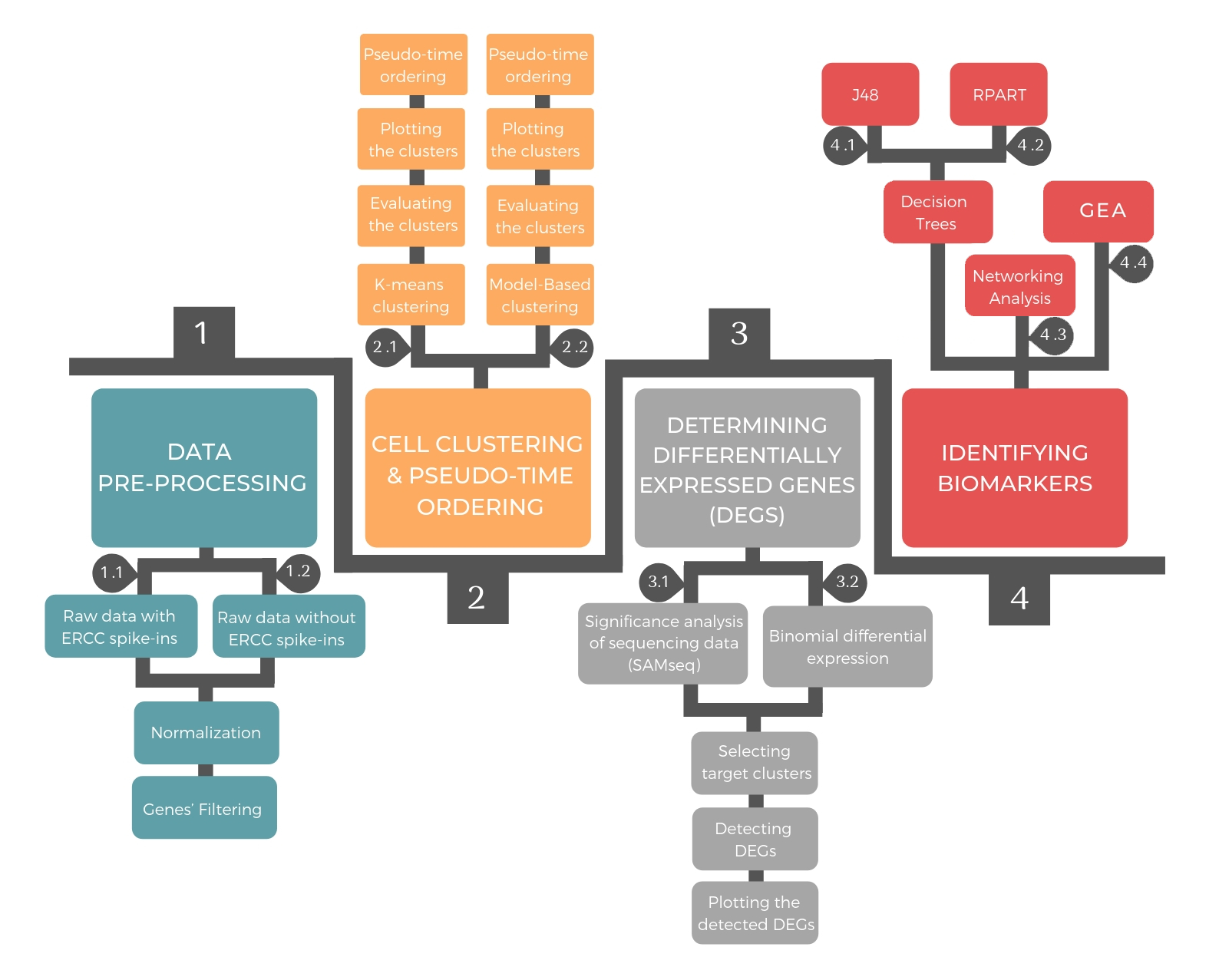"},{"metadata":{},"cell_type":"markdown","source":"# CTC Notebook [PART 4]\n\n## Running the DIscBIO pipeline based on a list of genes related to Golgi Fragmentation\n## [Data pre-processing and cell clustering]\n "},{"metadata":{},"cell_type":"markdown","source":"## Required Packages"},{"metadata":{"trusted":false},"cell_type":"code","source":"library(DIscBIO)","execution_count":1,"outputs":[{"output_type":"stream","text":"Loading required package: SingleCellExperiment\n\nLoading required package: SummarizedExperiment\n\nLoading required package: MatrixGenerics\n\nLoading required package: matrixStats\n\n\nAttaching package: ‘MatrixGenerics’\n\n\nThe following objects are masked from ‘package:matrixStats’:\n\n colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,\n colCounts, colCummaxs, colCummins, colCumprods, colCumsums,\n colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,\n colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,\n colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,\n colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,\n colWeightedMeans, colWeightedMedians, colWeightedSds,\n colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,\n rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,\n rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,\n rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,\n rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,\n rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,\n rowWeightedMads, rowWeightedMeans, rowWeightedMedians,\n rowWeightedSds, rowWeightedVars\n\n\nLoading required package: GenomicRanges\n\nLoading required package: stats4\n\nLoading required package: BiocGenerics\n\nLoading required package: parallel\n\n\nAttaching package: ‘BiocGenerics’\n\n\nThe following objects are masked from ‘package:parallel’:\n\n clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,\n clusterExport, clusterMap, parApply, parCapply, parLapply,\n parLapplyLB, parRapply, parSapply, parSapplyLB\n\n\nThe following objects are masked from ‘package:stats’:\n\n IQR, mad, sd, var, xtabs\n\n\nThe following objects are masked from ‘package:base’:\n\n anyDuplicated, append, as.data.frame, basename, cbind, colnames,\n dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,\n grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,\n order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,\n rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,\n union, unique, unsplit, which.max, which.min\n\n\nLoading required package: S4Vectors\n\n\nAttaching package: ‘S4Vectors’\n\n\nThe following object is masked from ‘package:base’:\n\n expand.grid\n\n\nLoading required package: IRanges\n\nLoading required package: GenomeInfoDb\n\nLoading required package: Biobase\n\nWelcome to Bioconductor\n\n Vignettes contain introductory material; view with\n 'browseVignettes()'. To cite Bioconductor, see\n 'citation(\"Biobase\")', and for packages 'citation(\"pkgname\")'.\n\n\n\nAttaching package: ‘Biobase’\n\n\nThe following object is masked from ‘package:MatrixGenerics’:\n\n rowMedians\n\n\nThe following objects are masked from ‘package:matrixStats’:\n\n anyMissing, rowMedians\n\n\n\n\n","name":"stderr"}]},{"metadata":{},"cell_type":"markdown","source":"## Loading dataset"},{"metadata":{},"cell_type":"markdown","source":"\nThe \"CTCdataset\" dataset consisting of single migratory circulating tumor cells (CTCs) collected from patients with breast cancer. Data are available in the GEO database with accession numbers GSE51827, GSE55807, GSE67939, GSE75367, GSE109761, GSE111065 and GSE86978. The dataset should be formatted in a data frame where columns refer to samples and rows refer to genes. We provide here the possibility to load the dataset either as \".csv\" or \".rda\" extensions.The dataset should be formatted in a data frame where columns refer to samples and rows refer to genes. \nWe provide here the possibility to load the dataset either as \".csv\" or \".rda\" extensions."},{"metadata":{"trusted":false},"cell_type":"code","source":"FileName<-\"CTCdataset\" # Name of the dataset\n#CSV=TRUE # If the dataset has \".csv\", the user shoud set CSV to TRUE\nCSV=FALSE # If the dataset has \".rda\", the user shoud set CSV to FALSE\n\nif (CSV==TRUE){\n DataSet <- read.csv(file = paste0(FileName,\".csv\"), sep = \",\",header=T)\n rownames(DataSet)<-DataSet[,1]\n DataSet<-DataSet[,-1]\n} else{\n load(paste0(FileName,\".rda\"))\n DataSet<-get(FileName)\n}\ncat(paste0(\"The \", FileName,\" contains:\",\"\\n\",\"Genes: \",length(DataSet[,1]),\"\\n\",\"cells: \",length(DataSet[1,]),\"\\n\"))","execution_count":2,"outputs":[{"output_type":"stream","text":"The CTCdataset contains:\nGenes: 13181\ncells: 1462\n","name":"stdout"}]},{"metadata":{},"cell_type":"markdown","source":"### 1. Preparing the dataset"},{"metadata":{"trusted":false},"cell_type":"code","source":"FG<- DISCBIO(DataSet)\nFG<-Normalizedata(FG, mintotal=1000, minexpr=0, minnumber=0, maxexpr=Inf, downsample=FALSE, dsn=1, rseed=17000) \nFG<-FinalPreprocessing(FG,GeneFlitering=\"ExpF\",export = TRUE) # The GeneFiltering should be set to \"ExpF\"\n\nGolgiFragGeneList<- read.csv(file = \"GolgiFragGeneList.csv\", sep = \",\",header=F)\nData<-FG@fdata \ngenes<-rownames(Data)\ngene_list<- GolgiFragGeneList[,1]\nidx_genes <- is.element(genes,gene_list)\nOAdf<-Data[idx_genes,] \nFG@fdata<-OAdf\ndim(FG@fdata)\ncat(paste0(\"A list of \", length(OAdf[,1]), \" genes will be used for the clustering\",\"\\n\"))","execution_count":3,"outputs":[{"output_type":"stream","text":"The gene filtering method = Noise filtering\n\nThe Filtered Normalized dataset contains:\nGenes: 13181\ncells: 1448\n\n\n\nThe Filtered Normalized dataset was saved as: filteredDataset.Rdata\n\n","name":"stderr"},{"output_type":"display_data","data":{"text/html":"\n- 97
- 1448
\n","text/markdown":"1. 97\n2. 1448\n\n\n","text/latex":"\\begin{enumerate*}\n\\item 97\n\\item 1448\n\\end{enumerate*}\n","text/plain":"[1] 97 1448"},"metadata":{}},{"output_type":"stream","text":"A list of 97 genes will be used for the clustering\n","name":"stdout"}]},{"metadata":{},"cell_type":"markdown","source":"### 2. Cellular Clustering and Pseudo Time ordering"},{"metadata":{"trusted":false},"cell_type":"code","source":"load(\"fg.RData\") # Loading the \"fg\" object that includes the data of the k-means clustering \nFG<-fg # Storing the data of fg in the FG \n########## Removing the unneeded objects\nrm(DataSet)\nrm(fg)\nrm(Data)\nrm(OAdf)\n\nplotGap(FG) ### Plotting gap statistics","execution_count":4,"outputs":[{"output_type":"display_data","data":{"text/plain":"Plot with title “Gap Statistics”","image/png":"iVBORw0KGgoAAAANSUhEUgAAA0gAAANICAIAAAByhViMAAAACXBIWXMAABJ0AAASdAHeZh94\nAAAgAElEQVR4nOzdeVyU5f7/8YsBRNbBHcXccEsQUVwTOq6nTBM1FTdSM5eTqZUdNUNtEbXF\nsjx6UDPNzBuXlFN+ExdQU9RAQFxzwSVzR1lF2WZ+f8w585sGHFFmuGduXs/H+WPmmuu+7890\nCt5cy33babVaAQAAANunkrsAAAAAmAfBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAU\ngmAHAACgEAQ7AAAAhSDYAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAH\nAACgEAQ7AAAAhSDYAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAHAACg\nEAQ7AAAAhSDYAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAHAACgEAQ7\nAAAAhSDYAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAHALJxcHCw+58j\nR47Y6CUAWA8HuQsAYEsOHz68devW+Pj4K1eu3L17VwihVqt9fHwCAgL+/ve/9+nTx8nJSe4a\n/yIuLm7NmjW//fbbjRs3CgoKatSoUbNmzYYNG3bs2LFjx45du3Z1c3PTd96+ffvRo0d1r9u3\nb9+vX79yXt3sJ5TlEgBsiRYAyuDMmTNBQUGmf57UqFEjKSlJ7kr/q7CwcNSoUaYLXrNmjeEh\nEydO1H80ceLE8tfw2BPa29vrOxw+fNg6LwHAhjBiB+DxduzYMWTIkPv375vudvfu3du3b1dM\nSY8VHh6+fv16uat4jGnTphUXF+te161b10YvAcB6EOwAPMbx48eNUp2zs3Pfvn0DAgLUanVO\nTs6FCxeOHDly+vRpGYs08vDhw6VLl+rfent7Dx48uGHDhkKIW7dupaamHjhw4LE5tQIsXrxY\nAZcAYEXkHjIEYO3+9re/Gf7Q6NOnz61bt0p2O3v27LRp03799VfDxp9++undd9/t2bNns2bN\natSo4eDg4O7u3qxZs2HDhv30008lT2I0b3jjxo033nijYcOGTk5O9evXnzRp0o0bN8pSc0JC\ngv48Li4ut2/fNuqQn5//448/6qcmQ0JCTPycrFGjxpN+nTKe8FHzpPn5+ZGRkb17965Xr56T\nk1PVqlXr16/fvn37119/PTIy8u7du+W/hM7FixdnzZrVqVOnGjVqODo61qpVKyAgYNq0aQkJ\nCU9UDAArQbADYIphQhJCBAYG5ufnl/3wNm3amAgfAwYMKCgoMOxvmEJWrlxZo0YNo0Nq1ap1\n8uTJx143NjZWf0j9+vU1Go3p/mUMSWX/OuVJXQ8ePOjcubOJw3fv3l3OS2i1Wo1G8/HHHxt+\namj06NFPVAwAK8HtTgCYsmPHDsO3H374YZUqVZ7uVB4eHq6uroYt0dHRCxYseFT/N998U7fx\n1tCdO3cGDBjw4MED09dq0KCB/vWff/45dOjQffv2PXz48MmrfqQn/Tpl9+9//9vwviRVq1bV\njQ6W/8yGwsPD58yZo19+J28xAMyFYAfAlNTUVP1rR0fHnj176t8WFxf/XsL58+cND2/btu3n\nn3/++++/379/PysrKzc399atW++8846+w9KlS7VabamXLigoGDBgQExMTExMTP/+/fXtFy5c\n+Oabb0yX3bRp08DAQP3bLVu2dO/e3cPDIyAgYMKECVFRUbm5uYb9V65ceenSpREjRuhbRowY\ncel/UlJSnvTrlPGEpdq3b5/+9datW/Py8tLT0x8+fHjq1Klly5Z169ZNpVKV8xLHjx9ftGiR\n/m3NmjWXLFmSmpp65syZ6Ojo0NBQ/UheGYsBYC3kHjIEYNWCg4P1Py7q1atn+NGNGzdK/khR\nq9WPPWdhYaGzs7P+kNOnT+s/MpwZ7NSpk34Ktbi42DCode7c+bFXOXXqVJ06dR71o8/Nze2D\nDz4oLCw0POTpbndi4us83b1IXnjhBV2LSqW6fPmy6as/3SX+8Y9/6Bvt7e2Tk5ONjsrMzHyK\nYgDIjr+0AJSVnZ3dkx6i0Wg2bdo0dOjQFi1auLu729vb29nZOTo6Gs6lXr9+vdRjX3vtNf0V\nVSrV2LFj9R8lJyc/dg6xVatWqampb7zxhru7e8lPc3NzP/jgg9GjR1fY1yk7X19f/eVatGgR\nHBw8fvz4L774Yu/eveaaTf7111/1r1955ZW2bdsadVCr1RVWDAAzYp0EAFNq1aqlf3379u38\n/PyyP1vi/v37ffv23b9/v+luRrOieo0bNzZ826RJE/3rgoKCzMzMklsrjNSpU2fZsmVffPHF\noUOHDhw4cPjw4fj4+JycHH2HDRs2vPPOO4ZjgSaU8+uU3eTJk1evXp2VlSWEyM/PP3jw4MGD\nB3Ufubu7T5w4cf78+eV8wodh+vT395e3GABmxIgdAFMMf+sXFhbu3btX/9bLy0s38i9JUqnH\nLliwwDAG+fv7h4WFTZw4ceLEiVWrVtW3ax+xxs6oXaPRPN1XcHJy6t69+9y5c3fs2JGenm50\nXzfDNWSmlfPrlF2TJk0OHz48aNCgkoEpJyfn888/nzZtWjkvYcj0QGwFFwOgnAh2AEzp06eP\n4duPPvqoqKiojMdu2bJF/3rKlCmpqanr1q2LjIz817/+VZaUdvHiRcO3ly5d0r+uUqWKp6en\niWOLi4tLDVhVqlR55513GjVqpG/RjUWVRTm/zhN59tlnf/zxx3v37sXHx3/77bfvvfee4Wzp\n2rVryzkNWq9ePf3r48ePy1sMADMi2AEwpWPHjob7Jw4fPjx8+PAyhiHD3RU9evTQv961a1dB\nQcFjD//222/1gUmj0axZs0b/Ubt27R51Azadq1evPvvss5GRkffu3TP66MqVK4aF1axZU//a\n0dFR/7rkHVWe4uuYPuGj6KOzi4vLc889N3bs2AULFiQmJnp5eena8/Pz9cU83SWef/55/est\nW7aUzHb6/4ufqBgAsmONHYDH+Oqrr4KCgvLy8nRvt2zZsnv37pCQED8/P2dn57t37xrd606v\nZs2a+gVtK1eu1N1w5Ndffx0/fnxZrpuYmBgSEqLbvxkZGZmcnKz/aPjw4Y89/OzZs//4xz+m\nTp3auXPn9u3be3t729vbp6WlRUVF5efn67sFBQXpXxsuKNy9e/fevXsbNWpkZ2fn4eFRvXr1\np/g6pk/4qKNmzZqVlpY2ePDg4ODgZ555xs7OTqvVxsTEpKen6/t4eHiU5xITJ06MjIzUDWoW\nFxf36tVrzpw53bp1q1KlyoULFzZt2uTg4LB69eonLQaA/GTajQvAlkRHRxsuIzPB8HYnU6dO\nNfzI3t5ed0dfR0dHw3Gmbdu26Q8xHIcrdTerEMLHx+f+/fumCzactzWhZ8+ehkc9KqFOmzbt\n6b6O6RNqH3EvEsNVa1WqVKlRo4bR+rYuXbqU8xJarXbWrFmP+sciDJ488UTFAJAdU7EAHi8k\nJOTIkSPt2rUz3a1u3bqGcWHu3LnNmjXTvy0uLr5//769vf2qVavKMsYTGRnp7e1t1FijRo2t\nW7e6uLiYPtbFxaXksUY6duwYFRVl2NK7d++SN/7Qe4qvY/qEZVFQUHD37l3DIUYvL69Vq1aV\n/xILFiyYN2+e6RntJy0GgPzkTpYAbMnu3bsnT57cpk2bmjVrOjg4uLi41K9fv0ePHu++++6e\nPXuMHvyq1Wrv3bv31ltvNWzYUPeA+ZCQkEOHDmm1WsM7lTxqxO7w4cO3b9+eMmVKw4YNq1Sp\n4u3tPWHChOvXr5exVI1GEx8f/8EHH/Tp08fHx8fNzU03xubj4zN48OCNGzfqNlgYuXv37pQp\nU3x8fAyfnKYf/XrSr/PYE5Y6nPbHH398++2348eP79ChQ4MGDZydnR0dHWvXrv38888vXLgw\nIyPjiWp+1IidzoULF2bMmNGhQ4fq1as7ODjUrFkzICBgypQpv/3229MVA0Bedtpy78wHAHNx\ncHDQ33n48OHDph8/DwAwwlQsAACAQhDsAAAAFIJgBwAAoBAEOwAAAIVg8wQAAIBCMGIHAACg\nEAQ7AAAAhSDYAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAHAACgEAQ7\nAAAAhSDYAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAHAACgEAQ7AAAA\nhSDYAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAHAACgEAQ7AAAAhSDY\nAQAAKATBDgAAQCEIdgAAAApBsAMAAFAIgh0AAIBCEOwAAAAUgmAHAACgEA5yF2AbUlNTi4qK\n5K4CAABYBQcHhzZt2shdRSkIdo939OjRDh06yF0FAACwIomJie3bt5e7CmMEu8crKCgQQuTn\n51epUkXuWgAAgMwKCgqcnJx08cDasMYOAABAIQh2AAAACkGwAwAAUAiCHQAAgEIQ7AAAABSC\nYAcAAKAQBDsAAACFINgBAAAoBMEOAABAIQh2AAAACkGwAwAAUAiCHQAAgEIQ7AAAABSCYAcA\nAKAQBDsAAACFINgBAAAoBMEOAABAIQh2AAAACkGwAwAAUAiCHQAAgEIQ7AAAABSCYAcAAKAQ\nDnIXAAAAYDaavLxLI0Zo7t83ai+8cUMI4Vi3rlG7ytW18YYNKheXCqrPwgh2AABAOeyqVHHt\n1Kk4K8uoPfuXX4QQLoGBRu32arVdlSoVVJzlEewAAIBy2Dk4eL33nmGLVqs9efLk3SNHhBBF\nI0f6+fnZ2dnJVJ3FscYOAAAo1vHjxwMDA/39/RMSEhISEvz9/QMDA48fPy53XZZCsAMAAMp0\n4cKFbt26NW3a9MqVK6GhoaGhoVeuXGnatGn37t0vXLggd3UWQbADAADKNHv27Hbt2kVFRTVo\n0EDX0qBBg6ioqICAgNmzZ8tbm4Wwxg4AAChQYWHh9u3bN23apFL9ZRhLpVK99dZboaGhRUVF\nDg5KC0JK+z4AAEDxcnJy7t+/n5ubm5WVlZ2dff/+/datWzds2NCwT3p6+oMHD5o1a1by8ObN\nmz948ODOnTt1S9z9xNYR7AAAgAwePnyYm5ubnZ2dnZ2dm5ubm5ubk5OTlZWV+z+ZmZm6F/fv\n38/IyNC/zszMNDqVu7v7jBkzwsPDjRqFECU7CyEyMjL0HRSGYAcAgDkVZ2efqFev5A1yH0Xl\n6tr6+nV7Dw+LVmU5Dx48yMjIePjwoe6F0etHfXTv3r38/HyjU1WtWrVatWrVqlVzdnbWv65T\np06p7fq3tWrVcnR0LFmYm5tbQEDAtm3bOnXqZPRRdHR0QECAm5ubpf6hyIdgBwCAOdl7eDSL\ni9NkZwshtFrt559/vm/fvl69ej1/5YoQ4teGDffs2dOtW7d3331Xdzc1lYeH7KnuwYMHZUlj\nJV9nZmZqtVrDU+mCl2EC071u0qRJyXCmf12jRg0nJyezf69Zs2aNHj06ODi4b9+++sbt27cv\nWbLku+++M/vlrAHBDgAAM3Pt2FH34l//+tfXv/0Wd/hwu3btrowdK4TotmZNv+Tk7t271zl3\nbvLkyWa8qFE4K/vr27dvFxcXG56qatWqpY6N6cJZqclM99rT09Oq7v0bGhp69uzZkJCQ7t27\nv3H7thDi9d699+7dO3fu3NDQULmrswiCHQAAlvL111/PnDmzXbt2ho3t2rWbNWvWV199VTLY\nabXajIyMzMzMnJwc3Xoy3eYA3evs7OysrCzdpoGcnJzMzEzd69zcXN2iMUOurq5ubm5ubm6e\nnp5u/6NWq2vXrq177eHh4eHh4ebm5urq6uHhoVarda8VtvJs7ty5ffv2Xb9+ffqmTUIIvx49\nFi1aFFjiwWKKQbADAMAiMjMzz58//8ILL5T86IUXXpg9e3ZWVpZarTZsnzFjxueff65/6+Tk\n5Orq6unp6e7urktdarXaw8OjQYMG+jSmC3Du7u6enp76MFetWjWLfz0rVnjzpiYvT//Wr1q1\nRVOmXLt6VQjx6pQpQoj8ixf1n6pcXBy9vCq+SAsh2AEAYBEPHz4UQri4uJT8yNnZWdfBKNiF\nh4e/+uqrrq6u1apVc3NzK3VPAEwrzs4+2aCBtrCw1E8zf/zRqMXO0dE/PV32ZY7mQrADAKBc\nHjx4cOrUKScnp9atWxu216pVy8PD48SJEy1btjQ65MSJE2q1umbNmkbtarXa6CR4UvYeHn5/\n/GE4YqejLSgQQthVqWLUrnJxUUyqEwQ7AACe1OXLl48fP37ixInjx48fP378/PnzxcXFw4YN\nkyTJsJu9vf3QoUMXLVr08ssvV61aVd/+8OHDRYsWDRkyxN7evsJrrxSUNLX6pAh2AACYkpOT\nc+7cuVOnTiUlJZ0+fTo1NfXOnTtVqlRp2rRpYGDgpEmTfH19/f39a9euXfLYjz/+uHPnzj17\n9lywYEH9oiIhxP79+2fPnn3v3r358+dX+FeB8tkZ3X4GJR06dKhr1675+flVSozfAgCU5/r1\n67oMpwtzv//+u0ajqVu3bmBgoK+vb6tWrQIDA5999lmjJ5DqFefknO3SRfvgge5tUVHR3bt3\n79+/r9vOkCGEq6trjRo19E8ptXN2bnH4sL2y9qIqW0FBgZOTU3x8/HPPPSd3LcYYsQMAVGpZ\nWVknTpzQx7jU1NTc3FwPD49mzZq1atVqwoQJgYGBT/SUAns3N++FCzUPH+pbGgjx4MGDa8eP\nCyEC/f11Oyf0VFWr2ivxEQiQBcEOAFCJFBcXX7lyRT+veurUqTNnzqhUqoYNG7Zq1apXr17T\npk1r1apVq1atnv5Gu3Z26pdfNmqrJkS98tYOPB7BDgCgZJmZmSdPntTHuJSUlLy8PE9PT19f\nX19fX92AXNu2bV1dXeWuFDADgh0AQDmKiorOnj2rn1c9ffr0xYsXHRwcmjdv7uvr26tXr5kz\nZ/r6+jZu3NiqnnwFmAvBDgBgwzIyMnQZTj8m9/Dhw2rVqum2OLz88su6F0bL2gClItgBAGxG\nYWHhuXPn9Bnu6NGjN2/edHR0bNasWWBg4JAhQ+bNm9ehQwevSnwbM1RyBDsAgPW6fv26fl41\nKSnp7NmzxcXFdevW1d12ZMiQIb6+vn5+fk5OTnJXClgFiwe7tLS0uXPnxsbGZmZmPvPMM6Gh\nobNnzy71wXklxcbGLl269PDhw5mZmbVr127Xrt3bb7/drVs3fYeWLVuePXvW6Kg6dercvHnT\njF8BAFAxTN8KeMKECSZuBQxAWDrYnTx5Mjg4OCsrq1+/fk2aNDlw4EBERERsbGxcXNxjlzu8\n9957ixYtcnJy6ty5c506de7cuRMfH9+6dWvDYCeEUKlUYWFhhi1GD1QGAFgtE7cCDgwMDAsL\nM30rYABGLBvsxo0bl5mZuWbNmjFjxgghNBrNqFGjJElavHhxeHi4iQPXrFmzaNGiLl26bN68\n2dvbW9eo0WgyMjKMejo6Oq5du9Yi1QMAzMrstwIGYMSCjxRLTk7W/SeakpKib7x27VqDBg3q\n1av3xx9/PGqreUFBQcOGDXNyctLS0urUqWPiEi1btrx8+fJDg7t7WwKPFANgY7TarB07NPfv\nGzUX3bkjhHCoVcuoXeXqqu7TR5j79h+mbwWsfzxXuW4FDMihkj5SLC4uTgjRp08fw0Zvb29/\nf/9jx46dO3euRYsWjzrw5s2bI0eOVKvVGzduPHnypLOzc6dOnXr06FHyP36NRrNgwYK0tDRn\nZ2d/f//BgwdXr17dQt8IAGxCcW7u1TfeKM7ONmrX5OUJIVQlVjnbe3i4nThR/meVcitgQHYW\nDHa6bQ0l01vz5s1NB7vExEQhRI0aNfz9/c+fP69v79Kly7Zt24zG8AoLC99//3392+nTp69c\nuXL48OHm+hYAYHPs3d39Ll8u2X5l7FghRMM1a8xyFW4FDFghCwa7rKwsUdpWBk9PTyFEZmbm\now68ffu2EGLZsmVNmzbdu3dv+/btL126NH369N27dw8bNmzv3r36nqNHj+7QoYOfn59arb54\n8WJkZOTy5cvDwsLq168fHBxcliJv3749bdq04uJiE31u3LghhLDcnDUAWFpMTMz333/faccO\nIcRvI0eGhYW9+OKLT3oSbgUMWD8Z7mOnS0gm/oDTxSw7O7vo6OiWLVsKIVq3br1t27bmzZvv\n27fv6NGj7du31/V877339Ef5+vouXbpUrVZHREQsXLiwjMHO2dnZx8enqKjIRJ/s7GwhRGFh\nIfdJAmBztFrtm2++uWrVqqFDhzZv3lwIkWBn179///Hjx//rX/8y8aOYWwEDtsiCwU43Vqcb\ntzP0qJE8vWrVqgkhWrZsqUt1Oq6urr179/7uu+8Mg11J48aNi4iISEhIKGOR7u7u8+fPN91n\nxYoVO3fuLOMJAcCqrFq1at26dfv37+/SpYtuKnb9mjWTJ0/++9//HhAQMH78eKP+Gzdu3Lp1\n6/Hjx8+fP19cXNygQQN/f//WrVuPGjXK39+/WbNmDg7c2R6wXhb871O3hK7kDYR1y+Z0fzia\nOFA3Y2tI12J6D6yuT35+/tNUDACK8+WXX86YMaNLly6GjV26dPnnP//5xRdflAx2586d8/T0\nfPPNN1u3bu3v71/yRzEAa2bBYNejRw8hRExMzIIFC/SN169fT01N9fb2NhHsevbsaWdn9/vv\nvxcWFjo6OurbT5w4IYRo3LixiYvu379fCOHj41P++gHA1mVlZf3+++8vvfRSyY9eeumlefPm\nZWdne3h4GLbPmTOnoqoDYH4WvJd3u3btOnbsmJKSsm7dOl2LRqOZMWOGRqOZNGmS4cKOtWvX\nLlmyRLdnQgjh7e09cODA9PT0iIgIfZ/t27fHxcXVrFmzV69eupbExMTjx48bXvHo0aOTJ08W\nQhg9iwIAKqcHDx4IIUq9vYjuJsB5eXkVXRMAS7LsUonVq1cHBQWNHTt269atjRs3PnDgQFJS\nUqdOnaZPn27Ybf78+WlpaUFBQfrH/y1dujQ5OfnDDz/ctWtXu3btrly58ssvvzg6On7zzTf6\nn1D79+//5z//6ePj07hxYw8Pj0uXLh07dkyr1fbv33/q1KkW/V4AYBNq1qzp7u5++vRpwyXL\nOqdOnXJ3d69V4mbFAGyaZZ++5+fnl5SUFBoaeujQoeXLl2dkZMyePTs2Nvaxm+Hr1auXmJg4\nZcqU69evr1y58vDhwyEhIYcOHQoJCdH36dmz5/jx411cXJKTk3/66aerV6/26tXr+++/j46O\nNpzABYBKy8HBYdCgQZ988klBQYFhe0FBwaeffvrKK6/Y29vLVRsAS7DgI8UUY8WKFZMmTcrJ\nyeHxhQBszrVr1zp27NiyZctPPvmkxtKlQoj0N9+cNWvW2bNnf/vtN/3DuAGUnTU/UsyyI3YA\ngAoTHx9f8mZP3t7e8fHxjo6OHTp0WL9+/fr16zt27FilSpWDBw+S6gDlIdgBgG3TaDRbtmzp\n3Lnz888/v2vXrpIdGjVqFBMTc+vWrd69e/fu3fvWrVs7duxo1KhRhVcKwOK4zyQA2Kr8/PyN\nGzcuXLjw0qVLQ4cOXbNmzbPPPiuE0BYXp//735r79436e6anCyG0a9bc+mu7ytW15j/+Ycd6\nO8D2EewAwPbcuXNn2bJly5YtE0KMGzdu6tSp9erV03+qffgwc+vW4uxso6OK0tOFEBmbNxu1\n23t41Bg71q60u6IAsC0EOwCwJWlpaV9//fU333zj5eUVHh6uuzmAUR+Vq2uzuDhZygMgL4Id\nANiGpKSkr776asOGDQEBAf/+979HjBjBY1sBGGHzBABYNY1G8/PPPwcFBXXs2DEjIyMmJubo\n0aOvvvoqqQ5ASQQ7ALBS+fn569at8/X1HTJkSJMmTU6ePPnzzz/rH6sIACXxBx8AWJ07d+58\n++23X331VV5e3ujRo2fOnGm4NwIAHoVgBwBW5OLFi1999dU333xTp06dmTNnvv76665sVgVQ\nZgQ7ALAK+r0Rbdq0YW8EgKfDGjsAkFPJvRFJSUnsjQDwdAh2ACAP3d4IPz+/wYMHN2nS5MSJ\nE+yNAFBO/EUIABUtPT199erV+r0Re/bsYW8EALMg2AFAxWFvBACLItgBQEVgbwSACsAaOwCw\nIN3eiN69e3fo0OHGjRvbtm1jbwQAy+EnCwBYRH5+/saNGxctWpSWlhYaGnry5MlWrVrJXRQA\nhSPYAYCZ6fZGfP311/fv3x89evTu3bu9vb3lLgpApUCwAwCzMdwbMWPGDPZGAKhgBDsAMIPk\n5OQlS5ZIktS6dWv2RgCQC5snAODp6fdGtG/f/saNG1u3bk1OTmZvBAC58KMHAJ6G0d6IEydO\n+Pr6yl0UgMqOYAcATyYrK2vt2rWffvppbm7umDFj2BsBwHoQ7ACgrC5durRkyZLVq1fXqlVr\n6tSpkyZNUqvVchcFAP8fwQ4AHs9wb8Ty5cvZGwHAOrF5AgAeSavVsjcCgA3hZxMAlKKgoCAq\nKuqTTz65cOECeyMA2AqCHQD8hW5vxGeffZaTkzNmzJidO3fWr19f7qIAoEwIdgDwX4Z7I6ZM\nmcLeCAA2h2AHACIlJeXLL7+UJMnPz4+9EQBsF5snAFReWq12z549L7/8crt27S5evLh169aU\nlBT2RgCwXQQ7AJVRQUHBunXr/Pz8+vbtW61atRMnThw8ePDll1+Wuy4AKBf+KgVQuWRnZ69Z\ns4a9EQAUiWAHoLK4fPnyl19+uXr16po1a06ZMmXixImenp5yFwUA5kSwA6B87I0AUEmwxg6A\nYpXcG8FzIwAoG8EOgALp9ka0bt3aaG+EnZ2d3KUBgAXxZysARTHaGxETE8PeCACVB8EOgEJc\nvnw5MjIyMjLSxcVlwoQJb731FnsjAFQ2BDsANk+/N8LX1/frr78ePny4o6Oj3EUBgAxYYwfA\nVj3quRGkOgCVFsEOgO1hbwQAlIqpWAC2RLc34vPPP8/Kyho7duyOHTueeeYZuUnTYbsAACAA\nSURBVIsCAGtBsANgG9gbAQCPRbADYO2OHTv2xRdfsDcCAB6LNXYArJdu5Vzbtm3ZGwEAZUGw\nA2B19HsjevTo4ezsnJCQwN4IACgLpmIBWIBGc++HHzQPHhg1F16/LoRwrFfPqF3l7Fx95Eih\nUmVmZkZGRn799dd5eXkTJkyIiYnx9vauoJoBwPYR7ACYnyYvL33FCs3Dh/qWwsLCzMxM+9u3\nhRDFtWt7enoazqiqqlb1HDhQ5eb27rvv7tq1a/r06ePHj/fw8JChdACwZQQ7AOancnNrfvCg\n/u33338/adIkLy+vuV5eQoiPVKqbFy5ERkaGhYUZHbhs2TIHBwd7e/sKLRcAlII1dgAsKy4u\n7rXXXlu4cOH58+e7devWrVu38+fPL1iw4LXXXouLizPq7OTkRKoDgKdGsANgWfPmzXvttdem\nTp2qUv33B45KpZo2bdrYsWPnzZsnb20AoDAEOwAW9ODBg0OHDo0cObLkRyNHjjx06NCDEhss\nAABPjWAHwIIyMjI0Go2Xl1fJj+rWravRaDIyMiq+KgBQKoIdAAuqUaOGg4PD1atXS370xx9/\nODg41KhRo+KrAgClItgBsCAnJ6cePXqsWrWq5EfffPNNz549nZycKr4qAFAqgh0Ay5o/f350\ndPSsWbP0y+kePHgwc+bM6Ojo+fPny1sbACgM97EDYFkdOnSIjo4eM2ZMZGTkoqpVhRAzvbyc\nnZ3/85//tG/fXu7qAEBRCHYAzK84O/u0r6/m/n3d27pCxGi1RcXFIj1dCBHr7OyQn283fHjq\n//qrXF1bnTplz6MmAKB8CHYAzM/ew6PhN98UZ2cbtRfdvSuEcCixYcLew4NUBwDlR7ADYE7p\n6enz5s2bM2eO1wsvyF0LAFQ6BDsAZpOSkjJo0CBPT0/2ugKALNgVC8A8oqKigoKCOnfuHB8f\nX61aNbnLAYDKiGAHoLyKi4tnzZoVFhY2d+5cSZJcXFzkrggAKimmYgGUy927d4cNG5aSkvLL\nL7/07t1b7nIAoFIj2AF4eqmpqQMHDvTw8EhMTGzcuLHc5QBAZcdULICnFBUV9dxzz3Xq1OnQ\noUOkOgCwBhYPdmlpaSNHjvTy8qpatWqzZs3Cw8Pz8vLKeGxsbOyAAQPq1Knj5OT0zDPPhISE\n7Nu3z4znB/B0WFQHANbJslOxJ0+eDA4OzsrK6tevX5MmTQ4cOBAREREbGxsXF+fs7Gz62Pfe\ne2/RokVOTk6dO3euU6fOnTt34uPjW7du3a1bN7OcH8DT0S+q27FjR69eveQuBwDw/1k22I0b\nNy4zM3PNmjVjxowRQmg0mlGjRkmStHjx4vDwcBMHrlmzZtGiRV26dNm8ebO3t7euUaPRZGRk\nmOX8AJ7OsWPHBg0axKI6ALBOdlqt1kKnTk5ODgwMDAgISElJ0Tdeu3atQYMG9erV++OPP+zs\n7Eo9sKCgoGHDhjk5OWlpaXXq1DH7+Z/UihUrJk2alJOT4+bmZpYTAjZKkqTXX3+9f//+q1ev\nZvoVQKVVUFDg5OQUHx//3HPPyV2LMQuusYuLixNC9OnTx7DR29vb39//zz//PHfunIkDb968\nOWDAALVavXHjxjlz5ixYsCA2NtYogz71+QE8Kd2iuldffZVFdQBgzSw4FXv27FkhRIsWLYza\nmzdvfuzYsXPnzpX8SCcxMVEIUaNGDX9///Pnz+vbu3Tpsm3bNv0Y3lOfH8ATYVEdANgKCwa7\nrKwsIYRarTZq9/T0FEJkZmY+6sDbt28LIZYtW9a0adO9e/e2b9/+0qVL06dP371797Bhw/bu\n3VvO8xtda9y4cQ8fPjTR59q1a0IIy81ZA9aMRXUAYENkuEGxLiGZWABXXFys6xAdHd2yZUsh\nROvWrbdt29a8efN9+/YdPXq0ffv25Tm/IRcXl7Zt2xYUFJjoY29vf+bMGXOt2ANsCIvqAMC2\nWDDY6cbSdONqhh410qane3x4y5YtdalOx9XVtXfv3t99950+2D31+Q25ubl99NFHpvusWLFi\n586dZTkboBjFxcXvv//+4sWL58+fP3PmTLnLAQCUiQWDnW6Jm24lnCHdsrnmzZubPlA3o2pI\n16KfNn3q8wMwjUV1AGCjLLgrtkePHkKImJgYw8br16+npqZ6e3ubCF49e/a0s7P7/fffCwsL\nDdtPnDghhNCv8nnq8wMw4dixYx06dEhPT09MTCTVAYBtsWCwa9euXceOHVNSUtatW6dr0Wg0\nM2bM0Gg0kyZNMlyytnbt2iVLluj2TAghvL29Bw4cmJ6eHhERoe+zffv2uLi4mjVr6n/TlP38\nAMpIkqSuXbt27tw5Pj6erRIAYHu0lnTixAm1Wq1SqUJCQt56663AwEAhRKdOnfLy8gy7+fj4\nCCESExP1LdeuXWvUqJEQokuXLpMnT+7Xr59KpXJ0dIyOjn6K85dTZGSkECInJ8eM5wSsTVFR\n0cyZMx0cHBYtWiR3LQBg1fLz84UQ8fHxchdSCguO2Akh/Pz8kpKSQkNDDx06tHz58oyMjNmz\nZ8fGxj72Qa716tVLTEycMmXK9evXV65cefjw4ZCQkEOHDoWEhJjl/AAM3b1798UXX/zmm292\n7NjBVgkAsF0WfKSYYvBIMSib7k51arV669atTL8CwGNV0keKAbB+LKoDACUh2AGVlOHjXzds\n2MD9hwFAAWR48gQA2XGnOgBQJIIdUOnoF9Xx+FcAUBimYoHKhUV1AKBgBDugsmBRHQAoHlOx\nQKXAojoAqAwIdoDysagOACoJpmIBhWNRHQBUHgQ7QLFYVAcAlQ1TsYAysagOACohgh2gQMeO\nHRs4cKCnpyeL6gCgUmEqFlAa3aK6Ll26sKgOACobgh2gHEVFRSyqA4DKjKlYQCHu3r0bGhp6\n7NgxFtUBQKVFsAOUgEV1AADBVCygACyqAwDoEOwAG8aiOgCAIaZiAVulX1QXExPTs2dPucsB\nAMiPYAfYJP2iuqNHjzZq1EjucgAAVoGpWMD2GC6qI9UBAPQIdoAtYVEdAMAEpmIBm5Genj5s\n2DAW1QEAHoVgB9gGFtUBAB6LqVjABmzYsIFFdQCAxyLYAVZNt6hu9OjRLKoDADwWU7GA9WJR\nHQDgiRDsACvFojoAwJNiKhawRrpFdc899xyL6gAAZUewA6yL4aK6H374gUV1AICyYyoWsCK6\nRXWpqaksqgMAPAWCHWAt9IvqEhMTmX4FADwFpmIBq8CiOgBA+RHsAJmxqA4AYC5MxQJyYlEd\nAMCMCHaAbFJSUgYNGsSiOgCAuTAVC8hjw4YNQUFBLKoDAJgRwQ6oaCyqAwBYCFOxQIViUR0A\nwHIIdkDFYVEdAMCimIoFKgiL6gAAlkawAyyORXUAgIrBVCxgWSyqAwBUGIIdYEG6RXXVqlVj\nUR0AoAIwFQtYin5R3cGDB0l1AIAKQLADzI9FdQAAWTAVC5hZenp6aGjo8ePHWVQHAKhgBDvA\nnFhUBwCQEVOxgNn88MMPLKoDAMiIYAeYgW5R3ZgxY1hUBwCQEVOxQHmxqA4AYCUIdkC5sKgO\nAGA9mIoFnp5uUV3Xrl1ZVAcAsAYEO+BpGC6qW79+PYvqAADWgKlY4InpF9Xt3LmzR48ecpcD\nAMB/EeyAJ8OiOgCA1WIqFngCLKoDAFgzgh1QJiyqAwBYP6ZigcdjUR0AwCYQ7IDHSElJGThw\nYPXq1VlUBwCwckzFAqb88MMPXbt2DQoKYlEdAMD6EeyA0ukX1c2bN49FdQAAm8BULFAKFtUB\nAGwRwQ4wxqI6AICNYioW+Av9orr4+HhSHQDAthDsgP8qLi6eMmXK2LFjP/nkk/Xr1zs7O8td\nEQAAT4apWOC/0tLSdu7cuWfPnueff17uWgAAeBoEOyiT5sGDq2+8obl/36i98M8/hRCO9esb\ntatcXZsuX37u3LkKqg8AAAuw+FRsWlrayJEjvby8qlat2qxZs/Dw8Ly8vMce1bJlS7sSvLy8\nnq4bKiE7BweHWrXsq1Uz/J/GzS332rXca9c0bm5GHznUqmXnwN85AADbZtnfZCdPngwODs7K\nyurXr1+TJk0OHDgQERERGxsbFxf32AVMKpUqLCzMsEWtVj91N1Q2do6O3p9+qn97/fr1KVOm\nREdHz9NqhRAffvfdgAEDli5dWq9ePflqBADAzCwb7MaNG5eZmblmzZoxY8YIITQazahRoyRJ\nWrx4cXh4uOljHR0d165d+9hLlLEbKrNbt24999xz3t7esbGxz6xeLYR4fty42bNnd+3a9ciR\nI3Xq1JG7QAAAzMOCU7HJyckJCQkBAQG6VCeEUKlUn332mUqlWrFihVartdylAUPh4eHVq1eP\njY3t1q2bg4ODg4NDt27dYmNjq1WrNmfOHLmrAwDAbCw4YhcXFyeE6NOnj2Gjt7e3v7//sWPH\nzp0716JFCxOHazSaBQsWpKWlOTs7+/v7Dx48uHr16k/dDZVWcXHxpk2bVq1aVbVqVcN2Z2fn\nmTNnTpw48d///re9vb1c5QEAYEYWDHZnz54VQpRMb82bNy9LsCssLHz//ff1b6dPn75y5crh\nw4c/XTdUWnfu3MnOzvb39y/5kb+/f1ZWVnp6OrOxAABlsGCwy8rKEqVtZfD09BRCZGZmmjh2\n9OjRHTp08PPzU6vVFy9ejIyMXL58eVhYWP369YODg5+0mwm3bt167bXXCgoKTPS5du2aEIK5\nYxulG6h78OBByY90jUYjeQAA2C4Z7u+gS0h2dnYm+rz33nv6176+vkuXLlWr1REREQsXLjRM\nbGXsZoKrq2v79u3z8/NN9LG3tz9z5ozpgmG1PD09mzVrtnPnzrZt2xp9tHPnzmbNmrGNGgCg\nGBYMdrrfl7pxO0OPGskzbdy4cREREQkJCWbppufm5vbhhx+a7rNixYqdO3eW8YSwQlOnTg0P\nD3/hhRcMs11KSsonn3wyf/58GQsDAMC8LBjsdEvodCvtDJ0/f14I0bx58yc6m24C1/TQWtm7\noVJ54403jh492rVr17CwsCHnzwshFkycuG7dutDQ0DfeeEPu6gAAMBsL3u6kR48eQoiYmBjD\nxuvXr6empnp7ez9psNu/f78QwsfHxyzdUKmoVKq1a9d+//33N2/ePH78+PHjx2/evLl+/fq1\na9eqVBZ/+AoAABXGgiN27dq169ixY0JCwrp161599VUhhEajmTFjhkajmTRpkuGStbVr12Zm\nZo4YMaJ27dpCiMTERCcnJ8NtjEePHp08ebIQwvAhE2XshsqpODv7jJ9fcW6uvqWpEB8JoSko\nEEL0PHBAHDiQOn68/lN7N7dnT5609/CQoVYAAMzEspsnVq9eHRQUNHbs2K1btzZu3PjAgQNJ\nSUmdOnWaPn26Ybf58+enpaUFBQXpgt3+/fv/+c9/+vj4NG7c2MPD49KlS8eOHdNqtf379586\ndar+qDJ2Q+Vk7+HRYOXK4pwco/ai9HQhhEPNmsb93d1JdQAAW2fZYOfn55eUlDRnzpw9e/bs\n2LGjfv36s2fPnj17tukHxfbs2XP8+PFHjhxJTk7Ozs729PTs1avXq6++OnLkSMNxvjJ2Q6Xl\n8eKLcpcAAECFsuP2bI+1YsWKSZMm5eTkuLm5yV0LAACQWUFBgZOTU3x8/HPPPSd3LcZYOQ4A\nAKAQBDsoX2pq6uHDh+WuAgAAiyPYQfneeeedjRs3yl0FAAAWR7CDwt24cWP//v1DhgyRuxAA\nACyOYAeFi4qK8vb2tsL1rQAAmB3BDgonSdKIESO4Aw4AoDIg2EHJ0tLSjh49Onz4cLkLAQCg\nIhDsoGQbNmxo0aKF4XPnAABQMIIdlGzjxo0jR46UuwoAACoIwQ6KdezYsVOnToWGhspdCAAA\nFYRgB8WSJKlTp07NmjWTuxAAACoIwQ7KpNVqN23axLYJAEClQrCDMsXHx//xxx+DBw+WuxAA\nACoOwQ7KJElS9+7dvb295S4EAICKQ7CDAhUVFW3ZsoV5WABAZUOwgwLt3r07MzNz4MCBchcC\nAECFIthBgSRJ6tOnT/Xq1eUuBACACkWwg9I8fPjwp59+Yh4WAFAJEeygND///HNRUVG/fv3k\nLgQAgIpGsIPSSJI0YMAAV1dXuQsBAKCiOchdAGBO2dnZMTExmzdvlrsQAABkwIgdFOXHH390\ndnbu3bu33IUAACADgh0URZKkoUOHVqlSRe5CAACQAcEOynH79u29e/eyHxYAUGkR7KAcGzdu\nrF27dlBQkNyFAAAgD4IdlEOSpBEjRqhU/FsNAKik+BUIhbhy5cqRI0eYhwUAVGYEOyjEDz/8\n4OPj065dO7kLAQBANgQ7KIQkSaNGjZK7CgAA5ESwgxKcOXPm5MmTQ4cOlbsQAADkRLCDEqxf\nvz4wMPDZZ5+VuxAAAOREsIMSbNq0iW0TAAAQ7GDzDh8+fPHixdDQULkLAQBAZgQ72DxJkoKD\ng+vXry93IQAAyIxgB9tWXFy8efNm5mEBABAEO9i6uLi4u3fvvvLKK3IXAgCA/Ah2sG2SJP39\n73+vWbOm3IUAACA/gh1sWH5+/rZt25iHBQBAh2AHG/bLL78UFBSEhITIXQgAAFaBYAcbJklS\n//793dzc5C4EAACr4CB3AcBTysnJ2b59e1RUlNyFAABgLRixg62Kjo52cnJ64YUX5C4EAABr\nQbCDrZIkafDgwU5OTnIXAgCAtWAqFjYpPT19z549MTExchcCAIAVYcQONmnTpk01a9b829/+\nJnchAABYEYIdbJIkSaGhofb29nIXAgCAFSHYwfZcvXr10KFD3JcYAAAjBDvYHkmSGjdu3KFD\nB7kLAQDAuhDsYHskSRo+fLidnZ3chQAAYF0IdrAxv//++7Fjx4YNGyZ3IQAAWB2CHWzMhg0b\n2rRp4+vrK3chAABYHYIdbMzGjRvZNgEAQKkIdrAliYmJ58+fDw0NlbsQAACsEcEOtkSSpOee\ne65Ro0ZyFwIAgDUi2MFmaDSaTZs2MQ8LAMCjlDXYff/99yUb3377bbMWA5iyf//+W7duDR48\nWO5CAACwUmUNdm+++eb27dsNW956660NGzZYoCSgdJIk9erVq06dOnIXAgCAlSprsJMkaeTI\nkQcOHNC9nTZtWlRU1N69ey1WGPAXhYWFW7duZR4WAAATyhrsXnrppWXLlvXv3//YsWNTp07d\nuHHj3r17W7VqZdHiAL2YmJj79++HhITIXQgAANbLoexdR40adffu3c6dO1erVm3v3r3PPvus\n5coCjEiS1K9fP7VaLXchAABYr8cEu1mzZhm11KpVq3379t99953u7aJFiyxSF2AgLy/v559/\n1v9bBwAASvWYYHf06FGjlhYtWuTk5JRsBywnOjpapVL16dNH7kIAALBqjwl2e/bsqZg6ABMk\nSRo0aJCzs7PchQAAYNWeYI0dIIuMjIxdu3b9/PPPchcCAIC1MxXsdAubUlJSsrKy1Gp127Zt\nX375ZRcXlworDhBCbN68Wa1W9+jRQ+5CAACwdo8Mdtu2bZswYUJ6erphY82aNVetWjVgwADL\nFwb8lyRJoaGhDg6MLgMA8Bil/7KMi4sbMmSISqUaPXp0cHBwnTp1bt26deDAgQ0bNgwePHj3\n7t3du3ev4EJROV2/fv3AgQMRERFyFwIAgA0oPdjNmzevSpUq8fHxbdu21TeOGzdu2rRpXbt2\nnTdvHsEOFSMqKsrb27tLly5yFwIAgA0o/ckTSUlJI0aMMEx1Om3bth0xYkRSUlLZL5CWljZy\n5EgvL6+qVas2a9YsPDw8Ly/vsUe1bNnSrgQvLy9znR+2QpKkESNG2NnZyV0IAAA2oPQROycn\np7p165b6Ud26dZ2cnMp49pMnTwYHB2dlZfXr169Jkya6ObXY2Ni4uLjH3rpCpVKFhYUZtpR8\n6kB5zg/rl5aWlpSUtHr1arkLAQDANpQe7IKDg+Pj40v9KD4+PigoqIxnHzduXGZm5po1a8aM\nGSOE0Gg0o0aNkiRp8eLF4eHhpo91dHRcu3at5c4P6/fDDz+0bNnS399f7kIAALANpU/FLlq0\nKDk5edasWbm5ufrG3NzcWbNmJScnl/ExYsnJyQkJCQEBAbrUJYRQqVSfffaZSqVasWKFVqst\nZ+mWPj9kt3HjxpEjR8pdBQAANqP0EbtPP/20devWn3zyyYoVK9q2bavbFZuSkpKZmRkUFPTp\np58adn7UuFpcXJwQwugxUN7e3v7+/seOHTt37lyLFi1MVKbRaBYsWJCWlubs7Ozv7z948ODq\n1aub8fywcikpKadPnw4NDZW7EAAAbEbpwU7/tPXMzMy9e/cafnTw4MGDBw8atjwq2J09e1YI\nUTJdNW/evCzBq7Cw8P3339e/nT59+sqVK4cPH26u88PKSZLUuXPnpk2byl0IAAA2o/Rgl5KS\nUv5TZ2VlidJ2PHh6egohMjMzTRw7evToDh06+Pn5qdXqixcvRkZGLl++PCwsrH79+sHBweU/\nv97NmzfHjh1bVFRkos+1a9fKciqYkVar3bRp0zvvvCN3IQAA2JLSg11AQIDlLqlb/Wb6Bhbv\nvfee/rWvr+/SpUvVanVERMTChQv1wa4859dzd3fv0qWL6Tuk2Nvbnzlzpixng7kcPHjw6tWr\ngwcPlrsQAABsiQUf06QbS9ONqxl61EibaePGjYuIiEhISDDv+V1dXefOnWu6z4oVK3bu3PlE\n1aKcJEnq0aNHvXr15C4EAABbYirY3bt37+DBg9euXcvPzzf66K233nrsqXVL3HQr4QydP39e\nCNG8efMnKlQ3wWpYiXnPD+tRVFT0448/Lly4UO5CAACwMY8MdgsXLvzoo48ePnxY6qdlCXY9\nevQQQsTExCxYsEDfeP369dTUVG9v7ycNXvv37xdC+Pj4WOj8sB67du3KzMwcMGCA3IUAAGBj\nSr+PXVRU1OzZs1u3bq17+Pr06dPnz5+vC1JDhgz5/vvvy3Lqdu3adezYMSUlZd26dboWjUYz\nY8YMjUYzadIkwzVwa9euXbJkye3bt3VvExMTjx8/bniqo0ePTp48WQhh+CyKsp8ftkWSpJde\nesno7jYAAODxtKUJCgqqU6dOXl7ejRs3hBA7duzQta9fv97e3j42NrbUo0o6ceKEWq1WqVQh\nISFvvfVWYGCgEKJTp055eXmG3XTjcImJibq3n332mRDCx8enV69egwYNatu2rS6l9e/fv6Cg\n4CnOX06RkZFCiJycHDOeE4/y4MEDDw+PjRs3yl0IAACl0y0Mi4+Pl7uQUpQ+YpeamtqvXz9n\nZ2ddotJoNLr2kSNH9unTRzeMVxZ+fn5JSUmhoaGHDh1avnx5RkbG7NmzY2NjTT/ItWfPnuPH\nj3dxcUlOTv7pp5+uXr3aq1ev77//Pjo62tHRsfznhzX76aefiouL+/btK3chAADYntLX2BUU\nFNSuXVsIUaVKFfHXnacBAQFLly4t+wV8fHw2bNhgus+FCxcM37Zt23blypVmPD9siCRJAwcO\ndHV1lbsQAABsT+kjdl5eXunp6UIIT09PNze3EydO6D+6fPlyxVSGSig7OzsmJsbw+SIAAKDs\nSg92bdq0OX36tBDCzs6uW7duK1asiI2Nzc3N3bp166ZNm/z9/Su2SFQWW7ZscXFx6dWrl9yF\nAABgk0oPdn379j106NCff/4phJg3b15eXl6vXr3c3d1feeWV4uLijz76qGKLRGUhSdLQoUN1\nCwAAAMCTKj3YTZgwQaPR1K9fXwjRvn37gwcPjhw5smvXrmFhYYcPH+7WrVuF1ojK4fbt2/v2\n7WMeFgCAp1amR4oFBgauX7/e0qWgkouKiqpbt25QUJDchQAAYKtKH7EDKp4kScOGDVOp+HcS\nAICnxC9RWIWLFy/+9ttvzMMCAFAexsHuu+++W7hwoe6WykKIuXPnNv2rd999t8KLhPJJktSi\nRYu2bdvKXQgAADbsL8Hu3Llzr7322tWrV52cnHQtt2/fTvurL7/88ty5c3KUCiWLiopiuA4A\ngHL6S7D77rvvtFptyTG5G/9z5MgRjUbz3XffVWCFUL4TJ06cPHly2LBhchcCAIBt+8uu2Li4\nuFatWjVp0sSok5eXl/6Fv7//vn37KqY4VBKSJLVv37558+ZyFwIAgG37y4jd2bNnfX19TR/Q\npEmTs2fPWrIkVC5arZZ5WAAAzOIvI3Y5OTnu7u6GLf/4xz9efPFFw5bq1atnZWVVRGmoHA4f\nPnzlypWhQ4fKXQgAADbvL8HO1dXVKLS1adOmTZs2hi1ZWVlG4Q8oD0mSnn/+ed1jTgAAQHn8\nZSq2UaNGSUlJpg9ISkpq2LChJUtCJVJcXLx582bmYQEAMIu/BLvu3btfunRp586dj+odExNz\n+fLl7t27W74wVAqxsbH37t175ZVX5C4EAAAl+EuwmzRpkkqleu21106fPl2y66lTp8aNG6dS\nqSZNmlRR5UHhJEl64YUXatSoIXchAAAowV/W2LVo0WLOnDkffvhhYGDgiBEjevfu/cwzz2i1\n2j///HP37t0bNmx4+PDhBx98wG0pYBYPHz6Mjo5etmyZ3IUAAKAQDkbv582bZ2dnN3/+/G+/\n/fbbb7/9S1cHhw8++GDu3LkVWB6U7JdffikoKOjfv7/chQAAoBDGwc7Ozm7evHmjRo1au3Zt\nfHz8jRs37OzsvLy8unbtOnbs2JL3LgaemiRJ/fv3d3Nzk7sQAAAUwjjY6fj4+Hz88ccVXAoq\nlZycnP/7v/+LioqSuxAAAJRD9fgugAVs27atatWqL7zwgtyFAACgHAQ7yEOSpMGDBzs5Ocld\nCAAAylH6VCxgUXfu3NmzZ8+uXbvkLgQAAEVhxA4y2LRpU61atZ5//nm5CwEAQFEIdpCBJEnD\nhg2zt7eXuxAAABSFYIeKdvXq1UOHDvF8WAAAzI5gh4q2YcOGJk2atG/fXu5CAABQGoIdKpok\nSSNGjLCzs5O7EAAAlIZghwr1+++/p6amhoaGyl0IAAAKRLBDhfrhhx8CUIUpdwAAIABJREFU\nAgJ8fX3lLgQAAAUi2KFCbdy4kW0TAABYCMEOFSchIeHChQtDhw6VuxAAAJSJYIeKI0lS165d\nGzVqJHchAAAoE8EOFUSj0WzevJl5WAAALIdghwqyb9++W7duDR48WO5CAABQLIIdKogkSb16\n9apdu7bchQAAoFgEO1SEgoKCrVu3Mg8LAIBFEexQEWJiYvLy8kJCQuQuBAAAJSPYoSJIkvTy\nyy+r1Wq5CwEAQMkIdrC4+/fv//zzz8zDAgBgaQQ7WFx0dLS9vf2LL74odyEAACgcwQ4WJ0nS\nK6+84uzsLHchAAAonIPcBUDhMjIydu/evX37drkLAQBA+Rixg2Vt2rTJ09Oze/fuchcCAIDy\nEexgWZIkhYaGOjgwNgwAgMUR7GBB169fP3jwIPthAQCoGAQ7WJAkSfXr1+/cubPchQAAUCkQ\n7GBBkiSNGDHCzs5O7kIAAKgUCHawlAsXLiQlJTEPCwBAhSHYwVJ++OGHZ599tnXr1nIXAgBA\nZUGwg6Vs2rRp1KhRclcBAEAlQrCDRSQnJ58+fXro0KFyFwIAQCVCsINFSJLUuXPnpk2byl0I\nAACVCMEO5qfVajdv3sy2CQAAKhjBDuZ34MCBq1evDh48WO5CAACoXAh2MD9Jknr27FmvXj25\nCwEAoHIh2MHMioqKtm7dyjwsAAAVj2AHM9u5c2dWVtbAgQPlLgQAgEqHYAczkyTppZde8vT0\nlLsQAAAqHYIdzCkvL+8///kP87AAAMiCYAdz+vnnn+3s7Pr27St3IQAAVEYEO5iTJEkDBgxw\ncXGRuxAAACojB7kLgHJkZmbGxMRs27ZN7kIAAKikGLGD2fz444/u7u69evWSuxAAACopgh3M\nRpKkIUOGODo6yl0IAACVFMEO5nHz5s19+/axHxYAABkR7GAeGzdurFevXteuXeUuBACAysvi\nwS4tLW3kyJFeXl5Vq1Zt1qxZeHh4Xl7eE51BdwcNOzu78PBwo49atmxpV4KXl5f5ykdZSZI0\nfPhwlYo/FQAAkI1ld8WePHkyODg4KyurX79+TZo0OXDgQERERGxsbFxcnLOzc1nOcOfOnfHj\nx7u5ueXm5pbaQaVShYWFGbao1WozlI4ncfHixYSEhMjISLkLAQCgUrNssBs3blxmZuaaNWvG\njBkjhNBoNKNGjZIkafHixSWH30o1YcIElUr19ttvf/zxx6V2cHR0XLt2rflKxtPYsGFDixYt\nAgIC5C4EAIBKzYITZ8nJyQkJCQEBAbpUJ4RQqVSfffaZSqVasWKFVqt97BnWrFkTHR29atWq\n6tWrW65OlF9UVBTbJgAAkJ0Fg11cXJwQok+fPoaN3t7e/v7+f/7557lz50wffvny5WnTpo0d\nO9b086k0Gs2CBQvGjRv35ptvrly58t69e+WvHE/k+PHjp06dGjZsmNyFAABQ2VlwKvbs2bNC\niBYtWhi1N2/e/NixY+fOnSv5kZ5Goxk9erSnp+eXX35p+iqFhYXvv/++/u306dNXrlzJ6FFF\nkiSpQ4cOzZs3l7sQAAAqOwsGu6ysLFHaVgZPT08hRGZmpoljFy9e/Ouvv+7atcv0TojRo0d3\n6NDBz89PrVZfvHgxMjJy+fLlYWFh9evXDw4OLkuRN27cGDt2bHFxsYk+165dK8upKietVhsV\nFTV16lS5CwEAAHI8K1a3us7Ozu5RHU6cODFnzpxJkyb17t3b9Knee+89/WtfX9+lS5eq1eqI\niIiFCxeWMdip1erevXsXFRWZ6PPbb7+dOXOmLGerhA4dOvTHH38MHTpU7kIAAIAlg51usE03\nbmfoUSN5OlqtNiwsrF69ep999tlTXHTcuHEREREJCQll7O/i4jJ9+nTTfVasWMGD7R9FkqS/\n/e1v3t7echcCAAAsuXlCt4ROt9LO0Pnz54UQj1qSVVxcnJqaeunSJXd3d/09h99++20hRERE\nhJ2d3euvv27iorp53vz8fLN8BZhWXFy8ZcsWVjQCAGAlLDhi16NHDyFETEzMggUL9I3Xr19P\nTU319vZ+VLBTqVTjxo0zajx16tSRI0cCAgICAwNNz7Hu379fCOHj41Pe6lEGe/bsuXfv3qBB\ng+QuBAAACGHRYNeuXbuOHTsmJCSsW7fu1VdfFUJoNJoZM2ZoNJpJkyYZrrFbu3ZtZmbmiBEj\nateurVKpvvnmG6NTLVmy5MiRI3379p0/f76+MTEx0cnJyd/fX99y9OjRyZMnCyGMnkUBC5Ek\n6cX/1969R1VZJXwcf86Ri0cREG8o9iLirQQ0KElDs8DUUPGCoOKNjIbeJtNxqqXZSh3B1muO\nznLGBi/plPpwUAHxmhcUHZAUEBSzVLxQy7uigoggh/cPZoxQEZLDPmef7+cv2zxn8+PRXD/3\nfi6DBrVo0UJ0EAAAoCjGvnli1apVfn5+4eHh8fHxbm5uBw8ezMzM9PX1rXZZ2/z58/Py8vz8\n/Fq3bl37yVNSUj766CN3d3c3Nzd7e/tz585lZ2dXVFQMGzaMmzQbQElJSWJi4ldffSU6CAAA\n+A/jvrLdw8MjMzMzNDQ0LS1t2bJlBQUFs2bN2rt3by1fFFszf3//iIiIJk2aZGVlJSUl/fzz\nzwEBAd9++21iYqK1tfWzz4+abdu2raysbOjQoaKDAACA/9DU5tVeFi4mJiYyMrKwsNDOzk50\nFhMSHBxsY2Ozfv160UEAAGhQpaWltra2qampffr0EZ2lOgHPsYME7ty5s337dr1eLzoIAAD4\nlXG3YiGrhISExo0bDxw4UHQQAADwK4odfg9VVUePHm1jYyM6CAAA+BVbsaiza9eu7d27d/fu\n3aKDAACA32DFDnWm1+tbtWpVy7fxAgCABkOxQ52pqjp27NhGjRqJDgIAAH6DYoe6yc/PP3To\nEO+HBQDABFHsUDfr16/v2LGjj4+P6CAAAKA6ih3qRlXVsLCwqq/6BQAAJoJihzo4efLksWPH\nQkNDRQcBAACPQbFDHaxbt+7FF1984YUXRAcBAACPQbFDHcTFxXHbBAAAJotih9r6/vvvz5w5\nM3r0aNFBAADA41HsUFuqqvr5+XXo0EF0EAAA8HgUO9SKwWDYuHEj+7AAAJgyih1qZd++fVeu\nXBk1apToIAAA4IkodqgVVVUHDBjQunVr0UEAAMATUezwdKWlpQkJCezDAgBg4ih2eLodO3YU\nFxcHBQWJDgIAAGpCscPTqao6bNgwe3t70UEAAEBNKHZ4irt3727dupV9WAAATB/FDk+RkJDQ\nqFGjQYMGiQ4CAACegmKHp1BVNTg4uHHjxqKDAACAp7ASHQAm7ebNm3v27Nm2bZvoIAAA4OlY\nsUNN4uLiHB0d+/fvLzoIAAB4OoodaqKq6pgxY6ysWNkFAMAMUOzwRBcvXvz3v//N/bAAAJgL\nih2eaP369c8995yvr6/oIAAAoFYodngiVVXDwsI0Go3oIAAAoFYodni8M2fOZGVlsQ8LAIAZ\nodjh8dauXevp6enh4SE6CAAAqC2KHR4vNjaW5ToAAMwLxQ6PkZmZeerUqTFjxogOAgAA6oBi\nh8dQVfWVV15xc3MTHQQAANQBxQ7VGQwGvV7PPiwAAGaHYofqDh48eOnSpZCQENFBAABA3VDs\nUJ2qqv7+/m3atBEdBAAA1A3FDr9RVla2ceNG9mEBADBHFDv8xnfffVdUVDR8+HDRQQAAQJ1R\n7PAbqqoGBgY6OjqKDgIAAOqMYodfFRcXJyUlsQ8LAICZotjhV0lJSRqNJjAwUHQQAADwe1Ds\n8CtVVUeMGKHT6UQHAQAAv4eV6AAwFQUFBd99911iYqLoIAAA4HdixQ7/sWnTJnt7+4CAANFB\nAADA70Sxw3+oqjp69GgrKxZxAQAwVxQ7KIqiXLp0KSUlhfthAQAwaxQ7KIqi6PX6du3avfrq\nq6KDAACA349iB0VRFFVVx40bp9FoRAcBAAC/H8UOSl5e3pEjR9iHBQDA3FHsoKiq2rVr1x49\neogOAgAAngnFDkpsbOy4ceNEpwAAAM+KYmfpcnJyTpw4MWbMGNFBAADAs6LYWTpVVXv16tW5\nc2fRQQAAwLOi2Fm0iooKvV7PbRMAAMiBYmfRUlNT8/PzR48eLToIAACoBxQ7i6aqav/+/V1c\nXEQHAQAA9YBiZ7kePHiwadMm9mEBAJAGxc5y7dmzp6CgYMSIEaKDAACA+kGxs1yqqg4aNKhF\nixaigwAAgPpBsbNQJSUlmzdvZh8WAACZUOws1NatWx88eDB06FDRQQAAQL2h2FkoVVWDgoKa\nNm0qOggAAKg3VqIDQIA7d+7s2LEjLi5OdBAAAFCfWLGzRPHx8Y0bN37zzTdFBwEAAPWJYmeJ\nVFUNCQmxsbERHQQAANQntmItzrVr15KTk/fs2SM6CAAAqGes2FkcvV7funVrPz8/0UEAAEA9\nM3qxy8vLCwsLc3Z2bty4cefOnWfPnl1cXFynGbZs2aLRaDQazezZs40xv6VRVXXs2LGNGjUS\nHQQAANQz4xa73Nzcl156SVXVXr16RUZG2tvbR0VF+fv737t3r5YzXLt2LSIiws7OzkjzW5r8\n/PxDhw7xXGIAAKRk3GI3ZcqUW7duff3110lJSUuWLDly5MjYsWPT09MXLVpUyxneffddrVY7\nffp0I81vadatW+fu7u7j4yM6CAAAqH9GLHZZWVmHDx/u2bPn5MmT//PNtNqFCxdqtdqYmJiK\nioqnzrB69erExMQVK1Y4OTkZY34LpKpqWFiY6BQAAMAojFjskpOTFUUZPHhw1UEXFxcvL69f\nfvnl1KlTNX/8/PnzH374YXh4eGBgoDHmt0AnT548fvx4SEiI6CAAAMAojFjsfvrpJ0VRunbt\nWm28S5cuiqLUXLwMBsOkSZMcHR0XL15sjPkt09q1a729vV944QXRQQAAgFEY8Tl2t2/fVhTF\nwcGh2rijo6OiKLdu3arhs4sWLTpw4MCuXbse/Xi9zP/QxYsXQ0JCSkpKajjm2rVriqJIsLcb\nFxf3hz/8QXQKAABgLAIeUFzZkDQazZMOOH78+GeffRYZGTlgwABjzF9V8+bNR40aVVpaWsMx\n33//fX5+fi0nNFnp6el5eXmjR48WHQQAABiLEYtd5Vpa5bpaVU9aaatUUVExYcKEdu3aLVy4\n0BjzV6PT6Z50y+1DMTExCQkJtZnNlKmq2rdvX1dXV9FBAACAsRjxGrvKq98qr4Sr6vTp08p/\nr4R7VHl5eU5Ozrlz55o1a6b5r8ruFRUVpdFo3nnnnWeZ3zIZDIaNGzfy+DoAAORmxBW7N954\nQ1GUnTt3RkdHPxy8ePFiTk6Oi4vLk4qXVqudMmVKtcETJ06kp6f37NnTx8enb9++zzK/ZUpO\nTr569erIkSNFBwEAAEZkxGLn7e3dq1evw4cPf/PNNxMnTlQUxWAwfPzxxwaDITIysuola2vW\nrLl169a4ceNat26t1WpXrlxZbaolS5akp6cHBgbOnz//d8wPVVXffPPN1q1biw4CAACMyLg3\nT6xatcrPzy88PDw+Pt7Nze3gwYOZmZm+vr4zZsyoetj8+fPz8vL8/Pzq2jxqOb+FKy0tTUxM\n/Nvf/iY6CAAAMC7jvlLMw8MjMzMzNDQ0LS1t2bJlBQUFs2bN2rt3r06nM4v55bB9+/bi4uJh\nw4aJDgIAAIxLI8Hj2YwtJiYmMjKysLDQzs5OdJbfIzQ0VKPRxMbGig4CAIAMSktLbW1tU1NT\n+/TpIzpLdcZdsYNwd+/e3bZtG/fDAgBgCSh2kktISLC2th40aJDoIAAAwOgodpJTVXXUqFG2\ntraigwAAAKMT8EoxNJjr16/v3r17x44dooMAAICGwIqdzDZs2NCiRYv+/fuLDgIAABoCxU5m\nqqqGhoY2atRIdBAAANAQKHbS+vnnn1NTU7kfFgAAy0Gxk1ZsbGyHDh169eolOggAAGggFDtp\nqao6duxY3pkLAIDloNjJ6ccffzx69OiYMWNEBwEAAA2HYicnVVW9vLw8PDxEBwEAAA2HYien\n2NhYbpsAAMDSUOwklJGRcfr0afZhAQCwNBQ7Camq2rt37w4dOogOAgAAGhTFTjYGgyEuLo59\nWAAALBDFTjYHDhy4dOnS6NGjRQcBAAANjWInG1VVAwIC2rRpIzoIAABoaBQ7qZSVlW3atIl9\nWAAALBPFTirffffd3bt3hw8fLjoIAAAQgGInFVVVAwMDHRwcRAcBAAACUOzkUVxcnJSUxD4s\nAAAWi2Inj82bN2u12rfeekt0EAAAIAbFTh6qqo4YMUKn04kOAgAAxLASHQD1o6CgYNeuXZs3\nbxYdBAAACMOKnSQ2btxob2/v7+8vOggAABCGYicJVVVDQkKsrFiCBQDAclHsZHDp0qUDBw5w\nPywAABaOYieD2NhYFxeXPn36iA4CAABEotjJQFXVcePGaTQa0UEAAIBIFDuzl5eXl5GRwT4s\nAACg2Jm99evXd+3a1cvLS3QQAAAgGMXO7On1+rCwMNEpAACAeBQ785adnX3ixInQ0FDRQQAA\ngHgUO/Omqqqvr2/nzp1FBwEAAOJR7MxYRUVFXFwct00AAIBKFDszlpqamp+fHxwcLDoIAAAw\nCRQ7M6aq6uuvv+7i4iI6CAAAMAkUO3P14MGDjRs3sg8LAAAeotiZq927d9+6dWvEiBGigwAA\nAFNBsTNXqqoOHjzYyclJdBAAAGAqKHZmqaSkJCkpiX1YAABQFcXOLG3ZsuXBgwdDhgwRHQQA\nAJgQip1ZUlV1+PDhTZs2FR0EAACYECvRAVBnd+7c2blz54YNG0QHAQAApoUVO/OzadMmnU43\nYMAA0UEAAIBpodiZH1VVQ0JCbGxsRAcBAACmhWJnZq5evbpv3z7uhwUAAI+i2JkZvV7funVr\nPz8/0UEAAIDJodiZGVVVx40bp9XyGwcAAKqjH5iTCxcupKensw8LAAAei2JnTtatW+fu7u7t\n7S06CAAAMEUUO3Oiqur48eNFpwAAACaKYmc2Tp48mZubGxISIjoIAAAwURQ7s7F27VofH5/n\nn39edBAAAGCiKHZmIy4ujtsmAABADSh25uHQoUNnz54NDQ0VHQQAAJguip15UFW1b9++7du3\nFx0EAACYLoqdGSgvL9+wYQP7sAAAoGYUOzOQnJx848aN4OBg0UEAAIBJo9iZAVVV33zzzRYt\nWogOAgAATBrFztTdv38/ISGBfVgAAPBUFDtTt3379tLS0qCgINFBAACAqaPYmTpVVYcNG2Zn\nZyc6CAAAMHVWogOgJoWFhVu3bo2NjRUdBAAAmAFW7ExaYmKira3twIEDRQcBAABmgGJn0lRV\nDQ4OtrW1FR0EAACYAbZiTdf169f37Nmzc+dO0UEAAIB5YMXOdMXFxbVs2fK1114THQQAAJgH\noxe7vLy8sLAwZ2fnxo0bd+7cefbs2cXFxTV/pLy8fN68eYMHD3Z1dW3SpImTk9OLL744d+7c\nmzdvVjuyW7dumkc4Ozsb7adpUKqqhoaGNmrUSHQQAABgHoy7FZubm9u3b9/bt28PGTKkY8eO\nBw8ejIqK2rt3b3Jysk6ne9KnysrKPv/8c2dn5y5duvTq1auoqCgzM3POnDnLly9PS0tzdXWt\nerBWq50wYULVEQcHB2P9PA3o559/TktLW7RokeggAADAbBi32E2ZMuXWrVurV6+ePHmyoigG\ng2H8+PGqqi5atGj27NlP+pStre358+erFrjS0tK333573bp1UVFRy5cvr3qwtbX1mjVrjPUD\niKOqqpub28svvyw6CAAAMBtG3IrNyso6fPhwz549K1udoiharXbhwoVarTYmJqaiouJJH9Ro\nNNWW5WxsbCIiIhRFOX36tPECmxRVVceOHavRaEQHAQAAZsOIK3bJycmKogwePLjqoIuLi5eX\nV3Z29qlTp7p27Vr72TZt2qQoSo8ePaqNGwyG6OjovLw8nU7n5eUVHBzs5OT0zNkF+/HHH7Oz\ns9euXSs6CAAAMCdGLHY//fSToiiPtrcuXbrUsthNmzatpKTk9u3bGRkZZ86c8fLy+vTTT6sd\nU1ZWVnVwxowZy5cvHzt2bH38BMKsX7++R48e3bt3Fx0EAACYEyMWu9u3byuPu5XB0dFRUZRb\nt249dYaVK1fevXu38teDBg1as2ZNq1atqh4wadKkl19+2cPDw8HB4ezZs//85z+XLVs2YcKE\n9u3b9+3btzYhL168GBwcXFpaWsMx169fr81U9Uiv17/99tsN/E0BAIC5E/CA4sqr62pz9VhR\nUVFFRcWVK1dSUlI++eSTnj17btu2zdvb++EBM2fOfPjr7t27L1261MHBISoqasGCBbUsdk5O\nTiEhIffv36/hmLNnzy5fvtzGxqY2Ez67I0eOnD59OjQ0tGG+HQAAkIYRi13lWl3lul1VT1rJ\ne6zK59KFhoZ6eHh4eHiEh4fn5OTUcPyUKVOioqIOHz5cy5CNGzeeNm1azcekpaVVuxXXqFRV\n7dOnT4cOHRrsOwIAADkY8a7YykvoKq+0q6ryztYuXbrUabbu3bu3bdv22LFjBQUFNRxWuc9b\n8wqcKTMYDHFxceZ+jSAAABDCiMXujTfeUBSl2qtOL168mJOT4+LiUtdiV1hYePXqVUVRrKxq\nWmVMSUlRFMXd3b3OcU1DSkrKlStXgoODRQcBAADmx4jFztvbu1evXkePHv3mm28qRwwGw8cf\nf2wwGCIjI6teY7dmzZolS5ZU9jZFUdLT06vtt964cWPixInl5eX9+vVr1qxZ5eCRI0eOHTtW\n9bCMjIz3339fUZRq76IwI6qqBgQEtGnTRnQQAABgfox788SqVav8/PzCw8Pj4+Pd3NwOHjyY\nmZnp6+s7Y8aMqofNnz8/Ly/Pz8+vdevWiqLs379/5syZHTt2dHNza968+eXLlzMzM+/du9e2\nbduYmJiHn0pJSfnoo4/c3d3d3Nzs7e3PnTuXnZ1dUVExbNiwqVOnGvXnMpKysrL4+Pi//vWv\nooMAAACzZNxi5+HhkZmZ+dlnn+3Zs2fHjh3t27efNWvWrFmzanhRrKIoQUFB169f379/f05O\nTkFBgZ2dnaen51tvvTV16tTmzZs/PMzf3z8iIiI9PT0rK+vOnTuOjo4BAQETJ04MCwsz0xc2\n7Ny58+7du0FBQaKDAAAAs6Sp4dVeqJSWlvbqq6/ev3/f2E88GTduXFlZ2YYNG4z6XQAAwLMo\nLS21tbVNTU3t06eP6CzVGfEaO9RJcXHxli1buB8WAAD8bhQ7U5GYmKjVaqu9WhcAAKD2KHam\nQlXVkSNH1nz1IQAAQA0EvFIMjyooKNi1a9eWLVtEBwEAAGaMFTuTsGHDBgcHh8pHOgMAAPw+\nFDuToKpqaGhozS/VAAAAqBnFTryLFy8ePHiQ+2EBAMAzotiJFxsb6+Li0rt3b9FBAACAeaPY\niaeqqvm+LQMAAJgOip1geXl5GRkZ7MMCAIBnR7ETbN26dc8//7ynp6foIAAAwOxR7ATT6/Vh\nYWGiUwAAABlQ7EQ6evToDz/8EBoaKjoIAACQAcVOpLi4OF9f306dOokOAgAAZMATcUXq3bv3\ngAEDRKcAAACSoNiJNGzYMNERAACAPCh2xmK4d+/n//1fw9271cbLfvlFURTr9u2rjWubNn1u\n2TKtTtdA+QAAgHQodsaisbKyatWq3Mam2vi9nBxFURo/8nyTRg4OGt4VCwAAngFNwlg01tYu\n//d/j45fCA9XFOV/YmIaPBEAAJAcd8UCAABIghW7hpOZmbl27doXdu1SFOWH6dPHjx/v4+Mj\nOhQAAJAHK3YNZN68eb6+vrm5uS1btmzZsmVubq6vr++8efNE5wIAAPJgxa4h6PX66OjozZs3\nBwYGVl5jt3v16q1btwYHB3ft2pU3TwAAgHrBil1DWLBgwbRp0wIDA6sODhkyZNq0aQsWLBCV\nCgAASIZiZ3SFhYU5OTkjRox49EvDhw/PyckpKipq+FQAAEA+FDujq+xtjo6Oj36pefPmiqIU\nFhY2dCYAACAjip3RtWzZUqfTnT59+tEvnTp1SqfTtWrVquFTAQAA+VDsjM7a2nrIkCFLliwx\nGAxVxw0Gw5IlS4YMGWLFCycAAEB9oNg1hOjo6KysrDFjxuTn51eO5OfnjxkzJjs7Ozo6Wmw2\nAAAgDYpdQ+jUqdP+/fvPnDnj6uqq1+v1er2rq+uZM2f27dvXqVMn0ekAAIAk2ARsIF5eXpmZ\nmbm5uTc++EBRlGNLl3p4eGg0GtG5AACAPCh2DUej0Xh6el5wc1MUxdXTU3QcAAAgG7ZiAQAA\nJMGKnbGU37nzw/PPG+7dqzZuKC5WFOXW5s3VxrU63QsnTzayt2+gfAAAQDoUO2NpZG/vFhdX\n8Uixe3DzpqIoVk5O1cY1Oh2tDgAAPAuKnRHZvfqq6AgAAMCCcI0dAACAJCh2AAAAkqDYAQAA\nSIJiBwAAIAmKHQAAgCQodgAAAJKg2AEAAEiCYgcAACAJih0AAIAkKHYAAACSoNgBAABIgmIH\nAAAgCYodAACAJCh2AAAAkqDYAQAASIJiBwAAIAmKHQAAgCQodgAAAJKg2AEAAEiCYgcAACAJ\nK9EBzICNjY2iKLa2tqKDAAAAU1FZD0yNpqKiQnQGM5CTk/PgwYN6mWr27NnFxcURERH1Mhvq\nasWKFYqicP5F4fyLxfkXi/Mv1ooVK5o0aTJ//vx6mc3KyqpHjx71MlX9YsWuVurxN8/Z2VlR\nlPHjx9fXhKiTvXv3Kpx/cTj/YnH+xeL8i1V5/n18fEQHMS6usQMAAJAExQ4AAEASFDsAAABJ\nUOwAAAAkQbEDAACQBMUOAABAEhQ7AAAASVDsAAAAJEGxAwAAkARvnmhopvlqOcvB+ReL8y8W\n518szr9YFnL+eVdsQysoKFAUpXnz5qKDWCjOv1icf7E4/2Jx/sWykPNPsQMAAJAE19gBAABI\ngmIHAAAgCYodAACAJCh2AAAAkqDYAQAASIJiBwAAIAmKHQAAgCRZoVnDAAAKHElEQVQodgAA\nAJKg2AEAAEiCYgcAACAJih0AAIAkKHYAAACSoNgBAABIgmIHAAAgCYodAACAJCh2DSQ+Pv6D\nDz549dVX7ezsNBrNmDFjRCeyIEVFRXq9fuzYsc8//3yTJk0cHBz8/PxWrlxpMBhER7MI5eXl\n8+bNGzx4sKura5MmTZycnF588cW5c+fevHlTdDRLtGXLFo1Go9FoZs+eLTqLpejWrZvmEc7O\nzqJzWZa9e/cOHz68TZs2tra2zz33XFBQ0P79+0WHMgor0QEsRXR0dGZmpr29vYuLy6lTp0TH\nsSwrV66cPn26jY2Nt7e3p6fnlStX0tLSUlNTt2zZkpCQoNXyzxvjKisr+/zzz52dnbt06dKr\nV6+ioqLMzMw5c+YsX748LS3N1dVVdEALcu3atYiICDs7u6KiItFZLItWq50wYULVEQcHB1Fh\nLNDMmTO/+OILW1vbV155pU2bNteuXUtNTfX09Ozfv7/oaPWPYtdAvvzyy/bt27u7u2/btm3o\n0KGi41iW5557btmyZePGjXv4N+kPP/zw+uuvJyUlVa7kiY0nPVtb2/Pnz1ctcKWlpW+//fa6\ndeuioqKWL18uMJuleffdd7Va7fTp0//yl7+IzmJZrK2t16xZIzqFhVq9evUXX3zRu3fvDRs2\nuLi4VA4aDIaCggKxwYyEtYoG0r9//06dOmk0GtFBLNGoUaPee++9qv8+fuGFF6ZPn64oSkpK\nirhclkKj0VRblrOxsYmIiFAU5fTp04JCWaLVq1cnJiauWLHCyclJdBaggZSWls6aNatp06YJ\nCQkPW52iKFqttkWLFgKDGQ8rdrBQlT3P1tZWdBALtWnTJkVRevToITqIpTh//vyHH34YHh4e\nGBi4ZMkS0XEsjsFgiI6OzsvL0+l0Xl5ewcHB1OuGkZycfPny5bCwMAcHB71en5ubq9PpfH19\n33jjDVmXWih2sEQVFRXffPONoihsizekadOmlZSU3L59OyMj48yZM15eXp9++qnoUBbBYDBM\nmjTJ0dFx8eLForNYqLKysqp/2mfMmLF8+XKuA2kAR44cURSlRYsWXl5eVbcIevfunZCQ0KZN\nG3HRjIWtWFiiuXPnpqenjxw5MiAgQHQWC7Jy5cqYmJjY2NgzZ84MGjRo165drVq1Eh3KIixa\ntOjAgQOrVq3ign0hJk2atHv37kuXLhUXF+fm5v7xj38sLi6eMGHCwYMHRUeT39WrVxVF+cc/\n/qHVavft21dYWHjs2LEBAwYcOnRI1sdTUOxgcf7+97/PnTvX29t79erVorNYlqKiIoPBcOnS\npdjY2JMnT/bs2TMrK0t0KPkdP378s88+i4yMHDBggOgsFmrmzJkBAQHOzs46na579+5Lly6d\nOXNmeXn5ggULREeTX3l5uaIoGo0mMTGxf//+dnZ2np6eCQkJ7dq1279/f0ZGhuiA9Y9iB8uy\naNGiDz74wMfHZ8+ePfb29qLjWJzKx3eFhoZu27bt8uXL4eHhohNJrqKiYsKECe3atVu4cKHo\nLPjVlClTFEU5fPiw6CDya968uaIo3bp169at28PBpk2bVv47h2IHmLc5c+b8+c9/7t279969\neyv/b4co3bt3b9u27bFjx2R94oCJKC8vz8nJOXfuXLNmzR4+GrfylvCoqCiNRvPOO++IzmiJ\nHB0dFUW5f/++6CDy69q1q/LfE15V5UhJSYmATEbGzROwFH/6058WL17cv3//LVu22NnZiY5j\n6QoLCyuvfbGy4m8hI9JqtZWLQ1WdOHEiPT29Z8+ePj4+ffv2FRLMwlU+aMnd3V10EPn5+/tr\nNJoff/yxrKzM2tr64fjx48cVRXFzcxMXzVj4KxXyMxgMkZGRK1asGDhwYEJCgk6nE53IsqSn\np+t0uqpPNrlx48Y777xTXl7er1+/Zs2aCcwmPa1Wu3LlymqDS5YsSU9PDwwMnD9/vpBUFuXI\nkSO2trZeXl4PRzIyMt5//31FUaq9iwLG4OLiMmLEiPj4+KioqDlz5lQObt26NTk5uWXLllLe\nP0exayDx8fFJSUmKovzyyy+Konz//feTJ09WFKVly5Zffvml2GzSW7Ro0YoVK7RarZOT03vv\nvVf1S56enjNmzBAVzELs379/5syZHTt2dHNza968+eXLlzMzM+/du9e2bduYmBjR6QDjSklJ\n+eijj9zd3d3c3Ozt7c+dO5ednV1RUTFs2LCpU6eKTmcRli5dmpWVNXfu3F27dnl7e1+4cGH7\n9u3W1tYrV65s2rSp6HT1j2LXQLKysv71r389/M/z58+fP39eURRXV1eKnbHduHFDURSDwaCq\narUvDRw4kGJnbEFBQdevX9+/f39OTk5BQUHlXWlvvfXW1KlTudIR0vP394+IiEhPT8/Kyrpz\n546jo2NAQMDEiRPDwsJkfUCuqWnXrt2RI0fmzZuXlJSUkZFhb28fFBQ0a9asl156SXQ0o9BU\nVFSIzgAAAIB6wF2xAAAAkqDYAQAASIJiBwAAIAmKHQAAgCQodgAAAJKg2AEAAEiCYgcAACAJ\nih0AAIAkKHYAAACSoNgBAABIgmIHAAAgCYodAACAJCh2AAAAkqDYAQAASIJiBwAAIAmKHQAA\ngCQodgAAAJKg2AEAAEiCYgcAACAJih0AAIAkKHYAAACSoNgBAABIgmIHAAAgCYodAACAJCh2\nAAAAkqDYAQAASIJiBwAAIAmKHQAAgCQodgAAAJKg2AEAAEiCYgcAACAJih0AAIAkKHYAUJPs\n7GyNRjN58mTRQQDg6Sh2AAAAkqDYAQAASIJiBwAAIAmKHQDUjcFgmDp1qkajGTlyZElJieg4\nAPArih0A1EFJSUlISMjSpUvff//9jRs3Nm7cWHQiAPiVlegAAGA2bt68GRQUlJqa+sUXX3zy\nySei4wBAdRQ7AKiVCxcuDBo0KC8v79tvvw0LCxMdBwAeg2IHAE/3008/9e7d++7duzt27PD3\n9xcdBwAej2vsAODpTp06denSpY4dO3p7e4vOAgBPRLEDgKcbOnRodHR0dna2v7//9evXRccB\ngMej2AFArcycOXPx4sVHjx59/fXXr1y5IjoOADwGxQ4AamvatGlfffXViRMnXnvttYsXL4qO\nAwDVUewAoA4iIyO//vrr06dP9+vXLz8/X3QcAPgNih0A1M3kyZPXrl174cKFfv36nT17VnQc\nAPiVpqKiQnQGAAAA1ANW7AAAACRBsQMAAJAExQ4AAEASFDsAAABJUOwAAAAkQbEDAACQBMUO\nAABAEhQ7AAAASVDsAAAAJEGxAwAAkATFDgAAQBIUOwAAAElQ7AAAACRBsQMAAJAExQ4AAEAS\nFDsAAABJUOwAAAAkQbEDAACQBMUOAABAEhQ7AAAASVDsAAAAJEGxAwAAkATFDgAAQBIUOwAA\nAElQ7AAAACRBsQMAAJAExQ4AAEASFDsAAABJUOwAAAAkQbEDAACQxP8Daan6YwwE+dgAAAAA\nSUVORK5CYII="},"metadata":{"image/png":{"width":420,"height":420}}}]},{"metadata":{"trusted":false},"cell_type":"code","source":"############ Plotting the clusters\nwithr::with_options(repr.plot.width=12, repr.plot.height=12)\nplottSNE(FG)","execution_count":5,"outputs":[{"output_type":"display_data","data":{"text/plain":"plot without title","image/png":"iVBORw0KGgoAAAANSUhEUgAABaAAAAWgCAIAAAAnwnOfAAAACXBIWXMAABJ0AAASdAHeZh94\nAAAgAElEQVR4nOzdaXRb9bnv8Wdr9CB5iOekCSUDhdOS5La0UEKgNKdddJVcTgOh0J50OKyu\nwuq9q+euDj4HkpN7ubfFqQslpLRNepqQlMmkJSnEYR56EghjWlKgNCaDhziW5UG2Zdma9r4v\ndrqzI8mObEuytvl+Xklb0tZfimGt/dPzfx5F0zQBAAAAAACwMtt0LwAAAAAAAGCqCDgAAAAA\nAIDlEXAAAAAAAADLI+AAAAAAAACWR8ABAAAAAAAsj4ADAAAAAABYHgEHAAAAAACwPAIOAAAA\nAABgeQQcAAAAAADA8gg4AAAAAACA5RFwAAAAAAAAyyPgAAAAAAAAlkfAAQAAAAAALI+AAwAA\nAAAAWB4BBwAAAAAAsDwCDgAAAAAAYHkEHAAAAAAAwPIIOAAAAAAAgOURcAAAAAAAAMsj4AAA\nAAAAAJZHwAEAAAAAACyPgAMAAAAAAFgeAQcAAAAAALA8Ag4AAAAAAGB5BBwAAAAAAMDyCDgA\nAAAAAIDlEXAAAAAAAADLI+AAAAAAAACWR8ABAAAAAAAsj4ADAAAAAABYHgEHAAAAAACwPAIO\nAAAAAABgeQQcAAAAAADA8gg4AAAAAACA5RFwAAAAAAAAyyPgAAAAAAAAlkfAAQAAAAAALI+A\nAwAAAAAAWB4BBwAAAAAAsDwCDgAAAAAAYHkEHAAAAAAAwPIIOAAAAAAAgOURcAAAAAAAAMsj\n4AAAAAAAAJZHwAEAAAAAACyPgAMAAAAAAFgeAQcAAAAAALA8Ag4AAAAAAGB5BBwAAAAAAMDy\nCDgAAAAAAIDlEXAAAAAAAADLI+AAAAAAAACWR8ABAAAAAAAsj4ADAAAAAABYHgEHAAAAAACw\nPAIOAAAAAABgeQQcAAAAAADA8gg4AAAAAACA5RFwAAAAAAAAyyPgAAAAAAAAlkfAAQAAAAAA\nLI+AAwAAAAAAWB4BBwAAAAAAsDwCDgAAAAAAYHkEHAAAAAAAwPIIOAAAAAAAgOURcAAAAAAA\nAMsj4AAAAAAAAJZHwAEAAAAAACyPgAMAAAAAAFgeAQcAAAAAALA8Ag4AAAAAAGB5BBwAAAAA\nAMDyCDgAAAAAAIDlEXAAAAAAAADLI+AAAAAAAACWR8ABAAAAAAAsj4ADAAAAAABYHgEHAAAA\nAACwPAIOAAAAAABgeQQcAAAAAADA8gg4AAAAAACA5RFwAAAAAAAAyyPgAAAAAAAAlkfAAQAA\nAAAALI+AAwAAAAAAWB4BBwAAAAAAsDwCDgAAAAAAYHkEHAAAAAAAwPIIOAAAAAAAgOURcAAA\nAAAAAMsj4AAAAAAAAJZHwAEAAAAAACyPgAMAAAAAAFgeAQcAAAAAALA8Ag4AAAAAAGB5jule\ngDW89dZbsVhsulcBAAAAAMA0czgcS5Ysme5VpEDAcXZvvPHGJz/5yeleBQAAAAAAeeH111+/\n6KKLpnsViQg4zi4SiYhIOBx2uVzTvRYAAAAAAKZNJBJxu936ZXK+oQcHAAAAAACwPAIOAAAA\nAABgeQQcAAAAAADA8gg4AAAAAACA5RFwAAAAAAAAyyPgAAAAAAAAlkfAAQAAAAAALI+AAwAA\nAAAAWB4BBwAAAAAAsDwCDgAAAAAAYHkEHAAAAAAAwPIIOAAAAAAAgOURcAAAAAAAAMsj4AAA\nAAAAAJZHwAEAAAAAACyPgAMAAAAAAFgeAQcAAAAAALA8Ag4AAAAAAGB5BBwAAAAAAMDyCDgA\nAAAAAIDlEXAAAAAAAADLI+AAAAAAAACWR8ABAAAAAAAsj4ADAAAAAABYHgEHAAAAAACwPAIO\nAAAAAABgeQQcAAAAAADA8gg4AAAAAACA5RFwAAAAAAAAyyPgAAAAAAAAlkfAAQAAAAAALI+A\nAwAAAAAAWB4BBwAAAAAAsDwCDgAAAAAAYHkEHAAAAAAAwPIIOAAAAAAAgOURcAAAAAAAAMsj\n4AAAAAAAAJZHwAEAAAAAZ3do5NCXwl+aH5/v0TxOcc6VuTfJTX7xT/e6AJzimO4FAAAAAEBe\ni0QibW1tDxY/uLtut3GwQzq2ytZe6d0tu8d5LYCcIeAAAAAAgDHF4/Fjx45Fo9HSgtLG9sYL\nRi8ojBc+OuvRe6vvFZEWaZnuBQI4hYADAAAAAMbU29v7vvL+z+f+/N3Cd3scPWElXBmr/NjI\nx/RHL5FLpnd5AAwEHAAAAACQ2ujoaE9Pz77yfU+WPmkc9Dl9PqdPRKqj1T+I/kCKpm99AExo\nMgoAAAAAKUSj0ePHj6uqWhovbWxv3NOy59zwucajBWrBziM7Z8dmT+MKAZhRwQEAAAAAKfj9\n/lgsJiIrAytFJKJE+h39ds0eV+IiMmobvbvm7t/afzvNqwTwd1RwAAAAAEAKQ0ND5rsOcdx/\n5P5Lhi8REUUUEdlTtqewsHB6FgcgCRUcAAAAAJCCXr6hW7VwVUvBqYEpi0cWt7naAvaAV/Pa\nbPxmDOQL/msEAAAAgBTsdrt+4+GKh9td7Xbt1N1DhYcC9oCIXG+7floW1iItN8gNi2SRV7xO\ncc6VuTfJTX7xT8tigPxBBQcAAAAApFBcXDwwMCAi+zz7Rm2jCY8ulaV3yB05XlI0GvX7/dud\n25uqmoyDHdKxVbb2Su9u2Z3j9QB5hYADAAAAAFKoqqoaHBzUNO0ToU/0OnrbXe0jtpG4EldF\n9WieA8qBAinI5XpGRkaOHz8ej8eLy4ob2xsvGL2gMF746KxH762+V0RapCWXiwHyEAEHAAAA\nAKRQUFDw7IJnjw4eXTa07Jr+a3odvdsrtz9W9piI/LPyzzlONzRNa29vj8fj8vepLroVgyv0\ngOMSuSSX6wHyEAEHAAAAAKT2TMEzzQXN91TfYz44LZtTgsFgJBIxH1EVtd3ZflftXSJSp9at\nt63P8ZKAfEPAAQAAAACpLZflPvEdkSPDMlwjNQtl4RpZc6PcmOPyDREZHT2jCcgZU11Ci7cH\nts+bPS/HSwLyDQEHAAAAAKRWL/X1Uj/dqxAR0TTNuB1RIn6n36E5YkpMRA4VHWrQGh6Wh6dv\ndUBeIOAAAAAAgHzncrlO39Zc+/66TxPtjeI3bvnwLWEl/GjRo9O4NiBP2KZ7AQAAAAAwY7VI\nyw1ywyJZ5BWvU5xzZe5NcpNf/BM9j9frtdvt5iOKKJ8IfaJILRKREinJ2IoBy6KCAwAAAACy\nZVd4V5O7ybjbIR1bZWuv9O6W3RM6j91unz179l3Dd/Xb+y8LXlYTrdGnuvTb+0VktbI6w+sG\nLIiAAwAAAAAyLx6Pt7W1xZ3xRq3xgtELCuOFj856VB/p2iItkzhhaWnpa57XnrQ/ualmk/n4\ntEx1AfIQAQcAAAAAZF5bW9vw8PBKWWkcWTG4Qg84LpFLJnfOz9g/0yM9+TDVBchDBBwAAAAA\nkGHBYHB4eNi4qypqu7P9rtq7RKQ6Wv2D6A+kaDKnzZ+pLkAeIuAAAAAAgAwzpxurFq5qKTi1\nJ2VxaPGmtk3l5eWTCzgAjIMpKgAAAACQYbFYTL8RUSJ+p9+hnfpp+VDRobtr7o7H49O3NGDG\nIuAAAAAAgAxzOE4lGi7Nte+v+w6+c3Drsa1uzS0ie8r2GI+mL1PjZoEZjIADAAAAADLM6/Wa\n7yqifCL0iSK1SEQ8qsfj8UzobIFAYEfPjiZpel/eD0owJjF93Oy35FuTWBtZCWYqenAAAAAA\nQIYVFRU9Pvvxk9GTlwUvq4nW9Dp6t1du77f3i8gXR75Y5JlAB46TJ0/29vYWlRU1tmdg3Gwg\nENgR29FU2WQc0bOSXundLbsnejYgrxBwAAAAAEDm7S/fv1fZu6lmk/ngR6Mf3Vi0Mf2TBIPB\n3t5eEVkZyMC42cxmJUC+yestKs8999w//dM/1dTUuN3uuXPnXnPNNS+++GLCc44cOfLVr361\ntra2oKBg0aJFa9euDYVC07FYAAAAADjtcuXyi+SiMq3MJa7Z8dmXRS/bHN38hvONWbZZ6Z+k\nr6/PfFdV1FZXqzFu9tborenvNzFnJVcNXHVO+JzqWPWKwRX6oxPNSoA8lL8VHP/+7//e0NDg\ndrsvueSSmpoav9//0ksvXXjhhZ/5zGeM57z99tvLly8fGBi4+uqr58+fv2/fvh/96EfPPffc\n888/X1hYOH1rBwAAAPBBVy/19VIvioiI2EXskzlJOBw2biePm62orfil65dNRWntN0nOStqd\n7aezErlVnJNZIZA/8jTg2LZtW0NDw6c//emdO3fOmTNHP6iqan9/v/lpN910UyAQ2LZt2ze+\n8Q39Cf/8z//80EMP3XnnnWvXrs39sgEAAAAgG4xxszElJn8fN/v/Ov+flEhjb1r7TcbPSqpm\nVxFwwOrycYtKJBK59dZbi4uLd+3aZaQbImKz2SoqKoy7Bw8efO2115YuXaqnG/oTGhsbbTbb\n5s2bNU3L8bIBAAAAILPcbrd+I+W4WVVVJ7HfxMhK9Lt6VpLNDwHkSD5WcDz//PNdXV1f/epX\nS0tLm5qa3n777cLCwosvvvizn/2soijmp4nIF77wBfNr58yZs3jx4j//+c+HDx/+yEc+kuul\nAwAAAEDmlJeXDw4OGneNcbNhe9ijnp41a95vUhOrWe9Yn3wqt9utF3HoWYkm2hvFb9zy4VvC\nSnhP2R53xJ39TwNkVz4GHK+//rqIVFRULF68uKXldG3Vpz/96V27dtXU1Oh3//a3v4lIcopx\n3nnnEXAAAAAAmAG8Xu/euXs7RjuSx81+buBz+nOS95vUzq8VV+Kpxs9KjFIRwLryMeDo7u4W\nkXvvvXfhwoUvvPDCRRdddOzYse9973vPPPPMDTfc8MILL+hPGxgYEJHS0tKEl5eVlYlIIBBI\n5736+vrWrl0bj8fHeU57e/skPgUAAAAATN1/lf5Xc2lzwrjZ80fP/1ffv8oYvTkeiD+QfJ5x\nspLVyuqcfBQgu/Ix4NDjBkVRdu/eff7554vIhRdeuGvXrvPOO+/FF1984403LrroonFernff\nMG9mGYeiKGd9ZjAYFJFIJOJyJaWgAAAAAJBNy2W5T3xH5MiwDNdIzUJZuEbWfLz14/a4XcbY\nb+KInXGh99fYX9fJuj/b/9xa2horjSVkJUu0JRtsG3L6kYDsyMeAo7y8XETOP/98Pd3QFRcX\nf+5zn9u+fbsRcOi1G3odh9lYlR1jvde99947/nM2b968b9++iXwCAAAAAMiMU+Nmz3TCc8I8\nYjJhv4nTeWogyvDw8MmTJ7cXb/993e8TzlAohZfIJWtkzY3KjQVSkNWPAORGPgYceu8MfaeJ\nmX5kdHTU/DS9E4eZ3rbjvPPOy/Y6AQAAAGBaVFVV/af9P3uV3uT9JqvUVWIXEQkGg62trZqm\nlRaUNrYnjpI9V859Xp6f3k8BZFY+BhwrVqxQFOW9996LRqNG9Cgif/nLX0Tk3HPP1e9+9rOf\nFZEnn3zyxz/+sfGczs7Ot956a86cOQQcAAAAAGYql8v1StUrT9qfTNhvcmH8wp86fyoimqZ1\ndnaqqqooysrASuMJKwZX6AHH+KNkASuyTfcCUpgzZ86XvvSlnp6eH/3oR8bBPXv2PP/885WV\nlf/4j/+oH/n4xz/+qU996k9/+tOOHTv0I6qq/vCHP1RV9eabb06zBwcAAAAAWNFn7J+5SC4q\n08pc4podn708tvw/1f98zf5amZSJyMjISCQSMV8WqYra6mrVR8nWxmvXS4pRsoClKXpLznzT\n2dm5bNmy48ePf/rTn/74xz/e2tq6d+9eu92+c+fOa665xnja22+/fdlllw0NDa1cufLcc8/d\nt2/fm2++efHFF7/wwguFhYWZWszmzZtvvvnmoaEhj8dz9mcDAAAAwHQLBAIdHR3G3YRRslt8\nWy4+9+JpWhqsLRKJuN3ul1566dJLL53utSTKxwoOEZk9e/brr7/+P//n/+zs7NyyZcuBAweu\nueaal19+2ZxuiMjHPvaxN99888tf/vLLL7/8i1/8or+//9Zbb33uuecymG4AAAAAgOWYazeM\nUbL63UNFh+6svHOa1gVkUZ5WcOQVKjgAAAAAWEskEjl8+LD5iHmUrFNzRpTIdK0NlkYFBwAA\nAAAgd1wuV0lJifmIMUpWREqkZIzXARZGwAEAAAAAea1FWm6QGxbJIq94neKcK3Nvkpv84h//\nVXPmzNlds3tL1ZZ3C9/tdfQeLji8bs46fZTsamV1ThYO5FQ+jokFAAAAABh2hXc1uZuMux3S\nsVW29krvbtk9zqvsdvsrVa80S3PCKNmlsvQOuSNbawWmDwEHAAAAAOSpWCzW1tYWd8UbtcYL\nRi8ojBc+OuvRe6vvFZEWaTnry5fLcp/4jsiRYRmukZqFsnCNrLlRbiyQguyvHcg1Ag4AAAAA\nyFNtbW2hUGhlaKVxZMXgCj3guEQuOevL66W+XuqzuD4gn9CDAwAAAAAy47B2+Hr1+ok2yxhL\nMBgMhULGXVVRW12td9XeJSLV0eofhH6QmUUDMwUVHAAAAAAwVSMjIz6fb1vBtp21O42DaTbL\nGEswGDRur1q4qqXg1J6UxaHFm9o2lZeXS9EUVw3MKFRwAAAAAMCUDA0NHT16NBgMlsRKGtsb\n97Tsee69577T/R390XSaZaQUj8f1GxEl4nf6Hdqp36cPFR26u+Zu41EAOgIOAAAAAJg8VVVP\nnDihaZqIrAysvGrgqnPC51THqlcMrtCfkE6zjJQcjlOJhktz7fvrvoPvHNx6bKtbc4vInrI9\nxqPTbnJTbIGMy5f/JAAAAADAioaGhmKxmPmIqqjtzna9WUadWrfetn5yZ/Z6vX7/6ZhAEeUT\noU8UqUVhe9ijerxe71SWnRHxeNzv92+3b2+qmvAUWyDjCDgAAAAAWEOLtKyTdW/Km13SNSqj\ntVL7efl8gzRUSdU0riocDpvvJjTL2B7YPm/2vMmduaio6PHZj5+MnrwseFlNtKbX0bu9cnu/\nvV9EvjjyxUJP4RRXPkXRaPTYsWORSKS4rLixfTJTbIHMIuAAAAAAYAHDw8P3h+9vmpXXlQJG\ns4yYEhORQ0WHGrSGh+XhSZ9wf/n+vcreTTWbzAc/Gv3oxqKN+u3D2uHb1NsOKge7bd05Dn1O\nnDgRiUREZGVgklNsgcwi4AAAAACQ7/r7+zs7OwtKCxqH865SoKCgwLitN8vQRHuj+I1bPnxL\nWAnvKt41lZNfrlzeLd1H5MiwDFfGKxdoC9bImjXONQVSoGlad3f3Nm3b72p/Zzw/Z6FPJBIx\nD3mRMzfm1MZr19snuTEHmDQCDgAAAAB5LRqNnjx5UtO0lJUCF2sXizJ9ixPxeDxOpzMajRpH\n9GYZBWpB2B6OS9wr3knXVtRLfb3Un7pjP+Oh9vb2wcHBkrKSadkeMjo6ar6bsDFni2/LvHMn\nuTEHmDQCDgAAAAB5LRAIqKpq3DVXClRHq783+j2Z1m6bNpvtQx/6UONgY5+tz9wsY8A+ICJx\niQclKJmurRgaGhocHJT82B6SvDHnpxU/3Sk7c7kGQAg4AAAAAOQ5cxfPhEqBTW2bKssrpzfg\nEJHi4uLXCl97wvZEQrOM2dHZd7XdVRWtGqu2YtJtUwcGBsx3E0Kf70e+L8WZ+Whjcblcp28n\nbcz5g/cP2X17IBUCDgAAAADWkFwpcHfN3b+M/HK61yUicoXtCr/4j8iRoBasiFXMi8xbGVj5\nhcAX3JpbxqitCIVCD8cebiqZTNtUvbunLjn0qamuyXbAcdR+tP6c+rfdb/c4esJKuDJWeWnw\n0u92f1efYlsiJdl9eyAVAg4AAAAAec3tdus3kisF9pTt2TqwdXqXp9ObZWia9t5778XjceP4\nWK03u7q6enp6HGWOxoHG80fOL1KLJtRBQ1FO9R1JGfpsVbL4nQwMDPh8vh3eHXvr9hoHfU7f\nrvJdBzwH9Cm2q5XV2VsAMBbbdC8AAAAAAE5pkZYb5IZFssgrXqc458rcm+SmaFnUZjt95aK3\n8CxSi0TEo3pKSvKoWCASiZjTjVULVy356JKrz7t6v2f/4tDi3x393TyZJyJ9fX09PT0isjKw\n8qqBqz4c+XB1rHrF4Ar9Vel00CgqKtJv6KHPwXcObj22Va8W2VO2p7CwMOMfTdfX19fe3h6J\nRErjpY3tjZ8c/qT50S5nl4gslaV3yB1ZWgAwDio4AAAAAOSFSCTySOSRJk/Slg1n7xUfvuJ4\n8PiyoWVGC0+9UmCVusrmzKNfbc3NUJNrK+6svHOZLBMRv99/xqsmPmC1vLy8t7fXeDsj9Anb\nwx7VY9S8ZFYsFuvq6tJv681NO12dIVuo1d0atAVFpE7qfiQ/ulFuLJCC8U4EZAcBBwAAAIDp\n19fXd/LkSVuprbE/xZaNWFGsuaj5nup7zC9ZrC7+qfOn07Te1JxOp3E75YYaEYlEIuaZsgkd\nNO7tuHfeeWcfsOpyuZ6Z/8zRwaPJoU/2tocMDQ2ZExwR+UbPN1YMrGiY3bDfs79OrXvF9ope\nogJMCwIOAAAAANMsGAx2dnbK2ENPz5PzfOI7IkeGZbhGahbKwjWy5kZb3lUKOByO4uLi4eFh\n44i5tsKreUU5S5XHXVV3LdOWGS02xvFMwTPNBYmhj1OcIVvIL/6zzmGZBHNnU0mKZrYHts+b\nTbqB6UTAAQAAAGCajbNloyZWs96xfp7Mq5f6aVrdxNTV1TX0N/TZ+i4LXpaytsLhOH0VlrLK\nI510Q0SWqcvaY+2HHYdHbaPGwahE75f7h2TorHNYJsG8sORopkFreFgezvibAukj4AAAAAAw\nzUKhkHE7eejp7IWzLXThUlBQ8FrNa0/YnthUs8l8fIm2pEFpEBGHw1FUVGT+yMlVHmcVi8Wu\nPXrtysjKx8sed2rOC0YvKIwXTmgOyySYe5cmRzO7indl402B9Fnn/xMAAAAAZpwWaVmrrX1l\n0Ss9jp6wEq6IVQQcgYShpxfHLzZXPeS/K2xX+MWvb6ip1qoXysKvKV+7UTm9oUav8ui39ydX\neVwn16XzFj6fT98wMtamnsx/KhGPx+N2u8PhsHHEHM2USB6Ns8EHk5X+NwEAAABgxtA0LRAI\nPKg8+EjZI+I6dbDb2S0iVw5euaZ3jbFlw6k5xztR/qmX+tMbalKVYxQWFr7meu1J+5PJVR4b\nbBvOen5N0wYGBsxHkjf1THbt41EUZd68eQ39Db1K76SjGSB7CDgAAAAA5Fo8Hm9tbQ2FQq4y\nV+NQY8IOizZ3m3noqc2eu0GwLdKyTta9KW92SdeojNZK7efl8w3SkPGenZ+xf6ZHet7X3g8p\noSq1aqEs/Lrt6+Yqj3HEYjFzp9LkTT1zFs0Re2bXe4rb7X6t5rW9yt6EaGapLL1D7sjKWwJp\ny6OR0QAAAAA+IDo7O/UmFCsDK68auOqc8Dl/LPmjXgggIueEz1k3Z51+91rt2rFO0iItN8gN\ni2SRV7xOcc6VuTfJTX7xj/X8swqFQg8GHmySpvfl/aAEYxLrkI6tsvVb8q1Jn3Ms9VL/urze\nr/SHJdxh63jR9uI35ZtpDoVJ2exTv6tv6kloU3pYO7w6vnqhtjAjX9TlyuUXyUXlUu4S11yZ\ne6VcuVW2HpADZVI2uRMCmUIFBwAAAICcikQiyTssnih94vXi1/W7z5c8r99YrC5udDSmPMno\n6GhTtKnJ22Qc0cOIXumd3ACRnp4en8/nKk1RUZKlnp2T5nA4nE5nJBJRFCXlHBabcuqX7Hg8\n3tXVtc2+7Xe1vzNePsUv6owNOEA+IeAAAAAAkFMjIyPmu+YdFnbNbhNbtVa9QFvwddvXv2L7\nSsqiBp/P5/f77WX2xsCpMGJXxa6fV/1cJhtGDA8Pd3V1SW57dk5FRUWFvmBdymafqqoeO3Zs\ndHS0pKyksT3fUxtg6gg4AAAAAOSUuX+EscNCH5sSV+KrhlY94n1knJf39/f7/X45M4z47MBn\n9YBjcmFEb2/vGSs09eysjlavs6/Lt839FRUVv7b9+mT0ZHKzz9XKav05fr9/dHRUrJPaAFNE\nwAEAAAAgp5zO01NRkndY7PacZd9Ed3e3+a45jKiN1663T2aAiLmoJLlnZ/W8aimaxFmzSFGU\nl2a91CzN4zT7HGfSijm1+Wvsr+tk3Z9sf+q2dWe1r2pKh7XDa7W1B5WDPsWX+3fHDEPAAQAA\nACCniouLHQ5HLBYzjqTcYZFSJBKJRqPG3YQw4t6Oe+edN28SSzKKShIqSvSenTvUHZM4Z7Yt\nl+U+8R2RI8MyXCM1C2XhGllzo5yaw6JpWiQSMZ6cnNrUnVsXs8dOnDix3bX993W/N545xQ4d\n6VNVtaura5tt287anbl/d8xIeVZoBQAAAGCmUxSlrq5u56ydW6q2vFv4bq+j93DBYWNsirHD\nIqWU21v0u4eKDt1VdZemacYTUo5ZeVVeTT4YLAzqL9ErSg6+c3Drsa1uzS0ie8r2uFyujH8J\nZ/U39W+r46sXqAvGGn2iz2Hpk76whNuk7Xl53jyHRVEUY5ZKykkrqqoePXp0aGioNF7a2N64\np2XPc+89953u7+jPyXaHDv3d+/r6SmIluX93zFRUcAAAAADItdLS0leLX33K8dQ4OyxScjhO\nX8KkHCCiX9XH4/He3t7f2n/bVJE4ZuXl6MvvOd9LOHhy9smfHP6JcdBcUeJRPTkOOFRV9fl8\n9yn3TXH0SUFBgb71JuUXNdg9qJd4TEuHjsn1B2mRlnWy7k15s0u62M+CZAQcAAAAAKbBlY4r\ne6X3fe39kBKq1qoXysKvKV8zdliMxeFwFBUVhUIh44g5jPBqXlEkHA4fP348Go0WlhUa00OM\nMSt90pc8UqTV1fqH2j/44r7knp3Xatdm+ZtI1NbWFgwGpz76pKKioqOjw7ib8EUNDg6an5zQ\noeM25basXiym2R/ELBgMPhR/qKk0Y4OBMfMQcAAAAACYBvVSXy/1ou+iUCbwwtra2g2BDf32\n/uQw4jq5TtO09vZ2vU9HyjEry4LLrhq4Sj9oLhl4ueLlvcrehIqSxeriRkfjlJOLV9UAACAA\nSURBVD7nBAUCgWAwKJkorCgrK9uibOkY7TC+qPsq7tO/qOtt14/foaN2Xm32LhbT6Q8i7jOe\n39HRMTAw4CxzNg5mZjAwZiQCDgAAAABWUlRU9KorxfaWxeriDbYNw8Fhfe+DIaE64JbuW5IP\nrrOva7I1dUu33rPzdEWJ7SwVJRk3fmnDD2M/lMIJnO2/Sv+ruTT1pJWTtpPxeFzG6Kv6oO3B\nqX+WcSiKondLSfnuDysPm5/s8/n0ryWDg4ExIxFwAAAAALAY8/aWKrVqgbbgG/Zv6GGEf8Rv\nfmZydcCs2KyUg2Dri+rrpf7UyyZSUZJZZymsqKudUMAxzqSVgaKBoaEhGaNDh9tcQZFpiqK4\n3W49h0r57k45PUhYVdXe3l7zyzMyGBgzElNUAAAAAGRdyoEm5pkgE6IPEOlX+sMS7rB1/NH+\nR2OAyPhjVu6uuXuskSJT+3wZY7OdukZLuU5jMEqaxpm0UllZaX6m0aFDRLyad6JvNFEVFRVp\nvvvIyIh5Ms6qhauWfHTJ1eddvd+zf3Fo8e623fNkMoOBMSMRcAAAAADIrng8vnN4Z5M0vS/v\nByUYk5jeHvJb8q2Mv5d54knqma+pDjqdzjHPmFuFhacqNFKu03h06oqLi58+9+lfV/86eVLv\n9bbrM/UuYykvL2/+UHPKOcEJ767vo9Elhz6Ns87okJLZHA2WwxYVAAAAAFkUCoXa2toUj9LY\nN6WZIGnyer02m81ckZEw8zXlQbc7izsyJqSioqK/v9+oWUgYfZLZIOb54uebi5vvqb7HfPCs\nk3ozZV/ZvmZJ3R/EfMT8kZP3szxW8pj+kL6T5X77/U2zGLPywUXAAQAAACBbYrFYa2trPB6f\n+kyQNDkcjtra2o2jG1OOWZkTmfNu4bvTPgh2HG63++lznz4ePL5saFnCOlcrqzP7XuN06Mjs\nG03l3QsLC51Opz4WR5c8GDgajR4/fjwcDheUFTQOM2blg0sxb2dCSps3b7755puHhoY8Hs90\nrwUAAACwEp/P5/ef3iCgt4dsmN2w37O/Olp9QDkw3zE/G+97VeyqpxxPJRwskZJBGUw4uFhd\n/EfbH8ukLBvLmJyr5epmaU44uFSWviAv5NU6c6NFWv4t+m8vKi8O2YfiEq+IVVw4cqFLcz1Z\n+qSIfFv79q+UXx07dmx4eDjxhQUtqxauEpF/kX/5jfxmGpY+Q0UiEbfb/dJLL1166aXTvZZE\nVHAAAAAAyBbzZWfyTJDK2ZVSkpX31cesGDNfFymL1siaTuncLbv1g1Vq1UJZ+HXb19MZBNsi\nLWu1tW/Kmz7FNyqjtVL7efl8gzRUSVU2Fj+9hRV5JRaLPTL6yKOeR40jfqf/eefz+u0l2pIG\npWFkZCQh3TCPWalT69bbGLPyQUHAAQAAACBbjF4YRnvImBKTv88E+U08W7+r10vqma+3yW2n\nbqU9biEQCGyPbn+k6hHjSLY7O5yx+A+wUCjU2tpq89oa+xvfKXzngOfAcffxsBIWkSIp+rn8\n/EblxgIp6BvpM78qIUfb1rdt3ocYs/JBQcABAAAAIFscjlNXHMntIfeU7XEO58vskrHoW2yK\ny4ob28frkNoiLetk3ZvyZpd05aDE44MgHo+bu7dcNXCVmHad3CA3fFO+qT/T3HUhOUdriDc8\nIo+keAPMRAQcAAAAALKlpKQkGAwadxNmlxQVFU3j2s5qZGREbyCSskPqxdrFem1IMBh8IPpA\nUznDOzKpv7/fPCDWvOukOlpdH6839uskDwY252i7PfwTfIAQcAAAAADIlvLy8l/Ef9GtdicP\nNPmn2D/Z3GlvFJkOgUDAfDfhGvsHkR9IsXR1dfX09LjL3I3BXAzBTZ/Vi0pCoZBxO7l7S0VV\nhRFwFBcX2+12cxpiztFKstTlBXmJgAMAAABAtiiKcqDywF5l76aaTebj58XPC7gDC7WFuWnb\nOb734u+t1db+yfanblu3eTGRSMR4TvI1dlVl1WB8sKenR8Yo8cjGENx0RCIRv9+/3bW9qSor\nRSUt0nKbettB5WBW/+2MjScpu7dsjm02nmmz2WbPnn3X8F0pBwNnfLYu8hkBBwAAAIAsuly5\nvFu6jdklC7QFX458uSPUcUfFHUb7z+na06GqaldX13bb9t/X/t44aCxmo2zUj6S8xv61+uve\n3t4zzmYq8aiJ1fyH/T/M/U1zIxQKHT9+XFXVs/YNmZze3t5tsW07a3YaR7L0b+d0nurPkrp7\ny8AZ3VtKS0tfLX71KcdTCTnaUll6h9yRwVUhzxFwAAAAAMiiM2aC2KSvr6+zs/Pxssezcfk9\nUW1tbcFgsKSsJOViCgoKhoaGZIxr7IJQgc/nM06VXOJRe26tuHP6cVRVbW9v1yfXZKOopLOz\ns6+vz1vmzcG/XUlJSV/f6fEoCd1bvF5vwvNTDgb+YM7W/SAj4AAAAACQI5FI5OTJk3K2tp25\nMTAwoDdAHSsLKC8v7+npMfZKJHdIHX8bxQPqA8lvmtXWGENDQ9Fo1HzEXFRSp9att62f9MlD\noZCeOOTm387j8eyZs6cz0pmye4vdbU94/liDgfGBQsABAAAAIEcCgYB5qGdC287vjX5PEn+Y\nz6KBgQHz3YTFfD/yfVex6+lznz4ePL5saFnCNfZ1cp2iKC6XKxwOyxglHi7VlfCO0Wi0abSp\nyZuteSujo6PmuwlFJTsGdsyrmzfpk4/fcvX74e+LZ9LnTm1/+f5maU7YdXJh/MK73Hdl+J0w\nUxBwAAAAAMgRPQ7QpWjbOasqlwGHudgheTE11TVSLM8VPddc1HxP9T3mFy7RlvzE/hMRKS0t\n7e7uNo6bSzy8mtduP6PKYGhoqL293V5ibwxka3+HOTxKLiq5Q7vjYXl40icfv+VqdWV1xgOO\n5bLcJz6je8tCWfg15WtfsX+FXScYS16PZQIAAAAwkyjKqc0DxuW3flff0zHRs70bfffa6LXz\n4/M9mscpzrky9ya5yS/+NF9us9nGWYz+6HJZfpFcVC7lLnHNlblXypVbZesryitlUiYilZWV\nu2t2b6na8m7hu72O3sMFh9fNWWeUeJjfKxqN6t0xVgZWXjVw1Tnhc6pj1SsGV+iPZmreitt9\nuueHXlRy8J2DW49tdWtuEdlVvGsqJ1cURQ9QUn5dxr9sBtVL/evyep/0hSXcYet40fbivyj/\nQrqBcSjmkA8pbd68+eabbx4aGvJ4Mp1JAgAAAB8kfr/f3JjTvKfDqTl7hnpKSkrSOY+qqidP\nntxk27ShbkPCQ9fINWlu9/D5fH7/6TQkYTHBaNDlStxjkuyL2hf3KnsTDi7RlryovKiHICnf\nS9/f0TC7Yb9nf3W0+oByYL5j/jjvclg7vFZbe9bJrPF4/PDhw/F43HxQVdTPnP+Zfnt/pVSm\nn/4k6+7uNperJHxdgZFAUVHRpE8OC4lEIm63+6WXXrr00kuney2J2KICAAAAIEfKysr8fr8+\n5kOS2nam/4PiiRMnBgYGSstKpzLOY9asWb29vSkX49W86aQbcuYQ3GqtWt9GcaOSOLwjFAoZ\nt5P3d1TUVUhp6vPrbVm3ubftrD37ZFa73V5XV/ez0M/67f3JvTkTikomSm+5Ota/HekG8gEB\nBwAAAIAccTqdT334qZRtO6/VrrXZ09pBHwqF9P6gKcd5fDL+SUmcsDHmYp6Z/8zRwaPJi1mt\nrE7zE6U5vMPIBVLOW/mN+puUrwqHw0ePHo3H4yX21INsk19SVlb2qufVpxxPJfTmXCpL75A7\n0vxQKWXk6wKyioADAAAAQO6M1baz0dGY5hmGhobMdxPGeXx38LtSke5inil4prkgcTFLZWmD\n0pDuKdLjdDpHRkZkjHkrzmFnyledPHlS328y1iDblK+60nFlr/TqRSU1UrNQFq6RNTdKYlHJ\nJKT8upZoSzbYEjcKAdOCgAMAAABA7phHY5y+/E7a0zGOWCxm3E4x/aSkZqqLyUQWkKCkpGRw\ncNC4m7C/o7i4OPkl0Wg0GAyaj5ijnJpYzXrH+pTvdUZRSUZN/d8OyCoCDgAAAAC5M/XLb2P8\nasrtHr+M/DKXi0lTWVnZvbF7fXFfcmuML8W/pNhTbG4xj2WVpCjnFyd+MW/RvBys3CxnXxcw\nOQQcAAAAAKykuLi4p6dHxtjusWN4x3QvMLWXK17eq+xNaI1xYfzCO113pny+efBqcpTzs+qf\nLZNlWV0wYDlpdfEBAAAAgOnSIi03yA2LZJFXvE5x/oP3H24/5/Z+R7/+qLHdQ0QmNIolxy5X\nLr9ILiqXcpe45qhzrlCv+I32m9fsr5mnyZq53W4j49CjnIPvHNx6bKtbc4vIYyWPTXol70Te\nuTZ67fz4fI/mcYpzrsy9SW6aygRZs8Pa4evV6xeoC/R/rMyeHBgfFRwAAAAA8pSmaYFA4AF5\noKm8yTjYIR07vTsPzj/44/YfW2icxxn7O9L4odlut5eWlgYCAeOIuXNHiZRMYg2xWKyjo+O3\n7t8+WveocXCsobMTpWma3+/fpqY10RbIBgIOAAAAAPlIVdXW1tbh4WF3mbsxmDgk9YjryJcX\nfNn8/KWydIaN86irq7vPfV+32p3cuWMSUY6qqseOHQuHw6WO0jSHzk5Ie3v74OBgSVm6E22B\njCPgAAAAAJCPOjs7h4eH5cwhqZ8d+Kx+zVwplXGJZ3v6yfSy2+0HKg8kd+5YKkvvkDsmera+\nvr5wOCwTHDqbpsHBQX1MTDZODqSJgAMAAACwthZpuU297aBy0Kf4RmW0Vmo/L59vkIYqqZru\npU1eLBYbGBgwH9GHpP6s7mciUhuvfdX+6jzJ9RiR3LtcubxbujMyyHZoaMh81zx0tjpa/cPY\nD6Vw8utM+Y+VqZMDaSLgAAAAAKxK07Senp5t6radNTOt68HIyIimacbdhCGpW3xb5p0789MN\nyehk1mg0atxO+D43tW2qraudSgYRDoc1TdO7oiafvLqmmoADOcAUFQAAAMCqOjo6fD6fN+pt\nbG/c07Lnufee+073d/SHrN71QFVV47YxJFW/e6jo0E8rfjpN67Iwu92u30j+Pu+uudt4VKcP\nQ1moLUxzGIrNZtPTjZQnt9m48EQu8HcGAAAAWNLg4KC+L2BlYOVVA1edEz6nOla9YnCF/qjV\nux64XK7Tt5OGpP7B+4fpW1q26NNw088UJqqoqEi/kfx97inbU1h4qsQiHA63trZu823badt5\nRDkSlGBMYnpZ0LfkW2Od3Hj5+CcHsoqAAwAAALAk8wBREVEVtdXVanQ9qB89ta9Bv2xeJIuy\ndNmcJYWFheaMQ0xDUkVkckNS81kgENjRs6NJmtLPFCaqsrLSXElh/j69mlev4BgdHT169OjQ\n0FBJrGRCZUGzZs3SKzhSnjzhnxLIEnpwAAAAAJakT8TQJXc9qKmrkQJRVfV3o79rKmoynml0\n6GiUxnWy7k15s0u68rM16ezZsxsHG/vt/RkZkprPurq6enp6isqKsjpg1el0Prvg2aODRy8d\nvDTh+7zedr3+nBMnTsTjcZn4MBS32/30uU8fDx5fNrQs4eTXyXWZ+gjA+Ag4AAAAAEtSFEVv\n62h0PYgpMfl714P7lPtGRkba2tqkWBp7Ey+b/6b+7bf9v22qSBF85E9rUo/H82rBq085nsrI\nkNS8NTw83NPTIzkZsPq0++nmquaNVRvNB43vMxwOj4yMmB8yD0Opjdeut68f5+TPFT3XXNR8\nT/U9CSffYNuQ/GRVVf8y+pf/4/g/bzne6rZ152fEBssh4AAAAAAsqbCwcHR0VP7e9UAT7Y3i\nN2758C1hJbynbI9z1Nna2hqLxVJeNl8wcEHhSGFW6wUy4h+Uf3hNe21AGVBFFRG3uD8ln3pQ\nHiyTsuleWsYkbzUyD1itj9dPdCBsPB4/NHLoduftbzne8tv95uxguSz3iW+sobORSMR8noSy\noF+d/NW8BeNNrhn/5GYDAwOdnZ0PlT20q26XcTAPIzZYDgEHAAAAYEmzZs3q7+837hpdD8L2\nsFfzBoPBWCxmPJpw2XxL9y110Trj0SzVC0xFNBrt6OhwFbj6605/xrCE98m+/yH/YyZdA+sp\nlS55q1FVTdWEAo7e3l6fz9dU3rS77vRXZM4O0hw6m1wWdFfVXTtl5zgvSXOi7eDgYHt7u4iU\nxkvzP2KDtRBwAAAAAJZUWFj41Iefah1uTW5RcZ1cFwqFjGcmXzbPis3S7yYEHz+I/kCKcv5J\nksTj8WPHjkUikVLnzL8GNnpzptxqtFXZmv6p+vr6Tp48KZPNDgoKTkcpyWVBaU6uaZGW8Xu7\n6CuUnGzJwQcNAQcAAABgVS94Xmj2NCe3qNhg29AX79Pvprxsvv3E7ZIq+Kiurs6HgKOnp0ff\nLvFBuAZ2u916GpVyq5F71J3meTRN8/l8+u3JfW9Op7OkpGRwcNA4Yi4LSmdyTSwW2xne2VQ8\nZm+XkZGRaDRqfok5YquJ1ax3jNfmAxgfY2IBAAAAq1ouyy+Si8ql3CWuuTL3Srlyq2w9IAfK\npMzhOPVbpn7ZfPCdg1uPbXVrbhHZU7ZHTMGH/jQ9+NA0bbo+i9nQ0JD5bsIE3H+P/Ps0rSsr\nZs2aZb5rHrDqUT2FhYVpnicUCukDUAzm7602Xrtezp4dzJ49e1f1ri1VW94tfLfX0Xu44PC6\nOevSnFwzMDBw+PBhpV8ZZ76seduUiKxauGrJR5dcfd7V+z37F4cW//7Y7+fJeG0+Jseik5Ix\nCVRwAAAAAFY1TteDcX6K96geGaNeoCA0wYaW2WH+kT+5zKT2Q7Xiyvoacjbmo7Cw8MlznmwL\ntaXcapT+eRIqIxK+t81dm+fNP3t24HA4Xql6Za+yd6KTa0ZGRjo6OjRNG794xGY7/RN7ytqi\nZbLsrIuckKGhoYe1h5tK8npgEDKFgAMAAACYgUpLS38e/Xm32p182fy5gc8ZT0sIPoqK8mCD\niojNZtOLEVJeAz9gfyDbCwgEAidPnszZmI8XvS82exO3Gi3RlvzE/pP0T2K3243bKVuEPiKP\npHOey5XLu6U7YRjKJ+WTN8vNb2hv+BRfyqzH7/eby3/G2nhSWFhos9lUVZUptPlIUzweb2tr\nGx4edpQ5GgdOdSR5rPqxjbM2yozr5AIdAQcAAAAwAymKcqDyQPJP8R+LfexC14VbqrYkBx/X\natcaDS+nV3FxsT48NXWZyQTnpsZisb+M/uV25+2HnIfSqcUYGBjo6OiQHI75+Ej8Iy/bXk6c\nhqtMbBpuUVGRoih6ypD8ve32pBvKJJcFDQ0N/ST8k6bKJvn7X0dy1jM8PGw8P0XRzfxTRTc2\nm62ysrK7u9t4sjliK1VK0/+8Z9XR0aGvylxUckXfFXrAMcM6uUBHwAEAAADMKMYYizalTRFF\nEUVEatSaRdqirylf+6rjq9d5rktuTbpEW9LoaJymJSeqrKwcGBgwKgISJuAqtgmkMD6fr6en\np2nWmGNTE56vaVoux3yoqnry5EmnzTn1abh2u72ystLvP91aYqLZQYu03KbedlA5aC7TqO+v\nj5yIFJUVjZ/16EUZMlbRTfx00U1VVdU217YT4RNT3JIzvpGRkeROLkZRSZ1at95GN9MZiIAD\nAAAAmDmCweD9kfubZp3uOKCJJiKfsn3KuFpeLst94kvYg3CjcuNEKyOyp6Cg4Jn5zxwbOrZs\naFnCNfD1tuvTP4/P59Mv+NOvxRgZGUlohGm+MK6N1663Z/LCuKOjY3BwsLQsM6Ui1dXV97nv\nGys7GGeAq6ZpPT0929RtO2t2nl6bdGyVrccdxzfKxpRZz8XaxUZNh8Ph0JuApCy6ccROX3gq\nirKvbF+zpJj+M36bjwkJBoPmuwlFJTsGdsyry3w3U0w7Ag4AAABghvD7/T6fr6CsoHF4vKvl\ncVqT5o9nC59tLmy+p/oe88EJXQNHo9Genh79djq1GH8Z/ct62/o/uf7U/Q/dYSVcGau8NHjp\nW0VvHXUf1Z+wOLT4Vyd/NW9Bxi6Mh4eH9UawKZf3yfgnxT7ma1MaJzv4sfbjLeEtTQWpe22e\nOHEiEAh4y7zJOUurq9V4iTnrqY5W/6/Q/5K/14V4vd6+vr7TKzmzt4vT6TSvJ3XEJpmM2IyK\nEklVVPJj9cdN0jT2q2FVBBwAAADATBAKhXw+n5ztYn6cn/GnZdljmfo18NDQUMLU27FqMaLR\naHt7+wOFD5hbivqcvl3lu5yaM6FV507ZKRkyzh6K6mj1vw79q8wa45VjS/m9rY6t9rX6VLfa\nqKVIvoLBoN7xJOVfzuLQYv1IcmeNqrIqI+Coqqr6te3XvUpvyt4uCYs8a8Q29b9SY0yypCoq\nebT40VWRVX+2/7nb1h1Wwnn7XwEmioADAAAAmAnMv59L0tXyrXKrOGVkZOTB8INNZRYYmTn1\nMpOEnSYJ1+fb+rbN+9A8EVFV9dixY5FIpNSVYp/IvMi8XS27Uo75mPpF+Fmm4ZbVTuJTp/ze\njrQeGRkZWTmSOvnS0w1Dwl/OLd23yBidNX4Z+aXxKqfT+Wr1q0/YnkgoHlmsLp5QbxdN0/r7\n+++XM7ZZTeKv1Ov1Gr1UdOaiEpfm2uXKxXwc5BgBBwAAADATjI6OGrdT/Ng+u6p3sLerq8tV\n6mocysVkkGk3/tjUn2g/eVgeFpHe3t5IJCJj1y+kbNU5MjJy/8hUL8KNFaaMDzbHNk/h0582\nODg4MjJi3E3IL34Y+6H+8XXJfzmzYrNkjM4a2wa3md/oCtsVfvG/r70fUkJVatUCbcHXbV//\niu0r6RfdaJrW2toaDAbPus3qrFwuV0VFxa+0X/Xb+5OLSi4cufC6vus+CP8VfNAQcAAAAAAz\nSuqr5fDmdDawzCTFxcXG7eTr813Fp37AT7lP5Idzfygis2Kzbui74XDB4YQxH/oc2YLSqV6E\nFxcX63U3KeODolDRVL8CETnbANfyWeXDyqknpPzL+b+d/zflOBuP6vF6veY3OlU8orcdtU1m\nqT6fT28OmpG/0tra2pcjLz/rfjahqOT80fN/1vYzb9ybcP5JND1BviHgAAAAAGYCl8ulF3Gk\nvFoOtYXMT074GX+tbe3Mu7QrKCgoKSnRu3jqzNfnJVKiHxxrn4iI9Dn6vrzgy8ZdvcVpLBY7\nceKEpmlnHSxyViUlJW63OxwOJy/Po3rMAc1UxONx/UbK/OLe0XsLCwv1ECTlX84y57K2UFvy\nOJtrtWtt9knFGGPQN6eYj5j/SmtiNesdE5tfoyjKP7r/sV/rPyJH9KKSc6LnfKHnC18IfMGt\nuSXpv4LvDn5XKjL3eTAdCDgAAACAmaC8vHysi3mP6jGuoiXlBpYPVYkn1wvOgQ996EMN/Q1d\nsa6UY1P159jtdj3jSLj+FxGn5lQUJaHFae9Ar3lCxziDRc5KUZSnz336yMCRSwKXTGUa7viM\ndpsp84vNPZtLSkp6e3vHKtN4wfNCsydxnM0SbcmEOmukIxKJGFmMJP2V3ttx77zzJjy/pl7q\n65W/dySxSYe/w2g4kqLpSelkmp4grxBwAAAAADOB1+vdM2dPZ6Qz5RgL4/I15c/427Xt07n0\nrLHZbC9XvJxybKoxbra4uHisyhcRGVFHbLYz6hTGj4oqSyuNgONv6t/Wamv/ZPuTT/GN1YX0\nCccTzRXNCYUDS2Vpg9KQma9AxOv1GuNyJdU2E7fb/fS5Tx8PHk8u01itrJ4v81OMs1EyOdJV\nZx55k/xXelfVXZfJZVN8i9w0PcE0IuAAAAAAZoj95fuTL+b1H9v7Xf16nULKn/FdEdc0LTnr\nzjputrKysr+/3yjKMF//ezVvQrphNtaODxGJRCKdnZ3b3Nt+V/c74/kpu5BOfRruWRUXFz8+\n+/GT0ZPJydfVo1cXFBeIyHNFzzUXJZZpLJWlG2wbyqRsKuNsWqRlrbb2TXlznJRH53Q6FUXR\nY46Uf6WTXoPB4/H09vamPP/jZY/b4ra35K08H5+M8RFwAAAAADOE+Wq5WqtepCwyfmxXS1Vz\ns8mEn/HdbveE3mjqE1Jz5qzjZp1O57MLnj06ePTSwUvT2SdifFcpL8J/E/hNOBw+evRoPB4v\ndaSYO5vQhXTq03DTsb98/15lb0Ly9dHoRzcWbdRvZylnGRwc3BHZ8UjlI8aRcWbN2O12j8dj\n7vmaEDal39xkLF6vt7Cw0JgpYz6/U3P+3vH7dNaJfEbAAQAAAMwQZ1wtn3k1WF5e/kv1l764\nL+UGlgm9SywWaxppavJOaUJqXnna/XRzVfPGqo3mg+ZtLGalpaU+ny9lxYdH9ZSUlLS3t+u9\nJPJnYM3lyuXd0q3nF5XxygXagjWyZo1zjZFfTDFnUVX1z8N/vt15+yHnoW5bd1gJ10rtFeEr\nvt357SJP0VlTHkNdXd197vt6ld7kv9LVyupJL8/s6XOffj/w/sX9Fyecf/HIYgbHzgAEHAAA\nAECu5b4CQlGUlyteTv4Zf7G6eELdIkdGRlpbW+0ee2NgShNS88qE6hccDsdTH34qZceKL8W/\npNk0fdCpYYqjQDLijPwi0+NygsFgR0fHI6WP/KHuD8bBDul4wP1A5+zOe9pOb3s566wZl8v1\navWrT9ieSN5mlammJM225uZZzTLrjINjDY6dkeOTZzYCDgAAACCn4vH4IyOPNHlyXQFh/hm/\nWqteKAu/pnztRtsEtiGoqtrW1haLxTJSm6Cq6tvht/+3/X+/5Xir29Y9jVtdJlq/kLJjxWJ1\n8Z2uO6MjUfPBjIwCmZC/jP5lvW39n+1/NiopsvqVhsPhtra2eDxeGk+xH6fN3aY/Lf1ZM1fY\nrvCLP+U2q4wsOGEb1+zQ7JWBlSkHx9bGa9fbpyGNwlQQcAAAAAC5o1dA2Dy2xv5cV0CMs4El\nTYODg3qnUl3CVev3wt9Lf9as3+/3+/0Plj+4q26XcdAqW11SV3zYbiyQglFl1HhachfSn1X/\nbOqjQMYSDoc7OjoeKHogI19pmkVG3d3dqqoqipIy81ocWiwpxxKXVY0V/c6GQQAAIABJREFU\ncEz9r3R85vMPDg22tbUZDyWsc3PX5nnzs5tGIePG7AkMAAAAILPi8Xhra6teAXHVwFXnhM+p\njlWvGFyhP2pUQLRIyw1ywyJZ5BWvU5xzZe5NcpNf/NO38FOM7owismrhqiUfXXL1eVfv9+xf\nHFq888jOWcFZ47zWzOfz6W0s9J/997Tsee69577T/R390dxvdXk3+u610WsXqAvS/MLrpf51\neb1P+sISbpO25+X5b8o39RIDl8tlDF7Ru5AefOfg1mNb9QKBx0sfz9JHiMVix44dGxkZmfpX\nqmlaIBB4aOChJml6X94PSjAmMT0o+ZZ8K+HJyftxWl2tRuZ1S/ctRsqjP0GfNaMoWYgupiZ5\nnXdW3jm9S8IkUMEBAAAA5EggEIjFYsbdhAqI+ni9FEgwGHww9mBTWWY2sLwdftvYsDD1PSBG\nZ81xJqSeVSQS6enp0W9PexvO0dHREydO7Cja8Wjdo8bBqXzhNputvLxcn0WqM3chLZGSzKw7\nid/v1/+0pviV6hlcKBRyljkbB08VGe2u3L2pcpMkBSWapuntVHXJlRqzYrNEJHnWzLbBbVP6\ntBliHh6UPBPnD94/jPNa5CcCDgAAACBHQqGQcTv5arCiqqIr0NXT0+MqczUOTXUDy+joaEdH\nx/3F92fq0l1EXC7XqRupJqRu6d2SzkmGhoY0TTMfma7GB6Ojo0ePHlVVtdR99nmu6aupqdnq\n3JpyYE2mRoEkM09XlaTs7Fa5VZxpnaejo0P/KzUHJVcGrtQDjoSgRFEUu92uZxwpM6/bT9x+\n6plnzprxer1T+KwZ43a7zVNjJVdpFLKHgAMAAADIkfErIDYGNw4MDEgm6hrC4fCxY8fi8Xhp\nQSYv3UtKSrq7u414InlCajonMXfxkKSgZ3tg+7zZOWp80NXVpf+LZLaQxGazHag80CzNCaNA\nxpo7mxHmbzU5O6ubV5dOwDE6OjpOUFKn1q23JWZPHo9H/6NNzrweK3tsbnRu8qyZa7VrbfZ8\naZUwZ86cDYENWR1Mi1wi4AAAAAByxOk8dZWZsgJiQ88G85MTxov+h/0/0u+56PP59N/VM3vp\n7na7987d2z7Snnw1+N8j/91V6ErnJEaLCkkV9GyQDQ/JQ5Nb3oTE4/HszXOd0NzZjLDb7foW\nlZTZ2UP2tL5Sc4WRJAUl9/XfN29OYvZUVVU1NDRkJHfmzMuu2O+pvidh1swSbcmExhJnW0FB\nQcrBtFlNo5A9BBwAAABAjpSUlPT19Rl3EyogIpGI8VCKH+Hn10laAYKoqjrO7/BTvHT/Y8kf\nm0sSaxMujF94d+HdaZ6huLjYuJ0c9Dxa9Og4r82g8QtJNrVtmvuRuZOe4jHRubNTV1xcPFYl\nxZ6yPa70/nSMnEJSBSUNakOTNCW8pKCg4NkFzx4dPHrp4KUJmdd/k/8Wl3hiypO5ga+ZYh5M\nm5s0CtlDwAEAAADkiMfj2TNnT2ekM2UFhLH1I/WP8Fq6dQ2xWMzc5CL50n3Oojlin8z6W6Sl\nXdrd4o5IRBPNLvYqrerf5N++bf92+leDxcXFGWx8kOY002TjF5JsrN14uXL5hFYyvaqqqgYH\nB1PuHiqRkjSnlhgVRpIqKNlVvCvlq552P91c1byxaqP54FJZ+qQ8WSZlk/1AuZP7NArZQ8AB\nAAAA5M7+8v3J3Rn0Cgif06eXFaT8Ed6ppdclUsRuP51epMxKltuWT2LlQ0NDTdJ0yHvIOBKX\neJfS9YK88F357oRONW/evIb+Br/mTw56rpPr0j9PNBrdGdnZVDyZiTMul8vhcOjbOpK/8OzN\nc80So5IiuedF+r0kPB6PzWYz13Gkkz3lfj8OMBYCDgAAACB3zFeDVWrVAm3B121f/4r9KwVS\nEC4L+/1+45nma0uv5jVXHIzPbre73e5wOCxjZCVp/p5viMfjbW1tw8PD9jJ7Y+BUy9LHqh/b\nOGujTKplqdPpfLX61Sm24ezv7+/s7FRKlca+SXZRrays7OrqMu5afYLGMwXPNBc0J/S8mNBX\narfbq6urN0U29dv702+6mbIC4v+zd9/xbdT34/hfdzotS7IkD8krgzhJs0hCCaMEAoH08y2/\nQgcECCspyYeSFAqlpaXNICWUNolL4zqUfkiTEAgFDGkIJYtgNmRQwirZw5Zt2bFkLWuext3v\nj3POF+kkS7aWldfzwR/WnXR6S7Gw7nWvMeDMGoQGAwMcCCGEEEIIZc85Z4PnhizKysrWEesG\nn9fAHcpsNvM3B3nq3tbW5vV64dyWpVfbr+YCHANrWTrIy/4ej4d7gYPpolpWVrZesl60Ymgo\nTtBISyZFWVnZvuC+t2VvDzj2xLKs3W7fDJsbSweSWYPQYGCAAyGEEEIIoYxI9SK2RCLZX75/\nJ7Ez6txyCjtlFblK9CHx6PX69ZL1ouNOUj119/l8CaaNVLFVy4mBtCwdZOMDi8USb0kpdVEV\nrRgaohM00tVL4jrZdQ5wnGRP+gifgTWMhtFziblJBkpYljWZTB6PR6lTJj+fOPEnJfnPEaaN\nIAxwIIQQQgghlH5er/eVyCuNxaldxJ5BzLCAhbsI33duOaDBE6LjTgZw6s7lbvCiWpZu7tk8\nvCJ6dGimMQwjHGga20W1ZmwNJFfQg/0jYvUGSrgyphTnyFgsFi4clnxmDcMwWwJbGovEPynJ\nf47sdvsLzAuNZZg2cl7DAAdCCCGEEEJp1tnZabPZKB1V50qtPcQ5F+EHOqOUk65T90gkwv8c\n27L0j+wfX4FXBrXQ1CWeZlpvrP9O5DtJtizBCRppxBWnCLf0m1nj9Xrb2tpADXU2kU+K2Wx2\nOBzCz9Hrpa8/Xf40xHyO2tvbnU5nka4o+bQRVJAwwIEQQgghhFBqEmfC22w2m80Gg2sPMXjp\nOnWnqL5ThtiWpVuLtg7+KVIlkUgIguBGoop2UaXwNCcXQqGQMBwWm1kz7FvDhGG7YDBoMpkY\nhhH9pEwNTHU4HHDu52imcyYX4BB+jpxOp9PphFx/4lA+SLYVM0IIIYQQQggAaJp+peeVRmg8\nCSc94AlDmMuEvxfu5e7Q3d0tvD9DMCaZibuIXRGpWA4D6ViRXodDh28O3TwqMkrNqqUgHQbD\nFsACK1hF76zRaKK28C1LASAn00YIglCr1fGWpGbUqY6JQWnHZ9ZwN7nMmqj7WK1WYTKO8JNi\nDBsXtC8Q3pnbu6ZyDcR8jhwOBxftij2OIWRYGlma5teG8hiGNhFCCCGEEEqW3W7v7OyktHFr\nT4LBYCgU4u8fdRH7b+1/Gz422x0rhBiG6ejoeEHywtbKvsyLxK0K5HK5Xq9fR6xLaXRophmN\nxudkz9lJ++AnzqCUHGePL2GWfE58biEtUelLUqmUJEkuZpHMfGJhe5fYdI+ScEm8vX/v+Pvw\n0X2fo0AgwB859jjG4UYoSv/7gPITBjgQQgghhBBKis/n6+zsZFk2QSZ84vYQfyn/y3R2eg7z\nC9ra2txut1anTalVQVVV1Sf0J+8o3smfaSMKheKA4cBuye6oJU1mJq+WrM7JkoaKAY8aYVm2\nq6vrOXhuS8UWfqMwOkYQRHFxMVctwhHOJ9awmqi2Mnw9i2gjlRXmFfH2PlX21BVwRewKRY/z\nIryY+puEhioMcCCEEEIIIZSU7u7uqEx4YQPFxySPAQFSqZS/QzIXsbPJ4/G43W6I06rgUubS\nePXrBEF8V/FdJ+sUjneZR87L7bSRayTXdEM3N820nCmvZWvnkfPuIO84nweg9Mvj8bwceblR\nO5BRI62trW63u1hXnCA6VlFRsVG6sRu6k0n2oSiKi3GIflL4AEfs3je1bwqPI5fLuak6oseR\nM/LBvGNoaMEAB0IIIYQQQklJPJrUMMIASpBIJCqVSph7n/gidjZx0Q2eMEBjCBlu8t90i+qW\nL8gvuoiu2Av7j8KjjxLpGe/CGXAeAe+caabYWjAJ3FASqU5a15PyqBGXy5UgOsY38qQo6oDh\nwE5iZ2yyz0piZdQx1Wo1TdP8TeEnRc2oFQpFIBAQ3Rv1OdLr9cLPZtQ9JRJJMm8OKgwY4EAI\nIYQQQqh/J+DEL6t+eUhxqJvqpgmaJVgSSAYYOJsJv5nZzN2zoqJilXNVHraHSNAcZG3r2p3a\nnVuKxUsPkjx+8jELhmG2BLY0Fg0kjwANjNVqjR1KkvyoEZfLJbwZFR37Tfg3oOzdNYOYYQEL\nn+wzhhgTbz5xeXn5OmKdjbDFflJ+HPlxZWXlatfqZDq/6PX6/2P/rzPUmW+fOJR9hDDLDol6\n9tlnFy5c6Ha7o3o1I4QQQgih8wHLsg6Ho56tf6L0iahdF/kuOqw8TBO0lJV6w16+PuX6yPW7\nJbuj7jyFnfI+8b4OdNlYtBjuAj4ABIngdeOu85AerlUBAPzY8eNLvJdIWWnUhf0JMOEQHErm\n4B6P56nwU7/X/T5q+w/hh1ExC5qmTSbTv4r+NZinQylhWfbYsWPhcJjfwkUoVlat/Fj9cUWk\n4oDkwHAQaX/LB606mA6aoMvCZVd4rviq6KvT8tPcHbjo2OSKyTrdQH6x/z/m/9tF7oraeGHk\nwg8lH+pA973w996i3oraK/o5ugFu2AE7krknGrxgMCiXyz/55JMrrhDphJJbmMGBEEIIIYRQ\nXAzDmEwmr9er1CljWw+4JC4+o17YfSOqPURvxwoilx0rAEClUnEBjsQtDyCVC/ucrq4uq9Uq\n08nq3P3UPnDvZzAYvDE4kDyC89Zx9vhSdulBOBg7uySZhweDQWF0I8nhPj6f7+XQ2YYdJABA\nl7Trdf3rUlYa1chzE7lpYK/ravJqK1i5T0pZpIxrpHKn5E7ukzKTmmkDWzKfo6vgqi7o4tJG\njGAcDaPvhrtz/olD2YcBDoQQQgghhOLq6uriGmpwif2vlbzmkDhG0iO5vWEizGXC38zeLHxU\nfraH0Gq1VquV73oQ1fKA2xjVOXU5tbzfw7rdbqvVCsnVPrhcrmAwyN+MqnR4NPIonpBGYVm2\nu7t7E7PpNeNr/MZUK3r6He5zJVwZ9RAuaCXasGN4cPjrJ14XRseUYSUMyDmflJheGcl/jnrv\nic57GOBACCGEEEJIHMMwXMoD7z3Nex9pPuJvtspaAWAyM7mOqsv24lJHEMSeC/accp263Hl5\nVKuC77q+CzEX9p9ue3rY2GH9thS12WzCm1EhkmXkMuGpqbD9amwfkJKyEgxwRDGbzU6nU6PT\npDTZN0q/w32i7u9yuRIErSb7Jkc18hQeH6EcwgAHQgghhBBC4oLBoPDSt/CEnACiPFQ+XjJ+\nHjnvdnLIZMLvonbtKN0BpedsHBcY94uuX8Re2F9jWHMVcVW/x/T7/fzPsTGLipEVwveGfz9j\nn67eWP/34N8H+QILjMfjcTqdMNDOoDyKolIa7tPd3S28yQWtfjPsNwBQEi6ZY59zXHE8wfxX\nhHIFAxwIIYQQQgiJS5DYzwJ7pffKf+n+lbvVDURsq4JZnbP+n/3/yVk5APR7YV8UP7VANGbx\nIvOi8M4U1XsCIppHsN6xPp2vdujjohu8qIqeR4KPgCrZQ1VWVq50rBQd7hMVoWBZNl7QCgDs\nlP222tv4m6LzXxHKFQxwIIQQQgghJE4mk/X9HHNC/qb2zRyubWBiWxV0kV1W1srfFF7YL4bi\nZI4pk8kCgQDEiVnIIjLhnYuLi+12u+jTqRm1RqMZ1MsrOMJ+JbHZMUaDMfkAh0KhOGA4sFuy\ne61xrXD7FHZKVIQiQVwPAKSslCCIvkaeYvNfEcoVDHAghBBCCCEkjqIotVrt8Xj4LbGJ/SzL\nHg4dXk4u/1LyZRfRNYAJF7llMBg2UBvOhM9Md0+vCFckuLAfj1ar5QIcnKiYBZ+ywVGr1W9W\nvdkZ6ozNI/hB8AdSJbZyOAdJ9vYvEc2O2QAbUjoaN9yHy98xsIbRMHouMTd21IhEIpFIJJFI\nBMSCVgAQYAME0V9rFoRyAQMcCCGEEEIIwWefwaZNkX37yLY2wumEqiqYMwdWrOhN7LcRttgT\n8lvJWx0OR1dX12bt5n9V9tWqJJhw8bX/ay4UYpVYaYLOh1AIQRB7S/fugB1RF/anwtQ/wZ+S\nOUJpaemz8KyFscS+RbNhduz9P9Z/vJPYGfV0k8KT6pX1g3gdhUmhUHDxNdHsGCWd2uySc/J3\nEgYoiouLhe11o+J6BInRDZSnMMCBEEIIIYTOawzDnDlz5ne/Uzc19VVkmEywahUwDKxeLT9g\nOLCL3BV7/v+I7RFzpxkAtBFtvxMuQqFQW1vbS8qXtlX2RT1SHfaZIbGNOVIqPSBJcl/ZvtiY\nxRR2ymrJ6tj7zyBmWMDCPV05U17L1s4l5t5J3YmVDrFKSkpsNhvDMFzGRFR2jEKRqXfMYDCs\nl6wXjeuJBq0QyhMY4EAIIYQQQucvhmFOnz4dCATM5pL77rPOmuXS6yPr1pW/+moJAOzZA6tX\nw9Xk1Vaw8on9Y4gxd8Pds0OzW8+0cgfpd8IFy7ItLS00TWtl/YdCsi+2MUeqhDGLBLUPIk9H\nxu5HfWQy2Z4L9rR4Wqa7pyfuDJpeUqlUNK43hZ2yilyVuedFaJAwwIEQQgghhM5fVquV6x+x\nYUOzRhPhNt51l40LcJSXA8RJ7Lf12PjpIRzhhItKpnI5uZzf5XQ6aZqGOKGQS5lLh/p5fvK1\nDyhV7xS9s6NoR4OhQbhxKkzNdKBBGNcrZ8pHw+h55Lx4Qat0OQEnlsGyg3DwDJzJXDubY8yx\npezSL8gvhmLTHJQYBjgQQgghhND5ix/DyUc3AGD/fjX3w803s/HO14XjLSBmwsXzzueHVw3n\n97rdbuGdo4Z9/sr/q+TGlaDz0SALiAYs+4k2PT09m0ObG0sb+S1pr+EKBAKdnZ3PKZ7bUrkl\nc8+CcggDHAghhBBC6DzFMEwoFIraePSoYu1aAwBcdJFv7lwKQCb20L7xFiA24WIVrHoZXubv\nwA2k4MQO+6woq0jTC8pr2bk4X3gGX0A0JHR1dVmtVqVOmbkaLp/P19LSwjCMVpqPlWIoLYZ4\nMhxCCBUqux1GjQKCAIKAJ57I9WoQQqgwxY66bGoqnjdvlNstueQS7zPPtEilccstlMq+ARbc\nhIvPD32+sXmjnJUDwNaircI7SyQS7gc+FMLd5IZ9coNUT8CJOTBnDIzRgEYK0mEwbAEssII1\nHS80x8LhcGdn5wvdLzRC40k46QFPGMLcZfN74d5crw6l03H2+K3MrbVMbUq/xj6fz2q1AsCN\nzhu/5/reCHqEIWy4ruc6bq+wnc1gmM1mhmEy/SwotzCDAyGE8kUwGHQ6nYFAgI1EKu65R97c\n3Lvj4otzui6EECpYBEHI5XKuOwbDwDPPGNetK2dZmD3bvmRJp0IhkUql8R6r0Wj4x/YeTTDh\novjcmhO1Wt3T0wNxhn2qwiqHw7GZyWxyfq4Eg8HTp0+Hw+EiXRFeNi9gLMt2d3dvYja9ZnyN\n35jkr7FwJC3E1HD9lvktyJNdRrxEIZVPJfy0Rj1LRaRiuWR5nEOioQQDHAghlBecTqfZbOb6\n1Rnr6+UffMAoFGQgAIABDoQQyqDS0tKOjg63W/LoozUffaSRStnFiztmz3YAQElJSYIHEgQx\nfPjwlY6VyYzS1Ol03d3dfNsOYShEw2o6Ozt7enoympyfQ21tbeFwGJKYNYOGNLPZ7HQ6NTrN\nAH6NuUa/nNgarvKK8iQDHB6P5xXmlcZikUDhpuAm4T2jnmVd17rhFwwHNPRhgAMhhHLP5/Px\n0Y3it98u37DBPXMmZbUqv/mGqawkjcZcLxAhhAqWXq8/dIi+++5Sk0lGUezDD58ZO5b++usi\npVKpVvfTG0Iul39q/HQnsTNqlOZUmPon+JNwC0mSb496+5Tr1OXOy6NCIT8I/oBL7ijI83+/\n3+/3+4Vb8LJ5QfJ4PFy/3kH+Gse2s6k31m+Ejf0+kGVZLsJC6ag6V2+E5fXS158ufxoATsAJ\nYT1a7LM8VfbUq/BqCi8Y5SsMcCCEUO5ZrVYuuiE/fbpm6VJ6xIj2FSvGzZoFAL7x49W5Xh5C\nCBUwgiB27640mQAAwmFi9epKfld9PTz0UD8Pn0HMsIAlmQkXu6hdO0p3QOk5G6fC1AfMDwi3\nCM//jWHjY5LHhvTUVeGVeYi5bL6he8Pw4XjZfOg5zh5fyi49CActpIUrA5lOTL+ful8f1nN3\niKoxeST4CKgSHVAul3OBMNEaLgXd/7yYrq6u2AjLta5ruQDH5XC5QtF3kNhn2aYe2oVgiIcB\nDoQQyj2fzwcApMcz/MEHAaC1oUFqsRA0DQC+CRNkwaBMJt7DHyGE0OB9/rn49m9/u//HJj/h\nQnTY5xx2zin/Kf4+scn5VbVVELcNyBDAhe85sZfNV+tX42XzBE7AiaXs0s+Jz/Nn7ky8RhuN\nqkZLlaWhtQHEfo2NBmPiAIder+cHNsO5NVxqRi2MTYiKRCI2m024JTZRSC6Xq9Vqj8cj+izF\nOKi5UGCAAyGEci8SiQDLDlu8WG4yta5ZQ48apd/WeyXBP2GCRjBcECGEUNrt2pWNZxENhTAs\nw/8smpx/OTu0S1Tk8r7eCXjZPHnhcNhisWyiNr1q6AsA5UPf2QSNNlrlrRDn13gDbEh8WJVK\ntWv4rjZ/W7/tbET5/X5hKC1ef43q6uo/2f/UDd2xz3ILccvA3hCUbzDAgRBCuSeVStWvvqp5\n772eWbOCNTWKo0dV+/dzu1iSlFosMGJEbleIEEIoE0iSlEqloVAI4iTnS4d0/gZAUVGRTCbj\nu6sCXjZPQjAYbG5uDoVCap06r/rOJm60Mdk3GeL8GitpZbxj8j4o/mBH8Y6odjZT2CmrJav7\nfSw3/JUTG2GpK6nbAlsAQCqVHjAcSKZpDhq6MMCBEEK5p9Foig4eBIDipqbipibhrpELF0Jd\nHTzySI6WhhBCKLN0Op3VauVvRg1YIcih3IEDgCCI6urq1a7VDokDL5snqaOjg4t55VvfWZfL\nJbwZ1WhjkWURvyvVGhOIU8N1OyHSziYWRfWd1cZGWP5d/G9+b/JNc9AQhQEOhBDKPYPBEDl8\nOO5uHBOLEEKFq7y8/B/kPyyMpVDP/1Uq1aeKT3dLduNl82QEg0FhnwjIp7kzwkyc2EYbe417\nO0Od093TB/ZrnHw7m1hKpZKiKG4aMScqUHiCOLEMlh2Eg3w3kx/Dj3PbzQRlCAY4EEIo9yiK\nivz3v6fa27kW4opjx0bPng0AwaVLZU88kevVIYQQyqDDocN/K/3bGfJMbHL+SmJlrlaVXtdI\nrumGbrxsngyapoU3o+IIz555dvionM2d4Setijba8Kq9e6R7GgwNwodMhamryFUZWs8JOBu2\nIM4ExgWKIkU1wZqHux4eExgTFWHZGtjaqGjkH5gP3UxQhmCAAyGE8oJcLq+trQ0EAoFAQPre\ne9xG2RVX5HZVCCGEMicUCrW3t7+oePFM5ZmoXRfBRXuJvQVz/j+Yi/Pns9g4wlNlT70Gr/X7\nwAxRKpVcdoloo43lkeV2sGctjMUwzJbAlsaivrBFj6TnsPLwvSPvFd5tMjP5py0/fUv2Vh2b\nR91MUOaQuV4AQgihPgqFQqfTqX7+c2BZYFm4/vpcrwghhFBGMAzT0tLi9Xq1EW1dW932E9vf\nOfrO/Zb7ub000AUT3YjnaOToLZFbRrOjNaCRgnQYDFsAC6xg7f+RmXc4dPjm0M2jIqPUrDrV\ntZ2AE3NgzhgYM7DXFTt35vNDn29s3ihn5QDwhuaNAbycdCkpKSHJvvNHvgwEANSMegm15D/w\nHzvYaaBbofVdePceuCdDv8aBQODEiRNgh9jPjgQkMpBVM9VXM1dvYDe8eOpFmU92o/PG77m+\nN4IeYQgbruu5jrtnDruZoMzBDA6EEMozdjtMmwbNzQAAK1bAsmW5XhBCCKH0s9vtXDFCvjWS\nzIJAINDZ2blJsWlL5RZ+Yw6rBvoqHeBMAALlkfLyUPnXRV8PYG1er/fl8MuN2oFXQ8hkMpVK\n5fV6+S35M3dGKpW+Pert0z2np7unG4IGu9Sek34xDMOYTKZQKCT62ZkH8zbABu46vsPhMNPm\nvgee2xV1MSwe4kOKkAgMcCCEUO6FQiGn0xkIBCKhUOX8+XIuugHYXhQhhAqW2+0W3ow69fp1\n6NdQlKOVZVggEDh9+jTDMFqpNh9moHq93kamsVHTF5LolHR2FnVO9E9c1b4qpbV1dXVZrVap\nTlrXM6jXVV1d/Sf7n2yELQ/7zr6teHuHYkc2G23Ecjgc3JQZTtRn53fs70DWu0vYrjW2K6re\noIeSrK0aZQkGOBBC52AYJhKJUD09xCWXYBJBdvT09LS3t3Mj3I319fIPPmAUCjIQAMAAB0II\nFSzhGVrsqVdlZWWO1pVxHR0d3J+8nKeusCxrNpudTqdEJ1ntWD2BniAMSQTIwAh6hHBtlzKX\nJqjv7+np4cb9Dv51yWSyT42f7iR25uHcGfFhrtntFytMb4n97JQYSviwRSQS4X4Q7Yr6bPjZ\nrK0ZZQ324EAI9XK5XCdPnjxy5MixI0e8P/gBYBJBVgQCgba2Nu6rXvHbb5dv2OCeOZMePRoA\nmMpKMBpzvUCEEEIZIZFIuB/4Uy/uJnfqxe8tMKFQyOfzCbcwBGOSmbjL78awcTlkbwaqxWJx\nOp0AcKPzxut7ro9q0DDZN1m4NkPI8Ev3LxMcrbu7m2VZ/uYgX9cMYsY0mKYHvQxkw2DYTJi5\nETbug3060KX6MtPrUXg0a402eMeYY8J2LZdVXfZY9WMOyiH62eGDGgBAUb27YruZbNdt5/ei\nQoL/qAghAACLxWKxWLifjQ0N6r17MYkgO/jvQ/LTp2uWLqVHjGhfsWLcrFkA4B03Ts2y/Eg2\nhBBChUSlUnGjwUUHUhSxhVmgEgwGhTejLr8/Y35m+JgszUBlGKa7u/ucLedWOnxZ9OWUiVP4\nta1tXWssiXvVgWVZv9/P/8kWScmprUyp3QPOneH4/f7Ozs7nlM9GLzZ9AAAgAElEQVQJ27Wc\noc68rn/dKXE2tDbEfnakrr43WqPRcDEsjrCbiZpRq9XqrL4YlBWYwYEQgkAgwEc3opIIwkYj\nazDkdHUFjruQRXo8wx98EABaGxqkFgtB0wDgnzgxwMWYEEIIFZzS0tJ4Ayk0rEa4q5AIo/ax\nl9/XGNZkbSU+n0+YcHHT6JumTJxyw9gbPlZ/PNk3+aXTLzkoR/JpNSzL8kcTTSvgUjVRSrxe\n7+nTp30+n+ikoVZ5K/dD1DAXYdiiuLh4m3HbuvJ1h5WHbZTtuOL4suplXDeTG+kbpVJsMVqA\nMIMDIQQOh4P7ITaJwD9hAuH1YoQ7cyKRCLDssMWL5SZT65o19KhR+m29Xdb9EyYUCdIsEUII\nFRKpVNpU23S65/QVPVdENZK8lbw116vLFIVCQRAEFwuITV35d/G/s7YSYcQhtkHD3wx/i00N\nUNGqeEcjSZKiqHA4DHFScqQsnkunhuuQwv2qiLY1kTPyw8rDUZ+dHwR/QCn7znAJgthXti+2\nm8nE0MR6ZX2WXgnKLgxwIISAG1MXL4mADAQwwJE5Uqm0eMsWzXvv9cyaFaypURw9qtq/n9vF\nkqTMagV88xFCqEDtke/ZUb7jr+V/FW7Mh0aSmUOSpF6vt9vt/JZczUAV9l8QDUmsMK8Qrk3D\nahSKRJ0mtFqtzWbjb0Y9tlBTcjLH5/NFFTQJa4jkrPyw8vBttbcJ7zApPCk2bDGDmGEBy0n2\npI/wlUXKatnau+Huu6V3Z7MrKsomDHAghIAgiARJBGrsAZE5dvsF110naW0FgOKmpuKmJuHO\nkQsXQl0dPPJIjhaHEEIos/JhIEX2VVRUPCd7rivSldsZqEqlks+54HAhCQkrAQKUrNJG2VJa\nW3l5+T/If3RDd+zrmg2zM/paClLidi1X0Vcd0B/gPjvlTHktWzuXmHsndWfsZ6e3mwn3ZbYw\nW/eic2CAAyEECoWCeuGFeEkESrsdSktzu8IMstth2rRsDsT1er0ejycQCEgIwviTn0hbWxPd\nGzu8IoTQUMAwzCef0P/8J3ngAGU2S5xOqKqCOXNgxQqQyeI+6vxsJEmS5L6yfTtgR25noBIE\nUVFRsca3xiFxCEMSPtIHAD1kzzXjrhGubSWxMvEBKYo6YDgQWw0xhZ2yilyVgVdQUE7AiWWw\n7CAcPANnAhCogIprVNf8lPqpPqwHsRqib0W+9R/4T++DMTkGCWCAAyEEOp3Of/AgxEkiYFev\nhl//OkdLSz+GYRwOh9frDQaDUomkcv58WbYG4rIs29HRwXc8MdbXS997j59Wc/T998OlpYpj\nx0bPng0A9JIl8j/8IaPrQQghlBZ2u72rq2vFiuqmpr4KC5MJVq0ChoHVq3O4tDwlTF0phdII\nRADgJJwsh/IKqPgf+J+VsLIcyjO9DJ1Ot1+1f490T1RIogiKZCDzgS/VtBquGoJ7XQbWMBpG\nzyXm3k4UeErOILEsa7fbXyRebCxp5De2Q/uLshfNVeaG1gYQqyHapt6WuyWjvIYBDoQQyOVy\n6vjxeHuJadOyuZiMCoVCLS0tXM8RANDW18vefz9rA3EtFgsf3eCn1VBWq/Kbb9iqqpGXXRYI\nBKTvvcfdQT59ekYXgxBCKC1sNltnZycAmM2y++6zzprl0usj69aVv/pqCQDs2YMBDhFc6go3\nqLWBaHiy/El+Vzu0b4SNNrBtg2ycwV4rvdYOdq5BQzlTPhpGzyPnDbhK6JyUHCzwTUIkEmlp\nafH7/Qqdos5bNz4wXhlRbi3ZyrURbVe2C++cq3YtaGjBAAdCCABAcviw1+u1Wq1+v196+DCX\nRBBZvlzy+9/nemnp1Nraykc3okIMUF0Nxrjz7QePYRi+91jstBp60iSFQqFQKODnP4ef/zzR\ngbJeU4MQQiieSCTS1dXF/bxhQ7NG0zv66q67bFyAozzjWQhDFX9mq9Kp6tqiz2xPwInsLOOc\nBg1Y6ZB1ZrPZ7/dDnDkp0yXTt5ZvFW1rks12LWhowQAHQqiXSqVSqVQAwO7dy22RXHZZTleU\nZh6Ph/sjCmIhhtDkyRkd4Obz+biJdKLTanwTJsS7VMQwjNPp5HqJyyjK+JOfSLNVU4MQQigx\nj8fDTxvloxsAsH9/7wCs2dhcMo6urq4EZ7aXw+U5WxlKk9i2GlHFR6FQqKenR/gQ4ZyUikjF\n45LHFxkWxbY1KexJQ2iQMFCJEIpGLFgALAssC9dfn+u1pBMf3YgXYvD7/Ux3N4waBQQBBAFP\nPJHGZ+/9Bnx2Wk37k0/So0YpDx/m9vrGjxd9VDAYPHXqVEdHBxfjkK9YIX3vPVap7N2NAQ6E\nEMqpUCgUu/HoUcXatQYAuPjiwIIFWV/TUMCyrNPpFG5hCMYkM3FntsawcTksz9HSUBowDNPV\n1fVC9wuN0HgSTnrAE4YwV3x0L9zL343/Ysa5afRNUyZOuWHsDR+rP57sm/xG2xvDYfgMYsY0\nmKYHvQxkw2DYTJi5ETbug3060MV79hNw4jb2ttHsaA1opCAdBsMWwAIrWDP4glE+wQwOhND5\nIirEEDUQ11lb6z5xYuSiRerM5EdQFAUA+m3bRKfVSGQyMJuhulr4EJZlc1hTgxBCqF8SSfTY\nyaam4iVLanw+8pJLvM89Z6eoYTlZWNb0e5VeVDAY5DNfIGYC6NNtTw//1vDMrhtlTDgcbm5u\npmm6SFeUuPhI+DsQOyelrqTuNXgtpUlDLMtardbn2OdeNb7Kb8xyVxeUcxjgQAidL6RSKcQP\nMbAkWbVypXrv3gz1HFUqlRRFqeJMq6m85x6oq4NHHhFu5KbJcj/H1tSEp0zB/4MjhFA2ffYZ\nbN4Mn3wCra3ADYK95ZbiO+7okEpZAGAYeOYZ47p15SwLs2fblyzpNBZ0GDre8ItkTiZZluV/\njj2zXWNYcxVclbmVo4zq6Ojgrs30W3wkE4xQjp2T8obmjZSel2VZk8nk8Xg0Ok0Ou7qgnMOv\nxwih84VGoyEIIl6IYeTChQDA50dEKiokaf1iShBEZWWl/NChuPeIiad4vV7uB9GamsCkSeo0\nrg8hhFBCkUjk8ccj27f3nZKZTPDnP0vc7mEPPNDqdksefbTmo480Uim7eHHH7NkOiqL0en0O\nF5xR4XC4paUlEAjItXJ++MXrpa8/Xf40JHEyKZPJCILgwhyxZ7bbddu5gyxll35OfJ5SbgjK\nrX7baiyX9BUfKZVKmUxG0zRB9I6cGcycFIfD4fF4IK1dXQaWoIRyCwMcCKE4Cm5ah1QqNRgM\nivghhpDRyOdH+MaPVzEMSaazUZFWq3X+5z/NnZ2RSAQAFMeOJZ5Wk7imhsYAB0IIZUsgEDCZ\nTKdOjYgdBLtvn+a227T33GM0mWQUxT788JmxY+lDhzQVFRVKpWTEiFwvPTPa29u5HMMfuH7A\nb7zWdS0X4Oj3ZJIkSa1WK2zDITyz1bCa1rbW5+RYaDD08JmnnKjio//r/L/htX3FRwRBVFVV\n1fXUOSSOwc9JcTgcwpvCwIohZFhKLoXoerJ+MAzzmv+1RlXKCUootzDAgRDqxY2jd7vdNE2T\nACMWLlQW3LSO8vJy2/79zRZLdIhBpyOCwZZ16/j8CP/EibJQSC6Xp3cBOp2uuLjY5/PRNK18\n/31uY7xpNYlrakTbdiCEEEo7hmFaWlrC4bDoIFiDgWhqGmYyAQCEw8Tq1ZX8A+vr4aGHcrHi\nDKNpmrtUzhOeTFYylcvJ/luEVlRUbJRuFJ0A+l3Xd3t6egq+0KDgswNii4+eKntqC2wR3ket\nVn+q/HS3ZPfg56TwPcsgJrCytnWtcbgRilJZfDDY0tJCFBF19kL+JSxIGOBACAEIOkJxN8vr\n65UffZShbhS5VVpaqtfraZqmadp3NhtC4nRG5Uf4J0zQnU2YTC+SJNVqtVqthgcegAceSHDP\n4uLirq6ueDU1ujlzYtt2IIQQ4uzdG9y8GfbtI81mictFVFXBnDmwYgUIqv6T5XA4wuEwxBkE\n+8MfhnbsEJ8z/u1vD2Tl+c/n8wlvRp1MbrRtHD6s/xahFEUdMByInQA6LjDuoTMPwXkwPvZ1\n+vVGeaFlBwivDMUWH/27+N+xD7lGck03dJ+CU17wGljDaBg9l5h7O9yuAEVKT83XucQGVuqN\n9f8k/pn8obh2HsFg8MZggf8SFiQMcCCEAAQdoSBmWgdbVUUUVps0kiSVSqVSqbTfcYeZZasf\neyw2P4KgKKnFAjU1OVynXC4vLS1NUFNTSIEnhBBKl0gkYjably/XNTX11fCbTLBqFTAMrF6d\n8gGjzudBMAj2oot8t9zif+CB0niPPRo5ugyWfU58biEtBXOVPvHwi1WRVa/Cq/Ef3WcGMcMC\nFu7M1gjG0TB6tn/2Zacvk7N9J8kDyA3Jf+FwuLW1NSKL1LGFlh0gk8mKioqEH5l+22qcMydl\nEJeWFAoF17xMtKuL8JeqX1w6M38zqtrlkeAjoBr4OlGmYYADIQThcJjvCBU7rSMwcaIyp8vL\nnJKSEoiTHzH8pz/Nh/yIiooKy8cfn+ru5jqx8TU17OOPE489ltu1IYRQHuIuvfp8PrPZENsv\nY8+egQQ4hOfzcO4g2IYGE0mKhyrC4XBnZ+cmatOWyr6c/MK4Sp94+MU2dbIvLXYCqMVtsbAW\n/mZUbsjzzueHVw358bHcr6jf77/RV5jZAVVVVaucq2yEbZBtNRKU8HANaA/CwS6ii9+1uHQx\nePseHtXVJaWuanyXdxCrdinRl2CAI5+ls39ehrz55psEQRAEsXTp0ti9p06duvPOOysqKhQK\nxZgxY5YuXRobZUcIJcZ3hBKd1uGbMCGXi8sknU6nPnYs7u48yI8gCMJoNI4bN27kyJFVVVXD\nLL1f+4hLLsntwhBCKD+5XC7uq+CGDc0PPNA1blzAaAzddZeN21tWxiZ8tDiuIxIAMAw8/bTx\nl78c7vORs2fb161rUasZfq9QJBJpbm52uVzaiLaurW77ie3vHH3nfsv93N6hfpVepVJJJOc0\nbORPJgFAeJX+a//XP6Z/PCoySs2qpSAdBsMWwAIrWOMdWXR8bO+hir5eqV2Z5leSC2632+/3\n8zcZgjHJTHx2wG/p3w7m4AzDfOH5IqX3PO0UCsUBw4G1xrW31d52zbhrbh598791/4YU22r0\n9PS85HypERpPwkkPeMIQ5oKD98K9drv9eevzrxKvniJOCXf9qvhXO2p2rCtfd1h52EbZjiuO\nL61aOrB+pXxMM/aXsN5Yz/VxQ3kr3zM4rFbrvffeq1aro1oZcb755purrrrK5XLdcMMNo0aN\n+uijj5588sl33nnn3XffVSoL9ZIzQunX+30izrSOwMSJuVxchlFHjrjdbofDEQgEZEeOjPzR\njwDybmqMRCJRq9UAAIsWwaJFuV4OQgjlL7fbzf0g2i/jxhtpSLGwHwCKi4vtdnvsIFgAIAii\n9//P57JarVyKe0E2kiBJsqqq6i/evyQYfhEOh9vb21+Sv7Stsi+ho98EFoWi718nNjfkddXr\nGX5l2SA8qYnNDtCX6WGg/c09Hk97e3ujtjH2PX8ZXr4dbs9abdTV5NVWsAqLj+6Gu5Nsq8Gy\nbFtbW09Pj0wnq3NHl/AcCR/p6OhQ6VSiDWjDuvAO2BHbr3QlkVpojI9aila7PBt+NqWjoSzL\n9wDHT3/6U5IkH3744SeeeCJ274IFC5xO53PPPfeTn/wEABiGueuuu15++eWnnnpKNN0DISSK\nyzWNN62DkssLe1qHRqPRaDQAAB9/3Ltp2rQcrgchhNCAhUKhqC3Cfhlz5vgHEOBQq9U2m37e\nvHLhINivvy4CgPHjdRQl8nXa5XIJbw5+YmW+0Wq1B1QH3qLeEh1+wVdhaCltSpNQNBoNRVFc\nS1dOgg4OQ3QKCX/9X7QX5t+Dfx/YYQOBQGtrayQS4ZKGjiuO/6P8H/xeP/izWRsVW3yUPIvF\nwtVNiwYHJ7onxtt1OVw+FsZ2QdfAAitCxcXFFktfqZTwl1DNqIuLRTqJoPyR1wGO5557btu2\nbdu3bz9xQuT/g59//vmnn346depULroBACRJ1tXVNTY2Pvvss0uWLCEyMwEBocIjl8uVSmW8\naR3GefPyoRtFNsyfD/Pn53oRCCGEBo4vnTh0SPnmm7oPPtCYzTKWBbmcnTTJRxAD/Or71ltV\nJhMBYoNgL7oo+s4sywrjLLFX6StHVg71AAcAzKRm2sAmOvzC1ePiqjBET0QvZS6NVyVPkmR1\ndfWf3X8WzQ2ZDbP5e4bD4UZ/Y6Nm6E0h4X9FRbMD1tnWDeywXV1dDMMQBMG95yEiVNdWp2SV\nDwzvm9eW/7VRDMPYbLZztgiCg8awcZFlkegurvfneNX4AQdWhBQKxc5hO9sD7bG/hDfSN8qL\nBppjg7IifwMcLS0tDz300D333PP973+/vr4+9g7vvvsuAFx//fXCjdXV1ZMnT/7yyy+PHz/+\nrW99K0trRWjoq66uJg4fjrs7D7pRoHg++ww2b4ZPPoHWVnA6YTCjEBFCaKgrKiriqlT+8Y/y\nd97pu9BK08TmzWXl5ZGnnhrIYb/4QvyymeggWK55HFf+KXqV/hXylYEsIs8kGH4RVVoedSL6\nsPdh0MU9rEaj+bTo092S3aK5IdzPPp/PZDJJNJI659CbQqLRaOx2O38zKjugN6U0RSzLRr3n\n33d9v03atrJqJQCUhEvslB2GQm0UTdPCnr5RwcGn257Wh/Siu9a2rjUajGns/fmh9sMd2uhq\nl0nhSX8t+mvangNlRp4GOBiGmTdvnk6nW7NmTbz7HDt2DABioxhjx45NKcDh8XhisxmFsGsp\nOh8oFAr6v/81nTnj8XhYlsVpHSmJRCKRSETqdhOXXALNzQDZ6+Lh9/uXLWN37y7itwxmFCJC\nCA11Op3OYrH09JAHDqgBgKLYBx/sam+XcVNU3nlngIkTu3aldn+lUsl9gRS9Si8Fkb6khURY\nYxJ7IlpRUpH44ddIrumG7niFBgzDcLUYQ7S/iUajeaPija5IV2x2wA2BG+SqgWQHMAwj7M8q\nfM/HBcZpI9oD1AFDyLCEWJLlk79Uy4gSTyBeY1izwrxCdFe9sX49uz6NK78KruKrXcqZ8lq2\ndi4x907qzlSrXVD25WmA46mnnvrwww/37Nmj1Wrj3Ycrboy9g06nAwCn05nME506dWrMmDHC\n/yPEk8x9EBrS5HL5iBEjWJalaVq6bx+3Ead1JGaz2Ww2WzAYBIYZuWiRmotuQKKcl88+g+ef\nZ/btIwefcOF0Os1ms8lUm65RiAghNNQ5nU6TSfqzn430eEiKYn/5yzNTpvgrK0Pc/xvLs9Wc\noaysrLW1lb8ZNbGSIIdGGfU39DfLyeVfSr60kJaUmlzwVRgDOxFN3MHB6XQKAyixdQr5P8Jz\nb+nencTOqOyAiaGJA84OIEkyXtLQUcVROBtaMtQYQKQlbqa43e5GSK2MSDiTSDQ4yAU4RHcp\nA+kcMXHOL+EQmDuK+uRjgOO///3vsmXLFi5c+N3vfncAD+c+20k24Kitrf3666+5NtfxbN26\n9Y9//CN29EDnCYIgFAoF3Hsv3HtvSg9kWZYgCLDbYdq0LGcx5Ep7ezsfSzU2NKj37mUUCpKb\nuSsW4AgGg11dXYsX695+uy8BdcAJF6FQqKOjg2XZDRua+WEBd91ly/KXeIQQyis9PT3btulN\nJhnE9MsAgB/+MARZyZ4oLi7eNXxXm78t9ir9reStWVjAIPl8PrPZ/KL6xa2VW/mNyTe5UKvV\n3N9H0RNRVXBQEQjhjNXY9JDSktL8D3DMIGZYwHKSPekjfGWRslHMqLvh7rnSuQPODiAIoqio\nyOv1wtn3nCboa8Zf4yF761a40NJmYnPaXkNCkUiktbXV6/VKdH1lRP82/PuvJX+FhGVEMplM\noVAEuK9SABAbHDwbx4GY6h6coYk4eRfgYFn27rvvrqqqqqurS3xPLncjqkk1xM/siGfSpEmJ\n7/DZZ58leSiEzkPBYNBqtXq93mAwKJVIRixcqEgii6EAuN1uPrpR/Pbb5Rs2uGfOpKxW5Tff\nhI1GymiMun8gEGhubo5EIu3t5WlJuHA6nVwmp+goxBtuCAxgUgBCCA11oVDoyBHx85wxYwJ3\n3pmlAAcAfFD8wY5ikYmVfCOJvBUIBFpaWhiG0SpTm4HC02q13d3d/Glq1DmqXD6oHo18FUN6\np5BkU292AHfxVAJp6ThrMBhaWlr4k385K//kyCdXj7vaKXESQLDAbtdtl0ey1B2zra2Ni7YI\ny4iutl/NBTgSlxFVVVWtcq6KN4H4rZFvmbym6e7pCRrQovNc3gU4IpHIV199BQCxLXaefPLJ\nJ598csGCBevXr4ez3Te4ThxC3MiVsWPHZmO5CJ3ffD4f9x2Iu1ny1FOKDz9MnMVQMBwOB/eD\n/PTpmqVL6REj2lesGDdrFgD4J0yQBgIKxTnxhfb2dm4yXLoSLoTXNzjCUYi33urFAAdC6Dwk\nkUj+/vcW/mZTU/GSJTU+H3nJJd6GBpNMNiJrKxHW8A9mYmUCGZqT2tnZyf1lH3CTC4Ig9lyw\n55Tr1GWOy2LPUQezNjg72x7ipIesd6SzEcMQolKp3h71drO7WXjy75T05tHQBK1hNaIjjdPO\n5/Ml6DJbxVYtJ5YneHhRUdGncpEus1PYKSuJlXep7tqh2tFgaIjatVqCdbmoV94FOEiSXLBg\nQdTGQ4cO7d+/f+rUqRdffPFVV13Fbbz22msBYPfu3X/84x/5e3Z0dHz11VfV1dUY4EAoKYOo\nKGEYpq2tjY9uRGUxQHU1xGQxFBKutI30eIY/+CAAtDY0SC0WgqYBwD9xIhsMCgMcfr+fj0eI\nJlzMTv3CQ1RjoKgv8RJJWb9HwPErCKHCU1RUxP3/mWHgmWeM69aVsyzMnm1fsqRTJiOymcSe\nuJHE4IXD4dfo1xpVaZ6TGolEuGvvvKg5ncupRGenvJ2SnTtKdkDJORunwtSVxMoBr42j1Wqt\nVit/M6pOobi4OMFjC1uTsmmHMvrkXxvRuiQuyGKOQ1R0I6qM6EX3i8ONwxMfQdhltm8CMXG7\nAhTicUMinXFDNNTlY4CDS9AQqq+v379///e///0//OEP/MZvf/vbl1566aeffvrCCy/MnTsX\nABiG+c1vfsMwzMKFC7FlBkLx0DTtcDgCgUAkFKr+3/8dcEWJcAJRbBZD8MILC/s0mSAIYNlh\nixfLTabWNWvoUaP023q/UPonTIj6MxubbQGChIuLLw4sWJDyH2Y+xTf2SzxFsf0mADscjiVL\npHv29HUbw/ErCKECUFZW5nQ6e3rIRx+t+egjjVTKLl7cMXu2AwBKS8tIskC6BbpcLrPZTBQT\ndfY0z0mNmi0Y2+Ri2LeGQRLfsjOXwKJQKHYO29keaI8tYfhh6IeUIjdnNxnKpkkJ955/BV+F\noO8fkYtuTIWpq8hV2VkGl6/KiS0jepJ98hXoZ0xyggnEmY4bogKQdwGOlGzYsOHKK6+85557\ntm7desEFF3z00UcHDx687LLLfvWrX+V6aQjlKW7uBnfx31hfP5iKEr7Ll2gWQ2DSpMIOcCgU\nCuXLL2vee69n1qxgTY3i6FHV/v3cLpYklXY7JLyIJEy4WL/eSlEjU12ATqfr7u4W/RJPUVRs\nlZ+Q2Wx2OBxtbaNx/ApCqMDI5fJweOTtt0tNJhlFsQ8/fGbsWPrrr4vUarVaXSDtl30+X3t7\nO8uymZiTKowBxZ6d/rXirzOIGckcZwAnosnHCD7UfrhDG93f5MLIhWsUa1J6xnTxeDz/DP2z\nUR83m+ZI+MgyWPYF+UWq82hSwr3nq2DVFtiS0dqoxBJPQtlatDXBYwcmH6JLKH8M7QDHpEmT\nDh48uGzZsqampl27dtXU1CxevHjx4sXYRBchUYFAgI9uRFWURCoqJAOrKImTxRC88MI0rjwP\n6fX60MGDAFDc1FTc1CTcNXLhQqirg0ce4bcIy1ViEy5KS3UDWIBcLvd6K2+/XR31JR4Apk41\nJrhK6XK5uAYiGR2/8tln8MIL7N69BNa/IISy7NVXVSYTQMwUlfp6eOihnK0qjSwWi7BKcWAl\nJPHIZDKpVMrlccSenb6pfXOQi4+HpumX/C816pKquBGmhxhYQy1bO5eYe4fkjmzWKfAn1Z1s\nZ0AVUDPqS7yX/KLrFxXBCmE2DTfy7HnZ8/+q/FcyL23wcp7joFZHj6IVlhEVQ5priEKh0Kv0\nq43qNNdqoaFraAQ4fvGLX/ziF78Q3VVbW/vSSy9leT0IDVHd3d3cV6LYihLf+PEqhkkpd5cr\ngtBv2yaaxSBVKMBshurqDLyOvKBSqcIxTY77nJsOo1QqubFnbrckNuGipKQkzlH68cYbJfG+\nxE+YEPdRfHtU0W4gN93EDHLgO8MwFotl8WJVWqbhIoRQqj7/XHz7t7+d3XVkjLBHRmwJSdXo\nqkF+wS8vL+/o6OBvZvTslOPxeFpbW2XFsjp3UhU30SUMWS9MDwQCr4bPnlQTAAAuies/qv+s\nL1vf0NrAZ9Ncxl7W3NwcDAa1ugHOoxmKFAqFXq9fR6yLNwkljc/V09PT3t5OFpN1jvPl7UX9\nGhoBDoRQWnBFJfH6YpJ+v0qVwux4jUYjkUhUcbIYtLfdFpXFUHioI0fcbjc3CU925EjtzTcD\nAPP735PLRa6e1dTUfPCB+b77hkUlXOj1eo1GOWJAff0H9iU+8fiVO+5gAVL4NYgSiUSam5sD\ngUB7uw7rXxBCObFrV65XkEkMw/DpG6JzUi+LXDbIYRklJSXrJetFm1yk9+yUE4lEuJ7lohU3\nl7GXZT9+kZjFYrFYLKRO5KS6Vd5qkpn4bJqf2X8WDAZhEPNohqiqqqpP6E/eUbyT0THJNE23\ntbVlqFYLDV0Y4EDoPMIwTIK+mMqz81CSJJFIqqqqFIcPx71HQY+J5Wg0Gq7bBbt3L7eFvPRS\n0XsqFIr337/AZCIhfVnTafkSHzV+haIGNUOxq6uLC6BktAOEpCAAACAASURBVP4FIYTOWyRJ\nkiTJjTATnZNKMWn4ei/a5CK9Z6c8l8slbEsprLgxhAy/9P8yM1kjA+RwOCwWC8SJWZySn7ph\n7A1wNptGQ2iE/T6jXtoSYkmhnooRBPFdxXedrFM4CWUeOW8wrUBiG21cGbnyZ5Kf6cN67g5R\ntVqPSR7Lt9AYyo4C/VQhhMRIpVL1q6/G64sps1ohYWfKWFqt1vvFF+1nzgQCAZZlFceOjebm\nnaY4cbYAEAsWQMyI6yhffile+pHlrGmFQsGNcBvY+JUEWJZ1Op3cz+mahosQQiiKRqNxuVz8\nzag5qRKJZPBPkbkZKLGEeYWxFTdlurK8CnBw0Q2e8KSaBJJkSWE2zRMdT/D3jH1plSMqC/hU\n7FF49FFCfBLKAIRCoVf8rzQWn9No45WiV7qquhpaG0Ds7S0fXg5Fg31eNBQV7qcKIRSjuLiY\nSrovZpJUKlVtbS3LssFgUHbgQO/WadPSsd5CkydZ03q93uPxiHYD0Wg0g0lsDoVCTEwekLD+\nZf58ZQ5KpRFCqLAYDIYN1AY7aY8tIbmZvTktT5HNRpWJK27+Fvhb5p461ekbwWBQOEY39qRa\nH9YLs2n+0PkH7tWJvrSXyZcz99IKic/nM5lMlIaqc4nUBEGct/cF5gXRo+HIlYKHAQ6EziOl\npaWhI0fi7h5ERQlBEHK5HJLIYkA5p9Vq//tf/113lUR1A6Eo6uKLq9L7XDH1L+PTe3yEEDoP\nyeXyA4YDuyW7o0pIJjOT66i6XK1qwPjMQdGKm/WO9Rl63nA4/Br9WqMqhekbwlIa0ZPqFeYV\nfDaNhtUUFRVxKZOiL00GOFesfwzDtLa2RiIR0Zqgyb7JEOftlQalsUcTzQTBkSsFBgMcCJ1H\nCIKQHD7c1tHBpbbyFSXBpUtlTzzR36NR4di1qyITMxSlUqlEIuG+/8XWv6hUMoLA9A2EEEqD\nayTXdEP3Sfakj/CVM+W1bO08ct4dZFbnpKaLVqvt6uri8ziiKm6Ki88pUEnX5XeHw9HR0UFo\niTp7b1LA66WvP13+NCScviGV9p0z8yfVf67480ulL4WJ8Hbd9rtsd/HZNLNhdllZGRfgiH1p\nGlZDkPg3sX9OpzMcDvM3o/qYLLIs4ndF/ebEltx6vd7W1lbRTBAcuVJIMMCB0PlFIpEMGzas\nsrIyEAhIPviA2yi74orcrgol47PPYPNm+OQTaG0FpxOqqmDOHFixAmSpXwHK0AxFgiB0Op3N\nZhOtf9Hr9YM6OkIIobN6S0i4E+RBjfbOPalU+tbIt1o8LbEVNz8K/4iS956tsCxrt9tfYF9o\nLBvs5Xe32202m+HcRqHXuq7lAhwJpm9QFKVUKrmZdBwCiGZ5M5fEESJCN4/uLRGawk5ZRa5S\nq9V7Lthj8pqmu6dHvbRbyVuTX3AB6zdiJXy3Y2uC3tO9ZyNssb85NzE3wbm9aCKRSIJMEBy5\nUkgwwIFQoaFp2u/3RyIRhc+nuvpqaG4GiO76SVGUWq2G+++H++/v94DBYDAQCBAEofD5pN/5\njugBz08Mw9A0HQ6HZR6PfPr0jL4zkUjk8ccj27f3BTNMJli1ChhmIINXM9cNxGg0HjkSmj+/\nIqr+paioSK0uLSvL1PMihBAaut5VvbtDFT20ZTIz+S/yv3A/syxrMpk8Hk+RrqiubbCX3xM0\nCq2IVCyXiMx651VUVKx2rXZIHPxJtY2ycbtIIKvYqtEwei4x93aityEr99IaDA3Cg2RoHs2Q\nk0yVUOIWLT3KnthhtBdGLvyz9M9Rz+VwOBIM6/lN+DegzMArRLmAAQ6ECkc4HG5vb+9NhmSY\nkYsW9Z5ywwD7awQCAbPZ3Bs7Z5iRixZJB3fAXGFZlqZpkiRlHg9Mmzb4SATLslartbu7m2EY\n7p2RZ/Kd4ZIqT5264L77rLNmufT6yLp15dzg1T17BhLgyBySJJuahplMBKS7/gUhhFChEg5t\n4UaKziXm3k72DW2xWq3c15vBX36PRCIJkgKebnt62NhhCdphq1SqA/IDb1FvxYZjPiA/0BG6\nBC8t0/Nohhan09nR0UEU91UJiUasZGfzVEUbbfzO/zsXuLi3t69WSyJSq+Xz+fifRYb1lJdh\ngKNgYIADoQLBMExzczNN09xNY0ODeu9eRqEguelrqZ910zTd3NzMR7sHf8CcCAaDnZ2dHo+H\nZVkuEqFORyTCbDbz81Az/c6Ew2EuqXLDhmZ+9updd9m4AEd5/vX8/uIL8S+GWZ6GixBCaKg4\nZ2iL2N8Qu90uvCm8/G4MG5dTiXIuoiRuFLrGsOYK5orEo3ZnUjNtYBM2QPmJ5CfCcEzcl4bO\n8nq9ZrOZZdl+I1ZardZqtcZr0bJMtuxxeLz3rglrtfgpb6KZIM+Gn03jq0O5hQEOhAqE3W7n\noxvFb79dvmGDe+ZMympVfvNN2GiUGAypdrI6c+YM/yUg6oCRigqJ0ZjW5WcETdOnT59Oe4zG\n4/Hw0Y3Yt5osL09vNbTNZuNeAh/dAID9+9XcDzfdxORb+XWeTMNFCCFUGEKhkLDNZOzl95qx\nNcn/JRROQxdNCpAQiaIbUFgNUHLFYrHwMQtIGLGSy+U7h+1s87fFNtr4YeiHlCLZk9nEmSCy\nnn76meFw2SEEAxwIFQi32839ID99umbpUnrEiPYVK8bNmgUA/gkTSJ9PpVIlfzSGYfi+36IH\nVKd7/ZnQ0dERL0bDVFaSA43RcDNoIM47w3o8US3fB8nr9UZtOXpUsXatAQAuush3221hgHQ+\nHUIIIZS3RC+/XwlXJn8EkiRVKpXwb2vUcJME9SkoLViWTVwwUj2mWtgi9IPiD3YUR7douTBy\n4RrFmuSfVKvVCvOAojJB1OpEX2x9Pt8rYRwuO2RggAOhAhEKhQCA9HiGP/ggALQ2NEgtFoKm\nAcA/caJccOkjyaNxkXXRA/omTChiGJLM68sWoVCI//oSG4kITJxYNIgjQ/y3mgwG07H8PnxS\nJaepqXjJkhqfj7zkEm9Dg4kkq9L7dAghhFBeoSiKJEnur6Ho5XeSSO0LidFoXOVcJWwUyicF\n3MzenJHXgAQYhkncOvTyyOXCKqGoFi1co43bJan1MVGpVNurt3cEO0SH9UjkcdN2Ojs7bTYb\npesbLpvMRGGUQxjgQKhASCQSYNlhixfLTabWNWvoUaP023qDyv4JE5QpBiN6gxfxD5jn0Q0A\nCJ4NNIhGIrzjxw84wEEQRIJ3RpPud4ZPpmUYeOYZ47p15SwLs2fblyzppChWmGqbD9I4yxYh\nhBCCszPI411+H0DORVFR0QGZeKPQ1ZJ8atxdoCQSSeKIFcWe890mukXLQFNsPtZ/vANEMkH4\nYT2xuru7bTYbnNvddqZzZr8ThVEO5fspCkIoSSqVSr9tm+a993pmzQrW1CiOHlXt39+7TyIp\ncjhSOppUKpVKpfEOKFMqwWxO8lAMwzAMA3Y7jBoFBAEEAU88kdJiBoYgCIC+GE37k0/So0Yp\nDx/m9tKTJg34yEVFRfHeGZYkU32r+8UVvLjdkgceGPHss+UUxS5fbl6+vIOiWIlEklLlUaY5\nHI5ly3wNDXDwIFitEAr1zrJdujTXK0MIIZS04+zxW5lbx8AYDWikIB0GwxbAAitYc7gko9H4\nuuH1deXrDisP2yjbccXxZdXLuMvvtxC3DOCAM6mZ02CajtVJWWk1U31V+KoN7IYD5AEdRI9B\nQZkQVRLCR6wAQM2oM3QV7Sq4ahpM04NeBrJqpvpq5uoN7IZPJZ/G+0fnRuYJtzAEY5KZ1lSu\nAW6iMKTQ3RZlTX5d+kMIDVhpaan34EEAKG5qKm5qEu4acd99UFcHjzyS0gHLysokcQ5Yec89\nUFcXfPBBmqYpipJ7veSll0bNXmVZtru72+FwBINBYJgLfvYzVeZHzDIMEwqFZB4PccklRc3N\nkwB6rr02UYymunoAz6LX6z1x3pmRCxcO4K3u9+m++MK9YEGlySSjKPbhh8+MHUt//XURAEya\nVNobx8k1lmVNJpPH4zGZRuf/LFuEEEKiaJo+c+bMc/LnXqt4jd+YD+0GJBLJ/vL9O4mdUZff\np8LUlcTKARzwnEahBF7zzTaj0bhRutFO2rNZJXROJkgS/+I0TQtn7kT1CnnG/MzwMcPTv0o0\naBjgQKhASKXS4hPxSwFTjymUlpaGjx2Lt7fdaHQePw4AorNXWZZtaWnhW2AYGxpUn3yS0RGz\nLpfLarUGAoGo9Uh8PhCLRBjmzh1wJIKiqPS+1YkRBNHUNMxkIgEgHCZWr67kd9XXw+TJ6X22\nlHE1Ke+/H25rq3G7JWVl4VAIamtpqZTN51m2CCGEovj9/ubmZoZhiiXFdW297Qa2lmzlJnfm\nvN3ADGKGBSx8I4YxxJi74e7bIbVGDChPyOXyA4YDuyW7Y6uE6qi6XK0qirAJWmyvkL+U/2U6\nTM/d6lBcGK5EqHCQhw5FwuFuq7W9ra2Tn9W5YgWwLMycOYADUkeOeNxuc3v7qZMn27dv5zYG\nly499M03zosu4m7ys1d7H3PxxQDQ3d3NRzf48SX06NEAEKmogHSPmLVarW1tbYFAIHY9Mmv8\nlNpBRCLIQ4dCweCZzs7m06dNb7zBbWQff3zAb3ViX34p/v/qb387nc/y2Wfw0EMwbRoYDCCT\nwciR8NvfQuKWqX6//7HH/A0N8PXXUoeDCoeJM2ekGzeWr11rBMEs29mz07lOhBBCmWA2m7kz\nuhudN37P9b0R9AhD2HBdz3Xc3qh2AyfgxByYk80ylkfh0f/Af+xgp4FuI9rehXfvgXswujF0\nXSO5hqsS4gpGZkRm5FuVUOxE4c8Pfb6xeaOclQPAm9o3hXc+xhy7JXJLLVObP4Vd5y0McCBU\nUCQSSVlZWU1NTWVHR++madPOuUeKvTDUanV1dXVtbW1NVxe3xTpiBN/7Oip4wVRWcsELvhOY\ncHyJ4sQJAPCNHy+cfD54NE1bLBbR9YSNRvLYsW6r9fSpU0cOH2452wp0MEEfnlQqraiouOCC\nC0Z0d3NbiEsuGcwBE9i1C1hW5L+rrkrbU0QikccfD6bUPsNut58+fbqlhbjvPutrr51sajp2\n6629/+5796qFs2wXLEjbOhFCCKXF0chR4flYDVvzm9LfOKi+NlJcu4G/VPwFYtoN+P3+l10v\nN0LjSTjpAU8YwlwZy71wbw5eydAXCAQ+c332o8CPRkVGnT+nx1zEykE4aKDbyfYPJB/MJ+bn\nVcRKJpMpFOesR9grRMNquI0Mw5jN5k2WTVskW06Tp/ETkXNYooJQgZo/H+bP5350u91Op5Om\naYJlq//3fxUD64Uxfz7Mn+/3+x2nTnEbYmev0pMmKc82woD4g1QV4bBUKk3XC3U6nVzEJHY9\n/gkTWK+3rKysrKwMAGDfvt7HRAV9BknwVqfMbodp06Lal2Sfx+Npa2s7deqC5Ntn+P3+zs5O\nlmU3bGjWaHorVPmaFJaFefNGcbNs165tpajxWXw1CCGEEgmHw52dnZuoTVsqt/AbzYT5df3r\nTomzobUBYtoNrOtaN/yC3nYDdru9s7NTqpWudq2eQE/IqzKWIYdhmI6ODqfTuaV0yxvaN/jt\n+dD3BJ2AE78d8dv3iffdEncEIqXh0gv9F8pYmbC7Lcuyzc3Nfr+/WJePhV3nJ8zgQKjAmc1m\nk8nkcrkCgUDxqlWKDz+MKidJSeLZq74JE/ruGmd8iV94n3SgaTreevwTJ3J7e82f35v5cP31\n6V1DSvx+f3t7+4kTJ44cOuT/0Y8g871XEwsGg62trZFIZMOG5gce6Bo3LmA0hu66y8btjdc+\nw263c3ElProBgpqUEycUPh85e7Z93bqWkhKMpCOE8tGR8JHZ4dnnW0p5JBJpbm52uVzaiLau\nrW77ie3vHH3nfsv93N5WeSsI2g1wG78u+vqpsqe4n/1+f0dHB8uyNzpvvL7n+sRlLEMIwzBf\neL7gcijUrDprvw9ms9npdAKA6D/HkDg9Pho5Wnifo1Ao1NbWtvHMxq3SrXbKHiJCDMFYpdZ3\ni9/drd0NAFPYKVx3W5vN5vf7IbnCLpQd+L0ToUJms9kcZ6eW8uUblNWq/OYbprKSTL0XRtTs\n1dY1a+hRo/RnSz+42askScpkMlVjo+j4EoKiqK4uqKlJywvsFWc9/gkTitL5NGngcDi4r4YA\nYKyvV370UUZ7ryaju7ubK7oWDVXcfDMrOnGe+3MudPSooqHBwP1MUezixR2zZzsAQKvVxj6c\na036ySfQ2gpOJ1RVwZw5sGIFyGTpeEkIIZRQJBLp7Ox8XvL8vyr/xW88T66Zd3d3c6H/G503\n8huv67mOu+A82TcZzrYbYIH9TPXZopGLaILepu59T2w2m/BoDMG0Sdu4MhZDyLCUXAqSrL2U\ntPF6vW1tbY3axjcqs5pD4ff7XS4X97PoP0eenx6HQiGz2bxJvqnAPkfBYPD06dPhcJhLyjik\nPLRPva9F3kITNAAUQdHT8PTtRG93W/5fkBP1iVgMiyFtKcsoWRjgQKiQdZ9tDyFavqFK/YBF\nRUUAoN+2TTR4IS8q4mav6vV6aZxBqsN/+tP0DlJVKBSS558XXQ9Lkkq7PX9meNA0zUc3ouJN\nkYoKSbp7ryaJbwfLE7bPuP12FkDkN0XYWhwAmpqKFy+u8ftJABDOspXJZCpVmfCeLMs6nc7H\nHlPs2qXkN3L9PhgGp8kihDKOG2vt8/m0Ou15mFKe+HxskWURv4tvN0BL6GIo5jYKo9tRZSxr\nW9eW15SDOuMvIb1omjaZTAzDcDkU2fx98Hg8UVuE/xwVkYrlkuVij8sL4XD49OnToVBISxXa\n56ijoyMcDsPZqNP3XN8DgBOKEzeNvgkA5sCce+Ae/s5BQTP22E+EscaIAY7swwAHQgUrHA4n\n6IXhHT++iGUJggiFQjRNkySp8PnISy9N3A+CoiitVquKE7wovfNOLnhRVlYWOnIk7srSmqqg\n1+u9cdYzcuFCdvVq+PWv0/h0g2Gz2eK1C/GNHy+jablcnv1VCWe8A0BTU/GSJTVc+4yGBhNJ\nDou6/2efwaZNkQ8+GGE2S9xuSXl52GgMffVVEd86NnaW7UMP8bvCLS0tgUCgpWU03+9j/Xrj\nK6/oIH6/D4QQSiOn0+nz+WBoXjMfJJZlE5+Pvad7z0bYrvRcaQwZbZTt+bLnhe0GQBDdjp2a\nWW+sf559PquvJx0sFgs/O4bfmJ3fB+4smhf1z/G88/nhVcMz9+yDZLFYuC+Z+fY5Os4eX8Is\n+YL8oovoCkCgAir+B/5nJawsh6Qud4VCoajAU+KoE0mS3Pco0U/Ei+SLaXthKGkY4ECoYPV+\nC4lfvuH1es+cOcNNVwWGGblokTqJfhBVVVWR/oIXBEFIjx61dnc7HI5gMKg4dmz07NkAwD7+\nOPHYY+l5eWdJpVLN8ePx9hLp7SeaqnN7iAbuvBPitwthAoGcBDikUin3HYth4JlnjOvWlbMs\nzJ5tX7Kkk6JY4Yw0ALDb7YsXS99+WwNns5A7O6WdnVKShNpa+uRJkfULZ9m2t7dzv2/C1qR3\n3GHlAhx5k2qDECpkPT09wptRKQyPBB8Ry1orEARBEATBhdpFz8f8xf7dkt1rjWuFj5KDfDNs\nXg/rK6DiO1Xf+bn55/qwPraMZbtuuyw49OoM3W638GbU78P/z96Zh7dRnmv/mRmNdsmSrcVL\n4iwkFJIQtqS0DdDSQq+ec0oXCJQAgZKUZilfgdIetiwQSmmStoTAARpOQiBAm0AJBUIghDUL\nkMMekjix8SrZ1mJZi7WMpJn5/hhlPJ5Fli3Jluz3d+UPeTSaeTWeyHrv93nu+27s7uLNlgii\nv59H+utYC2v/Af8o1rnzJnsp0EpiZWGdHhuhcQW74hP4JItswTBMd3f3k/iTL1T3u+cOtWVm\ngHebRHV6rPOx+mkDVCedTscJPbL/I7RsCYXCjB+QwIFAjFlUKhWGYZadO5W8MDr/7/+SJ+eU\nzo0bjQcP5uIHQRAE3tAQ6O2NRCIURelOnJh46aUA4qIPDMPsdrvdbmcYBvvoo8zG4gSpEkeP\nxuNxr9cbj8dVR45wYgq9ejVxzz3FOF12gsFgMBhMJBLAMJOWLtUJNCOWZVmGUdKb1AVNz80d\nk8kUj8cjEeL22yfs22ciyX77DJIkdbr+RhKv1+v1el2uTPFFLIbfdNPkSAQHAJstde+9bm63\nc86pOe00nfRE8XicXxWR9fuYP7847xCBQCAEcLMRDmkJQ7WzeswIHLJuR4sWGZLJPlCYj62B\nNX7wfw1fRyHqYB2qtKqVbKWAooACABe4njc976/1c0krMLCNxcgYR0WmzweGYYQdl9L7oW5K\nXfFmS0ajkc+5l/46XtS/WKwT5w3DMMLyT+l1q51aC4UTu/x+/1Zm6w7HDn6LVLZgGKYgaSYZ\nszkAkFOd/mb/23fgO8L9q6qqhJqp8H+EiTXhOAr0GAWQwIFAjFlwHDebzUrtJPW//nX3bbf5\nf/lLGLr/KIZhlZWVlZWVAAD792e2KtRK4DgOixfD4sX5v6Ms6HS6SZMmAQB74AC3hTjvvKKe\nUQrLsi6XKxQKsSyLYZjUQ1SdTGqfe07JLkTj94PFMsJjBoCqqqovvojeeGNdW5taaJ8BALNn\n9y+MUBTl8/lAUHyxcaOTUzcAwOslr7lmKvd4wwY47TSZE3E14SJ4v485c6jFi8vsmzECgShH\n+GVz2RKGJ/EnR3V0BSMSiaxcSbz+er/XNud2lEjU/upX/WWPovnYHfgdd8AdABAMBt1u98sV\nL5MsKZorHjEc2WTfJG1juZy9vLBv4Vj62Ap2xWf4Z17cS2HUUHsNcgHHcRzHOY1D9n7YQewY\n7BjyNELjSlj5CXzSDd1KFQd6vd5kMglLSGR9T0qQQUuBduDDvG5SXC5XMBg0WozZZQthmgm/\ncRgtM0KRTqo6vWx+WbS/wWB4Y/Ib7bH2eZF5So1diBEGCRwIxFjG6XSyJ/NZpXCJrVI/iMTM\nmXqAVCoVi8VSqZQmGjV+73tYayuAnDfHokWwaFGO42EYJpFIpNNpTTSqmTcvu9/H8MCKL6Yo\nEQgEuIpNDMNkNSNLJEKXnl0IQRBvvVXf1kaAnH3GrFmZx5xwA4Lii2PHZMo0YGBPihCRLykM\n9Pt48smASiX2+0AgEIiCo9frOXNl2RIGfbrU0reGQ2dnZyAQaGvrdzvatMm+Y0clALz7rrpu\nze6OeIdUobgSv5J7eSqV4rNg+WPyc0U1rn7Y+bCojWU2M3u9an2hxs+ybFdX19P40y9W91cx\nFCmew2g0civwsveDChvOXKm3t/cZ9pntldv5LUqDnzBhwtrg2u50t/TXMR9Kt6wRwzCtVssJ\nCgW8blLC4TAXozuobJG9ZSb3ViOVSmU2m5WKMmRVp3eM7+wy7tro2CjceBacxeXIIkYeJHAg\nEGMZtVpNHT7c0tnJfZnjvTDid9759dVXg4IfRGzGjGBnZyAQAMh4c2TUDRi+PyjLst3d3YFA\ngGVZ7piawfw+EolENBpNJpMkSRooSnfBBcUQRAoIH54n6yFqBDCZTKmGBqWXF8ouZBgJrF98\nIR/rJ5QqhL50HDff3P3FF1MiEeLss2MvvxyeMKE6+8BIst9JXOr3YbVaB3tnCAQCUQAqKyt7\nenp4yVVUwiD8pCpTgsEg9xdc6HZ07bU9nMBht8N75vd2mXeJFIqz4KwH4AHucSgUEkrSorni\nFcwV+zT7+DaWaTDtOuy6BXgmNbMguN3uYDCYZ69Bjtjt9kgkwp7sEs2zhoJl2Y6OjnA4rLVo\n10cHHzxBEAerDu6CbL+O0sRms3V0dPA/Fqn2hFM3eES34p3snXwjTHb33JpJNdJZbyM0cnak\noiqb2trap7RP+VgfrzptrdqapSjjArjAAx7uf4QTnNNg2kJYuAAK+T8CMSSQwIFAjHE0Gs2U\nKVPS6XQikSD37eM2ZrwwFPxHY6efHubUjaF4c2Snvb2dL8Ic9JgMw7jd7n4xnmEmL1sGORig\njiIMw3B/XJU0Ix1NEwShOnbM5/f7/X6apnm9KbVyJblmTUGGEQqFVq4kpTXJ2RNYd+8e/MjC\nrlSQhK2o1TalF/IYjUauEljW78NsLt1aXAQCMZYgSXLvKXubw83fCX9nTJaU8wnxSm5HwcHm\nYxn3cQCQmyt+w/qNvzn/lnl6wF+GwhCPx7Ms2s+l54K8Jj9MdDrd3lP2tkRaCnI/+P1+bvE/\n90aJMp0eV1RUPIE/0R5rz5K5kz9Cy0/preiodfACB/8tRbZl5jnsOeFhk8mkx+N5Uv3k887n\n+Y39VTaqlz60f/ga9lqOqtPtcPvtcHv+bxZRKJDAgUCMC1QqldFohGXLYNkyACBpGmtoUPIf\nZTCM9HpTDoeozyLlcDAWyzBsEsLhMK9uiI6Zdjpxu11kwTRA3SicyFJwWJbFenu5nBQcwHHT\nTd5f/1rJQ5RbHeK9V5PJJG9fQn772wUZDLdqJFuTnH8Cq06n6+3tBYWwFa128O9hKpXK4XAc\nOhRYvnyyyO/DYDCYTEajMa8RIhAIRI7s0ezZZd/1kP0h4caxUVLOsqxQnuDg3Y7OPjt23XWk\nLuf5mOxc8bHkYwUftpDssSY3h2+GqgKf8U3tm7u0MvfDMGoo+FpODuHgnWnnatVq6UvKd3r8\nrundXaYRqj2RvRW3wlZ+B51OxxmZy7bMaKD/2ytFUc3NzTRNmywmpRKhC7ELveAtO9UJwYEE\nDgRiPEIQhNVq1Sv7QXTfdlvkwgul3hxUJJKxXxoYgJq9Z4RXK6S9G/EZMyAaNZlM/M6JREKo\nbogEEba2FstqgDoCBAKB3t5eiqJYmp6yfLn+ZGkJNWuW9aWXlDJriO5umDCBP4harYalS2Hp\n0kKNyufzcatGSjXJeVJRUeH1ent7WWnxhVqtFv4Gs2Cz2fbuNbS1qUHO7+Pmm/MdJAKBQORC\nma6Z5wIrCeQSFdwRxLRBD8Jr1rJzxS2hLdKXnGBPr2zFSAAAIABJREFUcIagWTw1cyR7zI3T\nXPjvAIW6H1KpFBe7ziEd/MRvTCxGzctokf91+4r6ahW26nPicyUfWa1WyxVxyIewpvpPVFVV\nxSe1gaT1DMP7r7vL5eIiYLJU2ZSv6oQAJHAgEOOW6urq9LFjSs9SU6ZI+yziM2fGo1Gappl0\n2rZwISnbMyInfHBfVmR7N+IzZ6oEX2Vg4NKNVBChZs0axa+ffE4K96PzoYf0+/fzpSXqb39b\ns2oVKGTWwPr18PvfF29ggZNdRUVKYCUIIpWatGABISq+IAji7LOrRQ0sWRiqNSkCgUBw0DR9\n4AD17LP4oUOk203k6DEkyxieveA4rlKpuGm2tOCOJEGlGvzLP6doy9qUGBmjqKOQoqju7u4n\nNU8+Xy1X7T90Q9DsMTfFqB8Z3v0gzUm5mLh4kWqRNW1VGvz57Pm5/7ksffL5f8Q1Iz+jemZn\nzU5+o/S2qaysFC56iW5FteB/vslk2l0v754rbJlJJBKcN2r/SARVNtV09WpCpsoGUV4ggQOB\nGKfgOE42NPQGg5FIhKIo7fHjEy+9FAAif/hD28KFk26+WbbPIhKJRCIR54YN5Dvv8BP78PTp\nfZ2dFEWpcLz6hhukwgeO40p+H/EZMyoGhoTzyeqyggh1xhnZBY50Ok1RFEEQmmgUmzu3sL6k\nvb29/B9aUWkJXV1tmzmTVtaMitpZI1o14hDWJC9eXIBcgH/+U9fWBiBXfHHqqbkeJBe/DwQC\ngRDh9Xp9Pt99903cu7d/dp2Lx9A4pKKioqenR8HtqALH8UGPQJLkG5PfaO1rlSZfXsZchpP9\nR0gkEi0tLTRNm4mCGYIaDAbORkR20f7p6NPDOGbBoShqe3L7dtOAnJSt+Nb2uvaH2h4ChcHj\n2OAXf5zQ0dERiUQqLBXZbxuDwfD6pNdzdPpQcs8Vtp4JTT1AUmWzybOpfkp9Qd8oYhRA/80Q\niPELhmFWq7W+vn769OkTvV5uI/7Nbyr1WbA4Tnq9/MSemjYNANJOZztFBQKBaDSqve8+TvjI\nnODklF6n02U5pr63VziqzNLNSUHEdf/91NSpupNht/RZZym9nWg02tTU1NDQ0NLS0nTiRPQn\nP8nuS8qybDQa5fpNEp2dMHUqYBhgGNx3n9Ip+CoJYWmJtrERAGKnn47jOHH0qNvlOvLVV18d\nPtz0wgvczsw99wDLwkUXKR02f2QTWK+/fmokQsydG3300VaCEBctD4NPP5XfjoovEAhEUenu\n7vZ6vSzLut3qJUt8zz/ftHfv8SuvzHwg79kzhEM1QuNVcNV0mG4CEwnkRJi4GBb7wFeUcY8S\nDoeju9u4YMEp+/aZhAV3R46YksmawV8PAABv6d/a6Nj4i1N+8b3Tvnf5tMtftrwMALOZ2X8h\n/yLcze1289X+Pwr9aBI1yZF2/CD8A+5ZWU/NQTGZTDpdf60fv2gPAEbGWAp2TT09PU1NTUSI\nWNe+7tXGV99qeOs33t9wT3VoO4R7CgdvYnPq5RwP9PX1ceW6srfNXHqucOd3Te8+7HxYdCue\nyZ65Fl8rOuwFcMEcmGMFqxrUE2HiRXDRFtjyAXxgAQu/j7CChq+y4X78Uv/lX6r+AjmQ/WMk\nnU5/Gvn0Z4mfTaWnjuHPmVIGVXAgEAgAAFi0CBYtAgA9yzLz54OCN4f/hhsqt28Xm2gAgKSi\nAerq4KRZRmVlZVTZ70PUu2E0Gj0ej5IgojMawe2GujrR8Pv6+tra2vje40F9SSORiNvtzlQ9\n5JbSwrIsJ/wr9dpoUim1Wl1XV1dbW5tMJvEDB7gX4t/8pvw1LxxqtRrDMO7tS2uSdTpVQWpi\nUfEFAoEYeZLJJG/cmKfHUCwWe5Z6drt1wKr7sDspShaCIN56q76tDYc83I7k7RUGZsEWr9p/\nz5Q9TcGm83rPK8GYm0gk0tXVBQoODvOIeS/aX/SDv6jZIuWO0CwDJD6yt0Ruaazsb/+JQUwN\nagwwFtj+WxGTcfrIpWUmYyQHAHJVNv82/XvQwQeDwafTT2+3yX+M+Hw+r9e7o3LHv2v+LbvD\noMdH5A8SOBAIxAAwDDMeP670bMVrr4FkYg9yZhnJM87gOyNJkjQ3KtepDhQUdDqd2Ww2KAgi\nup//XGpmwbJsZ2cnr26IpBampgYf6EsajUbb29tzV0OEJ8rWa3PygBiGaTQaWLIElixRfNcF\nBcdxo9EYiURka5IrKipGZhgIBAJRcCKRCP9xnY/HUG9vb2dnp6ZCs76vMJ0Upcznn8vXaOde\ncJfLXJELR+cRVfv/vfvv9VOHWe2/C9+1q3IXVA7YeBacJV20H3l8vgHr8KKclHtV9y5zLJMm\njJ7JnlmkjB6pFciw7V1HjOxWrGa1eSu7dXtVv4KQhCQA/BR+mr9AoNFoDAZDNBrltwh9Pcww\nSGJ9V1dXT0+P3qKX7azx+/0ejwcAKuhBWm8QRQW1qCAQCDHYV1+xDBPs7e10u7tOrtp7b7op\nctFFpMcj6hmJz5ghW9EQO1nZwYEfOZKkqO6urpbm5vaXX+Y2svfeK9u7MWHCBENDg+L4JAJE\nPB7nv2ZJm0eoWbNE+3d3d0vVEK7jhq6uBoWUFk62yJKTQp5s8xkVampq3G6dtCb52LGKWEz8\nLeeDD1LLliXPPpu221m1GiZPhjvugIHfVBEIBKIkSA00oubgPYbmzKEWLx78IMlkktPBC9hJ\nUcrs3g0sK/PvgguKdUZptf9fbX8d9tFy6TUYFViWFRatXDbtsjNnnvnjU3+837h/dmz2jqYd\ntenaC7EL+cFPYCdcBBc9AA9Mx6bPhbkFb1iIx+PbI9u3w/YmaOqDvjSkuWKBG+HG/A9ePKQ+\nstyPnBVrLBbTxXXrO9aL2n8KJRDU1dX9y/avTfZNR3VHe1Q9J7QnVtSuyKXKpq+vj6smk/0Y\nOY89z3vye+A4+ZwpWVAFBwKBkAHDMIvFYrFY4PXXuS1EKKRkolF/223SigaprKBWq6urqwEA\n3nknc5a5c0EOHMfxhoZYLBaLxZLJpO7ECSsngih4hfLqhlLziDC3I5VK8d9OZGNrs3T3WiwW\nUqG0pCg5KUPJ4lWr1e+8M7mtjYCsNcnpdNrtdq9aZUVGfQgEoiyQmmIKc0+3bQurVIP7SvT2\n9grzU0Ul8f+d/m+Qz3dCZINPk4XhVvsrUbIxNwzD8DeSbE7Kt5hvDRg8BolEYh217oWKF/iD\nFKphobu72+/3ExZifTBTLLCzaucj9keg5IsFDAYDpxTIWrGuca/JEuCaP2q1+pDzkLTK5iw4\n6wF4IMsLewd6xok7a6K3iAzRUD7LaIEEDgQCkZVFi2DRou7ubu3SpaBkogEgFT4ItVrWLIM/\n5qBn1uv1er0eAHiRBebMkd0z8/VXoXlEJLXwhZFKhSdZBA6bzZYqck5KJBLp6+vjImmcv/yl\nfBavAl98Qchu52uSGYZpaWmhKMrtdi5Z4rv44pDVSm/aZOf62PfsQQIHAoEoOQwGA/9Y6jHk\ndMr9lZEgWnUXlcTbHXYkcAwDtVptNBqFfgpDqvYvRwiCwHGcm8fKTs75YgQOrjGKrCDXhwvc\nsNDT08NlzQi1gO+Hvs8JHCVeLGA2m3U6Hf+/UhT+yu9WPIHgQuxCL3jFFjMg4+shJME1MgOA\n3MeIzWwLQEBpB5TPMpIggQOBQAyOzWajT/akyCIVPmzXXluwiobBBBFuEUmpeUSt0wmlliGp\nISIwDCMbGjxeb29vbzqd1h4/Pm3+fABg7rkHX53v312GYbjUNO5HURZvLgLHoCagfr+f80nN\n06gPgUAgRgyDwaDX62OxmNRjSKPRDNVjSHbV/QnmieKMfexTV1f3QOCBgntqlrKvhNlsDgaD\n/I+iyTlO9BccxeNxvjGK31ioegSfz8eyLO8gXnbFAko+speELuF2EAkEm/2b6+sLJhDkWSIk\n+zHyWPKxLDv81fbXHbAj/5EjcgEJHAgEYnBUKlX6yy9PtLdzzSD8xD50222af/9b29Qk/7JC\nVDTkglqtzuJLWnXNNUKpRaPRqFQq444dsmqIRq9XLDwBAAAMw5xOp9PpTKfT2AcfcBv5nBSW\nZVOpFEmSWG9v7t0lHF1dXby6kSWSJh/C4TD3QNao72c/S6M/CggEogSpr69/7z334sU1bW1q\n3mPo6FFzTU2NWo1NmjT4EdTqjO217Kq7JqrJ/nKEEiRJfuT4aBjV/lmgKOqf1D+3m0s07Mbh\ncGxWbe7BeqSaznwYYHjb09OTpTFqBb4C5MsuB4eiqHQ6zasbIi3gUfej9dNLvVhA1kf2tMRp\nt3huATmBYJ113agLBFqtllslkv0YeSb+jN/v537j0h1eMo7+rTt+QN9lEQhETmi12unTp0ci\nkUQioXnzTW5jxQ9+0HPnnc0eD1euyQsf7L33YqtWjeTw6urq6JybRxwOB/7ppyCnhjiuuy7H\nwhOVSgU33gg3Zny8YrGYx+OJxWIsy2IsO2X5cv1QuktSqRTf2yl1BqFmzSrIt2+pVx9v1Hf2\n2bFrrmEAsjToIBAIxOigUqnefntSWxvAcHNPrVZrINBfPS5adRd2wSCGyvCq/ZXgejpUFar1\noRINoVCr1R85PtqN7xZpOrOZ2euIAX2egzRGTbAP+0+u0OtBtlhgHswb5qFHCmkO8ZXUlXO/\nnqthNVCqAoHVag2FQvyPoo8RvV5fVVXF9Q1Jd6jAUJ7dyIEEDgQCkSsYhpnNZrPZDLfeCrfe\nym2sArBarRk30Pfey+yp4B5aPAiCwI4d8wcCnIGFvrFx4qWXAshXT1RWVqaVo3CHUXgSDoc7\nOjr4hRrHQw/p9++Xdpd8/DE89RTzwQd4ezsEg1BbC1ddBWvWgFoNsViM20fJGaQgAgeO4zTd\nX7shNOrbuLFNo5lSiJMgEAhE4fn0U/ntOeae6nS63fW7O+Id0lX3y5jLMAIr3EjHHQU0BI3F\nYsXr6Sgg38W/6wNfE9sUw2J2xj4Npl2HXXc1frVI08luR7qV2TrsAZAkyT+WrSYY9pFHDOlt\nw6rZE6oTwpWYUrN0MRqNr018zZVwKTVkOZ3Op7RPuSn3oNU9iKKCBA4EApEvOI4bjUYAgN/8\nBn7zm4Ifn6KoRCJB07QuHtddcIFS3weO4zabzWazAQAcOJDZquBLqjp2LB6Ph8NhiqLUx45V\n/+hHssfMkDXNhKZpt9stzZ3lukvY2lrM6UwkEh6P5847FbNLMqsxCs4giZkzh3LBFNHr9dzi\ng9SoT63GhH74CAQCUVIM6jE0KO+Z39tl3iVddf8L+Zd8D40oEHyFP4ewp8OZdq4iVkFpKFGZ\nyTk3GHHITz8kSXKNvbIahJpSD3sAKpVKaNIJA7UAE2sqkQs1JDAMq6urWx9e30v0FtbSpYC8\nX/H+rgrxx8iZ7Jlr8bUAgGHYPsu+XSDeIZ+OLcQwQAIHAoEoXZLJpNvtjkajAAAMM3nZMsix\n70PBl5SmaYZhyEgE5szRtbToAGDNmn7HDYEaQtN0b29vIpFg0unqG25QK583EonwZRHS7pLU\n7NmpaLStrY1hmCzZJSqVCpR9UrNF0gwFm80WDofDYVxk1AcAVVU2vpsXgUAgxh7CkngH65iO\nTV8ICxfgw+ykQBQDvpgR5Ho6HJMc5RV2U1FRkfkCAwCSjoY8FxWqq6vXhdbJagHlWyxgNBoP\n6Q69TryuJBCMpPus7LnOgXNEnTULYeECrP9jRNp6k0/HFmJ4IIEDgUCUKDRNt7a2cqsfAODc\nuNF48OCQUkV4WJb1+/2BQCCVSgHDTFm+3CAULP7zP0VqSF9fX0dHBydbODdsUL/7bpbzco5T\noNBdkpg1q8vl4go0smSXGAwGgiCUfFJzdwbJjk6no6iJCxZohUZ9X36pNxgMRqMjz4MjEAhE\nKTOgJB7JuUUjnykoby0h29OxjdlW3KEXGqvV+hjzmIf2SDWIy9nL8zy4wWD4SPPRG6o3lKoJ\nypTvEd/zg18sRMICkiY9fs821bbtVUNwnx323UjT9I74ju1G8bl+Cj/9P/i/LC8cXsdWIzSu\nYFd8Ap94ME+pxQaVI0jgQCDGL+l0OplMkiTJVTQMKfJjBPD7/by6Ier7SDudqpxTRViWbW9v\n749f3bjRcOBAFsEimUy2t7dzX7NE52VqanCl8yp0l8RnzOAbSmWzS+bPBwDAcdzpdGqPHFF8\nGwWKpPnXv8z5GPUhEAgEAqFEKBR6lnl2u3WYASgkSWZJqSBT5KBHKBGOJI+shJWfE5+329pp\noKWNUetV6/M/y0Wqi3qgR2gFcj1+vbCaoByRFSIpimpqbUqlUjqLbn1Hxn12Z9XOR+yPgIL7\nLMuyPT0927ChCSIc8Xi8ra0NN+Lre4vudMuN8yn2qR32/oyYkooNKkeQwIFAjEfC4bDH48mU\nHkgrGkqDcDjMZbxL+z7iM2Zokkk+9i87wWBQKX6Vrq4mJIKF3+/n1A3peaOnnWZgGBwf0HHL\nVZkqdZdgKhXp9aYcA+oj+OySOXOoxYsz/qGVlZWBjz5q8Xi4yhE+koa55x58dcEC7fM06kMg\nEAgEQgrLsi6XKxQKaSya9X3DnBZWVFR4vV7+R1FPR45/9EcXzpZrG7ltZ81O0VM60J3Hnncd\ndl2hGqOUrEBGso9jBGBZtqOjg1srErrPfj/0fU7gkLrPciXA8XhcKIjkeDfSNN3W1pZOp0fG\n6bajoyMcDhsshqGOE5EFJHAgEOOO3t5et9vN/zhoRcNokUqlMAyT7fuIz5yJp1I5ftfJEr8a\nO/10PU0TxIAkeq5jVum8RCKh1+uF+5tMJpIks3SXMLfd5v/lL/ktwuySJ58MqFQT+acqKyst\nFkssFqMoSn8ykgb/5jdzeZs5kr9RHwKBQCAQIvx+P2dinc+00Gaz/R3+7mN9xejpGBk6Ojr6\n+voqLBXS+eoUmPIO9k5Rz84wjN/vfxp/erttmEU0JUg0Gk1wX1BPInSfrWFqVuPiRaCuri7O\ngXXQu1HaG3Jh6sKlsNQKVum5HCnHHcwdMKxYO9kmlDtDdybCiVzGiRgSSOBAIMYX6XS6q6uL\n/1E28mMUhyeEIAiGppX6PswDVYkscIUqioIFRYkEC5qms/Sb6AQxqxw4jk+YMEF19KjSAOIz\nZnAPpNklmcyXgUczGo1Go7FIkTQIBAKBQBScnp4e4Y+iAJTVqpzqEHEc/9D+4WvYa0Xq6Sg2\nkUikr68PFOar32S+mSVyJX9omm5paUkkEnqLvhzLAY4kj6zCVn2Gf+bFvRRG8YUnEB+wm8h9\n9qngU/W19cId0uk0p7XxCO/Garp6NbEaAFiW9Xq9T8KTO5wDekOe0z7XVdu1sX2j9FwPtz9c\n5agahsDR29v7VFqmCaWD7Pgr+1fe4l0kptyN3Y1m6sMDXTYEYnwRiUR4Ey9pRUPyjDOGJUwX\nBb1ej+/YodT3ofH7YcKEXI+lLFgYJfuSJGl6/nnZ87I4Tnq9YDKJXmIwGKjDhzu83lgslkql\nDE1NU37+cwCANWvYFStSjY2QTEYihCi7BMMwq9U69AuDQCAQCEQJkUql0uk0/6NoWvhIxyMT\nT52Yo7HrhdiFXvByHpP9vhJlEnbD98NyiOart0ZvBUsRz97d3c1VOiiVA5Rs60oqlXK5XNu0\n216seZHfyBeebGI38Rul7rMPMA9sh+3CoyUSCWHYsOhufLzr8fpT6lmWbW1tjUajJotJKga1\na9plz7XBueHv6b/DEJuAurq6enp6ZJtQmolmXt2QiinOiU40Ux8e6LIhEOOL7JEfVCkJHHa7\nPaHQ91H/61/nniqi1WrVzzwjK1gAQWj8fqgfoP2bTCa1wnknL12qdF6NRjNx4kQAYFkWO3Qo\ns3XOHAzDJkyY8P77nUuX1ouyS2w2m9GonjQpp6sxTvj4Y9i2DQ4cgPZ2CAahthauugrWrIFy\n6LxGIBCIcQq/cAJy08IHHQ/OY+flmEQ+wGOymPUOxSCVSnHeYSA3X62urC7eqRmGCQaDA7YM\nLKK5G+5+NvbsdrO4daWFbqlIV3yh+kJUNzFiqgfDMC0tLclksoKU6etphEZhP7LUfXanQex1\nIlQ3pHfjX21/fQFe8Pv9XD+yrBg0OzZb9lyvWl4lQ2Q8Hn82kauTbjQa5YqbZE90VuIspXFu\ncG7YBmUWG1Q6IIEDgRhfZL5hKFQ0pGbPHs3BDUSr1ZInTig+nbNXSGVlJasgWExaskQqWFRV\nVaWV+00GPS+GYbBokTB3Vq/Xv/vulLY2AlB2SVZoml6zhnnllX6f/LY2WLsWGAbWrRvFcSEQ\nCAQiGyRJYhjGTSxlp4U4Vm5axbAgCIL7liU7X32CeaJ4p04mk1nKFh7peIRiKJVJtT4kVhAO\ns4f9Gj//wpE37OAj85QKT0wmE47jQhFN6D5rBrPogNkFkZfNLwNAFjHIkXIs8y6TPZeRMTIM\n09zcrKnI1UmXt4GTPdEtoVuUxvmq5VUtUwZVS6UJEjgQiPGFTqcD5cgPtU4HbjfU1Y3qGPsh\njh5NpVK9vb2JREJ15Ejtf/4nwJCDbCsqKlINDYpPSwQLgiDor75qdrlisRgI0kwSd92lvf/+\nob8JAIAvvpB3DEHZJTyRSMTlcjU1TVmyJHjxxSGrld60yb5jRyUA7NmDBA4EAoEoXXAcN5lM\n4XCY3yKcFppYU479KeWO0WjkZs6y81UDZSjeqbOXLTzoeHCNe42sgjCJmnR71+2jaNiRva/n\nDuYOQkPU1NRsiG/oJXql7rNXYFeIDqjRaHQ6HWcyyiESRFiW5SQVDmmtzT7bPg/tkZ7rJ8mf\ndHV1sSybuycoXzcte6Kp5qkBCMiO08SaiJzN5hAikMCBQIwvTCaTWq1WivwwXXFF7q0fIwNJ\nkg4uY5Uf6pw5Qz5IQ0MwGOzp6aEoStPQcMrllwMAe++92KpVsvur1eqpU6fG4/FEIkG+/Ta3\nUXv++cN6BwAou2QwEolEe3s7y7KbN7eYTBkb12uv7eEEDrtcnSxqZkEgEIjSobq6+kn1kz1Y\nTy5T0LFKRUWFz+fj57Si+SoXKl8k1Gp19iKaNe413J6cgvBX518BwJFyPOB6oCaVKSwdlfwO\nLv+VQ6avp64aNGC1Wj8yffSG6g2R++xZcNYD8ID0mLW1tWuDawN4QPZuxAR6m2ytTdwcf514\nXXSuM+gz7orclWT7lRGRFvOH1B9ggGc9AF83rXCiLdiWbfptbsotHed8mD+064gQgAQOBGJ8\ngWHYxIkT8TxaMEaNgX0fQ8VisVgsFgBgP/iA24LNnZv9JTqdTqfTwW9/C7/97bDPi8gFr9fL\nfS3j1Q0A+PDDjAPsfMlf+b6+vlWriN27dfwW1MyCQCAQo4harf7I8dFufLdoWogDvhnbvAt2\nlYifZVHBMGzPlD1fh77+VvBbI6zyEASRpYjGyGT+nkoVhMp0JUjm6neyd8JIrRbgeKZ9SVYC\neIZ4hnv2ItVFPdDDuc86WMd0bPpCWLgA5N1ndTrdIc0h6d3ICyIajYYzZJUXg2CNH/y80+0p\n7CnX49dfTVztiXuSkBE4pFfSbrdLBQ6tVsuVA8s3oVDafdp9u2CXaJxnsmeuxdfmc1XHOUjg\nQCDGHTqdLn30aJfPF4vFKIoyNTdPvPRSgCG3fpQp2OLFsHjxaI8CMQAuV09IQ4P24YcdAHD2\n2bHrriMB+o05uru7/X5/a+u0JUt8qJkFgUAgSoTv4t/1ge9r+LqP7dMxujARBgAGGAaYkXd2\nGC12q3bvqtoFVQM2ngVn/Rn7c8HPJcrycE50OhKOc/vOvSR8iUheuSR0CSgoCGvca6Rz9ZqJ\nNSMmcBgMBq5hRF4CYDP6xQD32Rw6nvi7UVYQqays7Ozs5HcW1drcgd9xB9yReU7gHsP3Acle\nyU10f9oLj9VqDQTkm1CMjFGr1V4AF3jAw43TCc5pMG0hLFyAlUdsUMmCBA4EYjyiUqlqak5a\nXfKRIkNv/UAg8odlWaF5GADs3Wu+++4JsRg+d25048Y2DJvKCxyhUMjv9wNA7s0sCAQCgRgB\nbofb/8D8oaWlJR6Pv2J5hWTJUXR2GC3k56sKhQb5kEwmt1Pbt5v6szzcmNutc3+m++x/7f8r\n3HNWetYtnltAQUFY0blCOld/jniusKPNgs1mCwaDvHYg0hr4+o6hkl0QsVqtm7BNncnOIXVU\naTQabj1G9kpq+mRyCHU63euTXm+PtSs1oQwYJ6JAIIEDgRj35Nf6gUDkCYZhBEHQNA0ADAOP\nPurctMnOsjB/fuDuu7tUKlal6v9TxcWtQc7NLAgEIn+Q5Q0iR3p6ejhzx9xdGMcYIzNfDQaD\nbrebqCDWB8VZHjrQaUErlFcuoy9zMS4WZBQEI2OUn6uDzFy9SGg0mr2n7G0ON8+LzBNJAFfi\nVxbppBiG7bful/aGZK+1sVgs/JcQkFxJo9Eo+6p3Te/uMsk0oawjUMVpsUACBwKByAuapnEc\nx3p7Yc4caGkBGIOtLizLcp5hmmgUmzu3HN9mnvOTdDp98GDy2WfxQ4dIt5so+AzHZDIFg8FI\nhLj99gn79plIkr3rrs7583sBQKvVCgUOoS86h7CZZeFCFYxYWS0CMQ5gWTYQCKxYoX3jjf4A\nCGR5g1AiFAoJfxQ5O9yN3Y1mHvkTj8fdbrdSlscCWLAZNg94gQY2TdzUEe+QFhFwrSsgyRnh\nrTFHhje1b+7S7tro2CjcqGQgWiiGUWuj0+l21++WvZKXMZdhhPxFQ00oIw/6mEEgEMOBpmmP\nxxOJRFKpFMayU5Yv13PTfihhm9KhQ9N0V1dXKBRiWRYYZvKyZcZye5ssy/r9/hUr9MOen3i9\nXp/Pd999E/fu7U+bL+wMx263f/VVYunS+rY2tUrF3npr96mnUl9+qQeAs85yCt+LMAkPJM0s\nBHFKAUaDQCAAAIBl2ba2tr6+vvZ2ZHmDyIns6ZvV9dVo5pE/fr9f+KdQqCI5085VxCppR8Z7\n5vd2mcVFBLV0bSVTeVR3dNRTb0asr0fI8GoIIbLCAAAgAElEQVRtZK/kbGb2X8i/FPZEiHxA\nHzMIBGLIpFKp5uZmPtnL8dBD+v37Ga0WTyQAymbmPyg0TTc3N/N5b86NG40HD5bX26RpurW1\nNR6PD3t+wjl6AoDbrS7eDEej0bz99qS2NhIA0mls3boa/qkNG2DGjMxjDMNIkuRuPGkzC0kC\nSZJyh0cgEMPB5/NxDefI8gaRI9lDMZ/DR87ZYSwhMhO11di+bfz2rZ5brWmrVEVyTHKATnwE\noYLgYB1cLMjzxPOP2x5/3Pa4cM9i103IUkYSgOhKToNp12HXLcBROUZpgQQOBAIxZDo7O3l1\nw/zmm/bNmyMXXaTy+XRffcXW1mJOZ/aXlwter5dXN0Rvk66uJsrhbXZ3d3M9HcObnySTSb7d\ntNgznMOH5bWJc84Z8GNFRYXf75dtZjGZzMN2I0MgEFJ48/8cLW+KatWxf39i2zb48EOis1MV\nCmHIB6Q00el02VwYR9DZYWzAsmxPT882bNv2qn4z0W5V907rziAR/EvHX6Qq0mp29Z/YP30C\nn3gwTwIS1VDNBfQOcNzEAAC84OVzRkasbqLcGWqYC2JUQAIHAoEYGul0OhKJcI81zc0TVqyg\nJk1yrVlz2sUXA0B8xgxJCni5wvcSS99m7PTTjSw7wk2qQ4VhmGAwyD0eniVnJBLh62CLaur5\n0Uf0lCnM2WcTLheefV7ENbP86le1omYWgiDOPbdG7tgIBGI4pNPpdDot2ii0vLnhBq0wQZGi\nqNWr2dde658aFaqRjabpjo6Oe++tLF6XHKJQVFVVCWO/RYkYGF7SfzRLDZqmW1paEomEzqJb\n3yE2E23XtMuqSKd5T9uh38EfJEtAbxnVTYwkonoZXiGyAypaKxvQYheivGEYJhaLhUKhRGcn\nTJ0KGAYYBvfdN9rjGsskuAYNALyvr/63vwWA9o0bSa8XoygAiPHtBGUOTdPcl3vZtxmfOVP6\n1b/UoChK5FgBgvnJOefEFy8e5Ah8nY7sEebMoQY9wqAkk8nW1tYVK6KPPUZ+9hnu80EqlZm6\nrFghsz9BEG+9Vd/WpoaTzSzXXDP1mmumXnXVpF27UH8KAlEwpJ8ee/ear79+aiRCzJ0bffTR\nVoLo3yEUCjU1NbW0wJIlvuefb9q79/iVV2aqP/bsyXcknA8I1yVX8IMjCovJZHp90uub7JuO\n6o72qHpOaE+srFtZ7ESMRmi8Cq6aDtNNYCKBnAgTF8NiH/iKdLoRo7Ozk/vGdWnw0h+FfjSJ\nmuRIO34Q/gH37OzYbO4BryIBgJExmlKm9R3rX2189a2Gt37j/Q23z3gI6C0UO6md22F7EzT1\nQV8a0pxCdCPcONrjQgwBVMGBKFdYlvX5fH6/n2EYzv0Rys39sbxh2Yl33aVpa2t/8EFq6lTr\nS5mVgcTMmaMwmECg4BkumeoMhbcZnzGjsrTLN0BhfsJbcj7ySIdKdVr2IxAEkeUITz8dUqlq\n8xkhZ+aSTqfd7urcDT4+/1xemhc1syAQiHxQqVQ4jjMMA3KWN1otwX8+JJNJl8vFsmwxGtnC\n4XAsFgOFLrmqKgat1ZUasqGYxXN2SKVSO6gd2439HRxZahbKiHQ6nSWSxsyYK+gKqT/oJaFL\nxm1Ab/6k0+n29nZaTa9nxfUySCEqL5DAgShXuru7eXeAcnR/LF+0Wi0AWF96yfTOO+GLL05O\nmKBtaDB8+CH3LKnVgtsNdXVFHQPDMIFAIBqNJpNJkiBqFy9WF1rewnFco9Ho//lP2bdJqNUq\nj6fYbzNP1IIGD+n8xGQavMn2+HHT44/jn32m7+oiIxFCp2MjERwgc4Tq6nzffnd3N1cIM6R5\n0e7deZ4WgUAMDoZhFoslEAjIWt5YLBZ+z0AgwMmpso1sP/tZOp9vm3xHpOzB/+u/4gAGmZch\nRo+RTMTo6+trb2/Hzfj63rE2IxVloovMRE1g2mrbutW2VbjPaYnTbvHcwj0WBfTewdzB+58w\nDHM4cfhe1b1fqL7w4l7UhcHT3t4ei8UujQ1NITqSPLIKW/U58Tm6mKUDEjgQZUkikeDVDZH7\nY8rhgMpKVKpeJFiWZRjGaDQaP/gAAMx795r37hXu4Lz+eli/Hn7/++KNgWtq4LPoLBs2qN99\ntxjyVlVVFf7JJyD3Nif86lfDfpvpdDqRSGAYpo3FiPPOK2zhiRCVSmUymSKRyKDzE1lisdif\n/0zs2lXFb4lEMACYN69v9epOjUZTUVGRz/BYls0+dSmUwQcCgRgeTqfz6NGk1PJGrVafe66D\n3000E4OBVh2/+EUSYJBPmyxk9wG56qooEjhKjRFzdkilUu3t7QzDjMmaBWENpjSSZlZi1hyY\nw6lIdsY+Dab9h/8/LvZdrGE1IBfQ66hxcAJHKBTq7Oz8h+UfO2t28scfGzUveRKJRLhiMQ6R\nQvTf6f+WZtNQFOVyubbpt71Y8yK/McvFPM4cX8Gu+BT7FEkhxQYJHIiy4mQnghbAcdNN3iVL\npO6PiZkzk+FwVVXVoAdDDAmKorq7u/v6+liWBYapf+89xV2LXEHT0dHBqxsieQvq6kAYbpJf\n60plZWWqoUHx6aG/TYqiOjs7o9EoAHB9VcYi91XV1NQcP07/+tcTRPMTrVZrNFYq/S9hGKaj\noyMSiTQ3T1uyxDdrVuzPf65xuzP1IB0d6qNHzTU1NWo1NmnS8MeWTqe56nchwqnLL3+pARD3\nyCAQiBGDIIi3357U1oaBXH7zzTdnHvP/kY8c0b3yiuW990xut5plQaNhZ82K0XRe/4tVqgHf\nVIVdchs3tmk0ZZBmhSgSgUBA+EdENCO9C+6Ccl7sEtZgSs1Ej2uOJyHzRYhr0moKNyXYBCgE\n9G7FtgJAOBzu6OgAgAq6QupaWu41L3kiNMeVKkTWSqtI4EilUi0tLel0ukIz+MWkabqrq2ur\nausL1S/wG5GuVDxQ4yKi1KEoyuPxtLW1tTY3U5ddxhttxGfMUHJ/lHVGRORDLBb7+uuv+UwN\n58aNeCzGaDPlpq2bN2f2W7MGWBYuuqh4I+nr6+NXC4XylraxEQCSZ5yRSqW8Xm9bW1tzU5Pw\nhhmegkA2NISCwdaWluMNDS07Ty53DOttJpPJ5ubmjLoh6KvKZ3iDolar33lnstSS8/LLa//9\nb0UPEZfLxdVWbN7cctNNni+/1PPqBgC0t6t/8Yv6Cy8kX8rvL7I0g0ZkYahWo79QCMQo89ln\n8h8UQssbfib2xBP2Z5+tcrnU3NozRWHbttkeeCCvCguDIfNyhoFHHnH+7nf1sRg+f35g06ZW\no5ExGo35HBxRUgzVK1S43n7ZtMvOnHnmj0/98X7j/tmx2c9//bwtZhuRURcLrVar1Q5o6hGa\niZrBLNpfp8vMvzk15NMjn25p2cIVdLxqeZU7VFdXF7ePrGtpude85AlNZypJeYWI+5FTiPhn\neTweD1dfNujFZBimpaUlGAya02bk/zoyoAoOREkTCAS6uroyk+oNGzTvvcd3IsRPP13J/VGH\no3lRIWFZ1u128+skoqIJtrZ2El9IOWdOsQfDqxuy8lb09NO7Ghu5oYpumGErCBUVFZlejAMH\nMpuG9Ta7u7v5P5Cia0hXVxPOYq1DDtWSMx6Ph8Nh7jHXOXLsmKQuM+sRckSlUqnVaq4YR2oR\nYjRqSjyFF4EYD+RieVNRUREKhSIR4qOPjACgUrG//a3H5VJzZjrvvitJex4KFRUVfr/f50vJ\n9tlpNJpBj4AoC2Kx2A5maF6h/NcS2ZqFzcxm2VeVEXV1dWuDawN44Py+80VmoldgV4h2rqys\n7O3t5X8UBvQaGaNarY7H46L1P2HNizPtXK1aXfz3VLrwxWKy4bt/9/9dtD//TYlDVEC0kljJ\nVxH4/X4+DYfff8z0UpUmSOBAlC7RaLSzs5N7LJ0Qmvbtk3V/ZHFc39sLDofygRFDIxaLURTF\nPZb2BFGzZmkXL4b880LlSKfTBEFgvb18p4nuv/8bFi5UCjcJT5/OfeMpioKwaBEsWjS8lzIM\nw/tNSK9hfMaM4q1CDtWSkx8nz2OPtTY0aBctmhKJEHPnJg8eVKsK9Kejqqqqq6tL1iLEZivv\nxTcEYvyg0Wja29XLl0/u68NVKvZ3v+s+88x4TU2KEzhsNhZg+GIlhmHp9OSrr2ZbW0lhn53B\nYJg9GzWujwUYhnG5XOFwGLf0e4W+4nxlg3UDZF3fJkmSW/OQnZGS0XJuUAEAAJ1Od0hzaDe+\nO5dIGp1O9/qk19tj7bwasrVqq1ANEdnZiLowHnU/Wj+9vohvpuQxmUx+v5//UaQQmUwm4c40\nTQvbo6QtLbVTa+GktBsKhViW5ddsRFLICnwFasYtOEjgQJQuPl+mLlF+QvjppyDn/jh56VJ2\n3Tr4wx9GfsBjFV7dUOoJKrgxOteXFI1GaZrGAaYsX64b2GmilOHC4jjp9eJ9faIbJnb66bp0\nWlWoefnQSaVSXCGS7DWMzZihZxg8e+VREaJwZZHWYRY2GlZIVVVVQ0P6mmusIosQs9lsMg3q\ngopAIEYUhmH27Ys/+yx+6BDpdhOhEFZbC1ddBf/v/4VfeskqbIUTvuq//isOoM/nvNu2qVpb\nQXpwoQ8Ionxxu93cYrhwffvCngs5gSPL+rbZbBauootmpHxzU1nzXfy7PvBxZqIO1jEdm54l\nkkY2oPdM9sy1+FoAEH7HkK15mQfzGqFxJaz8BD7phu7x5oJpMBheqX2lK9UlrZf5ceLHWsOA\nCz7oxdyB7+jfIZnk1Q2pFFI9qVpqX4rIEyRwIEoXzq1AaUJoe/ttpRdixW+UGFdkPpcViiaS\nZ5xRqBNxU2uqq4v89rfrXS4A8N50ExaP6/bt4ztNtPPm4aGQQSHcZPLSpZ6bbrK88gpIVBg2\nFjObxT2rI0bmb6HCNYzPmCHbjhEOhyORSCKRIDBMGIXbaD73kZvhwAFob4dgELg5xpo1oM6r\nEjyDUAaSdo5YLAW2733tNWdbGwCauiAQpU0kEnG73X/8Y+3evf3zxrY2WLsWQiHtsWPyNRrT\npyeuvDKSp8Dx6afy2/PskkOUAolEIhQKCbcI17drmJrVuGLfhMVi+Z/0/3hoj3RG+rP0zzDN\nWGhyHBBJI/eGOEniY/ZjD+aJQUwNagwwFtj+gF4so4bodDocx7m6A2nNy79N/45GozvYoXUJ\njTH2W/e/hr0mUohmpmY+pH9ItCeGYXq9nnOBkS0gUmEq4c7cEpesFPIP/B9Ff2PjDyRwIEoU\nlmVZls0yIUyuX0+SZCAQiMVi+OHDEy+9FADYe+/FVq0qyNkZhiFCoZFZMy9xOG8qpaIJUqsF\ntxvq6oZ9fJZlfT5fb29vKpXiskVIl4t7CqMokd+Hqq7OqdVqjxxROppp/37ZG0YtCewYSUiS\nJEnSuGOH7DVU63RYZ6fwGrIs297ezneLOAdG4a5+ecY/BPoeN8dgGFi3rgBDNRqNHo8HAGQ7\nRwpu6YemLghE6ROPx9vb21mWdbvVS5b4Lr44ZLXSmzbZuSaU99/Xbt9+nN9ZFHRCkvm2mw21\nzw5RRgijK0Cyvv106On6mmx9EwerDkpnpGfQZ/xN87eCD7UE6evr20Zt2161ndc+uGiVn8JP\npZIEjuM2m83r9fJbRDUvLS0twi6hV6tffdDyIIwnF8wLsQu94G1im2JYzEbbTmFPWQgLF5IL\nZetlbDZbe3s7/6PwYoosYHU6HbdkKyuFqKEQa1OIgSCBA1GiYBimUqlMzz+fpROBqK+32+0A\nAG+9lXnV3Ll5njccDvv9/ng8ztL0lOXLDUVO8SwLdDqdTqdTKpqovPpqWL8efv/74R2cZdnW\n1lZptgg3k6965hlhp0nyjDM0AFVVVb2HDrWe9K/WHj8+bf58AOi99daY1Vq3apXsDaP2+WBU\nex6qqqpUCtew5oYbRNewu7ubVzdEfiIph+OzTqd0jrFnT2EEDp1OV1FRcfhwfPnyyaLOEZ1O\nZzIVWOJAUxcEovTxeDzcCuTmzS2c9zAAXHttD/fhYz9ZvS6t+VKpWFESBAIhRNgUKV3f/hPz\np+2wXfnVmRkp18FhZ+zTYNp12HVXE1fLzkjHGIFAoLOzU2fR5R74arfbn1Q/6abc0pqXS0KX\nwMAuoQv8F3ACx/hxwczUy3Bq0WC+GGaz+fVJr3fEO+ZF5mW3gLXZbPy3XBgohZhYE4aPhVKj\nUgMJHIjSxWw265U7EYQTwtTChdHLLqMoSqVS6d1u3QUXDK/swuv18tq2c+NGw4ED+WdwjA0m\nTJgAR48qPi17cXLzjPD7/fznvjifhSAAw4SdJolZszjHfKvVarFYKIpKJpPa/fu5l6u+9S3D\nc89BqTqzVFVVpRsaFJ8WXEOapgOBAPdYakCTmDlzy/2tWeYY+VNXV7duXZ9sRz3qHEEgxhss\ny/Kf0vwnDwB8+GFG7PzFLwiCIIJBkNZ8qdVqkTkfAiFE2BQpXd/eadiZ5bUg6uAYTwF6qVSK\ny3yVDeY4jz1Ptp8Fw7B9ln27QOzTcVritFs8t/A/CruEatna1di4TlfJAmd6stGxUbhRagFr\nMplE/q+8FHIlfuWIjnjcgAQOROlit9uZHCbVHo/H7/dzi0tcgwMMq+wiHo/z6oZopp12OlVF\nS/EsCzQaTfro0U6vt6+vL5lMGpqapvz85wADlAuWZYPBIBe5QhJE9Q03kDn8IvhUM+lMHqPp\njvvvV/L7wDAskxK/dCksXQoARpZNKetZo+7MgmEY2dAQDodDoVAikdAeP871VUnVn3g8nsWR\nND5zpuwcY/78gg0Vx/GmJnm/EtQ5gkCMN2iaZvks8JM0NGgfftgBAGefHfvlL8nGxkkLFqj4\nmq/p0xNffqnHcfycc2pQ3jMiC9KawCyl/gieYDAo/F8pCub4Xfx3SlfuArjAAx6u5oXz6fhp\n+Kfnd5yvYTOJy6Iuoeeiz9Xbx3W6ShakF1PJAlbW/1U2DQdREJDAgShdSJKkDh/+2uXiYsD4\nToTkihXq++7j9vF6vXzYCkgaHIYkcGSZacdnzCBiMb0+L5u0ckelUtXW1gIAy7LYoUOZrSdV\ng3Q63draygV9A4BzwwbynXcG/UWwLJtMJkFhJk+dcoqo00Sl0WTx+8AwjDh6tKOzk3Ms42+Y\nxF13ae+/vwCXoBCYzeaM1+m+fZlNEuUlkz2mbEDD78nPMc45J754cSFtuFHnCAKB4CAIgjfJ\n4xC5bGg0p23fTiq5BZ966sgPGVE2aDQaq9W6CdvUS/RK17dFpf4IHj7eDuSCOexWu5LAMaDm\nBQAAOvs6A2ymaFTaJfRH+OM/ALlgyiO9mErkLoUgCgISOBAljUajOeWUUxKJRCKRUL/7LrdR\n/Z3vcA9omhaqG6KyC6amBh9K2QX310JpzVyVSIxzgYMHwzBYtAgWLRJu7Ojo4NUN0S8C6uog\n+y9CYSav+frraVcM+HJjXbAgu98HQRATJ06srq5OJBLEyRtGe/75Q36TI4DkGvKo1WoYLAo3\n5XAI5xj/8z8uleobIzd4BKLIfPwxbNtWrKggxJDg8gK4LhWpy0ZFhR7H8RFwC2ZZ9uDB5DPP\nYB99pHK5cHRXjBlqa2sPUAfe0r4lzTd9AEPr24MgG8zxWPKx3I+QvUvoX7p/FX7Q44/cpRBE\nQRhP/WqIskWr1VosFv1NNwHLAsvCf/wHt72vr49fUxKWXWgbGwGAmjVryGc6OdN23X8/NXWq\n7mSDTHzGjMyJAgGYOhUwDDAMTlaRIKLRKN+hLf1FpGbPVnohhmEajUZpJi9PDlU5JEmaTCbp\nDVMuaLVatVrNu7pOu+KKaVdcYdm1i3t28tKl5l2vPfKI83e/q4/F8PnzA5s2tVZWyqvVH36Y\nXrYsec45jMMBajVMngx33AHJ5Mi9FwRiGCSTyXvuoTZuhE8+AZ8PUqlMVNCKFaM9svGKw+HA\nMCwSIW66adLf/25XqdjVq92rV3eSJDgcDgDYvTvzcSv6d8EFhRlAKBQ6fvz4PfdQjz+u/uwz\nHN0VYwkMwy7RXnIue66FtZAsWcfUXUhfuAW2fIh9aIHRtAYvZXjvXk6S+PTIp1tatnBtJq9a\nXh2Ss2+WLiEAQF1CI0wD3TA/PX8aO80EJhLIiTBxMSz2gW/wVyIEoAoORBnDhWiAQtlFYtas\nIZXsazQa9TPPKK2Z+7/4wuNwTF62TI9yVSRwSeCgXP9CKr/WarUqZYsAQPdtt8XmzZt62WUA\n4yupt7a2llQ2oPnzmxf9/bBd6ORXUVEh2ieRSLjd7pUr7Xv39n87KWygLAJRDAKBQFdXV3Pz\nKcWLCkIMFYPBkEzWL1igESYrHT5ssNvtRqPBYCju2YPBoMvlAgDZkFp0V4wBbofbb8dOrm8j\nz5YcsFgsXq8309AqCXwdkrOvXq83m82bVZtRl1ABOc4cX8Gu+BT71It7E5Cohuofwg//DH+2\ng6IhfCKR6Ozs3Krb+q+a/qoZF7i2wJYe6JHm/iKygAQORBlDEASAYoNDlsIBWaxWK6Uc2tJ9\n221EMKjfvx/lqkjJ7hmRvZSmqqoqdeyY0rP2H/0I5xq7QcarYgxjNBojn3zS1tXFeZTwfiKN\nC2+55P21bYcHpLdqNBqjscpm6385RVEtLS00TaP5AKK86Ovr6+zshMHiSBEjzwsvmJRcNoqa\nrMQwDJcWAeiuKBDpdPpw4vAacs2X5Je5z74QJYVKpdozZU9LpEWaUXo5ezlODK1Cf8KECfup\n/W/r3kYumPnDsqzX693Kbn2h+gV+46A6RSKRaG5uZhimQl2Re+4vQgkkcCDKGIPBgGGYZedO\n2bILrcGQxZBSik6nI48fV3oWj8cHJJjW1mLjO1dFCEmSoOwZQajV2Z1ByYYGf09PMBikKEp7\n/Pgpl18OAOy992KrVmViyBcvHpH3UVqYTCaj0UhRVCKR0J10JN0X/0Eu6a1dXV00TQOaDyDK\nDT7KqthRQYihMgIuG7JEo1Hu0wzQXVEIuOC57ZXbX6rpn2ihVeJyZK9u7y6dOKP0TPbM9ar1\nQz0UjuM/1P0wyASbseYoRB2sYxpMux6/HrlgDgOXyxUKhcwW85B0iq6uLm6xUDb391vwreIP\nfEyBBA5EGUOSpNVq1SuUXZivvDK7IaUU1bFj4XDY7/fH43FNQwO3Zu696abQJZecsmCBMFcl\nMXNmISMryhyTyYRhmEHhF1F59dXZfxEYhtlsNpvNxrIsnJRFsLlzizrmsqA/CnfZMli2DACe\nV7ATEc4xaJrmLVHQfABRRrAsy/e78QyMI9UAEKMxNMSoJSulUinpRv6uOPfcxOLFaAKWKx6P\nh7Nmr6DRKnHZIx/MgQ1Tkrgdbr8dR11C+dLX18cF+cnqFOex58le21QqxX9t4xDm/jrTztWq\n1UUd9tgDCRyI8qampibd0KD49NC7SLgUT5ZlEyczOBJTp0p9JaKnn44EDh6SJG02m/bIEcU9\ncvtFYBgGixePz3qNHMlljpFKpYSBjhzCWeKiRTr0/QVRgvD95DyiOFIcn44EjvEGhok/rIR3\nxZYtPSpV/agMrOxIpVJ+v597rLRKfCR5ZBW26nPic9S6UvqgYI4SJBwOC38U6hSOlOO2xG0g\n546SHOj9Lsr9fdT9aP109Ck3NJDAgShvuAaHeDze19fHNTjYLr4YIF9DSgzD6Ouv/+q73wWW\nnXTzzVJficTMmQUZ/5jB6XT6Dh5s9vm4KQrvGcF1moz26MYXOC5uvhXOBx55pIMkTxuVgSEQ\n2cFxHMdx7jNEGkdKkid9lxDjCWFAu/SucDjQxDtXhMFzHMLZVzVdfV37ddu0216seZHfAbWu\nIBBDQihViHSKh9sfdlQ5ZAUO4dc2ae7vg44H58G84o57zIEEDsRYQKfT6XQ6AIA9ezKb8jak\n1Gg0oOwrQWq1QzL4GIRAAObMAS6fpWyzQux2e2VlZTweTyaT+vff5zaiTpORhyRJlUrFZQxJ\n5wMVFfpBj4BAjAoYhhmNxnA4HIkQt98+Yd8+kzAqyGAwSsU7xJhHo9GYzWbZuwLH8crKytEe\nYNkgavaRzr4q05UVJGpdQSCGD/9HSqpTbHBueIJ5QvZVGo2GF/e53F8W2I8NHy+bvIzCqJfN\nL4/Y+McMSOBAjC0WLYJFiwpyJJIkjUajkq+E8/rrh2rwIYRhmEAgEI1Gk8mkCsdrFy/WjIn0\nWYIgMoHqy5fD8uWjPZxxCoZhVVVVHo9HdpZoE6atIBAlhtPpPHo0uXRpvTCO9Msv9RiGnXtu\nzeCvR4xF6urqmppg0aJq4V3x1VdGp9NpMKgmTRrt8ZUJwgIo2dnXGvcaZHCIQOSDXq/nulSk\nOsWrllf1CfkVJhzHrVZrT08Pv0WY+2sG8wiNfgyBBA4EQpHa2lr26FHFp4erRKTT6ZaWFoqi\nuB8tGzZo3nsPpc8iCojNZjt+nL722krRLLGystJkMpnkKiQRiFJAo9G8/faktjYS5KKCTj11\n9EaGGD0IgnjrrfpRCakdSxgMBv6x7OxrjXsN9+xQDQ5Zlj2cOHwPcc8Xqi+QeQdiPGO1Wn0+\nHx/8JNQpjIwxU2wuh9PpfFL9ZHe6+/y+80W5v1dgV4zQ6McQSOBAlA0Mw+A4PpLdHGq1On30\nqKu7OxKJ0DTN+0rked6Ojg5e3TC/+aYwfRbq6gClzyLyBsOw3bur0XwAUY4cPkzKbi92HCli\nxPj4Y9i6lT54EHO58GAQamvhqqtgzRpQqxVfMlohtWMJrVbLNfvwW0SzL27jUA0Ow+FwZ2fn\ncxXP7azZyW9E5h2I8QlBEG9OfbM53Pyd8HeGpFPgOH6w6uAu2PWw82Hh9rPgrAfggeIOeiyC\nBA5EqZNIJLxebzQapWmawLApy5drR7CbQ6VSTZgwAQDS6TTx0UeZrXkYfMTjcT4LStPcPGHF\nCmH6bPKMM5S/4CEQQwDNBxBlymjFkSJGBr/ff9ddmjff7C8ka2uDtWuBYWDdOsVXobuiIEyY\nMOHPvX+WXSW+JHQJDN3gMBKJdHR0sKTjTaUAACAASURBVCyLcmcRCI49mj277Lsesj8k3HgW\nnLUWX5v9hfK5vzDM3N9xDhI4EKMDwzCJRCKZTJIkqYvH8W9+U7YoIxKJtLe3877ftgcf1L7/\n/qh0c6hUqoIkmMbjce4B3tcnTZ+Nz5yZp8DBsmw6nVaFw9jcueXuWorIBzQfQBSKjz+Gbdvg\nwAFob4cc19sRCFncbndvb6/LNW3JEt/FF4esVnrTJvuOHZUAsGdPNoEDURCUVolPS5x2i+cW\nGLrBYVdXF/cNDZl3IBAcw9YpUO5vAUECB2IU8Pl8vpN5osAwk5ctM8oVZdA07XK5eHVjbHRz\nZN41y0686y7Z9NmK4R45FAr5fD6KoliaVrqkCAQCMSSSyeTq1cxrr/V/M8tlvR2BkNLX19fb\n2wsAmze3mEyZHvVrr+3hBA478moYEWRnX98PfT9CR/h9cjQ4pChKGIoJQzfvQCDKhWPpYyvY\nFZ/hn3lxL4VRWVxmkE5RCqDENcRI093d7fF4MvN8AOfGjcaDBxntyW/Pgtl4KBTifXqE3Rza\nxkYASM2ePaLjzko8Hg8EAj6fL9LWBlOnAoYBhsF990n3VKvVoJw+q9JowO0exgC8Xm9HR0ci\nkWBZNsslRSAQCCU+/hhuvhnmzAGHA9RqmDwZfve75LFjX7e0wJIlvuefb9q79/iVVwa4nflI\n7vzPcscdMHCWhBibBINB7gGvbgDAhx9mrB8uvZQahTGNJxqh8RfsL55gn2iAhghEHOC4BC7Z\nDttvgBvqHfWv1L6yyb7pqO6on/Cf0J5YWbdyUOMAae7smTPP/PGpP95v3D87NvtfLf+qh2zm\nHQglGqHxKrhqOkw3gYkEciJMXAyLfeAb7XGNU1iW7ezsfMr31Ivkiy1ESxSLpiHNuczcCDeO\n9ugQ8qAKDsSIQlGUMAZJVJTB1tZigqIM3olTtpsjMWuWvBPdyJJKpVwuV8ZWg2EmL1sGWUsn\njEYjQRBK6bNV11wzjPRZzqaEeyy6pGmnk3A4sCG/LQQCMb5gGGbNGvqVV/o/Vtva4MEH1cGg\nvYDr7QzD3Htv+tVX+5tbUD3I+CEp0bEaGrQPP+wAgLPPjl11VQJAMxrjGhf09PRspbfucOzg\ntwh9QDEMO1B5YKgGh9lzZ7ObdyCUCAQCTzNPb7dt57cgx9bRxeVyhUKhCgtymSknUAUHYkQJ\nh8N8y4m0KCN5xhnCnTN7nuzmcN1/PzV1qu5kbmspVHAwDNPa2sqbhuZSOoHjeHV1tfbIEcWD\nDr3ggiv6BblLGp8xgx8eAoFAyBKLxU6cONHUREsrNQ4eNBZqvT2RSDQ2Nn79NVPAehBEGYFh\nA8T2vXvN118/NRIh5s6NPvpoK0kiKb5YuFyurq4uA2VY37H+1cZX32p46zfe33BP8TO0C+CC\nOTDHClY1qCfCxIvgoi2w5QP4wAIWpcNqtVocz8wjOPOOT498uqVli4bVAEB28w6EFJZl29vb\nOzs79Ql9lt8UYiSJxWKhUAgALg1e+qPQjyZRkxxpxw/CP+CenUvPHYExNNANV9BXTGOnoYqe\n3EEVHIgRhS9olC3KoM44Q7h8o9VqQbmbg9Rqwe2GurqRfQcDCAQCSoGvWUonrFZr6OOP27q7\nuavBp8+y996LrVo1jGFwY1ByLSUoymg0Du8NIhCIMU8qlWptbWUYRrZSw2pN83sK19uvvDI2\npPV2hmHa2tpSqZTsWfx+5uyzWbebQCamYxidTscJ7gwDjz7q3LTJzrIwf37g7ru7jh/Xbt2q\nP3SIyT04FpEjkUiEaw76Segn/EbeB/QbzDcupy//QvWFB/MkIFEN1T+Hn8s6C0jBMMxut3s8\nnv4tuZl3IGTp6enhQnxL1rGVpukv41+uIdd8SX7pxb3cDaNkRTE2iEQiwh+FLjOOlOOWyC1Q\nWcSzp1Kpzs7OreqtL9S8wG9EFT25gAQOxIiSEfsVLDbTZ54p3NlsNnd3dyt1c5iuuEKxmyMQ\ngDlzRiBDhP/gkwa+xmfMwGMxg8Eg+8KKigqz2ZxMJpPJpGb/fm4jNjcPJVjhksZnzEDaBgKB\nyAJv+SxbqfHDH4a5B3v3mu++e0Ishs+dG924sY0ghvZ1NhgMcpKu7Fncbpx3H0JNK2VE9oSd\njz+Gp55iPvgA556tqXFeconquuv8K1bU7dtnIkn2rrs658/vBYD//V/73r39ehm6BwoIX+PJ\nIZqhVfdWP+Z4jH92qHMnu92+hdziptzS3Nks5h0IWbL8ppxp5ypiFYxqkROXD7Dduv2lmv57\nY8xPtoVGM5dNu6xRm6mjmR2b/XD7w86KImYdpNPp5ubmVCqFumOGARI4ECOKXq8H5aIMjV4v\nLMpQqVS1tbV8T4oMgm6ORCLR29ubSCSYdLruV7/SjkiGSDqdhiyBrwP9t0RgGKbRaDQaDSxd\nCkuX5jMMrVZLbtsme0lZHNcFAlBVlc/xEQjEGKavr0+0RVipcdllvdL1dpWKVQ9xbT0Wiymd\nRadjrr2254c/RKGhZUZvb++KFeo33ujX8Xlh4k9/Snd3d991V8Wbb5r4Z9vbsc2bbc8/bw2H\nCRyHqqr03/5W/cc/1lqtNE3DjTf60D1QDITWJ9IZ2gHjgTznTvss+4Zq3oGQwjAMXxEMcr+p\n2lNqYYjOc43QuBJWfgKfdEP3oNUWLMseThy+h7jnC9UX0uoMv9/PlepU0ONrss0bzUhdZjY4\nNzyeerx4p/Z4PJy8UrIVPaUMEjgQI0ggYJ4zZ9ZJ6UFalGG4/HJRUYbFYol+9lmn1xuLxViW\n1Z04ccrllwOI6zJ6enq6u7s5zw7nhg3a999ntFo8kQAorsBBEESW0gmdwH+rqFit1rhCncvk\npUvZdevgD38YmZEgEIiygw+r4hBVasRi+O23TxCttxMEMdTGN5qmWZblXRiEZ/nTn1zV1Rk5\nGIWGlgUsy3Z0dITD4fb2aUuW+C6+eIAw8cYb7KJFTel02uWySZ8NhwkAYBjo7s5M1/x+FQCk\n/z97Zx4fVXnv/+9Z5syeyTYzWYBgCBaCCyLU1lu98tL6s7fqrQoKihtUNrVWe1vaCiJ4vSre\nVsRWbxGpVi2CGyq4UFxBVFRUIMQQIEyWyWQms2T2MzPnPL8/zuRwMlsyyazJ837xx+ScM+c8\nkzlM5vk+n+/nEyGmTAkCvgeyRsIZ2tquteIBw5s7JcydnQ/zFaAY/MmYRCR8p34MP07rJCzL\nbglu2aobklmp1+vt6ur6Z8k/X69+Pf74V7lXRSf7sTbZVqvVQjaC4DKDAH2l/mrZxGUswe4o\n3aH2J5ZpjxyEkOD9IVJoip4CBxc4MFmE53mHw+H1ekOhkIyiahYtkovCimTE1SPUavVpp52G\nEIpEIvQXX0S3zpwpHuDz+bq7u4XHMUYYfHU1acyifkytVstffDGhdAIoSuV0glab8gSZQS6X\n00ePJttLSH5XGAwGEwNN00KNI16pYTbLli+faDIxNI3uvtty+unswYMqADjzzEoqzQKuTCYT\nqhsJ9SDiYWLTypw5GXuBmIwjmgUkdFTRaoOCvDHh3mTs26e55x4AyT1w9dU89sIfOQqFIhgM\nQpIZmlDgkM6dqriq1dTqoZ9/BaxYASuyNPixA0mSMplMWLFP+E7R6UzZnE6n2WyW6WSPugdX\nW/h8PpPJhBBKps7w+XxCG6PISG6Y4kKr1Yr/g2Cgy4yG1yTrQx85HMdJf+fxip7ahlo8iU8B\n/t1g0gYh5Pf7g8EgSZLKQEDxk58kdLsIhUInT54UtZFl69fLP/5YFFY4m5s9KlU4HFYfO1Z1\n2WXxT4+BIAiZTAaLFsGiRTG7ent7hQfxRhi+KVM0kjXDjFNRUeE7cAASSSfqliwZRuDrsKGO\nHPH5fDabLRAIyI4cEVxLudWrqfvvz80AMBhMkaLRaFiW9XioeKXGm29WmEwMAEQixLp11eJT\n1q+HgZlXg1NSUuJwOBJeRURsWpkxI7BokXLkLw2TJRyOaPxNQkeViy92pth7333myy7rE3e1\ntcmvvHIy9NvZStujbryRBKwCGDHl5eWCyahAzAwN4uZOT1ufnjBxQn7GOrYpLS212U5FY0jf\nKS3SEuRQv8oGg0Gz2YwQGqLaoru7W1BAJzteqFeKxNwwm2ybJtSN2huGIIhdp+065jr2I9eP\ncukyI+YTQRJFz78ROIM5FbjAgUkPt9ttNpujH3Y8P3HZMkjidtHR0SFWN2KEFVBbWzZlSpmw\n7+OPo08YrtBAaO1OZoQhY1mFIlvfkGia1iaXTmS1OyYetVot1JLRvn3CFuq883I5AAwGU4zo\n9fqDB/1LloyPV2ocPqxL+JQZM9K+ikajcTjKb7qpMv4qlZXhmpqwtGnl6aetNH3ayF4WJlvw\nPC+1dRCI8W1JvZeiEmh2Lr3UHdMexTANWXwZYwaVSvVu3bvt/vZ4H9Cf9v00fu70vxX/uw22\n5XvUYxG9Xv80+bSVt47QsdVutwsFC4EYW9mV5EqQyO/C4bAoT4g/3hgxrqZXp55s/2/F/26F\nrTB6eZt6e2fFThjoZTcdpj9CPpK9i5IkKZfLBVuWhIoeishRF3yRggscmDTwer0dHR3i56Zx\nwwbNvn0J3S58Pl8gEBAexwsrQmeeecqebuFCWLhwJKPieT5VhshAWV3GIZuaeJ7v6+sLBALk\noUNDUaNkGyKRzgWDwWASQtP0Bx/UmUxRH4QYpUZ/ylMGeO+9GpMJ4q/yu9919/XR0qYVgyGb\nyXuYkSGdOwnEJeygZHsff/ykdK9Y+Jg+3d/dLXvggRrxHlAoKJksTU9FTBI+0n60UxvrAzol\nOOXXPb+Onztt1ySIw0jLrhIzPEiS/Kzys7eJt2PeqbPR2Q8TDw/9PFJH5/jWBv04PUgMlMID\n7fBjjv9Lx18m/GBCWH3qmCHeMKOJfLnMVFRUmM1m8UecwZwWuMCBSQNRxgZxogyuqoqSuF2I\n1Y2kCSOZGxXDMKqXXkqWIcLYbFBXl7mrJYAkybKysrKyMnj33egmbHuBwWCKh4MHE38ZGIZS\nIwUHDiTe/u67uoMHVWLTCkmSFTj4qYChKIqmaUHIGe+oIpeTomttar8VsfAxY4ZfoeCfflov\nbVyqqKjIXnvpWCN+hvYf9v+YbZktR9FoXuncSUfE6rb8fv+W8FDtKjEj4ULiQitYhXfKgAyT\nick3wo3zifTm0uIX9YStDc/yz0oPTq3OeLzq8QvgAplMVlZWJk2xHVOT7Xy5zJSXlz9NPo0z\nmIcHEV+Jx8Twt7/9benSpR6PJ13T+FFGKBQ62t+OIT9xYtL8+WG9/sQLL0y55BKCZX0XX6yW\nmFBYrVar1QoI1d11l/bDD9sfe8x9ySVl27fXrloFALZnn9XffHOmBtbT0yNfsqT0jTcS786h\nEQYGg8FghgjP83v2dN98s15oWrnnHsvZZwcIgjAajZMmqbJcl8aMCIvF0tvbm9BRpby8nGVZ\nn8+Xwm9FWvi47LK+I0eU7e2n7gEA0Gg0Z52lnzgRFziyBcdxDzkeiumGeLP0TQBYCkufgqfE\nI3t6emw221ulb8mQLMZ+shEam6Apb68Bk4S2tjafzyf+KFVb0Ij+Ofvzg7KDNsomKnEWtS4q\nYUsSHi9DshARAgCE0MPOh7vD3YPeMJgMcjlcvhN2xmxkECMjZCyweRdShUIhuVz+6aefnn/+\n+XkZQAqwggMzVEQZW0JRhr+xUWolzDAMAJRt355QWEExDHR1QW1tRgZWWVnJNTcn3Z1bIwwM\nBoPBDAWSJHftqk3YtLJ+Pdx1VyavFQwGV69e/cUXX3z11VfCV/99+/b9+MfpxS5iRAwGQ1MT\nu2hRdYyjikwmO/dc47hx3EcfdS5ePC5mr0qlUqn6tFpeWvgwm5l339VB9u8BjBSKoj7Xf74T\nYvtWpsP0h+Ah8UeXyyXYXo61cNCiRqfTSQscUrWFDMneUJxaDhSUON113euOrkt4vKjOIAji\n0/JPB71hMJlFqr0qD5cziGln2kNEKAQhwEKqlOACB2aoREMBk7hdsGecIT1Yq9WSJKn++mtI\nlDBSfv31UmEFx3HBYDASiSj8fvm//VvCTJbUA0NNTaauLo/HAwCKlhYhQyS8apVs7drBno3B\nYDCY/JCsaSW+Nearr+D55+HTT6G9HVwuqKmBefNg7VpghtDuGIlEPvroo3XrTn2Dpyjq7LPP\nHvawMSRJvv/+BJOJgMSFCerDDyeaTGT83ttvV+zYoZIWPrZvT+y3MsT2qJHcGGOcoTgLSEM9\nIJH9ZK4HjRkCZWVlT/FP9XA98a0NZwXOmuOYE6PEMTGmt8e/3RnsTN0KkS8riqIDIXQwcHAN\nveZb6lupUmYYUguhO8blcnV2dgJAQiFVfO4vBnCBAzN05HI5RVElr7ySUJShUKulogyKoqqq\nqhRNybWL554LADzPWywWp9OJEBIyWeRJMllSQ9N0XV1dJBIJBoNUfyyLDK/OFSMOB8ycmW6R\nC4PBFCPvvDP4MQghp9O5apXi3XdV4kaTCR55BHgeJFWLxPj9fpPJZLFYli9ffuaZZ65evdpq\ntU6aNInBM+CR8c03iftHhMLEt9+SCff29emSl0XSHoPP51u9mnr77VPzq6HfGJhBnQU4jhNC\nHATi7SrHnT4OEr/PmHxCEMS+in3xZqVTglMea39My2mFH6VKnE90n+zUDaLOyJcVRXHh9Xo7\nOzu36La8Xv26uHEkUguEkMViER5jIdXQwZ9MmKFCEIRerxdFGQ1z5zbMnVu6M9obVrlgAWzZ\nIj2+vLw8+OWX3zc3Hz506PChQ8deeUXYjtasAYRg9mwAaG9vdzgcghGMmMkSfX76rSU0TWs0\nGuXttwNCgBD87GfDf7WYHMJxnNPptFgs5s7O8Jw5yYKHMRjMWIPjuBMnTpjNZpOJXLLE9vLL\nx3bvbrn2Woewd9euwZ/e3t7OcdzMmTOXLVs2ffp0YUV66tSpHR0d2R786Oadd6J/aWP+XXBB\nqr29vanKIkMHIdTZ2dnW1tbWBuKNcd11UZsP8cZohdZ5MG8yTNaCVgay8TB+ESyygS3peTES\neEkInWg/Kfwo2FXyWU6pwwybC4kLZ8LMMihjgKnla3/o++EDXQ+8cPwFobrBE7yJMZ1S4sDq\nC+AC8fjxMH42zN4Mmz+Dz0qhNN8vpZjw+XwmkykSieg43aMdj+5o3fH+9+/fbr1d2Ds8qUUg\nEBAcnUXi376Rj3z0gRUcmDSorKyMtLQk3R03HS0tLdXpdKFQKBQKyfvDBolZs4QHfX19Xq9X\neByTyRIxGimDAduLjQWE7GGO4wDAuH697MMPEwYPYzCYQuarr+DZZ7nPP6cy2ynQ3d0tZHI9\n80ybVhsN51iwwL5tWzkA6PWDdCi4XC7pV8MjR44I9fTGxkafz+f3+1UqVfxFMdljKJqdoWC1\nWl0uFwy8MW64oXfr1jIA0OsBADwezxYeZ38MH5qmCSKaRRAfDrqjdAcFVL7HiEmMVG3hC/ja\nxHWjOCXOXzv/OuH0CVidkREsFovw/yWDUouY6kbM2/eU+akJDROGP+LRC1ZwYNKDbm4O+P09\nFovp5EmLmIq6dq0oyoiBIAi5XK7VapmlS2OEFX19fcID+YkT41auZOvqOteuVbS2AkCgsVHq\nkIQZrQSDQZPJJFQ3xCIX29AAAKimBiTBwxgMpjBhWdZkMv3hD+6//pX6+muw2SAcjnYKrFw5\nojNHIhFhEgsA4iQWAD7/PJpo9otfRNasCW3YAMmu6/f7pSc8cuSI8GDatGnxezHFAs/zdrtd\neJzwxrjmGtTZ2WkymWQeWabWUccgBEFotVrplnamfWv51jARBoAwEZ5ATMCKmMIn6qAHAEmU\nOHka1/Dhef5b37dXsVdN4icVjjgrEokI5XiRjEgtUr99jxkeG8GQRzO4wIFJG6VSaTQa6+rq\nqrq6optmzhzGeYRYloSZLIFp00KhUMZGjCkoHA6orweCAIJgV64Uqt3xRS7/1Kn5HigGgxmE\nQCBw/Phxj8fT1cUMo4Vk0JPHb/z+e8UTTxgAYMaMwE9+0nL8OC9ed/78vpjrxkjom5ubAYAk\nyR/84AfxezHFQiAQiH/vpDfGlVfahNLYFa4rLuu7rI6tM0QMF7svFo7ELetDp6qq6tXKVzfq\nNx5RHrHT9tfKX3tP9x4P0V++oIi5DW7L7yAxqZHL5TQdnRILSpwDTQc2t22WIzkAvFnyZl5H\nlzZ9fX0tLS0vuV/aLt9+gjzhBW8EIoVwK4pZkwJXN1x99rSzLz/98r2avWf5z9p2bNt4NH4Y\np1UqlSQZna3Hv31vaN9I+eyxCy5wYAYHIeTxeGw2m8Vi6WtrQ6edJsxOoatrJG4XJEmKmSyd\nDz7I1tcr+5fXAo2N4v/nfBEOhwOBAGezibNxeOCB/A6peGFZtru7+8SJE60tLcGrrhJdNtyT\nJ0OSIpdv6tSYvxbCedxut8vlCprN+H3BYPJOZ2enMNV85pm2O+7omTIlaDSGFyyIrq7r0zOM\nj0WofkrZvbvk5pvrPR5q1izfX//aRlFIet35860x15XJZNKnNzU1AUB9fb1CoYD+OHNM0RFf\n3ZDeGBs3dvb12QccL1lHreKqcMv60GEY5gvDF08Yn7hu0nUXTbloc+VmYXtDsOGXtl8Kj7Ei\npsARHPQGbOkPggUAMQi2KHC73UJTcwZNLjJFaqnF41WPE8RwOu9JkqysrJRukb59OkI3giGP\nZrAHB2YQgsFgR0dH1Emb5ycuW0acPBndNzKLBJVKxbzwQsJMFkSSapcLSvNjbuRwOGw2Wzgc\nFl6vpvA8L3meZ1k2EonIfT7m/PNznTmSZtCJy+Xq6uqKWsmuX6/45BPRZcM3ZUqy4OFAY2MJ\nx4nzk3A43NnZGW1c4vmJy5ZhL1IMJr/4/X4xZCFhp8CcOSM6v7QAwfPw5JPGjRv1CMGcOY57\n7+2maTTodXU6ncMRlZP4fL729nbo708hSVKj0YxofJg8IS5HQ6IbQ6ORB4On7oqYlvUnu56c\nMBm3rKfBv5P/3sl2mmhTgAxURComhCZc4briZ66ftcvbN+k3AVbEFAMVFRWbZZs7Ah2pg2AL\nn0LOE2EYRiaTCStz8Z41b+neGvaZDQbDs/JnE+b4zoGR/ZUdveACByYVkUjk5MmTosONGHSS\nEQ/I8vJyX38mS8nu3dJdE5cuhUcfhf/6r5Gcf3hYLJbe3l7hcWZfb1LSqRcghHp6ehwOB8/z\nwjyfyck83+fz+Xy+UChEk2TljTfScReNRCIcxzFeLzFrlvS1sCwrVjdirGS5qipkMJQlCR5G\nJEn39EBdHQBwHNfW1iZ2LeXofcFgCgCE0J49gRdeIPbvl3V1UX19RKb8O0eONEJSROwUOPfc\n4KJFivgDho5CoZDL5SzLejzUihXj9uzRymToj380z5njHOJ11Wp1aWlpR0dHJBI5ePCg8EFU\nV1fndDpra2ul82RMEaFUKoWJRMIbQ6VSBYU/DZJ11AgRAYCDqoN/1v/53+Df8jn6YuM3kd/8\nvPXn0i08wXfIOk45C9BYEVMEfFzy8c6SQYJgC5xgMBjTvV5ot6LBYOgSm/clUguWYkeolBlK\nji9GCv7rjkmF3W4XqxsZDzqRyWQlrcnlZPmYtQYCAbG6Ef96Sb0+U20zwWDQ6/WyLEuTZMWC\nBfH1gmR0dHS43W7hcW7m+UIan+gIa1y/nv7gA6RUEoEAAKAZM3pttuh9kkjwYrfb4102plxy\nCQD4p07VaDTqIRS5bDab+Fct/n2hi8SLNBKJBINBgiAUfj913nm51t1gihCv19vZ2fnAAzW7\nd5/6biT4aPI8rFuXx6EBAMQLbnfvLrn33nF+Pzlrlm/TJhtNTxzhJWpqaj75xLxsWZ3JxNA0\nuvtuy+mnswcPqgCgsjJcUxOOv+7f/mah6UniGWpra88880zRrBQANmzYsGHDhv/4j//Y2R9z\njik6qqur9+61LF8+MebGkMlk06dXAkRlOyNcR+U47jB7eA295jv6OytpDUKwCqouhUsfhof1\nMLL+q+Ih3llAqoh5ov2JCVOwIqYIuAAu6IGe43DcBz4jGBug4Ua4cT7MV8CIytC5JHWeSCGI\ns8rKyjZRm7KhlBkFb1+OwQUOTCo8Ho/wIH52GmhspINBpVI5kvOTTU3hcNjhcAQCAerw4fFX\nXAEAaM0a4r77Rj74YSB+CU74epHXW1Iy0mZFhFB3d7eomo6pF6QuUrjdbrG6kbNgXbPZLFY3\nYi4KtbXtLCveJAkLLoJNYDIrWZVKJRqvJKD/tyG+6oTviyIcjum0LzRYljWbzdL+mgLse8IU\nGn6/32QyIYQE/85LLukrK+M2btQLCam7duW/wCGXy8XH8Z0ClZVlI7+EWq3+8MOJJpMMACIR\nYt26anHXihXdCxbY469bUjLgr1JbW5u0uiEyc1je2JhkpM7rTQ3P83v3Bl98kdy/X9bZSQ5F\nplRSUvLBB3KTiYG4G2P9erj8cqXUoVa6jqpFWhjaX0qHw2GxWLaUbXm9+nVx4xgMmk3oLCAq\nYh6vevxCuDB/o8MMlVEQBJv6VnzM8FghiLOypJQZBW9fjsEFDkwqhHJpstmpcmAxdXjIZDKj\nsAL/wQfCFmLWrJGfdngIMoFkr5dIpMdOF6vVKlY34usFqYNREwbrivN8wufLeEt5KBRyOp0p\nLipWN5IJK3ieT+GyUUrTkW++OdnZKawRKVpaGubMAYDgH/+oePBBcRipA3eowi5whEKhEydO\nCFG4MHb6a9I0asHEY7FYBPXTM8+0iU4TCxbYhQLHCP07M4JSqVQqlYFAIGGnQFlZBgocAHDo\nUOL/3Y2NwYTXjQm2rK+vjzcrxWSWYDC4ejW8/faptcSh64ycTqfFYnnggdp0ZUpNTfKE22fM\ngOrq6kdcjzgp57DXUZ1Op9lsBgDBy3BqcKqSU75W/prQ6j+mbDWz5yyAwaSFQqGgKEr4NhV/\nK2Y1DqYVWleilQeIAxawpFZy/B9emgAAIABJREFUYalFgYALHJhU0DQdDoWSzU41kmJqBli4\nEBYuzOQJ04cgiBSzcfWwDJClcBwntsDE1wtCZ56Zeq0rdf2FzkKwblR0kDzoJNlrCTQ2ykMh\nhmEYhlG99FIylw3GZlM2NJx++ukejycYDCr6u1QUP/mJdBgURUXC4aRepPkO3ElNd3e3WN2I\ndyGhiqS/ZlAikYjNZhOMWmQUNX7xYgVWqYwAjuP8fr/wOBv+nZli3LhxH3/ctXjxuJhOgfLy\ncq1WWVeXgUu8806Cjb29vfv3h+fPnxRzXZqmVaryQqj+jB2EWkBb26Rh6IwEC2oAGIZMKeGN\n0Y9qv3z/u9S7MeuoZ6OzHyYeHvQV8TxfyF6GuUev1wvlHoEMOgtgMEOHIIjKysqenp5TW7J/\nKyKEent7n4Pntum3iRtTKLmw1KJAwAUOTCrUarVyy5aEs1OCppUOB6hU+R1hZlEoFNRzzyWb\njaucThiY1ZQuPp9PWEtMVi9IXeCQBuvGz/N1WZjnR2fmSS7qmzIl2WsJTJsGLMswTElJCZHc\nZQOtWwe//S1BECUlJSUlJfDrX8Ovfx0/DJVKlex9IWha3tsL48Zl/LVnBJ7nvV6v8DhhGWh0\npDgEg0GpG3Hpn/8szcrBBY5hEEmkjxN9NM85x3/rrYpCCHqXy+UffjjRZCIhUafAXXdl67qV\nlZW7dimTdShk77qYGAKBgNlsRgil1hkJDSx796KODkJsYFmzBol1hIzLlC6iLuqF3mPomJ/w\nG5ChARpuIm6aTwxpHdXv94tVaQGpl2EVV7WaGlu2muXl5c/Qz4yCDA5MsaPX6//O/D1hnkg2\nbkWEkMlk8nq96lL1GFdyFR24wIFJhV6v9x04AIlmpxMWL85X0En2KCsrSxHsIszGR3L+1PWC\nQGNjakm3UqlMEayrcjozHqwrpAyUbd+erLgg6+mpefDBhK9FRRAAUFpaGm5uTnZ+Ymht8JWV\nlaEk70uB34fhcDhFScvf2KhGaHjR6IUDQkhIqRB+TLfxCpMQKk4fJ/XRfOKJdoaZmpeBxfPt\nt4nrLDNmZPe6LS3qvFwXI0W0kU6oM7r6ah6A9Hg8K1eS772nhn73C6EDhWUjixZFPzcyLlOK\nrqMKF0zzIza1rebGno0TThtztpqjIIMDMzrIZZ6I3W4X1qiwkqvowAUOTCpomtYePZp096hb\nmJXJZCle7xBn4ylIXS+gGAa6uqC2NtnTy8vL/bkN1tVoNARBJAs6mbB4cd//+3/JCi4Kux00\nGoIg6OZms8XidDoRQqLLRmjlSuaBB4Y4DJVKJS/O+zBavEhe0vr6a2LYznwFgs/nE+NCh9F4\nhUkITdNCQiok8u8sLU08t88LKTsFRuF1MVLERioRqc7o+uuRw8Gazeb29ob4DpTdu8lFi2JP\nKD59xozAokUjcjEfNqm9DP9U+adtsC35s0cn2FkAUyDk8lYUTegEYlJp76PuS7d4iskZuMCB\nGQSyqYnneY/HIwSd6H/6U4BcuwZGIpFAIBAOh+U+n/rf/z2rzoXUkSPBYLC3t1d4vfVXXw0A\n/P33k6szIElVq9UkSSarFxhvvjl1kYJhGDq3wbo0Tev1ekVTU7IDKK8XBiu4kCRZU1NjNBpZ\nliU/+UQ4gDn//LRGQh05IjieBoNBuqmp9uc/BygC90qGYWia1r78csIykEyhePx3x1/48FSk\nZeEkgA6doNCHkrxZiQHsOTocDAZDR0dHvI8mQRAGgyHfo8NgAAQbaQlSndGGDSaEarq7uyFJ\nB0plZaz5a8ZjhoeHSqUiCEJQpsR7GW7XjJX8FCnYWQBTIOTsVkQIsZJsgfiA5Or6asALOIVK\n/jt4MYUPSZI6na6qqkrf3h7dlKuAPYSQ2WxuaWkxmUzmzk40bx5k37lQoVCMGzdu8uTJ9f21\nW/KHP8zImUmSTF0vGPRFkU1NbDDYbTafOH7c9MYbwka0Zg0gBLNnZ2SQMRgMhr69e5sOHz58\n6NDhQ4eOvfKKsJ2//35ASGG3J33mwNdCUZRKpVIsXw4IAUJw3nlQXw8EAQQBQ5NyMAxjNBrr\n6upq+9u2c3YfjoSKigqxpNUwd27D3LmlO3cKu2oWLjyr6eUlS2wvv3xs9+6Wa6+Nxuvs2pW3\n0Q6DaERFv0ql88EH2fp6Mf3XPXny0e+/D/ziFzn4nzvK0Ol0gUDt/PmT9uzRij6ahw6pu7vr\nbLZRZX6EKV6YfrEZz8Nf/mK8554Jfj85Z45j48aTGg0fDAZTNLBccQUrNujFP91gyJs0gKbp\nmAwg0csQAHSELk/jwmAw+UFUcgk/HlQdXG9cn98hYVKDFRyYdMh50ElnZ6eYjZqHfM0svF69\nXm/99NPjNpvwtU9s2eBWr6buv38oZ5DL5dXV1QAAH30kbMl2sK7RaCwvLxcCMtQffihsFIo+\ndHOzx+Ox2+3BYFB25Mika64BALRmDXHfffHnCQQCvb29wWAwEgpNXLZMOewZbwEE7gydysrK\nyPffJ9v78/uq+H+PWoIXVALo0BFmOMkarziAyrVrlXv2YM/RYbB9e5nJBIB9NDHDQrD2zGoH\nnE6n8/v9CfN6FQpFjFUnDGxgufZaD0WV2+32+KeTJFlRUTG8ITVHmleild+Q31hJK0uwKQId\nU1BdXf2c4rnucHe8l+EcKIwEIwwGk00IghAbReOVXDtKd8jgVIR5K7SuglVfw9eD5shicgMu\ncGAKF5/PJ1Y3YpwLwwYDlJfLUj+/UDEYDGVlZV6vNxQKqT/4QNhInXde2ifK4TxfJpOVCg6m\nd94Jd94p3aXVarVaLQDAvn3CloQFF4fD0d3dLZR1jOvXF++Ml+d5n88XDAYpilL4/aoLL0zd\neUEQhOz7791ud19fXzAYVLS0jL/iCgCw3nGHdckS6ZHiwuY116C0bfHyh1arpSgqWePVxKVL\nAUD8nxsxGimDoQBfG8/zBEEQTmdBtdIcOJB4O/bRHAk5mPbnkmQvh6b5tWu5t9469XcyGx1w\n5eXl333nW7iwKiavFwBmzKgGcEgPjmlgoelyo7GqtRXddFOl9OmHD2uMRqNaLUs3Zhgh1N3d\n/Q/yH69VvSZulAY6Dn0SQhDEp+Wf7gRsq4nBjF3KysrEpCcYmEqrRVqCJAAgFArZbLbnmOe2\n6reKR6bIkcXkBlzgwBQubrdbeBDvXBicNi3s8ZSXl+d1gMNHJpNFFbC/+hX86lf5Hk6GSF5w\nCQaDYnUjplbFVVVROUvZGLETRF9fn9lsji5L8vzEZcuG2HkRzcEFgD17hC2BadOkB0gXNm+4\ngQDIj7veMCBJsra2Vt7fkxJP2GiUJuPybrdOVygab47jrFar2+0Oh8MEQqctX64qpFYa7KOZ\ncbxe76pV5LvvnurxKUbjGykPPBB5881T3+WElxMMhpYsOX7s2GlLlrhirD137crkKyUIYvfu\n8SYTAYl0RgsWRNtM4o1yaRopFAqCIN57ryZTMiVB8llSWpIw0JHn+ZcDL29VD3USgm01MUUK\nVhNkioqKio3ERkvEkiyVNhAInDx5kuM4nCNbaOACB6ZwEaLakjkXooFBbiMCOyBmGTFKML5W\n5Z86VR4KMVlbPHU6nU6nk2VZxHETly0byfTV4/F0dnZGLSeG3TO1cCEsXNjc3CwVb8csbJJk\nmguX+aakpMR74ECHxSIYjp5qvCotJUKhkxs3Sv/n8oFAgRQ4IpHIiRMnQqGQ8KPh8cdVe/cW\nqbAIMxR6enpsNpvJlCDRI7PT/twQiUTa29tbW2uWLHHGvJx33+V/+UsuobVnxjvgvvkmsSRr\nxgzQ6XRWq9XlgvgGFpqmhZpvpmRKouQzYaDjueFzW0+0EmriUcdQJyExXoaRSORg4OA82byD\nsoMjaX7BYLIHz/Mul+uf5D+3lmI1QQYgCGJfxb6ESq6HiYcRQh0dHcJ3OZwjW2jgAgemcKEo\nKkW+poockUVuX1+f0DJAAoy77TZFIS3bjj6EeW/SWlUwmKUCR2dnp8vlEh4b168f4fTVYrGI\n1Y0R6lBkMpnwRzHhwmb2yj3ZQ6PRNDQ0cBwXCoWC770nbKRcrvj/ubKBsQt5pLu7W6xuxLyh\nfHU1mTNhESYnuN1um80GSRI9isv4RqC9vd3v9yd8OWVlEUhi7Tkn0w4SKXVGNMuOnz+fiWlg\nIQhi+nQjSZKDPT0NPB6P9EdpoKMhbLjFdEs4HB72JKS3t7enp2db+bY3qt8QN+J5I6agCIVC\nJpOJZVmmlHnUEy3kvaF/Y0PFBsBqgvRphdaVaOXnxOckkDzwBBAqUJ0D5yyEhYKSy+1xi18h\nBKQfO9V89WoyA/GLmOGBCxyYwkWlUsHmzQmdCxFJavr6hvedVKi5iv0vxvXrFZ98gpdtswpC\nKEWtisnOjNfhcIjVjZH3xYRCITEwLF6HEmhs1KRzNp1OFwwGEzrzKZVKmaxI7WWAoiilUum9\n6aYuv7/2vvsS/s9V2O1QU5PfcQIAx3Gpm+BwTskow+GI+kEknPYXl/ENAHg8Hr/fD0lezqWX\nuqUHSzvgbryRhhxmG778siZZB8rUqZm8UFgi6owPdCyPnGpojal9rOBWpG46sdvtQh++jtNh\nFTqmMEEItbe3C99SpIW8i5wXCQUOrCYYOoFAwGq1/l3x923GbeJGBMgHvgqouBVuFbYIS3ci\nMR87z7mem1AzIWdjxsSAY2IxhYtOp9N+8w0kytecuHSpcvsw10xsNps4sRHnvWxDAwCgmhrA\ny7ZZgGGYZCkb0RlvFhDnM9Lpq6K1FQD8U6cKWgyEEDgcyQJrw+Gwx+NxuVxsdzd9+ulnnHnm\nGWeeaXziiYQ6lLTGVlFR0dOjjUkAPXhQdeiQOhKpzcjLzyM6nS5ZMu7EpUt1b7+d19FFYVlW\nuAcSCot8mZ17ZYivvoI77+RnzgSDARgGJk6E3/8eBi4gYZISCARitkin/QsWsPkY1PDxer0x\nW6Qv5+qrneL23btLbr653uOhZs3yPfnkSYBILseZM6NciqKEB6kDHa9uuPrsaWdffvrlezV7\nz/Kf9fLxl8u9qcy8eJ63Wq3C4ytcV1zWd1kdW2eIGC52XyxsxPNGTCHg9Xpj5ts8wZsY0yk1\nAWA1wZBwu90nTpzweDzasPbRjkd3tO54//v3b7feLuxNVtCM/9h5WPdwjkaMSQRWcGAKF5Ik\ntUePJt09LKkFQsjeP51OuA6fg2XbSCTi9XpZlqUoShUMDhrDMQooLS3lU6RsPPoo/Nd/ZfaK\nCCFhKSNZX0zvyZMsy0ZCofrbb4835ohEImazOVoI4/mJy5bJhVVIAM3nn8frUNgzzkhreCRJ\nvv/+hGTOfD/4wbBfd0HAMAzZ0pJsL/XDH+ZyMIOQRFgUTLNilW1CoVBPT88f/1j6r39pxY3F\n7o6ZY8T+MoE445uJeRrXMIlJYI15ORSFIEkHHE3n9ItfzoxyVSqVUNROGOi4pnMNQRDiJCRC\nRKC/9vF/4f9LcVq/3x/zq5YKQKq4qtUUnjdi8o+g5xKJURO84HlhgjF3aoLidTnlOK6rq0v4\nYzFoR5u0mzj+Y+c11WuAyR9YwYEpaIjDh3mOc9jtnR0dXf2LwGjNGkAIZs8exglDoZDwZSVf\ny7a9vb0tLS2dnZ02m81iNvPXXTfEGI6ipqSkRP3990l3Z+eFS/tiOh98kK2vV/YnfQQaG30+\nXyQSMW7YIBhzSEfC8/zJkydPNTH1m4kKPyoPHozXoSjUaujqSmt4KZz5RFiW/eAD9223+adP\nj+j1qIgW7enmZofd3nzkyOFDh4698oqwMfCHPwz7f27GkcvlBEEkExbJFIp039DsEQgEjh8/\n3tfX19kpW7LE9vLLx3bvbrn22qhAadeu/I6uaBC/jPI8/OUvxnvumeD3k3PmODZuPKnR8EVn\nfCPWKRK+HADweKg77qj729/0NI1Wr+5avdpM00gulxfdKx0iOp1OLpeLP4qBjgCg4TUEQUD/\nJORA04HNbZvlSA4AO0p3pG4JjEQGCF5iBCCvm16fAFiFjsk/vKTVN15N8D/a/8nZSDiO2+bd\nthW2HoNjXvBGICK41dwGt+VsDMOmr68vvqCZTAij1WpF4ZiA9GOnBEpyMmRMYrCCA1PokCRZ\nXl5eXl4ufpEnZs0a9tmii3hJlm39WS5wiK28AoPEcIyubBe6udlqs9ntdo7jxJSN8KpVsrVr\ns3E5giAYhlFv3ZqsL0ZmtSq/+05qzBExGimDgQBwOByi1DPWe1KhIIPBeB1K5YIF6epQUi9s\nIoTMZrPT6XzooQm7dxdlpGV5eXlZWRnLsvDJJ8IW5QUX5HdIUiiK0mq16iTCoqpbbsmGsGh4\ndHZ2Cl+5Ro07Zkb46it4/nn49FNobweXC2pqYN48WLsWks3fUxjfaDSaHOsaRo5Go+nt7U34\ncgCgq0u5ZMn4GGtPAJg+fdT2YBIEseu0Xcf7jv/I9aOYQMeruKsGHNk/CWEpVsNrNJpUBkrS\nCUy8AOTP+j9vg23Jn43B5AhpnS5eTfCq8tXcDMPv97e3t5Ma8lFnrFvNh+jDSWiSlbQWsqZD\ntFoTiBHCPO9+fkLVqYImRVG1tbV/8v7JSTmT5chi8kWR/UXHjGkWLoSFC0d4DplMRhBE6euv\nJ5z30nI5dHVBbVZMEBBCYisvJE9t8Pl8fr8/FAzqb7qJKXhxB0IoFAqFQiGaphV+PzFrVrKK\nDEEQBoNBr9eHw2HYu1fYKPvxj7M3ttLSUiZ5X0zvLbeUb9sW06BE+v1qtTqF9yQMrNYPIKPv\nUVdXl+CQ2tXFFG+kJUEQCoUCli+H5cvzPZYEVFdX8/2ingQUxn86v98vfuXKTShGUeD1elev\npt9++5Q55KC1v4qKikOHAgsXVsVM+0mSPPfc/LvepotGo7Hby26+WR9fxaivV37wgd5koiFR\nB1xjY97GnG3eod/ZWbETKgZsnA7T/8T86amap7rD3fGTkCtDV8qUqRQcarWaIAhhaSR+3rhd\ng/NTMAWBVqvt6emRNuJJC3m5URNwHGcymTiOS9jc4SbcbiL65aooEojiC5oPwUNbYIv0mJKS\nkv3q/e9S78bnyD4ED+V0uJiB4AIHJlsghAiCKDQZAkVRGo0m2bJt7aJF2Vu2lbbyJkxtkHNc\nR0eHYB1nXL+e+eijAs92cblcFoslquDl+YnLlmkGq8gIwgpYuhSWLs328CorKyPNzcn26t55\nB+KMOahgUK1WC7lfCZuYem+9lbr/fp7ng8Gg/PvvDZdeCpD5GzsYDIr5L3jRPnvIZDKuudnc\n0+N2uyORiCgsKoRPKpGYBSUB0U7y3HODixalTIAYdYgxWG1tDWLtb9Mm40svlULK2h9Jkrt3\nj09mfHPXXTkZfUZ5772ahC/nscfQwYODd8CNPi6AC3qg5zgc94HPCMYGaLgRbhQCHfeW7X2b\neDtmEnJG5Iz1yvXJziZAkmRlZaUQMCwgnTfqCF1WXgkGkyZyubyiouIp/qk8qgmcTqe0v0Pq\nVlPCldzZc+ePfT8u8AQiaZvbEG01LqIu6oXehB87ORw4JhZc4MBkGI7jbDabx+MJhUIkwMRl\ny5QFJkOoqqqCfCzbiq28yWwve9rbfT4fZCLTNAc4HA6z2Sz+OEi7TT4gSZJubrZYrcIfXXH6\nar39duWRI9oPP4wPrFUjBAAURUXC4WShtpUKhVqtBgD417+iV5o5M7Mjl+Yj4EX7rEJRVE1N\nTU1NDcdx5BdfRLdm+g3NLFI7yWee6aXpunyPKKd0d3cLGitp7e/6621CgSN17W8oxjdFRLKX\nc+65xK9/neOxFAQrYMUKWJFw14XEhVawCpMQPa+fhCbdRNx0A33DUCYhBoPhOcVzncHO+Hnj\nHMCfxZhCwVPl2chvtJCWfKkJhG+wAimimhMadhYIOp2up6dHWqYZVAiT4mMHk0dwgQOTScLh\ncFtbW6jfArFy/Xrlnj0FNekFALlczh461GY2C5/F4ryXv/9+cnUW7dCjrbxJ7D/YM84QxhMv\n7vBPnUr5/SpVDgJehgrHcVIzkZiKDKqpIQqjIkOSZFVVVVVVVTgcJvbtEzZSbneqwNrKSpVK\npXrppYTHAEUpHQ4QChyZ6JlKSIytnYC4aD9zJrtokTz+AMxIoCgKFi2CRYvyPZBYpAtK8aEY\nFRVleRxb7olEIk5n1GZiGLW/nCV65IbCfzkIoc8+C//jH+iLL+iuLmooVilZYsAkJE17fYIg\nPtF9slO3E6vQMQWL1+t9MfyipcwSs/0cOGcf7MuNmkA0Ok0YV7S2a61U02EIG37P/x4K7LuM\nsObxZ9+fEwphcEGziMAFDkwm6erqEqsbBTvpBQC5XH7aaadFIhGWZWX9fhBkltMrVSoVSZK6\nV19NOHMmaFpmtXIqVUJxB3i9BVXg8Hg84l+y+IoMe8YZhabMk8lksHgxLF4cCASo+fMhiTEH\nWrcOfvvbioqKYJImprolS3LgPUnFOX1IF+2ff95N09UJn4gZfahUKoVCkcwds6xsbBU4/H5/\nTNorjO2GnULG5/N1dXWtXl21e/epZc9BrVLS9Y7NDSmaX/I5LAwGAAAsFktvb6+8VP6oN9ba\nkwU2Z3epaHSaMKr5sPJwjKajqraq0AocAKDT6fZrsK1G0YNjYjEZIxQKidJ66aRX0doKAMFp\n0zJ7OY7j/H6/x+MJWSxQXw8EAQQBDzww9DPQNK1Wq5mlSwEhQAh+9rPMjjAGoZVXtP9omDu3\nYe7c0v7s2+pbb9Xt3Jks0zThkn4eEctYCdtt2DPPzOfgUqJUKjUtLcn2EjNnAoBCodAePZr0\nFNkXIkld/eMzIKur1dkeAKagGDduXFeXcv78SXv2aKV2kh0dtVarMt+jyykxAX4AsHt3yc03\n13s81KxZvk2bzCmyUL78Et1xB3fuuaisDFEUyOVIpSqm6OXiwufznTx5MhQKCTbJQ8k2RgjZ\n7faVK30bNsDXX4PNBuFwtCCycmVOBx/PCljxJXzpAAcLbDu0fwAf3Aq34uoGphBwu929vb0A\ncIXrisv6Lqtj6wwRw8Xui4W9uWwD0ekGWNLERDXHJNeuN64nyQKdhF5EXTQTZpZBGQPMeBg/\nG2Zvhs2fwWelUJrvoWGGClZwYNIhpWOomKyZzGMiU9/EeZ63WCxOpxMhJHhbFn7aiIDBYIgk\nn13TTmey1gnGZoOaAvL5j/5ZStJuEyrgAgcA0M3NLpfLZrOxLJsssJY6ciQUCjmdzkAgQDc1\njbv8coDceU8qlcqSkhK32x2/aC/sysEYihdhBXjvXtTRQRTOCvBIUCgUH3440WSiYLS4Yw4b\naRRifMNOWVnivE+WZS0Wyx//WCaVEoRCUQOLIopeLiK6u7sFrc0QbZJ5nj958qTf729vbyje\n3CgMJvfY7Xbpj9I2EGPEeB91HyT26sk8Wq32rZq3EsYV/bTvp6vMq2I0HUpUoAV6bKsxCsAF\nDswgsCxrtVoDgUAoGKy//XZV8lJCVDmcZNKbKQUHQshkMolWRgXobZkaurk5GAx6PB5hdl0p\nhI+uXeu75x76hhsgSetE+H/+B84+Oy8DTohSqQSAsu3bE1Zk5CpV9tJ2E5NmWE9paWlpaSnP\n83y/MUd8YC3DMEahqeqDD6Kbcug9OW7cuD17umMyIFtayqqqqmga6saWrWQa9PX13XsvtWuX\nBmBUTV+/+y5xPnGRumMOG5VKRVEUx3EJG3a0Wm38U4LB4IkTJ3ie7+oyCjPnP/xhPMPwR44o\nAaC0lHO5KMBT6IwSCoXEBY8hWqV0d3f7/X7AuVEYTJoEAgHxcby1Z9VpOW0DSRhXNCU45dc9\nv4aBhp1apC1YBQdmFIALHJhUeL3e9vZ2wW3BuGGDau/eFKUEwQwv2aSXlsszMul1uVxidSPG\n5iNiNNIFY/ORAoVCoVAoACQ63Zkz1Wp1KHmmqexHheU1rVarlUplsrTdkmuvzYFRhd/vt9vt\ngUCAC4eHF9ZDkiS5eDEsXjzIcVkzE00BSZK7dtWaTABjftF+6FitVqvV2tExSlaAeZ7fuzf4\nwgvE/v2yri5KJiOSqVFYlv30U3bLFvrLL5muLqqvL+mRxQ5Jkkaj8fPPe5cvnyit/R08qJLL\n5RpNWXl57FO6urqEP2HizPkf/zjR20tfeeVkAKirY10uFQBUVPC4aTdThMPh+I2iVco55/gX\nLRpgKcVxnBiMjXOjMJi0EG2JElp7/hP9M5eDiY8rquVryz3lHUxHjKbjWvLaXA4MM9bABQ5M\nUjiO6+joEL4axpcSSL0+5sugQqFIMek13HRTRia9fX19woN4b8tAYyMdCAjignwydDXBwJkz\n3dxs6ujweDwgyXbp+81vtOvWpapzpyleyBTjx49H+UjbFejt7e3p6RH+rhsLMqxn5Bw4kHj7\nWFu0HyKBQMBqtcJoWQHu6+szm80PPFCb2qARIdTd3e1wOB56aMLu3aoUR44aysvLd+9WmkwM\nDKH2FwwGxeVN8a7QarkdO6Kt1DU14e++AwD4+c8DANjdJjPE/8GS2iQ/9ZSZpidL97Ism8I7\ndsaMwKJF+f6zjsEUKgzDsCwLSaw9Zbxs0DNkkPi4osupy7fot/xV/1fpYdiwE5NtcIEDk5S+\nvj7B0S1hKYFzu0tLY+12amtriaxOeh2O2gsukHV2AgBXWgpxNh/yUCgvBQ6O4wS/hkgoVLNo\nkXxYniAkSdbV1QUCAb/fT//rX8JG3cUXw8Avi+Fw2Gaz+f3+UCjE0PT4xYuHd7kRwjAM//33\nNrvd5/OFQiH1sWO1P/85QC6KLIFAQAypjSm9cVVVVDGoeIZC4WdAFhSp00OvuIKFArRrT4LH\n4+no6AAAwaAxhRpFqG4M5cjRRHNz4g/5+NpfKJF3qDhznjIluHevBgDOOcd/3XVeXODIFAqF\ngiRJYXUkUbZxrIuQmMklIi2IPPlkF02fnqOhYzDFhk6nE4r7AjFtIPGhbDkGJxBh8gIucGCS\nIvTQJnMM5fs7bKUoFAqDoMRTAAAgAElEQVT20CGTxeL1ehFCogxhJJPeQCBgs9mE2sFpy5er\nOjuF7ZTLFWPzEWhsVKbT0cfzPMuykUhE7vMx558/bBFEIBAwmUxC0Ilx/Xr5xx+PRE2gVCqV\nSiXcfTfcfXfCa508eVKMEtCtWzfCy40EkiT1er1eWBzvT9vNgVGFaKkVX3rzT52qjEToFFEK\nmFGKsIQlRSqJnzcvWEQFju7ubuFBajUKy7JCdWPQI0cZI6n9iTPnhgbWZGICAXLWLN+GDSa5\n3JC5AY51CIKorKy0Wq3xVinCrpjjhf5WgfiCSEkJlm9gMEmprKzcSGy08tZ4a885kP/mLmzY\nickLeBqASUkSx9BAYyMTJygVkMvldXV1CKFQKCTrN+AY9qTX5XJ1dXVFOxEef1xqAuK58ML4\ntBGlwwGJfObiXhbq6emx2+0jz2HhOE6sbmRbTYAQam9vF6sbMZeD2lrIo3ghh0YVguY8WekN\nAoGEXoOY0Q1BDHCKl64Ab9hgoumqfA0sXViWFXUHqf0IxFjuQY8cs0TdjgBg4Mx56tTA998r\nxSk0TSNpNjNm5Oj1+qNH+QULymOsUvR6vUajjLFJlslkKpXK7/cn9I6NyZ7EYDBSSJL8rPKz\neGvPs9HZj5CP5GtUGEx+wZZamKQwDJPMMTRaSkgOQRByuZz85S8BIUAIfvazYQwgFAqJ1Q1x\nMs82NAh7tZ980jB3bsPcuaU7dwpbJi5dSr/88lDO3N7e3tvbG62b9OewRPelWeBwOp1CdUOq\nJlC0tgKAf+rU+FXlkeB2u0XztvjLDSWcleM4obcFHA6orweCAIKABx7I4CBzAEJILL11Pvgg\nW1+v7G+MCjQ2xvdyY8YC4iIwz8Nf/mK8554Jfj85Z45j48aTGg0vnegWOMLnSQwD/QiGdOTM\nmax45JiFYRihcuHxUHfcUfe3v+lpGk2eHGxuVtI0Wr26a/VqM00jnU5XRHdIUUAQxDvvVEmt\nUm64of6GG+ovvVTbv1AygJqams5Oxfz5k/bs0UoLIkePlns8cc6x+QYh9J3/u6vYq+q5eg3S\nyEA2HsYvgkU2sOV7aJixyIXEhTNhZhmUMcCMQ+MuQhdths2fE5+XQmwjOQYzRsAKDkxSdDqd\nL4lj6MSlS/lHHoEMJb8mw+l0CpPV+E6EpAyhPOF2uwUjT0hknkoZDGlFhguRLsnUBBG/Xyq+\nHSFi8F7CywXPOCNFYILX67VYLNEz8Pxpy5er8+HckREYhlFv3Zqs9Cbv7YWS2B7vUc9XX8Hz\nz8Onn0J7O7hcMFpDNFJQVlbmcDjcbjJ+BVjwP873AIdKaoPG//u/bpqOFnnjm6ulR/7jH300\nXZOLERc2tbW1H33UedtttSYTQ1FIo+FaWxUUhebNc5SVcQcPqtRq9VlnjdJmnrySlk2yQqH4\n6KPTTCYKCj43KhAIdHR0bNFu2V59qlTTCZ2bYbMd7NshUf0Gg8kmA9pA0voKi8GMUnCBA5MU\nmUymPXo02V5y1izpjzzPh0IhmUxG9fVlKtQjhQmI9Y47fPfcAwDU4cMTrrwSANCaNcR99w3l\ntKlzWAifLy2tMs/zKRp5SI5L/fS0iGoTklwuhYLD7XZ3dHSI0gbjhg3qTz8t3tiR0tJSSF56\ny0FCbaERDAbXrCF37DhVzBjFIRrJUCgUgUDt/PmqGEk8SZIzZlTHNLAUMgqFgqIooRMt3o9A\nrz+1Iif9pIo/srp6XB5GX3jIZLIPP5xoMhEAwHGE00kLD55/vuL55yuEYwptCj06SNcq5bvv\nErshFlRuVCgUEmywdJzu0Y5HpwanKjnla+Wv/dXwVwBohdZ8DxCDwWAwuMCBSQl15IjH4+np\n6QkGg6JjaGjlSkbS1BAjDai//XZVhqQB0k6E+NqBVqutrKyEDz8UthADCy4pEPrbk2kuZP09\nIENEJpOlaOSR9/ZCnKHasGEYBgCSXU6mUEBXF9TWxjyL53mx0wfiRCuopoYottiR0tLS8Pff\nJ91dbPWaEdLd3W23248fbxBDNDZtMr70UimM3hCNZLz+eqnJBJBoBfj04glhIAhCr9dbLJZB\nDRoVCkVJSYnb7Y4/UtiVx1dRUHzzzSDlrYKaQo9ZiiI3ymq1CsXHK1xXiBsvdl8sFDh+BD/K\n28gwGAwG0w8ucGAGQavVarVanuf5ffuELcz554t7XS5XZ3+sCQAYN2yQ+oCOcKopl8tlzz+f\nrHagsNuhsnIY3pYkSaYyT01zpbekpIRP3cgzZUpaJ0x9LYvFok5yOd111yUUL3i9XtGXNF60\nwp5xRjG2ntPNzT1Wq91u53leLL1F7ruPXrMm30PLKVarVciUkYZoXH+9TShwjNYQjWSkJYkv\nZCorK1tb0fz5Oqka5fBhjcFg0GjkUoPGcePG7dnTffPNeumRLS1lRqNRJiNirBzHLEUxc8YU\nBWJ/qwBP8B2yjj9X/RkADGHDvcS9+Gs1BoPB5B38SYwZEiRJkosXw+LF0o0cx4lxhpAFaUBp\naSmbvHaA1q2D3/52GKdVKpXMCy8kq5uoXS4oTcOWqaSkJNTcnGwvOWRdyVCgabq6ulrR1JT0\niEQVJdHoNJlzRzEWOAiCMBqNBoMhHA6LCbX0j8bW6hnP8729vcJjHKIBo2seu2OHPpkaRdpM\nQZLkrl21QzkSgxnj7N/PP/ccv28f0dVFDc+oCCHESdpOr264ulUR7Uk5y3/WE+1P1EyswV+r\nC4RWaF2JVh4gDljAEoRgFVRdCpc+DA/rYYwV/jGYMQlOUcEMH7fbnVAaMPRQj9QolUpNS0uy\nvcRwo2fLy8tFEUR8DovslVfSPSHd3NzZ0XH40KHDhw4d63964A9/AIRg9uzhDTIZZWVloa+/\nbj16NOZysHZtsstFPQuTxI6EzzorsyPMJQRBMAzDLF06krCe4sXv9/M8H7NRDNE499wgDtEo\nXoauRkl25FNPgcEADAMTJ8Lvfw/9ybMYzJiD47jOzs577/U++ST97beUzQbhcNSoaOXKNM5D\nEIToARwiQjaZjUbResZB1cH1xvXxvr+Y3IMQslqtz9me20ZsOwbHvOCNQERwgb0Nbsv36DAY\nTC7ABQ7M8An1f2VOKA1gR1zgAAC6udnpcBxtaZFO5sOrVo2kdsAwTElrciew9NtqSJIcN27c\nD37wgwkTJlSbzcJG5QUXDG94g1JSUjJ58uSpU6fW19fXO53RrcnLPUL84SDOHZgihIuzsN29\nu+Tmm+s9HmrWLN/GjZ00XkssWt55J1q1i/kX/7kScyTLhi6/PAQALS0w7IkcBjNqQAidPHnS\n5XJ1dTFLlthefvnY7t0t114bzbnftSu9s6nVauEBg5g9zXsONB3Y3LZZjuQAsKN0h0wmy+jY\nMWnD83xbW5vValWz6kc7Ht3RuuP979+/3Xq7sBe7wBYmLXzLXG7uZJisBS0OXcZkBFzgwAyf\naDBBEmnAyBUcAmVlZaeffvrUqVPr+tX4sh//eITnJJuaQizbbTafOH7c9MYbwka0Zs1I6iYy\nmaykpER95525URNQFKVSqchf/nLQy6nVaoVCkUy0UjpvHmzZktWhYrIELSlg8Dz85S/Ge+6Z\noPC7etQT93+pmXHuZJCYAWPGAr29va2trceP8+JE7rrrojXQdCdyGMzowG63BwIBAHjmmbY7\n7uiZMiVoNIYXLLALe9M1KjIYDNJIJgKIc/3nqngVAJQAtvXNPzabze/3A8AVrisu67usjq0z\nRAwXuy8W9mIX2ELD7/efOHHi7z1/f4V6BcttMBkEFzgwwye1NIBRKtOQBjgcUF8PBAEEkXBW\nRlGUbMmSDNYOGIaprq6ur68X6yZDz2HJKqFQyOl0WiwWu90e6OpK/WsZIhMmTBALTwkYY7Ej\nowaVSiUooj0e6o476v72Nz1Dc/snXW3wmaJH4Hd2LOFyuSwWC0JIOpG74Ybo51u+HGe/+gru\nugtmzsT9Mpj8IAbDJzQq+sUvImmdTalU7p60+2nD00eUR+y0/aji6KraVU7KCQBzibkZGjJm\n+DhFWSsAAPAEb2JMggusMWJcDavzNC5MAjweT1tbm9/vF0KXhy63QQgdCh66JnxNA2rIquij\nFVrnwTwsLSlGsIIZM3y0Wi3DMMlCPUquvTZhqIdIMBjs7e0NBAKRUKhu6dJMhcumTfo5LFkC\nISTUNaI/8/zEZcsgE78WhmG45uae3l6v1xsKhTTHj4+/4goAgLVrYdWqEQ571BCJREKhkNzn\no847L/prL+zfD0EQFRUVX33lWr58ohCi8e65v570+ccRRkmHAgC4wDG2sFqtwoPCcZwNh8Nr\n1qAdO065OAr9Mjw/tgKMMXkkHBf9LhoVnXOO/4YbeABNWif8l+JfOxU7Nxg2SDdOh+kPwUMj\nHCpmhEQikUjkVMUq3gW2dnItYJuUwgAh1NXVhRCCNEOX7Xa71Wp9sfTF16pfEzcKog872LfD\n9kyN0Ov1vsS/tLVka1avgskSWMGBGVw9kQyCIMaPHz88aYDL5Tp+/LjL5WJZtvKxx4Rw2UGf\nNboZUN0AMG7YoNm3L1O/FoqijEbjpEmTpk6dOr5/IpTCuWN0gxDy+XwOh8PpdAa6uviJE4Eg\naJnM+7vfBX7xi4wUlXIAQsjtdm/fXmYyMQDwn5HXLvr8L2/Cld+EzgAAr64WRhZjhCkiQqFQ\nKE4XIU7kZswI5N5x1uFwHD16FPfLYPILMTD6XWpU9OSTJxkm7a/BF8AFM2FmGZQxwIyH8bNh\n9mbY/Bl8Vgpp5K+lBV5GHgYJXWDzOySMFK/XK61GwUC5TRVXlVBuY7Vau7u7OY5LV/SRFgih\n9vb2kydP0m5avMqdvXdm9iqYrIIVHGOUvr4+r9fLsixNklW33soMd0anVCrDTU1mm83n8wnS\ngLr//E+ApEvfPM97PB6/32+326m+vknXXcd0dQEAe9ppvFqtPHw4YjTSY3JWFg6HHQ6H+GNM\n5i5fXU1m8NdSMKKVvOD1eru6uqLLejw/cdky0hTt6ZC3tgpFJTIYBCj0AofX6w0Gg83NVQAw\nFZqfhVuOwukLYXMnjAMA/9Tp6a1LYoqZhI6z9947zu8njcaw3w81NTC8aMzh4fF4zGYzADzz\nTJuoKLnhht6tW8sgf/0ymDzCcdy+faEXXyT375d1dpI5uxtVKpXQpcLz8OSTxo0b9QjBnDmO\ne+/tlsminbZpsQJWrIAVWRhpYhBCrwZf3arEy8iDQ9M0TdPCtFlwgUWAvlJ/tWziMpZgd5Tu\noAis30gPhFBTqGk1ufpb6lsrac1g4C7LstIfY+Q2m+2bJ4yfEPOUUChks0XremmJPtKlu7vb\n7XbHXGW2a/YTlU9k8CqYrIILHGMOnufb29u9Xq/wo3H9euajj0Yyo5PJZDU1NdEfvvgi+iCR\nNMDj8XR2dka/iPP8+N/9juk36bAuXz5u5UoACDQ20oGAUqlMdxjFjtfrFaR6MDBzd8ollwAA\ne8YZY+43kh38fr/JZBJ/1aJMRrj/dbt2iUWliNEIFRWF/BEpWKk99dRJ0uudNG+ezMYTWx79\nPPKx4pogACgvaMz3ADG5Q5pPKZ3I1daGurqYnp5ouEPOOkR6enqEB4XTL4PJIzabzWazrV07\nbvfuU06cubkbKyoq+vr6PB5qxYpxe/ZoZTL0xz+a58xxAkB5eYUY+1qYhEIhk8mElOhR9OjU\n4FQlp3yt/DVhLoeXkRNSVlYmzoFB4gLLUqwWaYFI8VRMLF6v12w2v6B9IRvNIFJplSi3iRAR\nADioOriOX7cVtsY8xe12i1/eBHiC75B1CKKPar56NZkBj5VIJCJdbszSVTDZppC/vWOygtls\nFqsbMTIBqB2Opj0QCASDQYSQwu9X/fd/R7d+9VWMFajf729vb4+ZWAJBAEIAECkvF8JlA9Om\nyYLBMVfgcDhKzjmnrL0dAGyLF5e89x7EZe6Osd9Ituju7hZvwtj7nyDYCRPEolKgsTHi8ZSV\nleV1vKmI1gr7Y4zaH3uMra8v2x79zpGpGCNMUcAwjFwuZ1k2ZiK3bVvF5ZfbLrmkr6yM27hR\nv21bOQDs2pXdKWUkEgkKFXMJUuODRYtUWbw8psCwWq2CQYwQ1Jrju1GlUrHs+PnzlYJR0d13\nW04/nT14UKVUKtXqgpaLIoRMJhPLslewWVysHmXo9fpN1KYerucn3p8Yw0Y7bX+u8jnsAjsM\nvF6vsBokNINkvL4mFU/Fy21eV78e/5QYP50Y0cc/+v4xoTpW9DEMfD5fiqsklJZgChBc4Bhb\nhMNhl8slPE4oE5CnczaWZTs6OqJfZAdzxOzp6YmZWEYqK+neXgAIGwyM2SzsCjQ20gMLtKMV\nlmUFCxLEcdULFzLt7cJ2zeefx09Ww2edlb+Rjh4ikYiQFwgx9//FFwMAIklpUSkwbRoq7LAH\nmUwGyWOMKIaBri6orc3rGDG5o6qqas+e7mXL6trb5eJE7p57LCoVX1LCCdGYwpQy2x0iHMc1\nNSnfeqv0m29U3d0yj4cqKeE9HjIcJmbN8m3YYCLJKdgCbIwQDofFFXVpv1LO7kYAeO01ndCG\nGIkQ69ZVi9vXr4e77sr61YdNX1+fVMYvXUY2hA0ruBWQdnvN6Ickyc8qP9sJO58wPiHdPh2m\nP0w8nK9RFSPialCWmkHUarVCoZCWwqVyGx2hi3+KVG8VL/p4GB7eAltGOCoY2O8Zf5VHuEe2\nwbaRXwWTbXCBY2whaNoBgPR6J/zqVzBQJuBvbBx6gSMcDre1tYkWQTFS/5gCB8/z4qWFiWWk\nooLu7QWSBJ4P1dWJszJEkvLeXigvH9HrLHjsdrsQ6IgQqnr8cWmXkPLgwfjJqlKjyfhkVZjt\nh0IhhmGUgQD9ox8VRXTISBBv1wH3f08PEQoBgPvSS6VFpUBjo5IoaD1rSUlJT09Pshij0nnz\nUscYYUYZWq32/fdl7e1yiJvIrVjRvWCBXewQ+c//DAPIsjcSnuc3bdJLmxEcDgoAfvCD4MaN\nJxmGKPC+AIzI/v38c8/x+/YRnZ1kXx8xDNcMj8cjrm3kq1/pwIHE22fMyPqlR4J0JTk+EKS8\nshwXOBJyAVzQAz3H4bgPfEYwNkDDjXDjfJivwL+vIRMMBmM8MqT1NWPEuJrOQJvG+PHjH3Y+\nbCfs8XKbOTDgc6EVWlfBqi/1X1r0FpZgKyOV53vPf7P1zdJIqSj6eE31WpLrpIewdCQQLy3Z\nrsHGN8VBIX7J8Hq9W7dunT9//tSpU1UqlU6n+8lPfrJp0yae5+MPPn78+A033FBVVaVQKCZP\nnrxy5UpxIo2JJ/o77Ne0dz74IFtfL8agBKdN4zgOITSUXBWbzSZOF0WpP9vQAAARozGm1SV6\nWsnE0n/22cKAAED95ZelO3cKR05culT1xhsZf+EFhcfjEUvjut27pb86ITOlZPfuhrlzG+bO\nFX8tmjlzYEsGKtMCCKGenp6WlhaTydTd3W1qawtedVWxRIeMhOi0auD9X/Hii8Je3/Tp0qIS\nIknlwD7MQkMul1dUVCiampIeMXrfSkxCjhxJ/A2+sTEo7RC59lpP9sbgcDhOnDghNCM8++yJ\n884bIPelaaTRYPfbIgAhZLVa773X++ST9LffUr29RDgcdc1YuTKN88SntIKkX2nmTDYH+T7v\nvAMIJfh3wQVZv/RIEFeSEwaCxPsKC0/5zv/d1aGrJ/GTxmzkygpY8SV86QAHC2w7tH8AH9wK\nt+LqRlrEN4OcPe3sy0+/fK9m71n+s15te3UCZKBNQy6X7zfuf8L4xHWTrrtoykXXNFzzZumb\nEBe6HAgEXnS+uBW2niBP+Ek/R3A9sp7Xy15fXbNaFH0AQAmUJL1SOqjV6pgSfDaugsk2hVjg\n2LRp07x581577bXS0tLLL798+vTpX3zxxW233XbVVVfF1DgOHz48c+bMLVu2/PCHP1y6dGlJ\nScmDDz548cUXiyp0TAypNe0hjjv28cfNTU1DScoUHIZhoNRf0doKgnnBwPCnqAGeZGIpdmTE\nQ4z27FJRrxv/qwMqucV35iarFovFZrPFG21m/EKFBsMwMpks9v7/8kthb81DD0mLShOXLtW8\n9VYuhjXcnGYAqKqq6tu7t+nw4cOHDh0+dOjYK68I29GaNYAQzJ6dheFiChdxItfWdvLQocPi\nP4eDkkZjkmSC1YKMIISnIISeeabtyiudq1eP++ILNUVFP2pkMnTokJplq7J0dUwGMZvNVqtV\nKFQJKb/XXhst+KaV8huv1pEGtT7/vJ3GSuIkiCvJwjLygaYDm9s2y5EcAHaU7qDjfnF2u72l\npWVL35bXmddPkCe84I1ARLCEvA1uy/XoMcVMwmYQ4ccUgbut0Hoduq4BNQy9uHYhcWHq0OW+\nvr4TJ07IfXIxrvUiz0XCrjZF21HF0VW1qxKKPoYNSZJGo/Hl8pc36jceUR6x03bpVbCTS7FQ\niH9Yxo8f/+STT15//fU6XbQF68iRI7Nnz37zzTcFZYd45KJFi1wu19///vdbbrkFAHieX7Bg\nwZYtW/70pz+tTGuJYcygUqkoikqmaa9bssTym99QLpdyz57UuSoIIaGEkbDVJTBtmiwSkf71\nJUlSpVLJX3xRnFh2PvRQ5bPPCpNJ88qVNf/93wAQWrmSSXOCl3FCoRBFUVRfH8ycmY2WDYSQ\nIDJK+KvrvfVW1UMPCZ0jipaWiosvzvgAWJa12+3ijzFGm6imhhjVMb0Gg4FIcv/Hk71aG0LI\n7XYHAoEwyxpvuWXYOc0EQRiNxsrKSuGeUe/ZE90+a1ZmB4wpLpj+FoL4aEyaRkzWYjnF6q1W\ny/3975UmEwMAHBdt9Tp8WHn99acVuPEBBgB8Pp/T6YQkrhmVlQiGHEehUp0ylI2/G43GmhTP\nHeNotVrpH2upQ4GG12i1WunBvb29FosFALJkCYkZUyiVSoIghGWw+DaNN7QJdNZ2u/1Z7tlt\nhlP+FDF5K0fR0Xv5e78hv+kheqRxsylClyORSFdXF0JI6gPiIaMKxJPMyWsarhEex4g+RkhF\nRcVnoc/+xfwrxsnlbHT2Q0TGroLJKoWo4LjmmmuWLVsmVjcAoLGx8e677waAjz/+WNx44MCB\n/fv3T58+XahuAMD/Z+/M46SozvX/1tL7Pkv3LDADIyogoAHRa4xejTGaG43RoJEYJYoSQG40\nJvdyoyBLbq4LMRL0Z6JGjKhBAnGJIKiICwJqAGOAGZaBWXuWnp7u6emteqmq3x+npyi6q3p6\npvfp8/3wR3dVd9WpXoY+z3nf5yFJcs2aNSRJPvvss3xpGFWOFJIkq6qqktS0k8GguGOCr6mR\nzFUhCIIkSblWl+DUqYmLNjabTRBW4vovaoayV5Rf/3ralzgCotGo3+8Ph8PgcvETJ6Il9IFf\n/KLpyBH/976XpZaN5F1CwalTlUplZWVlbW1teWtr7DkZnWYLpTcgVUIy5qM3LBaL/tgxub3d\nq1ejG9zKldmrgAiHwydPnuzo6HA6nepf/xo5sMT2jerDRlGUXq8vKytTLVoUW8E/M8MIU2qg\n/0C9XmrJkvpnn62kaX7FCvuKFV00zaNPSzZOKqi3iKYm6einAjc+wACAx+NBNyRdM667Lj4i\nJwk6nQ5pHImfRq1WIf6lh4lDr9e/XfO25DLy9aHrxTIly7IopwYArh+4/lrPtfWhemvUetXg\nVWgjjlzBjAiSJMvO9MJL3qZht9u7u7v1Yb1QZ3Gv41606wSc4Diup6fnxd4Xt1BbThInU68t\ncrvd4sp9juDalG39in40HlQkklj0kRGuUl41i59l5s1KUNZytf/O/ft6WP8Z8Vlmz4LJHoVY\nwSEJ+l9QpTptgrlr1y4A+M6Zv+Nra2tnzJjxz3/+8/jx4+eee26OB1kUWCyWgX/8o6W7GzVw\nqo8dmzRnDgA4lizxXH31WXPninNVwtOny9mO6nQ6esMGufgGZV9fnCOmTqdjjx+XHdauXbks\np/d6vT09PTELJY6buHixbkhNCE6dalu3TrdnT/IallFDURRJkqa//U3ypQOKont7Yfx4AIC7\n7oK77srgqRHSRpuiPNoRJekUI3RTE8MwyKBe2dRUde21ALEymer169FjyIsuytLZheQ/SCif\nYauqqDFdPoPJGTqdbmCg4sc/LouLxgSA6dMrsuTxGWcK8Ic/tO7caXzooXGBAInCU2bNOlvs\n34YpWBKNM8QeLj/8oR9gBMHldXV1H39snz+/WvxpbGw0VldXq1RkfX0mR54XjrJHl8Pyg8RB\nB+kQL01XQroJMZ9aPn2HeCduGXladNpazRk9Aj6fL66DW2wJWcVWraAyYAmJKSmqqqr+rPpz\nd6R72MBdn8+HCr7k8lba29t9Pp/RbBxpbZE4YyXRZ3dy2WSr1ZqxCz6TpbB0KTFUWlKIxQCY\nYSgOgYPn+Q0bNgDA9def/vIcO3YMABJVjHPOOQcLHMkxm81GozFmkvzuu2gj09Ag2WwiN92t\nrKwMy5T6j7v7bsn4BqqxMRqNDg4OMgyjbGqq+Na3APKQ2eHxeDo6OnieJwgCAOLkDLqvTzzn\n5KqryUzPOQ0GQ5IuoWwnX8QZbcbl0UaR+etYR61WxzLYhdcflclkR1QSIyT/JeY0B6ZMUTCM\nOBwegxk127dXyUVjZqlOi6Iooao5sRlBoYBE1wBMYUKcGSAVJ1QplSP7P5Gm6V276osxqHVY\nGiONS8NL96j3oIkfIq4yPx0uJy53gAMFglRylQ1cwzxy3m30bXGWmYmWkOKp4AvOF+rqMmAJ\niRkdKAHkABzogZ7M6l9ZhSCIPWV7JAN345pBkLohEJdnvGRwic/ng/TiZhPjWtfa1v4x8sdR\nXhumBCiOXxurVq367LPPbrrppm+hKTEADJVQJtY3ms1mABgYGEjlyIFA4A9/+EOcI2Ycn3/+\n+YhHXPAgUwytVuucN+/wNdcAz9ffd1/idDdJw4JWq1UlqciQqXqgaTpW9rZjR2xTbi1FOY7r\n6uqCoR9wcUvo0X/Rir4AACAASURBVPLy6sceE885mfPO0w5zyBFjs9n4oZ4UCbLs8anT6fr6\n+uSMZtU6XcbzaAua7CsacaDkPznzmpDPhwUOTBL27Yts2MDv20fa7VTy2M7cR2MSBGEwGAYH\nB71eaunScbt3GxQK/sEHu+bMcQOAXm8gCjt3GSOg1WpRM6Okh4tGM4LyDUSRBrUmgef57u7u\nDcSGrdVbJR+QEduLpbD0tEMBKbuSTInsyROngmvK1myCTekPBjMKAoHAK8wrm8pOv/4Z1L+y\nTYqBu6FQSFgyTKyzMNAGBk4XYsTJH7/ifwXyllBC2X6iD8hW81bV4JgvOMaMniIQOJ5++ulV\nq1bNnDnzxRdfTOXxaPkoxR9SHo9n586dyQUOu90uHHbsgWZTctNdhVqdZLpLNTaGw2GPx8Mw\njKKxUVzqP/yJcz6xRHi9XqGOOnEJnQwEgCDEc07/lCkZFziUSiXzr381d3ai6juhSyiyfLli\nyAMie+j1eo1GI1dCor3ppmyXkJQ4LMvKlc8Ep05VSyX/YTAAwLJsV1fXww+bdu483f+MYjs5\nDh5/PP7x27fndHgIm83W2BheuLAurjWGIIhZs3B4StFgNpv7+voGBiBRqEJLIyM9YF4+jVml\no6NjcHDQZJZw9ETk0vZCp9MJtxOngm/qC30inYSj7NFl/DLkTBkiQsVS/oDweDydnZ1qk3qN\nvyhtX8/Q1+QhCALNuSTrLH7T8xvhkYnyh63WlkTgMJlM4si/5D67GIyYQhc4nnjiiV/+8pez\nZs16//33jcYzXG1Q7YZghSUgV9khSXV19fbh/uN99tlnFy5cOFaXnnQ6nVKplJvuWubOTT7d\nRY6YAADvvx/bVNghr+FwGN2QXEIng8G4OWdo2rRsDEOtVk+aNCkYDDIMo/roI7RRcckl2ThX\nInV1dXxTk+zuAomJdbmyFGST32HQNC2nJ/IkqXI6JW19MSUOsm4JBAJ2e+VPf9r3rW95LBb2\nuecqUarFe+9JCBzpsH8/vPwy7NkD7e0wMABJ6kTiUKlUu3bVt7UpQKoZ4ZxzMjlITPagaToS\nqZ87l44TqmianjUL556Az+dDFS7iqvsrB68UBA5rxPoQ8VCmfmIP2+OgVCpNJpP497B4Kpho\nCVkU8Dzf19f3EvfS36r+JmwsovIHyQQQoTXjYv7ilJOICh21Wh0MBkGmzuLRvkfRsqKk/LGB\n2JDkyCqVakf9jjZ/W6IPyI3sjaQSe2NgZClogWPlypWrVq265JJLtm/fnihYIIuNYwmBCCdO\nnACAc/AvqdQgCGLcuHFU+h0TearIGCkxoUpmCT1wwQUjqmERwzAMwzAEQagDAdWll6YyJdZo\nNBqNBpYsgSVL0r6yEaBQKPijRwc8HhQio2tutn772wB51REAACAQCPT19QWDQTYSmbh4sTY7\nQTbJ4Xne5XJ5vd5wOEwRxLh77lFldBgGg4GV0RMnLFzIPvoonHde+mfBjDE8Hg8KKEk/tnNY\nfD7fihX0O++cLkJOUieSyKFD0jaixduMUJps2qSVc8249NJR6l9jhrilNY7gbph0Q6uqFd2d\nEpzyx7Y/VtdXZ+Qnts/n28Rv2mQYpsehtrZ2g2ZDL9ubOBWcA3MyMI6c09PT09/fPwpnygLB\n4/HEJYCIWzN+Hvg5jJUEobKyMrENR1ydhVarRSuLkvKHmh2mJ/dD/Yfb9PE+IDO4GU8on8jG\ntWDGDIUrcDzwwANPPvnkFVdc8fbbb0tm2n3zm98EgB07dvzf//2fsLGrq+urr76qra3FAkfq\naLXa0KFD7b29fr+fZVnN8eNn/eAHAPmf7mYQnud5nicHBspnz64YCkxJXELX/vOfk24+wx3a\nNm/esC0bwWCws7NTyGSZsGhRZqfE2YAgCLPZjAxr4MMPY1vzWn3jcrm6u7tRLaLt97/Xfvpp\nloJskhCNRltbWwXjbtvataqPP87sMAwGQ1i+fIbKWnoLpqjxer3ohmRs5/XXhwAyYN3C8zwq\nvG9pmSTUiTz/vHXTJgukXCcy9poRShM514wZM6IrV7Lbtp3ufh+R/jU2EJt6iqvuEU2aprW2\ntRvJjWmeheO4jo4Or9dLmak1A7FJ/luVb60rXwcJk3ySJPdV7EvFErIoYBimv78fZJwpZ7Oz\ngZJ9boEQ+00IAFKtGd5y7y3cLV+SXxaX86gkGo3m3QnvStZZ3EzcXFZWJnZFFMsfBt4g6Twt\nLlkKQEAJSgIIDjgbbzubOPt2uH0uGe8DgsHEUYgCB8dxCxcufP7556+55po33nhDzs5q5syZ\nF1100RdffLFhw4Y77rgDPfG///u/0dPHakdJllCpVMhkOxqNUoKpamE3m6SIx+NxOp0Mw/As\nK46DBakldGmSTmsZhmlpaRF0etu6dfq9e3M/Mx8FPp8vGAyyLKu+4grThAlEayv8x3/kS9UK\nhUKCuhHv/Gqz0bnq2rDb7YK6ETcMvqaGyNAwqMbGdrsdVTgLDize//ov/WOP4T9cGEmSx3be\nemswIwJHT08P+liK60Ruu82JBI7K4vvhjRk9kkKV1+vt6Og4daohB31ShYwQtBxXdQ8AFE+x\nBLvVvFWZxFogNex2O1I2xZP8K9xXIIEj0eMjRUvIogD9IRKIK3+4b/A+KM/TyFJGcI6QbM04\nhzlnM7lZeHARtd5IIllncT5//mPkY1qt9r2J77X52y71Xpo8bhbBcdwWZssm7emSpTCEAeAG\nuOFNoihfHExeKESB44knnnj++edJkiwrK1u0aJF41/Tp03/xi18Id1944YVvfOMbd9555+uv\nvz5x4sTdu3cfOHDg4osvFj8GMyJomob582H+/HwPJDP09PQ4nU50Oy4OVo7W9es5s7nhppsA\nUqph6enpEdSNuCkxW1VFFaSfQjgcbm9vj83kOW7CokWEoPvkSZFxu93o10Ci82tw6lQqENBq\ntRzHRSIRpc9HzJ6dDW+OcDgsrJMnDiM8fXqmDLspiqqrqwuFQoFAgBqS2AxXXglY3cDIIE5J\ngITYTpUqA7YILMu6XC50W7JOZE5R1rljzmD/fnjpJW7fPnIU3SWRSKSjo4PjOMk+qZLSv4SU\nmcSqe5ZgAcDAGwgyrb/noVAosRFGmORXc9UryBVxT0nRErIokKuRiTlTGgvxl1UcQiCaZGvG\nKvuqIm29kURaXCNi4tou3a5tum3rrOvET7kALniUeDTuOD6fr6OjAwywpn/svDiYvFCIAgcq\nS+M4buPG+AK/a665RixeTJs27cCBA8uXL9+5c+f27dvHjRv34IMPPvjgg6PIMMOkSTgcDoVC\nNE2r/H7yoovy7w0J4Pf7BXUjsSig/6uv/H5/KBTSnzw5/vrrhdHW8zwh5PUMV8PCsixK/QSZ\nmblEb1W+4TiutbVVMFstkJITpLbIhadGBwa6u7tRGc6ERYv02ekAQh4HcsMITJ2a2UQylUql\nUqng/vvh/vszemDMGESr1SL1TTK2cxSpFokEAoHEsDChTmTWLGb+/OJbB8YIRCKR3t7eBx80\nvf/+6eiBEXWXuFwupOZL6l/f/360MH9SZgOLxeJ0OqPR6OayzW7K/Q3fN+pD9SpeFSJiXQm3\nkLekeQrhpwUibpL/0sBLdTV1aZ6ikJGrkUHlD8+Ensnr6FLCZDL19vYKC2BxzhSSrTe5TN7J\nLMnFtdTjZtvb2zmOG2MvDiYvFOL/Ro8++uijj8arenKcddZZf/nLX7I6HkxyfD5fd3e32IFi\nFPPPWIZ2RhMrhNVISenBbDbbUHnFvn2xJ1x4ISAX0pQNU6PRKJoSyE2JdUPZ4IWDy+US1I1E\n3YesrMyLLTXP80nCU71Db2VW5ZjY7E5mGAz2/sTkj7Kysv7+frebT4ztNJvNyky4O4r98BDi\nOpFnn+2h6bPSPwsmL4TD4VOnTkWj0c7OilF3lwgSsIC4T+qHPwwDmLMw9kKEoqj3G94/6Tm5\nW7f7oPZgNmwvWFFkeOIk/xHukU2wSf7ZRY9Op0OLnZLlD3/2/jnfAxwemqbfnfBuq681sTXj\nas/V6DHiqpwqtmoFFV+VMzZIsbbI4XDI2bLaorYV9Nh8cTBZohAFDkwR4fV629vbhXW/kc4/\nWZZ1OBwosYIEmLh4sSZzi/NIc5ErCuBDIZVKBZBW/ktskUF+Zl5o6gaI3AoldR/w+/MSLa5S\nqZSvvCIXnqpwOCJWa6IcQ1RUZNBoDM0S5TJcaZUqxTwdDCbjUBQVjU6YO5eMi+3UaDQXXJCZ\nam2x2VtinYjZrMvIWTB5oaurKxqNgkwKT4rdJXESWFyfFElWZXjQhc27yne3VW6L26gH/aPw\n6HyYn77thfj7mDjJf0P3RprHL3AMBoNKpRJ8OuPKH/LyK2UUfKD9YJs2vjVjMjP5/t77IaEq\n54/df6w7ayxX5QyLz+cTbif2JVU1VKVta4MpIbDAgRk9PM93dXUJ6kbc/JOrriaTOlBEo9FT\np04J1QSVa9dqdu/O8OK8vPSgSijGHgUKhUKhUOj/+lfJKbFKqy3AKTFaF5LTfRTRaF5GZTab\nw/LhqT2/+IX38ssT5Zjo4KDFYsnUGLRarUKh0MkMo/L224fN08FgssfGjWq52M777svA8bVa\nLU3T0WjU66US60SKZUaBSSQSiQgzh3TcVRQKRTAYBJk+qYyUERUR2Xb0NBgMBEGIu8bEk3wj\nGDNyloKFIIj3Jr7XPNB8ieeSuPIHM2GexE9yEI7CDx+J+5BMZCde3XP1dwa+o+JViVU5v6v8\n3WbYPOwxxzBC1ZJkX9Kr7Kt5HR2myMACB2b0BAIBwQgqsRwgNG3aGVYoCe0nXV1dcr0SGUms\nUKlUmo0b5YoCNC4XmDKQQl5ZWUnKTImrfvKTApwSUxSVRPfRSEV25QCtVqs8dkxub2jiREk5\nhhPFsKUPQRA1NTXKxkbZRxRwJg5mzCMX2zlzZmaOTxBEVVXV3r2OxYsnxNWJKJVKvb6svOBj\nCzCShKT+TgrdJTNnBufPT8m2zGQyDQ4OSupfNE3rdKVV45NtR0+apisqKp5hn0EeH6nET4wx\nttPbt1VsW1uxVrzRGrG2KFqEuwUePhL3IYlwkWPu2O+cxKqcNw1v3hS+6Sv6KwdZBNpNNkAK\nO8j0JdFRPGPFjAD8ccGMHkHdkCsHCPT3DwwMhEIh5M2hFbWfRKNRIQYsURxhzjsvfZ9Yi8US\nlS8KyJT0UFZWFpWfmRfglFiv18s1gwBFad1uyNNSLd3U5Ha7+/r6wuGwEJ468MADnT/5Sf19\n90nKMepMdwAZDAbfwYPtQ54ywjD4VauIhx/O7LkwmBEhGduZWcxm8wcfqNvalJC1OhFMbuA4\nbvfu4Kuvkl98oejs1Ho851mt0WuvHViyxKFQ8OLukj/8oYumz07lmCaT6Z//9M2bVxmnfwHA\njBmVBdiPWezYbLa9ob07VTsT0zcfIdL1+Ch84sofagI117mv44HXcJoizddQKBQGg0FoE4Yz\nq3IUvOIN5enOowLXbrKBwWBwu93C3bi+JIVCkcexpckJOLEclh+AAz3QU5rqVe7BAgdm9MR+\n0MiUA/gnT/Z0d6PbtrVrtZ9+Km4/YYayWiXFEf+UKekLHHq9PjfSA93UFAgEPB4PwzCqo0dr\n/uM/APKcIJOEsrIy38GDIKX71P/0p/ktObFYLBaLhWVZfu9etIW86CI5UwyeJLVuN2Q6iFev\n15999tnhcDgcDis//RRtJGbPzuxZMJjCpLFRusA+U3UipQzP8/v2RV5+GT7/nO7sJEea0po6\nXq/Xbrf/7//W7Nx5uqqiq0uxfn0lxxEqFS/uLrFYRhD29e67NW1tBEjpX9OmZfAKMDG+pfqW\nm3ejSb6Vt06CSfPIeUL65thGXP7Q3d2NPEfFFGO+Rk1NzSOuR5zgTKzKmRGcMcc1p0i1m4xQ\nWVn5PPm8i3Qlvjg3cTdBBh3XcovX693IbdxkOu0KXILqVe7BAgdm9KBsQrn5Jwsg6Q0Z8+ZA\nLcFZTqxARQH9/f2hUEh19ChajWdXrKBWrszI8QW0Wm0sqfHDD2ObhouYzRcURRlPyP+XWQAl\nJxRFwYIFsGABAGijUf7mm0GmDId//HGYPDkbY1AqlUqlEhYuhIULw+FwIBCI9PWp/H7DlVcS\nra0AhStgYTDpkIM6kdLE5/N1dXWtWFG1c+dp94QRpbSmSDAYRM7fdrsyMTBl8+Yyv58Ud5eU\nlZWlfvAvv5Qu08D6V5ZYCkuXEkM9DiVcIuPxeMR3xfka1oh1ObUc8hL/NnIUCsXn1s/fId6J\nq8qZzEx+sv1JAxurnxW0m6kw9Va4tURW/pVK5efWz3dQO+JenOns9N8qfpuvUaUDx3EdHR1e\nr1dhVqwZXIPUqzfK33i68mkoMfUq92CBAxMDeVkRbnfqQa0KhcJoNMqZMsp5QwamTNEDoAQT\nOXFEoVZnyp4TFQXwPM8PxcFSF1+c/mFlSSOTJWcQhw/zPO/1eoPBIHnoUOXVVwMU6IydpmnD\n8eNye4ksq0gcx3V1dQ0MDKA7ExYtiqkbUBBKEAaDKQr8fn9bW5uc6JBiSmuK9Pb2ov/NJQNT\n/H5S3F1iNBoNhhEYyGL9C5N7eJ6PiuzPE/M1qidWgypPgxs5lxOXO8AhVOXUBGquH7geOY9C\nQjZqra/29+bfC88d8yv/V1BXOMHZzDcHiEAFW3EWf9Y8ct5t1G1FWrLU09ODOpKuH7he2PhN\nzzeRwFFElUfFCBY4Sh2O4/r6+jweTzgcJnh+4uLF2pEEtdbW1rJNTXJ7Jb0hA1OnajlOoVDo\n9Xo5ccQ2b15meyUIgiDuvhvuvjtTByx2CIIwGo1GoxG2DQXdFWrJCXnkCMMwDocjEAjQR46g\nMpzoww/Tq1Zl+9RIeke3RxqBjMFgMAghbiydlNZU+OILbt06w5df2rq7FV4vJfhuCIEpgN1V\nMMUGQRBCoIxkvsYmctNwxyggxK03A56Bzs5OYVeidrNHv2eNd00G+1ZybwYxojPGXhxUrFS0\nPSmIaDQqthSBM9WrKrZqBbUiT0MrCbDAUdKwLHvq1CnBYt36+9/HOWUMewSKosijR/tdLp/P\nFwqFtCdOjLvuOgBwLFniWLBAzhsS/UdVU1PD48SKvFMMJSdqtbqurg4A+D170Bb637KufPt8\nPkHdiGuzilitvMVSWqGIGAxmVDAMI/wnm05Ka3JYlu3t7X3oIf3OnafTbpDvRn+/Ytcu2SIN\n3F2CKXw0Gk0gEACZfA0FUaz2kyR5urVGUrtZbV8tPCB9z5FwOLw5snmTLkdmEDzPDwwM/IX4\nyyZzKdpPBAIBccxznHr1p74/1dXX5WloJUGRdK1hskP3UFoEiKZwoUmTAICrrk7RvpEgiPLy\n8vr6+nPOOWdcby/aGJo+Xa79hFQoqJ4eAFAqlVRjY2dHR1Nj4+FDh5q3bIkdcfVq4Hm48sqM\nXitmLEDMnw88DzwP3/lOts8lqBvilB/1iRMAwJx3ntgIHYPBYOQQ4sbEnJnSmu4potHoyZMn\nXS4XaoHZvLl5585jt9ziQnvfftvs9VKzZ/s/+6yJ43j0F1T4d9ll6Z49x3zxBXfvvdGZMzmr\nFZRKmDAB/ud/YChxHjM2qaioEN8V8jUAwMDnJ/ctI2i1WiF+CGk3B48cXN+yHrWrbDVvRbs4\ngmtTtgmeIw9GHhzFuRwOx4kTJwg3saZjzdYTWz84+sF/Ov8T7cqGGQT6o2S325U+ZW7OWGhw\nHCfcFtQrdPdf2n89bslcXyJGClzBUbpwHCf4NkkGtWpHcdChcgC6u1uzaBFItZ+Mv+ceof2E\npulx48YBQCQSoT//PPaIQu2VKEpcrtRNVTBi0LRELgKZF7UEYzAYjBziRVqEOKX12Wd7aPqs\nNE/R29sbDofhzBaY226LtcBwXCwwxWTSFnWYK8uy3d3dDz1kzLZRK6bQMBqNO+p3tAfaE/M1\nbiFvyffoRg9N0xaLxeVyCVvislFBqm+lsqYSRliz0t/f73A44EwziCsHrnyq4inIjhlEe3s7\nSkvM2RkLDXGubWLl0VuGt/I4tlIACxylSygUQtVTckGtoxE4hrBarUm8ORLbTxQKBcyfD+mv\nZGEAwuGwy+ViGIaNRGrvvls9ElMVjABFUXIpP8GpU3VUkbeHYjCYnKDRaAQHAY6DZ56xiVNa\nKypMaR5fvFYhqBteL/XAA7H65+uuG1ixoosgCKvVmua58ojQUWu3V2TbqBVTgHxk+GibYVtc\nvsYFcMEj8Ei+hpQRqqurX1K/1B3pTtRurvZcLdm38iL/4ohOwfM8UjcEsm0G4ff7UUuR5Bmr\nueoV5Ni3n9BqtTRNi/1xxeqVEYxJnotJHyxwlDzyU7h0jkpRFDQ2dvX2ejwelmXVx44hb0h+\n1Sri4YczMGyMDAMDA3a7Hf2Ytq1dq/7kE+yLOTp0Oh2sXy/ZZsWTpN7jgTOLZjEYDCYRkiTL\nysr6+/u9Xmrp0nG7dxuElFaCICrS/jMSiUTEtdAA0N6uvPvuid3dCgCorQ3fcovr0CFdZWWl\nXq/T6dI8W97o6+tDHbXZNmrFFCaXwWW90IvCR2xgmwSTbofb58LcxHyN3PtopgNBEHvK9myD\neO1mMjP5/t77JT1HVKGRZcYwDMOyp91/4kpCnu15tq4hw2YQcepG3Bk3eDbUVddBsb1Tkhzj\nji3jl31JftlL9MZdAkEQVVVVTwaedFPuRPXqZuLmfI99jIMFjtJFqVQSBGF+440sBbVSFFVT\nU1NTUxOJRMihiFZi9uwMDL0YiEQiyHiVJEktw+ivuCIHfSIMwwjqRpwvJltVRaVmqpJ3eJ4P\nh8PhcJimaZXfT150UV5abEwmE/nllyATgZzZlB8MBpMK+/fDyy/Dnj3Q3g4DA1BTA7feCqtX\ng7KwLX+rqqqOH+fmzatsa1OKU1qtVqter66vz/DpnnyyCqkbAGC3K++4owHdLurAlMQqFRAZ\ntf7gBzxAEXffYIZFHD6ShFAo9FrotU3GYnK1TNRubmZuvujkRciJAxL6VtTqkWWmiosIEktC\nflv+2y2wRf7Zo0GspySe8RH+kY38Ro/Hs5HYuMlUTO+UmGg02t3d/SL94pbq069e3CWYzebP\ndJ+9p3gvTr06nz//EaK4K48KHyxwlC4URRmNRrmg1uo770wyheN5PhQK0TRNDw4Oa/GgUCjg\nnnvgnnsyfw15R8bhwul09vb2xvyTOW7CokWQkz6R/v5+dNJEU5XAlClalqUKvrHC4/H09PTE\nbPk4bsKiRfo8tdgQBGE4flx2Ny6HwWBySDQadTqdy5YZ3n33dBFCsfgvEATx3nu1bW0AWUhp\nVSqVJEmiIg7UAiO2qBBTvIEpPM8nerUKRq1f+1rg9tspgJEta2PGHi6Xq7u7mzbRazyZjFbN\nNhLajRoeq33MHrInrvzPgRGnLtH06bleYknI341/T/sK4kluP/G69vWWlpZAIKAwK9YMxt6p\nNyveRPYchfxOCUSj0VOnToXDYZPZtKYj2Yftm4pvusDVzDcHiEAlVzkJJs0j580lJCqPMJkF\nCxwlTXV1NTvCoFaGYbq7u2PpRxw3cfFiXYlZPPh8Pr/fHwqFaJKsvOMORcLlu93unp4e4fG2\ndev0e/fmpk8ElQXK+WISwaBer8/e2dPH7Xbb7Xbhbi5fOkmIw4d5nvd4PIFAgPjXv6pRdAv2\nasVgcgvDMC0tLSzLtrebBf+F55+3btpkgSLxXzh4UHp7+qIDQRAWi0WyBQYAampqysrK0j1H\nASD4mCDERq3r1rUpFJPyODZMIeD3+7u6uuBMV8v0o1XzxW7z7sS+lfP58x+nRvzHTq1WJzGD\nMPCGjBc/GY3Gnp4e8Rc2rggF/VgtXv9Rh8OBfJ2H/bDF1Cv0CuPk0hyCBY6ShqZpoqnJ3tMz\nODiYilOG3+9vbW0V/mbZ1q3T7dlTOhYPHMd1dnYODg6iu7a1axUffshrNEQwCBC7fJ7ne4ey\nciHnfSIcxyUxVdGI/rMpQJBDvnA37qXja2qIfLTYEARhNpvNZjPs2BHbhFN+MJgcwvN8e3s7\nqnk+MyXEiQSOwvFfSNJBs317Fs9rs9mamiJ33VUV1wKj0+n0essY0DcIglCr1cFgEKSMWtVq\nSrxijClNnE6n+K7Y1dIWtT1MPVxcPUzivhUrbz2bOPt2uH10K/8EQdhstrXBtZJmEN8mvn0r\n3JpZIwyFQlFRUfEM+4zkGa/2XC1+cDH6jwodc4hi/7CNSbDAUepQFFVbW1tbWxuNRqmhoFZJ\npwye5wV/ByiY+Wcu6e7uFtSNuMuH2lqw2QCAYRhBJpcM3020eAsGg8FgMBKJqPx+01VXEa2t\nAKMsE1AoFPq//lXOF1PZ1weGwk2M9/l8glVe4ksXmjYtz/V8QxHIGIwAy7J79oReeYX4xz+U\nnZ2kx0MUiytEEeH1etFaGcj4L8wZccl2VuA4btWq6Natp994cQfN/v3w0kvc3r1ERweRcfcQ\nkiQ/+KAuSy0wBUJ5eXlnZ6dklUpZWVlRx99iRsEJOLGMX3aQOChMyy8yXXR/8H5L1AJS0apV\nE6uKq4fpjL6VtD/dFotln27f+8r340pCprPT6731j5kfE7ZkygjDZrPtC0uccVp02v299wt3\n496plwZeqqvJsONpxolGo0lMW59qf6p2Ui2eXucd/A5gYtA0nTyoNRAICL8yJafumhyNND+E\nw2G3241uJ15+ePp09DNVUDfk+kTEAgfLsp2dnV6vFyDmNxFTN2CU5TAmk4mSMVUpfF9M4dMl\n+dIxeRc4MJgzcTqdDofj178eJ7Y8KBZXiCICrdvHIfgvzJ4dnj8//2JSKBRqa2s7ebJOMsH0\nl790PPig5v33T+vLGf+cZK8FpkAwm81NTZG5c01SVSoFU8ODyT4cxzkcjheJF/9q+6uwsRM6\nO82dbtK9rn2dZLTqK+wr+RtyQXCV8ioX7xJKQs7iz7rJf9M3Or7xnum9Nd6sWJYIZwwQAStv\nnQST7iDuzgDGLwAAIABJREFUuHrg6gF2AD0g8Z16CB4ygKHAc1VI8nSrieSH7VLi0vyNDhMD\nCxyYVEk+/wyOdYHD7/ejG5KXH5g6Ff3Ejpk5yfSJhKdPFx+zvb1dOGxG/CbKysoiTU2yuwu7\nhyj2f4bMSxeZMSOfg8NgzsTpdCKrHbtdKTmnxQJHpuATeuvE/gsbNnhouiYvAxPgeb6trS0c\nDksmmBoMjMPh6OyclNXPSVZbYAqErVsrx3aVCmZYOI5raWkJBoMGsyHR3LFd1Q5SrpZbzVuV\nbP5l0PyyFJYuJU6XhPj9/pb2FkjDsmTYkNe4MyIG6IEBiAkcie/Uu5p3WThdHFGYuSokSapU\nKpRaLflho4hCt/MvBbDAgUmVWAloalP3sUese0Lm8kPTpqEbarWaoijjli2SfSJqnU4I3/V6\nvYK6EdfwErFaObN5FNWUBEFQjY0dXV2oP1AwVQkvW6b89a/Tuv7so9FoAMDy5puSL51Kq00z\ntxiDyRQsyzocDnRbck5bOK4QYwClqIsj0X+hvNyax7EhPB4PWgCQ7KD55jddgD8nmWDMV6lg\nhsXhcKCSLslp+YzA6YWQOFdLcZIIBgD6+/vFd8UuEtaIdRm5DOQn6TzPu1yuV4hXNpWNOORV\nr9fHGQaL3yk1r17ZubLwE3DKy8uRoy1CfAlGkA6xwuQY/IXHpApK3pabfyo1mrE9/0T/O8pd\nPqlQoMtHZk6kTJ9I2Y9+JPSJxDpTZPp9wj6fSjWahlGKosaPH19dXc0wDPXxx2ij8utfH+Vl\n5xCtVqvVauVyi4233FLgLTaYERGNRsPhsFKpTCVqutDw+/2CX0whu0KMDZAhP8dxif4LBEEY\njfn/NYkSAcSIE0xvuskNMp+TG29kIck0AnMmpVClgknOwMCA+G6cueNEduJzlc8lulr+gP9B\nfoZbwIhb/xJdJKzjrZDoGAcAACzLtrS0MAyjNqvX+Efc20LTtNVqfTrytKT/6HcHvnut51r0\nyEJOwCkrK3uOeK4r3JV4CTcTN+d7dBgALHBgUketVieZf4qn7mMSpDrLXX7l7bcLl19WVhY9\ndkz2QEN9IsitQ67fhxcFeo0Cmqb1ej3cey/ce286x8kx48eP54u2xQaTIoODgz09PbGWN45r\nuPdebbFFTUcikcSNwpx21ixm/nzsGJMxaJquqqr67DPn4sUT4vwXLBaLXq+qr8/zCMWGc5CQ\nYEpRZ7TYiLWPH/2IAyjo6G4MpnCIRqPirNPEafkjZz+yg9oR52o5g5uxhl6T04EWA0INhaSL\nxAZ+g9wT7XY7wzCQRm9LZWWlpP/oZGYy8h+N062Wk8sLMGD1U8uniTm+F8AFj8Aj+RoSRgwW\nODAjYNy4cXxjo+zuIpmcSOByDbuGTFGU1WpVHzkiexDR5dNNTeFw2OfzMQyjOnq0/KqrEo9M\nUVSSSFcdVYrLegqFgmtqcrpcfr8/HA5rT5yo/e53AYpmYR8zLP39/eIwYNu6ddpPPy26qGkq\n4espntO++KKLpsfnZWBjlbKysl27dG1tSihI/wUhozSxg4amz1A34rQPhSLf2kwB88UX3Esv\ncfv2kZ2dZMZzZzDFiDgrR3JafjlxuROcyEezkqucBJPmkfPmkqOJVh3zqFQqpBZJW5ZEpL9m\n4XBYCBNEiMWIKrZqBZVSyOtVyqvc4G7mm5H/aG2w9jr3dd8Z+I6KVyXqVrZ6GxSeyZ84x9cG\ntkkw6Xa4fS7gD1uhgAUOzAhQKpVsU1NPX5/P5wuFQrrm5gnf/z5AUc4/g8EgugqKICrvuINO\nYQ25srKyb+/ekw4HUr4Fhwtu5UpyRfzfdKVSWVZWBgCwY0ds04UXih+g1+th/Xq5SFfD4CBU\nVGTgOosNkiQrKioq0LXv3h3beuZLhylSIpEIMuZEFG/UtE53ung3cU5rK5KrKC6OHJFu2SsE\n/wWj0eh0OiUTTIVu88TPiUIRa/zExMGybHd390MPGXE+EUYMRVEKhQIV0ElOy18nXv8V/AoA\nmqJNy2H5l+SXP4OfLYAFhRnGkV9MJpNgAwcJliVKGR0xLtMqToz4U9+f6upTCnmNheASsXM3\ntjairk9J3epl7uVRXGC2OSPHF1N4YIEDMzIoiqqqqord+fzz2I2imn/yPN/d3e1yudBd29q1\n9K5dKa4hV1ZWWiyWQCAQCoV0H32ENpIXXZTsfHfdBXfdlbjZaDSS//wnFGeka46QeekwRcrg\n4KBQFptoPROZMaNYlmYVCoXFYnG73YlzWpqmLRZLvgc4Bilk/wWtVjswUPHjH5fFddAAQEOD\nRq/vl9Q+jEZTYikQBsVkMAxjt1fgfCJMHBaLRTB4hjOn5QbeAASEw+Gurq6XVC/9rfpvwsMK\nM4wjv1gslj/yf+yJ9iS6SMwBWRMpsTloohjxuOXxzbB5FINRKpWo7UW6nCRaLD8NhmHY3BlM\nBsECByYN8jX/TKGjJAkOh0NQN+LWkKG2FoZbfaVpOmZrt2QJLFkyiuEjCILQp2DVgcGMGVCs\nGshYz4SmTy+iXzHV1dXNzTBvXqV4TtvYaKyqqtJoqLy7QmByzPbtVXIJphdfrJg71xinfSiV\nypkzq2UPN4bYvx9efhn27IH2dkil08TpdKKpDs6dwSRSWVn5PPl8L9ubOC2/hbwlGo22tLRE\nIhETbUoMkS3AMI48QhDE3vK9iS4S5/PnP07JiohCOx5IiRFvGd4a3WBMJhP61sfGdmY5ifik\nxUswGNwY3rjJNOLcGczowAIHpjhAjX8MwxA8b503TzFaV0KO45xOJ7qduIYcmjZtpMklgUCA\nYZhoNKoOBAxXXkm0tgKkKrsQhw/zPO/z+QKBAHz1le2aa1J/LgZTdCSPmo42NEBDQ7HEqZAk\n+d57tXJz2ry7QmByTJIE07ffrijNzwnHcS6Xa/ly7Y4dWmHjsJ0mHo+H53mCICRzZ266iYMC\n9BvE5AqCIPZV7JMzd+zt7UUNLKP2vywpxC4SVt46CSbdQdwxl0jmIqHVaoUuIURGElLLy8uf\nhWcdnGNE5SRFhMvl6u7uVpgUawax7pYjsMCByRXJyy6S7u3v7+/p6UGlcba1axUffjhqV0K/\n34+OI7mGHJg6NXWBIxKJdHR0xAICOW7CokUxdWMkoyIIwmAwGAwG6OyMbSqqfp8ChGVZhmEI\nglD5/dTFFxfLhLkU0Gg0IJ+1rHn6aSiqOJUkc1pMqZGkg+b//k96+9j+nITD4dbW1nA43Np6\nltBp8qc/2V57zQxJO03C4bDYSxLOzJ358Y8JKEC/QUwOSWLu2D3YLX6k2P/SGrEupwoxjCOP\nnOEiQSR96BAEQdTU1PzW+1vJkNdRJ6SSJLmvYt87xDuJCThJykmKhWAw2N3dzfM81t1yCRY4\nMFmEZVmPx8MwDBuJ2H7yE+WZs5dgMOhyuRiG4aLR8QsWqGXmNoODg0LsQvquhLE8P/n4khS7\n5zmOa21tFUrubevW6ffuTSsMAvtNpA1qvvX5fAAxyUlfVBPmMY/RaKRpWi5rWX3kCK9QEGhp\nqBjer0J2hcAUDqX5Oeno6EBR0OvXtwq1GD/6UR8SOJJ0mgi2rIi43Bml8qzsjhtT8MiZO7Is\nKw5sTgzjqGmogSJqgyxUDAbDF9ovEuN400xIvZy43AGO+HKSMZGA09/fL/6bFqe7LSOXAfZi\nygJYzMRkC7/ff+LEia6uLpfLpf71r5UffcQJjvGzZvX39586dcrtdgeDQfOaNepPPhHvFR9H\nMJQSd5SoT5wAgODUqSMdFU3TIL+GTCoUYLenchyXyyWoG4LsEpo0CQCiNhuX20ZhjuM4jgOX\nCxoagCCAIODXv87lAAqBSCRy6tSpmLohkpxiu4thwjzmIUmyrq5OIx81HR43DgDYqqphrXAw\nGEzB4vP5hLQFyU6TOfJV56jOCwA4Dp5+2vbAA3WBADlnjuu551pNJkIu3AGDIcnTMxrB/xLd\nRWEc4gcAAM/zXwW+ujF0YwPboOf1ClCMh/HzYX4f9OV03EXIFdQVF8KFFrAoQTmOH3clXLke\n1u+DfWYwj/qYS2HpP+AfLnCFINRBdHxIfHgn3DkG1A0AiBV6AwDATZNuOv+8868757pP9Z/O\nCMzYfHKzlbHmcWxjGCxwYLJCJBJpa2tDIdtx83+uujpgMKB6rcS9cXMb1G4AMh0lvsmTxbJo\nKuh0OoqihDXkSTffPOnmm83btqG9VT/5CWzcmMpxhIm0pOwi/nOWPXiedzgcx48fb2xsbDx8\nOHDDDTmr8GdZNhAIhMPhglBVXC5oaFAolZOnTLE++ywM96HCZIDRvu9arZY8cqTLbm9qbDx8\n6FDzli1oO2s2N+3erezqAoDAlCmCeojBYIqOuCxJhNBpMnt2eP582eeWl5cDgNdLLVlS/+yz\nlTTNr1hhX7Gii6b5srKyuO4VDEaAIAhBHUP+lwePHFzfsl7FqwBgq3krWt9CBIPB5ubmjZ6N\nb6rebKFa/IQ/ClFk+ngP3JOfCyge4sSIXbBrzIgR2UCYp0jqbigfF5NxsMCByQpOpxN9aRPn\n//7Jk/v6YgJ54t7AlCnib3tcR0nnb34TamgQVoCDU6eO9E8DQRA2m0195IjsI1KTBpB2Iym7\nBM87D+3NKihLz+FwoDJg27p12k8/zUHBgt/vb25ubmpqOnXq1PGjR/3f+15efBMYhrHb7SdP\nnjx+9Gjw+98XxhCcOjUjlT6YODiOczgcJ0+ebGxsPH70KHPjjaN+3xUKRU1NDVqJ1X75JdrY\ne9994m+Q2FAdg8EUF4n/L+/caZw3r8HrpWbP9r/0Uh8t3x5tNBp9vqq5c8/avdsgzp1pbq4I\nBvFSJyYZFRUV4ruC/yUAiP0vI5EIajE2saY1HWu2ntj6wdEP7nXci/Zi08dRczh0+AeRH5zF\nnWUAA66IERDqziR1N1yVliWwBwcmK/j9fpCf/6PlHbm9FMNotTHTdZqmCYIwv/GGZEcJQdNU\nTw/U1o5obGVlZc59+0729iJVVX3s2KQ5cwCAXbGCWrkyxYNQFJXEyENDZb2jrq+vT6gTibMm\n4aqryewULAwODnZ0dAhqtG3dOt2ePWk5j4wKt9vd1dUlmM5qdu8WxhCqr69fvBgSvGO1HBdX\nnopJHZS9J1RVWH73O9RTls77zvM88Lxh714AaH/yycFvfUv8DVKOsDILg8EUDuJYR46DZ56x\nPfdcJc/DnDmuhx7qtljKkz+9ZHNnMGliMpmeI57rCHYk9790OBxo8QybPmaKUChkt9tf0bzy\nevXrwkYcg4owmUxoToSIC8FVqUYa3ohJCSxwYLICy7JJ5v8cxyXZqxUt/pAkqdVq5VwJ6xYs\n4FauJF96aaRJGRUVFWaz2e/3h0Ih7a5daCN18cWpX6BOp1O+8oqk7AIUpXW7wWBI/WijwO12\noxuJYbeBKVP0WTgjx3GCrACZMHwdHQzDCMOIG0PEaq3+7W8lP1S4sDkd7HZ7ouNMmu+7SqXS\nbNwo+Q3iSVLd3w/m0XfzYjCYPGI0GlEXqtdLLV06bvdug0LBP/hg15w5bgAwmUzJn47ziTCj\n5mPjx9uM0iGywl2hxRgRZ/r4EPEQnhuNCLQEEo1GTUrTmg4cgxqPxWL5A/eHXrY3UXf7Af+D\nfI9uzIK/xJisQNO0/q9/lZu9aN1uxa5dcntVTifoT8/Qq6qqSHlXwtDmzZpRlcrTNB37mfWz\nn8HPfsaybCgU4nw+dSBA/9u/DauYlJeX+w4eBCnZpf6nP4U1a+CXv0x9MCOFZdkkPTKBqVPV\n0SidpAh4VPj9fqH1JlFVCU2blpv+S8GPOnEMbFmZ5IdKoVYTXV0jrfQpcUKhEMMwJEnSg4PV\nl15ab7cDQP+PfmR5803xa86cd97oMhvNZjMrI1xOWLgw298gDAaTPWiattlsX3zhWrx4Qlub\nUtxpYjQaDQZNfX2yp5dm7gwmIyQJkRUeI24iTgxbqa6vxnOjxkjjclj+Jfmlg3SEiFAVVH0b\nvv0oPFoJEg76DocDvaS4IkYSgiD2lu+VDMFdQ6/J16jGPCX/JcZkB4PBoJSfvfhXrox8+aXc\n3ri5jUaj8R082NLZif6ACh0ljiVLiGCw8oUXhi+Vd7ngwgvlNItoNNrd3e3xeABGkC1KUZTh\n+HHZ689Ns4Z8FczQfp5lWXpwMMnlpw4y+wD53qLcCByoMUdyDIjED1XNXXfhCXPqBINBu90e\nc8HguAmLFmmGooXQCxv3vo9O4DAYDJGjR2V349QbDKaYqaio2LlT19amBNxpgsk0h5hDK6mV\n/6T+6SAdDDDi6bdciKwYiqLQ70nB9DFKRGHI9PE16rVj3LFl/LKDxMHE4+fi8vIKqtXdQG1I\nvdlkcHDwjCPgipgExnAIbsFS8h86THaoqKiIyJddaC+7LPKXv8g+OWFuo9frzz33XJ/P19/f\nrxg6LBEKiUvl2aoqSlQqHwqF3G43wzBcNFp7990qGc2CZVmxuYCQLZqKuQB55AjHcYODg8Fg\nkDx0yHbNNQBpKQipQ1EUTdOGzZvlwm6Dzc29NB0KhXiWnbh4sS4TPqCxLg8ZVSUyY0Y6V5Q6\nyLtBcgzkmf/LngGeMKcGwzAtLS2CR2DcN0LhcMS95sx55436XIqjR/v7+51OZyQSEYTL6MMP\n06tWpX0dGAwmzzQ1SYufM2fC/v3w8suwZw+0t8PAANTUwK23wurVgO32MMkJBAJ2u/1V/avp\neD3odDq0poVMH3ng9+v2L5qwKESEtpq39nX1/Zn685aqLaM+flHT0dHh9XpN5lSbTXieT14R\nUzOhBs81z9DdcMN0TsCue5isQJIkcfjwqZMnDx86JA6DDP7qV8DzxDe/STU22js7jxw+LN4b\nWb4ceB6uvDLxgARBGAyGcDjsvvHGw4cOnXjrrfJXX5WLX3G73c3NzU6n0+fzGR59VPXxx3Lx\nIk6nM9FcIPVsUZIkzWZzdXW1rbMztunCC1N6gdJOV7VYLHJht+Pvucf/pz8xDMPzvOADGnta\nGvN8FMBmefNNuTYQGFrnzypKpVJuDF0PP+w+dKi9re3E8ePtf/977AmrV8t9qDCJdHd3C9+j\nuG8EACS+5rRKlc77Xl5efu65506ZMmVCf3/sgP9W6uWsGMzYYPt24Pn4f9Eoe845vcuW+det\ngwMHoK8PIhFoa4PHHoNly/I9YkxhEwwGM5J+UllZKbblEoet6Dm92+02Ro2lma7i8/m8Xi8A\nXD9w/bWea+tD9dao9arBq9Dei7iLEp9CEITg4C4Zg1ri/u6hUGi/Z//3me83sA04XCaXlLyq\nhskaSqWyoaEhGAwGg0HFBx+gjZrLLkM3KIqqra2tqqoKhULU7t1oo+KSS5IckOd51CUh1yKh\nCIfVanUgEJAzoUyMF4l1pshYdWpZlkoxD+Wuu+Cuu5I/JBgMer1ehmEogrDOm6dIr6qisrIy\n2tQke66pUyHh8qM2G52GD6hGo0li+GqZOzc3bSAmk4lI6t1gQWMY+lClKjlhAFiWFby+E78R\nIPWaW++4I/33naIoWLAAFixI5yAYDKbACYfDLS0tkUikvd3005/2fetbHouFfe65yr/+tQwA\n3nsPHn8830PEFDA9PT1If0/T60GtVu88a2eLt+Xrg1+PM3282nN1+scvXpI3m/zc/3MQeX9H\no9FDzKHVitVfnv1lH9UXIkIV0YorB6+8v/f+k6qTQkWMkijRuiye57u6utxu95byLW+Z3hK2\nl1RBUB7BAgcmu2g0Go1GA/fdJ9l0S1GUVquFRYtg0aJhD0UQBEEQPMfJGU+YSRIAnE6nnAml\nf/JkPc+LlftIJALyigkdDqOyhfTp6upyuVzotm3tWsWHH6aZskmSpOLoUUdf38DAQDgcFir8\nB3/5y/Z580Dq8oNTp9LBYDpXNG7cOF6+8yg3bSBmszkir+ycHkMKkhMmDvR1gKQWJxLg9h8M\nBpMCHR0d6I/MCy+0GAws2vjjH/cjgaNy7PsbYEZPNBoVZ23CmdNvW9S2gl6R+tHeV7+/Tb3t\n95W/F2+cEppyf+/9kscfS14SSQxEI5EIP/QjObHZpKqsSjhIb2+v0+ncVLbpzerTs/ReRe8b\nljcGqIG1HWtRDKoRjDm+usIBqRsAgAqOcLhMjhkTX1ZMNon9sUvq05kzNBqN6tVXJdsTCJpW\nOBwwblwSE8rgeecpQiG1+rSpT3LFxJShyjqHwyGoGxlMVyUIwmq1Wq1WjuOIzz9HG/1TpkCS\ny2eYdAQOpVLJNjX1Op0oYVd/8uT4668HyOlHgiAIuqmpu7fX5XLxPC8oO5HlyxWrV+dmDGOV\nWCmpjMVJ2zPPeC+7DERGv/yqVcTDD+dvvBgMpmhA5ZzotqBuAMBnn8VC0+bMycOoMMWC2OgB\npKbf488dn7q7gWTYytdavkaztNzxbeNtxT5nGtZAlKIopG5I2q8+xz6HntLT0+N0OkE0df/A\n8MFew97PdZ8DQLO6eXntclQRczNxc+4vsxBAPoDodskWBOWXIv+yYrJGJBJxOBx+vz8cDtMk\nWb9w4ejSWDNLeXk5L9OeULdgASqV5zguSbyIjmXFz9JoNMpXXpFTTJR9fTBuXJpj5jgO/U8A\nWUtXJUkS5s+H+fMBIHjqFPj9cpevJNJ1N6IoyiYoMvv2xW7ktg2EJMnq6mqbzRYKhcjU+psw\nqaBUKmmaNm3YYPjwQwCo+/nPXbfeSnq9aK9apxtvMoUrKxVD7zsxe3bexorBYIoKQd0Qc/So\n+qmnrABw4YWh+fNVOR8UpmgQWzkkTr9/X/X7y4nLUz+aZNjKUTgaBdl0lVfJVzNwGXmlvb3d\n5/MlMRDV6XQDAwMgY7+qY3QAEA6H+4c8s4Sp+4GqA0jdAIAOZUeHsgMALoALHoFHcn6VBYHP\n54vbIi4IqmKrVlAjKDjCjIKStn7ByMEwTHNzs9vtRp4X5b/7nWb37owYVaaJyWTSHzsmu3vW\nLABQKBRyJpQ8SaqGtAZERUWFnFVn3YIFxGuvpT/mYDCIukYlqyoCQ5GumUKlUiW5fPXQf0uZ\n4a67YsZx3/lOJg+bGiRJajQa1aJFeRzDmCEYDHZ2djY3N3PRaOULLwjby157TfhG2ObNIzdt\nUqvV1D334Nccg8GMCNQ6KmbnTuO8eQ1eLzV7tn/9+h4ar7hh5EH6O/oUoen3wSMH17esV/Eq\nAHjb9Hb6pxDqWxOPv9W8VaXKpwAXiUT2e/bfELyhgW3Q8/pReFV6vV40605iIGoymZSiKCOx\n/aqBN6DXx+fzxX2XOYJrCDXoOT16Sg3UXAlXrof1+2CfWWzaUUokFhydf975151z3af6T2cE\nZrzT804d1OVrbCUCFjjGAgzDuN3uvr4+b1sbP3FiOtkcAMDzfEdHBztU6RCXpMDX1AybLZJV\n6KYmn9fb3tZ2/NixtreGbHtESRlGo1FOs5iwcCG9ebP4aAaDYVjFJE1ir+RQUUnnb34TamjQ\nDDlZBKZMSf8UYpKkq0xYuFD9xhuZPR1mDOB2u0+dOjUwMMAwTOXatbTDIftQbLeBwWBGhXji\nxHHw9NO2Bx6oCwTIOXNczz3XWl6uEPbu3w//+Z/czJmc1QpKJUyYAP/zPxAO52PQmEIiSfpJ\nRrweysvLxXfjpvd5TAPp6+s7fvz4lsCWv2v+3kK1+Al/FKKoteQeuCfFgyQaiLYp2wSHkQe8\nDwAASZLvN7z/QtULjZrGfrr/uPp4YrOJ4NWFQFP3P1f82Uf6ZgRmHLAfsIN9F+y6E+5UQ/oF\nyoXCUfbonOics7izUlSXxAEFieEyj1uwnXLWwYJ5ccOybGdnJ0p1Ao6bsGgR0doa2zfaqQiy\nV0C3E1sqwtOn572KVK/X6/V6AIBPP41tErVIVFRUREdihEk3Nfn9frfbzTCMsqmp7nvfA8ik\nqYRCoQD5dFVKqQS7HWprM3IuANBqtcosSzaYsQTDMHKpQ1GbrXf9+trvfhcgn847GAxmDKDX\n62majkajXi+1dOm43bsNCgX/4INdc+a4AcBkMgEAz/NOp/PBB9Xvv28QnohCZDkOZ6yUOuXl\n5S/QL3Qynd/wfSMu/SQjXg96vX5H/Y72QHuWjj86+vv7e3t7IQWvyhNwYjksPwAHeqCHAUbs\nHgpn1hRIGIiWxwxEt9Pbt1VsW1uxVjyGC+CCR4lH0W3JqbvQy/M4PL4RNmbhZcgbPM/39fW9\nxL30t6q/CRuHTUKJTVIAQKrf53Xt65LPwmQQLHAUMTzPt7a2Cn2ttnXr9Hv3ppnNAQAMerq8\nUWXeBY7TSCVlUBTFHTnS1tWFdB/BEDG8bJlSpqpFp9PpdDoAgE8+iW3KnKmEWq2maVouXbX6\nzjuhrAyQBWmG5pB0U9Pg4KDT6USSDbp8dsUKauXK9A+OGWMgr1aQid2psttjj8NpuxgMJg0I\ngqitrf30055Fi+rb2pQ0zf/85z3nnBP617+0RqPRYNDpdNDZ2enxeDo7J+EQWYwkn5g+2Wba\n9pTtKfHGDHo9fGT4aJtB4vjC9D7H8DzvGKqpTO5V6ff7N0Y3bjJtEh4TNwkXKlAkHUae555H\neyXtV+fCXKEcI/ZrGQBKY+re09PT399vNBtHlISiVquNRqO4akYoCCrxcJmcgQWOIsbj8Qjq\nRuLSK1FRQSV/fnJkfDrD06enO+7so1Ao6uvrw+EwwzD0Rx+hjcqvf334Z2YhW5QgiKqqKvWR\nI7KPGApYyWB5hdFoNBqNPM/ze/eiLdTFF2fq4JixBPobIqdm8j/8ofGeVCtgMRhMsbN/P2zY\nwO/ZAx0dxMAA1NTArbfC6tUg6i8ZPQaD4cMP1W1tCgCIRonHH68Wdq1dC3feOejxeACHyGLk\nGXb6XeDHHynBYJA90xpfMhy3q6vL5XIpzIo1g7KTcJ1Oh75f0gaioZhsIWm/Kkaj0ej1erGJ\n5hieujMMgxxVkbrUpmxbU73mK+1XaG8rtM6H+UKNTBzjxo17bOCx7kh3YkHQHMCRUVkHCxxF\njHc4xSIOAAAgAElEQVQo3UBy6ZXz+VDN50hBRkpyLRUKtTqzLRXZQ6lUKpVKWLIElizJ70jM\nZrP7iy9OdXcjt1GhqCQwa5b2wIH0i27kIAiCuPtuuPvuzB4WM5ZInjqk5Lj8Dg+DweQMl8v1\n0EPK9947XVyd8faQQ4cUkttnzgQU3wAyIbI33BABkH4upnQYdvpd4McfKcnDcf/Q9Ye6SXVO\np9PlcsFwJR5ms7mvr09w0BCrEgbeoFaPQMEZP378o+5He9neMT91jzMu+cT4yQ7TDuFuAAJJ\nGlVIktxTtmcbZLHgCJMELHAUMegPn9zSK3Xmn8XU0el0SVoqLHPnojTWdEdfOrhccOGFlpYW\nC4D3v/6r7Y47BIdR7YEDQtENX1ND5NW9FVOaKJVK7WuvJYvdMZeoCzoGU1LY7Xa3293Rkd32\nkO3bZXc1N8f7iAohsl/7WuDWW8NQqokMmJKFFmULJbaWPGl98hL+EueZ4YBycaTIQPSk5+Ql\nnkvSdBihKGpfxb5SmLqLHVXF6tKU4JRL/Jesr1gPSRtVCq0gqKTAAkcRQ5JkkqVXEzXKDhWS\nJMeNG6cYiU8nRgzP8x6Px+/3h8NhBUXZfvITRUsL2uU95xwAcN94Y+D888+aOzdSWSkuutHm\nc9SYEsVsNvMyauaEhQuxmonBlAJer9ftdkNe20PEARkAsHOn8aGHxgUC5OzZ/nXr2pTKcbkY\nBAZTSGg0GoqiUJdKYmvJW4a3wuFwEvfQZ+zP1J19Oo50h2JHcgPR1CmRqbuccUmTpqkmUoN2\nCTUyiRRaQVBJgWNiixidTifXSMKTpG6o2nMU6PV67quvWk6dOnL48OFDh5q3bIntEKWxShKN\nRn0+3+DgYKi7Gxoa0gysLUai0eipU6c6Ozvdbrff71etXq348EN+KFx98OyzQaboxjd5cly0\nOAaTA0wmk+7oUdndWM3EYEoApG6ATHvI978/yoLQESEUySeGyOr13IhK6DGYsQFBEFar9Ywt\novBaE2HiRG2kiXGkT1Q8IX7uZXDZhXChBSxKUI6H8VfCleth/T7YZx55bdRSWPoP+IcLXCEI\ntUP72MuFRWi1sZVHpC4dPHJwfct6Fa8CgF3GXYBqZGBFPoeIkQFXcBQxZWVl3oMHITtLrxqN\nZuLEiTzPh8Nh5eefx7bKJymwLNvV1YUcjFBgrWqobKGk5kgdHR1yzq98TU20vDxJ0Q3HcdRo\n624wmFFDNzX1Ohz9/f0cxwkGMZHlyxWrV+d7aBgMJhcI2fAC4vaQuXOjkH3vwLKyMrfbLRki\nq9frlRmxOcVgio3y8vIXlS92BDsu9V6aaHihUJw2ppF0DxUfKpcFBckza4sFo9GoVCrD4Vj3\nHAHEI9WPhIgQAPDAzwjM+H+d/6/unLqkx8DkByxwFDEkSRpPyLZ+ZURWIAhCpVLB/Pkwf36S\nh3Ec19LSIuTLZiqwtujw+/1+vx/dTnR+jZ5/PkEQ5jfekCy6IWia6ukpCvdWzBiDIAibzWa1\nWkOhELF7N9qouOSS/I4KUzrs3w8vvwx79kB7O2Q8uQMzCuLaQ2g6F+0hGo2GYcbNnauNC5Gl\naXrWrJocDACDKUxQeO066zrxRmR4QdO0VqsNBALC9jj3UCASDpd9QqHQa6HXNhllM2uLBYIg\nttdtP+k5iexUexQ9bao2Ye+/tP/6XeXvLuUvjWuvwxQCuEWluCEOH+ZY1u1yddntPTuGrH2H\nayRJFZcrxTaT/v5+Qd0QyhZCkyYBQNRmg5LxzhT+j5FzftXr9YJ766Sbb550883mbdvQU+oW\nLICNG/M0cEze4DjO7/e7XK7BwcFwT08eG7sIglCr1apFi4DngefhO9/J8QAwpYnX6125MrRu\nHRw4AH19EInEkjuWLTvjYZ99Fl20KPy1r7GVlbxSCRMmwP/8D4TjXSkxo6RA2kPeeMPc1qaE\noRDZ225ruO22hh/+sG7r1vjVuM8+iy5eHJk5k7NaAX8eMGOb5K0l1dXVW8q3PFf5XKOmsZ/u\nP64+vrx2+ejcQzOCx+Npbm6mB+k1HWu2ntj6wdEP7nXci3YdgSO3wq1nw9kGMChAMR7Gz4f5\nfdCX+0Gmznuq956yPfXDs354xeQrbj3r1jARBoC6cJ2SVwLAVvNWrG4UJriCo+ghSdJisVgs\nFhAEDvlGkuSwLOt0On0+Xzgcpkly/IIF6tTaTIQgJcnAWmUohKJnxzyxuHKZJpTQtGlVVVWA\n3VsxQwwMDHR3d8c+Nhw3YdEiZUk2dmFKE5Zl29vb/X7/qVOnkzvWr6/+y1+MIEruiEajXV1d\ny5ebd+483SWR8fjSEsdisXg8Hsn2EJ1Ol7P2kIMHpbfPnHn6diAQ6OrqWr7cij8PmBIheWuJ\nRqP5XPn5DmpHXKbJ+fz5o3APTZNQKNTZ2cnzvGRmrSFs2KQssrKOy4nLO0IdbXRbkAyWR8vr\nwnXXD1x/jeeaa869JkyF81UjgxkWLHCMIe66C+66K9UHu1xw4YWAZlOrV8Py5ZFIpKWlReg0\nq3jiCfUnn6TYZoKeJVe2QEQiJSJwoGZIOedXWqVSOZ3Br7460dmJGp4Fv4Poww/Tq1blceSY\n3DMwMNDZ2SncLdnGLkzJgtQNODO549Zbe5HAgZI7OI5rbW1lGMZut2Y1vrTE0ev1Xq9t7lxT\nYnvIzJnVORtGkhBZhN/vb21t5XneblfizwMGg7iCusIJzma+OUAEKrnKSTBpHjlvLpGHTJP+\n/n6xX744s9YasV7df/WPoj+awkzRsJrXy15HqkeSmNVCYCks/Rn3s8ccj7lIF2pU6af7/7fm\nfwUblHwPECMNFjhKC6/XGwgEIqGQdd68uLXijo4OQd1IdMckkraZkCTJRqNy3pl6slQ6oQwG\nQ09Pj04mdNMydy6sWaP55S/PPvtshmEYhlF9/DHaRf+bbMoUZkzC83xPT49wN+4bx1VXkyXT\n2IUpTcSORZLJHXPmAIj6H/MYX1oibN1a2dYGMNQeImxfuxbuuy9vo4qjq6sLTZ/w5wEA9uwJ\nbdjAf/45bbdTHg+BzWtKlliJByolyOsvbsFlHxIya59qf6osWibsFco6ksSsFghJamQeIx/L\n16gwySmVmSeGZdmWlpa2tra+vj7V6tXKjz7ihK7aWbOCwaDgHyFuM1GfOAEA4enTkx9cq9XK\nlS0QNK3u78/WVRUYSqWyoqJCfeSI7COGluXVarXZbNbcey/2OyhNAoGAkF2f+I1jzjsvr6PD\nYLKOz+dL3Cgkd8yeHUbG1kL/o1gEeeMNC7rx2WfYjyNjpNIeAgD798N998GsWXzuzS8YhhHS\nXpKIYqVANBptaWlZuTL03HPqr76inU5CbF5zjDt2M3vzJH5SETkdYMYGQmxtYmbtWtva2GMI\nrk3Zhso6iiVm9QrqigvhQjNvVoKylqv9d+7f18P6z4jPRpGwi8kNuIKjVBCKgePWitmqKspm\nY9xu9DC5NpPkHSYVFRUhmbKFugUL0gysLS5sNptjz55TTif6Ky80ofCrVhEPP5zv0WEKhUgk\ngm6QPl/dkiVkMKhqa5ty2WVoY/C887T5GxsGkwNi1jMixMkdGzZ4aLoGRN8UgaNH1S+/XI5u\n+3wEYP+FDDFsewgA+P3+Zcvg3Xd1MNR3nssXPyylowii2MyZwfnzNVkfRAHA8/xQ31Z1Yp/O\n1q1h8ucvbqneIjy+KJwOMGMDpVKJVEjJzNrV9tVxZR3P2J+pO7sIYlYLp0YGkyL4XSoJfD4f\nUjcS14oDU6b4fL5Yy9yQO2bnb34TamjQDNlhhqZNS358jUZjOH5cdneJuQlYrdZzzz13woQJ\nNTU143p70UZi9uz8jgpTOEQikb6+PgAAnh//q1+pOjpA1LMKAJEZM/IzMgxmJKDF/AsvhFEs\n5tP06fWVxOQOi4VCu8gzOxx37jTOm9cQjRI1NZENG07t3HnslltcaNd772XmojByDAwMtLa2\ntrdTP/1p3+bNzbl/8cmEdlf0efB6qdmz/X/6UxddGmt2AwMDQt/WkiW9kyczNlvkxz+Olcqa\nTGETa0oMsChwp4N8wXHcV4Gvbgzd2MA24IKX9DEajeK7QmYtAOg5fWJZxxMVT+RhlJgSAAsc\nJQFSN+SqM/x+P/JIT+KOCXZ78lNQjY0hhunu6mo5darj7bdjWzMVWFtsUBSl1+vLysrUixfj\nJpTUYVnW7/cPDAwwXV15DEzNKsg0ES1xWN580/DRRwDAn+nCq9Jqh/3GYTB5hOf5vr6+hx7y\nDRvvKofBYEA3vF5qyZL6Z5+tpGl+xQr7ihVdNM0Le7XaWDGTWAS54YaBbduOf+1rAfG8rtT8\nF3IMyrLheV5yUl1RwSd/euokUc00Go2QyJgoilVWloSXOQB4vV50Q7JP59vfHrx+4PprPdfW\nh+qtUetVg1eh7YXvdJB7PB7PsWPHNno2vql6s4Vq8YEvClFU8HIP3JPv0Y2ME3Ai4wmsozim\n2Wx+q+otyczaqz1Xo7KOg0cOrm9Zr+JVALDVvDWdEWIwcpSG3F3yRKNRuezS4NSpCpbV6XQK\nhULOHbP8tttSaTNRqVTV1dUAAB9+GNs02sDaeBIyX1J8Xjgc9vv9kUhEoVBoGUZ16aWjOEhp\nghwi6MHB0b3yI4Xn+d7e3pj/NsdNWLQIxmhgqsvlEtrIzUNSIDG0BYH8aEunsQtTXHAc19LS\nEgwGOzomjTrJQqPRmEymQ4eCixdPiEvu0Ol0BoO2vh4AoLy83OPxDA6SifGliBL0X8gLHo8H\n9V1KTqq/+90gQAb66rxe74oVinfeOR39IG6BoSjKbDa73e7EOFuCIMrLy9MfQFEg+DcJCH06\nX/ta4KabYt8OcYCFLWp7mHoY51mK8Xq9KNAUFbwUV7SHGI7jBgb+P3tnHudUee//71myJ5Nk\nlmQ2tgEVZkC8CnYTq9X6s95SK4tX3IUWRGn12vbSioDS61LpQkerlSrX6r0K1bpUUMuiVSoI\nom3ZBpiBmclMZrJMkpnsJ8k55/fHkzmEJCeTyZ6Z5/3ij+EkOefJyXaez/P9fj6Dr5CvbNPl\nMoGV5/k3mDe2yUe3T4Ig9lXte5d4N86Pc3pw+v3W+6P3GS7rYCgGx6xi8gQWOMYFNE2LVWfw\nJCm124n6+vr6eulwT0oSRjXJHFVgrQgsyzqdzmAwGAmF6pctk41yusvzfH9/v9MZLaBFc+bR\n7mQcwrKs1WodGhpiWRY4bso996gKctLMZvPg4CD6e2wHpgqLb7IzZ5CkmJyx9awxYwmLxYKs\n8rNMsmhsbNy40dvdLQXx5A6FQsEwE5YskceJIABQXR12u6nx5r9QLFBPRCyxk+pFi4ayFDg4\njuvp6fF4PJ2dZ1WzF16offVVLcSoZnV1dadOcXfdZYx7P9TU1KjVCiSKjXkoiuJ5XihmiTWv\naW3tpigekgVY1E+tB0nRxlyCWCwW1J09f3C+sLGMoj0Q4XAY1YRKddKNnqhM83bN261VrZCF\nTBMKhUwmEyfnNvKjln4uJy63ge00nPaBz8AbpvJTl0SW9Az2vFr5qhCz+sfqP6KyjsXE4sxG\niMGkBgsc4wKNRhMSqc6YfPfd4cceg9mzNRqN7x//6B2+chXcMYtS7ODz+UwmE3KhM27aJPvo\no9FOd/v6+lyus6t8Y3vOnCtYlj1z5oxQX2BsbVV98kkBThpqS0F/x5ngRoxGqKoaS99TaPEN\n9YtxcvnpV18lIpFpCxeevQcuL8KUMGi1EP2dZZIFQRDt7ZqkN8Umd/z5zxVJ40tvuMH1179q\n0bzu2Wf7aPq8tJ8EZtQI02lE3KRaIqkUe2Ca9PX1IfE3VjVbssSGBA5BNSNJcvfuCaUfZ5tX\nVCoVOlccB888Y9y8uYbnYdEi55o1/TTNQ0yARYSIwHCAxdfIrxV53KUEwzDMuYWTcQUv6+ky\niPYAAJPJhJ5IrExzhesKJHBkJtPwPN/d3c0wzPxgJtJP1I8TQQAQAFL4Vs23ksasPkE8kcEI\nMZgRGUsTB4woSqVScuKE2K2SL0e/rVQq1dSpU1mWZRhGum9f9OZctZmkTSQSEdSNpJkvI+6B\nYZhYdSNuJ3x9PZHGTsYhVqtV+MmPO2lcXR2Zt5M2NDSE/og1wZ1+9dUAEGhuZr1enW7sBHFR\nFCXWLxal4J84DCZ9QqGQEAQoELuYv2zZKFby00nuEIsvfestvTCvq6xMLpRgcoVs2Cco6aRa\nJsvK/yIUCqWvmqUZZzuGqaysdDgcTicn1reVNMCCIqgijrnUiGvziSt4+V3v7yaeXwbRHl6v\nFy1JCsTKNHVc3XoyE5lmcHAwVv2J3achbPgZ/zOQjnqfV1BXDMCAUNYxDabdTty+hFgiB/nI\nD8ZgRg8WOMYL1PHjvX196BpCqM5w//jH6l/8Is6ZnKIopVIJy5fD8uVFGarT6UTqRuJ01z9j\nhiwUQpaoKRC6AJLuhJk5E3+hJsLzvHCJmfTMq/N2aJQEKWaCm24qQ5mgUqlk//d/SfvFup97\nrvHaa6mJZXBdhRm38Hy8nWTsYv7TT/fQ9PTcHlEQQfr7+x0Oh+C/QNNn53Xjx3+hWGi1WqvV\nOjREJE6qKYqKi04YLcgHPQ5BNZszh1m27KyAko4oNrYhSTISmbxkCRHXpyOTyTQab319NFk5\n1umgArJ6gcYeFHVW7kla8HIZXFa80aVL3AcnTqZ52f3yxNpMLidid5vY61RpqITRF2zFl3Vg\nMHkGCxzjBZIkGxsba2pq/H4/NdylUvGNb0BC7lrR8fv9ID7dZf3+EQUOQZtPuhNm1iwscCQS\nDofRwmzSk+ZvblZyXGJKX04gSTKFCa6q9N6i2VBVVeX74gtI1i82acUK7C1asnAc9/e/B//3\nf4mDByVmMzU0RNTXw003wYYNMNIX0phCKpUSBIFkjsTFfLU6j0kWRqPx5En2zjsNcfO6yspK\njUalUuXsQDzPo9f6wAF6PL/WAocOwcsv07t2XXDyJMlxBEHAxRf7mpqiZigXXWSMnS5mAFrS\niCVWNXvhhQGaHh/uGmnzyiuypH0687dvmlxzBjsdjIhMJqMoCr3xEgte/lLxl2IPMC1ii+kS\nZZrH+Me2wtYMdit8HpNKP78P/z4XY8dg8gsWOMYXMplMJpPB/ffD/fcXeyyisCybYrpLJ1RH\nA0AgEGAYhiAIud8v+9rXajs7awFs996rOH48cSfsRRcBQDgcjkQiUq+X+tKXcLQKCC3W4mc+\nrgc7hyiVSvLFF8VMcFWDg1BdnadDFx6apjWnTonejN1hSpKhoaG+vr6f/7xh9+6zC6Gx+Q7j\nB4qiNBqN2+1OTLIAgLx2k5EkuWtXY779F/x+f29v74YNtfi1RgSDwfXr4d135QBRFYPn4cAB\n9YED0aq+TZuguTmrQ0gkZ90vE1UznQ5XH8Qj1qcz8JXd71TuiHM6uAguehweL8SwygeCIGpq\naiwWy9ktZVjwEvvBSZRp3lC+kdluaZoW2+d23XbahWeOmDJgTC2NYsoIjuO8Xq/D4XA6nQGz\nGZqagCCAIODnP5dIJKkzX2L34/f729vbT58+3dvb29PdHV60SIgXpYaGxHbS8dFHJ0+ePN3e\nHvjud8dqHOlooWmaoiixM0/LZERfX54OrdPp1MNFDdMWL562eLFuxw500+S771a8lWHIWclC\nHjvGsazL6ewzmy3vvx/dumED8DxceWVRh1Y0OI4LBAKhUAiczthvg2KPC2A4TZBlWbNZumKF\n/bXXOnbvPnnjjdGEpp07izu6IlBXV9ffr1qyZCrqExEqKU6dqvR689sqkm//hWAw2NXVFQqF\n8GuNcDqdp0+f7uyEFSvsF13kT3qf7E++Wq1GArrHQ61aNem552poml+/3rx+fR9N81n2v4xJ\n3nsPeD7Jv+sr582BOXrQS0E6ASZcCVdugS37Yb8Oxo6PVa6orq5+b+J7m2s2H1ccd9COU/JT\naxvWllfBS0VFRdzKkyDTAEDGMo1afU5Hcuw+1Zw67lYMpjTBOhymCHg8HrPZHG0k4bjJK1fG\nqgwVFRUgnvnCP/kk/OQn6L/oYlQo0ouLSqEHBpLupOqWW8I/+lHwzjtxtEosBEHo9Xq5yJlv\nWLYsf90TFEWNt6IGkiT1er1erwdB4Bhz3qI8z/v9flRapQgE5JddlrRUKhAI9Pf3o8a0QiYT\np4+QJphlKuqYQSKRfPDBpO5uEgqeZJFv/wWr1Yp+UPBrDQB+v7+vrw+Gz8aqVQAAnZ2y73zn\nPAC4+mrYtSs3B6IoymAwfPaZ6557Jif6SqjVFVptbg405jnH6QAzEh9VfLSjoowLXqRSaXV1\n9TPsMy7KlcO+JK1W+3T4aStrTdznfGa+VDku+/Qw5QYWODCFBkXACk51iSqDVqsNt7WJPZyI\nmQdaLBZB3UjMW1GcOSO2k0Bzc2IcKVlTM84rmgwGQ0T8zOd1tkkeOxaJRFwul9/vJ48cmTB/\nPgDwjzxCrFuXv4OWBEuXwtKlxR5E7kktYgp38/v9cRplYZKJ0yc2TTDLVNSxxD//mfzLsqyT\nLHie93q96O+kr/XChfy48scbGBhAfxTgbNTU1OzaperulsI4zn/FFJh5MM8KVhTtYQTjNJh2\nG9y2BMop2sNoNO4P7d8l3ZVbmWZf1b53iXfj9tkSbtmk2JTxPjGYQjLOJ3SYItDf3y+oG4LK\nwEybBgBsbS0YjQRB0G1tvT09R48cOXrkSMfrr6M7B372s9gafpZlhYvR2NQPeXs7AASam6Un\nT3o9HqvF0tvT49izB93TtmrV0SNHItXVifcX9lZgQqFQKBTiHY6iV+aTJCk5ccJus3W0tx87\nevT0n/+MtvOPPFKA7gmapmtqaiZNmjTBZkNbiLlz83pETJ7wer0mk0nw+hVEzOjNMbKF2WxO\n1CjRtwFXVwclEOcclyaIODcVteBjKgHEKuTnzSv2yLKAZdnEjJjY1/rOO+O9MMc20bqqGGLP\nxi23BHN7uBMnkgcMl7VqhillVsPqz+AzJzgZYExg+gA+uAvuKiN1A3GV9Ko5MEfH66QgbeQb\nr+CvyL4v6XLicmGf9Wz9vMi8zZHNhySH9IQ+7p7t0H4T3HQenKcBjQQkE2DCMlhmB3vS3WIw\nBQNXcGAKSjgcDgajV0WJWaSB5ma0NoQyX4xGYzAYpP72N3R/xbnXzqmjUvzNzSqeV6uH2wWH\nm6cDLS1i+SxETO53AeA4zmq1ulwujuPQEre6BCrzkfNWTU0Nz/MwbMBRaKFhjBY1jB+SiphC\naRU1LFsgb2D0d9I4Z0Wex3noELz8MnzyCZhMMDgISZMyEuMhYvMdnn22j6bPy/MwMQUiMSUq\n9rVube2WSnOcgFvicOdaesedDZLMcbIJzn/FYDIg2peEqqlyVFN1zj5FIpJYlh0YGHiJemlb\n9TZhYy/0boEtDnC8BWPNPQ1TXuAKDkxBCYejCe1iqkTsnSUSiUajUa5aFV0c/Na3Ym+NXowO\np370Pvoo09SkOH4c3Rqf+rF0KfC8qbvb87WvpXX/PMNxXGdnp8PhQFeQKZa4iwVBEMSyZUnP\nPAaTglAolFS2QKVSwZaW2HuiP9L5Nsg5wWBw3bpAayt8/jnY7RAOR5MyHnronLvJZDLBUp7j\n4OmnjQ88MNHvJxctcm7e3GU05luEwRQOkiQViugLmvha19TI8pSTXbIIieyJZ0Ot5mITHDCY\n8gLXHWRJKBTq6Oiw2+3KoHJjz8bt7dv3nNhzr+1edGs7tBd3eBjM+Pq1xhSd6HKoiCoRmjUr\n/V1JJBKpVCqW+iFTKsFsjnuIQqFIkc+idLmyfHbpY7fbA4EA+jtpn07BRoLB5Jb0Rcy4ZOK4\nbwNm5sz8DdLtdp8+fbqrixgxKQPVNEGyfAeJBGrGm+3kWMdgMIBIlge6aVyB4kuSng2FQiGV\n5thr8NAhuO8+mDMHDAaQSmHyZPjpT2FYBcVkSyQS+Yf3HzcwN0zlpo7zWb3f73916NVtsK0D\nOrzgjUAE1R18H75f7KGVDb29vei3fv7g/GuHrp3ETDJEDFe5r0K3fhm+XNTRYTC4RQVTWNBy\nqOa115KqDFKFAsxmaGhIc2/V1dWkSOpH7Z13JqZ+6HQ6X3r5LPlmcHAQ/ZFYme+fMUPN84Us\nJ8FgckiciGn6zW+Ypib9cNZvrGyBVstH0CjT/jZIn3A43Nvby/N8mkkZVVVVp05xS5ZoY/Md\njh5VGwwGtVo2Kcd1+phiotFoGGbCkiWKuCyP6upqjUaj0RR7fIWlurr68GH/975XH3c2AODi\ni+tGfPioCIfDDz/M7dghE7agiiqOgyefzO2hxiNWq3VgYGBb5ba36s42DozPbgKbzWaz2SQ6\nyUb3xhnBGQpW8UblG78z/A5w3UHaBAKBOIMejuB6JD2/rv01ANSyteup9UUaGgYTBQscmEJj\nMBjEVAnD7bePKou0srIycvKk6M0JjR4SiaSiXfQHjChUTifP80j5FnMDkYXDOV8cw2AKg0wm\noyiq4vXXR5QtUA+aSuTboOqWW/KUTBw1vhHJhpg/nwGQxT1k+/aa7m4AnO8wDnjjDS1+rREk\nSe7ZM1EsD3h67gxJULbamTNTVqywX331kF7Pbt5cgwTHnTuxwJEtVqvVbrcDgJbVbuwZ17N6\nt9tts9kAYP7gfGHjVe6r0KnAdQdpIljpIRZMW9Auj76LLvRfuMWxZeKEicUYFwZzFtyigik0\nlZWV6tGoEqmh29r8Pl9/X19XZ2e/4FG2YYNY6gd57BgTDPb29LSfOnXmjTfQRu7hhwuQEhJP\nmu4hmBLE6Sx65E3Jgno6BNli2uLF0xYv1u3YgW6tue02ePVV4c4NDQ3K/CcTx5W+X3xx5W9+\nYwyHz37KYrMhbrzRk7iHL75IvudC5jscOgQ/+AF3ySU8LuDPK6XwWheLxCaRP/0p73nALMua\nTCaWZV94oXPVKuv06UGjMXzrrQ50K24Cy5JwOCzE/eJuAuFUIDiC65Z2o7oDY8S4HnDdQaRy\ntrEAACAASURBVFrEpk2FiJBdYqf56Hr5YeXhX+h+UaRxYTBnwRUcmCJAt7UFg8GhoSGGYWQn\nThj/3/8DANiwAdauzWBvSqVSqVQCAHzwQXRTyloMmUzW2NgIALB3L9pCXnppBsfNGIIg5HK5\n4tVXky5xEzRNW62ARogpGYaGhtxuN8MwBM83fv/7shKIvClZqqur0yytommaa2uzOxwej4dh\nGGV7+6TrrwfI/NsgkWAw+PDDRGzpu9lMb9lSw/PEAw9YICEbgqKSzKiKm+/A87zdbn/wQcWu\nXWd7JHABf54Yt1kewWBw7Vru/ffPZrWiSpaf/CS/7zGXy8WyLIhUVH3nOyEAXM+YOR6PJy78\neDx3EwjeZ5BQd/CU6anaplr8XkuH2BJjKS/d27aXB/6Q6tDKySsZgnlLPY46njAlC67gwKRF\nJBIJhUI5XLiWy+VGo3HixInG3t7opnNViVAoNDQ0NDAw4DWZ+ClT0jro0qWjS/0Y7f1HA8/z\nDMNEIpGkJ62yslJsiXvi8uXE1q05H4/YID0ej91ut1qtQ52d6Z7ncQbP8yaTqaenZ2hoKBgM\nVvziF7KPPiqpyJsSBJVWWS2W7q4uy/vvR7cmK60iSbKmpqapqWnGjBmThOW1HPWL9ff3d3R0\nnDnDC2aiN90Utb/Zt0+dNBtCJovvTykuPM93d3fbbLbeXsmIlqgYTGYg293ubrLw77G4Zn5I\no6IKkz6RSCT2vwumLZjdMvvb53/77+q/X+i/cEf/jomQs26CPEWT5Gq3PM8LWk9i3cEm46Y4\nJai84Hn+n75/3sDc0MQ2qXl1Xk1kVSpVXIgSAcQl/kuUnBIAKqAi6fAOBw4vCC3AHreYwoAr\nODCpQCuHTqczEokAx01euVKd84XrpUth6dLYDRzHmc3moaEh9J/JK1cSXV05Pmg+YRimv7/f\n5/PxPA8cN+Wee1QJJ02v10dOnBDdRUGeZjAY7OnpiSZ6luF5zhikPQGAzOcj5s4F9OqIlwzY\nbDa3243+FiJvaLtdcfQoX19P4MgbEc6WVu3ZE900omyR8G2QDTabzeFwAECsmejNN9u3btUB\ngEbDrlo1ae9ejUTCP/hg36JFLgCgaVqtVudqADnB5XJ5vV4491mksETFYEYLy7Jms1nMdre6\nmgfIY9dk3KwyrqKKJPFbPCuirs8AEDOrjxARADisPPwk/+RWyM2CCsMwrwZf3abdJmzJiYmp\n3+//E/enbeoc7JYgCIlEghzQEusOtuu2S/hyTT72+Xw9PT1btVsLYyJLEERDQ8NG90YX5brM\ne5kxbHTQjj9W/9FFuQBgMbE4cXhms/kVzStv1r1ZgOFhMIAFDkwKeJ7v6ury+Xzov8bWVvW+\nfZxcTiJ7obzNgU0mE7qgL+RBc0UgEOjs7EQWhgBgbG1VffJJ4vgJgpCcODE4OOhyuYLBoOzE\niaYFCwCAf+QRYt26AowzEol0dXUJaztld54zIxKJ9Pf3u91upD2lI9jxPI8myZAs8ibY0qIo\nyMjLm5FkC47jSJIEpxPmzBlRb0oTjuOEduukpe9nzsicTjouG2L2bANJllZho2s4vjrps1iw\ngMOVmJgsGRoaStEk8u//HgBQJn9kLhCWgjkOnnnGuHlzDc/DokXONWv6aZqPWyjGjBaVSiX8\nnTirf1P1ZorHpo/b7e7p6ZFocxlNwvO82WweHBwkdeRGV3S3b9e83VrVmvFuKyoqhB90iKk7\nYChGw2tK7cs/TYLBYHd3N8dxhTSRVavVBxUH36fef8r4VOz2i+Cix+Hx2C2BQKCrq4vneexx\niykkWODAiOJ0OgV1I27hmq2tpfKzcO12uwV1I+6gYYOBqKoq1luWZVmv18swDEEQymBQ9fWv\nJ52Jmc1mQd0YcbVfp9PpdDoAgP370RZi7tyCPBtwOByCuhE3zojRSBkMY8/mNBKJnDlzJjTs\nypimpsMwDHpBk0beeKdPxwJHxoTDYZvN5vF4IpEICTDlnnsUuSsQ8/v9widRQCh9r62NWCw0\nJMuGaGnJ8sg5JpTgIxpbwH/LLZDXySdmPBCXiQDnvscWL3bn9T2m1WqdTqfHQ61e3RhXUUWS\n5LjL5s01crlco9F4PGc7fWJn9Um7CUZLKBRCwdu5jSaxWCyDg4NwbuLJFa4rkMCR2W4NBsMf\nyD8MwEBi3cFCfmFm4yw6VqsV/dgVOBrmCuqKARg4Dad94DPwhvOI826D25bAEjnIY+9msVhQ\nlRZOrsEUEixwYEQRVg4TF679M2YoWTa29DFXCD/DSVfLWa83qggUFpfL1d/fH50vcdzklSsh\n2UwsGAwKV4qjW+3PaWV+OqQ4z4HmZjoYVCjG2szdarUKc8X0Bbvoiz4ceWP6zW+Ypib9W9GK\nykBzc0HGPgZhGKazs1NQ2Wo2bVLs3ZvDGiK0Ih1LbOm7REIggSOO0k/KiCvgl0imFPLoBw9y\nL73E799P9vQQg4NQXw833QTXXw9bt8Inn4DJBMLGDRsAR12XC6mbRChKn9ejq1SqwcHqW2+t\n7O6WxlVUtbRU5uMyY7zR2Nj4hOsJK2u9zHuZIWRwSpzCrH4RLMp+/0LwNiLWxNQQNvyU+2lC\n7vbIRCIRp9MZuyV2t3Vc3XoyE29UiqIOGA68S7wbV3dwIXfhk1RZ2jXzPC8sCiLizv86el2e\nOsxWw+rVsDr6H5FDsCwrLJQmDs8YMa6nx5HHLaaQYIEDIwqaDSZduA60tNChUD7mwKg9Uuyg\nRDic8yOOiNvtNpvNwn9TrPxH/SxExu9vbi4dzQBNLMXOs+JcW7IxAM/zUVcXEcFObJUQuYXr\n33oraeQNJZWC2QwNDQV4CqMmpx0fOae3t1eshignziY0ffbXLbH0XatVTplSUGkgYxQKBbp+\nTXwWEgnk1RL10CF4+eWzyoXBEJFI2K6us0dESS6vv86ePk3FbcTxLmWE8C5K2iQil8tTPzx7\n3nuvFiW2JFZUzZ6d74OPfSiK2l+9fwfsGLGbIDNSR5NUG6szEDi8Xm+s7ha32/9x/s/Exgy9\nUS8nLreBTag7mAbTbiduX0LG1x2UCyzLpjhRT5meqp9aD8Vr8wqfe9GeOLwJF0zIp8MPZvxS\nlv1mmMIxvHDd++ijTFOT4vhxtDl/C9cURaU4aFEWcywWi/C3MBNjpk0DALa2FmJmYgRBAIie\nNGbmzIKOOyU0TfMcV1LnOa+wLJui0yTQ0pK44I+gaVqpVIpF3jQsWwavvlqg55AG4XDY6XT2\n9/db+/sjixcnrTMqBYLBoHBNHKs3ydvbIUefFKVSid7GHg+1atWk556roWl+/Xrz+vV9NM2X\nmpNoCiorK0HkWeh0uvw1jXu93vXrg62t8PnnYLdDOAxmM93VJZs92x+XstHbSwjRGzffHLXj\nxfEuZYRWqyVJMul7jKKoioocdDGk5osvkm8v/YqqcmEezJsDc/Sgl4J0Aky4Eq7cAlv2w34d\n5KAkVijfyGE0SewvcuJun9A+kfFoV8Pqz+AzJzgZYHqIng+JD++Cu8pU3QARE1n0X3T+i3s5\nl3p4v639bfSyGYPJNVjgwIiiUCh0b76ZdOGaoGmZEOgoQvT3aZTJsiqVSmy1nCdJ9fAifA5I\nb2AMwwh9DYkzsTihB610iY1fqlBATCVIcVGpVJVvvy324irOLQ0dA6TWngLNzSl+Zevr64V7\nJqFktIOBgYFTp0719fU5HA5y7Vr6gw/ymmXLsmwgEAiHwxmkRwudXGK1TtkPjyAIg8FgMkmX\nLJm6d68mtvT9+PEKr7cq+0MUhoqKCo/HmPgsTpzQMUxtPo6IcpG7uro6OyExNDQYJKdPDxqN\n4VtvjXr1zZoVWLXKijbedJMVbcTxLmWERCIJBBoS32OHDytZtrEAE6T33osmtsf9mzcv30ce\nL8TO6k1g+gA+yOGsXjrcjcYH+QXLF1z0pYtk18ogCADwjvYdaUa9arHmssgb9YtjX2zp3CLj\nZQDwlnrUuRvHw8cXhheOvYxSgiCiaWXJTtR23fbiOqdKJBLhDZA4vHe07xRxbJixDW5RwYii\n1+vRwkrF7t0Vu3fH3jRx+XLYuBF+/OPERwUCAZvN5vf7WZalCGLyypWjMg7U6XSUyEEn3323\n2EHTBPUp+P3+MMPU3nWXLI2BCVX0Yiv/sQvBUqlUrVYLq/1x46++9dYsx59Dqqur/aN/ccsX\niqKkUqlq27akmg4tk5H9/WKdJnK5PPCvf53u60NFB/KTJ6ctWgQA3MMPk+tLpX3U5XIJpUb5\nzrL1+XwWiyVagiEShJwWIs4muap1qqqq2rVL0d0thWSl7/fdl5ODFILt22vECvjz8Sz6+vpQ\nLnLS0FC9PvqVKKRsXHfdWd1Z2LgoB639mMLxxhtasfdYKZUeYkoRnU6H3EBPnjy5ZcsWAAAK\nwAsgBzWnjo1xSR+VSkUQRGz1R8beqKFQyGw2vyR/6Y26N4SNpZxRynHcv/z/2iDZ8C/6XzbS\nxhBMLdReA9c8AU/UQBLl2GAwdHV1Cf/NuYlsltTU1MR2eZfa8DBjFSxwYETR6XThEydEb042\nk0FRYcJvUvVvfjNa40CSJCvaxVOjsliIDoVC3d3dyCbDuGmT7KOP0hlYtJM/7ZlYQ0MDVw6r\n/RKJRHPqlOjNJTPOHFJZWUmLaE8Ny5al1nQUCsXUqVNDoRDDMNK9e9FG8tJL8zrg9OF53mqN\nrpwnOowwM2fmsPrW4/GYTCbhMy4WhJwa1PMvVuskkctz5Wxy4kTy9IfyKn0vZAF/OBxOHUx7\nzTVuODdlY8GC6P2FjXPnhpYtwxaj5QRuEsFkjFqtfsv4lo2zVWoq71x9Z+Mljb/kfxmsDgLA\nAm4BQWfSg0BRVE1Nze8iv3NRrsTEk8XE4jT3E4lEOjs7w+GwVpI8o5Rl2cOBw+mrCfkGmb5t\n0217q+6s8pJajlGr1buadnV5u77m+VrGJyp/6PX656nnewI92byOGMxowQIHJhWSEycGBwdd\nLlcwGJS2tU1duBAA+EceIdatS7wzy7Jms1mY+cQtI0NDA6S3jEwcPcpxHCq1II8cqfvWtwCy\ndUnkeV5QN0YVeSuTyWia1rz2WtKZmEKtjpuJSSQStq3NOjDg8XgYhlG2t0+54Ybsx58PyGPH\nOI5zu93BYJA8csRwzTUApTjOXFFVVRUZpWAXh1QqlUqlsHIlrFyZzhGRfzjDMBRFKQIBxbx5\nebL8ZBgmhWusb8aMXAkcPM/39fWJfcbTLxVRKBRyubwAtU7vvZf9PopPIZ+F3+9P3BgnZySk\nbPBwbvTGli0Oms7QAhBTFMbGJwVTFFwu1y75ro81H8N8gLNJoKDt1D5a92jGuzUYDPuYfbtl\nu+O8UWfzsx8n0vVGtdvtyOcyaUbpxaGLT3ac3KYfhZqQV7xeL1oj1LLJ5RixB+5R7tmh3NFq\naI3dmCsT2ez5qOKjHRX58rjFYJKCPTgwI6DT6aZMmTJjxoym4WU9Yu7cpPd0u92CL1SiXUVo\n1qz0D0qSpF6vb2hoqOvri26aMyfjpwAAQ0NDSN1IHJh/xoxYD/BEjEajmMdk1S23JHpMUhRl\nNBqnTZvW0tIyRTCzyG78eYIkSZ1OV1tba+jpiW6aMycDS4WygCAIyYkTgy5XV2fnyRMnuobL\ncGDDBuB5uPLK3B7O5XKdPHnSZDJZrda+3l72xhvzZ/kZbaQScRjxz5iRqwP5fD7BFD1Lc9DG\nxsaycDYZbySN173jjiaPh5o71/e733U9+6zhgQcm+v3kokXOzZu71GqO4+Dpp42xG3U6fGmB\nwYwXbDbbJf5LWgItFWyFhJdU+irhQ4ClsPx/lodsoWz2fLXs6kv4S3S8TsJLGriGy9nLt8CW\nT4lP0/dGHTrXuI0juG5ptxChenvn7RzHITVhe/v2PSf23Gu7F90zhZqQPywWC1o/mD84/9qh\naycxkwwRw1Xuq9CtX4Yviz0wryayo+JI8MjC8MImtinW6+RiuLhEhocZP+AKDky6EMuWwbJl\nKe6Q2jiQmTUrk5LlpUth6dIMHhcHCuIW89Egfb4Ukbd6vT5y8qTorlPPxHI0/nwztHCh+5vf\nZBiGBGhYsCAdd5Iygud5giBQbKqus1MHABs2nK27yYP2NDQ0lGa0cE5AjVRiHR+kRJKrjo/U\nQciBlpb0S0Xkcnno6NEeq9Xr9bIsqzh1ChWIjeEaomxgWfbjj/2vvEIdPCjp66OHhoj6erjp\nJtiwATKy8BMl1tsvLjT0hz+0rV49Ye9ejUTCP/hg36JFLgDweKjVqxvjNpZRSA0Gg8mGUCgU\nDoeX2pcutUcvdV588cVf/epXADD7f2ejS6+MWQ2rVxOro/8ZfacLz/ORmMz7xIzSykgliBR3\npFAT8kQkEhGuohEcwfVIepAcY4wY19Oinl+rYfVqWC12a2FgGKa3t/f/lP+X6HVyPVz/GXxW\nxLFhxiFY4MDkGhG7ivCFFxZxUCzLig0s0NwsF4kIFaDb2kKhkNvtZhhGduJE9dVXA4yRmRjP\n8z09PchTEEbjTlL6hMNhu93u9XpDoRBNkpPuvvscv9vrrsuf9pQ0Whj1cXB1dWROLT8BQC6X\nSyQSsY6PER1G0icujCbuoxSaNUvoyiFJUhEIKC+/PEVXjlQqnTBhAgBEIhHqwIHo1pKsdSou\nqCv7sccadu8+aynS3Q2/+AVwHDz5ZC6PpVKpSJLkOC5Oubj0Ut9ttzV1d0tjUzasVsmvfmU0\nm8/ZKJPJ1GqtDq/MYTDjgMSar7a2NgAgSfKCCy5gWTa6wFAMCIJA32YQk1EaISIwHKG6wbxB\nuHP6akKeiNViIJkcM+GCCRmoPIUhHA53dnZGIhGtbHTNNRhMnsACByZnyGQyamjo/PnzKZcL\nANSffhpqbIwNqsjVMnIGSCSSFOmzsoGBEf1BpFJpdXU1AMBf/xrdNCZmYjabTVA38pi+4XTC\nnDl5cqBISjAY7OzsFK69qn7969H63WZz6KR9HMjyM9DcnImn/EgYjUb5sWOiN+fo+aJCJ7GP\nEkHTZ/buZdDHhOMmr1yZZlcOTdMwUoHYuMXv96OubLNZumKF/eqrh/R6dvPmGpRpsnNnjgUO\nkiSNRuOBA4577pkcK2ds3mxImkeDKOuQGgwGkzGxNV+IY8eOAUBTU5NcLqdpuljqBkKpVHq9\nXhjOKOWBP6Q6tHLySoZgtuu2CwJHnJrwjPmZiecV2kUoNo85UY75be1vLycuL/CQ0sdutyOB\npkTKYTAYLHBgsiUcDjudzkAgEGaYCf/1X9SwVUfltm2V27YJd6u48cYiho9qNJqIyPr25Lvv\nZp94Alpa0t1XmXSdpAPHcQ6HA/2dOBUPtrSI9u2MRCQScTgcfr8/FApJKKrhe98rZNsLz/O9\nvb2CupGucJMjFUZQN8T6OPIhcOh0Osenn54e7uAVsmzDa9dKNmwY6dHpolAolEqlWKmI4fbb\n2QceYO66C/LflTN+sNls6DVNGtpakweb/6qqqt27lSnkjBHB0RsYzDiBpmmFQiEYmfl8PpPJ\nBAAtLS0AoNFoijk4gOrqaiRwIGIzStVctJMuUU34jeE3X4OvFXioEolEKpWGQiFIJse8o32n\nwOMZFcI6GSK2HMYQNjxEPgSUyCMxmPyABQ5MViDPZzSTNG7apN63DwgCYqLLz6F4kxy1Wp0i\n8pYqmbzPrBj9/JxhGFS9KZa+kZnAEQwGu7q6hHrLyo0bC9z24vf7hV7WROEmNGuWLGaoKO+G\nBKi5/XZJ1ioMz/NoOkoNDl7wzW+ip6zdtSs8LKmMym13VFRVVWm12mh8z7D0IPnKV3J7lMbG\nRl7cHDTY0gKjDCrCpIDneaGJPWlo68KFfCa96SPR1pb8o//xxzBvXs6PhsFgypja2tquri63\n2x2JRA4fPox+ASdNmuR2u43F/uZXq9U7p+zs9nUnRqhe474G3SdRTfhLxV+KMlqj0dgjOL6f\nK8dUQEVRhpQOI3qd1E2uwwIHpsBggQOTOZFIRFA34qY0YYPh5J49yvb2pgULAErCroI6fry3\nr29wcBBi1re9//VfqieeKG4JZTYwDONwOILBIBsOT1i+XD7K+TlSN1K4k2QwJJ7nTSaT8GuX\ncVpwNqT2uw20tCCBw2KxDAwMoHsaN22SfPhhNioMz/M2m83hcHAcBxzXdNdd5PAw2IoKoY9D\nplTmr1eLpmm9Xg8AcP/9cP/9+TiEVCpFQcjI3ETV0THxO98BANuqVbYVK0CkFCgfRSvjAdTB\nHrcxNrT1jjskAPEl4tmDQ0MxGEyaqFSqiRMnTpgwITaypLW1tbW19brrrtsxnDo3WtqhfS2s\n/Rw+t4AlCMFaqL0GrnkCnqiB0dWtfaD6YIcqSYTqo9yjATiboBerJmgJbWZjzhKtVvs89bzJ\nb0qUYxYTi4sypHQY0etkK7m12GPEjDuwwIHJHJfLhdSNpFOaKVOmqA4ejN61BOwqSJJsbGw0\nGAx+v5/aswdtVF9xBZStuuFyufr6+tD8x7hpk/zjj0c7P5dKpSBuqUBJpRlMxT0eD6qxhGRv\njPCFF+Z+NiaGiHCD0kwHBgYEdSMn5iMmk8nj8aC/ja2tso4O4aa6xx4T/q665ZYi9mrlBBSE\nHF2a278fbQy0tICIouTPj+3IeICiKIIgYjWO3bsr1qxp9PvJuXN9Tz1lkskuKOLwMBgMBgDs\ndntcICtiTqbXfoFAYGt467aKs23OKI/DAY634K0UD0xkHsyzgvU0nPaBzwjGaTDtNrhtCSyR\nV8k3Sjb2BHou814WpyYsgkWZDTt7PlR/uEOdRI55HB4v1pDSQaVSoeufpF4nUiKnWV8YTBpg\ngQOTOX6/H8QXyWmGUZWeXYVUKpVKpXDffeVughcMBgV1I+OOAIlEolAoxCwV6pcuzWAqLvTi\nJn1jBGfOLIDAIZPJQFy4kcjlfG+vfViMSFRhmJkz0487RQwODgrqBno5OJWKFEvIG0uGFEuX\nwtKlwWDQ09GRIl2luGOM49AhePll+Pvf+Z4eYnAQ8pS3mhMIglAqlahLJS60dc2afq1WSZJk\nsceIwWDGO01NTYm1Zhljs9nsdjutpTcO5SCPI0WE6kcVH+2o2PGU8anYjcVVE0TlGBjtVUlB\nQV4nwnugXJprMGMYfG2EyRyO44QpTe+jjzJNTYrh5vxAc3O0/QGTHxwOB/otiZ2fy9vbAcA/\nY0ZicpsY9fX1CnFLhQym4nFtL3FvjMLMdVUqVWxs6rTFi6ctXqwbLpStvPnmyMsvo1MkZj4y\n2iMKi1fCy3Hy/fd5mQwAPFdeaXn//ej9NmwAnocrr8z+OY6M0wlNTUAQQBDw85/n9VAymYym\naTFFSa5Sgdmc1wGkj8vlWrPG29oKX3xB2O0QDkfzVh96qNgjE8FoNBIE4fFQq1ZNeu65Gprm\n1683r1/fJ5FA0fvbMRgMJre43W7krDx/cP61Q9dOYiYZIoar3FehW3ObxzEP5s2BOXrQS0E6\nASZcCVdugS37Yb8OihZzvRpWfwafOcHJAGMC0wfwwV1wV2mqG+3QfhPcdB6cpwGNTqW7rPmy\n/5j2H5+qP3XQjlPyU2sb1pZ+cw1mDIMrODCZI5VKpamzV1FgZNbwPB8KhUiSlHg8BU4bLVlS\nl88QgYBarU5nPwqFwv/Pf3b09SHfCsGdhHv4YXJ9JjnwqdteCpMWTBBEY2MjLS7cRGbPBhDt\nYfHPmFE1yiOirhyxl0MrtKvkuVeLZVmXy4U8WWrvuqtgyTUEQdTU1FAipUBVt9wClZXgdAIU\n+WPb19fndDp7eqYVIG81VyiVynB40pIl0tjQ1qNH1TU1NWq1ctKkYo9vjILKfD75BEwmKPEy\nHwxmLGG322P/G5vHYYwY19OZXJaIkaK4A5OaYDC4NXROD9EgMTgoH/z+5O/H3q30m2swYxUs\ncGAyp6KighXPXuWffBKmT8/yEOFw2GKxuN1unueB46bcc4+qgGmjpQzP8ynMQeWjKZ9RKpXT\npk0LhUIMw0j37kUbyUyTZSoqKiwWi1jbi/Y//qMwDhQqlSrwr391Wa0+n4/neUG4QRNsIhiE\njg4xFYaUSEarwhAEkeLlUNx6K9x7b66fYjwon0+INCpwck1VVVXk5EnRm5G6UZCRiOH1ep1O\nJxQwbzVXvPaaursbICG0ddOmcu+0K1HC4fDDD3M7dghpS9EyH44rURUMgxkb8DwvdLlCsjyO\nhvMacB5H0XE6nf39/Ul7iCigKKDKqLkGM1bBLSqYzNFoNCrx7FUi68XqUCh0+vTpoaGhqI9m\na6vqk084+fB35fgWOCQSidj8PFo+M0qkUqlGo5GtXAk8DzwP3/pWxgMzGo3yY8dE71GoF06h\nUEyePLm5ufm8886bOjgY3TpnDgDI5fIUPSwNy5bBq6+O9lgpXg6FML3PG5FIpLu7Oy7SiJk2\nDQDY2toCJNcAAN3WxgSDVoul7fjxjtdfRxv9l1wCAKXwsXUOvwpJ81a/+91IkseUBl98kXz7\nxReLPuTQIbjvPpgzBwwGkEph8mT46U9h2PwXkwq3293e3n7mDL9ihf211zp27z55443Rd87O\nncUdGgYzxoltbRbyONB/UR5H+u23mDwRCAT6+/vFeojugDtKv7kGMx7AAgcmK+i2Nkt//7Gj\nR48eOSJMaYIPPihmNMAwzODg4MDAgNdkGtEgoL+/PzFtFM3ZuLq6wszZCgbHcQzDcAMDafom\naLVasfn55Lvvlr3xRiEGLUJ1dXXg4METbW1HjxyJfWMU1IFiGIIgZDIZsWxZnHBTV1eXQxWm\nsrIyxctBv/ZahqNPm2g8bdaeLFkik8kYhmFZVnBdUX7+ufCx5evri/ixZRgmbkts3uqSJf5i\nDAoAYO/ewPLlgdmzwzU1fFIx4r33om/euH/z5iXZG8dxAwMDDz3ka22Fzz+HsvAZKR0Yhunp\n6eE47oUXOletsk6fHjQaw7fe6kC3lnKZDwYzBqAoSjBORnkcXxz7YkvnFhkvA4Dt5XjGsAAA\nIABJREFUuu0SSeFy2DBJGRgYiDWU5QiuW9ot9BCt5cZv5zimpMACByYrCIKora2dPn16U1NT\ng8WCNsovuyzxnizLmkym9vb23t5eS18fLFkCKZtNWJb1er3o78Q5W6C5OR9Ppyi43e6Ojo7j\nx4+3nzzpv/761KdFQK/XK9vaRG8udnmLXq+fPn36+eefP3ny5MmO6PSgFNKCBSoqKgIHDx4/\ndixOhYmsW5eBCqNQKDSnTonenP+XAwVtiJmAIMeWAhAOh91uNwC4brih/e23OaUy9mMbbGlJ\n/VifzxcKhQpjj7p7d8UddzR5PNTcub5nnumii9GviYrUNmwI/+EPisOHJQMDRJZiBMuyZ86c\nsVgsJhOFCxAyQLh2T1rms3BhznIiMBhMUioqzgndEPI4AEDDawiCKNK4xg6x5qASkEyACctg\nmR3sIz8SAIYN4BALpi2Y3TL72+d/++/qv1/ov/BPHX8yBA35GTUGMzqwBwcmB1AUpVQq4d57\nUxgNmEwm33BqprG1Vb1vX2qDgFAohC40k87Z/M3Nqjw8kcLjcDj6+/vR3+mcFgGCIOi2NnN/\n/+DgYKzHROihh6R5jsxIn2go7/LlsHx5sceSBL1eX1FR4fP5GIZRfPAB2kh/OUOTdur48UAg\n4HA4AoEAfezYlBtuAAD+kUeIdetyNmJxWJZNYQIiLVQFB7KqBfGPrSLZo9xut8ViQUat+bPa\nkcvlqIgjMW+VpnkULVxIOI7r6uoKhUJmszRXpqdmsxm9BGXnM1Ii+BKinWPLfG6+GQCURRgW\nBjNuMBqNL9Av2Dhb7/O93Z90n3CdYFYx0AIAsJBfWOzRlT0Oh+Ml/qVt1WfNQXuhdwtscYDj\nLXgrnT0IbURCD1GEiMBwD9FL3Ev5GDYGM1qwwIEpBENDQ8KFo9BsQtvtiqNHwwYDUVWV+EaM\n6vQic7bUS8HlQigUsgyXvcSdlojRSFRXp/bSIkmyoaGhtrY2GAxSH3+MNkq/+tU8j3pMQVFU\ndL3ohz+EH/4wy70pFIrGxkYAgGGvVmLu3Cz3mSYSiUS5davmww89X/963eOPT/zP/xRu4klS\nareDroC5d6P52LpcLnNMiKxgtZNze1S9Xj80NOTxUKtXN+7dq5FI+Acf7Fu0yAUAKpWq8AKH\n0+lEmk5SMaK6mgcY3VqlUD4DIgUIyGYXk4K4dPPduyvWrGn0+8m5c32trd0EMaFYA8NgxgkS\niWRf1b6dkp1w7tLALHbWkxT2+M0cnud7enrcbrdSp9zYE28O2g7tae5HKpUiI1jUQ8QDf0h1\naOXklQzBbNdtl4Zx0BSmJMAtKphC4PF40B+JzSbBlhahFSUWqVRKkqSYcaNELoeYSVGhyVEJ\nveCfmrQHR5irpIaiKJVKJb/nnizNQTG5ZOnSgr0cHMc5HI5IJIJMQDQffSSx2WLvMPnuuxVv\npbUykz2pQ4ITP7YsywoVTJBgtZNbzw61Wu3xGJcsmbp3r0bIWz18WHnsmIZlG3N1lPQRvveS\nihHz58c7hoxIbPqAgFCAcMklwWXLMhlnmuzfH165MnTxxVxZ25qiDv9jxxSPP173jW9M/8//\nnOj3k0olN3NmQCbjcf8/BpNXWJbt7OxscbZMHpgs88tojqb6KPgQGtY2HKQO6qCAMv2Yw+Vy\noavKpOagX4Z0a1dT9BCpObUUJ2ljSgNcwYEpBOFwGMQNAshIkvwCkiR1Op1SJG209s47C5M2\niuB53uFwuN1uhmFIgIkrVihyUUKPCubFTguX4ImIwcQRDoe7urrQGymFZ2r2kUZpIpPJFAqF\nWEiw4fbb4z62brdbWDOPlfmmX301AIRmzcptWcX27TWlk7caSfjeO7cbgoFR+s9zCeHQsQUI\nmzdbabop/b0dOgQvvwyffAImEwwOQn093HQTbNgAiZevPp+vr69v3Trj7t1nL3zLNFeVpmkA\n+P3vDX/7m0bY6PeT//M/1RRF/eEPOBEgOYcOwUsv8fv2ESO+WzCYFPT09Ph8vqW+pUthKQB4\nvd6vfvWrwMOXF34Z53Fkicvliv0vR3A9kp6z5qDk2jSXvKuqqp6D52yc7TLvZcaw0UE7/lj9\nRxflgvR6iNqhfS2s/Rw+t4AlCMFaqL0GrnkCnqgB3EKJySVY4MAUAoqiUhgEVFDJWzFqa2sj\nJeCjiVrlBV+l6k2bFHv35qSEniCIFKdFjs20MCPR09MjhIPY7rln4gMPCF1OaGPgZz9TPPZY\nIYfU0NBADEeoJOHcz0toeIk/qcwXnDkztwJHBnmr+YM693svrhtCLh91N0Rsl02iz0hlpSbF\nY+NgGGbdOu69985apogJFj6fr6uri+f5HDqJFAuXy+XxeEwm6b59agCgKH75cvupU/I9eyoA\nYO9ebbEHWIrwPG+329esUe7cqRY2lqm8hSkuPp8vrp73+PHjqMr1/PPP93q9arVa5KGYkREc\nsgBgwbQF7fJoT8qF/gufMj1VO7k2TQWJJMn91fvfJd59yvhU7PYLuQs30htTP9br9W7ltm6r\nyNwBBINJE9yigikEKpVKrGqdJ0nV4GDSR5EkKTlxYsBuP93RcfzYsc4334zeUNi0UYvFIqgb\nuS2hl8vlKU6L8ly5HZNHChLbkXP8fr/wzkzsckIokkaJ5hO5XA5HjnR3dcWlR/OPPJL4sY2z\n2ul99FGmqUmImA1feGFuxzaqvNV8I1yscxw8/bTxgQcm+v3kokXOzZu7NBpeqRy1maVCoUDl\nwR4PtWrVpOeeq6Fpfv168/r1fTTNa7Xpzs+9Xu/p06e7uoh0clj6+/vRDKTcc1V5nrdarQDw\n1lv6UIgAAJYlnn3WgNQNAOB5fL0UD8dxnZ2dNputp4fGqT2YLEnsVj4+/FvQItLLjMkAwRwU\n/ReZg45qD5cTl8+BOXrQS0HawDV8nfv6FthygDyQooeI53mTydTV1UW76Y09G7e3b99zYs8P\nBn6Abk3fAaS4ZBlAgykkuIIDUwj0er3niy8gWdX65LvvTtFsQhBEdXV1dXU1AMDw5D9p2mg4\nHA4GgzzPy/1+6Ve/Gg1b3bAB1mYVys1x3OCw/pJYQh9obs7GUl+r1dLip4V/8kmYPj2L3WNS\n4Xa7PR5PMBikCKJu6VJZHmI78o2gbiQtfxj60Y+0v/xlUQYmk8kmTZrE83woFJIMf2yT+q0q\nFAoQ9+yQKhRgNkNDQ8FGXkgqKysdDofLxSeanlZX11AidW2paWho+PjjvpUrJ3V3S2N9RpRK\npUaTlsTBsmxPTw/HcelYnzIMI6wKlrutaSAQQE1DbW1Jo37gO98p7IDKAZvNhr6FcGoPJnvY\nhLSvtrY2ACBJ8oILLki8FTMqZDIZ+rpObg7Kj6KdbDWsXg2ro/9JT/jt7+8XHECEjVcOXvlU\n9VMwGgeQIsIwzCuBV7bpcPlJeYBXJDAAkPcVbJIkK9rFBdo0p5Qixo2hUKizs/PkyZPd3d2m\nrq7QwoWQu8kqwzCosz3pHNI3Y0b0fhmdQJqmNadOid1aMN+EMQ/LsoODgxaLxWKxDHV28lOm\nAEFUaLWSJ54IBAKqxx6TffQRJx+uziwfgSPquSBS/sDMnFnMwQEQBCGTycjvfS+F36parZbJ\nZIJnx7TFi6ctXqzbsQPdWnHjjfDqqzkYSklW6FAUxbJTEk1Pu7pqAwFDZvtUqVQffji5u1sK\nwz4jt9zSdMstTTfcUJumz+zQ0BCaSCQVLP7938/xMQ0lMxEVnEQuvjiQV1vT3CJYojz7bNeR\nI0fRv9de60Dn4eKLA48/XtTxlR48zwtd/eUub2FKAeSAE8uxY8cAoKmpSS6XJ96ab8bYcr1e\nr4/9b6w5qIbXkGQe54ORSCTRAaRb2o0cQOq4uvWwPn9HzwmosFHqlQrlJ/fa7kU3lUv5yXgD\nV3CMXzwej8vlYhiGZ9mJK1bI87yCTRw9iqohAoEAeeRIHZrtZF1hEQ6Hz5w5I1ybGltb1fv2\n5TBjElVfizll+GfMaG9vl1BU/bJl0oxOIHnsWDAYtNvtfr+fOnp02qJFAMCuX089/HCWI8cg\nPB5Pb29vdPGH4yavXEl0daGbAs3Ncem8fH09kbvYjnyTOrKElslKv/yBIIiJEydC2p4dacKy\n7MDAgNfrDYVCNElOWL48399vmfHKK7Kcm54eOZI86SNNn5HYPm1ErPXp4sVugLNVa4nXxLFO\nIr//fT9NT0t/5MUlcfqUjUVriXDoELz4IrtvH9HbS+bc+zMSiSQuqse+W+66S45X0TDpo9Fo\nbDERYD6fz2QyAUBLSwvE9PQVBoZhXg2+uk07dpbrKysrn4PnLBFLojnoIlgUDAaPBI/8t+y/\nj0iP2EgbQzA5tP/0+/3Ra2kASHAAed7+/MRJE7M8RF4RChtjy0+ucl+FEna/xH9plJHumEKA\nBY5xSl9fn9MZbZQ1btok//jjHIoCYpAkWVlZCQDw179GN2VdpGCz2QR1I26yytbWUllPVlPP\nIXmS5Hp6VK+8Iv3b3zI+gXK5fMKECQDA//3vaAv1pS9lOezCw/N8MBgMhUISiUTu95OXXpqr\nLqFsCAQCJpNJ+GWN079YlWryj38c23MUbGlJXp5ekqjVapIkxSJLKm++uZBJQxkjk8kix49b\nHQ6fzxcKhVQdHRPmzwfI/J0TCoW6urqE4oLqX/2qMN9vGZAP09P33sv8sSBIusPEWZ9S1Dlr\ngAqFgiAI9JBEW9Pq6nJy5VQoFBRFoRl74nOpra0q9gBHB8/zNpvtZz9TFDLaJu7dQtO4xRIz\nChQKhVarHRoa8ng8kUjk8OHD6Ltl0qRJkUgEhfEVhsHBQbPZLNFKNro3zgjOULCKNyrfQLPZ\n8l2uJwhiX9W+HbAjzhx0Nj/7fuv9HY6OP1f9+S+6vwjbcyjoxCZ8CQ4gESICAIeVh5/UP/ka\nvJblIfKKUNiIiA2gMYQNDwQegArxB2OKBBY4xiMul0tQN+JEAa6ujizACvbSpbB0aU72hJr6\nIJlBhn/GDDXPE9llkdA0rdFoxOaQk+++e3D+fN0775yNrmhoyNh2lFi2DLIv6XY6Yc6cAosL\nDofDZrPFVkmoS2PB3Gq1ChO2uLd6uKamYd06OLfnyN/cXEYCB03TRqMxRTpsSU3mU4CeSPQ/\n+/ZF/8hU/ezt7RXUjRKv0MlSjMgHQhRL4iSfpvnYoBYY1qwdDofHQ8U5iRAEUVVVTqIAQRA1\nNTUWiyXxuVAUFfWBKiDpx/QmxWQyeTwes3la/qJtaJpOIQkpFHRei94xY5KGhgaCIC677LKh\noSFhY2tra2tr63XXXbdjuHsxrwSDQbPZzPN80uX6WLeIU/ypNdyaL4gvbKStLBJP58E8K1hP\nw2kf+IxgnAbTboPbrrBc4XP4AEDLajf25EXQia2PS3QAeVvzdvaHyCupA2iqtFVY4ChB8M/P\neGRgYAD9kZi84BdMJcoBlmXR1VVSg4xAS0tOJP+6ujqFeAm9dufO2BMYmjUr+yOOFr/fb7PZ\nenp6+np7w4sW5dCCJB3sdnt/f78gbwtVEoUcQ1J4nvf5fOjvxLc6wfOJvhWB5uZijTYzqqqq\nAgcPnmhrQ54BqSNLyoNzrXYOHYL77oNLLuENBpBKYfJk+OlPIZnzQ5RAIJAiWaYoH8/yQqvV\nEgSRNIeFJMmKivjrOKPR6HDoE51E+vom2mxlpBYCAFRXV3u9tXHP5fjxCqt1itlc0NUglmU3\nbAi3tsLnn4PdDuFwtPjioYfSevjg4KDH44E8R9sQBKHT6UAktSeu4R9TeILB4OrVq6+44gq1\nWk0QBEEQ+/fvL/agRoAkyVAoFKtuCMwplCuZw+GILWSLdYswhA1rImuEu71oe/F16vUz5Bkv\neCMQQSUP34fvF2acGbAaVn8GnznByQBjAtMH8MEtoVuQugEA8wfnXzt07SRmkiFiuMp9FdqY\nE/tPlUpFUVTsWY11AKkoH3kg+wAaTMHAFRzjDo7jGIYBEVHA39wsj0QKb+aUGdEFIhGDjEBz\nc2UuVpCkUmno6NHu/n6v18vzvPzkSeSUYf/+9yt27pTY7bEnMDhzZi66m9OF53mz2SzkvBg3\nbZJ8+GEhq/HD4XBs02xJLZizLIt+UJO+1emBgcSeI0oqLX3fijj0er1Op2MYhmEYxXCXU9LI\nkrLD6XSuWSPduVMtJHeMWGMvrLSIiZ6y5I/DRJFIJMFg45IlirgcFgCYNas68aeBJMmdOxty\n7iRSLN55p7roz8Xr9fb09HR0TFmxYjCz4gvhFyGp9+f114cBkhu1jBaj0djWFl66tDbu3SKT\nyc6cqd6zJ/MKFEyWRCKRv/3tb0/GvF0oipo9e3YRh5QmTU1NcY1yBSYQOGulnLhcX9NQAxqw\nWq12u12j0+Sp5KFgJIbvxvZf1LK166kc2H8SBFFbW7spsMlFuRIdQBYTi7M/RF6RDn9tJQ2g\ned71fHGHh0lKecxjMTkktWtmoLm5uD8to4IgCIVCIX/lFTGTRdpqzclkVSqVothLhmGCO3ei\njeoDBxJPYIGjK6xWq3AtGycuZNMskz4ej0d4wyR2CYVmzSrifJKiKIIgeI5L+laHZD1HdXfd\nVRa+FXEQBCGXy+VyOdx9N9x9d7GHkxt6e3sHBwd7ekZXYx+XLFPcj+cnnzAvvcQfOECbzdTQ\nECHM8f75T+7FF9n9+0mzmSrBud8bb2jFJvlJK2DEnESeeAL6+0voeaVDPlxRRgXDMN3d3TzP\nZxO8mphuE+v9efPNkVwJHCRJ7t49obubgIR3y8yZ7NGjZ++Zb/sPTCx+v7+7u9tisdxzzz2z\nZs1av369zWabOnWqRJKb131sIxhGJLpFbDJuepF/kWEYVAedtIflUu7SMiqOj/MJjhN0tji2\nTJyQG/tPvV7/qfrTnZKdiQ4gjxOlnlCl1WptNpvwxhDKTxiKUXPqxMJGTCmABY5xB0VRFEVV\nvP56UlGAoGnaaoXGxuIOMn2qq6t5EYOMhmXLcjtZRdNI/513Hr3mGv2bbzasW1fc6AqWZR2O\naNVxorjAzJxZAHFBuJJOumAeLMgYxCAIQqVSSV5+OelbXZQy8a0Y2wwNDSHlbrTTPGQSUfRk\nmXA43NPT8/DD1YkWj0NDno4OvpDWj6NltJN85CTi9/sXLoT33z+bsWKxlNbzSoeiu6LYbDYk\nGWcTvEoQxLFjinfe0f3jH8r+fonbTXEcwfMwZ47vqae6JZIJORzwP/6R3OXK42FXrHDmyf4D\nkwKO40wmE8uyc+bMmTNnjtfrtdvtADBjxgyTyTR16tRiD7DUkUql6MIm6XK9JChxu91xPSyx\nlpP3ee6DyqINfrRQFCX8nSjobOQ3boWtuTrWNyTfcIKzg+/wE/4armYaTLuDvGMJsUQO8pEf\nPEraoX0trP0cPreAJXt7FIlE8v6k97u8XYnlJ9+NfJeW4al0KYJflfGIVqtViogCE5cvTy4K\nFMO6Mh20Wm3k5EnRm/MwWa2oqOjv7xezHdUvWVKwEgCfz5eiBcPf3FwAcSF1l1Ck2AWxRqOR\nEXmlAMDyox8N3Hmn0HPEPfwwub7Uw9jHCS6XC/2RdJq3YAEnZiClUqkkEonYx7PqllsK8PHk\nOK6rq4thGLNZmlh+8uGHEpKE/Fk/Zk8Gk3wUOtDdPbWUn1dZINgGCcQWX9x2Gw0wQj1MMBgM\nh8PPP98YK6IhWloCajWnUOTSGyXu3cJxXHt7ezgc9niojCtQMNkwODgoRMsBwPHjx9F1QnNz\ncyAQ8Pl8KpWqeKMrAyoqKmIbN+KW6xUKhWDSD8l6WIzaEvKxHpHY8N1EQedN1Zs5PNZqWL0a\nVkf7TfNW5MKyrM1m+yP9x201uYz4/UD1wQ5VfADNhdyFv5b9OqvhYvJG+dRRYXKHwWBI4Zop\niAJDQ0Mmk6m9vb3j1KngDTeMzrrS6YSmJiAIIAj4+c+zH3MK6LY2v8/XZzafOX265513ols3\nbMiTyWLpRFfEVeMXxS9TqVSC+IK5TKkEs7kAwxBDoVBUtIs2xFZ+85sTJkyYMOwhQl56aaHG\nlS4sy4bD4UJ+mkqEWNNyxLk19oFkDwIAIAiivr4+ne+3/OF0OpHPUVKLR70+klfrx0Jy8CC3\nahV78cXc1Kmaiy5qdrmocBimTmVin1d1dVo9j8hNds4cSNNNdqwSVzG+e3fFHXc0eTzU3Lm+\nZ57pAoiIPC5KOBzu6uriOM5slt5558DFF/sAQLCi+vRTdUVFRV49tpxOJ/L2Fpcmi4zT6bzv\nvvsuu+yyMrLeHBWCyzLi+PCXYUtLS+KtmET0ev3btW9vrtl8XHHcQTtOyU+tbViLlusXcAtA\nWNcRsZzMMrmvwEil0srKcwpOytT+ExEKhTo6OhwOh4pRbezZuL19+54Te+613YtuzcYeZR7M\nmwNz9KCXgrSRb7yCv2ILbDlAHtCBLkdjx+QYXMExHqFpGo4fN/X1oYxVYQWbXb+eevhhAOB5\nvre3VzCyNm7aJP/449TWlTzPDw4ODg0NMQxD8PzEFSvkBczyUCqVaKYNf/tbdFM+Dberq6ud\nBw50Wq3oYlQ4gfwjjxDr1uXvuHGg61QxcaEwfpkqlUoul4stmKsXLSq6pQV57BjLsh6PJxgM\n0seOVV99NUC0CkmKFkNXroSVK4s4wkR4nh8YGIhOFUopdrdY7N5dsWZNo99Pzp3ra23tpulJ\nKe6s0Wh8//hHr8WCvOKEj2fBSs9QgAWIzPGuucaddPt3vxspo19kj8djtVrXrDHElglYLJIt\nW2p4nnjgAYvwvL797SDACCUDwWDw4YeJHTvO1pyVWttOwaBpGgkESWN6R9Qm7HY7Wr1/5JHe\nn/xkIvL+vOOOgRdeqAEAmYyIRPL7i5DoWRgrTd54YxhAm9cBpCAYDPb29h48eLC1tVXYWC7W\nm+kjOAUg2traAIAkyQsuuAAEFzaMOARB7Kva9y7xbuJy/S8lvwQAoQYqaQ/Li54XCz/mbKir\nq3tZ+bKZMSf2XyyC9JriSgaz2Yy+P0eM+B0t0fITRDlJWOOXsrmcwuQWmqYnTpzIsizDMPRw\n8gL1pS+hPwYGBgR1I51cDJ7nu7u7UcgIQRDpCCL5YulSWLp0tA/iOC4UCtE0TbvdaXbiVFZW\n6vX6YDAYCoWKFV2hVCopihITF4x33FEAcYEgiIkTJ/JFXTAfEYqiUJwhvPtudFOhAucygOd5\nk8kkTJKF2N0ifJqKh1wuRzOlpNM8ZLSRApVKNXXqVPT9Jt23L7q1UC96bH04InaOt2CBK+n2\nW2/ly+UX2eVymc1mAEjag7Nvn/q6684+r8WLPakFjv7+fofDcebMWTfZ5583bt2qg3HZ3qJW\nq10ul8dDrV7duHevRiLhH3ywb9EiFwDIZDLpSH6twvfGrl3a7m4pAEQiBFI3AODwYdk77+Q3\nDiaxAiVWmiSI2jweOyXhcLizs5Nl2VAoFGu92dTUVKwh5Yk4J9Fjx44BQFNTk1wuT7wVk5TL\nicttYDsNp33gM/CGqfzUO8g7lpBRt4iKigqJRIIm0pDQw6LRaIo69lFDEMRe3d4dEN9/cRFc\n9DiUuv1nLAzDoBa/bmn308anjyuOD9ADDMHQQAOAgTXkJBEGUy6Ux+UUJk9QFKVUKuOSF3ie\nT2FdGWhuVibsx2q1otkIQRAlFRQ6IsFgsL+/3+/38zwPHDflnntUaS+VowwXhUJRrOgKkiQN\nBkPRm2WkUil34sSA0+n3+0OhkKqjo+5b3wIoLa+WKBnpXwXG5XIJs5S4TxNbW0uV8Kcph+j1\neq/Xm3Sal36NffT7bflyWL48z+ONP27sf+PmeBTFJ90ukUwu5CAzJhKJ9Pf3o7+TWsDyPNxx\nR1NMuU1Vir3Z7Xb0cxO7q5tvtiOBoxzbdrLEYDAcPRpcsWJCYkzvRReN/NkXxLW2tuSiUr7j\nYITPZmYVKPnDZrMh8SXOerO5udlisYwlmUOr1QqXcD6fz2QywXB/CkmSZTf9zoZgMLh+/foD\nBw4cOnQITX337dv3la98ZcQHxi/Xn7tiT5LkrqZdHYMdX3V/NbHkobxaVBDzYJ4VrEjQMYJx\nGky7DW5bAnmx/8wfQr7vXs3e97XvC9tZYAFgOjN9ojI3iTCYsgALHJh4wuEwukhKal3pnT49\nTuDgeV6wXEpTECkRfD5fV1eXULFpbG1VffJJeS2VV1VVDezff2Y4vyqx26gwkCRZXV0d/c/H\nH0f/KOEqiVJG8NdM/DT5Z8xQsmzc/HlMotVqjxwJ3HprZdw0j6bpSy6pL/boRkCtVqNG96Rz\nvKTbpVICra+WPm63W6iBT9prc+qUHCCtchuO49AkU2xXaYaGjCUkEskHH0zq7qYhWUzviMZK\nFEWhn+9nn+2CBBHt3/5t6ojVT1mi0Wg8Hk9SaRLFWuX16CkQVGNErPWm3+9nx9D3qlKp1Ov1\nJpMpEokcPnwYPc1Jkya5XK6GhoYiakyFhGXZgYGBTz755MmYGrActiO9L3l/R82O39b8Nnbj\nbH72k1RZlpydI+iUP1pW+3jv44/XPe4n/SgRBgD+Kf9ncUeFKTDj4psOMyrirCvjcjECzc2o\nD0W4fygUQg9JKoj4ZswoTYGD53mz2SyoG+VVeBJLdXW1Tqfz+/0Mwyg//BBtFLqNikA5VEmU\nMsifMumnKdDSQjFM1G5mrPPee7Xd3QDJpnl5rbHPnsrKSofDMTgIiXM8AEg699PrKwXjuhIH\nvT/jOHFC3tpqQH/HPi+apisqRG3qAoFAnF8AxLTtXHJJcNmy8hB9csvhw8kvzNIpvlCpVKi9\nNFFEUyjofKsbAKDX67/4wv2979UnVqDMmKErlojA83xc41ic9WY4HB4zAgcA1NfXz5w5EyVt\nI1pbW1tbW6+77rodO3YUcWCFIRQKdXZ2hsNht9sd2450/vnn5+rXM3nJQ34STzFpIkxMkAHH\ntwe/zQP/lv6tdQ3rAMBLxtsDYcY2WODAxCOVSgmC0L35pph1JdHXF2tdmVr9hSOcAAAgAElE\nQVQQ8c+YUfBnkBaBQCA07NGfuFQebGnJZZJenjk7i/jBD+AHPyj2cDBZQRBECnlRU4blr5nx\nxRfJt+e7xj57aJpm2SlLlpBxczyNRsPz0h/+UBG3XalUqtXlIadCzEWkwO7dFQ8+2BgIkACQ\n2FWRQriJ82uAcysOnn22j6bPy/Xwy4AMYnoFampq3G63200mimg1BWn4IQhiz56J3d0kJJMm\n/+3fCjCE5KMiSTJWTYuz3hxL6gYAdHZ2xqobAnPGR1llb28vMsiIa0c6//zzQ6HQiEY26TDG\nSh7GBnFVWhzB9Uh6/qr9K/qvltdic9BxBRY4MPGQJKlWq8WsKxu/970468rUgggtkxUgyyMD\nhHVIsaXyXAocTmea3qWYMUYoFPL5fAzD0P+fvTOPb6LO//97jtxH0yNND0qhLVrKKaKIiovf\nRV0P1gURrYviyq4c8lXXPVjBgrDqKuwqW/y6iouux/4QcUVdERVEkRu5FLkK9EzaJm2u5pxk\nMvP7Y9phSDJp2tztPB/9I50kk8/knM/r836/Xjgu93rlN9zQ69tAKpWK3nkn7KcJMEzS2Qkl\nJckafiqJZZqXcjZulIYtP7npJsjQshQWbisNt0yAoU9dFVy/w9CKA5VKWAvtM1KplCSHVVeL\ngkS07OxslSr70jjIRHH8eHhJK7XSpEKh4M5/uNabYrF4gFlvlpWVDdq0FI/HExqUy7YjWSyW\ngoKUOd0KJBTmdd+cs9mKWT/O/rhZ3My99q7AXcKUd1AhvNoCYSgsLIw+FwPDsAiCSPG8eSkP\nCg1L9zokz1K5b8yYWHZOUZTZbHa5XD6fT4Rhxb/+tXhwx3wOToxGY2dnZ/eJJkUNW7gQongb\nZGdnM9ULoZ+m0vnzU/5pOnwY3nkH9u6F5maw2aCoCO69F1atgnisig0c+MpPurrCb0//shQW\nxuSVJElur01pKXH+fBg9IvJxSaXS0F2xFQeDyg0xjrz/viK1Ilp6SpNarZYJeoMQ683k1LYI\nJAcv46HGgduOFHrtQOIcnKuBmiNwpB3aveAtgIKb4ebn4XktpMU7nKKo793frxKt+h7/3oSa\nCISI7wiZksBvVN98q/o26KqsQNbTxNPClHdQIbzaAmEQi8XEiRMNra2M7zRrXUk9/TS6IkzM\nUp8EkTSBWYfM/uijsEvlIqm034UnPp+vsbGR7X/JXrtW/M03meVdKhA7JpOJNVCEvqS9ajQa\n/5kzvPtN6fvH4/HU1NCff36xjbmpCV54AShq0MV5RiY953hxAUXRkpKS3bvbFiwYypYJjBvn\nQRCkoKCgrExWWsp730OHqLfeog4cwFpaEJsNioqQGTOG33hj86OPloa6ycrlufn5STywgULm\n9nYlFLlcXlxcfPbsWZ/Px7XexHE8E2MvBPgItfXhtiMN4MIWj8ez0bdxU9Ymdose9G/AG2Yw\nfwQfpXBgDC6Xq6WlZVPWpo8KLw4mviMUiUSBQOBK95Vm3NwibvGgHlVAZcNtFFBu1J2H5/W+\nC4EBBDKAP+3x4rXXXluwYIHD4VAqlakeS7IhSdLr9YrffVe8cCEAwGefAZMACsFtF74lS1pb\nW5mw2F4FkTShoaEh+7e/1Xz8cfir+7tUfuHCBTatSr19+9AnnmC9S6G4GPT6fg9YIFMIBAJn\nzpzhWthy3wZUYSHa2hp5DzabzWKxeL1eyZkz5XfdBQD0ypXI8uUJH3rEIRkMhrvvLr/hBse0\nafbs7MD69VomGXTcODguOJQPJv70p8ALL4SxLeArE/B6va2trfPn5+3YEew5OnEiefhwmLWW\nDGrbEcgUcnNz2dA3lkFivTlIcDqdjY2N3C133HFHU1NTRUXFli1bsrOzi9OvYzp2Ojo6TCbT\nJ1mfiGjRSO9IWUD2Yc6H/5f/fwBQBVUn4WRqh+f1euvr6wOBwKfZnyZuhCaTyWQycbdQCDW1\ncqoVs2YHsi1Y8AdfIHZ8Pp9EItm7d++1116b6rEEkxm27QKpAsdxpVIpXrAAaBpouuu66wwG\nQ0NDQ3Njo++uu7j19mKxeNiwYVVVVeXl5aWdncxm9OqrUzb0KCguLpbFu/DE6XSy6gbXu1R6\n7hzE3PkikCm4XC5W3Qh9GxCjR/e6B41GU1ZWVlVVVdaTGotcdVXiBtwrPp+PSR3asKFh8WJj\nZaVXp/PPmWNmrhVKvAcb338f3pQxbJmAx+Opr693u90Gg3j+/I7Nm8/v2HF29uzu082zZ/sf\nGiIgED319fWh6gYAfPbZZwiC7N+/P/lDShwkSXZ0dDz66KPXX3+9UqlEEGTgHWNYFAoF11El\nqB1Jo9GkbGQJw+FwGI1Gmqan26b/zP6zUqI0n8z/addPmWuvgWt63cM5OHcv3DsCRqhAJQJR\nCZTMg3kd0NHrHaPEaDRSFIUgSL9HGA25ubkfaj9cr11/SnbKjJvrpHU1xTVWzAoAM6mZcXkI\ngQxCEDgEooKiqMbGxubmZqvV6nK5ZM88w7RddF/dowWgKCqTyUTz5zOCyMVyj1AsFigrAwQB\nBIE//znW8fVrbw6H4/yWLT+eOPHjiRMd8+Zdcl17O9x4Yz8GwqobYb1LvVHMbDOJ+L6IA2Mk\nAADARhLyWdhGvytk3rzeP02Jx2azMZKNSnUx+eLAge6itunTw0SHCgxgtm3rflcG/U2ZEubG\nBoOBqRsPq45NmtSHXQ1s9u/3L1zomzCBys8HsRiGDYM//Ql6mh0FYoWx3gwEAnq9/t///jf3\nKgzDxo4dm6qBxR2j0Xj27Nnt27evW7du7969TK8xhmHjxo1L9dASDoIgRUVFCII4HA6r1Xr4\n8GG2HQkAmHSVgQGrShQoCsaPGj/t8mnLi5dbcSuFUE3iphcLXgQAHalbAb2UUXs8nve63tsE\nm87DeSc4SSCZzpHfwG/iMk6appnibhbuCPP9+TVUfHz3MQw7mH9wnW7dPeX3TK2celfFXZ9o\nPgGAMYExfxX9NS4PIZBBRPLgoChq06ZNu3btkkgk06dPnzZtWtAN/va3v23fvv3zzz9P5AgF\n0gK2/QQA1Nu3azdsuKTtQhdVxiFJkna73ev1Bvz+gl/9KkbfTZIku7q6vF4vHQjkz50r6uPe\n7HZ7W1sbc5k5Im9lpfTMGQCgc3OR6I4olMihudEs3aczgUDAbDa73e6Um6fSNM1obcxIYn87\nxZfu0EGetwGZgWeZHo+Hpmlus/qZM9J16/IB4Ior3LNnuwEkqRudQPpCEARr7BdWHZs1KwWj\nSjc8Ho/BYFi+PJ/bwiMY3CQCg8Fgt9t9Pt+iRYvGjBmzYsUKk8k0fPhwq9WqUChSPbo40N7e\n3tnZCQBBx1hRUSGXy3u9+wBApVINHz68uLiYm5VbW1tbW1s7YNqRSJL8wPfBJvkmgO6laqPI\nuCV7y5dZX7pQF3Obse6x65rXFZYXAn9GENPbgmfha+xrgjpHzsG5uAyVoiiuGcLMipnnpOe4\nIywqK4I4mZTfgNxgAtMFuOACl5bSltPlc9G592H3SUHI5Bp08AocgUDgzjvvZL8IamtrZ86c\n+eabb6rVF399T5w48cUXXyR8jAKpxu/3s78T3Hr7ymnTAIAYPTqamY3D4dDr9YzLsa5/vpsc\n1w9i2bIL993HqAm6tWtFX3/d170ZjcagI2p4/fXKadMQgvBOmNDvjFgmYp3PuzRtQ3OjgSCI\nxsZGdgEkheapJEk2NjaysyZ1+tm4KhSKCNnJUoUiE98GXHVjxw71smVD3G70qqtctbVNOC64\nQQ5wmPScPXvoHn/QaNNz2EBuLqw6NmGCZ968eEZyZyIej6ehoYGiKKaFJ8jg5ssvBYEjbng8\nHrvdDgATJ06cOHGi0+lkrKBHjRpls9lyc3Nlsox8N7rd7pqamu++++7o0aNMvca7774bdIyV\nlZVOp3OQeMm1t7dz1Q2WiRMnJn8wccdutxsMBlDDGnOwKuFFvDiNkwgJAD/If1irWzuJmsS3\nn66uLuZMeLptOrvxp10/ZXYVr84RFEURpNvw0Yf4OkQdQSPcjG2OywMBwBJYsgSW9DxwvPYq\nkJHwChyvv/761q1bdTrdb3/7W7Va/a9//evDDz9samrasWPHgOxhE4gAGyoett7eXVXVq8BB\nEERzczPzBRdUAEIVFqL85RIul8vlchEEIcKwvPvvx3tW6duLixl1o097Y/H5fEzKSdgjco0c\n2e9zHJVKhaIoX2hudnV1ymM++wdN083Nzay60e8qnrjQ0tJyUd1I6Uj4wHE8NzdXyvM2UM+e\nDStXwr/+xXr0Qk18SjQTh7hnIktR8MoruvXrtTQNs2ZZli1rw3FaIhHKNwYyNptt2TL8yy+V\nAN0iV/TFBSgafJrJVcdee60dx8sTMeYMorW1lW3hYYtc5swxMwKHYHATR7ouDWo+deoUc1pS\nVVUFAA6HI+MEDr/f39bWtmfPnhdffJHdyCSGMJe5xzh4zPKZdqRUjyIhuFwuvV7POG6wG1lV\n4ue2n680rDysOLxw2EICIT7VfCqiees3uEFvAEAhVIuo5WJvCx6fiAAEQRQKBVMDLqbFu0/v\npoHmjhBDwjs6CQjEAq/A9fbbb+M4vmvXriVLlixcuHD//v3Lly8/cuTILbfcEvQLITDgCWq7\n0D/7LFFWxtpzeqMwFOjs7GR+bEINF12VlWF/hyiKam5ubmhoMJlMdrsdW74c37mT7jn58FRV\nhd2be+TIaI6o2yKB54hclZXR7CQsOI7rdDrpSX5H6KTUF/j9fofDYbPZvK2tcTGncDqd7Eps\n6NPuT2L3stvtZlaowo4kfWxcdTqd8uxZvmupjz+GdOqp6RVG13Y4sMWLS197TYvj9IoVhhUr\nWnGcxnF8YJR2C4Slvb1dr9e3tOCh/qBfftn73aXSi7XBFAUvv6x74omhbjc6a5Zl/fpGrVZy\n+DA89hhMnAiD03jC5/Oxzk1CC0+iYd2RGE71/OgzDpRB16Y/fr+/vr6+q6uL6Ub5xz/+kZ+f\nDwBlZWXs5457jAPJgWLQYjKZuOfMQX4WC00LEUCudF8pp+QAoKSUoRIzA03T7DcPAMysmDlu\n1Lg7Lrtjj3LPWPfY98+/X0QWxWvM+fn53ApQ7gjVEJyrJSAQF3grOH788cfrrruO1YBRFF25\ncqVWq/3f//3f22677YsvvhDOaAcPjCU1X9sFJhb3Wm/PzEj5DBdxrzd02aS1tZWV0oJW6Umd\njszN5S0n8fu5HtphwXE8whGhIlEsHQS5ubm2775rbG9nzpbY0NzkxHySJHnxqaOoYQsXxmUi\nHbmKxzt6dC/PePzodSRx6uWMFQRB8NOnvV6v0+n0er3UsWNDf/5zADAtXox4PNoNG2iZDGFO\nLzJB4JDJZE5nQXW1uqlJjOP0b3/bftllxA8/yAFg/Hgd3ymUQKbjcrmYfv5+FxfgOJ6VlWW3\n2x0ObMmSIbt3q0QieunS1lmzrACgUCiefprYuvViBdBgM57whdNyhBaeBNHtjtTD6dOngVPv\nEHRt+mM0GhnNIrTjhr0N9xgz7gBTyI/EjyvQFcex4ybU5AVvARTcDDc/D89rIZUlVRRFsQs8\ncKmfRYG/YJVhlZgW10nr3sp7i0kPmQW8+mj3yiUA8HSOXEPFp0UFAORy+fay7Q2Ohusc1+n8\nOjNuZkd4N3J3vB4lds7BuRqoOQJH2qE9fV50gf7BK3D4fD5GCeayePFir9f7hz/8Yfr06QPD\np0cgGuRyOYZhfG0X+Q880GvbRSAQ4DNc9FRVyQOBoNsTBBHB9cNTVRVhb+pAoFeBQywWSyQS\nviMq+c1vYmwk0Wg0WVlZTCOMdO9eZmMSYj6ZsBu2fUNXW6vcty8u5hTdKwb85qmqGIceNZFt\nXL2jRqXVcoBUKpVKpS0tLdjx48wWhCDSsKcmGv7737ymJgAAkkRWry5kt69dC1VVKRuVQEKx\n9qQUhy0uuPNOP0Twr+uhqKioro6aN68wSB2TSCStrab6+lLWeOKf/9S9954GBpPxROQWnn/8\noxXHR6RkYAMShULR2RNjDwAnT54ETr1DZq3bURQVueOGgXuMmXWAqYIgCL1e/6783Q8LP2Q3\nMsEiZjB/BB8leTwul4sxWDl27BhrsDJu3LggVaJd1P7wsIe5dxxLjV2N8X6NYhiGYRjjixe2\ncwSnI8VQ9JUdsh1bZVtr82u5G8fD+L/AX+L4KLHg8/k2EZs2qTaxW1L4ogvEDu/bt6SkRK/X\nh27//e9/73Q6V65cOXPmzOzs7ESOTSBdQFG0sLAwlrYLHMdVmzeHLZegUVTc0QGX9oWyEjVf\n0Qdf8QWNorjRCKWlvR6UTqcTJ7KRBEEQiUQikUhg/nyYPz/GvUWJxWLhM6cgdTo8hol0+pin\nRq4nSkMbV6/Xa7fbYcYM64wZkvr68upqrlrnHzs2acUvMXL0aPjtEyYkdxwCSSTUIpSbnnPv\nvT6A3j25MAzbubM0rDq2ZEkbtzbkvvs6GIFj8BhPSKVSFEUZ3TbU4CYnJ2nS8aBApVLJ5XKm\nDNDlcjU3N0NPvYNCocgsfwqSJLmL8BDScQOXHqNEIsnKykryIDMOv9/f0NBAkmSWJGtNS6KC\nRaIkEAi0tbXt3r37pZdeYjeyBUdcVeLhYQ8zMocYxFpKWwEVc9G51Wh15PQQtVrNStjA6Rwh\nMEJFq+JbmDkFphjByOSb6EBXARX3w/3V0MsIkwbj24qpsTW2FL/oAvGCV+AYP378J598Yrfb\nQ78Qn3766a6urpdeekmodhs8aDQa23ffNbS1MXIv23ZBPf00uqJ3IyKVSiU9ehTClUsMW7Ag\ntFyCeRS+VXrfmDHKzz6Lfm9hUavV1kOH6tvamFME9oj8NTWiVat6vXt6wq7nhC17EXm93H74\nPqFSqRAE4at50dx7b9LMUyOPJOe++9LNxpXNV055d0+MbNsWxY04UUcZYZ4q0CdC0nOibdLm\nU8eqqryD3HgCRdHs7Gyz2Ry2hScvLy/VAxxoDB069MSJE06n84cffmDqHUpLSwmCKCgoSPXQ\n+gbX1ICB243icDhIkmSPsby8XKFQ2O12ISUgMiaTiWkuTmiwSDTQNN3U1OR2uwmC4Mb9cg1W\noEeVUFEqK2bNDmRbMEv06SH5+fn/xP5pRszXO68P6hy5i74rvodzSb5JmuH1eiP4tibzRReI\nI7wCx4wZMz744IONGzcuWLAg9NoXX3zR6XS+/vrriRybQHqh0WjUarXX6yUIQrprF7MRvfrq\naO6r1WrJnrWFMISUS0T2yBDLZPIffoh+b3xkZ2erVCrGIkHeM08WTZ4c5d3TEKYdl6/sBfz+\nfgscIpEoheapJEm63W6fzycSiWQeT+Vtt2HNzSkZST+IrNZ5R43K9CVan8/X1dVFEARC09oH\nHhBllHkq9ASg7t0Lzc3QpwDUwYBUKmWM6MKm50T/fRKkjjmdzsbGxqDbsLUhV17pnTcvLZb1\nkkNBQUFdHTV3rjaohSc3N1elkkdRj5ippOSjh+P4tGnTLBYLu6W2tra2tva2227LrM5rkUiE\n4zjXGJXbjTJt2jQmEJfhxRdffPHFFzPuGJNPUNcPN1gk359fg9UkLXzUarUypUahBiubczZb\nMWvsqoRIJDqYf3Abum2dbh13e+TelriQVm4XZrM5yLc1VS+6QBzhFTimT5/+0ksvhdpwsLz6\n6qsjRowwm82JGZhAGhCyGIuiqFwul8vl8Mgj8Mgj0e8JwzDyxIkLej1zrsyWS3iXLpU++2zo\n7ZVKZYRV+vwHHvA/91zdXXcx9mzs3ohlyyTPPNOnQ8RxvHtB4/HH4fHH+3TfvuLxeNhZutLn\nk15/fdwXulEUjeBOooit4DAvL8966FCj0RhknprohXqTydTR0dH980NRwxYuVPaoG02vvOKY\nMiXJNq59JaGOtinHbDa3t7czr45u7VrR11/HxfMlOQQCgc7OzqeeUn7xxcXW9MFmchmZ7Oxs\nq9UatrhAJpP1WzANhPgucWtDXn21DccrYh165oAgyJdfFvMZ3Dz2WMoGllDsdvtTT+HJ/+jV\n19dz1Q2WiRMnJvBRE0NOTo7JZGIuc7tR9Ho9V91gycRjTARer3fFihUHDx48fPgw0xC9b9++\nyZMnUxTF/WriWniOdY9d17yuqKwIkiV8B72CXIOVb1TffKv6NlSVWIOv6dNDnINzBtQgAYkP\nfDTQGGBaWvsk8uTD6MMJ7Rzx+/3vE+9vUqaL2wVrXQ/hXnRtiRYE75oMhFfgUKlUj0ec76Eo\n+oc//CEBQxJIJexiLAqQd//9cVyMlUgk5eXlbrfb6/WKd+5kNkqvvz7sjUUiUW5uroy/TEN0\nzTUjRoxwuVxer1fy1VfdD3HddWFumgY18xRF6fX6iysD8Qs3CUIul8vfey/sRBowTGaxQGwe\nY9nZ2RqNJsg8FXrOmSiKYtJkEQSRut3KqVNjf9qNRiM3qj3INjX/1luzNBrZ7t3MtUmwce0H\nTF83n1qX+8tfpltPTfTY7fa2tjbmcpDnC11UhKS3eSrTa+3z+Zqbs1iTy/XrtUw+yOAxuYyM\nXC53uQqrq1VBxQUYhk2YUNj7/XlgVD+G0NqQrCx5PMaeSQw2g5v29vbOzs7m5orkf/TKysrC\nJtNnIlqt1uPxtLa2crtRSktLi4uLrVar0I0SFpIkd+3atZrzJsMwTDFecS/cexg53FbVRiBE\nHpl3jfMak8gUFCzyPvp+0sYZlOnLNVjxur0OuaMBbXCBS0tpy+nyB7EH70Pv65MqQZLk+973\nf1BePM0OQKAdad8JOx+FR+NxBOFh3C5QNbrGmi5uF6yqFTZN5i3qrZSMSiBGkAHzRZ84Xnvt\ntQULFjgcjswyoOoHHR0dbMK2bu1a7YYNFxdj29uTkPVA07TNZnM4HARBYAhS8KtfyffsYa5q\nXbq06LnnIDrXD4/HYzabvV4v6fOVLljAToBh61a47bZEHkF4mpubuXWPiXtuCYLw3HOP5uOP\nw1+dyIm0w+EwGAzd5bJMncW+fd3X9fdp9/v9dXV17HeUevv2oU88cckU2mCIz+gTjMFgyJ06\nVXr+fPird+6EG29M6AD8fr/X66VpWup2i6+9Nl5637lz5xgTSsY81a/V1r/7buW0aQhBeG66\nSfbll3EZfIJoaGhg1u4cDoy1gWhokPz85yMAYNo02L49lcNLH5Ytg+eeC7M9luICmqbPnDkT\nCATC1oYUFBQI3hMDGLZBqdePHrPSfuDAgSNHjnBX2mMfg9frXb58+aFDh4LW8GPfc/LJycnh\nWkUyCN0oYenq6tLr9YcOHfruu+9YV4vLL7989t7Zf879c9CNb+y68e/Nf2eDRUS0yIeECXVO\nEOfPn2c94wFgyZIln332GYqiBw8elEqlw4YNi2VK4na7m5qaPlJ9JKJFQSpDFVSdBP5m5Njw\neDz19fVBE89z0nMzK2YCwEPw0AbYkKCHjkB9fT23iIObJiOiRV1El1QqTauemvTB5/NJJJK9\ne/dee+21qR5LMPEMARLIaKxWq9FoZC6nZDGWiThlv2V0a9fK9+xhVQBNT+lBr64fFoulra2N\nlWlku3entmbe7XZz1Y2EPrcSiQSvq+O9OmGHzxTHsj9a8YqndTgc7D5DbVOJ0aNT2abfl8qg\noqKi1m++Yc9B++rRGws+n6+1tbXb6JSihi1cKI5T6RBJkoy6EdbzxVlZKYtt5HGBr8mfpgk2\nqmmQm1z2SiKKCxAE0el0Bw50Llo0LKg2BMdxhSJH0DcGMGyHSNiP3owZAQAMAGw221dffRW0\n0j5u3LgYHz0QCBiNxj179qxZc7GePy57Tgn19fWh6gYI3Sjh8Hq9LS0tNE0HuVpUVlbKPLLQ\nzJRmSTM3WEQNSc2gl8vlXIGDa7CCIIhM1v8f2EAg0NTUFAgEku+p2dnZyed2oSN1y7HlEOyc\nmwzUajVX4OC+6EpKKZVKLRbLO/Q7m3LTpadGIBoEgUOgG1bdSNVMsrW1lf2KCVUB5EuXwtKl\nve7E6/Wy6kbQTgIFBVgqaubZEA0I99z6xoyRxPXhsFOnfD6f1Wr1er3Yjz8OueMOgIT35hiN\nRm6dRbyedrZEM+wUmhgzJskCh8vlYoJ46UCg5OGHZVErBQiCFBcX5+bmMo7oqp7Ckyg9evsN\nSZINDQ3s0xgv4YndOQCveap75EiapkN9/pOJx+OpqaE///xivwPb5L90qSf09pllcrlvn+/t\nt+kDBzCDAbPbkcQZNEaVntN3cnJyduyQNTWJYTAZTwhAb9nD1dUBAFVbW5vZbHY4HEH5ETGG\n97GNaW63m7vniooKuTwjG6MGUsdNorlo5gUAl7pacKf6AaRbd7vce3mdtI618LwbuTuZo83N\nzbVarcwIgyKNNRpNLB8Ei8XCtRoJ8tR8kn4ycT4j7LoChHO70A3TpSQ0Nicn51X6VRNlCvVt\nnUnNbDY0d3V1yTRhJDAhQTadEQQOAQAAr9fLTFf4FmMT/Z1DkiTrqBSLwsKaIYfuxD1ypNTv\nF4mSHcrJmpzzzdLjK3AAgFgs1jGaQo87CSRyMScQCLDKVNinvd8pId0/4TxTaDK5C25cN5D+\nVQZJpdJuU8ZHH4VHE9jjymIymVh1I+56X2TzVATHkdbWFJqn2mw2g8HQ1FQetsn/ySeDpwRc\nk8vXXzfh+PBUjDoqSJLU6/UrVuTs2HFxRTETvVFPnw6/CDlQjScEGIIm5CHZwyVOp5MxsA9a\naa+qqtLr9eXl5f1+6La2NsabPHQN32azCaYVAxvuKj1c6mrBXGCm+u9ndxttfJb12WdZnzGX\nx8P4v8BfkjVSAACJRDJkyJDTp0/7/X6uwYrf74+lfAMA3G43u/wQqjLk6nITJ3BQFMVcCOt2\n8S71bqIeOCIoiu7P2/8Z8lmob+uTXU8yJdhCgmzGIUTfCACEJFnqn32WKCuT9Xz7u0eO7PMe\nLRYoKwMEAQSBPwd3Nobi8XiYr++wKoC7qirKh2X2w5eTymS4JJluOyC/Q+AAACAASURBVD2e\n5zYwfnwCH/uhh4Cmgabh1lsT9yCRFRzPqFHsT1pfUSgUwD+FlsjlkCwPjq6uLlbdYJUCoqIC\nAKjCwiTY0/QPtjeKKzxJz50DAPfIkf1+XRhwHJfJZKx5asXdd1fcfbemp+t76MMPw8aNsQ2/\n//j9/tbWVpqmN2xoWLzYWFnp1en8c+Z0Z35ptSCRXNQVKQpefln3xBND3W501izL+vWNubnJ\nlkGjh6bpxsZGp9NpMIjnz+/YvPn8jh1nZ8/urvlPb9uTYLZt6/5+CvqbMiXVIxNIJOynL/Sj\np1RSEokkqOeCu9Lu8XhCC0CihCRJh8PBt+ewjR4CA4mg8KbTp08DAIqil19+OQDMrJg5btS4\nOy67w4JbZJRMFVCJQVwCJTfCjW/AG/thvwaSrX9lZWXdfvvtN9xww+LFi5kttbW1EyZMuP/+\n+2PZLUVRjLrBqgzMdkZliPHEIDKsvbSYFu8+vfvoyaNvNLwhoSUA8KnmU675dJK5AblhIkzM\nhmwxiIup4p9QP9lAbziIHgyYL3nPUAjVJG5ie2pWQGJbjAViQRA4BAB6W4zFxOJoZpJ+v7+j\no6OlpaW5sdF31119SgnpXtLhUQE8UQscNE0jAHw7SVwlp9/vdzqdLpcr0NERpOwwRlB8z61U\noUjaLD1BBNVZhD7taH/jaWUymVKp5JtCK2fNStoUms3DDlUKXJWV6VkhTFFUhLIsz6hRQSbt\n/aCgoIB9ocPA/eD3UfGMEavVypylhW3yv+MOr0wmE4vFAOBwYIsXl772mhbH6RUrDCtWtOI4\nnc4LuUz3GQCE1W7y8tLxrSjQD/bt882f7x03zq/V0mIxDBsGf/oT+JJncdh/Dh+Gxx6DiRMh\nPx/Cjjw7Oxt4PnoKhUIsFgdJGEEr7Vxjgj5BEETQdzV3z/3WTQQyhaACXq6rRdBU34N6pnVN\nM3QamqF5J+z8FfwqobGpfCQo0ph9HsKqDAktc1arL/ExYd0uAEBJKcVxb7CMmiWw5Dv4zgIW\nAgg9qv8G/eYh5CExJfZxvrlYCWyPcs9Y99j3z79fRBalasACvSK0qAgAAEgkErFYzJdkWTxv\nXq8BHEzyEzOp0K1dK/7mmz4V8DNfqXwqAC6RgMEQTbm7WCzmy0mlUVTS2QlZWb3upE94vV6D\nwdBdG8Kkh1yq7MjlcpVKxffcymbMyNyUUAYcx6VSqWzjxrBPu0ypjPK1C0tJSUng9Gneq5Pl\nGsu8vnxKgdjn41YEpAkoiiIIQlNU2AYfT1VVdn+FJxaFQtF1+HC9wcCsjLHmqf6aGvqppzo6\nOlx1dX6/X4RhQ+fPlyYgF5mP0CkQt8l/9mwXgkiLi4u//bZ14cLSIJNLlUqlUqVvZBa7BB1W\nu5k+nYCUNDELxA8mVnzFCk0mtiDRNL1qFfnf/16cI4WOXK1W22x5c+bkRJk9HLTSHkcSt2eB\nNESlUrEyVpCrBTPV58ZnfKr5VOJM8c96ggxW1Gq1zWZj/w3y1FT0OPonAq1Wux5Z3wmdoW4X\nd9F3Je5xYydsT81kOiNzlwYJUQkcNpvttddeO378uF6vD130O9AzkxHIaAoKCsQn+aOhIs5J\nPB6PXq8Pa+1JFRaiURTwMwuqfCqAbu7cKFWArKws4NnJsAUL4i4leL3e+vp6tqKPz8SxpKQk\ncOYM715Ske0SX/Lz8ymepz3/gQdiedoxDEPPnOlyOFwul8/nk9XV5d98M0DCbVODoGmazwrE\nU1WlSmRJZyzIZDLJv//NV5YlMpli98hQq9VKpdLpdBIEIdu5k9kYuOKK+vPn2c9F9l//Kv32\n2xSGGYU0+WsBQKFQfPPN8KYmHDLK5DL0J5ir3dx3nyBwZDwtLS0Oh8NgyA9rH7N6NRw+DG+9\nRe3fjwZlA6Vu+bMbn8/X3Nx8/vyQ+fNtYUfOsm1bQVMTAM9HTyqV8uVHAEC3k1HfkUgkCIJw\nZ4zcPaehSC0QX/Ly8ux2u8ViIUmS62phtVpxHFepVEFT/fTVuWNDrVZ/UvhJO9keqjL8gvwF\nJonJxzcyGIYd0B7Yhm4LdbtYg6/hu1eqQFEUx3GmEjasBIYLVQJpTO+vzeHDh2+66Sau2icw\nIFGr1dZDh+rb2phpCbsYSy5fjq9cGfm+rDd1WI/JKH8iiouL8SjL3fnRaDT+JEoJbT1PF0RU\ndlAURc+ccbvdLpfL7/fL6uqyb7wRINmz9MShVqsDCYunRRBErVZ3VzYmxTY1lAiVQYBh4o4O\nGDo0meOJkry8PD7hacivfx0vvQ9F0e5X59FH4dFHKYpqOneO4jE3heLiJFiWcJv8X3lFt369\nlqZh1izLsmVtOE6z1/7wQ/hfwHQ2uQwyzw/SbiSSIakamEBccDqdTJHOhg0NbJHOnDlmRibI\nyQno9W1Ll2Zt337RuzlNijtomm5qaiIIIuzItdpLbhw5ezg7O5s95wxaaZfL5f1WIphJLOtM\nFLRnpnFGYACD4/jw4cOvueYa7oymlqit/aL2CuMVL/3upaCCgr6mgJ2DczVQcwSOtEO7F7wF\nUHAz3Pw8PK8Fbe93Ti57c/aGemqOCYx5UfJioh/6J+hPOqDjPH3ejbi1lLacLp+Lzr0PvS8l\nTUC9kp2dzfqvwaXVLipahaCpzIkTiEzvAsfjjz9us9meeuqpBx98sLi4OIUeMAKJJjs7W61W\ndy/G9swk8Wt6dwlmvKn5/EFFBBHNGYlCoXAdO9bc2srUELIKC71yJbJ8efRHgZ8+3W40WiwW\niqK4NfOiVaui30k0kCTJRl6FKjueqqqgOj+5XN4dRPf5592bBlBSPRNP29XVRRCE+PRp7U03\nASRAwXnoIXjooXjuMDqysrLEPEpB6fz5adtkpFarybNnea9OTCWFw+FgqwzC5iInYZlZo9F0\ndnZ2daFLlgzZvVslEtEPPtjpdqO//GVZe7vY4cCYRe+PP079ondfUSgUzNdOqHYjEkGGRl0K\nsLCx4mFbkG64wWSz2fT6PL7ijhRit9uZ3+6wI7/zTj/Axb6VyNnDCoUiLy+voaEhaKXd4XDk\n5+fHMsjCwkKv12s2m4P2nFBjRYH0Qa/XB6/XTge4HY7Bsakwld02jh7X14KCrq6ud8l3N+Vs\nuvhYoH8D3jCD+SPorvcMBAInvCdWiVZ9j39vQk0pFEFuQG4wgekCXHCB66LKgCVDZVgCS5bA\nEmCUgbT3gdRqta+jr4dNkE1ybLBAX4mqguOOO+74c+Jt4QTSAQzDshiXiscei75KOxAIRCjg\nV17qXB0BhUIxYsQIn89HEIRkzx5mI3LVVX06BARBCgoKdDodQRDI7t3MRtHk+HfKseZDYZUd\n18iRvI2MKZqlJxqxWJyXlwcA8MUX3ZsGioKTl5dHpsQKxGKBiRO7/Xr7pRbhp097PB7GmVJ0\n6lTJ9On93lWUsLXlYT8X3tGjkyApSCQSl6uwulrJNvl//bX6u+8ufiL7uuh9+DC88w7s3Qsp\nbwrIzc01m802G7DazdKlrbNmWQEgJyd3kKxApM/LEXfYUCoWbgvSjBkW4Cnu0KZ6kZiVZli4\nI7/7bg9AbvR7KygoCEo2qa2tra2tve2227b2+Ez3A5FIVF5ePmnSpEvW8OOxZ4GMINTV4gV4\n4QP4gJnq60BXARX3w/3VSHWfpvqtra0Wi0Wqka5xrRnpHSkLyD7M+ZBJEj0H3SGsJpOps7Pz\nvez3thRuYe8YKoIkh26VgSF+KkMG1bBE5pT/VA3UHMeOm1CTS+uigQ6qdhlHj3seeT5VwxOI\nht5PhtRq9dC0rL4WSB9EIpFi0yY+a09xRweUlgbdxeVyeb1eiqKkbrfqxhu5UzixWCwWi2HB\nAliwoN9DQhBEKpXCwoWwcGG/dxKZ7nAQHmWHGD06QY+bAQw4BQdFUezUKX1bm91up2m6Tw1c\nfcXhcHR1dXm9XhSgaN48SczenDKZTCaTAQB88033pkQKT0GJSEGfC//YsYl7aC4ff5wTtsl/\n0SJTZyfep0VvgiCWL6e2bZOxW1LYFIBhWCAwvLoaDTJoVCgUSmVMi9uZgsfjqamhP//8Yq1K\nmvRoxIXILUgYRgNPicSsWckcZhiCMjiDRo4geX3aW319fdjc1hjzIwCgqakpbM917HsWyEQu\nmer3C6vVymSdTLdNZzf+tOunjMBxDVwDACaTyWQyAUBWIGtNC68IktE4HI6N1MZNWZFqWNIf\nkiQNBsPb4rc/LPww6CoU0CK6aAQyoh8SmEDy6V3guOWWW/bv30/TdF+70QQGDyqVSha1tSdB\nEC0tLd3LvBQ1bOHCPgXKpg8SiQTDMPUHH4RVdiRyeSzpIQLpBoZhQ4YMKSoqIggC7akMiqaB\nK3pomjYYDOzJt27tWsmuXfH05kyK8MT0o8WeiMRAURSKov2oZOFr8p80yZWdTUa/6O10Opub\nmxsby9KnKWDjRmkEg8aBTVdXV0tLS1NTefq8HPFFqVQyodRh7WOCbsyWSEyY4Jk3TxZmd/Hj\nwAHyrbeoAwcwvR6125HQqhm2eijsyPtaW5Sg/IiE7llgcBKU5EohVIuo5cWCFwFAR+qWY8v9\npJ/1cYgggmQuNE3r9Xq73S7SiNZ0dcs3W3K3vKx9GTJKvqEoqqGhgSCILE0YHaoSKk8i/FEM\nAmlG7z85zz///KRJk37/+98/88wz3cuAAgKXotVqA9H5g5IkyTTWMv/yxY5kBAiC5OTkSGJL\nDyFJkiAIFEUlLhd69dWxNCMIJAEURWUyWYIqgzo7O1l1I8ibky4qQhLvzRkX1Gp1W1sbXyJS\n1j33RPO5CAQCJpPJ4XD4fD4UYNjChfI+yqDbtoHdbm9paQm9auPG7lL5Xhe9A4GAXq+nKCps\nU0BeHg2QAt0/skHjAIYkSSauK61ejviiUqnkcrnRSIS2IAXBLZF47bV2HC9P0JACgUBbW1tN\njTpybK1KpbJarQ4HFjpyBEEGaiCFwCCHpmlu4s/MipnnpN3z+bHuseua1xWWFTpdziBNjSuC\nFAQKVmArkjnmuGMymex2O1wq3/yP/X8YgSOD5Buz2cwYCQ1IHWqw0bvAUVxc/PXXX1999dVv\nvvnmqFGjug0aOHz66aeJGZtAxoDjeODEiQt6vcfjAY4/KLFsmeSZZ7i37OzsZNWNoCkcqdPh\nUUzhCILweDx+v1/icqluvBFpbARImSKQn59PRghtuewyKCvj0yw8Hk9rayvzjDGVLMrMrGQR\niAs0TXd2djKXQ705vaNGZYq6jGFYUVGRtL+Z0wDg9/sbGhpYjxvt2rXyPXv6IYMyZypBsIve\nV13lmzevF8+Grq4u5vsqbFPA7bd7AFJg6hnZoHEAY7fbGTPIhL4cKTf48PtL77uPamwUcVuQ\nJBKJSuUsKvJDuBKJnBxVr7vtH0w2itvtNhh6cTZVq9WdnZoHH8wPap4CgBEjlOIB4I8iIBAO\nVrzwIb4OUQdO4yRCAsAP8h/W6tZupDcGZXsHiSBvWt4cOiSDfQAoimKKzi5uyVj5ho1YYuAe\nSL4//0/Un0DIks4cehc46urqpk6dyqwr7unxfRQQCEIikZSXl3s8Hq/XK9q5s3vjddcF3YwJ\nwAOe2BGR1xsh4j4QCLS2tjI6MaMIdKsbkDJFAEEQ0ZkzdrudSQ8RnTpVeuedAGBavNj0m98M\nX7RIwaNZuN3uxsZG1rk9oytZBOICQRBME3tYb05nZWWmCBwAoNFonMeOXTAagxTPS2Q+/q6T\n1tZWVt2IpZIltBCdu+j99tt2HC+KvIdu/ZED1zdx1ix7SgSOQQt3pZThUhvLrthfDrfbXVMD\nqTX4ePttrLERg5AWpJoa8+zZbWFLJHJychI0GLvdzqSkReNs+uWXxU1NSOjIX3qJ7qNXuIBA\neLxe74oVKw4ePHj48GEmT2rfvn2TE+AiHyUIgohEIkbCENPi3ad300AfVhxeOGwhgRCfaj4V\n0aJuvzYACCeCrKZXvwfvpWr8scO46bH/Bsk3r7a9OrQ8Y+QbrhQVWoxTUFwgCBwZRO/muY89\n9lhbW9v8+fP37t3b0NDQEkISRimQKchksuzsbOWjjwJNA03DrbcG3YBZDg07hfOMGhVqIM+l\nubm5W93gKALd16VUEcjKyiopKcnLy8O//57Z4hk1Sldbq9i7l2+Era2t7E8CO4UjKioAgCos\nhAxpRhiE+P3+rq4ui8Xi1uuhrAwQBBAE4hEy1f1+6PHm1D/7LFFWJuvp/HKPHBn7QyQTpVJZ\nXl5eVVU1YsSI8p6+G8fllzc2Np49e/Z8XZ13xoyw5jskSYaVQaXnzgGAd9So6MfATaemKHj5\nZd0TTwx1u9FZsyzr1zfm5ooi3DcsO3ao584tcziwq65yvfJK4+BILElTTp6UPfJI6T33VDgc\nGIJAW5vohRdUPbJYP7HZbA0NDU1N6Pz5HZs3n9+x4+zs2d3d9V9+GYcxRwlfC9LUqeq2NkV1\ndfnu3SpuiURjY4HZnKgGEHZJM2zVzPTplxRJHTsWvkXoyiszvnVIIB2w2Wzbtm1bvXr1rl27\nGHUDw7Bx48aldlQajYb7LwLIle4r5ZQcAFS0CkVRheJigBcjghw9efSNhjcktAQAtii2QCbD\nVTdY+Yb59wf5D3/L+1uKxtUfWCkq9EDW6tYGOUCfg3P3wr0jYIQKVCIQlUDJPJjXAR3JHrQA\nD72foO3Zs2fatGmvvvpqEkYjkGJizqTsFQzDAiTJFyiruvTrg0tXVxfzewYhi7r+/HzIyenz\nZCWuMF3KgRkzrDNmhI4waNmZIAh2KTJsM4KwKJyGhBYQxdcclynhjpc3Zy8k/pPOgKKoRCKB\nefOoX/2qsbHR7XaD0wkAOWvXSr/9NmzJUuSUWdfIkdFXsqhUKgzDAoFA6KI3giCh7ZahsHX1\nYX0Thar7JMMqVhQFTz45pKGh+1+ahvZ20csvi2Sy/tdZ+P1+g8HAZ/CRzBBW/hYk0ZNPljY1\noZBEf1m/3x/kMc+tmqmuJoCzpjlom6cEkoDBYLBarXa7fdGiRWPGjFmxYoXJZCovL49Q9psc\n8vLy1iPrO+iO653X6/w6M25+K+8tK2YFgFkwCwBkMplKpWKFe+CIIARGqEHNu+tMQCS6ePYd\nWsPyifqTFI6trygUCqZ6NGwxjpTufqfRNG21Wt+FdzflZHZkzMCmd4FDJBJdfvnlSRiKQPKh\nadpsNtvtdoIgUIDSBQtkCbaBUCqVNE+gLILjUrMZhgwJe8cIvS3eUaNIpzM7OzsRA44Sp9PJ\nhuSFjpAYPZr7C8yW34edwrmrqgSBI91gG9GZfxPRUoTjuFwu5/PmLJ43Lxpvzgj4/X6Hw8Gk\nz+bdfz+eXMOXtrY29tmLquuEJ2W2TxUcGIYVFxfv3WtcuLA0yBcgNzdXqRSHpFcHk5WVZTQa\nu7rQ0KYAFEWjkUgE4khWVpbJZLLbkSVLhjQ0SFAUfvMb0913W+MSpGK1WpmeprClCnfc4YU0\nCAU8fjx81W3i/GVRFOWqG0Hhr2KxEBMmkAxsNhsTGzxx4sSJEyc6nU4ml2TkyJFGo7GwsLC3\nHSQQDMMOaA98hny2TreOu30cPe4F9AXm8pAhQ563Pm8MGPlEkMxFIpFIJBKu4xVXvlHRqgyy\nfs7Ly7PZbGxza9CBMPUdzNmg0+mUZEnWuAZm4u/AoHeBY+rUqUeOHEnCUASSDEVR3WuqAACg\nXbtWtnt3om0g8vLy3DxTuKEPPxxhChe5twUu9XBKPpE1C++lAkf3+SLPFI4YPTqpQxeIArYR\nHcKZ46Jabe/NflFQWFiIRpdG1Fc6OzuNRiPzs61buxbfuTOZhi+BQIBNh+nVPzVyyqxIKu1T\nJYtarf76a1lTkwj6tegtEok8nuLqanmob+Lo0bl9Tb4UiBGRSOR0FlRXK5mX44kn2seN8xiN\noquucjECR24uFU3jbVhCLWm5pQqzZ7vSQeBIfomEXC5naifDFjHJ5fFU49PNXkEgfQiysTx1\n6hTzc1ZVVWW1WgsKCrgyXPK5AbnBBKYLcMEFrnw6vwIqHkAeqEaqpT1fGhiG7c/bvxW2Bokg\n42H8X+AvqRhyPCksLFzTtcaKWUPlm7uRu1M9uj4gkUi2l21vcDRc57gu6EBmo7OZ2xiNRqfT\nCQA/t/+cvaOQtJKG9H5+tnr16smTJ69atWrZsmUYfweBQMbR3t7ON2ejCgvRxNhAiMVi/By/\nwMk/18IwjE8R8FRVyRHE5XLhOC52OpGrrkp+2GpkzcI/diz3xjKZDEEQzZYtYadwErk8ns0I\nAvGAbUQPa44LLpdKFYcUA5lM5j5+/JzBwMy1WG/OwIoV2NNP93u3Vqu1vb2duZyS9FmPx8Oc\njPKVLHEFDpFIpFQq+SpZdHPn9rWS5cSJ8O1rUS56b9miaWoCCKePXPqxFkgGGzdKm5rEEPJy\nMNx2mxugn24UQZa0QaUKOJ7EHpV0Ijc312w2M1UzQUVMGo2GW50eIwRB7NixYzWnAicd7BUE\n0oQgg+FTPSsBo0aNoijK5/NxHZeSzxJYsgSWdP/Do7RMgSlGMDIiiA50FVBxP9xfDRdFkMxF\nqVQelB78Av8itIbleeT5VI2qf+yQ7dgq21qbX8vdyOpQNE1bLBbuVZkbGTPg6V3geOaZZ5hu\ntzfeeOOKK64ILcr917/+lZChCSQSiqIir6kmrksCPXkyEAjY7Xav14v9+KPullsAehcjFAoF\n8uabYRUBGkUtJ04Y8/OBoiIElyQUmUwG/MvOYpmMq1lgGKbRaPimcHlz5sTYjCAQdxhvbb4C\nIjx+BURyuXzEiBGMS4vk22+ZjdikSbHs02QyMRd6bZ5KEN1TR36BMuj2RUVFdPwqWWJc9OZz\nfExcU4BABL7/nneVpSdIpZ8CB9fgI7RUIbXTpxSC4zhJDquuxoOKmGQy2fjxcdNGu7q6Wlpa\nOjs7ufYKZWVlwqKaQFhOnz4NACiKMh30oYFZacglIsiA40b8RjOYz9Pn3YhbS2kroGIuOpdb\nw5IpRNahCIKIEBnzf/r/G3pZxkTGDHh6Fzjeeust5kJTU1MTs5J1KakVOC5cuLB8+fKvvvrK\nZrOVlJTcc889S5cujW/Z5IDE5/Mxn1I+J7+EPoMYhnXH2n3+efemiRMj3yUrKws7dgzCKQLD\nFixo/93vOh98kA0u4a29T5i3okKhkEqlfJpF1j33BGkWhYWF5OnTvLsTYmLTjMgFRFloXDpU\nLsI0tcKiRbBoUYy7IggigjrjrKxMwtlHX/1TxWIxeeqUvr3d4XAEAoHwKbOxceAA+dZb1IED\nmF6P2u1IURHcey+sWgWhtqGCb2Ja8e9/W7jF6iGWEP1PS9VoNJ2dnWH9VnAcVyoTFVOS/mza\nJOcrYoqLsylJknq9nqbpIHuFqqqqlpaW8vLy1HYfCKQDYrGY20R28uRJACgrK5NKpQiCCH7P\nKadbvmE+qXE+IUoqkXUorpQWmvj7ovbF6+H6ZIxSIAp6FziOHTuWhHH0jx9//HHKlCl2u/2O\nO+4oKyvbvXv3s88++9VXX+3cuZNZURfgo69rqjE+FmK1hlcWHnoIHnoomp2gKKqqq+O71lNV\nxVd7z1aLAEVpH3hAlLD6jqFDh0a/7IyiqOjMGYvVyvg+ys+dK5k+HSCpbTUC0SOXy0XvvMNX\nQKS02+HSoLj0oTt6meeTnpz0WYlEEkH+K3jwwdCSJRzHhwwZwowfO3iwe2tvMmg0kCTZ2tpa\nU6PZseOid31TE7zwAlBU/y0qBZID+8sets4ilt99iUTidhdVVytC/VbGj9eh8RYxM4hEFzFZ\nrVbuoijXXsHr9brdbm7KpsDgRKPRGI1G5rLL5WpubgaAUaNGAYBarR7MH8/EcSZw5in6qWPo\nMRNq8oK3AApuhpufh+e1MEj79YATrAY8SSspHJtAEL0LHOPHj0/COPrHvHnzbDbbm2+++eCD\nDwIARVFz5szZuHHj3/72t6eeeirVo0trxGJxBBuIuGRS2mw2s9lMEARFkmWPPCKPWVlAfvyR\npmnG7hE9caLgZz8DANPixab58yX19aX/+7+htfddXV16vZ45edKtXSv6+uvEeSuKxeLA6dPG\nzk6Xy+Xz+RTnz0fWLBAEycnJ6a5k2bu3e2s8pnACcScnJ8d19CjwFBCFzs/9fr/P5xOJRCKH\nIyWmMCyMESZf9QQmFifH8KWoqAjrV9cJjuMwbx7MmxeXYTDOyl6v12DInz+/Y9o0e3Z2IC4Z\nHALJQa1Wi0Qii4UKrbMQi8VqdUyBix99lM1XqhBXzT/DSHQRk8fj4f7LtVdgrhUEDoG8vDyH\nw2E0GkmS/OGHHxgJrLS01Ol06hJvIzXYoCiqra3tLeyt/xT8h90oxKB+7/7+aezpo5cd7cQ7\nCYTII/OudV77W+NvMzQyZsCTwarn0aNHDx06NH78eEbdAAAURdesWYOi6GuvvZYRLXkpBMMw\nrpNfxd13V9x9t2brVubaoocego0bI+/B5/O5XC7SZIKyMkAQQBD485/Za1tbW/V6vcfjoShK\nV1sr37OHYrPKY1AWEATRaDRFRUUFBgOzxTNqFF9widvtbmlpYdQNtr6DqKgAALqoCLg/ihZL\n2KPoKxiG6XS6srKyysrKkh7jg6g0i4ceApoGmoZbb+33owskDhzHIxQQcd/SXV1ddXV1Z8+e\nbWhoqDtzxn3nnZAKUxgWiUQiFov5PunF8+axn3S3293Z2dnW1ma9cIEePjz2jwMXuVxOff/9\nhfPnfzxx4scTJ85/8AGznV65EmgabryRveW+fb6FC31XXBHQammxGIYNgz/9CXpCimLFYrEw\nZnUbNjQsXmysrPTqdP45c7pbHrSDd10qY0BR1O8vra4u371bxa2zOHlSBVAaYy9D9KUKhw5R\nixb5J0yg8vMh7u/SwUbQ2Vom2isIJBoEQYYNG3bbbbfdcMMNixcvZjbW1tZOnjyZnQL0Fa/X\nu2TJkqlTpyqVSgRBEATZv39/3EacyTQ3N1utVjWpXtOy5tNzRq6mugAAIABJREFUn3515qtH\nTI8wVw3OGFSfz3fhwoWN9o0fST5qFje7UXcACRhFxi3ZW35X8rua4ppMjIwZ8GRwyt3OnTsB\n4NZLJ4TFxcVjx449fvx4XV0d8+sowEdhYWH/nPwsFovJZCJJEihq2MKFypD5W1dXF+szHNQ5\nEigowOIitz/0kOUXv2htbQWaLn3ssdDae9+YMTaTiTk3CvVWdI8ciXo8TqeTIAgcRfPuvx8P\nOorY3Tqi7r4RyAgYc1yr1erxeNATJ4pvvx0g+L1htVoNPdIbAPRuCpMUdDqd5ORJ3quvvDIQ\nCLS0tDDJZ8yHGmlsZK+N1zBkMll5eTlJkgRBiPbsYTYiV13F3sDr9RoMhhUrtIlrHmHTcFSq\nALvxwIFue4U77/QDxC0VQiBBbNwo5auzuOyymPYcTakC0+K0bJnQ4hQ3ggwUuPYKwDF/FRjk\nNDY2stb4XCb2q+7V5/Pt3LlTSO0JxW63MycD023T2Y1sDOoketJAKlI4B+dqoOYIHGmHdr42\nHKbw0+fzZUmy1rSsGekd+UzhMweU3WWw3ym+Yy6IQORCXB3QMZhbeNKK8ALHL37xCwD4y1/+\nMnLkSOZyBD76KDXVSmfPngWAUBXjsssuEwSOaBCLxf6TJ5vb2hwOB03TrJMfvXIlsnw5372M\nRiNjAAYAutpa5b59ofM3Vt0IqyzISJIpm4+RXoNLbA0NoNXyuaiaLlzoPoq1a/GdO2mZDPF4\nAKAlP5+oqxvym99IU7rwLpCGYBiWl5cHALB9e/cmzqlVIBBoa2tj/01JIGtYsrKyLAcP1re3\nM9VM7CfdX1MjWrUKAJobGlwuF3Njvg91vMBxHMdxWLAAFizgbvf5fA0NDYFAwGAQJ655xB+S\nd3PmjHTdunwAuOIK9333kekmcBAEsXcv8d57ou++E/fqhzpISGGuTSAQqK+v9/l8g6HFae9e\n4p134MABzGDAEvrG02g0TDfrK6+8cuzYMcbM/vz5899///2ECRMGs72rAJeysrJ4lfM4HI6W\nlhaTycRN7SkvLxfMSoGzDMDAjUHN9+f/wfcHGCgdY4FA4APig03yTeyWsG04VqvV5/MBR/GZ\n7JrcIeq4ILnA3Zsf/O/Cuw5wDNoWnnQj/Dzz448/BoDf//737OU0xG63A0BobK1GowGAsEJv\nKI2NjZMnT+aaM4fCXDsg6yRFItHQoUNpmvb5fNi+fcxG7ppqEF6vt7Ozk7kcNH8jdTokL4+J\ndGOeMb5MTZQg4iVwyOVyPudCzb33en/3u865cyO7qAYdhT8/3y6V6lavln77bcoX3gXSl3Dl\nOQ6Hg7XKC5X2fGPGpHAhMicnR61WO51Or9cr7/mkiCZPBgCHw8GqG6Efh0BWVnJC3oxGYyAQ\nAIANGxrY8oo5c8zM1DFezSMYhnE1jpAMjjQKeKMoqrW11Waz/eUvQ3fsuOidKRQLpDDXxmQy\nMWe6CX2XphyCIPR6/dNPJ7CWiguGYSKR6MCBA2+88Qa7EUEQnU6nUCgE/0iB+OL3+5nm5aDU\nnpEjR+r1+mHDhqV6gCmG+xMZFIO6rnmdLl83MAQOh8Oh1+tBBWvMa0Z6R8oCsg9zPmSqVILa\ncBwOB/dfCqF+av/pd4rvLkguqAPqtc1rS4lSvvsKpJDw88yWlhYAyM/PZy9nEIwSEWUvbklJ\nyauvvuqL2Du7ffv2119/fQAHlSEIIpFI4OGH4eGHI9/SbrfzNX14qqrIrq7s7GxgXgL+fJY4\nfjeWlJRQ/F02xOjRfPUdNIqKTCbU6Qw6Cu+oUYnqqREY6LBfI3ymMKmttMZxnBF/4fHH4fHH\n2e3dnSnhPtTeUaMIp1MqTbjEQdM0ew4RtnnkrrtoiEddrFwuZzw4QjM4RCJIq+wtg8HAiPgJ\nLWkR6BORW5zi9S5NLSRJNjQ0kCSZnDeez+err68nSfJxzpcSANA0fdNNN912221bewyDBATi\nQoTUHqZteZB3RbGSYmgM6lrd2jeQNyLeu//U0XXLqGXH0GNGxJjo0BaCIJqbm2maDtuGcw1c\nw70xs/TCEKr45JA5Ee4rkELCCxxMPl/o5bSCqd1gTgG58FV2hAXDsDvvvDPybSwWy+uvv96v\nMQ40mCkcX2kG3TPBk0gkqs2b+ZQFSWcnxMkUXSQSBU6fNpnNLpeLIIig4BKkuVnx6KPAk3xh\nXLxY89//Bh2Fv6gocT01AgObbg2UR9rzjx2bysHxw+TI8n2oKSZlNsEEAgHuGScDt3nkwQfF\ncXGMys3NtVqtXV1oaAZHTk4uhmGxP0Rc8Hg87E/bwC4WyCBomo7c4nT//RhAxk+Nug22kvXG\na29vJ0lSr9ezdWRcGHsFiqKsVuuqVauOHj167Ngx5pb79u2bPHlynEcjMAhwu93cf4NSe9xu\n9yAXOORyObPsETYGVe6Xx/0R/X5/W1vbm+I3Pyj4gN2Y0NCWjo4OblU+tw1HR+pW4Cu4N2ZP\nDMIqPk+3Ps1t4fkj+UdIo4WSQU20p4xtbW0dHR0Igmi12oKCgoSOKUoYiw3GiYPLuXPnAOCy\nGN3GBMKBIEiE0gxZT5FLdnY28HSOhM3UjAUMw5hSIwCAni4bxhkhPz8f4a/vUO3ZE3oUqu3b\nIVS4cbtjTB8UGAz0agqTnEDWvoLjeIQPtTwpc/7QKvSQ5pHKuDyQRCLx+0urq8VNTWJuBodC\noVAq83u/f7Lg1sSGLRaYNSvZQxJgcha4p8VB71KRqCKFw4sXkatUZs6k4pi+R1EU81YfMmTI\niRMnAODQoUPz5s0DgCeffHLu3LmXX3652Ww2mUzHjh2rra1l7yj4QQ5avF7vihUrDh48ePjw\n4f5JXUFielBqT6jUPtjIyckxm81s2QICCDcGNe42JX6//8KFCyRJqjVqxsIzQrdIvODKqWHa\ncIbruEq1UqnkU3w+yv5oS/YW7n21+VpB4EgTevmhslgsS5YsKSkpKSoqGjdu3NixYwsLC0tL\nS5ctWxalyUXi+J//+R8A+Pzzz7kbW1tbv//+++LiYkHgSAQymSxC04fcamUuazQaxZkzvHtJ\nnKXFpWGrUqmUPHbszOnTQcmUpsWLDatWyY8fDz0KkcnU/sc/EmVlsh5lxFNVJfzgJQ6fz0eS\nZLxielOLQqGQSqV8gazq2bN7jV5OCQqFIsKHWnWp31iCQFGUbYShKHj5Zd0TTwx1u9FZsyzr\n1zfm5Ynj2Ie/ebOyqUkMPRkcv/xl2S9/WfaLX+g+/jiNmgvIcIUzbLHAVVf55s1L+pgE4GIT\nU+i7VKNBB4ZDYeh7j1ulMmdOJMOyvuL3+4PczbjL6X6/v6Ojo62tLRAI+Hy+RYsW/eMf/2DW\nMyoqKuTy+K8kC6QVTOXO448/PmXKFDbJ9ZVXXlm9evWuXbuYOWo/pK7IqT0D41McCziOby/b\n/k/dP0/JTnVinXXSuoTGoLa1tTHfOdNt039m/1kpUZpP5v+066fMtQnq+GDlG7Yog/mXKcoI\n+g7MyckRiS66j7OKD/Nv0H2F+UL6EKmCo76+ftq0aQ0NDQAgFou1Wi1N0x0dHc3Nzc8999zm\nzZt37NgxdGjKXNkmTJhw9dVXHzp06O23337ggQcAgKKoP/7xjxRFLViwYABbZqQQjUbjPHoU\neEoz6NWr4Q9/YP4VnTljNpvNZrPP52NTG6inn0ZXrAjdbeJQKBSXX365w+EgCELSM2Dv6NHq\nbdvCHgUASC5cCNNTw5gX9AZN04jVGmu+7CDA7/e3t7d3u3JS1PBFixSZn1mDIEhJSQn0K3o5\nhajVauT4cUhKvVUE8vLy9Hq9w4GFNo9o41oWn8IMjugJbZbhFgv8+99OHBeMgVJAXl5ec3Nz\n2Hdpbm5uqkcXH1AU5Z6jB1WpSCTxrFIJPU/jLqcjCMJGtgX5QVZWVjqdTiFgZQBjsViMRuOx\nY8f+/ve/sxsxDFMqlUHRJ33tKMnKymIXaF0uV3NzM/T0p2AYpohTA3VG86Xky63arX/X/p27\ncTyMfx55vt/7POk7WQM1x7HjJtREIARjsfEc9RxbrtgkbnpZ9/Ip2akOvMOLegFARskWo4tj\nORA+cBxnOu7DtuGI/JeEqaEouqN8xwX7hWts1+j8OjNufivvrW7Fx3L3U61Pce8rcQ7q/qa0\nglfgoCjql7/8ZUNDw3XXXbdy5copU6YwuiZBEN9+++2KFSv2798/Z86cXbt2pVBK2LBhw/XX\nX/+rX/3qww8/HD58+O7du48cOTJp0qTf/e53qRrSwAbDMFVdHd+1yKVp5Lm5ubm5uRRFQY9Y\ngF59dWLHF3ZUCNLdYNLjrSjv7JT+9a98t899553cd95h/w0SbsLicrk6Ozs9Hk/A7x++aJE8\n8+fqCYV1lWP+1dXWKvbuzdzMGpqmvV6v1+tFUVTqdkt6jqvz0Uc9N93ENYVJ5Sgjogxp9LtI\nsl4OjUZz9myguloV1DySnZ2tUkVpqRQVKczgiB6FQvHNN87//ldz7Ji8rU1kt+PMlHPmTGtN\nTWthYWmqBzhIUavVTmdBdbU66F2qVCqVyryBYYyiUCiYKUeoEa9Mhsd3fVskEuE4zl0v5S6n\ni0SiIAN4rh9kV1eXIHAMVDo7O9vb2wGAqdxh5Yzhw4dfe+211157LTf6xGAw9GmpVaVSqdVq\ng8FAkuQPP/zAvKNKS0utVmtpaamQ2gMAU2CKEYwX4IILXDrQVUDF/XB/NVRLoT+O40wi2DvY\nO1sKt7AbGYuNDuh4jn6O2bJbtfvzrEtK8j2oZyWsTIQHh0qlMpvN7L/cNhwlpQz9ltuGb9ua\nuxUuFbErvZWPGx8Puq8gkKUPvALH9u3bDxw4cMstt3z66adch0WJRHLTTTdNnTr11ltv/eqr\nr77++mumVSQljB49+siRIzU1NTt27Ni2bduQIUOWLl26dOnStDLDH2CgJ0/6fL7Ozk632439\n+OPwGTMgYmkGiqLw61/Dr3+d3GFGIi8vr2PfvgsmE/PDxhaY8BEk3ARhNpvb29uZXen+/nf5\nnj2ZO1dPDq2trewZbVBmDVVYiIZm1lgs/SuKYdYhUZstcTU1LpfLYDB0n4VT1LCFCyU98lbe\nLbdAe3v37SK+hVIO8uOPAOB0Oj0eD338eP7NNwOkQJT55JPcpiaAnuYRdvvatfDYY33b1eHD\n8M47sHcvNDeDzQZFRXDvvbBqFWRE9TFN0y6X65//vCSkkyErK6BWy4R5XQr573/z4vUuTU+0\nWq3T6QxrxBvfWioAQBAkJyfHZDIx/wYtp8tkslCBg7nANLDEdzACaQJJkuxbIqhyh3ljQIjU\n5fP5+iS9lZSUjBs3jttoX1tbW1tbW1ZWNmTIkCNHjgwkI9tzcK4Gao7AkXZojzKdZAksWQJL\n4jWAlpYWh8ORpckKtdg4j5xnb6agFHJK7kN8jIUnO/h4DYNLXl7eemS9GTFf77w+qChjJjUT\nQpzHghQfMSke7h4+t3OuD/HVSeu490VFgkCWLvAKHP/5z38QBKmtrQ2bHyESiV5++eWRI0e+\n//77KRQ4AKC8vPz//b//l8IBDELEYnFRUREAwLffMltSUpoRC1qtNjs72+l0+nw++c6dzEbz\nO++0X3EFTdOs5EEsWyZ55pkI+yEIglU3gubqpE6HC/myIZAkGTmXVA5AUVRnZyfTWISj6ND5\n86V9KYphOulsNpvP5wOKKnvkkQTV1Hg8nsbGRraHXFdbq9y37xJ5S6eDhx6K4yMmFKVSqVQq\ngc0FT7ooE5fmEZ/P19HRsXSpevt2FbuxqQleeAEoKgOiVWmabmpqcjqdBkPFvfdadu1StbWJ\nUBSYCo6vv86aNy8bw6BUqOFIERnR4hQLcrmcIEqqq6VBVSoajUalysnJifPDabVar9cbupyO\nIEiohMFtYBFW2gcqTqczyMiAK2ewW5gLbPRJnwSOhoaGsDaC9fX19fX1zOWBYWQbCAQ+ID7Y\nJN/EbkloOkkoLpeLqQgLG8h6NXU12xM3wzpjhnUG0y0yf9h8P+KHhHlwiESig/kHt6Hb1unW\ncbePCYz5qyhMfXeQ4nMHfsdW9dYd6ktaesdSY8PeVyBV8P5CHDlypKqqKoJVZ2VlZVVV1ZEj\nRxIzMIFM4FJTz8wCx3GNRpOfn6989FHmKHLnzKmsrBw+fHhxz8K75LrrIu/EYrEwv7vcubr0\n3DkA8FRVeTyeRB9FxkEQ3R51YXNJ3VVVJEmeP3/eZDJ5PB6KorL/+lfpt99SPQ6UvSoUFEU1\nNDSYTCZm6U9XW8vU1ER59z7Znba1tbHqBitvERUVABAoKIAMlbdS96Hetq37kYP+pkyJdg8e\nj+fChQtWq1WvF82f37F58/kdO87Onm1hrv3yy0SNPI5YrVZGAWRCOtvaRADAnu03Noquvx77\nKBnnpQLhif1dmv785z/qUCPe22/PScQbD0GQoUOH3n777TfccMPixd399rW1taNHj160aFHQ\njbkNLILJ6EAlqGwHQuQMCIk+8Xg8S5Ys+clPfsJ6ke7fvz/CQ5SVldE9MOcMJ06cePPNN7lG\ntv1w90g3HA5HXV0dWGBNy5pPz3361ZmvHjE9wlyVoMqIsGPg/kshVJO4iQ1Vfdz+eJDLAY3Q\nef485jIK6FOBpxI0sJ+gP5kIEzW0RgziYqp4Cjnln9Q/D2GHNNC7494UmDIRJmZDthjEQ+gh\nU+mpb8AbB9GD0dxXIGnwVnA0Nzf3WppRVVW1a9eueA9JQCBldFtMPfIIPPJINLf3er3AM1f3\njBqFezwDu1uKpmmfzxcIBCQuFzZpUh/aQHhySb2jRun1evb8Jqgohi4qQnpTDZjmqbB3D9v/\nQtO02Wzu6uoiCAJDkJKHH5ZFV+5BkiT7QKGlKJ6qKqGLIMnQNK3X6xl3dEYdYLbPmWN+//0c\nAMgIiwR2XVGlCpw+Hf7bY8AUCwikJ0muUqmvr7f2RLBxmTRpEhvQCJc2sOA4np2dnZDRCKSa\n0NqcIDkDQqJP9u7du5pTnten4guz2cy8x4LaYUaOHGk0GrurlTMQgiCam5tpmg5bOpGgyohQ\nuA47YQJZs3QWyrI5Z7MVs17v/P/snXl4E+e59p9ZtEvebXkB25jVJkASDG3akENO+/VqSdKT\npiQNWU8hDUtocyVNSk4JcaGnXxtoczimJQ0NOaXt16xNctrQbGQlISWHkBPAGGK8SrItydp3\naTTz/fHKw6DRCNna7fd38YcYzUivNLI07/Pez31f+W8z/q1P0cfvf53zujplnbhhJCPERBmo\nukJOLPz6AkEHDrQoVCRPqdvtjlkzSlNWVubOSYggZvrAcVyRhYaOz9WNP/tZXL5sfseVVTiO\ns1gs3d3dPT09fefOBa6/HlKrCyiVSoIgpHJJKbk8NC4QFYtiQpdcctGB2e12qcP9ra1xO0ej\n0b6+vtHRUb/fH41Gy3/5S9XhwynKPXj5tJQU5aJDxWQWn8/H64P46gYA/OMfsVpTUqedQkG4\nevn44wMnT556/vlz6OVcdpnf6fRNMbEApgDJsUpFuJwuZPv27TNnzlSpVB6Px+FwHDt2DCnm\nZs2apdVq8cXnVEWszYkrZ8R5tQCA1+sVii9aWlpSt2iJK64J22GcTmdcjHGB0wM9N8PNc2Gu\nDnRahfYr877ySMMjDtoBF0on9Iy+A3IUaMgngiUMZKUoiiCId3Xv7tHv+c7s7wirGwAQhSgO\nxMRMGkkFRzgcFifVxUFRFH9BicGkQyAQsFqtfr+fCYezZ5qQcRQKhfxPf0o4V+dIUmmzQcZb\nlgsDg8HAX18m8J6QhqKo0tJSzSefQKJc0rrvfpf44Q/H/vVfpaoGCSy8BRak7E9+wnz72yBd\ndFAyjNBUaHR0lG8jmqhaJPb1KC1FSXJs4cAwTDAYjEajSr9f8eUvF3W8MZJTxXHmjHLPnhoA\naG8PrVtXBHrjuOu5uJBOmsbeG5hpBEVRLS0ty5YtE85Cd+/evXv37lWrVh08eDCPY8NkCbVa\nrdFokM0nXFjO8Hg84ugTmqbjxBdtbW0mk0mj0ST0EBTCcVzcLEbYDsOybDgcLopGFaRF/RP5\np2crztttmGXml8pfclJOo9wYL52YpYecvCyNRoPyShIGsmoDWq/Xu9S/1EbbDHJDgAxUMpVl\n0bJzinMMwbxe+rocisEYHFOQXOSPH4PJAW6322AwxIJIxk0TiiKIpKysLCwxV08lX7ZIcbvd\nfHVjEtaqdXV10e5uqXsDbW0XrRpEo1Gn04nKYfXr1iniymESh8dpatCDoNsJ7U6TNxfJ5XKa\nprXPPZewvKXUaMBkgoaG5G9FHolGo8PDwy6XCyA+/6XA/+hSR1gd+K//stP0zHyP6OIolUq0\n9igO6ZTJQKmcTEofJn3+53+4AwfYI0cIo5EsulyeCfHBB8E//hGOHqVNJsrlIvL+Svv7+xM2\nsLQXdjQVJh1mzpw5MDBgtVoZhvnss8945c43vvGN2G8WAIxHn6xYsWLv3r1wofiCZVm32z0J\nU9y4dpiiUBBEo9GBgYFAIKAsU+7yxSeVDCoG7bSd5miUToKkE39k/pibwo1Op1OpVPxKUlyo\nqlqtrqysXGtYu9Z63o6dJdiVC1Y6KIeO0xFkEbz/mMIkWYHjv/7rv5555pkkO2APRUz6MAxj\nNBqlgkjI6upCtkpXq9Xys2el7k2eL5sVJpuoOiGS1AUCbW2U35/c/o2iKKK722y1opwUdU8P\nChuGHTsCDzzg6+0tf+mlhFUDmVIJJlOgomJwcBA1dup371a89x5fDiOXLaMdDt3zzyc8nJTJ\naLOZLzoEg0H0qUso9/C1tl7UPaW6upo6fhwSlbdq7rgDdu2CBx5I9T3NLRzH9ff385KHCWlw\nChbh5Zq4OlBZWZnk2MKhoqLC4/F4PJQ4pLO0tOyiskpMxuE4bmxsbOtWZZHm8qROJBIxGAzb\nt1cJ84nz/kpRA0t+nhuTJ2ianj179vLly+OUOwl3FmbHCrekIjAnCEKhUAj3FLbDkCQpk8km\n+yJyx/DwMJqOJbTbWOJfssO0I046IYvk6HURBPHGrDfOOc99wfGFuEDWm8ibAKC0tPQJeMIY\nNIoTW28kbszNIDFTkmQFjnA4LHYzxmAmicTc2+l0ooyohLNlzuu9qBdMfqG7ux0Ox9jYWCgU\n4vNlmUceobdvz80AGIZxu93BYJDguKrbb5dlZx0e+YmGw2GapqNW67x/+Re5yQQAnEwWZ60q\nD4cv6m9PkqRer9fr9RzHEUePxra2t6tUKoVCIdXAUn377eyjjw5+85uouhFXDovW1lJ6fQVB\nyCUOn/m97wmLDrGL5jR6TCorKxnp8lYhlwlsNhtf3Zgy8cZarVYmk4XDYa+XjqsOEARRVlYc\n9uY6nc7rrV2zpiQupFOhUFx2WW2+RzcdGR4edjgcRuOc9eutX/2qq7w8um9fNbKtfeONqVPg\nQFkS4XDYZJJP7VeKKQqklDuPPPLI9vGLq+HhYd51CxJ5kaZCRUXFyMgIuh3n7lFWVlb4Cg6G\nYYSqFgBgCdYgM/BJJRstG0EknZhQqm6aHCQPHqw4CBeKaS6FS39B/ALdfr/0/YOlB+MSW4U7\nYDCTQLLAgdUZmPRBXQCBQIAJh+vWrk2ogU8eRMIFAikWOHw+XyAQYBhG4fOVffWrxMAAQI7c\nBMrLy8vLy6PRKHfkCNpCfzFHDtUOh2NkZARViPS7d8veeScb6/Aul2t0dDTm2sWyzT/4Aapu\nAMDYd78b1waiFFmgJ4EgCFi7FtaeVyc2NDRQ4+swYrzz56Pqhrgc5m9tlQWDVVVVjHT/i/A9\nQQv+SexOU+kxobu7/X6/y+UKhUKKM2fqULRqwXtY8B1GCauKsmCwGFshCIJoaGg4fHhk48am\nuOpAeXm5VqtsKhL/ir/9rWpwEGA8pJPfvns33Htv3kY1PfH5fGiWlTCXp6qKmzIe+na7HS1o\nTflXiikKUlHuxP1OxXmRpvgrVlFR4fV6h4eH49w9AoFAUbhvxE3WhEklM8Izvu76uoN2yDm5\nUBlxA3tDlqJJErICVpjB3Au9PvDpQT8H5twOt6+BNUpQprgDBjMJJAscxXiBiykoAoGAVCtB\n/Nxb2jQhlU8hwzBDQ0OxzE6Wbd64MVbdED9RNqEoCu6+G+6+O2fP6PF4TOOFhkkkqqaIw+Hg\nnwXG2xk4mYyIRADAt3x5nLWqym6HNEQ3arU68Nln/aOjyGOMF8Vw27cTjzziHBoCt1uqHBbx\n+5VKpezMGYvV6nQ6w+Fw3OHCJ5LJZBqNJondaYo9Jmq1OqZYefvt2KaCbw5HtSqptxEikSL9\n/tdqte++O2twkIZirg7kOKQTkwR+aTRhLs+11wYBMhAE/vHH7IED7EcfkXk0+ODTWBO+0m9+\nMwy58STEYFKmtLTUbDajaPA48QVFUSmujREE0djYeNlllwkFI8jdoyiMbIVlID6pBNltGOXG\nJ6uffLL6SeH+i6KLfin7ZS5HeEGo6qR2wGAmQSH7G2CKmGg0ylc3+Ll3aM4cAIjW1oJeDwAs\ny6Lac+XTT+veeQcAGu+7r/7f/z1+tpwUjuMGBgZi1Q2Bm0Ds7kwXODiOQ07dZrPZ1d/PzZqV\nSpxtJBJxu91jY2OewcEUD0kFi8WCbogjUTOV4hGNRkdHR/n/8qeS1WjQlua77ppz441l4xcB\nzRs2yF54Ic0nValUs2bNamtrmz17drPNhjYSy5YBAMuySXJ50YUOQRA1NTXz5s1ra2ubPW4X\ngg6Po76+XiWtFpnwh2ft2liaItJxFDAkSSZ5G8mJaHAKjRMnElfti6g6kOOQTkwSxF26fC7P\nZZf5v/Mdb5qPz7Ls8PDw1q3evXvpTz8lrVaIRGK2Fw8/nOZjTwz0Yy1E+EpvuQVLejF5JhgM\n/uhHP7rqqqu0Wi1BEARBfPzxxw0NDV6vV5gi3NTU5HQ6dTpd6o5FRW1kK2w2QUklx7uOP9X/\nlIJTAAABRBlXJgd5A9uwglnxJPvkx9THZVAcDZsYTDrGp+ugAAAgAElEQVTgFBVMVnA4HEla\nCeShkMvlGhsbY1kWWHbB/v38gRXPns+4at6wgX30UWhtTfJETqdTyk0gUlNDVFZm8CMeDAYN\nBkPMjyo1qQjHcSMjI7E20YyqS6LRKCoPJVyH98yfn4GFRQCv14uqBnDhqWy96irJYzJUVCJJ\nUqVSxYliZDKZVFMJR5KKsTGorhY+AqxbB+vWST2FQqEInTw5MDKCVi+TyD2KEmnHWbVarX7m\nmYRvI1CUym6H8QJW0fHqq/keAWYKEVfsi0vtlcvTUslxHDc4OOjz+UymirzbXsTNBuNeqUJR\nBAlEmCmMy+V68803d+3axW+hKGrJkiVqtXrVqlVpii+K2shWqVQqlUphSrrQbqOCqxgjxgAA\nSLyijZle4M87ZmJEo1Gv1+t0OoPDw9DSIiVGQP0FUhr44eFhi8USc47o7KTHlQhiyEQL70J4\nYW1CFQN/b/owDDMwMMC7bacoFTEajbwJVmbVJbEFN+l1ePT2pgm/gBl3KoHjAGDsBz/o+fzz\n/pdeQvtw27cDx8HVV6f/vFKUlJTwTSVzbrwxTjyi/dvfJvqACoWiubk5oVqk6OA4zm63Dw0N\n9fT0DPT1hb/9bZBwnK2srJR6G5vWrycFRcbi4tgxuPdeaG+HmhqQy6G5GR56CFJxyj52DL7/\nfXYSB2KmNipVrFDMsvDrX+vvv7/R7ydXr7bv2zeg1bL8vZPD4XCgH8r9+/s3bzYvWBDU6yO3\n3Rb7FhKUanOBZrymKX6lOh13Ud9ozHQgGAxu2bJl5cqVvIbio48+ysHzWq1Wg8Hg9Xo3bdr0\n+OOP19TUAEBLSwvDMH19fcUrvsgU9fX1L1S+sK9632nVaRtt+1z5+baGbTiIBDPNwQoOTKpw\nHDc6Omq32zmOQ2IEqekTXNhKIHbWQFd1kEhzMbh3L1pFDz/8sDyFJg40z5eqpJAi2e2ksdls\nvIhXHDxB1dSIHdh8Ph/fwi1+pWxZWTo9zTRNg7RHJkHT5MjIRT0yL0psAVPiVHKXXz537lw4\nfDj2pNmvC+h0urC0h+hFy2GSByZSixQXqADHL+OU7d4tf/ddKdcbpVIp+/xzyccq4PwXKViW\ntdlsDz+sfv3189qTVBIuI5GI2Wz+8Y9Lp3wIKGYSlJeXW61Wl4sQp/aqVKo0p/3JDT6uv57J\n5RVaZWWl3W53ODjxK62qqsb5xNMcjuMcDseHH364U/CdiDQU2X7qUCiEunHb29vb29tRjzAA\ntLW1mUym+fPnF6/4IlOo1eqjiqOvUa+Jg0h+Dj/P16gwmPyCFRyYVDEYDDabDf2WXFSMQNN0\nklYCmcUCEpoLvdGIdpN/6UupjIqiqCQqBlQFyAgejwfdEA870NYm1Aemckhw4UL+3slBUZRa\nrZZah2+8+26YMSN9pw90BS91KhVqNZhMk/GesNuTyH+SQ3d3m4zGUydPnjp58ty434f/oYey\nLR4pcIxGo7hXC7neQEMDiBxnqdOnQ8Hg6MjIQH+/gVe+7NhRjG9jNBrt6+szm81DQ9T69dbn\nnz936NDZm26KKafeeEPywFAodO7cOafTaTTKJnQgZppA03Qk0rRmzezDh3XCXJ7Tp0s4rjHN\nB09u8JFj2wuKoqLRWeJX2t+vDwRqcjkSTKERiUR6e3uHh4dtNptQQ7FgwYIcSHucTqewhHH6\n9Gn037a2tmg0muZ1VLbJmeZlJbWyHdrLoVwO8hncjKvh6qfgqY/goxzYbfj9/h/+8IcrVqzI\nsa4Hg0kOVnBgUsLj8fDRkmL9AlFVFbe+U1JSwknkUzRv2DD6wx/aV69OqLkgbrlFN5G0A61W\nSx04IOUmoHW5oCwz3+/JpSKqRFKR5IewaatL9Ho9ncQjE5HeajxapZSKGim56aYUo0YAIBgM\nOhyOUCjERaP169YlzAxOBZIkGxoaampqgsEg+c47aKM6iSfIROA4jiCIJO4VhUkwGEzYq4Vc\nb0KXXJJQK6RQKGprawGKKf8lIcPDw6i4kzDhMonU32QyIYuZiR6ImT4895wmS6m9BHGB7C/O\n9kImS7eAMlH+/GcFzifGiBkaGkJfsHEainnz5sV+MbMJ3xeMOD1+zYPSUuLuLSj8fv/bb7+d\nG83LBUEkuQp0Zll2dHT0vffee+yxx/iNudH1YDAXBSs4MCnBVzcS6hfEVhelpaVq6VaCQGur\nlOZioj+W5eXluk8/BQk3Afr55yf0aElILhVJqODNdlaFRqMJHTt2uqsrTs4AABnMkZk5c2aS\nU5ni41ut1t7eXpvN5vV6tT//OcoMTmeEMplMp9Npvv/9jASXRCIRk8n0+eefd3V1dXd1Ba6/\nPkn71UUJBAIOh8Nms/kMhkkLVSb6jOiGZPJrcoon/0UMwzDJpf6rVyc+MBQK8elLEzoQM63I\nXmovv/qd0PYiTYOPSYDziTFivF4v//uC4DUUCxYs4L97s0fcNWF3dzcAkCQ5f/588b2Fg8Vi\n6evrGxsbE2pe5syZk/u/6yyBPJLtdnsoFIrzRsFNQ5hCACs4MCkRiUQgidVFJCI+hO7uNg4P\nO51OEORT+B96yP/DH8off1yqe0XjdEJVVeoDIwhClxM3Aa1WKxU8QdC0ym4HkVZTo9Fw+/dL\nvVKtywU16Up/tVotf/t89WTxYmBZ1alTXH09IepNmCgymYzt7h6z230+XzgcVvf0NFxzDcAE\npA0ej8dsNqPbcfIftq6OTHuEaRIKhfr6+viwmKr/+A/V4cNS7hXJCYfDRqMxNm2+mE9NBokZ\nykpYpYQuuSR7T513EnaH8VL/yy8PrFuX+IIy4dJfKgdiphXZy+WpqKhwOp0eDyW2vSgrK8+9\n7QVOIMKI4avAPLyGYt68eVu3bj116tQnn3yCjNWOHDlyxRVXZHYASqVSWEbp6uoCgJaWFqVS\nie7N7NNlBI/Hk9A3ZGxsTKvVorc0G+9VLnE6neikJ/RGmTdvXr4HiJnuYAUHJiUmIUYgSXLG\njBnz589vbGysGx5GG9VXXVVeXq49fhwkgjBU47Oy1CFOneJY1uN2W8xmC983n2k3gaqqKqlh\nN959N/HMM+JDSkpKpNQlzRs2qP/7v9Mfldfr5dNS/EuWsGp1qKlp4De/QeKaTM1sSZKsqqpq\namqaO3duw+hobGvK7QzoZw8SyX/8STOAc4PBYOCrG3HuFVx9vdi9QopoNNrf389fDmY2NCc5\nMpkMpK1SKLkcTKbsPXt+ES8WHTpUcuedLR4PtWyZ73e/M6XuwyM88MknhzNn4IPBJECtVgeD\nM4S2F3PnBk+cUJ89Wx4O1138+HyAgoqWLuWEeUNud37CNTA5QBzHxmsoAGDv3r3vv/8+muim\n0ptwOnL625Fvt0RbtJxWBrKZMHMdrLOCNckh5eXl/BWmz+cbGhqC8f4UuVwuXOMpHGzjiWwI\nXvNis9nQFUIR9XEEg8EHHnjgqquuivvrjhPvCL1RwuFwnOoHg8k9+AoOkxJqtVrK6iImu6io\nSHigTCaTyWTw/e/D97+PtlAAGddcEASh0+l0Oh0YDLFNmXYTkMlkEx12DtQlUjGuSFwTvOSS\nzK9urF0La9emvjvHceinLuEI/W1tSoYRe8GyLOv1etEauyoQ0K5cmSVHDL/fz0sAErpXpP4G\njo2NRcalTGKfGjqbQhWtVktRlJRVStVtt6VulVJ0yOVy/jbLwt69+n37qjkOVq+2b906Ulam\nkTpQuPQnPrCysiS748ZgAF56qayIbC9cLtfWrdQbb2j5Ln+UN9TbO/LCC7kO18DkBlQ9F8Jr\nKABg06ZNixYt6ujosFgsLS0t4p15WJYdGRn5A/mHF+te5DcawfgUPGUD28sgubJF03RDQ0N3\nd3ckEjlx4gSaRTc1Nbnd7paWlsJsUYmTvfCalwcffPDAgQMWi2X27NkFqD3pgZ5tsO0T+GQU\nRoMQrIXaq4JXrXxt5a9+9St+H/6vO84jOc4bJRKJTJlmHEyRghUcmJRILrtQvPhi0qPjIbu6\nIuHw6MhIf1/f0F//ijZy27cn1FxwHIc8FMOjoxd3NMimmwDZ1cVGo06HY3RkJEWpCFKXOB2O\nYZNpZFwBLPVKJ0Hs111CXBNetCj9p0gTjuM4jksi/xEvELnd7rNnzw4NDZnNZvPICKxZk36j\nRzgc9ng8wWCQs9mEnyK+ujFJ94oLh41uJPSpyaoXGkmSdXV1yq4uyT2KMPk1RRQKBbqQ8nio\nzZubnniimqa5jg5TR8cwTXNl0h7Dcrlco9FIHVheXp6714CZrhSR7YXZbDYYDAYDLc4bOnq0\nVNiEP3v2bGHZEVPU6HQ6YRFBqKFob2/fuHHjpZdeyvcmDI9rdcWYTCaHw1EaLd1l2PVKzytv\nnXnrHss96K4e6Ek+htLS0lWrVl111VWbN29GWzo7O7/85S+vW7cuzVeXJeJ0hbzmZdWqVei9\nam1tHeXFsIVBKBR6zvvcs/DsOTjnBS8DjBGMf1b++fHLH4+z2ED7xwm3i8UbBTN9wAoOTEpQ\nFJVZMYJMJoslOIwHYRDLlsXtw3Hc2NjY2NhYNBpFjgbyjDsaTDAvg3Q6y/j9EReTihAEUVZW\nVlZWBq+9FtsieqWTBk3tpHoT5CoVmEzQ0JCpp5sEJElSFFXywgtSSTe02QwzZ/L7+3w+g8HA\nXx/wjR6TcMRAuFyu0dHRmLaCZWdt2qQRf4ok3CsmVCFKHppDRCIKRcIwk8xQVlbmOnZscPyV\n8q433PbtxCOPZO95C4H6+vr33jNt2NA4OCgXJlyq1WqdLkmJAxoaGt5913j33TPiDiwtLdXp\nClP7jJlSFIvthd/vRxOzhHlDDQ3yjRs38k34ra2tQ0ND/EQIU9TI5fKqqiqr1erxeBiGEWoo\nHA4HTdPd3d18bwLaRyzJ9Pv9qKPhOud1/MavuL/ym5rfAMCy6DJIajjT19fncDjE29sLNfZL\nJpMJBQ685qWvr49/rxwOh16vz73VTkLGxsbMZjNZSu5y7GoNtqqiqhcrXkRnx613C/+6kcXG\n3LlzVSqV0ABL6I1CEASWb2DyDi5wYFKF7OpiGMZutwcCAfLkyZnXXQcZmT5JtzyMjIzY7bE1\novQnujwcx7lcLr/fHw4G69auvWheaSgUstvtwWAwGonM+N73lPz+Bw/CqlUTeOIJNnekglqt\nVqlUUr0JZTffXAi9CSUlJVIjbFq/Pm6Eo6OjfHUjrtEjWltLTbDRw+FwmAT2E/rOTs2HHwo/\nRajoIFUhohWK1CtEFEVFGSZhoSTQ1qbL/nVMaWlpSUlJKBQKh8PKDz5AGzNYTStYVCrVO+80\nDw7SMEGpv1wuf+ed5sFBcqIHZg+O444cCf/pT8TRo7TRSDqdUF8PN98MO3YAXhTH5At+epkw\nb+hrX3PDhU34fr/f5/MhhRSm2NHr9SRJXnnllULbhc7Ozs7OzhUrVixfvhxtQb0JwWBQXBv2\neDzC/7IEa5AZHqt9DABqIjX3uu+FymQDKLpgjtLSUt56TKh5EfZxoO7dQqijo0UgkCg/LfYv\nhgv/ukOhUCAQqKysdDqdaGOcN0ppaam4yIXB5BjcooKZADRN19TUNDU1zbRY0JbsTZ/8fj9f\n3YizfmT0+uhEklaERCKR3t5eo9Fot9s1//f/XjSv1Ol0njt3zmaz+Xy+0p07le+/nxvbyNSZ\nOXMm3/GRgAIYZE1NTYojZBiG96ZK35E0Go2OjIzw/01oIKpWq+VyOV9/iWu/qrz1Vnj6afRQ\nfr8/HA6D3S7VJ6VWq6UKJQRNKy90HcsSBEEolcqSkhL5hg3Fm/w6CU6cSHw5dVGp///+b+If\nwbz0CNjt9jNnzvzkJ6Hf/lb+6aek1QqRSMzm4OGH8zAeDAYh7rDj84Yuu8x/ww0OEDXhi9M3\nACAYxHakRYnH40mYCLtw4cJUehOQvBFxw5wblixccu28az/QfrDYv/j53udrw7VZG3h+qKqq\nUigUHo/H4XAcO3aM17x89tlnIHivCqRqYxm/nkewBDsoH+TLTxstG0H01x0Oh5VKZX19vdfr\njXuNoVBILYoUxGByD66xYSZFFsQIcfC/pmLrx0BbW9TjSSY9lwAFdyNZXSo2kMFg0GQyoS/u\n9NUEWUIul0e7uy02m8/nC4VCmnPnkLgm436ck0Ymk4VOnuwzmdAlL989Edm2TbZjh3BP/jJI\nqtFDy3Gp93Z6PB7e4EPKQJQgiBkzZlDS9ZfQJZcYe3tjZRepDhcAAKiurg5ICFUa7767EKQ0\nU5hJS/0Lp0fAZrOhepzJJF+/3vrVr7rKy6P79lWjLoA33oCdOy/2EBhMTjh0qGTr1hl+P7ls\nma+zc5CiOBA14YvNlYLB4JtvvrlzJ7YjLT6EGoqBgQGv18vfde2114IgtzVhJybv1xAmwlaZ\nleZohmAA4IT6xG797t9Gfpvt8ecYiqJmzZq1fPlyp9PJb+zs7EQ3+PcqiSdrzmAYRli+vGHO\nDT3KmCXKYv/iPUN7KpgKkLDYKC8vX7VqlbB7COl6Vq1adXB8lQiDyRdYwYEpUFAHo9REl4+r\nmBDIZhIkbCDjVJQAYLPZ0I96QjUBny2adyiKqqmpmTVr1oIFC3hxTcZzZNJBoVC0tLTMnTu3\nsbGRD5qViULgY7JGCUfS4MKFE3KuumjEDLpXrVbDyZODAwNdp06dOnny3AsvxI7fscPn9Z6b\nOZMXlfAdLrEdLixwKBSKkh5ps7QCkNJgChaGYcxmM7q9f3//5s3mBQuCen3ktttiwp/q6vwN\nDjPt4RMfWBZ+/Wv9/fc3+v3k6tX2ffsGtNpYIUPYhA+iyZvL5ert7bXZbHGGhVjKXnRUCCLz\n4noTdDpdwhPKN2LIOfnh7sPHu44/1f+UglMAwCtlr0zJVqahoSFhdUMIeq8UCkUhBKkIr2P5\n8hP6Lyo/odtxf93IYqPovFEw0wr804IpUEiSlLJ+DLS1aSZl0YwUBFJFE87v1+l0qe9PFEb/\nZDzZF9dMGoVCoVAo4J574J57Eu5A07RCoVA/80zCRg+lRjMhz9TYqpHEpyiyeLFwYE1NTRzH\nRSIR2dGjaCO3dCmv3wGRhIerrydEEh7kU+N0OoPBIHXqVB1qDykYKQ2mYBGqjRLaHKxenYdR\nYfLFxx+zBw6wH31EFogPS3l5ud1u93ioLVtmHD6sk8m4H/94ePXq83ObuIkuCm7n7w2Hw0aj\nkeO49vb29vZ2oWGhwWCYPXt2zl8QZvKUlJSUl5cPDQ3FeY56vV69hKxVq9WqVCp+qYAAYql/\nqZpVh6iQltVOyQIH0rxwHNff3282m9F7hVJgmpqanE5ngbjw9tP9D8588LTq9Bg9FiJCVUzV\n1e6rV3pW/mjmj0JE6JWyV3aYdojLWKh8WXTeKJhpBS5wYAoUlUpF/v73CSe6HEmqHQ6YuA1H\nNBpNUjSRiRQZwnxT8f5KkQQXkz56vZ6VaPSoueOOCTV6JI+YUajVceUSgiDkcjmsWwfr1gFA\nwO8P9/XFdhZ1uAQXLkzoEk7TdBX6ZPJBwgW/msFxHLImlclkCp+PXL489VyhbHDsGPzxj/Dh\nhzA0BIUwu8sBCSVpvM1Be3to3bosRvBgCgeWZUdHR7du1R46VMJvRD4sLJu3NiWVSuX3169Z\no43LGwIAlcpZVeWLC9eora0VKjgcDodwIiQ0LAwEAn6/HzftFxcNDQ2LFi1KvTeBIIjXm1/v\ndfV+wfEFfURvo20Hqg44KAcA3EjcmLtx5xyCIJqampYuXRrXq1IgfRwej+fP4T+/Vvkav8Us\nM79U/pKTdqLykyaqcTgcwr9uv98fC0DEYAobXODApMYE41TTp7y83Hv8OCSa6DZv2DA5RwOZ\nTCY13eVIUjE2BvX1cftrnn022f4lJfHPgUmPkpKSaIYCiTUaTZKImZKbbkr+KUre4RKQKHCc\np4ClNELsdrvZbI7pVFm2eeNGbcbDmFMmEolYrdatW0veeOO8Nirvs7scwPeo8whtDg4ccNJ0\nPsOeMbmB47iBgQG/328yVRSaD8t//3fF4CCAKG9Ipfq3QOAX/H8TTt6EcZIgMiwMBoO4wFFc\nTKI34e/U3w9WHISKCzZeCpc+Sj6a8eEVFIODgwl7VfLexzE2NjY6OqoqU+0y7BpQDASIgIfy\nPF/xPAAcUx/zUB4ACPwxcNXaq/hDCqc0IyQYDHZ0dBw9evTYsWM+nw8Ajhw5coWoAxoz3cAF\nDowkHMe53W6/3x8JhfT/+q8XjVOdGBermFAUpcvQRJdHp9OFJKa7zRs2MD//OQjaFgCgtLSU\nlN4f20ZmCer06Ugk4na7g8GgvLu7+v/8H4BJltVmzpzJTTZiJub3ISHhCS9aNNHBFCBWq5W3\nfoCMhjFPgkAgMDAwEI1GDYaCm91lW1QinOCxLOzdq9+3r5rjYPVq+9atIzU1eMVsWmC321Ff\n5P79/Xyn0m232dCfQH59WI4fT7w9EPhQvDFu8hanY7+oHSmmwJlEb8IKWGEGcy/0+sCnB/0c\nmHM73L4G1igh/z4UWaUw+ziCwSD66UfRsPc03fO+7n3+XlTdWBBccOb+M+Jj816aERIMBl9/\n/XVsXYwRgwscmMREIpGhoSHUM6nfvRvFqaYz7WFZ1mazeTyeUChEEUTj+vXKi1VMyK6uSCQy\nNjYWCATIkyebr78eANif/ITs6JjUawKVSkWfSfB9jaC/8IW4LeXl5ZHubsmHw7aRWUMmk1VW\nVgIAvP56bJPwNzVlPZFcLmfPnLEKImZmXHvtRY9CIDMtKcmPTKmckCFIARKJRIT5cKmYjGQP\njuMMBgMSkhTU7I5lWavVunWrJquiErVarVar/X6/2OaApulJJEalzzRsFMo7fHZYQh+W669n\n8njNJp039L7UHTzyCz80cYaFCXM3MFOMLbBlC2zJ9ygwMeK6xi73Xz4iG+lX9DMEQ3HU5dHL\nN1AbblHeonQUdPnJ7XYbDAaHw7Fp06ZFixZ1dHRYLJaWlhZcM8UALnBgEsJxHF/dyEg8ajQa\n7e/v53WqVbt3K99/P5WKiUwmq6urAwB49120hVy+fKLPLoTu7h4ZHbXb7RzH8Xml3h/9SPOL\nX4gTOgiCoE6fNo6MuFwu4f7hhx+W//Sn6QwDkyqCRg+fz4f0RNV33CFLWU9EkmR1dXU1mh9/\nOL7YmMIShEKh0Gq1Uh0ulbfemg0JD4dycHPSEebxePhLHLHJSHjRolxOOzweD98TVDgum/wX\nl8FQmm1RSWNj47vvGu+6q15oc9DVpdPr9SoV1dSUsSdKBb/f39FB/v3v569up0OjUN7h/wR4\neB+Wyy7z33orC1B4ttYpUFZWZrfb0e04w0KapqekxyQGU8gkj4a9pOYSYVZOYRKNRpENvNi6\n2Gg0zps3T9z4iZlW4AIHJgFerxdVN8TTHn9rK+nzTfSKZHh4mK9uTHKhOEOOBgRB1NXVVVdX\n+/1+8u230UbtypUgEctCUdSMGTPq6uqCwSD53ntoo/xLX0p/JJjUYRjGYDCg7kr97t2yd96Z\npJ5ogp+iGTNmsDmR8IRCIYvF4vf7I5EITZLNGzdeVN+UPheN0c1lgSOuSx/Bz+6WLQuvW5cH\n5cDIyAgaWA5EJTRNv/120+AgASKbg698Bf7+99xJJ4aHh+12e3//HL6m87vf1Tz7bDnku1Fo\napAkISWuyC70YensHJTJclvlyhxqtbqysnJgYCAud8PpdDY3N+N5CAaTY/i1DT4aliEYGI+G\nfZJ7Mq+jSwmXyyXMuBVaFzMM43a786J8xBQO+HcFkwCv1wvS3oponpk6DMPwylthxUTZ0wMA\nwYULMzz6FKBpuqSkRPuDHwDHAccBivOUhqIojUajuueeFPfHZBCO4wYHB9Gnjq+OhebMAYBo\nbS1ks42Cpmm6u9tiNvf19nafPj3017/G7tixAzgOrr46I8/i9Xp7e3tdLheK0qh87DGkb4rd\nnbUCR1yMrvFnPwu1tKjGLUuY3HaxiruUDx0qufPOFo+HWrbMd+CAlc55NT4ajSZvGci4qOST\nTxK3ar/1Fjz8cIafSwqLxYIW2/fv79+82bxgQVCvj9x66xi6V1jTYVn28OHAhg2hyy9na2pA\nLofmZnjoIRCpEDAxUKF261bv3r30p5+SVitEIjFpDDq/KPgJAFgWfv1r/f33N/r95OrV9n37\nBnQ6jr+3GKmrq7vmmmuuuuoqlJQJAJ2dnStWrFi/fn1+B4bBTEOU4xcYck5+uPvw8a7jT/U/\npeAUAPBK2StF0TV2UeviPIwJU0jgAgcmASzLSk17Am1tUVGcanL48POEFRN/W1tGx46Zang8\nHrGeCFXH/K2tQqVlNiBJsqampqWlpbW1tdFqjW3NnMkWy7JGo5FvGY2r4EBDQ/YqOMjY8iIx\nurlCeEUlnt1VVsqSHJslQqGQuOzCi0ouvzywbl0mny4cDu/effbkyVOtrcH1663PP3/u0KGz\nN90UE/bzocNZheO4sbFYLSN5TWdsbOzMmTM7dkSeeEKRcK6eY44dg3vvhfZ2KNhSC2p3crlc\nJpNc6vyihGmPh9q8uemJJ6ppmuvoMHV0DNM0V15eXtRKh0nkbmAwmCwRp24ggFjqX6pm1QCg\nZbVFkWqU3Lq4AI1dMTkGt6hgEkDTdJI4VbnVCnV1yR9BSOyLRiKNIi8KDkwR4fF4QFpPFPZ6\nc7fakIXkV7fbzTAMup3QCCN7fQkajUapVEqZjGhXr85lTpBOp6MoKhqNil02CYIoLS3NzTCS\nI2wZ+M1vjDQ9P4MPbrVa8+6xGggExPZsF9Z0VCAI30Fz9bzn3QSDwe3byVdeOf+3UoCOIVar\nFVVjk5xftVodDM5Ys0Yt9GE5cUKtVCovu6z244/Z3/8+inpbXC6iuGxfCzNLAoOZnqhUqtea\nXhvyD13pvVIf0dto24GqAw7KAQDf5r4tNqTLFD3Qsw22fQKfjMJoEIK1UPs1+Nov4BfVcP4X\n7kz0zMPcw5+Sn1pIi9Q+IDInjrMu5iUqmGkLLuXb9qMAACAASURBVHBgEqDT6cITiVNNDvoa\nkqqY0ApFsadRYLJKNBqVqo4F2tqU49WBIoUXUias4ISyWeAgCKKxsXHSMbqZhaKo+vr6Dz80\nb9rUHDe7q6io0GoVOXPZFKSHqB2OhTU1zNe/7ty0yfK739UIo1tLSjLcL+B2u9GNhNKJG25g\nc6C4FFc3hDWdvXtNND2PYRg+fCfveTccx42MjNjt9t7e844h+/fXPv10KRSYY0jydqdvf5sD\nIADgpZfKBgcBRD4sO3a4338fDh0q4bcUYBEHg8EUC+/q3j2oO7hHv0e4cQm3ZBe9KxtPxzCM\n1Wo9QB94tvpZfqMRjE/BUzawvQwvA0AkEhkeHv69/Pd/qfuL1D48paWlFosF/WbFWRdTFFVS\nUgKY6Q0ucGASoFarZROJU02OQqFIslCsv/POXC4UY4qOi+uJcphmmnGS65vCixZl9dlRjO6Y\n3e7z+cLhsObcufpVqwCyGN2ShNLS0nfeUQ0OykE0u9u9G+69NxdjcLvdHR1yQXoIMTwse+qp\n6jfeKDUa5byoBEQS3zThOE7c+idM0Lj9dhIg60tStMDphGVh7149qumUlzPnzimuuGJuQwNc\nfz1z550gkwEUQN6N0DGEH8yaNRZU4MhXtLAYjuOQw46QC88vBaAAgOPHEz9CU5PNZKorBL0M\nBjMJgsFgR0fH0aNHjx07hhy1jhw5csUVV+R7XNOXFbDCDOZe6PWBTw/6OTDndrh9DbFGmYUf\nmnA43NfXxzCMpkyzy7CrNdiqiqperHjxNzW/AYAe6AEAhmH6+voikUhpWanUPkJkMlltbe3Z\ns2fF1sWNjY0URWX8VWCKC1zgwCSGOn3aNDKCmmb5eFTPgw9qH310Euq1hoYGsjAWijFFh1ar\nZZPriS65JE9DmxQXRsAqN28GaX2TTKnMtr6JJMmqqirU/A+HD8e25qkx/tSpxIKVyy/P+lNH\no9GhoSGfzydMD3nySf0zz5QBgNF4gahErVZrteUZzNEjCIIkSaGAIi5BQ6GYm7Enk0apVMrl\n8nA4zDcKkSRwHDgcsUuFwUH4z/9Uut36++8fFR7Iz9Xb20Pr1uWoZSwajaboGJJ3CIIgCELY\noyFKSJmDtr/6avyxPp+vv78fJPQyVVUx6QcGU7CEw+G33nprp6AUR1HUktz6WGPi2AJbtsCW\n3DyX0WhErbjXOa/jN37F/RVUvPgifBEAzGYzqgIn2SeOioqKa665Rmju09nZ2dnZuWrVqoMH\nD2brxWCKBFzgwCSGJMmGhgYUp0q99RbaqLv6aqk41eSoVKrgiRP9IyOocs9XTLjt24lHHsng\nsDFTj5KSkrB0VutE9UR5weVyOZ3OUCgELNu4fr0wArakpGRkZERK31Ry00051TdlwWRkQohn\ndznDZDKhbyfhNPKWW6yowAHZF5VoNBpkNyOUTqB2GLVaJpPlyGa1rq7u8OERvlGospK54grv\n8uU+nY597bWSgwfLAODIEe39958/RDhX/8MfXDRdn5uh+ny+JC6wuSy1pIJKpfL7/ZDo/CqV\nVJLzm7y35ZprAgBF4AiImbZ4vd6hoSGr1bpp06ZFixZ1dHRYLJaWlha8xp4DUvG8yDaBQAB9\n9fGwBGuQGR6rfQwAaqO1HVQHx3H8F514Hz2jf4R6RFzIxdbFmCTgAgcmGXK5XC6Xw733pn8t\nr1QqZ82axbJsKBSSHTmCNhLLlqU9RszUgWVZgiAIh0OocYBt28iurn6DIa465n7gAd3Ondlz\nw8oURqPR6XSi2/rdu1EELImsN5YupSiqoaFBifVNeSUUCiW3wBCTcVFJdXW11+t1u8k4j1UA\nqKmpyfCTSaPT6d56S8Y3CpnNspdfLn/55XIAuOuuWIpQeXnM+EY8V6+pmYD/dJqIm3qEpZZ9\n+8w03ZKzwVyUyspKv98v9tBFdyX5Hkve2/Kd73hxgQNTsESjUYPBwLJse3t7e3u71+u1Wq0A\n0NbWNjQ0NGfOnML/BS9SOI6z2Wx/JP74bKWk50VuiEtsvWHODT3KWL/JYv/i/WP7GxsbmSgj\nFDDG7bNnaE/DnAbxhBVbF2OSUMSpY5hihCRJlUpF3303cBxwHHzjG/keESb/cBxnsVg+//zz\n7u7u06dO+f/lX0CgcQAAmqZnzZo1a9YsvV5fPTSE7in553/O6rURwzBgt0NLCxAEEAT89KeT\neBCbzcZXN+IiYNm6OuQeUlpaynz6aV9vb9epU6dOnuz9y7i91o4dwHFw9dWZeT0YaVDtLA6h\nFiASiX1j8f9WrMjwGNRqdTjcuGbN7MOHdcJ2GKNxhsuVSb+Pi3LiROKVj1AodsFw3XUhSJRm\nqlLRmbUmSY5wBTguWviJJ/rLygrr8qa0tNTj0YvPb29vtc9XleTAuHTYQ4dK7ryzxeOhli3z\n7d07IJcX1svEYIQ4nU5hIfL06dNoRtrW1hYKhbxeb/6GNpWJRqN9fX2jo6OqgGqXYdcrPa+8\ndeateyz3oHvFfhZZRViDCBNhq8xKc7GfmBPqEzvLdwKA8FpOvM9u/e6iDsnG5AWs4MBgCheG\nYUiSJJ3OOEVDvseVSViWHRgY4BWM+v/8T/UHHwg1DvyeGo1Go9HAfffBffdlbzx+v99isfj9\nfpZhmjdu1F5YapkoNpsN3RBHwAba2jTju2k0GrQWEYlEZEePxrZOG5klwzAcx8k8nnx9zpOn\nh+zfP0bTuQhxeeEFXcIEjZx5rCKefdY9PDwct/HMGeXatbMAYOnS4P33lx454lu7tlaYd3P6\ndEltba1SSeYs70aj0SDjkoSyCJ1Ol6NxpMwrr1RP4vyq1Wok3hbrZWiaU6uxfANTuAQCAeF/\nT49rFVHaRSAQKMC/0ynAyMgIeudT97PIHsI8VzknP9x9mAPumObYxuaNISL0svZlAKAoSqFQ\noCBt8T6vlL1CErjAgZkYuMCBwRQckUhkdHTU6/VGo1Fg2ZZ77lGnN80uZKxWK1/d4DUOtNWq\nOnWKrasjc5uQ4nK5jEYjWnDQd3ZqjxxJWGpJkWg0Gg6HQSIC1tfaqmJZ4boEQRByuRzWrYN1\n6zL0ggoalmWtVqvD4WAYBlh21qZNmjx9zoUOCOJpZHl5aW6GIZWgkQOPVSHi3nhhueepp2xK\nZeNbbzXmvRZDUVRVVdWxY05xtDBN0xpNeWVljkaSIpM7v+Xl5Var1eHgxEUcjUaDCxyYQiau\ng6C7uxsASJKcP3+++F5MRmAYJomfBfK8yOV41Go18q7mtxBALPUvVbPqEBUqgViea2VlpbCw\nnnAfDCZ1cIEDgyksQqFQX18fr+rUd3ZKKRqmBrxHlFjj4G9tlbRAyAIMw5hMJnTJFVdqYfR6\noqpqopZoySNgA21t0/nyLl6509mp+fDDfH3OtVptEi1ASUmOrq7y6LEqRKPR8JEf4nJPba0e\nCqYWU1NT88YbmvxGC6fO5M4vSZIM07xmDRlXxJHL5ZddVpvpMWIwmUQuvyAYq6urCwBaWlqU\nSqX4XkxGCAaDwkuLOD+L3478tnF2Yy7HQxBEQ0PDTtdOB+W40nulPqK30bYDVQcclAMAbiRu\nRLtVVFT8jvydKWRKsg8Gkzq4wIHBFBZGo5GvbsRNs7n6eiK3ioZswzAMCg9LqHHwt7WpotGc\nea273W7UqpCwnSTiclVMMBeUoiiSJEv/8peEEbAETVOjo1mNgC1kxsbGpJQ70dpaKrefc4qi\nampqPv7YLtYCqFQqnS5nJY6CgKbpiooKm80mLvfIZDL0V1AgtRgAOHtWk3B7jkstWeXpp5W5\n18scOwZ//CN88AFnMBBOJ9TXw803w44dgOekmNQpLS1FrqIA4PP5hoaGYLw/hSTJ6fXFmisS\nel4wBAMAJ9QnflX1qxfghRwPSaPR/I/qf14lX92j3yPcfilc+nP4Of/fw2WHD8LB5PtgMCmC\nCxyY4objOIIgwG6fGi4VoVCI71kVT7ODCxeq8jq8bCGtccjlKJDXd8JSS2DhwmgoNNEHJAii\ntLRUKgK28e67cxoBW2Dw3quJlTvo7zqHVFVVHTpUNFqAbFNbW9vXR956a3mcy0ZdXV0uXTZS\noXBKLdkj93oZt9u9dSv5xhtaGM9mHByERx8FloWdO7P1pJiph1KprK6u7uvrYxjmxIkTaO7d\n1NTkcDhmzpyJk2KzgVAXI/az+GvJX/Myqn8i/8kK1l7o9YFPD/o5MOd2uH0NrFGCkt9nBaww\ngzn5PhhMiuACB6YoiUQiFovF5/OFw2GaJJs2bFBNCZcKPk9LUtGQz9FlHpqmaZrWPf98Qo0D\nKZPlWuMgXWqZ3LKlXq+PJomAfewxePBBgOKuyk0ClmWTuJMEFi5URCK5Vy93dyf+85pKWoAU\nIQji73/X591lA4NIsYiDNBcffghDQ5CO5sJqtZrNZoNhzvr11q9+1VVeHt23r/q55yoA4I03\ncIEDMzH0en1rayvfiwoAnZ2dnZ2dq1atOnjwYB4HNlVRKBQqlUpo71oIfhZbYMsW2JL+PhhM\nimBbWkzxEQwGz50753A40Byp8rHHVIcPs8rxEm8xFzhiq9bj02zjz34WamlRjc+QQ5dcks/B\nZYfy8nJe4zDnxhvn3Hhj2fhFz8zvfQ+efjpnI1EqleUvv5yw1MKRpFpwfZY6NE2TXV1Dg4On\nTp48dfLkuRdi0lDLPfd4v/QlGBmJ7VfMH9pJkPxzHmhry7F8A/Hqq/FZsFlKhC0KCsRlA5Mi\nHMft2BHp7IRPPgGrFSKRmObi4Ycn9jihUMhisQDA/v39mzebFywI6vWR226LpUFVV6c1yGAw\nuGXLlpUrV2q1WoIgCIL46KOP0npETMHT19fnSPTr2T5tYsJyT319/QuVL+yr3ndaddpG2z5X\nfr6tYRvys2AIRgc6Gchmwsx1sM4K1nwPNusEg8EHH3zwn/7pn/DXzrQCKzgwRQbHcQaDYUq6\nVLAsiwwp0DSbVaka77sPAILz56Md5CoVmExTzLWhurqa6e6WvDuHM/+SkhLq+HFI1E7SvGED\n++ijMKmWGZlM1tjY6Pf7BwYG+Dm84ty5NCNaihqCIBQKhfqZZ6TcSWizGWbMSP0BWZY9ciT8\npz8RH38sMxpJbBmQPsXV+nHkSPgPf+D+8Q/KZKJcLmK6nf1wODw4OHju3Mz1651pai4cDgfq\nI9DpovzGf/wjZvd87bVBmJRcnGXZsbGxDz74YKdgNBRFLVmyZBKPhikiUAJ6vkcxvVCpVB8r\nPhZ7XgCAC2IBK0YwPgVP2cD2Mryc8wHmCJZlLRbLe++998tf/pLfiL92pgm4wIEpMvx+f2jc\nDUHcvR9etEiR9PCCxeFwjI6OosKN5tgxACDHFYbKs2fRjarbbpt6rg0kSdLd3War1el0RiIR\n5dmzc1avBgBu+3bikUdyORKapnWffy51L7lsWToPbjabWZZ1fOtbjm99q+TNNxvvvz+PnpqF\nQEVFBZUhdxL0t7N9e8OhQ+fFt9gyYJoQiUSMRmNHR+W0PfsokCgcDu/f389XJW67zYYKHBPV\nXIREZkNnzij37KkBgCVLvD09D69c+b/Hjh3z+XwAcOTIkSuuuOKij8kwTH9/fygUcrvdmzZt\nWrRoUUdHh8VimTt3Lo65xWCygdDzoipaNSM4Y2Z45lLv0sXBxaqo6qXKl35d/WsA6IGefI80\nW/BJbYFAQPi109LSguN7pgO4wIEpMpK7VAQWLizGAofT6TSZTPx/tYcPS+46FZf6SZLU6/V6\nvT4ajRL8Mn56BYVJjqSrKxAImM1mv98v7+5GpZbItm2yHTvSedhoNIrmAyAR0ZLLNNwCoaKi\ngjlzRvLulD/nNpttZGQEAEwmObYMmG6gS9hQKGQy1U3bs+90OlG3ZkLNxfXXMxO60kMJwXyP\n2KFDJVu3zvD7yWXLfOvXv3rXXf/J75n6Qujw8DCqm7S3t7e3t3u9XpSsMX/+fK/Xq9VOw+8/\nDCa78H4Wdrt9eHg47t5/dv0zKnB8Eb6Yh8HlBD6pLe5rp62tzWw2N0wtKTRGDPbgwBQZMa3j\nRF0q7HZoaQGCAIKAn/40V4NNCY7jRkdH+f+WvPkm7XB4rr46cMklAMDw0aQ7dgDHwdVX52WQ\nuYGiKPKuu2LOB9/4Rl7GoFKpmpub29raWsbbhmUpLFEmJxKJoBtS3rHTUMFLEITszBm7zdZ7\n7lzXqVO9f/kL2s5t35765zwajZrNZnQ7G5YBmALHbrejmfN0Pvt88ZSH11xcdpn/pps8E3o0\nhUKBqhssC7/+tf7++xv9fnL1avu+fQMU5du0adPjjz9eU1MDAAsWLEhFf8EwjNvtFm45ffo0\n+sZra2uz2+0TGh4Gg5kQfEwvgiXYQfngY7WPAUBttLYDOvI0rqzDJ7UhhF87LpdrGl50TTdw\ngQNTZCgUChh3qRB378uUShiXQvh8PrPZPDQ0ZDIYIqtXQ6HGrAQCAWS9ARcu7yt7egCAra2N\n7YcduZIgKGCFt20LBoOczZZmSSuDpZZYGF6BeWoWAhUVFbNnzxaWkyak3PF6vSzLotsJl69X\nr87QQDEFiccTm70nPPvXXTfhaOdihHekQhw6VHLnnS0eD7VsmW/v3gEAZkKPVl5eThCEx0Nt\n3tz0xBPVNM11dJg6OoZpmmtvb9+4ceOll16Kpkzz5s1LZZLAiy55To9/9S1cuFB8LwaDyRTh\ncJhfXwGAG+bcsGThkmvnXfuB9oPF/sXP9z7fCI15HF724DgO6dp4hF87LMsK3xbMlAS3qGCK\nDK1WK5PJNBLd+xW33AK7dnE//KHRaHS5Yl5K+t27Ze+8U7CGjnx1I+HyvvurX63q6srn+AoV\nn8/n9XpDoRBNktV33CEbL2CN1Nd7Pv+8eeNGbcGUtGQymUwm0z73XMKqnFKjmXresROCIAhY\ntw7WrZvogQmvUfjl6/b20Lp1xdiyhkkV/suTRyheuPXWCMDU/wDQdOxCjmVh7179vn3VHAer\nV9u3bh2haU4mk03o0eRyeTA4Y80a1eCgnKa5++4bnTcvdOKEGgCqqiL19RF+IXTBggUul6us\nrCz5A4qLIN3d3QBAkuT8+fPxOioGkz34BQAACBNhq8xKczRDMABwQn3iserHvsx9eZqsrwi/\ndvI9FkwuwAoOTJFBEERDQwO/+p2ApUtHR0f56gYfsxKaMwcAoKEBMm7omF7/S/Ll/fCiRZkd\n7BQAJen09/dbrVa32y37yU9QAQvdG2hr03d2ooyS2AH5LnAAQHV1tVQarv7OO3OZhjuVIMn4\nnzDh8vWBA1Ya1/CnNHEfgDjxglw+La5wdDodACTUXBAEMQmHixdfLB0clAMAwxA7d9bdemsL\n+vf22yVw4UIoanFPDhJdCunq6gKAlpYWpVIpvheDmQQ90HMz3DwX5k63DNTk0IKfQDknP9x9\n+HjX8af6n1JwCgB4peyVoqhuTCJemiAIpfKCvCfh1w5FUROt/GKKjmnx84+ZYmi1Wvazzwb6\n+7tOnTp18uS5F16I3bFjB3Acs2IF39Yr7viQNOmYIG63e2Bg4MyZM91dXYHrr0+n/0WlUpEk\nKdV0E1vezxqBQMDhcNjtdr/RWLA2JXGMjIxIFbAiNTXq48eFW9i6usyXtBIRjUb9fn8kEklY\n8KqoqNCOp+EkoABKMHnh2DG4915ob4eaGpDLobkZHnoILhSWJkNoASC2DKipmUyeJaaI4Gfv\n4rOv03HTJKGjpKTEYilZs2b24cM6oebixAl1MKinJ17kO3488fa2tiBcuBAa1x2TELlcLjwR\nPp9vaGgIABYuXAgAFxWAYKYGk5ijpk4gEPh/jv/3LDx7Ds55wcsAgzJQvwffy9RTFCk0TatU\nKuEWAoil/qVqVg0AOk6Xp3FNAIZhDh8+vHPnzvfeew/5DaVob1zBG9iJvnZQI17WhowpCPDy\nFqYoQU6QHMdFIhHZ0aOxre3tAOD3+5HqVcrQMf0Fo+HhYb6Got+9W3X4cDr9LyRJVlZWKpI2\n3WQjGjYUChkMhlgLNMs2b9xYsDYlQiKRiGPcryFBTvCsWXFbggsXZnuW4/V6R0dH+Xdy1qZN\nmkTvJN3d7ff7XS5XMBhUnDlTv2oVAMCOHbBtW5YHWKAEAoFt27jXXjt/fiaa7qlUKrVardfr\n9XioLVtmHD6sk8m4H/94ePVqB0VR5eXl2Rr6hRw5Ev7DH7h//IMymSiXi6ivh5tvhh07AEfR\nZZvKykq73e50QtzZB4DKyqqYOG6qQxDEm2/OGBwkYVxzwd+1e/dkvstfffX8bT6liEe4EJri\nKmh9fX1/f7/T6WQY5sSJE+gHuqmpSdxhhJmSuN1uNEflt6QewZPKgxsMBkWpYpd3V2uwVRVV\nvVjx4m9qfgNTOgM1derq6h51PuqgHFd6r9RH9DbadqDqgINyAMBqKHSTKrfbbTQaTSaTMOd1\n3rx5qRSvy8vLfT6f0WiM+9oJhUI4JnY6gAscmCKGIAi5XB7XvR9bUxrv+Bj6j/8ItbSUv/wy\nujfQ1pbmpMflcvHVDV4+QFutqlOnorW11KTEAjU1NRmJzEydSCTS39/PX1/yPR2FaVMiJHkB\nS3n2LIhKWlktcLhcLqPRyHeS6zs7NR9+KPVOqtXq2A/zO+/ENk1X71h0VTo4ODvNdM+ZM2e+\n955p7dpaoWVAV5dOr9er1VRTUxZfAgAwDGM0Gjs6Kg4dKuE3TrRMg5k0NE0zTPOaNVScYYRW\nq9Vqp0eGCgAA/O//JlbjXn55uo+s0+lGR0f577e4hdCSkpJkB4+jVCpnz56t1+uFuQadnZ2d\nnZ2rVq06ON6sh5l6sCw7ODjo8/msVqtwjjp//vyMCKzQNzDHcdc5r+M3fsX9FVTg+AL3BZj2\n6/Rqtfqo/Ojr9Ot79HuE25dwSx4lH83XqFIhEAgYDAaO4+JyXufNm+dyuUpLS5MfThDEzJkz\nlyxZwi+JAf7amU7gAgdmqoHWlKQ6Pii5PE1DR5vNxnEcQRBi+YC/tVXLcZNQvqHITL/f7/F4\nQqGQ8uzZmq99DSCLy/tWq5WvbsSVaRi9ns5JT8fkSF7AopzOuC2ZakpKCMuyw8PD/NV/3DvJ\n1dcTUu/k2rWwdm32Blbg8Fel+/f38/kXt91mQwWOqioOUr4spSjqrbcaBwfRw8YvX997b4ZH\nLoTjuIGBgWAwaDLVplmmwUyaZ55R5eXsFxRCzUVmkcvllZWVY2NjHo8nbiE0Go2Gw+EUp6lG\nozEutRHRPl0rvNMEo9GI2grEc9RoNJq+xsrpdAp9NFmCNcgMKAO1JlLzQOgBmLAFzRTkavpq\nG9jOcef8hL+arZ4Dc+4k71xDrFFCQXdxWiwWoQmxMOfVYrFctMABAN3d3cLqBg/+2pkO4AIH\nZqqhVqspipKKWdHfeWeaHR/BYJAgiITygcDChfJweNKuaeeX9/kxZ+1b2O12oxviMk2grU3F\nMMk6t+12aG+P9bPkvMMCDUyqgOW/7DJWrRZukatU2cso8Xg8fBe6+J0MXXJJQV8+5A+Xy4Wu\nShOme15zTQBgAot7UpYB6S9fJ8dut6O+pIRlmuppJCDIJ/k6+9OH2tpagiCuvPJK3vkIJr4Q\n2tLSggNTphuhUIi/0kDwc9TW1laHw1FVVZXmUwhjhm+Yc0OPMtaTsti/eM/QnsrySlzgAIAt\nsGULbImtGhSP9SIqjfEI7Y1DoVAkEknSIheNRq1W6+kL4wgoinK73dPEmwmDCxyYqQZJknq9\nXpkkWjW9/guO45L0v5Rk5Bouy8v7HMch+YZUmUYmKnAwDGO32wOBQCQUmvG97ynz59ah0WhI\nkpQqYKk//bR5/Xrhlurbb8+SiQkA8EHrCd/JIC5wCGBZNhgMMgyj8PnKvvjFyqEhALBs3mxZ\nvx4uTPe86SbPhAoc2Vu+To7H40E3EpZprrsuNB0ySvNOvs7+tMLn8wmrGzx4IRSThLgJKkw8\ngid1xBmou/W7Hw8/nsGnmOaciZ55mHv4OHHcQlpCRKgWar8GX/sF/KIaslLL5zhOqM0BUc5r\nNBqVKnCEw+H+/v5IJOLxeCZh3oGZGhRPKQ8zJUkvYFWKiooK3z/+cbqr69TJk8KYFfYnPwGO\ng6uvTufB5XK5lHwAKEpmsaQ5+BxAEARJklLBtIG2trjwRZ/P19PTY7FYPB5P6c6dyvffTyuB\nNe1U3ZqammQFLDFZq8LE2pEk3snI4sVZet7igmXZkZGR7u7uvr6+oYGByOrV1NAQuivQ1gai\ndE+KKo6V3kgkErdFWKa55ZZAPgaFwWQepL8Qs23bNovF8oMf/ODKK6/MRjoGpqiJm6DCxCN4\nLgovmE2YgYpDiDMCx3EjIyMHrAf+Qv+ln+r3Eb4c5NQQBBHXwSS0N4YLE3DjMJlM6Ne5vb19\n48aNl156Kd8YJZT8YKY2uMCBySksy9pstsHBwZ6env7e3tANN2QpuaOysnL+/PlNTU21tbX1\n4ybw5PLlFz0wHA47HI7R0VGbzRYwmcRT8bKyMl4+MOfGG+fceGPZuEa3af166rnnwuGw0+m0\nWq3ugQFu1qzCDF7VaDRJbErk1vPp8dFodGhoCF2LxGWyRmtrU0xgdbvdw8PD/f39hsHByOrV\naZ70qqoqz5EjKCRYWMDyP/TQQH//2TNnDH/7W2zXHTvSL2klAQWwSb2Tse6Yac/Q0BByrgGB\nnS26y7egLS7dU6tli+WqNO7yK65Mo1BMiwgPzLTFYrGcPXv20KFDe/bs+fDDDyeU4IiZDogX\n2CcRwZOc0tJSoeuZMANVy2p1uiKIQeXhOG5sbKwAy4Umk8lms5UwJbsMu17peeWtM2/dY7kH\n3ZXVnBrh6YuzN1apVFIFjmAwKO5t4c07UESA1+u97777Cu19xmQW3KKCkSALPgsMw/T394dC\nIWTSWbZ7t+K997KX3EFRlE6n0+l0sHkzbN580f05jkN1jdj/JZJTKysrme5uqQexNjaaP/+c\nP5wYGIg7vECorq4OS3R5zLjrLmFPh91uLzZ88QAAIABJREFUR9WNhKaqsmBQqbywD+PCTw67\ndevQ0JDX60V36nfvlr3zTvonvbq6ury83O/3h0Ihzbvvoo3qq65qbm4GAPjww9h+SRTUmfiE\nq9VqpVIp1S9T+p3vZK87plhwuVz82Y8zYQ1X6zd0XB6X7klRVIrRDHlHo9EglTXLwt69+n37\nqjkOVq+2b906IpMBlsJipjBmsxktiobDYaEIfPbs2fiTj0FotVqSJHkdx+QieJIjl8tfb359\nwDsgzkD9VvRblLxoqswej8dkMh0/fnzPnvNZJ4VQLvT5fMgeOGFOzXJ2efYWymtqatxut8vl\nirM3djqdFRUVUkeJNRrCxqhgMGgymd5///3du3fzOxTC+4zJOLjAgTkP+ioJhUIkQMNddyky\nra0wGAyhUAgACIKIm+pAQ0OKWoDsYTabz1c3pJNTSZKku7tHzGaHw8GyrPLs2TmrVwNAtKPD\n+N3v8m35BR68qlarFagQkxDBaNEUTsqtI+L3K5VKv9/v9XqDwSBNktV33CETfHKGh4el5rdp\nnnSapmNXSOICloSJCcdxLpfL7/dHQqHa7343/U84yiGDC42sLqDAznvu4Vv3xQWy97zLDh/W\nCdM9AWDx4ur0rfVzQ2Vlpd1udzphy5YZcWWaiorKYnkVGMxEiUQiY2Nj6HZcOkZra6vb7S6W\nGiUmq6B+0tHRUXEETyQSETewTI63NW8f1ByMy0BdzC7+lfxXGXn8HIBKPxzHxZULW1paMqJz\nSQf+mhYRl1Nzn+8+KMvWU8vl8ubm5vr6+gnZG4vNjIWNUaFQCC2MCd/nWbNmpR4IhSkWcIED\nAwDAcZzBYOD9rvVZ0FYEAgFeNiae6oQXLZKn/xxpEIlEhNWNuKk4W1dHCqbiJEnW1dXV1tZG\nIhFiXCwQXryY/yWIOzxSU8OWlRWa7J46fTocDrtcrmAwKDt9uvbrXwdIoGVgWTaJqao6GjWZ\nTHwQV5xAI7J4MR8NmDBkJJfvSTgcHhwcRCW2DH7CFQoFc/r06NiYz+cLh8Oac+cav/lNgDzk\nyxQm4XCY4zjK5xMXyA4HlkOidM9sBvtmEpqmGaZ5zRpqcFAuLNNoNBqttibfo8NgsoXX642b\nRQhF4B6PBxc4MAiUk5JmBE9yVsAKM5h7odcHvhquZi4x93a4fQ1Z6BmoQkZHR9GfT1y5sK2t\nzWq11tfX53FsQqspcU6Nvjy7C5Ojo6MTtTcWt7gKG6OQHjnufV64cOHIyEhJSUmc/RymqMEF\nDgwAgMVi4asbcZNzrr6eyIS2IhCIWe5JBqym/xxpILxiSzgVV4kOIQhCLpfD+vWwfj0AOEdG\nwGZLeHhw4cKw11uAzgJyubwapVm++WZsk+hnQy6XyyU8JjiSDPf3O8bL3mKBhl+nA6cTpE96\nzt4RjuP46kbcOKO1tVR6n3Capmtra2P/4Ts5cb4AAACQJEkAJCyQfQIJ6krFle75zDOqwUGA\nRGWae+/N26gwmKyCQriECEXg4nsx0xkkDRZvz1QETywDFUEk3bUgYRiGvzxGCMuFbrc7vwUO\nfs6fMKfmCeaJrD77JOKlVSqVXC7nE+7iGqOECN/naDTq8/mKy7QFkxxcrMIAx3G8eEE4OVf2\n9ABAUPSlMDlickSJvIlMPcuk4a/JEk7FQ4sWpfgIkq0cosCFwmLtWuA44Dj4xjfi7ikpKZEy\nVW3esIF+/nl0W/zJCV1yScwmXTquJSevDQDA7Xaj6oZ4nP7WVnGa3eSRfienJ2q1WsqE9Te/\nNYT7B9C7xf9bsSK/450Yx48n3l5cZRoMZkKI1zmFInDcnDU1CAaDDzzwwIoVK9I0YpSK4Nm+\nfXumhhqNRj/zf/at0Ldms7N1oJOBbCbMXAfrrGC9+MEAPdBzM9w8F+ZO4tj0EV8cJikXIp/+\ne++9N/3zkiJ840bCnJo02zqCweCWLVtWrlyZwddCEERDQwNBEB6Px+FwHDt2jG+M8ng8wo4b\n4fsMiU4EpqjBCg4MhEIhVH1IODn3tbaKxQuTQC6Xg3TeBK1QgMkEDQ2ZeKrJEDNklujFSCXv\nk6KoZK0chXnNl4LRpk6ni5w5I/UA/tZWkP7koPbRJHEtOTvpqIQhVX4i/H6NRpODYUxDKioq\n/BImrM0bNhS7Ceurr+Z7BBhMzhF/WwpF4LiVfQrgcDjefPPNX/3qvI1FwRox2u320dHRp8uf\nfrnuZX4jCjG1ge1leDnJsQDg8/meZp5+tvTZSRybEcQFQalyod1uN5vNn376aWdnp/DwrJ6X\nsrIyq9XKCyL4nJoQFdKy2nQunBiGef/993fu3MlvydRr0Wg0LS0t9fX1fIs0jDdGrVixYu/e\nvWiL8H2GRHVbTFGDTyfmItoKNINNH61WS1GUlBag8tZb4emnM/JEkwN9TUtNxVVa7UXzPpME\nr3IkqU0k0UwCx3Fgt4tDatOHYRiHwzEyMjJiMqWY2Ep3d48MD6NYVj6TNfjjH7ucTt/y5UkE\nGhqNJslJr7njjpyd9Gg0mmScMaUJJgvI5fKSHukkuWlvworBFB1KpVLosiEUgcvl8rKyrLkO\nYnKCxWIxmUx+v3/Tpk2PP/54TU0NAMyaNUucT5F3HA7H8PAwy7Kl0dJJhJiazeb+/n6ZR5bj\nAFQhcrk8zklUWC7kKwg2m214eDgajSIjUv68ZDu3iCCIN1ve/P/snWl43OS59x9pNPvifcZ2\nHO9JHCchARJKKaSh5bSFtoctaQlha9Jma6BAaVPCYhJOW5ocXhzDgdOcQkt73i6Uc8rbC0oJ\ngRACgUBYstqJE2/j8diz7yPNaHk/PLaiSKPxeFZ5rN+VD2NppHk0Uka67+e+///nq58/pT3l\nxtxnNGcemfUI9KlZhaxKe7eBQODMmTMjIyPcY5kzZ062jsVut3OzGyzcRhXu9wyAbHxWbMgV\nHDIgR9PsDMOEQiHYF6CNRvVf/vJ8NpZOSEFDHZVKVVZWJub3aVi5ctKpZpPJhH7+ecLNU5+p\njkQiTqczGo2SsVjzD3+oy7aRjd/vt9lsMKWVumMrgiA1NTVmsxnHcfTAAbhQc+WVFIaBpFcO\nardXV1drTp4UHVC+TrpSqUySflI5nYAV0ZDJNujJk7FYzO124ziuOHECirAy27cjjz5a6KHJ\nyMikQ11d3dDQkN1u57pjNDc3GwyGQCAg5zikjMfjeeyxx44cOXL06FHoknbo0KEvfvGLcC1B\nEFB5USjEODo6ajKZxstdJQBN06Ojo/B1QhPTy8HlSTYPBALw0NLYNruYzWbbxBQaN12IIAhU\nSaMoamxsDL4h/75Fr2Ovv1b5WmdlJ3fhErDkV+iv0tshjuNWq5VhGN6xzJs3z+fzZeXXI6F4\nB0mSZ86coSgKQRChaTEsM5cpGqTyOyVTQJRKpU6nE4vtZ61bl0YZeSgUGh4eHu8epOnGTZvY\nSoHhX//ad8UVAADWYFUioU5tbS0p3osxaSiOIIjh9Om0NwcTcxHwR9nS1aV7773sGtmEw+Hh\n4WG4/zSkZBUKhV6vBz/8Ifjh+BSHjmG4BRq8K6f6rrvArl1lDzzg/eij/tFRWCXBnnT6scfQ\njo7MDypFjEZjXLxRgvzlL0EKMisyaaNSqWpqagAAYP9+uARZtqyQA5KRkckAFEUbGxsvueQS\n1kILAPDUU0899dRT2XLHkMk6FEVZrdZDhw49/fR5U1WFQrGIc/vz+/3cyJArxEjTdCAQKC8v\nz+eYkxCJRHjVl1wT02qqukOR7BmDtToWbmshLR1Y/p5PysrK4vH4uXPnuOnCxsZGrVZLEIRW\nqw2HwzxXXZ4QaU4THFyfGguwtILW28Htq0H6PjUOh0PsGhsbG8tdehTDsIaGhpMnTxIEwTUt\nJghCblIuPuQWFRkAAKipqWEr9hMwxeg6Go0ODg6ygj2Wri7DoUO0ZvyncNa//uucOXPq6+vr\nneMaThIJdRAEUfb0RCMRp8NhGx52v/XW+IodOwDDgKuvnnwPJ04wNO33+ewjI6P//OeUNo/F\nYmx2g80+EK2tAADSYmHMWXCdZG8qQqHN9PQ+EQRJpUCjrKxs3rx5TU1NtbW1sybmW9DLLkvj\nE9NGr9frxbNX2Be+kM/BzGhkEVYZmaKgr6+Pm91gyZY7RhrgOP7Tn/50+fLl+dFfTJtQKHTf\nffddeeWVeR7n0NBQKBTi9Tg0NTVxi/lZtQUIT4iRt7aw8FQhb2q9afGCxd+a+633DO9dFLno\nlaFX6kF9ks253iW8bV86+1JNvCbJtlnHbDZ/85vfXL58+ZYtW+CS3bt3t7S0rFmzBiQVIj1+\n/PjKlStzeiFtBVs/Bh97gIcAxBAYehu8/T3wvUxceHmy7txrLB6P5/Qa0+v11157Lfd77urq\nWrp06R133JG7D5UpCHIFhwwAAGi12ujRo2dtNthjmeE0+9jYGMMwCIKAhJac1dVqaFW9cSPY\nuDHrx5IhWq1Wq9UCAMAbb4wvmsrjGoIgJSUlJSUl5+UHU9vc5/MJsw/QZTba3o6EwwaDIfVh\nCKFpOonQZqitTTtxyqZEWVmZ5/DhvtFROL3AXjlUR4fiscfYt6EoqtfreQUgeUZx6pTNbodP\n5Ow4Qz/9qf6JJ9I4cBkZGZmZTBoOjjklEAjs3bt3165d7BIJ6mLCrooDBw50dp4v+M/POIPB\nIHwGEPae+Hy+yspKqETAuxvyhBglda/kCnAKTUyfrHzyJfCS2LbQyUVs205L5xfovE57JE8X\nJvEtevXVV9mFErzgE8Kru+FdYznVROvr6/N4PMLlBUzLyuQIOcEhM45Wq21tbY3FYgRBqA4e\nhAvTmGZnA2mQKFaPzJ9vSCuQLgBr14K1a/O2OUwtidl8oDieYYKD59gq9HmhKCq93try8vKS\nkpJwOEwQhG6iAUEhvZoIFEVnzZpVVVUVjUYVb78NFxpWrADT4mqUkZGRkRHB7Xbb7fZQKLR5\n8+ZFixZ1dHQ4HA6oiykp7UCr1RoMBgmC4I0zHA7nepyhUIj7J7cvAK6FCQ6tVsuNtHlCjOPT\nP9JAp9MhCAKPApqYMoA5oj+yqXETgRCvGJJ5oCAIgmEYbKMWbvtq6atKRplk86yTPF0ovDbg\neampqbn++uvZC6mlpUVSJyghvaD3p/U/Pak56cJcBEJUkpXhO8PgY9BcMn6N5VTkRWppWZnc\nIbeoyFyASqUyGo3qTZvSLiOnKAr+fIjF6rJjRUIYhkli85H5LzKGYQiCiAltAoVCMdE8kgYK\nhcJkMlVVVenvvlviDQgqlaqkpMRwzz0SH6eMjIwUOHQotnEjsWQJWVXFqFSgsRH87GdASnX6\nMiAej0OxyaVLl27atGnJkiVcXcxxLTAJEAwGg8EgSDROh8OR63Hy9s/rPWHXlpaWshEmT4gR\nPh8CAHAc37p164oVKwrbCoRhWFlZGXcJa2IKAChBSpJvXlJywRu42xoZo6RMQ9VqdULfIt6F\nNH/+/IRlINKBoqiXQi+9bnp9SDUUQSMUQo0px0LfCYH/Gr/GNBoNz1Mmk89yOp333HNP/nvB\nZKSAXMEhk2XGiwbFKwWEpt8yAAC1Wo2J23xo3G5QVZXJ/hEE0ev1YoKgDRs2pCElKyMjI1Os\nkCRps9k6Osr27TsfWgwOgl/9CtA02LmzgEOTOY/f73/wwQc//vjjkydPQlWFbdu2cXUx/X5/\nRUVFoYcJAAAwu8HCraGArnM5dZ/hTYzz+gLYBzMURWfPnn3ixIlYLMYVYgwGg83NzSRJOhyO\nQ4cO7eT8ByhgZ0RNTc2LmhftcfuVoSstcYsbc79Y+SI0MV0JVibf1mw2/xf6Xy7gEm57M3Nz\nPkY/FWbNmkVRFEzYcc+L1+s9evQoeyH5fD7pqMDywHF8YGAANaC7vLvm4/PpAP1K5SsvNLwA\nAABzQENDg8/na2pqyspnud3usbGxzz//nKenOy1aeGSygpzgkMkyKIpqtVrNH/+YMFZX63TI\nyEgaprNFT2lpKS5u88Hs3Ana2jL8CLPZrMielKyMjIxMscIwzMDAAI7jNptlwwbnNdf4y8qo\nPXuqXnqpHACwd6+c4Mgfhw9TL75IHz6MWa2Izwdqa8Ett4AdO0A8Hh4ZGfn444+fe+459s0o\nirJNslLTxeRJRbI1FHPmzHnqqad6enqOHj0KB8/1bc0WBoPB7Xazf/J6T7g9sHq9/rrrruNK\nFXR1dXV1dX39619/6qmnKIoKh8PcFpt58+Zl2F+D43hHR8fhw4ePHDkypW8AQZD3y99/Dbz2\ntOVp7vIlYMkvwS+Tb6tQKA6bD/8D+Qdv28XM4p0Kyf33VigUjY2Nl156KbdGA54XNimwYMEC\ngiDyNqRj0WMdaMdR7KgDdRAIUQ2qvwa+9gR4ogokmI1jGGZoaIgkSdaU98qvXumv94NjAAAA\nPhw/lqx4MHk8HrvdDgCAerrTq4VHJlvICQ6Z7GM2m6mk1qFypYAQjUaDibvMIpkJIJEkqVAo\ndDhOTzzqObZsCaxYAYU2iYceUv/bv2WyfxkZGZliwuv1Ql2k55/vNxrH2ypvu80NExyZldPJ\npEooFBodHX34YbOwiIYg4t///gDDMLwYprm5ube3F0hSF5PX9cDWUCAI8sILL7DLczTPbDQa\ndTpdJBIBgt4Tk8nEDfzEhBhbW1thizFPpnTOnDkkSaYtnRAOh9988820S0IyMTFdjix3AAfc\n1syYW0HrHcgdq5ELtu0FvY+ARz4Bn4yCURzgycP4nNLf35+wAwV+8/CCz8/VHo/HrVbrH7V/\nfKXmvNDJMBh+AbzgBu5XQAL1E7/fz001DtmG/GY/gOfcBsD28eWZi31CHV92b9wLFbbwSLbC\nRSa7yAkOmexjNBqpM2dEV2evUmDcq8XjAUuXgv5+AADYsQM88ki29p9nsO5uv9/vcDgIgmBt\nPuKPPKLcsSO9HeI4PjY2Bh3UUQCaNm/WDg7CVaovftFstcLX6i99KSvjl5GRkSkOAoEAvL+w\n2Q0AwIcfjs9yf/vbBADqAg1tphAIBKxWK8MwNptKWETzz3/S69YxIJEnyOeffw44tQkaTfp+\nltlFp9P5/X72T7aGAgDAzdHMnTs3R4Kj9fX1J0+eDAQC3B4HkiR5EuYJhRiDweDgxPMDhNti\nA31Y0hiS1+sdGRnx+Xy8LFXqe9gKtm4FW9P4aP62iTIDkUjkL9Rf/mL8C7skeRifU+B5oSiq\np6eHe4K+9a1vgYkLPg9XO6xuIwiiRFWyy7prPj5fS2n/t/x//8P8HwCAXtCbcCueKW/vwl5w\nBgAALopc9HTw6bmH51ZXV2dlePCJl7uEe6EGg0E5wTFDkJCIjkwxoTh1Kh6LuZzOYavVsXfv\n+NIdOwDDgKuvznDnNE2PjY319vaeOnXq1IkT0RtuGM9ugGnfZ1FSUjJnzpz29vbGiVJSZbp1\nqqFQ6Ny5c8FgEP7WV3V2ag8epCdufqVf/arp3nunh9CmxwOamwGCAAQBjz9e6NHkhPGHlYRH\nOgMOPxeMuwDK355MWsTjcd5caE+P5umnzQCAiy+O3HprVGQ7mexA0/TIyAj8YXz++f4tW8ba\n2nCLJX7bbeN3xtJSfrsHfHNrayu3NgHDMK46Y2EpKytLqN/Jk4qcO3cu22WTLaAs6DXXXHPx\nxRcvX758y5YtcHlXV9fFF198++23T7oHboAK4cqUCtemQiwWg2eZ9w20t7cPDw8X1u2CYRib\nzdbX16fwK3ZZd73a++pbPW9tcY5/b2JhfB5QKBRceVReMU4e5Ga8Xi9shPm279vf8H+jgWgw\nk+avBr4K115GJ/ZeZJMOrCkv/BOa8kaj0WzJ1grFetkLtb29ndcmJlPEyBUcMhmQtHRCqVSO\nZ/TZBEc2jKYpiurv74fFwwAA80TcjsIleUhw5L5gBEVRdP16sH592nugaZr7fGB6882q558P\nXn015nRqT5xgamsRiyVLg80+DMN4vd5AIIDjOApA/YYNmmJJYPGgadrlcgUCAYIgUAAaN23S\nThyps77e19uLAlD3gx+oi/TwcwFN0w6HIxAIxGIxhGGaNm/Wyd+ezNThiWHv22d66KG6SARd\ntizc1TWoUtUVamAzhHA4zAYqCYtovva1APf9pzjyUmxtgt/vb25ulo4dBoqijY2Nx48fx3Gc\npxOJYVh3dzc7z+xyufR6fbY+1+fzHThwYKe4bEx6fQE8mdL0BsbNYnBn2mOxWCgUgqYtBcHh\ncMB+EFYzAgDwFf9Xnql6BgBwObi8UAMDANTU1OA47nQ6eYKjCILkwaaQ5zdMI7RVaf0/1f8H\nAGCOmx/AHwCJTppKpRp/kciU9/Z3bs+WbK3Qx4C9UNva2mSXg5mDnOCQmRpQkzwajVLxuOWu\nu1SpBA9r14K1a7M1ALvdzmY3eHE7XVODTha34zgeiURisZhKpdIThPpLX0olVUHTtNfrjUaj\ncYKoXbdO+gFnMBhknw7VfX11Dz9MNDQM79jRds01AID4RRepCjq8JNA0PTg4yM5fWTo7Ne++\nm9cEVr6gKGpgYICd+Kq8MFXnbmwkCcLS2ak+cKAoDz8XkCTZ39/PqqyZd+/Wvfee/O3JpIFe\nr4dqBcePa3/xi9oTJ7QAABQFw8Oq3/zG/PTTWQs+ZRKSUCuRLaK59FL8ppsuECOAMQwA4Mkn\nn4QvsqhZmEU0Gs03vvENoX7nVVddddll41PfCxYsyFYFB8MwVqs1EAh4vV5uD0hbWxv7jaWI\nWs3vyeLKlLLh65Rgn+UgPOdaHMcLleCgaZoryAouDOOrqeoORUdBBgZRKBTNzc3Lli3z+Xzs\nwrxd8NwSiZtab+rVjBezXBS56Omhp6urEneamEwmh8PB5rNYU15CQRhog9/v516fra2taauB\n6nQ6BEG4iTPuhZrFvKGMxJETHMVJLBaLRqPxeFwdDhtWrEAGBgDIQrlBNBodGhqCJV6Wzk7V\nO+/kOXigKIptYRXG7fiCBUn6Vmmattls5ztgabpx06ZUUhU4jg8ODrJHPS0CTva5AQ2F6u+5\nBwAw1NWldDgQggAA4AsXSjbBAUVD4OvpVXgyVUZHR9nsBu9I42YzWVFR3IefC+x2OxsXpZH9\nlJFhqaio8Hg8Ph+4554Gl2v8SYmmgd2ufP75yvJy2UUltwjLLrhFNC+84EYQwO1gOHr0aML9\nZK5ZmF3E9DsXLFjALYjgiQikDawQBAKlknnz5vl8vim50hqNRoVCwRYIcDsjEATJisFt5iUh\nSZiSUUskEuGeAl4Yv2dsT31TfdZHOCUGBga42Q2WPFzwbBEE22xCIiSYaDZ5AXkh4VZqtfof\ns/9hjVqFprz/4v8X3vXZ1tbmcDgsad2yMQwrKytj/5dxL1SFQiERx2iZPCAnOIoN2Lk6/sNH\n042bNo1nN0Cm0ThFUYODgzB3ywseqOpqRV6Ch1gsBvOyCeP28Pz5SRIcw8PD8E4PsXR1GQ4d\nmjRVAQsKYHajUEedBuPd4wwze9s29eDg0FNPEc3NZa+Ma2LFFi0q5ODEgZUy8LUwgRVtb8+J\n8FohoGmafTpJmKpLuFD2N0sCRVHsf/CpZj9lZHhgGBYImFevNrpcGIqCO+5wXXpp5JVXSt96\nywRkm9jcw9VKpGnw7LOWPXuqGAasXOl56CG72VxFkhg3hhkZGQEAXH/99T//+c9bWlqkIyzK\ng9XvJEmyp6eHu4orFalUKrPycbwyBG4PiNvtTpiVEEsEoCg6a9YsWA/C64zAMCwajQpLPCaF\ntwnPuTaNHYqB4/hbb72VehMEN7shDON3le96GbycrbGlR0Ih2PxgMBiCwSAQaTYxUAaxDQ+Y\nDrxm4hv6tuFt947dC1/zrk+z2ZyeKUxNTQ1JkjabjSTJo0ePwn02NTUZjcZgMJiVZJyM9JET\nHMWG1WqFPz0g5Rg+RdxuN8xuCIOHyPz5OorKQ2/b+A+6SNyOL1ggtmEkEuFmN1KfG/f5fDC7\nkfColTguzQcp+GRQ9sorxv37A9dcE6ur0/T06D/8EK5VajTAZgOzZhV0jAmIxWLwwSKNBNb0\ngiCIJKk6vLVVuDDU1iYnOJKQ/CstpotHJg8QBPH88/TgoAoAQNPgd7+r/N3vzq+VbWJzjU6n\n02q10Wg0GFRs3Vp38KBRqWS2bRtZudKLomhZWZlCoYjFYna7nRtsNzY2ajQaXKr3ZRaYRDhw\n4MCxY8dgHd9vfvMbnm9r5p8Sj8d5gos8WdBxH7oL+fDDD8USASaTqbGxsa6uLludEaWlpS6X\nC547nlgmhmE8b5e0cbvdo6OjTqcz9SYIruWtMIz/u+nvWRnYNKWsrMztdrOer9xmEyNjTOIW\nzDX0NTPmFqbla2Nf+7rn62pmPJPFvT5pmsZxPL1GFQRB6uvrlyxZwnXV7ezs7OzslFrPmkzu\nkIr2kkxWCIVCbHaDjeGJ1lYAQNxsjpWVZbJzmMtPGDxEs9cymhy1Wo0giFjcjqnVwGZLuCH7\ntYALUxWa3l6QtKJBCkedBkajEcMw/SefAABM+/a1rlrVumpV6cTPesl3vwv+9KeCDjAx49Mm\nEwms4Z//nGhu1k7c86Lt7envWppuGiJHajh8OMuHP3MQ+UqTZD9lZIS4XK7ubtFn65tvLqS/\nwwxh9uzZdrt+9eqWgweNGMbcd9/o3LnEsWO6gYHqTZueWL58+cKFC6+44gquJ8ju3btbW1vX\nrFlT2JEnJxAI/OMf/9i5c+fhw4dhdgNFUWhLDABoaGgIhUJZqeAQ9rnwekB4JQBQwX1gYGDz\n5s3PPfec2WwGALS2tnKzRWNjY1nsjFCr1RaLJRgMer3eI0eOsN+Az+czGo1ZUYcNhUJ2u11o\n1NLW1uZyucS20mq1vECdDeMBAEamYNKnUgBF0Teb33yh5oVT2lNuzH1Gc+aRWY/AZpPvoN9J\nsuFWsPVj8LEHeAhAWBHrO+g7N3hvYLMbQHB9ZtKo1dfXx81usEitZ00md8gVHEUFG8YnrNCO\nBYOZtJ9RFCVWOhFtb1fmXroZAKDo50YmAAAgAElEQVRQKIxGIxu3m/bt466t+d73wK5d4IEH\nEg8eACCSqohddJFYKSRN00mOWpelRtmMSOTqgqJoXV2dkiMsz0eS6iFQqGySBFbKhScEQfj9\nfoIgEIax3HWXUkrSsCqVCkGQ0r/9LeGRao8dC151VYaHP9NI/pXK357MlIhEIs89d8HzcU+P\nZu3apmBQcfHFkdWrGQBksbrcolKp9u9vHBxEAAAkiezcWcNZ6QbgfbENpRzDOBwOaPPErSag\nafr++++Hb8iiVKRSqUyitohhGC+DMDQ0FA6HhWoIVqu1oaEBvifrnRGVlZVz587lxqLZFcvk\nZTG4TRAul6uysjJhEwSCINXV1U9FnvIqvELNiFXIqswHNq15HXv9tYrXwIXxxBKw5Jfgl1Pa\nj0ql4grN8nqUMknzFbCFR0YiyAmOogI2U4iVGzACd+gpgWGYWOTJoKjK6QSZVYikSE1NDT31\nuH28fUYkVUEtWSK2v+RHrXa58l+sDPW9Q6EQQRAYis5evz6hVKrBYMCPHRtyOMLhMEVRut7e\n5ptuAiBX1rZZAValiiWwateuhQkshmEikQhBECiKaqPRhFY4DofD6XTCO5yls1O5f7+kpGGT\np+oAAMaDB40HD3KXsIefv1FOKzAMSy/7KSMjhOe2yLOJRdHZhRrYjOKzzxJ34N94Y8PXvvYc\n22vw/vvvw3IDiROJRBwOBxBIfgrJSo4GRVGTycQKq/N6QHhKBKFQiFuRyk0EBIPBSCSi0+Wk\nyS/XM+3QC4mF2wRBUVQsFhNT+igtLf1Q/+Fe5V6eZsRiZvETyBNZGdv0hdtsYgGWVtB6O7h9\nNVitAVNrDTOZTGyCg3d9arXa9Kx5ZGQgcoKjqFAoFEnKDfSZaWQYjUaFSPDQuHEjs3Mn+MlP\nMtn/pNA0TRAEgiCKgwfJZcswqxUA4LnllvI//xkAwGzfjjz6qNi2er3e6XSKpSrUOp3Y7K7J\nZKLEj5r+1a/A/PlZPMZJ4RlhViR1ddFoNPX19QAAiqIUH300vlTCs1sAgJqaGpA0gRUMBkdG\nRmAuT8wKx+v1wudIIGErlklSdQmRQGpGyqSX/ZSREaJUKqF+gVDhEsOYbGlAyiTn9dfPv45E\nIn19fRN//QubHViwYIHT6SwrKyvUSWEYxu1279ix45NPPvn8889hRJ3QoYMXybNJhAcffPDW\nW2/lNYNkherq6nA47PV6ebKgkUiE93WFQiHe2OALGG2GQqEcJThyPdPO2/mUmiC+ovyKB3jO\nMmcjSKSKrmoFrXeid65GphzGFx9bwdatYGvm+6msrPT7/S6Xi3d9+nw+2e5EJkPkBEdRodfr\nwQsviJUbGPx+UFmZ9s7LyspIceN0JJdhcywWGxkZGb8B03Tjpk0Gq3V8VBPzNsiyZUn2YDAY\n9Hq92Oyu9sYbxWZ3jUZj/EKpcy5o0g/NBcPDw2JGmElCd4VCAdauBWvX5nGkaaJWq2MnTgxM\nnG7N6dOtK1eCiQRWOBweGhhgH1nEZHTZaTEpe5EolUry1Knh0dFAIEDTNHuk8UceGbrzTtib\nzS6kH3sM7ego8IglD/xKbWNjgUCAoijexVPo0clMJ0wmU0KFSwCASqWSuIZlUcKVCQcXlhgw\nDBMMBsvLy/M/KhzHrVbrxx9//PTT5+f5xRw6uNX4QJBEyIU2qlKpbG5utlgsk8qC8kqWeIkA\nKi89yLlAqVSycpggtSYIr9f72GOPHTlyJHm6SiZzUBRtamq67LLLsiVbKyPDIic4ioqSkhL0\ns8+ASLlBhhXaKIoiJ070Dw/DOkY2eIj87Ge6X06t725KEATR19fH3l95MS2ybRvYvTuV/dTX\n11PiCZoks7uKU6dsdrvP52MYJm9HnRCCINhpFmHoTixcWBwP3SqVqrGxERbsYO+PN1rDBBZU\nC4NLxFx74/E4fKBJ2KslKS8SDMPq6uoAALFYDGM9br74xZaWlng8ThCE6r334EL0sssKNspp\nBYZhs2bNmjVrFkmSisOH4cLnPr70+UsZqxXx+UBtLbjlFrBjB5CrX2WSUFFRcfRo+Pvfrx0c\nVHEVLgEAS5ZIogRspjFetTcBLzvAW5u3IfX398NOB66mRlNTE5lCRzAviZAjhoeHU5EF5bng\n8RIBefDIyxEmk4mV4eA1Qej1ep6SKEmSVqv1gw8+6OrqYhcmN5SVyZChoaEsytbKyLDICY6i\nAkEQ45kzoqszrtBWKpVNTU3RaDQajWITCRTd8uUZ7jY5drudzW7wYlq6pgZNud1AoVAoTp8O\nhUKRSCQWi+l6e8u/8hUAJtekgPbvZrMZx3F0/364MNdHnRA4qw/EZVaKI8EBQVFUq9WCDRvA\nhg1wSTweZ2fAhPmdaHs79JQbv1pEerUi8+cn9MYrLCqVCnz/++D732eXKJVKpVIJNm4EGzcW\ncGDTFwzDwrfcMrpixcaNVftePe+5ODgIfvUrQNOA44QoI8MHRdG33qofHESBQOGysxPIjkb5\nh6eIycsOZMVxY6o4nU54u+FpaixYsGB0dFRo9apWq9mbOBAkEcTEIDIkxR4Qo9EolgiAa3Mx\ntjxQVVUVCATcbjevCcLv91ddKKDGMMzAwACO48J0VTgczlGHjoysBiqTI2Sb2GIDOXGCoWmf\n1ztis9nZBtYdOwDDgKuvnmTj1Kw0tVpteXm56d57AcMAhgHXXpu94fMhSTJhzQK0d03DONNg\nMJjN5rq6unJWuCG1PLFSqTQajfq7787DUYsxfhuYvkaYmXm1snN0CfM7kYmLAc7JiOmtoEol\nMjKSlaORkTKBQGBgYCAajdpsqg0bnH/969l9+05/5zseuHbv3sKOTmYa8PnniR+QLrkkzwOR\nAQAAXoTJyw4UJP4U6lawXTOxWIxtJmUp4wix85IIGo1Gqy1kcaFerzcYDELHVqgbUtixZYJC\noWhqarruuuu4dsJdXV1XXnnl2gubdr1eL5xB4RnKQpGX6dukIyMzM5ETHEUIgiClpaW1tbU1\nbCAnHsOHw+GxsTGr1Wq32chVq4CUrDQBAGzzZPKYNh3Wri1gqiI9UnJRlRgkSY6NjfX395/p\n6YnecEMmF9j4HJ1Ifie2aBF8gWGYVqs1fPABAMC0b1/rqlWtq1aVTjRzzv7BD8Cf/pTpUcnk\nniNHwN1305deypjNQKUCjY3gZz8DnGbqZFAUZbPZ4DP688/3b9ky1taGWyzx225zwzfk3ftI\nZvrx+uvjtwjev6uuKvTIskc0Gn3ggQdWrFhhMBgQBEEQ5IMPPij0oBJTUlLCKiYIDRf0+gK4\n9vL6UHhdM8IuFb1eX1lZKUwiBAIBKZRIzJ49W5gIWL58+U9/+tPCDixDrFZrKk0QwWCQ+yc3\nXUXTNNdiRiYVcBzfunXrtPhtkSlK5BaVoiapriTDMFarlRXusnR2Ym+/zWi1CCyhlEaCgxfT\n8toNpkHNAovHA5YuFbqZTgmdTqdUKsWkUqtuv11qRpjRaHRwcBA+51k6O7UHD2bi1apWqzEM\nM/71rwnzOyqtlrZavTpdJBJhKMp44IDojqRxbcuIQVHU2NjYtm3GN988/9A/pdaSYDDITrgZ\njedn3j78ELYxgZUrszlgGZlpB5zbOHz48JNPPskulLLcAIqiDQ0Nx48fx3GcZwhiKZAxlkKh\n4Npw8LpmEupWVFdXt7e3c+1UpCOpODg4mDARsCzvYurZJcUmiKmmq2SS4PP53n777Z2cu7WU\nf1tkihK5gmPmMjIywmY3WG0LvKUFAABmzQLSsNJUqVQoiorVLKi0WgnWLEBomnY6nf39/T09\nPb2nT+M33ph5dQyCILNmzdJK3giTYRgcx4PB4PCxY83XXLNw0aKFixbBC4xobQUAkBYLM2F/\nkzoIglRWVrL5HV5phvmOO9zPPGO32/1+f8nOnWgkQk8o0vf/5jfwBfnooyn1ak2VRK03489D\nmXXlzEAoiurr6/N4PMPDyrRbS2KJKj16ejRPP20GACxdSqxbl70Ry+SMI0fAj34Eli4FaVTx\nyCTB7/cPDAxEIhGCIDZv3vzcc8+ZzWYAwNy5c6WsNaDRaL7xjW/wSgy+8IUv3HXXXQUZj8Fg\n4P7J7ZrBMCyhJUpfXx/PLBZSQElFmqbdbvePfvSjO++8ky2EOXToEDPB9u3bCzW2fMJLSKWS\nrpJJyPDw8PDwcCAQ4P62tLS0yOZTMvlEruCYocRiMfYuK9RrjC1aJBGHARRFy8rKtCI1C5Y7\n75RazQKEJEmoVgX/rOjs1Lz7bibFCywGgyH82WdWux3uXGpGmAzDOJ1Ol8tF0zSg6cb771dN\nZKDiZjNXEJQJBoUabJNSWVlJnj4ttjY0bx4QKNGSFkvVRHEpdvnl6RxVIgKBgNfrJQgC0PTs\n9eu1E9mr2KJF9sHBSCRCURQKQNPmzVqJtX1JnLGxMdi7/vzz/WzxxW23uV96qRyk3FoiFJHd\nt8/00EN1kQi6bFn4xRd9GDYrm4POjCNHwB/+AN5/HwwNAdnnhSUSiTz2mOK1184rL84cgdgj\nR8CLL9IffIDm4pKgKGpkZATOafPUMefOnRuPxxN6Z0oBmPoULi9UdqCystLv98MiDl7XjFhR\nidQkFT0ez9jY2GeffSb7huj1eq6oCjddhSBIQXqgphE4jnd0dBw+fPjjjz+G3rr//d//vWnT\nJva3Zf78+aOjo7W1tYUeqcxMQa7gmKGw/YRifhyZfkD2Jq4tFosuLXvXAjIyMsJmN9h4GxYv\nMLW1GVbH6PX61tbWtra2pqamRve4pgAijSJSm83mcDjgAx9r6AtXWXft4l5g8BaYBlh3dyQc\nHrXbBwcGRv/5T7gQ37btxPHj4csuS6hEy3zve9nVW7HZbENDQ8FgMBaLlf37v8PWG7iqv7yc\n7Y+omujKGd9Mktdq2uRidp1hGLZGOpPWEu5MEU2DZ56x3H9/fSSCrlzp2bNnoKoqJ24F6UHT\n9I4d8a4u8MknwOkE8fh4GP/ww+Nr9+8P/uAHkcWL41VVzAypYqBpemhoqK+vr6+PYat4br11\nvN6wuAViCYIYGhp68MHAM8+gCS+JzAkEAjzFRK7cAK9JQVKN9DA7IKRQJQZqtbq+vj4ajXI1\nNRobG5VKpdSMuhLi8XhGRkZYm1vuZLuUC3mSkMnlWl5ezrrG8tJV3FUyQiKRCGxIOXDgAHy0\nYytfeL8tksruyRQ3coJjhsKz0syKHwdJki6Xy2azWQcHYzffnC29UhRFlT09Hrd7oL+/p7t7\n8P/9v/EVQmuYXLYDUBQVj8dT+Yh4PM72/gjjbWLhwqyMB8MwvV6PrV8vHanUUCjEPhyPp3VW\nrGBUKgAAVVISueQS9gKLtrdncp/T6XTV1dUNDQ3VE+UhUG5WLFuXdjIlIR6Ph61+4mWvSIsl\nXl6ecFXmiS1JkTwsT5t4PM7taYewrSUXXxxZuzaly8ZgMEBF3mBQsWVLw69/XYVhTEeHraNj\nRKVCSktLMxpl9giHw2fOnDl7lkrYjBMMBk+fPv2LXzC/+Y3u2DGly4VkPdaVJmz7JFcg9pZb\nxuDaIhaIjUaj586dCwQCObX+Ebp7cOUGuGuj0egbb7wB4xY4KTIz5/aTYDAYeF0zu3fvbmtr\nW7NmTWEHNik0TY+OjsLXPN+Q+fPn+/3+go4uHQiCOHDgQNqXq0KhaGxsxHGcJwFL07TcW5GE\nkZGRvr4+h8PBzZGx9kbc3xaapoU/PjIyOUJOcMxQYA1qEivNVLQtoBEa43bDmB9TKunt271e\nr+bxx1XvvJPFiWsEQcrLyxsbG9va2homrNrB0qUURTkcjnPnznV3d2dL54IHwzAul+v06dPd\n3d2nu7tD3/72pB/BGt1n3/lF2rCPRGxaJ3T55YpAAACAz5nDvcAYFFWz5zETJqxwoitWiGXr\nou3twpg5E9wTVTNJfIuFq1iTlyIgeVieXfbtM915Z3MwqFi2LPzsswMpzqIhCDJ79mybTbt6\ndcvBg0YMY+67b3TuXOLYMZ3d3mCzSWIuLhaLQQnehD4vFRX00NAQRVEzzeaWIIisVPFMOxiG\nGR4ehj9WObX+ERYXcOUG2LU+nw8KRnDjlsbGxgceeOBLX/qSXq8veEGHFJi0awbH8Z/85CcS\nKYHhEgqFeHdG7mQ7O0kzXXA4HL29vXa7nXu5zps3b0qlKBqN5utf/zpP5GXx4sW33XZbTgY9\n/XG5XPD6F3rrwjfwpExkZPKGJJ7zZPIPfDpJz4+DpumxsTGv1zuus7Bpk2Ei5o+2t/MUEOia\nGjS7E9cT1jDxeLz/3DlWTbDyySezpXPB5QKjmYmei+QfMV6bUATOL1MEngtuWqe6sxOu0h85\n0rpqFfvOxo0bqSeeANmL+ZVKpVi2bjyZUlOTlQ9ipyCS9HYlXIUvXCihvogMgGE5TdMJNTL6\n+4HZnL5qgFKpVCgUsL6MpsGzz1r27KliGLBypeehh+x6vSr1wm+tVvvOO02DgygAgCSRnTvP\nXwCdneBHP5rCqHKE0+mEAUbCMP6rX/WyNrdpa5FMRxLaMbJVPMuWxdatK05tEij5CV/nNLMj\nnI7myg3AtfF4HOp08EQ6amtrn3vuOXZDuaAjiaYGjuOjo6Mffvjhv//7v7MLpfONxeNx3hLu\nZLtwrZTxer0OhwMINGXmzJmD47hY/QWO448++ujhw4c/+eQT+LPz8ssvS0rkReJAzTXuEm6O\nDC7hSZmoZriylEwekSs4ZigYhlVVVWlOnhR9h0j0TtN0f3+/2+1OqLNA6fW8ievI/PnZHz0A\nAACr1cpmN9JrByBJkqbpJF0nXq9XaDQDP4Kqrhb7CPgLLhZvY2q1ZJ1fMgRBEF4ZRZILTHHZ\nZVn8aJPJJOau0rhxoyl79nvjU17i1SJiq4qmgmNcQVYkBgsEMupYQRCkrKwMJGotwTCmfKL9\nJ0U+/zzxDe6SS6Y2qhzBFbSDcJtxvvWt8Y6MhN/zzTcXbSez0I6RW8Xz29+OFWsvfMLibfaS\nuPRSPFvWP0ajkSsowJUbUCgUJSUlAACfz8ed3mfjlvr6el4humQVSQtLKBQ6d+5cKBTCcVya\nPjUoyv955E62Ty/TEJjdYOGG2bxVLF6v99VXX921a9e7777L9rNce+21khJ5kTgEQQjVfOAL\nWMHBkzIxmUzCq05GJkfIl9rMxWw2B95//+SJEyeOHz9x/PjZl1+Gy+nHHktipelyudgWDL4A\nQVXVrEcfBYKOjISWjRmC4zgrrDDVdgCSJG02W3d3d09Pz6kTJyLXXy/WdcJWSgs/IjJ/vpgv\nularVavVSdxMwZ/+lO5xSxqdTsdL64QnpE8H/vM/B/bsga+jDz6Yda9WjUZjEHdXwb7whWx9\nEIZhCoUiSbVI5e9/n3CVUqOZNLHFMEw0GuW2fUnQXDZJWF5dHf/zn8/t23f6llvG/+Ok0Ulh\nNpudzhJha8m5c1XB4NQSHK+/Pi5Qw/t31VVTHlUu4P2A8JpxUDSZFskdd0yn+dUpwY29hQKx\npaUz6KElR5kdFEVnz56NomgwGOSpY2o0GvgfnBXJhrBxy7XXXsstRG9vb+dN4coAAGiaHh4e\nZn1quN/Y3LlzJSJDILQF4U62SyQLkwqxWIxXb8INsxNWhNntdpvNFgqFeNm6aSENKx142Q3A\nyZHV1tbypExCoZBWqy3AKGVmKkU6FSKTGmazuby8PBwOEwRheOcduBBNOrWeMOaH3p+AYYQd\nGdH2dqPgRzBzkutcRBcsEGsHiMfjfX197O3Q0tWle+89sa6T5M0IKEGICWvPmjVLMXGLTUBx\nuWmwlJWVhUWanho3biRvvBG+1uYmvlScOjXmcLhcLoZhWPfc8Nat+ieeyO4HlZSU6MQPE3ap\nCFeV3nJLkravWCxmt9tDoRDDMLy2L6ldLbxnGtZ+9ZJLIv/xHwMGAw0AuPVW55//XArS6qRA\nUXTfvtmDgwBItbUkW2AYBn+IhM04GMYgCMItfefa3HZ1DWo0bYUbeG6BUgUMwwSDiq1b6w4e\nNCqVzLZtIytXeuHaQg8wV/Csf3iXRGXl1LJ7ydHr9S0tLdXV1VzPlN27d+/evfu66657TVDy\nxmuk586QCzOeMqFQiJe+5HlJiJnI5hOVSmUymdgaVV4hz1TL5QpIkjB73rx5QgWucDgMhbR4\n/Szt7e3Dw8Otra1ymiNFhNVbbI7sxhtv5OrUdnV1dXV1JfxtkZHJETNoMkQmIRiGlZSUmM1m\n3ZYt8ViMIknwhS+IzR4zDCPUWWBjfszlSjinrRQpEcwEns5F6i4wdrudzW7wKlDompoEXSdJ\nmhHE0el09NGj586e5VXHMNu3Z714QToolUpTb6/YWuzuu1M1fEnLDQdBEIvF0tbW1tjYWGu3\nw4X6L385xc1Tx2w2a8WzV1gS8XmRVEUsFuvr6wsGg/Cq5rV9SS3BwZYu82bXn3++H2Y3QMaq\nAZ9+mni5RFpLknPkCLj7bvrSS5lJ3XNhrJ6wGUelUrHzq8IqBrNZU8SFviqVqry8fGhIJazi\nOX26zOs1FXqAuUKn08Ech/CSUCoBbN3KIjabjecIC4FyA2r1BXME3Ll9cOEMuVgl40wmdZ+a\nwlJXV6fX63mFPE1NTQaDIRgMFnp0qSKcZ+JersK1vMuem3siCIKdPJOZFJVKxf2hYHNkjY2N\nCV14ZCkTmXwiV3DIgHg8PjY2FggEoGho0+bNepHZY6gBztB0QvlMIDKnnWTiOm2S61yMtwPM\nmsXbiiRJ9rYtrEDBFyzgFWVqNBrlX/+a8COAQqFxu4GgyJNFq9W2tLRQFEUQhOrQIbgQmWjZ\nKFbQkydjsZjb7Y5EIooTJxpvuAEAwGzfjjz6aPIN4/G43+8nCALQtPnOO5Xp1i8oFAqDwQC2\nbAETKuhZB8MwcOrU0ISTJVstQj/2GNrRQeH4yNhYJBKhKErX29t8000AALBjB3jkEbEd2u12\nNkjIuUZvxhiNRrfbnXB2HcJ2UixdSqxbl46y6uuvZ220+QSaOm3bZnjzTSO7EGqR0DTYuZP/\n/qqqqhMn8A0bZg8OqrhhPABg8WLz7NmKcDgcCKC87xkm8vJ5XPmnurr6jTeMg4MqUOxVPDzq\n6uoOHLAJL4ny8nKDQdPQkOZujxwBL75IHzqEWK0IRwBYVB0TAFBSUuJ0OuEbeI304MIZcrEy\nRhku0vSSQFG0sbHx0ksvZY3PAQCdnZ2dnZ3TaLJdqVRqNBq2qYp3uRqNRt77eQkmnmwEQRDT\nqD2n4NTU1AwODgYCAZIkjx07xqaKHn/88Tlz5lRUVBR6gDIzF/nONNOBs8dsfGXp6tK//34S\noxCNRqP54x8Tx/xi5GAKWq/XK5VKMReYijVrEmZVCIKAv78JK1DC8+fzbmvl5eW0yEc0bNiQ\nSuJGoVDodDqwfj1Yvz6d45yGqFSqGmhZcuAAXDJpWsfj8djt9vH6hc5O5f79WXfDyS4YhtXX\n10NHFcV778GFsLdLo9E0NDQAAEiSxD76aHwD8YkLiqLYGm9h0i3a3i6aQisQlZWVx45FEobl\nlZXxU6e0bCfFb3/rwbDZhR5vnqAoqq+vjyCI4eGyDRuc11zjLyuj9uypgqYne/cmSHCoVKq3\n3qofHFQCkTCeJBtXr1Zyv+fjx/VVVVUGg7B9vqhAEOTMmcStKNOiiidtNBpNdq1/oOXZgw/q\n9+07X/mSJOnGolarLRZLb28vN25paGjwer0YhnFnyIu4aShtUvGpkQj9/f3c7AbL9Jpsr66u\nFobZDQ0NgUBg0nSwNHNP0wWDwVBfXz979my5IUVGasgJjpnOyMjIlGaPy8vLEZGYHwAw+uMf\nu+66i53TJh56SP1v/5aLYSMIUltbq5qizsV4a6WIgSuxcCHv/SaTiRSXrpRm7C0hJgx9kxMM\nBkdGRuBr3hXI1NYiEpusJkkSx/F4PK6JRLRXXaWFlSaJCjQwDEvlG4jFYsmTblKLZJVK5dtv\nNwwOYkAQg11xReiDDwysakDWi+qlzNjYGJwYnJKr6/HjiR0oYBj/0kv6maBFkhApVPEcOQL+\n8Afw/vtgaCh95+OpkkXrH5qm+/r6cBy32VJNunGprKycO3cuN/qFccsVV1zBzpCjKFpZWTnl\nkRU7BoNBpVKx8urcmgIEQUpLSws6ugtIYnM7jTAYDLNnz66vr08lzNZoNKxEPRC0X0kq/TQt\ncDqdckOKjASRExwzGpIkk8weC1s2AABlZWXxnh6xHZZ+9auq2lrV22+P7/NLX8rFsCFGozH8\n2WfDo6OwZ5LNqiRpiFCr1QiClP7tb8kMXC9sbMG6u30+n8fjwXFc3dPTcvPNyT9CZqqMjY3b\nYSasX5BOqShN06Ojo16vl9UBFTPfmRLJk25J1GTyh8cDli4FnFTOsWOJbxyHDhm4HSvC2uBi\nhWEYtq87oaurmBZJ8jB+WmuRSIFMMhSRSOSxxxSvvXa+xyqVwofMyWJmx+l0wrr9KSXdWPr6\n+hLO7ZeUlMCQuLm52Wg0RiIRVU5TPtMQBEHq6uoGBgb8fj+3pqCxsVGtVsvfWCbgON7R0XH4\n8OEjR45Ah5RDhw598YtfdLlcKYbZ5eXl4/dxQT+LVquVnT6mSnHkyGSKDznBMaNhexETzh5H\nRMJLZU+Pz+fzer0w5ocqAzDm1wCgAQDccw+45548jB9Kwaeuc6FQKEwmk1hjS/VddyXsOikt\nLYVTLswHH0z6ETMCQcSb9p4oioKP4AmvwFBbm5ZhJCJpbrVaWQEXVgc08z4alUqFomjJ//xP\nwqSbSqtNqCaTByKRSCQSieF41R138CRRuDGY0+n8+GPv5s2NvI4VnU5nNM6UFEc8Hhdq9XNd\nXdeu1QIw5ctYClUM0xeCIDo6mH/84/x8bIoZCpqm4X/2vr5WtvDht7+t/b//1whSKHyQDmx6\nImHS7eabmeTXpDBuqaio8Hg8r09cl0899dRTTz0lF6InRKfTTcmnRiYVAoHAe++9t5PzP1Ch\nUCxevBhMJczWaDQJ26+CwZhXWXoAACAASURBVGBVGr5fMjIykqRoldhlpsDUvUhKS0ubmprm\nz5/fPPEIVcCYH+pcYOvXp2LSUVNTk8QCI3mkiqxbl6oPSNGB4/jw8HBvb++pEyci11+fleIF\nwBq8iVvVCOPGghAMBtnsBs98h7RYqAyKtFEULSsrY5NuratWta5aVTrx+Ft9113gT3/KePhT\ng6KowcHBvr6+0dFR5WOPQUmU8XWC011VVfXmm3VcPcg1a5rXrGm+8cZqjvrwjGPfPtOddzYH\ng4ply8LPPjsg6zBmSOreNJBAIHDu3Ln+frBhg/Ovfz27b9/p73zHA1ft3TvJZ7GpzOef79+y\nZaytDbdY4t/97ihcO10iIJqmhf4m3KTbnXdOzf2kr6/P4/EIl8uF6GIk96mRmRIkSfb19Q0N\nDTkcjs2bNz/33HNmsxkA0NbWloYmaGVl5Te/+c3ly5dvmRAjh71Xa1Noqi1ufD7fvffee+WV\nV0K7bgRBPpiY2JORmV7Ij10zGo1Gk6RlQ8yL5AJS01mQDhiGId3ddocjGAzGYjHtmTOw6yTD\nSoTixu/3Dw8PsyKguvfey5YIKFTgF3PDQTBMMTpakPoFHtAwBYj00dChUElJSdo7t1gsZHe3\n6Oq8S71YrVbYucaTRKGqqxWJJFF6ehI/X86cTgqlUqlQKGC2jqbBs89a9uypYrVI9HpVVqqQ\njhwBv/898/774EIvjNxKQhQchmFcLte2bZoUvWkAALFYbHh4mKbpNFozIpEIm8qcUreR9Nm3\nz8QKAHd1DSqVc6e0uVyILgbDMH6/f/v27Z988smnn37K7ZuQv7FsYbVaoXDG0qVLly5dGgqF\nnE4nACA9ZVCx9quZnHsiCGJ4ePjw4cO7d+9mF7IFMjIy0w45wTGjSd6yYbnzzlw4vBYchUJR\nU1NTU1ND0zRy+PD40hl8Y0tOPB632WzwQY0X8ZIWC5aZCCiKonq9XuwKrF+/XiJXYDweByJ9\nNNEFC9B4PJOdoyiq7OlxezyBQADHce2ZM9BetyBJt3A4DLMbwlROZP58NBwWGnjInRQIgpSV\nlblcroTuueXl5Rnun2EYt9v90EOavXvPO1bkRxKi4AwNDQWDweHh1tRlMr1eL6z8SpihuOEG\nMsmTDytKxYUtfFi2LLZuXf7ySdFo9JFHHvnoo494YXMq26IoqlarYReqMOmm0Shke9epQtO0\nx+PZsWPHp59++vnnn8Mz8sYbb9TX13/66aednZ3sO6UfFmZyaeWfcDgMB8ly6tQp+Ewyb968\nYDA41WZIOVvHA9opUhQVi8U2b968aNGijo4Oh8PR3NysUCgKPToZmXSQ73AznZqaGlpKs8f5\nBEVRsG4dWLeu0AORNGy0kLB4QRGJZGgab7FYFOk2DeUNFEXFdECj7e1GNNNePwRBKioqxk3j\n2YrQQiTdYIAnlsoBiRIcMgAAs9nc3R3/3vcsQi0Sg6Ecnti0sVqtgUDAap1CkF8c+Hw+tlsk\n9VoMrkUChNua8d3vxgAQtbEQtnVwCx9+/3s/htWmeTBTgaIou91+8ODBJ598kl041bC5vLzc\nbrfnKOk20/D5fHa7/bPPPnv66afZhQqFory8nCRJXljY2tqa4W0xd0C17HfffTeTSyvPCNOO\npyaeGRYsWBAKhYpA7SkQCHR0dBw5cuSzzz7Lf8rJ4XDACkRegUx7e/vY2Fh9fX1+hiEjk0Xk\nBMdMB8Mwurt7zOkMBAKwZQOKhuZu9hjHcRzHKYrSRCL6L385K1qVMrkDmtSIRbyKaDTDJzmd\nThf85JN+mw2GFqwbTuzhh1WPP56FA8gGOp1O8eKLCftoGBTV+3wgiwFDQdu+SJJMkspRCsI/\nGQiKovv2zc6Fq6vP54MdUul5YUxroNkBgiAJazFuvJECIMHsIm9ulteagaLVST6RW9cgLHwo\nK8uHJSpFUX19fQRBEATBm01Vq9WTbz9BeXn5yZPEHXdU8pJuWq3WYCjq6ybbwCZNAAAvkdHU\n1ARdRXlhYVtbWyTj1H8uYJ2DcRznXVpSNnYRSnF1d3cDAFAUnTdvnkSEutImGo2OjIwcPny4\ngEVAbBMuhC2QaW9vDwaDjGS03mVkUkdOcMgAFEUtFovFYmEYJqctG/F4fHh4eLzUMHtGmzI5\nhWGYJBGvPht1nkajce7cueFwGMdx7YTHsOqKKzLfc7YoKysLffopSNRH07hxo0T6aLIChmFi\nkigMiqqcTlCbjxns6UiOXF2Te2HcdBNdxGLhBEHwHqy5tRi33koDYBBupVKpYBGHMEOBYUzy\nQM5gMDgcDgBAwsIHgyHBx2Udh8MBW0sSzqbWpvwfEEGQN96ozUXSbUbBMIzdboeveWdkwYVC\n7Nyw0O/3SzDBwToHCy+t0dHRurq6Qg8wMcJ2qpMnTwIAmpubNRrNtG62wnG8v7+fpmle7qyl\npQUAsHXrVqEhbtbHQFEUL0nELZBhGIYkSaVSmfXPlZHJKdP4d0Em6yAIkrvZY4qi+vv7YxPC\n91k02pTJHIqiotFoLBZTKpXaaBS7/HK2ska1bl2SiFfjdoMMPERYUBQ1QlvRfHkMTwmFQmE8\nc0Z09bS6euGzlEKhUAaDQrtfg8EQF5FEady4Mf6LXwAJVzIXlhxpkbBm3iwXBvkMAEXbNMTL\nbghkMhsSblVSUuLz+RJmKDAMS95jpdPpTCbTiRO40PlYq9Uajfno0OJZb/DC5tQTHCBnSbcZ\nRTQa5TUucc8Ibzl80d7eHkvi8VM4klxagUCApmk043bLXGA0GsfGxtg/w+Hw0NAQmEgwTev+\nFLvdDpMLvJTT/Pnz9+7dm9AQN+sITzq3QCbhG2RkpI981crkCbfbzd7yhUabIDOtSplMcDqd\np0+fHhgYGBkZGezvx2+8kVtZU1paKmZi2rhxo/7vfy/UsPMMevIkRZJOh2NocHD41VfhQmb7\ndsAw4OqrCzu2FIGTRd3d3WfPnj3d3R3+138VllDp9XqduCiP8vLL8zDOxHg8oLkZIAhAEJBa\n7xLDMO++G1m/Prp4cbyqikloL/rhh+TmzfGLL6bE3iA1ZpQBrWbCn5imwTPPWO6/vz4SQVeu\n9OzZM2A0MmL9Gkaj0eMpX7265eBBIzdDceyYjqLqJq21rqure/31aqHz8U031eTB+ZgkyXHz\n7Am4s6kURQlVQpLw+uvjzua8f1ddlc0xFzfCL5x7RrjL2bCwra1NgjEhwzDxC/WwuQdC03Q8\nM7Xs3KHRaMrKygAAwWDQ6/UeOXIE5mUaGhqgLmahB5gmJEmKiae2t7f7fL7MDXFTAUEQrVbL\nXcItkFGr1cl1RnEc37p164oVK2RnWRlJUdQPRzJSIrnRphLH2WdZmXzicDhgSTZEWFmj0+lU\np0+LbY5k3srk8QjrCKSJQqGogpoHE6UNyLJlhRzQVIhGo7AUFv5p6erSv/9+whIqxalTg8PD\nUNyRlUTx3X+/adeuPD+1kyTp8/lwHKfi8ervfU89lY42giCGhoYef9yyb5+JXch1HiFJ0maz\nPfJImdgbJIJGo4Eae8KGC6USTEmUYdpRXl4eDAYT1mKYTCVJStOTtGZcGJMmAEXRs2dNCVfl\nofBBmH/hzabKzfB5ZtL5bRZuWCjB/hQh0+jSqq2tRRDkyiuv9Pv97MKurq6urq7rrrvutYlJ\nl+mFMDXDTTktXryYW9Mxd+7c3GlhVFVVwaIYICiQqUqq80QQxP79+/NTaSIjMyUkl2OWKVbg\nNIiYVqVkpw6Km3g8Du+dEF5lDVNbCytrsO5ut8vV09194vjxsy+/DN8ce/jhtIsXaJr2+Xyj\no6OjIyPxlSunnxTL2rXjM6HXXpvlPU+9TiFFbDYbm90QO9EQhULR0NDQ3NxcXV1dNfHQU3rN\nNZNmNz76iP7hD8lLL2XMZpB5NUQoFOrt7R0dHfX5fLqf/1x94ADN5kAnu05Ikuzv7ycIwmZT\nbdjg/Otfz+7bd/o73/HAtXv3jus4BoNBsTdIB2h4EQwqtmxp+PWvqzCM6eiwdXSMYBhjNBqn\ndf/5pBiNxmDQIqzF6O4uicVqkmyYYWtGAQsfFAoFTyWEGzarVCrZtTHP6HQ6XlTJPSPsQm5Y\nSJLk448/vnz5cknNaSMIwptG4h6IQqGQss4CgiA4jnOzGyxLU5hl8fl8995771VXXSWpM5JK\n7oyt6Whra2P1mLKOyWSyWCyhUIhXIJP8/uJ2u8+ePTs2NsatNJkzZ860yO7JFD3F/GwkIymS\nG20a5Ie2QgD1seFrYWUNsXAh+zQETUzj8Tjz3ntwSdoioKFQyGq1wjJsS2encv/+GS7FwjBM\nIBDAcZwmycrbb1fmIN0DZfPha+GJxhcs0Ao20el0Op0O3HcfuO++SfdPEMTIyMhDD1Vkqxoi\nHo8PDQ3BjAybjsGcTu2JE1R1tWKyjjaXywUzqmLOIy6XC86eSd+axGQyBQLmW28t5UlCYBh2\n6aVFJfh65Aj4wx/A+++DoSHg84HaWnDLLQCAqjRkMnOkh5IfysvLR0dH4WvebKps75p/UBSt\nqKhwuVzwT+4ZQVEUwzC3202S5LFjx9iw8PDhw7t372b3IJ057YqKCpvNBl8LLy0pV3AAAJqb\nm5mpi5pHIhGbzfbxxx9L8IzA7g9uS5owd8YzxM3dL0BVVdW8efO4OZTkBTKhUAiK7/LUQ+BO\nYEuRjEwBkSs4ZPKEXq8X06pEMEzr8RR2eDMTtnAmYWUNsWgR7/1KpVK1cWOS4oV4PB4Oh2Ox\nmFglAo7jg4OD8I6evI5ghhCJRM6cOWO1Wp1OJ9bRAdM94+uyl+BghSoTnugIVypv6iUk0Wj0\n3Llz4XA4i9UQLpcLZje46RhNby8AIDJ/Pk+kQAjs6QAiziPXXx9nZwLFrUkkxGuvmYWSEN/9\nbv2rrxbJFAXDMG63++GHw11d4JNPgNMJ4vHxBNlLLyXepIhlMisqKkwmk1BugCRJuXyjIFgs\nltLSUt4ZaWpq0uv1lZWV3/zmN5cvX75lyxb45q6uro0bNzY0NEhwTrusrEx4IA0NDfF4XMo2\nsWkTiURgKR/0KGHPSEtLixR6ohEE4SYseCknCLemY9IbX4okVM3o6+tLWCEiViDDLf4FF6qH\n8FbJyBSEInk8kskD8Xg8EokQBKFSqbTRqPpLX5qSbkJlZWVUxJ2hfv36YjLanEaMPy6LVNbE\nL7oo9V35/f7R0dHxjAlNN23erE9UieBwOOBdUFhHEJk/v2jdIESIx+PCdA+sU6BratDspXvG\np+bES6iGhoYIgkABqPvBD6YkdQEAGBkZgcmILFZDQJtPsY42JBpNbtgp1AXkOo+sWUONjPB7\n4rhvuO02BABhUUvBKG4vDJqmBwcHw+Hw0FDrhg3Oa67xl5VRe/ZUwevHZALZsKKeTiAIUl9f\nv2TJkuSzqQkLXnbsAMUUqOI43tHRkQenzOQgCFJXV3fRRRdxz0hnZ2dnZ+eKFSsShoXXXnvt\nkiVL2DltXDIqY8IDme5KFkmw2+3weUPoUeJyuWCyo7CYzWaCIGw2G68IyOv1YhhmNBqzbogb\niUTefPNNoWqGTqdLvUCGYRh4j2bhVprEYjGSJIu7fVJG+sjXn8zkMAwzOjrq8XjGf/5ounHT\npqlGQWq1WtnbK7p6RvYmFBw4rSRWWaPR64HNBmbNmnQ/brcbFitCkghYwqn1hIFreP58db5u\ninAuFPF6C6tv6nQ6YXYjYbonWQQ/ReCztdiJJhmGOHs2bjZbOjuh1EXqHUMEQUSjUfg6YTXE\nypXpDJiiqCTpGPVkE1kKhYKb4+DZi2o0TSiKcmfDBP6jzekMOmdM64aLSXE4HDB2lX67UN6Y\ndDY1Ho9v3868+ur5ZIYE9XEzJBgMvv322xLRLxQ7I8uXL9+/fz8Q3AQBAB999BHXhFUiCY6p\nTtRPCxImwpYuXcremyA8x2UpJDiSZDOvuuqqXbt2cWs6kqf1U8Hr9Y6MjHi93s2bNy9atKij\no8PhcDQ3N0/1uYthGF42hKceQlGUnOCQKSzy9SczOXa73cNpIREabaS4H/TkSWiLEI1GsZMn\na2CPg7SNM4obnU6n1+v1IpU1hpUr8ccfD27YQNO0JhIp+epXE+YC4vE42zEOBJUITG0tMlGJ\nwDAMTdNJAtdcZ/1xHB8bG4tEIhRFoQA0bd6sLai+aZJ0T6S9HcvepJ9KpTIajWInunHjxtEf\n/zg2a5bYiUtCQos+thrikkui69alUwqhVCr1f/lLwnQMg6IqpxOUlCTZ3GAwwK4cofOIWo1C\njwNoE5PwDcVtTSIpGIZhn+yzmCCb7iSXG/B4PHa7/dy5Frbg5b/+y/yXv5QBAPbuLZIEx+jo\nqMvl4kViLS0tPD/LvDGpAERyRwzh2kIVp6SnZCFZ4A/IwYMHhYmw5GdEOsL2YimnlpYWbhsR\nL1mTBlAqi2EYXj1Le3v78PBwc/MU0vooiiZRD0EQRMqCtTIzBDnBITMJBEFwsxtJwtdUwDCs\nsrISAADefHN80XSeNygCZs+eTXd3i62119aGx8ZgzY6Y10kgEEiiVBq/6CJ2khFBEBRFS/7n\nf8QCV2xsDDQ0ZPcAWcLh8MDAADvUqs5O7cGDhdU3TV6noBP0WWTCrFmzkpxosqwsFeVRIUJd\nOm41xH/+px3DWtMYrdFoxMTTMczOneAnP0myeWVlpdfr9fsRob2o2WxGEKSyslLMf7SiokLi\nYnvFRCwWEzaWc9uF1q2ThHiBdAgEAiMjI+DCgpc1a1wwwVEcBS9+vx+Kego7C8bGxqqrqws9\nwAQkd8TgrQ2Hw++8845EilOmLyRJDgwM4DjOS4TNmzdPp9OxutosSc5IAeGlnBiGsdvt7e3t\nv/vd7373u9/BhVlpIzpfhQ0AuLCeJRKJTLWLymQysXkZnnqIXq+XztcrM2ORExwykwDnOSHJ\njTamxtq1YO3a7AxRJgMwDGN6enx+PxQH1fX2Wr7+dQCA8+67x9avh+9JXrPDzpMkrETAFy7k\ntoQbDIYkdQS5k2KhaXp4eJi9u2eYp8sWCoXC9PLLYukepcMBMi5JZcEwjO7uHnM6Q6EQQRC6\n3t7GG24AADi2bHGtWdNyyy1AUEKSSoKD+0gkrIaoqDhvqjIlyYDy8nJSPB2DTJYVVSqVFNW0\nerWC5zxSWlpqNJZXVAC9Xo/jdatX63hvMBgMBkNRxIjTBOFkMjdB9vTTQwpFGwByvuk8DocD\nvijighfPhaLj3EjM4/FYLBYJpiCFMqLcOW1u4Yndbne73U6nkxuTS0eIdLoQCoV+/OMff/rp\npydPnoTVDXv27IGJsDlz5pAkmdyjRLLfdoaGuEngZXy49SwAgGg0OqUEh9lsDgQCPp+Ppx7i\n9/uriiPPKjPNkRMcMpPAtrKLGW1Ioq+Uh8dTWG2F6QWCIKWlpaWlpQAAsH8/XMg6a/ByAaTF\ngl2YC0guYEleOCtlNpuRidtqAnJWRhEKhdiSVGGeLrZoUUF6EoxGozaP6R4URS0WiwWevolM\nSrS9XayEJJV9YhhWUlLi9/sTVkOwEvE4jj/yCP3Pf55/pkwuGYCiKHry5MDwMOzi0Zw+3bpy\nJQAA37ZN8/OfpzKwP/9Zm9xe9G9/K03Df1Qmu6hUKgRB4MMxL0G2bduIXq+SYChbQEiSFM5L\nF1/Bi1A6Ab5YsGABTdOxWEyCTWQGg0GtVrN+Vdw5baVSWTLRUufxeNxuN0hkrhkIBEwmk8ju\nZc4TjUZHRkYOHz68Z88ediGUVeImwsxmc0VFBZsQ5J4RWMRXkMGnQo7aiJKrZkz1E5VKZXNz\nc3V1tc/nYxcWsWCtzLRDLiKSmQSe0cbwz39ONDdrJx44qCVLCjk4DiRJOp1Oq9U62N9P3HST\nWD+FzCSsXQsY5nRPT/DKK0Eik85oezuvtRXOTYkJWKq0WmCzsW/WaDTxTz/t6e4+cfz4iePH\nz778MlyOb9sGGAZcfXWODouNCsTKTHL0ucmprKzUFiLdAwAAa9fi0eiJ48cxlyvhicPUau6J\nS0Jtbe3YmHH16paDB43caojh4TqXSw8ACAaD586dGxxEp2Qii2FYY2PjnDlz6urqqidGorny\nyhSPb1LnkeK2JpkuoCgKg7pgULFlS8Ovf12FYUxHh62jY0SpBONZV5kJhO08+/aZ7ryzORhU\nLFsWfvbZARSVlsNxeiSPxKBnk9SAapFKpTKhm2wgEIBvg603LNziFN4qmYREo9H+/v5oNMpz\nfm1ubj579ix8z4IFC6DHR1VVVUlJCe+MNDY2qlSqhOpRxQ0vLcitZxGuTQWbzcbNbrBMa8Fa\nmaJBruCQmQSDwTA2NiYWvqp1uhSNNnJKMBi0Wq3wuScNJ4hpRu7rU2DZjphJp4IkVZy+ApPJ\npFKpkiiV8ioRDAbD3Llzg8EgjuOaCSmW1APXjBApM4ktWpSPTxegVCqjR4+esVrhwxZbpxD5\n2c90v/xlrj9do9Go1WqxE1d9110plpAoFIq33qofHERAomqILVso2ByU0COjspJJ3oCgVqvV\najW45x5wzz1TOrpJnUeK25pkGlFdXd3dHV+/vo7XLqRWq/V66c6yFgSuBrOwI0ylQoqj9V2l\nUrGlEECgX6iSqhGuWq1ubW2tqqoSusnCOW2SJHlxNbc4JRqNMgwjlywlh3Ul55XALFiwgJsI\ng7kMBEFmz569ePFi7hnZvXv37t27Z2CVQWlpqZhqhkqlSqNnp8gEa2WKjGK4F8rkFK1Wy9VN\naF21qnXVqtKJG4P+5pvBn/5U0AECgiCGhobgPY/tpyBaWwEAdE0NKIS2QtZhGMbn842MjOSn\nPkWhUIjV7ETb23lGJ/AZYkqVCCiKlpSUWCyWkvvuAwwDGAZAS52ckdwnVanRpFitkHW0Wu2c\nOXPq6+vNZnPV0BBcqFu+PD+fXltbm5USks8+S/xQfsklwO/3w2nnhJIB3/xmpsrwYhw6FNu4\nkViyhKyqYlQq0NgIfvYzMOmk3Xvv4evXRxcvjk9pK6lx5AjYsoW65BLabAbSPwqlUrl/f+Pg\noApMJMjWrGles6Z55cpZf/+7/IhyAQqFAlbMCQteMIwxGo2FHmB2KOF4JPEiMYPBMF5VKkkG\nBweTmLAKa094MbkcLiYnFoslcX7lJsJYF4+itMVND71eX15ezqtnaWho8Pl8BoNBzqzJFBly\nBYfM5MyePZsSF/yDURDsjMUwDAsE8qx/4XK54C+1UFsh3Namp+npPqkVj8cHBwdhk0V+6lMM\nBgPy29+KdS6onE5ezY5WqyVPnRp1ucLhcDwe1589O/vb3wZAQgIoBoNBqVSKVSuU3nJL7vRN\nJwVBEJPJZDKZwH33gfvuy+dH6/X6yOefnx0ZgVcXW0LCbN+OPPpo6vtJUg0xMpJMMmDlSj8A\nU5g4OnQo9vvfMx9+qLDZFH4/klCplKIom83W0VG6b9/5hvbkkh8AAJIkrVbr9u0VU9pKatA0\n7XA4HnxQN72O4vPPE/9Ey+1CQqqrq999d2TTpgZewQuCIEuXFkM2HwAATY4cDgdPvzAYDMJ+\nBMmSfE4bwzBWcQbCjckVCsV0f1bJNUmcX1taWriJMDZHJlcZcKmtrV24cCE34yNl1Yxe0PsI\neOQT8MkoGMUBXg2qvwa+9gR4ogrIIqYykyMnOGQmR6FQKE6fDgaDrNFG5TXXADAevkYikdG+\nPtjxCGi6afNmfX71L+BHi/ZT4Lhk5bJTgWEYNrsxqd5ntqiqqoqK5AJmrVuXMBeAYdh5975D\nh8ZfSGaSBEXRuro6rFCCFxJGp9O1trbG43Ecx9XvvQcXIsuW5ejjuB4ZXV2DGFaW4oYkSdps\nto6OsklDd6vVGgqFbDbzhg3Oa67xl5VRe/ZUwY6YvXsTB/nwf1k0GrXZalLfSmowDDMwMBCJ\nRGy21ul1FHK7UOro9fq33qrnFrywq4pGHxdF0aampqVLlxaZfiGKogaDgXWm4xWnyAqjk5LE\ni5e1q29oaCBJUqhWIwOmTz0LwzAul+v3yO//UvkXduEwGH4BvOAG7lfAKwUcm8x0Qc4Wy6SK\n0Wisrq6ur6+vhN4DAIClS0OhUH9//3h2AwBLV5f+/fdp1msqL0EjRVFJ+imm+30OalUAEb1P\n6DGRddRqtam3V3T1pKd17dr8NJ5MCb1ezxw7NjQ42H3q1Injx/v+93/HV+zYkVN90yng8YDm\nZoAgAEHA44/n85OVSqXRaFRt3Jj1E8dKl9E0eOYZy/3310f+P3tnHidFeef/p6r6vuaengvm\nQmBGUeTQEIHVTTYmutk1iscEEhUMl6g/SVY3GhkH1xwmrONocNUQE93VGJNo4oExHCoySDII\nAgMMAzPTPdPdM33fXX1U1e+PpymKOnp6+u6h3i9fL4eqPp6qvur5PN/v5xNEV6xwvvjiiEZD\nJmlsBgUIn89nMskSO5X6fD74odixY3jTpom5c3G9PrpqlQPurajgtyf0eDyw8pn3XpWVxbEA\n6HA44Fcx71GIyX3Thv5+/k/NdCp4GRkZmZb+hTU1NRiGcdsEfD5fAUbDFBrQhIW5BZbAkCS5\nefNmuKWnp+fKK69cuXJlHsaXBIFA4MEHH1y6dCnsCkEQ5MCBAzl7dljPwqWrqytnY5gUkiSH\nh4cnJiZUuOrnoz9/d/Dd3ad232u9F+4dBMKXpiIiDESBQ+Q8BEHEYrHJZ1nnpq/U179uMpno\n8j+W/wVVV5cb/wupVCrkrUChqMxmy8EYskcgEADC9SlwbzZA+/tJgnC7XOMWi+2cFWgBaQEJ\nEH4DKxSKmTNntrW1tbW1tdDrGNm7Yk5CsCAIwuFwmEwm48hI5JZbpl/6T0lJCYqivJYBdILG\npLjd7iQFCHp1dEqWH7RQmGOjkMxCTwh5j+Lmmwsxe0IkBXbujOuQrP+WLcv3yDJHUczEAACh\nUOgHP/jB8uXLE89Xg8Hg97///WXLllVUVLS3t3/5y19evnz5pk2b4N6enp4vf/nLd955Z27H\nXnygKFpWdr7ojy6BHgI0+AAAIABJREFU4VKAQhhFURMTE3/5y1+6u7v3798PL94wDLviiivy\nPbTCwmq1QqX+m+5vft3z9cZwY3Ws+iver8C9XwJfyuvoRIoGsUVFBFAUZbPZXC5XNBqdUo+J\n3++PRqPwb67/RfiyyxQJ7pw5tFqtTKCfomn9+ng/RfaTR7IESZJC2R+h9nZZNgPzUBSNxzS+\n/358U+FdNAAAKIryer2wf0qCovq77pJO9gbGMAysXg1Wr078yOFwGMdxFEUVwaB0yZJJ3z8U\nRTkcDlh0gyHIzHXrFAlH4vf7R0dHYZGRvrtb9tFH0y/9RyKR4HhDR4eCZRkAAJg3r5JlWCtE\nYtnim98MAxD/soEBQEyYlh+33+4HQM19/MT3uuOOwJSMQvIFt0GdeRSrViEAKPMxLhGRaQi8\ncNqzZ8+2bdvojdz5KkVRVqt17969//3f/534AQtwTl6A1NTUhMPh8fFxpj/LAw88sHHjxurq\n6kIOlrZYLE6nMxwOb9y4cd68eZ2dnVartbm52ev15r6NGsfxzs7OgwcP9vX1Qamlt7d3yZIl\nOR4GF4qiWE00JEKOSkf/u+a/AQD6mL5T0pmnoYkUGaLAcbED27bpQgC6xySZWRZ9Pc1bX4Dn\nSuCorKyMCnsrWOrq8LNn69askRfn2rhEIpmkPqW2NvEjpA951134HXfEYjF5ICBvaSkonYgg\nCIPBcL5Jqrtbundv+jJBMBg0mUzxqEKSbNqwYVLRhCRJ6IAA/1nZ3a345JMEI4lEItz0H+iu\nQtbWotMi/Qfyxz/qYFsb1zIgyXzeyQSIEC1wsBIWWJYfcjm/QyFLZ+Hcq4b3XoUGq3ibdRRS\naUu+BiYiMv0YHR31er04jjPnqy0t7E/Z2NiYx+Nh3WzWrFn/+Mc/Cnk2XrCgKNrU1LRw4cLi\nSn4NhUJOpxPwpds6HI6ysrJcNigRBPHxxx8/xfBkKpxCkmg0yuwrv3nWzYOKeE/K5cHLnzU+\nO2POjITJ8iIiccQWlYsdh8NBqxusHhOipiZxj0n8elrA/yJ82WXZHfo5UBRF+/uHh4aOHzt2\n/NixM3/4A9xu3bTp+LFjjssv1/zkJzB5JH6HohI4dDqdUEZv0/r1JXRtRXagKGp8fPzkyZND\nQ0PGkZHoihWF1kMxNjZGawqsNzCor0+tSSoYDA4PD8fVDQD0PT2a3t5J3z8Wi0VoJLztWna7\nHaobXHeVwNy53EDB4uXzz/m3J28ZwJUt7ryzxefDFi8ObN8+Ipef36tWxws0eC0/6L0sEt9L\no9EkO9C8ojj3FuUehU4HZMykGRERkTTweDxerxcAsGjRog0bNsyfPx/OV9vb202MxHG/3+/x\neLg3u/TSSy0WC/dLniAIq9V63333XXPNNWq1OvceDUXB8PBwUThlMoHvFhpmui1FUXSJYg4I\nBoODg4MWi2Xjxo3PP/88jCVqbW2lfz4KhwgSsUltEiq+/HBUdbRb353fIYkUEWIFx8UO/TvB\n7TEJtrUlzliFX4hC9QUypRKYTKC+nqIoBEGy2iQilUqbm5txHMdxXLp3L9wYgs7kRb42rlQq\nJadOCe2VXH11Vp/daDTSP730PL9weihCoRA9PO4bODJvXmpTOovFwnWWSfz+IQiCdkDgjgS/\n9FJub0BidxU0FBKajRcd6WdkqNVq+EKTJNi+Xf/ii1UUBVascD76qEUioZgnqqSkxG6322zR\nhx9u2LdPK5VSjzxiXrHCBQDQ6XRKJX+PRmlpqd1ut9tj3HuVlZUVizRQXl4eCAR8Pox7FKWl\nZWICpYhIpmAZoDLnq+FwOBQKwa8aqG7w3owgCL/fzzQhcrvdFovl8OHDzz33HL2xcJbWC4di\nTH6lu7khdLotDNBh7c0esVjMYDAQBMEqJGlrazOZTDNmzMjNMBIglUpRFIXan4yS7Tu5jwJU\nn7pvQ9OGMBJ+t/RdVqGiiIgQ4hXPxQ5sMxGaZXGbupmoVCqlUilUX1DW0eF78cXBwcETJ06c\n7O8P3XRTthf/FQpFaWmp+r77Bk+fPn7smG/pUt7kkWw8NQ1FUeFwOBgMEjZbphIxJCdPmk2m\n/uPHmfUp/oceokgyq36fXq+Xlg9YJQkxvZ4sgFQGumJCqEkqhceMRqPQzxIIJNfw3iscDsNL\nLt6RBNrauHdJnP4znSo40qe8vFwikfA6lZaVlUmlUvqWCILEYk3f/vasffu0TMuPwcEKgmgQ\nenwEQQiimXuvs2erotG6nBxiBigpKfH59B0drayjGBgoC4eLo8tGRKQoYF0asear9N4kbwYA\n8Pl8Y2NjBEFEIhHm0npLSwvz+02kSGEVIdLptnPmzAF88bdZwul0MhtAmIqbx+NJfMGfGxAE\nKSkpuWALQBYGF6pIFQBAS2nzNC6R4kOs4BABCTwsSybTShsaGoCw/4W9sRHW+Vd2dyv37cvN\n4j9BEPBJhaaayoRlKSkDLbKdTidJktC1QZMhQQdBkLq6uqqqqlAohO7ZAzdqrr0WZFnJposq\nuSUJofZ2EAhotXn+sYn/VAu8gfFLL00qouNCEjvLBNvbecsq4gtKwh8l7l2kUqn2zTeF3FWk\nVivI9+nNDX194NVXwf79wGgEbjeoqwN33AG2bgXMsgkURQmiuaMDYTmVqtXqK65g22q8+qpk\nZAQAPsuPBx4QHEZq9yo03n23SsjuJOWjSOYFEhG5qGAtI7Pmq/Re1pVGgmnt+Pg4/IO1tN7e\n3m6z2erqikZmFeFFpVI5HA76nzDdtqWlBdZB58xklF4TgrAUt2AwWAjlijU1Nb+W/tpG2Zb6\nl+qjeofE8dvK37owFwDgVuTWfI9OpGgQBY6LHYVCoXjtNd5ZFiKRyGw20CC47AkAkMvlsRMn\nLDZbIBAIh8Oas2cb//3fAQCe739/9K674G1YRf5UXR0i3CRCUVQkEpFKpajbnVpLS3zpW3iq\nSWZB4KAoymAw0GGT2ejmkEqlUqkU3H8/uP/+9B8tGRJX90hyVVSZALi0JdQkhclksElqSo8Z\nX2kReP8IOcvAywKhkUjkcu5Ikkr/me643e7HHpN98MH5yzuDAfzsZ4AkAcMBDQAAXntNnuTU\nPTXLj/SNQgqBjB9FLBbr6iLffff8Va/QCzRVPvss9tvfkp99ho2NoR4PIuomySCKTQWCUqnE\n4Y87AIAzX6Vb4ZRKJdNeQWhaG4lEaMsnCHNpPZcGDSJZQqfTyeVy+CrT6bZQVlAqlTmzeWJV\nhrIUtwKpG8Uw7LOqz95H3n9W/yxz+3ww/6fIT/M1KpGiQxQ4LnbKyspQgVnWzLVrk5llSSSS\nWjrI4+BB+H83dHlM2o8AABAMBsfHx0OhEEVRgCRb7r1XlVIFhEQiQVG05I9/FFJtsPHxxKpN\nCrjdblrdYAk6Mb1eUlSuHzQoiibQiXQF0Amp1WoRBFELvIErVq5MQSaQy+UYhun+8IfEzjKs\ne0mlUrVaLTSS2rvv5o6koqIiJlz9VAgWJ1mFoijo8GIwzFq3zvbVr3rKyogXX6z6/e/LAQAf\nfsiePyc/dU/N8iN9o5BCILNH4Xa7zWbz2bMtybxAyROLxUwm02OPle3adb7EKlO6yTQGx/Et\nW6idO8//foonLV+Ul5fT/mWs+apOp6ObSsrLy2kzadbNNBoNbevITYliLq1z94oUHQiCzJw5\n89ixY6FQiE63bWxsDAaDer0+Z74SrHYnluJWOM1Qy5HlVmA9C84GQEAP9LPArO+A73SADgUo\nOCdUkYJF9OC42CkrK1MLe1hOeZa1ejWgqIDf71u6FEzFj8Dr9Q4PDweDQfi9r+/pUX36aWq5\nJwiCaLVaIWeQmWvXIr/73dQOKgl4DSZp1wbW4kw2wHHc7XY7nc7g2FimvD+USmWChFr1hS5r\neUEikVRVVSn6+wVvMXWZAEGQiooKofdP9Xe/C15/nfeOdXV1yqkIFhiGIcePD509y0r/wR95\nBFBUVt1VCgGr1QpXJnfsGN60aWLuXFyvj65aFS/i5Rq87NwJKAr84x/g/vvBwoWgqgpIpaCx\nEbz3HiiAxuFpSCAQMJlMJEkm+QIlCUmSw8PDPp/PZJKtW2d7880zu3YN3HabE+798MOMjH0a\n4na7z549OzKCiCetEFAqlbW1tX6/3+Vy9fX1MeerzHYDiUQyc+bMQCDAvRlz0Z5l0AAuXFrn\n7p1m4Dj+8MMPX3vttRqNhhUck2BXNqAoyuFwPPDAA0uXLs34M8rl8uuvv3758uWbNm2CW3p6\neq6++uq5c+fm5ugAAMy2YpbihmFY4fiaPwwe/gf4hxM4wyBsBMY9YM/d4G5R3RCZEmIFx8UO\ngiDSU6ecTqfL5cJxXDEw0HrLLQAAqqsL2bIltcdM7EeAX3op6/YkSZpMJqHcisQtLbzo9Xoq\nt2vjibs5QCSSvZDzaDQ6NjYWz/olyaYNGzJl5lpeXh4o+B6K6upqW2/vkM0GV8kUAwOzVqwA\n6b2Bq6qqYlNX/eRyefjYsRGLBdbyJDMSmUzW0tISCoVwHJeec1dRLF2a2rCLCHgdCf/Was97\nnn32Wfyif8UKnnsFAoEtW9AiXcEuuuYCq9UKv5OTf4GSwW63Q8F3x45h+pFXrXLAwpACcC4u\nRMLhMPyJFE9a4VBRUXHJJZcw80p7enp6enpuuOGG984J4gAAjUZzww03OJ3OBDeTy+VSqZQZ\npcFcWi+WmOrUCAQCe/bseYrxDc4Mjjlw4IDQrozj8/nMZvOhQ4d6enqy8YxDQ0PMtwENvTEH\niTmlpaUul2t8fDwWizELSVwuV0NDg5i0JTKdEN/NIgAAUF5e3tra2t7e3nLu1xpZvDjlR4OT\neaHFf6lCARhB8QAAr9dLGztzKyAi8+ZNdQAymQwcO3b2zBnW2nhsy5YsrY0jCJIgESN79Ydw\nOTSubjC8P+K7OVPxSCTi8/l8Pl9kfHzSQg+pVKobHBR87kzoROFw2OPxuN1u3GxOufCkqqpq\nzpw5TU1NtbW19ees2tJ5A0PVz+f1msbGzp45Y6KvVrduTfz+kcvlTU1N7e3tra2tTecm8JOO\nRKlUlpWVae6/H1AUoCjwjW+kPPJiIRwOc9t9T51SPPtsNQBgwYLQmjXsu1it1uHh4SJdwfb5\nfJ2deE8POHQI2GwgGo1LMz/6Ub5HJgBFUSw7OsB4ga68MnjXXQTf/SaHti7m1U1uukmsxufB\n6XQmEJvEk5YXhoaGmOoGzaJFi1g3453Wsm4GM1MgzKV1FEWrpoWCFQqFfvCDHyxfvpxZrQC/\n1R0OBzM4ZtasWUqlMhaLDQ0NGQwG5q7W1taMrxXBIpFly5bp9fq5c+euXLny5ptvZqbYZOoZ\nYbotJBKJnDlz5uWXX+YeeEaeKwGNjY033HADq5CE+U8RkemBWMEhch4EQcCaNYA7t5giif0I\n9HfeyVr8T5xbgV92WQo/L3K5vLW1NRKJ4Dgu/fhjuFHypS+ldkSTolQq1W+8wSvoAAxTOBwg\nO4swDoeDPnuJvT/gGmB80kKSTRs2yJIo9ED7+yORiNPpDIVC2PHjM//t30B6xREJxpNO4QmG\nYRqNRqPRgHvvBffem+bYIFqtNl7Pee79Ay68JBUCRVGlUgnWrgVr12ZkJNMPrrqxa5fu0Ucb\ngkF08eLAs88aJZILGtn8fr/VagVFuOxPEITRaAwEAsPD561Gduyoef31EpCek0VWIUmSLqmD\nMF+gnh4DgswCIJXK+SjHnJipm6xaRYmXJVyYfpYQ5km7/fYIAKX5GNdFDZyvZupmZWVlsVjs\n7Nmz0WiUXlpvbm5Wq9WhUCh7FaBZAsfxxx577ODBg59//jlcgOnq6tq2bRt9AwzDWltb4bc6\nKzhm7ty5NpvN5/OFQiHWrra2ttHR0aampgwOlVUkgqLoD3/4w1gsxkyxqanJZMw29KTHcZx7\n4GazuX6KtuhTZWRkxM3XX7woucsbEZFiQazgEMkcTie9CD/jN7+Zkh8BAECoAiJ6+eUpj0gm\nk+l0OuW992Z7bTyBa0PjunWSN9/M0vPyJrnS3h/0ZXEkEoEWJ/CfkxZ6MJHJZDU1Nc3NzTNt\nNrglneIISDQaTXk8eWD16ountiI3MLPoSBI895x+8+aZwSC6YoXzxRdHysvZU1y73Q7/4F3B\nvuWWyecP+WJ0dBRe3zOdLDo6rHBvwUozGIbR5crcF0irpSSSFGUIVhX0rl26O+9s8fmwxYsD\n27ePyGRFfE0SjUY/+si/dm3oyiuJqipKJgNNTeA//zMDHjEsQZB10jCscN//IslTVVV14403\nMtfSu7u7m5qaVq5cmd+BTRWn0/mXv/zlF7/4xb59++C3H4qiNTU1zGqFlpYW+uoFwgyOsdvt\noVCId5ff76dLVtMkGo2ePXuWVSQC24KymmLj8XiYkiXzuVwuVyTLnlLMQhImXV1dWX1eEZEc\nU8QXEyKFAEEQDodjdHR0+OzZ8M0304vwkquvRvv7R43GE/39zCYRqquLW+QPDZyFWlriuRWF\njUql0p4+Lbg7azN2uBwq5P1BL5ZOTEzQTux0oUd41iwAAFFTA5K0OMncPN9ms2VgPCJFi0Qi\ngX5mPh+2aVPjCy9USSRUZ6eps9MskVAlJSWs2zMvdiHMFeyVK9nr2zQEQcA55/z5sczOOZMh\nGAzS4UqZdbLIAbDzn36BEATU1kZ379YtXtx+/fVzHnkETe0c0haMXN1EpwN0rkTRMTExcfr0\n6SefJF96SXnkCGa3IxlsRKIX8LknTaMhZQXr4yIyFZJsZilwLBaL2WwOBAIs1eDLX/7yhg0b\n5s+fT1dGsOqSmMExLEWPuQsAkBGBA5ZRwCIR5sDgUzCfkVt0lib0jwKEdXSsvVOCIIiJiYlN\nmzZdc801arU6N8alIiKFiVgLKpI6oVDIYDDAmaq+u1v+8cekQoHCH62FC6VS6YwZMwAAkUhE\nQqe08i3+a7VaqVQq1NKiu+22AvGzTAx24kQwGHQ4HDiOS/r7m7/1LZChbo5ET4phsWhUKMlV\njaIAAIqi6CUIbmpvsK1NQ1E5SymD8Bae0ONREcS0940Xqa2t/fhj07p1MwwGmURCPfjg+OzZ\n4aNHVTKZTK2uZN04cbsEhjXxPoXL5bJYLE8+2bBr1/kGsVz6kvJeiNPSzOLFkTVrCndqqtfr\nT5yIrF8/02CQIQggSWCxxEMETSZJyuewsrLS6/V6vejDDzfs26eVSqlHHjGvWOECAFRUVOb4\niyhTTExMwNkRjIbJYKQupKSkxO12+3wY96RJpVJmbIdIIYDjeGdn58GDB/v6+uCXQG9v78KF\nC51O53/9138dOXLkyJEj9PYlS5bAeyXZzFLIhEIh6B69ePHixYsX080Xl56zlmdWK7DuywyO\nSbyLtmxLB6/XK1RGwXrGjFtvssbPOroEqcC87yv6/QNTvY8cOfLLX/6SvksOjEtFRAoTUeAQ\nSRGSJGl1g+X+QNTUYIxFeJlMBu65B9xzj9BDIQgyY8YMLLe5J9lApVLFrzX37YNb0u/mSIxa\nrVb97ne8lS+IRKJ0OoFaTRAEXA8RKvRQEkTKBecpQFEUfNsIjUcWi4kCx7RHoVDs2dNoMEgA\nALEY8tRTtfSu7m7wwAMX3Fgmk8GLUZIE27frX3yxiqLAihXORx+1SCQU7wq2x+MxmUwga3PO\nZOBeiDOlmd/+1i2RZLfdOh3kcvnf/tZgMMgAALzTrtTOoVKpxPGGjg4lS9hSq9UaTfXk9y88\nYrEY3UKVJY8YrVbrdleuWlXOOmkAgPnz9UWqCk1XAoHArl27WNkf1dXVp0+fPnr06PSefLLM\nHbhyBqtagQkzOCbxroxcriQuo2A+Y8bDU1mXN0keXTAY/Pjjj4UyZXw+39jYGAAgEols3Lhx\n3rx5nZ2dVqu1paWlwK+mEqs2IiIpIwocIinicrngNJV3EV6K41MqNlapVJHjx8es1mAwGIlE\n1GfOwAoIsHUreOyx7BxBNlm9GqxenYPnqaysDApUvsxcuxZWvsTXHwRSe0Pt7TnOBoNlkxRJ\nCo2nLIPjcTrBokXxzqkifS9NX44e5f8BWrCAvaWkpATHcd4VbLVazXtFOH4uTyePvqTMgXGl\nmbKyilwMIg0OHUq0npzyOfzTn0oMBgCSELaKAp/PRy+8Z7wRKRaL7dsXeu017C9/qbZaUcB3\n0jhr4SJ5w2q1Wq1Wl8vFmmTCJjvW5LO1tXWaVd+w/CO4coZQmQYzOAYAoFQq6bZE1i4AQNz8\nOz0SlFEwnxFBkMpKdkVhmmi1WloJ4h4dbyqw2Wx2Op0TExPM98+cOXPo98/ExAT8g2VcCk1S\nGxoaMnsIGYGiKKfTmUC1ERFJB1HgEEmRuHeUkPtDMDjVbmqZTAa/hSmKQv7+9/jWouo+zT0y\nmUwyWZIriqJyuVyo0EOqUKAWC8iycTcLlUol+9//5R0PJpNJrdZ0xuP1et1uN47jCEXNWLtW\nkUY4SwYQFRZhdu5M9pYVFRXHjoVWr65hrWCjKLpwYR339jiO043TeTS/0Gq1UGfhlWYycpme\nPWKx2HPPneVuf/31ih//uBakcQ4//5x/O1fYKgp4S8rpRqRFi8Jr1qQYgQF7rH7844ZduwSn\nwUV60qYlgUCANxaErl/gZoL4fL4C/x6YEqxiIq6cwapWkMlkDocjFovRwTGNjY1er7eqqkoi\nkZjNZtYul8tVUVGREaceliwOB9bU1BQKhehnbGpqUiqV4XA4s+mtOp1OqVRarVbu0VVVVXEL\nEm02GzRnYb1/LrnkknA4LJfLo9Eo19AkeyapGQF6oPj9fq/Xy1RtcpOVK3IxIAocIlPk3IRt\nJgDWTZuU/f28i/CqNJokEQTJWQXENADt74/FYm63OxQKSfr7a6ED6IVz6dLSUqlAoUfd6tW5\ntzipqqqKCYyn4Z570hnP2NgYvTai7+5WfPIJ0xcmrUEnB1yU8Pv94XAYQ5CG731Pnl+FZVqA\nouiuXTMMBgQkt+yfeM65YEFozZpcXELJ5fLy8vIjR/wbNzaxpBmVSqXVZrz2OZPwWusxvV3X\nrElx8Tl5Yaso4FaAZ6QRKXGP1RVXgCNH0h+7SCaB9hM0Qn4TrMnndBI4lEolMxuFJWewqhVK\nSkrq6uquvvpqZmNLT09PT0/PDTfc8M4778yfP59313vnIurSQaPR0Jau9MDGxsaWL19O3+aZ\nZ5555plnMvWMNAiCNDY2Llq0KJmjoyjKdi7ADsKKm6mvr+f2QjJrZwiCoHLuszYpNpsNdglx\ns3KdTmdFRaGXN4oUPmKKisgkwAnbyMjIwMDA4MAA/q1v0VEpmNvNuwhPoajcbsdx3O12O53O\n4NgYHR8Lnngif4cybZFIJJWVlTNmzKg1m+ObGJUvFotlYmJC0d8veH++iXc0GoXrhzabzW80\nZvYV1Gg02QidcTgc9BUDK5yFrK3NQTgLQRBDQ0MWi8Xn80UiEd3Pfgadd+O7RYEjDQ4f5r8+\n413B5p1z0smav/qVOWeeM7W1tX/9ax10soDSzMqVLStXtnzrWzXnBOEChdu5xjyH//M/xhz6\n9uSZvj7wwANg0SJQXQ24QTxMmYqbcqLXpyil0TXnzIDhVaviU+iCDRjOLIFAYPPmzcuWLdNo\nNIUfCcEKexLym2BuT+AoWYyUlpbS3xtMOcPn87lcrr6+PrpaIRQKqVQqg8HAsu2ALFq0aGRk\nRGhXRoaq0+lUKhVrYLwRrdlIsTEajUkeXSgUSpApAyupub93zNoZDMMKTd0AALACg5iqDW+W\nkIjIVLlorlBEUgI6iQYCASgAs5bEJXY74FuEb1q/3vGf/2mE4e0k2bRhAxAXsXMDp/JlYmIC\nLiudeestAIC+u7tqxw5SLkfDYQAAeOgh8LOfcR/GarXabLZ4Y3l2XkHsxIlwOAx7SaQnTtTd\ncAMA6TZx0AtoXF+YUHt7DhbLTSYTfY3Lct6l6uoQMf42Daa07K9QKDAMg+taXPOL6uqybI2S\nA4Igp0/z9FSDgm8ukMlkCc5haWkhFp/09YFXXwX79wOjEbjdoK4O3HEH2LoVpJOjGgqFOjuR\n998/XxXPCuKRy+WlpaW8KScymay0tDSFJw2Hw/Rcq+gChjMCQRAWi+WTTz55+umn6Y0F3p/P\nykAR8ptgTT5zOcJsI5VKGxoaTpw4EY1Gmc0XX//615mVHcxqhQTBMdnOlGlsbFy4cCFXaMh4\nyQaX5BNzWOoGuPD9A/dKpVK5XB6GF3UAgAtrZ3hNPfJLNBplSXtM1SYcDpMkmWNvOJHph/gG\nEknE+Pg4VIgRBGEtiVN1dSp60svBe8kl8A99T4+mt1dcxM4LTHt/wJhyx2jTrGuv5d7Lbrdb\nrVb61zd7r6BcLtfr9Y2NjXUWS3xTGkslBEHAKQGvL0ygrY17oZBZotEob/ytYnAQABC+7LKs\nPnsBQpJkLBYDTmfuC7gQBKmurgYA+HzYpk2NL7xQJZFQnZ2mzk6zTJZ507jE7NwJKIrnv2XL\ncjmKKUO763HPoURC5fgcJkM4HO7qivT0gEOHgM0GotG4ErFmDbVpE7FwIcVbf5GY8fHxs2fP\nDg+Ddetsb755Zteugdtui68ufvjh+ZvV1dU5HGUdHa379mnpRqSTJ0smJppHR1O5ykrcY3Xl\nlcE1a1J41KKBJMnh4WG32w1dOZ9//nn4cW5paSnAtWgalnuCUCxIgU8+00Sn0914443Lly/f\ntGkT3NLT08NUN2iyURkxJRLUj+R+MEJwLbSZ7x96r56xfMKsnUFRFH52uOA4/h//8R//9E//\nVAjlUQlCgkVEUkOs4BARhCRJl8sF/+YuieOXXir/4AOTxQJvoxgYmLViBQDAvXnz2N13w3ux\nFrFjer1EXMTOIbD0Bv7NU9TwL/+ihIYdDEiShDZpENYrSNbWotl4BTNhuRLXL4TDYrK9HETX\nbvAqLMH29gwYo+WXpA1THQ6Hw+GIRCKAJJs3blTno4CroqJicJDq6NAxzS/6+7XV1dVqtayx\nMWcDKWIqKyu2fCj8AAAgAElEQVRPnYp95zsVLAOR8vJyrbawpmYTExM2m+3s2Vm0XcWvfqX/\n3e9KAQB//jPp851fJ2fVXyTA6XRCgXjSIB4URT/8sD6D0TDcWQ3T1+P5580SySVTftDJKJy8\nRofDAU0TuW6dZrO5tbU1l4NJ/rSUlpYGg0H4Nzcdg7tdoVDodLqsH0BuGRoa4m0x2LJlS1dX\nV+7Hk4DkyyjyiEKhkMlkdD0X631Fv390Ol1dXd3p06eZtTPNzc1qtToUCsnlF/gcw8u8Tz75\n5Be/+AW9MZflURKJBEVR5poTS7URyzdE0kcUOEQEwXEcfksKLYkrUbS+vr66uhrHcXTvXniv\n4Dk/Ld42AXkkwvWIFskS9DIg7yuIX3YZtzuc2fDJq2oVbKgd/FEs+eMfeX1hEIkEGx/PalhM\nYoUFv/Ayt1igKMrtdgeDwWg4XLt6dTKGqRf4vPb0qPfvz7HPK80771ROpzjS3IMgyAcf1Bb+\nObTb7XACzFQivv1tGxQ4SBLhmnR++OHkAofNZuvvV77zTunhwyqLRerzYdXVsZkz43XgrCaR\nzEbDyOVyqVQKfV65/UFVVam0vSQGx/G9e/fmMa+RqyP87//+LxwAsz8/FApFo1GpVJqDIcES\nyN7e3iRPS1lZmcfjGR8fZ6VjeDwejUYTDAaZ22fNmqVWqz0eT2pNTAVLUagGxUVNTY3RaPT5\nfKz3VSAQYLY4lZeX33jjjUx1qbu7u7u7m9VxQ5LkyMhIMBgMBoOs0GKWDpI9EAQpLS3lmrxC\n1aasLHc9pCLTGFHgEBEk/islvCQO/5BKpVKpFNx3H7jvPgCA+8QJQJJC8bFoNCoKHDkj/uMn\n8ArG+K7SEmsioXMCB0mSPp8Px3EEQZShkPa661JPQs1QkCqCIDqdTi0QzjJz7dpsh8XAN3bZ\n22/zKiwSuRyYTDmO402TSCRiMBhgZ6++uxsapiaWKjweD9fnFZb/EDU1WG4LuKZZHGleKPxz\nyEwZ4LWruPde63e+E+/U462/4CUSiUSj0V/9qnbXrvNr7Gaz1GyWAr4QmYxHw1RXV5tMJq6v\nB4qiVelZjMZisU8+Cb7+uuQf/5CZTJjHg+j1xNe+5luyxLZhw4bLL78cTnhmz56tUuVI0I5E\nIrt27WLqCMxidZZbZyQSyYHAgeP4yMhILBZjxVjOmTNH6LTAdIwFCxbwpmMcOHCALokFAGzb\ntm3btm05sHsQKXZ0Ot2MGTOgUkZv5KauCNXOsDpu7HY7rDPihhZPTEzU1fHErmcDvV7v9/u5\nIcGhUCg38qXItEcUOEQESW3ChmEYSRBCmohWLDzLIbC1svStt3hfQYVazX0F46XRAppI9PLL\nAQBer9dkMsWTyVKyII3FYg6HIxQKRcPhhu99T5mhFga9Xk+euxTmIcvlA0qlUiaTCSks1d/9\nbu7jeNOBoiij0QjVjeR7zRI0tQXb2hS5WnqFFGwcaW9v5NVXwWefYWNjqMeDZMQLM0sU7Dmk\nCYVC3JREpl3Ft799PrwzeZNOn88HLoxo3bKlvrdXAwBQqcjt20cwrA2ALPpBlJWVnTkDOjrU\nme2xcjqd4+PjP/lJw65d52fpY2PYr39dRVE3bN68gJ7wzJ49OxAI5CDK2O/3G41Gu93O1BGY\n1hWs/vwclK9TFDU6OgrlftY8cPbs2bFYjNtDBBHK/mhtbX3//fe52wvK7kGkYHE4HEx1g4b5\n/kmydoapsoELy6PcbndtbW1ubG4wDGttbRUKCRZVP5H0EQUOEUGkUqlKpRKasNXefTfvhE2t\nViO//71Qm4DC4QANDbkZv4hEIikvL1cKvILaW2/lvoJKpTJBo4dCrQ6dOTMaDnMtSJPvQQgG\ngwaDAc5J9N3dyn37MtXCIJVKo/39BrMZTk5oXxiisxN7/PF0HjkZEASpq6uT5k9hySywQgcI\n9Johfj+vBwMURITKf5Bw+CJfnIlGoyaTqbOznFkXkLwrhAgXrrrBtKvo6TFgWPzLilY9Fi7E\n16xJZIkTjUbHx8fBuZ4X2CRy4ED8DT9vXrC0FM3BNODtt8sy2x/k8XjMZjO4ULih23Z6ezWb\nN18w4bHZbNkWOAiCGBsbI0mSpSMwrSuY/fkoiuagkN7v9zMzKQBjHtjW1uZ2u4VMdhNMMnt6\nejI+TpGLhEw1/pAkCRvfaJjlUXBvzoqsi8LkVaR4EQUOkUTU1dUhU5ywVVZW4vlrExBhUVNT\nEzt1SnA35xWEntsSgVew9I47nD/8IfXtb8N/ptCDQBCE0WiEc5JseNBKpdLGxkaCIMLhsOTT\nT+FG7Oqr03zYJNFoNIHDh41mM7w4phUWqqsL2bIlN2PIFLANXkiqoAIBXoEDQZAETW0Fa+CS\nG2A2RCQSMZlqUnOFEOHCXEvn2lVIJPFZAVP1eOklq0TSnOAx3W43nE5otQTdJIJhFEEgAIDr\nr/fmxhsy4/1BExMT8A9e29Syshi4cMJDW2ZmD6/Xy4yMYcorcAurP7+8vDwHFRxQ22XCPC3c\nvSIiRQFXlmWVR6Wm24ZCoccee+zgwYOHDx9O3p9YtGsRySpiv4BIIhQKBXX06NDZs8ePHTt+\n7NiZP/wBbqe6ugBFgeuu472L9vRpwUcsqkXsaQCCINJTp/BQyG6zmU0m55498R1bt17wCjKy\nPCuff14zMCD0gJ5Zs+Af3CTUYFvbpD9XbrcbXs5y7x5qb4c/jemDYZhKpZKtXx8P5OSExWQP\ntVp9ySWXzJkzp6mpaeY5awBk8eKcDSBTkCRJSxVjTz4ZbmlRnrvED7W3C2XuyuVyoaY2CkUV\nDgfvvSYlHA7v2eO9557A/PmxqipqqmGfBYLdbodm+Dt2DG/aNDF3Lq7XR1etip+T9HwVLl7o\nrETeOFsAAEmC557Tb948MxhEV6xwvvjiSGXlJEuUdCKS0SiD4a8YRsFHmz0bnz07EgrxJy9m\nlswGDEciETqLgdes5Gtf84ILJzwkSeYsfArC1BF8Pp/L5err66P786PRaG58ELlHzTwt4qxM\npEhBEIT1CWKWR2EYJtR7BfF4PP/v//2/a665Rq1Ww2TZ3t5eq9X65z//edu2bZ9++im8hMux\nP7GICC+iwCEyCQqFoqWlpa2trbm5udEet2pLPGHDTpyIRiI2q3XUaDTTfaesGbVIDlEoFJWV\nlXV1deW028WiRV6vd2RkZGBg4GR/f+imm5g+GpKTJyPhsMNuN5tMjt2749u3bo2Ew4GrrgLC\nC/tC814aeDkrdHfWxW46EAQRi8WYwg144olMPfikSKVSjUaTF4UlU0gkkgRSheycdsOivLyc\nbmqbdeuts269tfRcM23T+vWSN9+c6jAoijKZTIODgz/5CdixQ/3FFxK7HYlG420dP/pRqoeX\nD7xeL/yDd3p5000xnvuITAaCIHq9nlYimHG2R4+qBgcVXNWjvLw88WPSM9i33y4zGGQAAIJA\nwmEUAHD6tGLVqqZ33y2+6ldmoQQN06zk5ptdgJPXmO1OHJZYwNQRvvGNbyxfvnzTpk1wV09P\nz4IFC1atWpXV8UC4VfrM0yIapYsUL8yMEm55lNDnnSTJsbGx995775lnnunt7YW1XRiGVVZW\nWq1WHMc3btz4/PPPV1dXAwBaWlp4v21ERHKJKHCIJAWGYWq1WrpuXZITNqlUWlVVNWPGjDqL\nJb4pE211sVjM4/FYrVaHwxEcG5va3DVPc93CYvVq+AqaLr/caDT6/f5oNFr59NPQCCN+m4UL\nAQAymayioqKurq5iZCS+fdEilgUpd2F/0uLh1OoCkoeiqImJiVOnTp08efLUiROBf/u3qRqg\nikCYkTRcqaLk/fcB3dDL+GRpu7sTlP+k8BJYLBZoigZdA95888yuXQO33Rb3iv/ww6kfWBKQ\nJPnJJ8G1a0OZLRhhNT+DC6eXK1fmue69rw888ABYtAhUV4PiqpEpKyvbtWsGVCKgXcXKlS3w\nv3vuaWapHibTDKuVG5B9AfQi54kT/LcsnBCZ5OGuze7apbvzzhafD1u8OLB9+wiGUawJj1ar\nzfaohNaT7Xb7pK6K2UOr1TIDOJmnBUGQkpKSHIxBJOPgOP7QQw9de+210H8dQZADBw7ke1Cp\ng+P4ww8/PNXDqaio0Gq13PKocEKHrNHRUbfbHYlEmEJGc3MzLApbtGjRhg0b5s+fDw102tvb\nzWZz+pdzYNq9ZCK5pBBXIfx+/3vvvff2228fOXLEYDBIpdJ58+bdddddq1ev5k6fzp49u2XL\nlt27d7vd7hkzZtx+++2PPPJIzrLNRCZn9WqwenVGHsnhcExMTMS/NJML78Bx3O/3RyIRKYaV\nr1yJiXNdAAAAbrebdtJmGWGQtbUoywiD8QqiAKhUKvn//Z+QBSliNidOQpVKpQnqAuR2O6hO\nvfaboqjh4WG6b1zf06Pevz9TDqYXG0qlUiLs3hJsaxs/fToSiQCSbN64Uc0s/3nsMY/H43A4\ncByXnTwJXUjIxx9HOzunOoZIJELn3vG6BmSjrcPv94+NjT3xRF3GfUAxDGM6YrK8MGWypjRG\nnRYURTkcjh/9SPnXv553lCwu69OTJ/mVCKcTA1M36SwpKbHb7T4fhqIUAICOaAUAYBg2Z86c\nHNhAZByZTCaTyeCEhGtWEgp5Xa4L8hq9Xq8++7nOJSUlExMT8EmZOkJDQ0N/fz+MLOns7Dx4\n8GBfX18gENi6devXv/71Sdv70wTDsNra2rGxMZ/Px4qxRFEUx3E64SUBOI4zRw6SMyYQyQbh\ncHhiYuLAgQM///nP6Y1F3Unh9/v37dvHTFZO8nBgmPGVV17JjFNJHF/i9/uhcXsCJ2BwoYFO\nLBYLBAK8Cin3c3HbbbeZzWaWf0c0GrVarfv37582L5lIjilEgeNXv/rVgw8+KJPJFixYMG/e\nvImJid7e3v3797/zzjtvvfUW88Li+PHjy5Yt83g8//qv/9rS0rJv374nn3xy9+7de/bsUSon\nWaIRKS6cTqeFLgZJIryDoiiz2Ux/g+u7u7HduymlEoFNEBf3XNdxzgqBG5ARmDtXQ1EJypKr\nq6tjAhak+jvvnNREtqSkJCJw96b168mf/Qyc85ZLATrgHaRkgCrCQnLypGV83Ol0UhRFG6b6\nH3rItWmTx+OBi/u8KlJJSQlc5KTOLbagV12VwgD8fj/9N29bx6Rhn1MlFAoZDAaKonhjJtL0\nAVWpVHB6eeyY8sc/rjt+XAkAQFEwNib71a+qf/nL/PxmQevTUChkNM4qXutT3jjbb3wDfPAB\nz/ZJ6y+USqXPp+/oKGFGtB49qgIAXHFFdTGqGxC9Xj86OkrbptLCDYIg1177DWbFRM7yGqVS\naU1NzenTp1k6gsvlmjFjRjgc3rNnTwqzuPQpLS3FMGzGjBmpnRaPx7N37968jFyERSAQMBgM\nJEmGQiFmFPHs2bOLcTUUOrUHAoGJiQnm4cydOzfJwxkaGmKFxUKEyqOgukHDcgKmzl0xMg10\nAAARvvI/iqJ6e3uZnwsURX//+9/T/4QfExzHh4eHCYIIBoPT4CUTyQuFKHDMmDFj+/bt3/72\nt+k6wBMnTlx33XV/+ctf3njjjY6ODvqWa9ascbvdL7/88l133QUAIEly1apVr7/++rZt235U\nXM3ZIgmBfQf0PycvOmBUtnNvD+rrwcU914Um8EJGGPKEOWEajYZIw0RWpVJJhesC0PTMOF0u\nF/yt5Qo3wba2xMKNCBcEQWpra6uqqkKhEHLOnha96ir6in9SFQlZswasWZPyABK7BixaFF6z\nJsOOg/RicjYKRiorKz0ej9eL3n9/o90e//ElSWCxSHfsqCwvz5aU0NcHXn0V7N8PjEbgdoO6\nOnDHHWDrVgA/5RaLBXrf5KxGJmfwqh5J8u67VUIRrRcuWxYTJSUlg4NUR4eKKdz092sJgvB4\nSgFgt4Tkph+koqLixhtv5K4nf+UrX+nu7nY4HMwZzqxZs3K2fGWz2VJrkzGZTC6Xy+1252vk\nIjTQPAJW/rIKEGbPnh2JRIrOUWV0dBRWOrAOB8agJMNU40tYP8QsIYO+rJo0kAXH8dHRUaPR\nyPxc1NbW/vu//zvrY3LmzBlY7ch9ycLhcG6chkWKnUJciLjllls2bNjA7HJsb29/8MEHAQAf\nf/wxvfHzzz//+9//Pn/+fKhuAABQFP35z3+OougLL7wg2lxPJ4LBIF3azU3fwDnXm8zKdu7t\no5dfnsOxFxwURVEUlcAIY9LPDnbiRCwadTocFrPZTldhJG0iKzl50joxcaK/n5nLE/rhD9P0\noKUoKhKJIAgiJNxwHRBEkkEikWi1Ws3990P3Ftu5i3veGB3eRZuUYbbBQ5iuAa++6kjo+D5l\nSJKkc3x4C0ZuuSWtnxWFQuFyVXR0tNrtEhQFd91lf/ZZ41e+EncezYafCEVRHo+nsxPv6QGH\nDgGbDbD8WQmCcLvd8Ma5qZEpFjIe0VogvPVWKcus5I47GleubOnuHqE4dHV15WBIQuvJl1xy\nCeC098+dO9d+zuw828B54FRPi8vlgofDHblNwJtZJHtAozHmFmYBAv3tVywEAgFmYSNgHM6c\nOXMsFsuDDz64bNmyzDpWsH6IWUIGDdOIFwDAkvMIghgZGQmHw6zPBfdj4nA4wuEw7zG2t7fz\nfleIiHApRIGDF6h3MHW7PXv2AAC+caHbZX19/eWXXz42NnY6wSKzSLFB68dCc1fW7elZSpK3\nv6hAEEQmkwkZYQAMk1qtkz6IRCIpLy+vra2thKucYAomsgiCVFdXz507t7W1tX58HG5UppZ8\nyEVYuMnM41/0wCsPoU8W67okTTQaDf03N+yztlad4L4pQBAEV91j+oDedRfBd79kwXH8lVcw\nOL0kSfCb31Ted9/M3bvjTh8Zr5UgCGJ4eHh0dHR4GND+rB0d8RVpqKeEw+EEh7xgQSiN+pvi\nZkoRrX194P77qaLwZy1A4YZXRzh79uy9995L34Y5w3E4HIW8guW4MAybOXLY7pencV2kwHpV\nJswChMz+YOUA+uKWhnk4e/fu7e7uZsW1puZFykStvuCnliVk0ANj+hOr1WqWwOFwOJiVIKw+\nF+Y/WR0xrGPkvqAiIrwUh8BBUdQrr7wCAPjmN79JbxwYGAB8RVmzZ88GAIgCx3Qirh8LzF0j\n8+axbh//Gk1/rjtNg1dKS0uFAjIa161D33hjCo91LpZlqkmoKIoqlUrlvfdmKkgVBrwLCTeI\nRJKMcCOSFMKfrMw2Acnl8tLSUgCAz4exwj41GrlOp5v0EaZE4oKR5583yOXsG0wJl8sl5IUJ\nAPjmNzN8qT06OgotaXbsGN60aWLuXFyvj3Z0xD8FUE/hTreYh/w//2PMbI3M9IMkyYmJiUce\n8T37LMJbI1NoTEm4ySOsyHDmDCcWixVsOR5FUawJGGvkYnxm3hEqQCgKmDbVEObhsFJOWlpa\nMAz75JNPnnrqqY8//pipekzpSXU6Ha1lsIQMhUIRCARYgSyBQIC5OAHhFp7AP+Dj0P+EBqUJ\njnFKIxe5mCmOi5eurq7PPvvs5ptv/upXv0pvhL2R3LwueEGcZOFZLBZ75513Ev/kHDp0aMoj\nvoiJRCKRSATDMHkggF51VTzoZOtW8NhjKT+mSqVCUbTkj38UCu8AJhMzvAPm4QnNdTGZjHV7\nGoqi7Ha71+sNh8MoAI3r1yunY/BKZWVl7ORJwd1Fe6Tl5eWYgIPpzLVrJzVAFUkGhUKhfuMN\noRwchcMBOFc26VBfXz88jH3nOxVM14CBgbKamhqpFGlszOBTxUU3OLPixkyUlKjSlG/C4fDz\nz1+wunvqlGL16mafD7vyyuDtt4cAyFhrcTAYpC8oE/SeMPvPuYes04mWAYL09YFXXqH27o2a\nTBVut0SrJb/yFc+6dbaXX64sLn/WAiTeR8mANcPJSAJlbmCNXKzgyDHcvBtmAULRuTlww56Z\nh8NyrGhvbz9z5ozZbGZ6XlxyySVT9emEwSvHjh0LBAJMJ+BIJFJbW3v11VfTLeFA2IiXNc9i\nfS7of86dO1cikbAqa4r6JRPJF/kUOEiSvP/++5lbNm/e3NLSwrrZc88919XVtWDBgpdffjmZ\nh4WfvSSvRE0m08MPP8zVRJl4vV4g/iwlQSAQMJvN8S8mkmzasEGTIXUARdHKykqZwNy1rKOD\nNXeFlXhqgdtXrlrFO9eFLYL0wlFVd7dy375pGTKKoih24oR5YgK6ctIBGURnJ/b44/keXeqU\nl5fHhB1Mp9MrmEfKysoI4RycjKtICIJ88EGtkN1j4rDPFKiurjYYDLwxE9VpBBjzwoqJlUgy\n6XxMxwkxoXtPFi+OrFkjAwBIpVK1Wh0IBLiHDPjWD0QgBEF0dRHvviujNSmfD3377bKyMmJ6\n+LPmFwRBpFIps0yDOcOBe/M3ukQkHjmKotwJqkhW0Wg0EomEnl0zCxAQBIELokWEVqtlOu6z\n6ikgzHYPiqK4XqQul6usrGxKzyuVSv/lX/6FK2Rce+21zI00XCNeiUTCtOhi9bkw/6nT6aLR\nKH1j1jEW3Usmki/yLHD88pe/ZG654447WALHtm3bfvCDHyxcuPBvf/sbqyAZXntxPa6FKjt4\naWxsnLSZ5YUXXli/fv00yF8gSRJBEMTlAosWZaSwgonf74fxivCfk8a4TpXq6urYwIDg7gsf\nXyqVlpWVKfr7k7w9ZHx8nFY3WPEQVF0dMr2CVzAMq6urq62tjUQi6P798Y1XX53fUaUJgiDS\nU6ecTqfL5cJxXDEw0HrLLQAAqqsL2bIl36MrUGKxWCgUikQi8kBA/U//hIyMAJDom0Gr1Sb/\nScwIuXQN0Gq14fCMjg4ls2Dk2DF1VVWVRsPqRJ4ycrkcVlVwayUkEoq70jgpBw5EX3mFOnhQ\nMjaGsuJRuKo9U0/5zW9cEkkD3F5XV/fRR2Pr1s1gRaIqFAqNpryiIq1Dnpb4/f7R0dGzZ5u5\nqbq9vZra2vjk9qL1Z80IJSUltJkoa4aj0Wi43WRZgiRJl8u1devWzz///PDhw7DIv7e3d8mS\nJUJ3KS0tpc1EWSPX6XTFmzFcpKAo2tDQYDAYvF4vK4pYJpMFg8HiSlFRKBSlpaVut9vn83GT\nlaEjOKv7A8JUPaxW61QFjqGhIV4hY/ny5Xv37k3mETQaDS27sz4XzH/Cq3elUjkyMuLxeFjH\nCIs7xDQikWTIp8AhkUgSl0U8/vjjXV1dS5Ys2blzJ1ewgHVNA5xL7cHBQXDOiUMEAECSJEw7\ni0QiCEU1b9yoynTbBUVRZrOZfjWTiXFNAcnJkziO+3y+cDisGBiohP1KAjOx2tpa80cf0X7L\ndJEC+fjjaGcn9/bMNAFuyGiovX1aRm9D3wqwbh1Yty7fY8kY5eXl5eXlFEUB2oAjvfTZ6QpF\nUePj43HfO5Js2rAhrm6ASb4ZJCdPejweh8OB47js5MnEn6z0SSfsMwX+9KeSLBWMlJWVOZ1O\nrxfl1krIZLIplQ3DcrktW/S7dp3X/aH1A0mCp54CzCVurp5SVlZO75XL5Xv3NhkMGMhJjUyx\nE4lEjEYjSZK8qboyGcmwpJUDkKN5eI5JHDycEaqqqnw+n91uZ81wvF5vFaM2Bsfxzs7OgwcP\n9vX1JaM+TAmXyzU+Pn748OGenh5646QWBnDkNpuNNXK/319TU5ORgYlMCY1G09raWltby2xd\nF+qkKHzq6+sRBFm6dClzfRcezrJly7Zv387rWMFUPaLR6FTDVqeaLMuloqICLj4xPxc1NTUj\nIyOnTp2C/2xpadFoNF6vt7S0tKWlZdq8ZCJ5oXCL5TZv3vz0009fe+2177zzDteuBgDwz//8\nzwCADz744Mc//jG90Ww2f/HFF/X19aLAASFJcnh4mC5MqH7mGdWnn2a87QKuAMO/uepA+LLL\nMiW3KhSK+DonnakoEN6BIEh9fX1FRUUgEIhEIpqxMbgdveoq3tvTaQK88RDBaSpwsKAoKhaL\nSbxeZPHijNf4pILTmXK1EYIgYM0acNGGQCSB2WymFcCpllyVlJRA0Zk658cu9MkqOrJXMKJQ\nKAKB2o4ODatWAkGQBQtqky8SDAQCIyMjFEWZTDJuEQG0foBtehRF8faeaLVa5gN+8QX/PLzY\nI1Gzgc1mgwYQvM4mAwPKSAQ513M0Pf3wwuHw44+D9947PztiKmuZAsOw5ubmq666KsEMJxAI\nfPTRR08xnjUFA0UhXC6XyWQCAEDjRtrCoLW1NfECMoqizc3NixcvFudmhYPZbOY15uN2UhQ+\nCIKEw2Fu9To4VxDBm3KSdxcb+In+0pe+xBz5Cy+88MILL9D/fPrpp59++mn4MZlOL5lIXihE\ngYMkyfXr17/00kvXX3/9W2+9JfRbsmDBgquuuurvf//7K6+88t3vfhfe8aGHHoJ3nwYdJRlh\nYmJCqO0iU4UVAABa3RAKj8x8Pdnq1WD16klvdV4QeeCBxGuRcXH6XDyE8emnwy0tZW+/DfdO\n+5BRv99vtVpDoRBFEBk0T0mBSCTi8XhwHKcIoubuu2XT0eSVBUVRfr8fx3GSJBXBoO6f/3nS\nPpH0wXGcVjdY3wzR6mqipCTJfglk2qlIWS0Y+fOfy4XKQ5K3h7dYLPD7ireIAC5vy2SyysrK\nQ4c8Gzc2sfQUtVqt1V6wapDjGpmihhvTSDubAAAiEQTWyCiVkpy1UWS1ioGF3W6fmJgYGmoV\nUtYyiNFoTDDDsVgsDofDZrMx1YfZs2dP1UCRF5Ikx8fHKYpCEIRlYdDW1uZ2uxNX+BsMBnFu\nVlCkX4BQULAOx263j4+Pw795XTkAR/XIixeMyWTi1WVYwI/JNHvJRHJPIQoc27Zte+mll1AU\nLS8v37BhA3PXvHnzvv/979P/3LFjx9KlS+++++4//elPzc3N+/btO3To0NVXX828zcUMRVEJ\n2i6CbW2ZSjuIt5UKqAPhyy7L0PNkEVitJxS8IpHLhYJXpgFOp9NsNsO/UzZPiUQioVAoGo0q\ngsFkrLSZ/2IAACAASURBVBx4cbvdJpMJ/qrpu7tlH300LU1emeA4bjQa4xLhVPpE0gR6JwO+\nbwb80ktxny8FSwiRSUm/PCQcDtM5lAniUQAAer3+ww81BoMMiL0nmYNlbrJrl+6RRxpCIRQA\nwKyRmWqLe8pQFNXb25ulKgYWXq8XzqMSKGsZJMEMx+FwOBwOAABLfZg9e7bf7+et+Z0SwWCQ\nIAjmOhnTwsDr9SZ+fcW5mUguqaysDIfDRqOR2f3R1NREu3Jws13zYtPL+lxEo1Gn0/nEE08c\nPnz4iy++yLY4K3KxUYgCB/zdIkny9ddfZ+26/vrrmeLFZZdddujQoccee2zXrl07d+5saGh4\n5JFHHnnkEdGBBhKLxeDVmGDbBUlmxPIKnnAhdUCmVBa+OiCRSDQajVDwSu3dd0/XkNFoNGqx\nWODfrJX8mF6PVFZOugpJkuT5YsI0pujBYJBWN7JXbVRQxGKx4eFhesqUcWveBECff6GSqxgj\nBSBT5KBvv/BJv1aCaURPc+qU4pln9AAAlYp87DH0vvvip/fkSX5bVLH3JGUkEgn8wDKdTQAA\nHH/WXGSowFmN0WhkxUBm6RLIarXCPxIrazmA9h+FMNUHu92evsAR5XwBsiwM0nx8EZHMUl9f\nP2/ePLoqEwDwzDPPPPPMM0uWLPnZz37G9IJxu93Nzc35G2kcq9Vqs9m++OILZtZE9sRZkYuQ\nQhQ4fvrTn/70pz9N8satra2vvfZaVsdT9GS/7UIqlep0OiF1oGLlyqJQB2pra8G5ixgepmkF\ngdvthr98vNaqhM83aSiX0WiE2RAgvSm6zWYTGkkGq40KCpvNRqsbXHVJkk1NB8OwBN8MikxX\n10cikc5O8v33z1eFZKNvPzfkV6nhStJ0PAoAIBhEoVE9PL3/8R/gr3/N+pDC4fD+/eHXXsP6\n+uQmE+bxIMmfk6KTvXQ6nc1mo51NUBRAgSP3NTIkSY6MjESjUW4M5MTERMb9LAmCoEuHaOj2\nnCuvDK5erQQgF63BkUiEJTEw1QfegOSpwm0vYloY5Kz5SEQkSYaGhpjqBs3nn3++fPly+p8F\n4gVjt9uhWsoyuGH5hoiIpIMYWDWdkUgkGIYlaLtAzy3dp099fb3q5EnB3cWgDsjlcuT4ccPI\nSP/x48ePHTvzhz/A7VRXF6AocN11+R1elgiHw0B4JR/uTYDP56PVDXqKHp41CwAQra6Olpcn\nvPcFwAJFoWoj3oXrYsfn88E/mJqOYnAQABBqb+dOJzKISqUS+magUFTN10CeMh6PZ3BwcHgY\nrFtne/PNM7t2Ddx2WzxwjjYLLhYIgujqivT0gEOHgM0GotG4lPCjH+VoAEqlkq6cJ0nw3HP6\nzZtnBoNoaSnxve/FT+/tt8evdHNwesfHxwcHB3/yE7Bjh/qLLyR2O5L8OYlGo48/Hs7jyUyB\nyspKi0Xd0dG6b59WIqEaG/m/IXNQI+N0OpnzfGYVg8PhiMVimX06rivhrl26O+9s8fmwxYsD\n27ePoGiObAu5I2GqDyRJpt8ewjXyYFoYZMTmQ0Qkg8DuDxbHjx/nvYTLrxcMSZJ0LdiiRYs2\nbNgwf/58KM62t7fzhtGKiKSAKHBMZxAEKS0tpQsrZt1666xbby09J9zWr1kDOE1AKYNhmPTU\nKYfdbhgZOT0wMPrOO/EdW7cWkTogk8kaGxvb29svueSSJocDbiy0kNFwOOxyuaxWq8vlClss\noKUFIAhAEPDEEyk8GoIg9Er+2JNPhltalOdWw5Kp8UkwRccvvZTWPiaFoiiSJBOMJPem3zkg\nQQdZ6NJLWd3+mUWr1eqOHAF83wxN69dr6M9v2uA4PjY2BmM1N22amDsX1+ujq1bFP1wZ79vP\nKqFQaHBw8OxZMo9KDXSnAgD4fNimTY0vvFAlkVCdnab33z99//3x07tyZbyAP9un12q1wmYB\nmOcypXPi8/kGBweHhqjikr0wDNu9eybtbDI8fD5MpLsbUFT8v2XLsj4Slt0ps4qBoiiuGWqa\nYBjGq6ytWOF88cURnY6n6iFLcO0DmOqDRCJJ32NeIpEwXTaYFgYYhlVUVKT5+CIiWSUajRoM\nhqNHjzI3YhgWCAQoiurq6srXwAAAwWCQdTnHFGfpS0oRkTQpxBYVkQxSXV1N5KqwAkGQioqK\n+G//p5/GtxahbTiCIHK5HKxdC9auzfdYLoCiKGa0JzS8kKeXMyKXyxOs5KtcLpCwUSKxlQNI\nulcZQRCJRKJ9802hkUgmJkBjYwoHWMhgGEbEYkJ9IrpsThgQBNEMDAjuztw3g8PhgGEEvH37\nt9xC5aasPX0IgjAYDLFYLDcOiwmoqak5fZq8884qZjwKnGlXVkbr6qK5sUUgCAIuu4Gpu05G\nIhGj0UhRVN5PZgoUSKouq0aDFQOZcXkURVGNRuPz+QSCh3WZfboEYBimVqtpBYdloKjTZWYk\ntbW1sVjMbDYzjRubm5vVarXf75+0c1NEJF9EIpGhoaFYLBYOh1kJx4XQAMItLmOKsxkvPRO5\naBEFjmkOhmHIyZPjVqvH44lGo4qBgVkrVgAAqK4uZMuWLD5xcjGu0xCnEyxaBKDokOmYT5PJ\nxAyfy4gnZWlpqV/APKVp/XrqqafA3LkJ7p7YykEzlSm6VqsVsnFpWr++KGxcpopGo6HeeINX\n00GlUrndDhoasvfsyPHjMGgpGAwiR4/W3XADyMI3A7clntm339FBAcBvhJkbkveAcLlc8NqL\nV6m56aZYzn5PEQT58MN63rjZhx+2LFoUoE/vmjVZrKWHi4Hw76m6TtrtdnjfYpS9CiRVlxX0\nmIMYSL1ef/JkdP36mazgYRRFFy7MqQl0TU3N8PCwx+Nhqg+NjY1+vz9T5iMoijY2Nl555ZVM\na4Pu7u7u7u68WxiIiCRgfHwc/lRxE44dDkdVvvXjxAY3ecmvFZmWiO+k6Q+KojU1NTU1NQRB\noAcPwo2F1nZR1AQCAbvdHgqFiGi0eeNGVXolFUKEQiGmusHypKTq6pCUPCklEon29Gmhvchk\nBThqtRp5+WWhsguNxwOSruatrq4mLzKT18rKyqCApjPje9/LgaaDIEhZWVlZWRntRZnxbwZW\nMSrtiLl4caCnx4CiMzL7dFMamN1uf/RR1YcfnnewTWB9yq35Zyo1t90WBiBHyaBAOG42GETv\nvLOFcXrnZq8RlXepjT4nCxfia9YIrhYmlr3uuIMEYFraCmcSrVZLl3OzqhgQBFGrM68bKhSK\nPXsaDQYpyHfwsFKpbGpqqqur83g89MaMGygKGTfm18JARCQBBEGwujyYDSButzvvAodKpUIQ\nhGmUwxRns/HFJXJxInpwXERgGIasWRNvEf7GN/I9nGmC3W4fHh72+XyxWKz6mWdUn35K0kWA\nGZ2TM3+0uIYXkXnzUn5k7MSJUDBoGBk5eeIEba0a27IlGfOUkpIS7eHDQMDKQf6nPyU/DKlU\nSh09emZw8PixY0yT18iPflRENi5TQiaT6QYHBXfnUtNZvTpL3wx0wzy3b1+jIbnt9LmBIIih\noSGr1Wo0Ykl6QHCVGqbDIoJk0TCFy86dgKJALEYcOwY/Lse/+OL4unW2557TM20RMpICLgR3\nqY15Tn7zG2uCpThWA0W+7CqLmtLSUrlc7vP5XC5XX18fXcXgcrlkMlmWHDGOHeP/wGajPcfr\n9T744INLly7VaDQIgiAIcuDAAXrv+Pg4U92gyaD6wGvcmHcLAxGRBESjUZbJLrMBpBDM2jEM\nY4osTHFWIpGIBjcimUKs4BARSZ1QKDQ+Pg7/ZpVUEDU1WEZjPun1Ul7Di/C8efJE954EpVLZ\n2NgIAKB6e+EWyZe+lMwdURRNUAAy1Sm6QqGYNWtWKBTCcVy2dy/cKPvyl6f0IMUF2t9PkqTX\n68VxHD12rPprXwMg881NeUSn0wWDQd6+fZlMlq+W4PHxcRhS8+tfjyTpAcFUarZv17/4YhVF\ngRUrnI8+apFIqLwoNRiGKZXKUCiUF1sEtVpNL8Rxz0lNTaJvP6lUCu17CudkFh0oijY1NV11\n1VXMyr5sx0Dmpj0nGo2OjY0dOHCgu7ub3ohh2BVXXEH/E6oPuRiNiEjxwHXYZTaApO+/mxGq\nq6sJghgZGWG2mLW0tMCqNNHgRiQjiBUcIiKpQydacUsqgm1tmRXL4ytyAjkjxPz5GXmWFGp8\nkOPHSYJwOZ2msTELff2bRnqOUqksKytT33ffRVJthKJoaWlpTU1N9ehofNM0KoGuqKiYmNDS\nsZp03/7Ro6pYrD4vQyJJkp4TJm8eAc0LWdklnZ1miYSC/otZHzcfer1+dFTOPb3HjqlxPLu2\nCBiGVVZWAr5zolJJyxNGRCc4mVKpVKlUZnXk04bR0VE3X6JzznooQqGQ2Wy+9957r7nmGt46\nixQgCGJ4eDgQCEQikY0bNz7//PPV1dUAgJaWlgyNWkRk2iKTyViFe3QDiFwuLwSTUUhtbe2N\nN964fPnyTZs2wS1PP/10Q0PDypUr8zswkWmDWMEhIpI6oVCIoigsEODNEKFwXMb1KkwVtVpt\ns9mEEk8UajUwmUB9fmaMKIqyrBym0xQ9d0xHa14EQXbvnmkwIICvbz+JJOLME4lEuGu/TA8I\nXm9OnU7ncJSxskuOHlUBANrby3KWkclCo9EwU0vTtEVI3nIVotfrh4bQjo4S5jk5dapUr9fL\n5WiC1KPy8vIjR/z33FPHPZlXXFE9tUFfxOSxioEkybGxMa/Xe/To0e3bt9PbWXUWKWCz2eDa\nAMsisb293WKxtLa2cu+C43hnZ+fBgwf7+vqgV05vb++SJUvSGYaISDGCIEh5eTlM7wYXNoDA\noMO8ju48osGNSLYRBQ4RkbRAABDKEJFn9NJTo9Go1WqhnBHlt75VEDkj03GKXnBkM6knGxw+\nzF8Wm+NYTRrunJBpffrcc6MSCX9y0F//Wiek1GSogioV+vv5u9OmdHqj0ajdbn/0UW2Slqs0\n775bxZvnklheQVF09+6ZBgPKe8dLL53CyEXyAlQ3AACwzoKOopw1a5ZKFdcHU9Md4MPSMC0S\nQ6FQLBZjmb8QBPHxxx8/xXiPpi+yiIgUL9XV1bB7mpUxxLL2zC9ii5lIthEFDpFCgSAIv98f\nDocRBFEEg9rrriv8KZxMJlO+/rpQhojcbgclJRl8upkzZxInTwruno45IyKQWCzmdDpDoVAs\nEqm/5x5FdpJ6skSBxGrSyGSyBOYRWq1gEW+hKTWQ9E8vjuPDw8MEQYyOlq1bZ/vqVz1lZcRL\nL1W/8UYZAODDDxMJHEJ5LpOekyNH+Dtk83syRZIhGAzSMgSrzmLu3LmBQECtVvv9/tR0B2jO\nQsO0SAQARCIRpsARCoUMBoPFYmGKLK2trYlrJ8WKD5FpDHTnWbhwIbNEItvuPCIihYYocIgU\nBB6Px2w2x631SbJpwwaQwykcQRDhcBhFUXkggCxenLywUlZWRgiUVDStX5/xkgoMw7CBAZ/P\nFwwGI5GIanCw4itfSXKoJEmiKFp0i/8iAIBgMGgwGOCnQ9/drfjkE1KhQHEcgOIQOAoNDMN0\nOp3H4+H15kzgcFZoSk1GoCjKaDTCd9eOHcO0KcnKlXYocCROFdy5k7+x5eqrJ3neaXkyLxL8\nfj9rC7POAga7uN1um83G1B1mz55NF3ckAEVRZsgO0yIR0F5UAAAASJI0Go2xWIwlsrS1tRmN\nRiHPjkgksnv37sxWfIiKiUhBMTw8LDaAiFzkiAKHSP7x+/1jY2N0uZq+p0fT25ubKRyO4xaL\nBV6RQGFFMxVhRavVRk+dEtydnZFrtVqtVgsAAH/7W3yT8I9WLBazWq0+ny8ajaIANG/cqCyq\nxX8RgiBodSOtpB5R22JQU1Nz6lTse9+rZ3lAKJVKjaa8YPqUc4Hf76ftkJO3XKXxer1btkh3\n7jxvC5pMY4tIUUNHetEw6yz8fj+MKGLpDrNnzw4Gg5NqHCqVipmJTlskKhQKDMOYpRlut5tZ\n7sEUWYLBICwkYT24z+cbHR1lKS+tra1yeeopZGKPjEihITaAiIiIKSoXCyRJRqNR4HSClhaA\nIABBwBNP5HtQcSYmJujvYnoKF541CwBA1NSAjIatMsFxfGhoKK5uMISV+O4k5v/RaDR2+LB9\nYCDa0MDcHtuyJeUMkSmwenXinJFIJHLmzBmn0wmvAqu6u5X79k3pAEXyjtPphOoGb1IPnEgI\n4fP5xsfHDQaDeWwsduutdFVUqL2dsNkK8KsgZ0il0j17GpnenCtXtqxc2XLzzbV//nNBBOnl\njFAoxN1IW64uXhxZs4b/jhRFjY6OGo3GkRFk3Trbm2+e2bVr4Lbb4sFSH36YtRGL5BuWCwa4\nsM6CFR/G1B1o78MEVDFKhpgWiXAXM+cyGAyyngj+AW/M2gsAiEajo6OjJEkuWrRow4YN8+fP\npys+xsbGJh0YL8FgcHBwEPbI0IEvaSomIiIiIiJpIlZwTH9cLpfdbg+HwylUKOSAWCxGX2Ez\np3Bzv/pVAECwrU2btac2m80kScK/WWvjZG0tmlBYcTgcNpstFovBsyq98PJI8qUvZXCcsVgM\n9qTIZDJlKCRdsiTJdXiTyUQvtbEOkKqrQ7KmHIlkEHiZjvr9vEk90WCQN/gNFm/TleT67m7J\nnj10VZShvLzhppsK7asgx4geEJDElqu//a1bIuHPZrLb7R6PB1zY2LJqleP3vy8HkzW2iBQ1\n3MoIZp0F/asKYeoOXNGBi0qlqq+vHxgYiEajTItEiqJYwgrriVjNLKy9AACn08ncyGqrgb+w\nkw6PCV1ex+2RMZlMM2fOnNKjiYiIiIhkCrGCo/iIxWLhcJhyOJJZgLVYLCaTKRwOg5QqFHIA\n3W0rNIXjXqZkhGg0Sl9scdfGQwnjK81ms8VigdoB+6yOjycoqZgqFEWNj48PDAwYjcbx8XHj\nyEj45puTdCcJh8N0cQr3AHExqKBIIEkSUBRM6hl78slwS4vy3IQh1N4u9OkYGxuj1Q1WVVS0\nurri//6vAL8KcszOnfH6J9Z/y5ble2S5hbnUTP5/9s49PKry3ve/dZn7JfcZkkASAggEUNDQ\n1v1YWvexF+XpttV4QdhqwUqhVFt6s6AG6e7p2bqfAqnFitp2S09ra9vT3dY7BS0FtEZFhBBu\nSSbJZDKZZO63NZe1zh9vsrJYl8lMMplL8n4eH59hrTVr3rUymcz7fX+/75eFJ5+0bt9eFw6T\nLS3uAwd6KipUss/iOG5kZAQ9nkRjC6aoMRgMo22SAHB5nYW0A0WoO6T517ysrGzt2rVr1qzZ\ntm0b2tLW1nbllVeuX79eeJhKddmbUyiyAIBUrci04mNCPB6P0C5EqJj4/X70vQuDwWAwuQcL\nHEUDx3Eul6uzs7Ozs/PCuXOhf/u3CSe6wWCQ/w4qmuQkrNbpa/3IiFHPMIUpXHTZMpKclncp\nX0YrK6yElQWOUCjkdo+WYUvvKmexZHGQDodjeHhY6k4yunsigQM9UFKOsjhOzPShVqvL/vSn\nVEk9EqLRKJ9xINW2ElVVwjctV1NTIB8FmLxgMpnQh3AgQG3bVv/001U0zbW22ltbB1QqKFHI\ngYrH41IjBr6xZdWqsFJjC2ZmMHfuXJPJhPxE29vb0R+pRYsWaTQaoYMGCHQHjUYjkiSU6Orq\n4v/IChFZJArfnKJmFoIghBIMItOKjwnhlxAQU1dMMBgMBpMVsMBRNPT19TmdTvmqAYWJrtfr\nRQ9kKxRSd+/nDJqmtVqt0hROazCA3T4drzvayqsgrDDLlys9MfVdFX3jmQrRaFT4JU8kpqQ7\nL1VWjiKRSCAQiA0OzmYvhsLHbDYbxpJ6Ft5228Lbbisdi3lr+OpXDX/+s/Qp/JtQVtvSnj8v\nfNPGVqzIzYXMbNrb4YEHuOZmsFhArYaGBnjoIbjci6BAoSiqurq6t1e9bt2Co0dNQsvV3t6a\nwUF5KwHpbPDQIfM99zQGAtTq1aH9+3soClvczWQoiqqvrxfVWfzXf/3X3Llzd+zYwR8m1B0I\ngjCbzemcHFkkSnnssceEh+n1+tLSUpHIUl9f7/F4NBqN1ChEVNMxYcXHhKRWTITFHRgMBoPJ\nJdiDozjw+Xz8kqzITCFhtdIKE10kYSgt4CejUdnu/dxjsVhYhbBVy913TxC2OtlsCK1WSxBE\n6f/7f7LCilqnA7sdamWaz1Hph9JdpRjGaDSmOYbUCNfBpO4ksRUrUpuYocpzJeUolkz2njgR\nr6xs2LJFPbu9GAqc1Ek95OrV0o2jX7vHtK3ePXuYxsayP/1p7Dmk8E3LTPRGwqQmmUwODQ3t\n2GF8443xFePiShIpLS09ckQvtFzld+3dCw8+KPMUlUpFEASaUrIs7N9vPXCgiuOgpcW9c6dD\np6OFZpCYHBCJRB5++OF33333/fffz01SaVdXl2wU5bXXXksQhN/vTyQSQhONUChUXV0tPX4q\n1NbWrlixgl91AIC2tra2tjb0WHQHSkpKkGsMSCo+aJqWGotMSOoemTTLVTAYDAaTdbDAURzI\nVg2giW6kqUnNMLKW3QRBKE1yIk1NGtkvoPnIkjSbzcnz5xV3S2bdfr8fRRvSJGm9917VpObn\nJEmWlZXpMxdWUt9VU/a+1vMBeLJiyoTzUo1Go9PpDAoX2PDVrw5+61uU15uzRF7MpKE6OgYG\nBz0eD8dx2nPnFra0AEDk+9/X/e//LXs8WrpU0rZc997LGo3G48fRPxM5jDOMx+ORSCSZTGpC\nIf2aNTMgszaRSHR1dcVisf7+ss2bXTfc4CsrSx44UIWMNl9/vTgEDgA4fVp++VrJcpWiKIPB\nEAwGAwHqe9+be/SoSaXiduwYaGnxACg2tqTgnXeSv/xl8sQJ0m6nfD6ipgbuvBN274bMl9Vn\nHYlEwuFw/OMf//jxj3/Mb8xBUmmKKMpQKFRbW8urCTCmO9x0000vjRWgZYXu7m6huiFEegfM\nZrPJZBoYGBApLx6Pp6GhYRKSnNls5l9dpJiQJJmtpQ4MBoPBZAoWOIoD5KegVDXAKQgcWq1W\n++tfK3Xv69xuKCnhOM7n84VCoVgspqKoSesFU4Tq6IjH48iXS332bOUNNwDIzHxYlrXZbOPB\nrnv3qo4cmfT8fM6cOYmzZxV3K5xNp9OpDh5Uuqt6jwfKyzMahhIidxKRmJLOvHTu3Lkw1pMi\nhYxE0iwFwuQXkiRramqsVmskEiHffBNt1CmbYRqNRoIglLQty9NPk+EwPdb9pNHrlYqVUsOy\nLMdxlM+XjiSaTCYHBgZGJzws27BlS5peuQDQ3g4HD8KxY9DbC14vFNTU1+FwoJKuYk8SeeWV\njJ9SXV395pv9mzfPs9nUwsYWmqabmzO4cmSl/PDDhkOHxvsXiqsEJo/wEhvDMFu3bl2xYkVr\na+vQ0FB+k0qdTqdQ3eARmWhMncbGxmQyefHixVgs1t7e/u677/J3YP78+VKbmLq6upUrV0or\nPianvJjNZqPRiBzHRYpJbW3t6F9wDAaDweQcLHAUB6mrBnQKKw9lZWUx5QV8eOKJ5De/2dPT\nw6e0TlEvmCIqlaqiogIA4NVXRzdJvgz19/fz6oaoVQdqazP1SiRJUtXZ6fZ4kLCiO3++7t/+\nDWCCJeWysrJwyrsqW/fBcVwsFovFYiqVShMKEatXTzgnNBgMLpdrAneSlPNSjUYTP3Omb3Aw\nGAwmk0nd+fMLbr0VAIa2bfN95jML1q0TlQLRkYhOp0txQkweoSjKaDTCtm0w1vGuhEqlqqys\n1J45o3RA5cGD/GPDrbdO0AUmYWRkxO12o+Tp+Vu3GiaSKjiOu+xzZsxCKJ3PGZZld+9O/uUv\n48Xek5n6Tk9hWjKZ5DsHpzVJpL0d/vu/2RMnyIKSeDQazeHD9TYbDWk3tsjS39/v8/ns9uIu\ngckXTqcTSWzSpNKhoaGst4SkSYrijqwzMjIieweWLVvmcDhErh9KFR+TVl7q6upWrVqVLcUE\ng8FgMFkBCxzFgVar1b/wQqpaDIlhOADo9Xr1uXOKJ73mmr6+Pn7WMXW9IE1YlmUYhiRJdTAo\nP8/fuBE2bpQ+kWEY2WwI3pNiEt/2CYIoLy8vRzUXx46Nbk35XUetVlOZNNQAgNfrdTqdoy0n\n6c0JAcBoNOr1eqV1eP0tt6QzL1WpVPPmzQOARCJBvfMO2sgsWCBbCqSNx7HAMTOwWq3Of/zj\n0lgED9/YIk8mUmZvby//a2htazMcOzahVOH1epU+ZxJWK2WxKJWGB4PBvr6+ixfnb97szXTq\nGwgEvF5vNBrlksm6zZu101CYxjCMdBbHJ4lcfXVk06ap/jYlEgmn07ljh7kwDT5OnZL/CqHU\n2CIlHA6jpX7ZEpiKChZboacAFWAKtwiTSr1eb74EjlzCfxwhhHcA9cQJ/6hlXXnp6enJrmKC\nwWAwmKmDvzoUB+Xl5SmSFOgXXxQdn0wmg8Ggz+dL/O1vbH092ui+8070gN21Czgu8olPBINB\ntEUaCDId2QqxWKynp6ejo+PSpUtpJt0KSZ0NkYXc040bgeOA4+DGG1MfSHV0RMLhvt7e8+fO\ndf3xj2gjuqtw/fWig91ud39/P2+owc8JR3envPC6ujp95k00stA0TWzadPHChdOnTpX++c/S\nXJVIUxP2BZxJWK3WxYsX19XVWa3WGocDbRx+/vmuS5ecr702etDu3bJvWiU8Ho/U7RjF+iTn\nzFGSRFNk1qYIHopGozabLZlMPvdc97ZtziVLolZrfMOG0dzr1N0fDofDZrP5fD6GYcr+67+0\nf/97mr9xU0SYJPKzn/VKYhxkaG+HBx8E2fiVeDx+6dIlj8fT36/avNn14osXDx06d/vto71F\nr78O//wnu3VrfNWqZFUVl5folldeGf3IFP2n3D4lJnUJzNq1kayOd6aRSCREQR7CpNJkMjkb\ngjxEfSiirFZpl0p2STPwBYPBYDC5BFdwFAcGgyGRshaDf8iyrMPh8Hq9HMehXnetzYZ2lY1N\nmIvUawAAIABJREFUP8iPfQwm0guiy5dnt/yZYZiuri7++1ZGZer8pQEoelJEly3L2NduCuh0\nOlQZAf/4B9qC7qqIRCIxODjI/1O0fM3V1BApy2RomuY6O70+Xzgcjsfj+gsXqj7zGYDJl9nr\ndDrdb34jWwoEFKVUCoQpUmiaHq3QHmtsqQSoBIAxI4/UxUpS+NAEaQlVeOlSfTIp23aO1D0l\nXVI1pv2JcLlcaK01RfeHrD3H9u3ekZFRHUT0G8dWV5PZK0zTaDQpkkTM5onLNxiGaW3lXn55\nPM1KWJ3hcDjQrZOtbigpie3cGc2Kb0UeXU7ikp8+XwKzalX49tsDABlnW8wepJK0KKl0NmjW\nJHnZQp3oDoj2YjAYDGY2gD/6iwb67NlgINDT3d159uylP/wBbeQee0y0AGuz2VDaAghEBLSL\n+P73hRUKIr1AtJ4fnXpBxOU4HA5e3RCt/bLV1em0w6DQNSVPCkqtBrs9u2NOi5R1H4FAgF9h\nm1yZDEEQpaWlNTU19fX1Vb29o1snW/5aUVGhVApUv3mztBQIMzNJu1hJRGq3Y7R3HLcbGhuB\nIBYuWmT52c9kP2ciTU1KVnx8fRmPcOp7991xhmFaW6NtbfDee+ByQTw+OsPfuXO0BF36Gxde\nujSj600NRVFIPwoEqG3b6p9+uoqmudZWe2vrAE1zZWVlqZ8+MjJy8eLF7m6Qrc5IJpN8ULSs\nxPPpTw/b7WrZ52YEwzC7djHS2/jww5mdZ3KI5p/CEpj9+3s0GmzTmAqaplMklWo0mtkwvRfF\nuwrvAEmSuOkSg8FgZiEz/4/fTMJoNDY0NCxZsmTBWM8nsXq18ACv1yv14EQiQsJq5SwW4cG5\n1AsSiUSKdphIU1M6JzEajSRJKs3PKzdsgN/8JlsDzhapo16jy5dndrrJzkt5tFqtKUMDEQzm\nMhQkUfRbHI/Hh4eH+/v7+2y22K238j1olM+XKnhI5kU4UXW9aOobDHqV1IGjR3Wg8BsXbmqS\nlgxMherqaofDsG7dgqNHTcIkkUuXqvz+VAJHMBh0OBwcxyk14KQ2+Fi1KnzLLR7Z51ZWZmAx\ngESWri5u6kLJ5OBnpywLTz5p3b69LhwmW1rcBw70GI2sXq/PxSCKmVFnbgCQJJWWZynPq8Cp\nrKzkdRzRHaioqJgNEg8Gg8FgROAWleJkzIYzFotF/X6WZbXhsPa660q7u0vHMjJEBeSRpiYi\nFBIGs5tMphRZkhXr12earZCC2FhfuNKsI50qZIqiLBZLimyIApyfj1YIK7TVxK+8MvdDojo6\notGo2+2ORCL0mTP1N98MANxjjxGPPpr7wWCKC61Wq/7Vr5RanJiuru6xlg3r3r3qN9/ke9Do\n4WHIJHiIIAiKopDGIe3+oGluaGgIqQPS3o2yskSKzKnSrFoM0jR95EiDzUZAhkkiw8PD6EH6\n8SuHDpl37pwbDpOrV4fa2mwUxSn7VqSlCwQCAYfDAXnNuC0pKRkaGhoZSX7ve3OPHjWpVNyO\nHQMtLR4AMBgMosX5NDl2jDl4EN5+m7LbKZ+PKJDQmWmioqIiHA7b7XZRUinLsrMkplStVtfV\n1XV0dDAMI7wDHMepZ+SPHIPBYDATgQWOYiUej9vt9tGyCJZt2LKFXyxlGhvT6XWnaTpnekHq\neX767TCVlZXDJ050DQ2hvg8+G2Iq83OO4wiCmKYgSbQCqVQmo9bpJox6nQ60Wm1NTQ0AwFtv\noS2iUiAMRpaysjJQkETrN292fvvb3D33gJzXjInvrpKi8DljMpm8Xm8gQEmnvrz2ITvD//zn\ng0q/cUBRtNMJyD0nS3zwgbzNQeokEam16uUNOCq1OpXBB0WxAITsc++4I5imwIHSNGGaM25F\n/POfLIq87e8nvV6oqSE+85kFb7zB2mwqYQmMRqNZuTJjt5RYLNbf379rV2VWrEmKAoIg6urq\nVq5c6RFUQqGk0muvvfb48eN5HFvOMBqNN954o9vt5rfgrFYMBoOZzeDivaIkmUx2d3fzTR8i\nr42yP/xBtoBcWqtZVVUVPHGi48yZ0x99dPqjjy7+/vdou9TaY4qgZuAJ5vkpGGvmB4KofOqp\nxYsXNzQ0VFdX1475d05ifp5IJBwOx/nz5zs6Os6eORP54hczinRJE4PBoNVqldpqzLffnrqt\nJpFIBAKBkZERv98fdzr5mwA/+EF2xjflnhfMrKK0tNTQ2am0FzlcyDpfkGfOJOJx19BQX2+v\nnZ9ypMxwqaqq6u/XSrs/Tp3SO51a0cHCGf6GDUwKoxnyt7+d4k0QMYkkEZSzINwiasAhiGRq\ngw+heaTouWp1Wn/WOY4Lh8OijcLbeO+9WQ7gYBimp6dn587g/v30Bx+QvN/Hs89SNpsKxkpg\n1q9vXL++saWl9q9/zWwBBv1ZDIfDWbEmKSLOnz/vkevz+vSnP53zseSHrq4uobrBg7NaJ4Rl\n2ZGRkQcffPC6664zGo0EQRAEceLEiXyPC4PBYKYEruAoSoaHh/mmD9FiadJsNh47ptjrXiJO\nGqmsrCwrK4tEIrFYTP/3v6ONWV/PJ0mytLRUr7D2a73nHmmZejKZ9Pv90WiUSyar7r5bJVAf\nKIoyGo1GoxG+9jX42tcmMZ5YLNbd3c1341fu3as7ejSjSJf0qaur48ZkJhmUX2toaIgPkkBF\nOqppkGAwmIxQdXa63W63280wjKazE5VQsbt2nW1p4ThOtgcttHSpNpmkaboKtT288cbouVJO\nPzQazeHD9cKpL7/r+98fvuuu8QoIUe9GWdkc3aR+43IGQRA0TaMAS9kGHGSQVF1dfe5c8r77\namw2tai6wWQK1tTEZZ+bpm+FKF4UJLeRIBYCZK3HgU/RstvnbN7suuEGX1lZ8sCBKtQOI8vV\nV0N7Ozz/PHfsGPT1ERPGu7hcrhShM7npuMk9qJTpo48+am9vf/fdd1esWNHa2jo0NLRo0aKH\nc+MTWwCgrNZ8j6L48Hq9Dofjgw8+aGtr4zdSFHXVVVflcVQYDAYzdbDAUZT4/X70QBrWyOr1\nlN+ffq87ACC9AABg61bYunWaxmyxWNizZxV3Xz7rCAQC/f396Kubde9e1ZEj2VUf+vv7eXUj\n0+jWTFGr1Wxnp2tkJBQKxWIxw8WLtWvXAkzQCON0OvkCcphUqi4GM02Ul5eXl5dzHAd860dz\nM8dxKZwvksIE2TELodSwLPvuu0kAlXTXmTO6sWNkZvharZbq6OhzOHw+Hwga2ZKtrdSuXVO7\n9KxhNpvdbrdsA47BYED3iqbpw4frZQ0+vv995xe+4JZ9bpoCB0VRJEkimUN6G1UqoOlsfj3g\nU7Rk1YcbbhhXvXg8Hs+dd6pef33cNyp1s0nq0JkvfjExI7/w8Ksdzc3Nzc3NwWAQ/eFoampy\nOBwLFizI9wAxBYrX6+3v7weAWCx2//339/T0vPnmm7FYLJlMGgyG48ePX3vttfkeIwaDwUyS\nGfj3fjaAJueyi6VEioyA/M2KR0ZGhoaGkijdlmUbtmwx8r3BGzbAwYPCg6PRaG9vL1qQEakP\nbHU1OWX1IRqN8rXZUoWIWb5cXP4+ZUiSrKqqGl2+/sc/RrcqL1/HYjHegxCmX4LBYCYBQRCw\naRNs2gQAJADZ0VHyhz9k0fmir6/vJz8J3H77wjVrAqIF/64uLQDIqgMqlUqn0xEEMW/evNra\n2mg0Sh09OjqKj388e1c/VSwWy+nT0fvvnyuqziAI4ppravjDlAw+liwpW7euTPRctVq9atWc\n9MdgNpuVXE6MRlMW4yeEKVpp+n04HI6RkZG+voXSco/XX5cXOKT5OJc3LnEz8gsPv9qB6Ojo\nQH86m5qaIpFIPB4X5chiMADAcdzgWIdvc3OzWq0+cOAAvxcXcWAwmGJnBv69nw1QFMUmk7KL\npfYf/KB8w4ZwOByJRKjTp+d94QuQ74wMl8vldDr5f6JiBE6lGtVibrpJdDzKRwA59SGSXt5K\naqKoCEI50iXrAsdlpLF8HQwG+YLb3EgwGMwUMZlMSpFM9Zs3ZxrJFAgE0IK87IK/1UqOjJTd\nc0+VaIYPACtXWnl/CpIk9Xo9bNkCW7Zk4xKzyVh1BgVy8StXXDH6+JVX5J++c6faZgPZ56aI\nbhFhsVjOnGE2b54nJ7JkIJRMCN9QKUSoPmzadFnVSTgcHhkZgQybTSiKEvbdiDpuNJrG7FxM\ngSGSdTrGmrNQTioWODCyhMNh1CKHiMViN9988//8z/+gfzY2NuKEZgwGU9RggaMo0ev15O9+\nJ7tYStC00eczzZ0LAHD48OjG/GVkxOPxoaEh/p8yxQjr1omegvIFlJr51dn6xqZQTs8sX56F\nk08N/jur7E1gVqyQETimJwUGg0kTi8UC2XO+SN1usHZt5LXXapTCWZuaMnqpvPHhh/IOF6nj\nVxDvvy+/PZ3n8qjV6sOH6202GlKKLFNHaImKEKoPP/1pP00vFu71er3ogexP/wtfYAA00lcx\nGAzoidKOG42G1GhknjID4PuMEGfPnkUbFy9ejB7kbWSYAkakizU3N58+fZr/Z1NTUzgcxhoH\nBoMpXvAfv6KksrJSKSag7v77iRdeGD2uADIyAoGAbDECileQqgkcxyWTSV59kGbBoEbuqYC+\n6SpFuqi02gkiXaaf0W+lCjchMVY7yrKsx+Ox2+3dly4xt9ySZgoMy7KodFmYTZO1WBbMbEWj\n0SRPnjx/7pwokin28MOTiGRK3W5wxx3B9MNZ29vhwQehuRksFlCroaEBHnoI5EoKcs0k4ley\n8lwhp07JL3JkJJRMCErRQo9ZFp580rp9e104TLa0uA8c6KmqEksPDMOItgh/+nfeKU7YRVRW\nVhIEIRs6U1VVJRVZZgaiWeiZM2cAoLGxUavVUhQ1U2UdzBQZd0Qa46zAIq2pqQkZGGEwGEyR\ngis4ihKdTqc+f15xdxqLpclkkmEYgiA0oRD5sY9N38p/psUIBEFQFGX+/e9l1QeOJGmnE+rr\npzIknU4njG4VldNXbtiQaTl91jEYDKAswWj0erDbY1VVNpsNzQSse/dq3nprQgvScDg8ODg4\n6j/CsvO3bjXgWBZM9tDr9YsWLQqHw9FoVDNWPqb+l3+ZxKlE379F7QZqtUWpd0NEMpnctSvx\n0kvj07zURpWzjTRv4xQhSbKsrGxkZETW76OiokJ0vEiMEP30aVq+fUar1cbj9evWqUUdN2az\n2WSqrKycpovLM5WVlbwNRygU6u3thbH+FKT45HNwmEJFr9cTBCGMnkHSGGLZsmVSiRmDwWCK\nCCxwFCtUR0csFvN4PNFolD5zJp1gDkQ8Hh8YGBitAEd+n9M5yxUVI4j6QeJXXil9Sopm/hRZ\nMBlRW1tLFnCQpF6vNxgMSjfB2NLCPf5475e+hNQNUddPwmql5SxIg8GgzWbjv9BY29oMx47h\nWBZMdiEIwmAwGAwGeOABeOCBSZ8nRbsBTXNIAZyQaDTa09PT1dWQvlFl1mlvh4MH4dgx6O2F\nCYNOC5OsXILVaj13LnnvvRaR+lBaWmoymUymyw7WarXIlFT2p6/T6ZRe5cUXjVO3Jiku9Hp9\nbW3tuXPn4vH4qVOn0Cd8fX09y7LZzcHBzCQoitLpdLzbOi+NwVh/E5bGMBhMUYP//hUxarXa\niqayf/vb6CblYA5EPB7v6uritfkchI+iAtrUxQhQWyt8isVi4aZZfdDpdMxHH9kGB5GdJx8k\nWTjuFXV1dUnlVN1IUxOySpX1YYVAQDRj4DjObrfz6gaOZcEUOCUlJS6Xa2QkKV3wN5lMKaa4\nPCzL2my2RCKRkVFldgmHw62t5Msvj5epFV39SDwef+wx7q9/HRczJncJJEm+8cbcNNUHVO7h\n95PSn75Op0vx08+KNYmIwpeoysrK1q5d63a7+S1tbW1tbW033XTTS2O9qxiMCIvF0tPTEwgE\nEokEL40BQH19fTweT+czFoPBYAoWLHDMCNII5kA4nU5e3RDNcpNz5lDTMMs1GAx6vV6pGMF0\n223Sigy1Wh0+efJcXx8aKq8+RHfs0P7wh9kamEajqa+v5zguHo/TfJ7lRApRzqAoiuzs9Pn9\noVAoFovpL1ywfPazAKMSjM/hgJER2a6fyLJlXDgsEjhCoRD/c8exLJjChyTJRKJh3ToQLfhr\nNJo0k1B9Ph96z2dkVJlFnE6ny+Xq7s4g6LTQ8Pl8drv90qXGrFxC+uqDRqOJRueuW6cT/fQp\nirr66mq5cwDLskePRubNI6+6SmW3Uz4fkRUlIhKJtLYSBS5RdXV1CdUNnuaC+XOGKUCMRqNO\np7vuuutEdhvd3d0PPfTQ4bEeQwwGgylGsMAxi+A4jm/Wlc5yw0uXmlI+fdLMmzePUy5GkK3I\n0Ov1V1xxRTAYjEajmjFNRHvddVkfG0EQarUa7rsP7rsv6yefIgRBlJSUlJSUAIwH4iAJhmVZ\npa6fSFOTSuLDysc0KmkiWODAFBr/9/9OKQmVr77mERpVtrQEASxZHK0In8/ncrkgw6DTgiIS\nifT393Mcl61LyMjv449/LEn/p+/3++12+3/8R+2hQ+O9S1NXIlwu19DQUHf3ggKXqBobG4Vm\nClkE+Vjv3r37vffeO3nyJAo4O378+LXXXjsdL4fJMYlEQtZMdNWqVYFAoLS0NPdDwmAwmKyA\nU1RmEYlEAuXJKc1ypx5QIotKpVJ1drpHRvp6ey9dvDj46qujO3bvVoxXcLuJBQtMZnOVxWIO\nBPKeBSNDjiNILg/EUalUSl0/HElqhodFzx7tp1WIZYmtWDHt48dgMmSK7QbC7EwAOHTIfM89\njYEAtXp1aP/+HpJklZ6ImGL2yvDY76Bs/QjqhytwXC4Xmjbn5RLS/+kHg8G+vr5kMmm3qzdv\ndr344sVDh87dfvtoRcPrr09yAH6/3+l0In1n2zbnkiVRqzW+YcMI2lsUEtUU8fl858+ff+21\n19ra2o4dO4bUDYqirhqL8cIUO4sXL04kEmVlZaLt+/btW79+fV6GhMFgMFkBV3DMIkSzXOnK\n//TZShEEUV5eXl5eDgDw1lujWy8voPV6vcFgkGEYiiBqNm1Sy1qfut3Q3Dx9mS+pYVnW7/dH\no9FkPG655x7VNJizxuNxlmXVwSCxenWKyzSZTIyyD2viRz+Cy91bUT+tkiai1umkTigYTH6Z\nYsCHeqwzQdaoUp2yb8Hv97e2qifdmMBxXCQSEW0U1o/ce68GQBzTWGigCa0Q4SVs2EBNa49P\n+j99JENAtotlkL5DEETxSlRTwe/39/X1AUAsFtu6deuKFStaW1uHhoYWLFig1eKCv5mDzWbz\neDzS7bi/CYPBFDVY4JhF0DStUqmMv/ud7CyX1mhC588zlZXqYNB0/fVETw/A9IgIEscQlmV7\ne3uRbT4AWPfuVb/5Jm99yq5a5fN4IpFIMh633nuvvPAx/UQikd7eXtTVb927V3XkSBbNWVmW\nHRoa8ng8yWQynWgbnU5Hd3YqnY3++MdFW7RabQonlLJ16/KejIuZ+eRWnTSbzS6XSzaXlCAI\ncW7HGPxnkdA749lnrS+8UAppNyaIikdAEnRKkosKX+CQlsBcfgnz8zUwIYlEgteSsqhEIIlK\npPgXnUQ1aTiOczgc6HFzc3Nzc3MwGEQtV0uXLnW73TM2dHf2MX39TRgMBpNHsMAxu6ioqKAV\nZrm1mzYNfutbw3ff3bBly6i6ATkSERwOB69uSAM+Lvj98ZERkAgfuRQ4EolET08PauERjZCt\nrianZs7KcZzNZuPXS9OMtqHPnh10OkdGRoQpMMHvftfwf/6PbCXO3LlzpzubBoMREQ6Hg8Fg\nLBajCKLq7rvpHKqTOp3O77fcdVepyKgSAJYuLVWpVLLPGhgYQJ9FwnKAu+5yIYEjzXIAiqIo\nikIfF9L6EZUKiiK/k6ZppOfKlsAUyCUkEgnpRqESsXGjDiDjysQJJSqCWDiDBY5IJMKbUiM6\nOjrQNLipqSkQCGCBA4PBYDCFTEF8R8HkjIqKioTyyn+kqSkHwbEiEokEXyEptT4NLl6Mvmnl\nJvNFieHhYTRdkY4wtGSJgWVJcvJ2NiMjI7y6kf5lEgQxZ86cysrKSCRCjFmQGj/9aVDoM1Kr\n1cmzZ53Dw6gPyHDxYv3NNwNMci2d47hoNMqyrCYUoj/xiXw1DWEKFo7jBgYG+F9t69699OHD\nOVYnX3rJomRUuWqVzPGxWMzr9aLHUywHMJvNHo9Htn7EZDJPXzNgFjGZTG63W/YStFqtkkKU\nYyhKrDIIlYif/rRfpVo8udOSJIlkjuKVqCaNVDbqGBPHly1bJtI+MBgMBoMpNLDJ6OyCIAhV\nZ6fP67X19Jw/d872P/+Dtg9t23b6o48onw/NrpmFCwEgbrFEUYTHdMKHHShZn8LlsoL2wgUA\nCC9dKrt2N02gRV2lEUr77TOCn1PJXmZq51eapk0mk/GBB9LxYaUoymq1LliwoKmpqZ43Is2w\n1ZZlWYfD0dHRcenSpe5Ll6Jf+hLkqWkIU8g4nU5e3eBlO/TBArW1kBN1MlObUmnwCgjKAZqb\nmU2b0n1pi8UyMKBft27B0aMmYf3I6dPGWEw+6LTQULqEU6f08XhNvkc3ikqlEpqtPPmkdfv2\nunCYbGlxHzjQY7XqJn1ms9kMAIEAtW1b/dNPV9E019pqb20doGnOZDJNRdEWEQ6Hv/Wtb113\n3XVGo5EgCIIgTpw4ka2TTw7p1Z09exZtX7x4cRavHYPBYDCY6WAmr0LMIjJsbh8PH/3HP9CW\nyLJl0tqE6LJl0UBguh3FRouBla1PlWQFLhxG30FzQDKZTDFC7dTSZxiGAWX1hI7FkEVolpE4\noaTD5LppMLONZDI5MjKaNyH9YGGWL59Gd0oBmdqUSsVEYTnAs8+6aLohzVOpVKrDh+ttNgom\nG3Obd2iaTnEJS5fmb2SXY7Va+/r6pJUmBEFUTSHsxGq1dnTE7r9/rqjFiSTJa66Zk5WRcxzn\ncrkOHz784x//mN9YCDElOp2OIAihNcOZM2cAoLGxUavVGgwG5adi8kA0Gn300Uffeeed9957\nD0f5YjAYDGCBo0hJJpMulwv1GpAADVu26Ca3ir5xo/1zn/N4PGQwuODOO0Eyu05MfzEqWn9T\nDD0liLpvfUtWVlBNT6itLDRNK5mzciSpGhqCKUgtBEFwLKuknpgLqZrd4/EoddMkrFY6h01D\nmEImFAqh2ZGsbBduasqNwJEpwrYLaWNCWVlmv+Mffijv0ZBmzG0hUBSXUFJScv48u26dQahE\nnD5trKqqMhp19fWTPG0OJCqHw+F2u6PRqDCmZP78+aFQSK/XZ+EFJgtFUeXl5bxGGQqFent7\nAWDZsmUkSVZUVORxbBgRw8PDR44ceeKJJ/gthaCRYTAYTH7BAkfxEY/Hu7u7Y7EY+mfl3r26\no0cnvYpOUVSq2gRJh3PW0el0KpVKKeCjYcsWAFCUFST57dOEyWRSK8eyco8/Dt/5zqRPrtVq\n1b/6lax6QtC02uWCefOmMvgs4vP50APpsnykqUnNMBpNYU5dMTlltBRC+YMlR7+3GWIwGJDz\ngoJ3hnzwihJTjLktBIrlEv70pzIls5WpKBFK+s6KFZM/J08kEnG73SCJKVm2bNnQ0FBpqaIP\nbm6YM2dOPB632+2JROLUqVNIr5w/f75erw+Hw6lTljE5o6+vz+fzBYNBoUbW2Ngo9abBYDCY\nWQUWOIqPgYEBXt2QZo4QGa6iGwyG5DPPKNUmGH2+6e6WJwiiurpakyLgY3pkhYyoqKhIKI+Q\nmFpifEVFBff++yB3mXX3319QAa7ojafolhKLYYEDAzBqwahUlkWp1WC3Q21tXscoAzKpeeed\nka1bG0SNCVqt1mgsKS3N9xAxcmRqtpIOHMe98IL/rrs0L78sbtJ89VX413+d/JkRfr9f+E9h\nTAnHcYFAoLy8fKqvMQUIgqirq1u5ciXvpAMAe/fu3bt370033fTSSy/lcWwYhM/nQ0sOq1ev\nXr16Na+RNTU1DQwMzJ9fEEHOGAwGkxewwFFkxGKxQCCAHsu6ZmTq1mA0GuHkSVAQEXIzuzab\nzf729h6HA9mz86Gn7K5diV//Wn3+vOyzpigrZARFUYmPPrrY1xeNRoUjDD/0kP5HP5riyUtK\nShLnzinuLiRjC4IgUizLG7D5HAYAxkohlMqyrPfcU1CynZCKiopDh/Q2mxqK1jtjFpL1SpNk\nMtnb2xsKhbq7F27e7LrhBl9ZWfLZZ60oLfj11+Hxx6f6EvwqBUIYUyLdmxe6urqE6gZPcw7/\n8mKU4DjObrfv2bPn1KlTZ86cETqdNzU1hUKhWCyGC20wGMysBQscRQaaYINyc3umAgdBEMYC\nmF2bzWaTycQwTDQa1R09ijaSH/sYuXNnd18f8n3IrqyQKRqNZuHChcFgMBqNqscmbPo1a7Jy\ncvrsWZ/Ph/qxNZ2djbfcAgDcY48Rjz6alfNnC51OZ/jtb2WX5YGitCMjgP3nMAAkSVqtVu2Z\nM4pHFJJsJ+LsWfkP0YIynphu2tvh4EE4dgx6e8HrhZoauPNO2L0bZsmMqb+/H/3Ree65bj4w\n+K67XEjgmIJ16TiiLBJhTIl0b15obGwU+oxiCodoNNrX13fy5Mmf//zn/EbeFxZpZAzDYIED\ng8HMWrDAUZworKIzy5dP4mTE6dMcx/n9/lAoRJw6VX3jjZCP2TVBEFqtVqvVwpYtsGUL2kgD\nzJ8/PxKJRCIR1d/+hjZmS1aYBEaj0Wg0wje+Ad/4RnbPPB5tM5YRSKxend2XmDoVFRUxhWX5\n+s2bC3ZZHpN7KioqXMePXxoaQt+5eXUy2dpK7dqV58GlpFiMJ6aPZDK5ezf7l7+Me0DYbPCf\n/wksm4XKBSXa2+H557ljx6Cvj8ivpBKNRvkySV7dAIC33zaiBy0tWXgVvV4vrI8QxpSgvVl4\nDcxMJJFI9PT0JBKJWCwmtN4wGo2BQIDXyIhCsifHYDCYHIMFjiIDeRwoNbfTGs3kmttjgqPc\nAAAgAElEQVQJghidYL/66uiWQppd63Q6nU4HDz44K2rEJxXgmhv0er1GoWMIoKCX5TG5p6qq\nqqysLBQKMQxjOHIEbaQ+/vH8jgqTmkAg0N/ff/Hi/M2bvag148CBqt/9rhyy1JohheO44eHh\nHTu0b7wxbuOaA0lFCT4oSkhnp/YnP7EAQHMzs2lTFpyGSkpKhoaGUFemMKYEAHAUKyYFLpcr\nkUiAxJ4WScm8Rob+j8FgMLOT/JdBYjJCo9HodDq+uX3hbbctvO220jHHL8vdd8NvfjOlF9i4\nETgOOA5uvHHqo8XMPKiOjkg43N/Xd/HChZ6x0iHusceA4+D66/M7NkyhQdN0SUmJxWIxfP3r\nBfLBcvRo5P77I1ddFa+sZNVqaGiAhx6CAnA8KAii0Whvb28ymXzuue5t25xLlkSt1viGDaNx\noVlpzRDBcVxvb6/T6ezvV23e7HrxxYuHDp27/XY32vv669l/xQlhWVa05dAh8z33NAYC1OrV\noV/8wklnY2GIJMm6urpIJOLxeNrb29HstL6+PhQKmc1mvPyOUYIvL0Lw9rTBYBDGNDKTyURn\n5W2KwWAwxQkWOIqP2tpaXYrMEbyKjplmdDrd3LlzFy5c2DAyOvMpqHqfWUV7OzzwANfcDBYL\n4Ol6ahKJRHd39+7d8Wee0Z06pRoZIePx0UqBhx/O9+DyR3s7PPggoLeQyaT57Gev2LPHqtWO\nT/Kz25ohwuv1oglbziSVCRHms7IsPPmkdfv2unCYbGlxHzjQU1aWtQBOnU73+c9/fs2aNdu2\nbUNb2traPvGJT3z5y1/O1ktkRCgU2r59+6c+9Smj0UgQBEEQJ8b6JTGFAyrfQHAc98EHHwj3\nIo0MVwBhMJhZDpZ4iw+tVst89JFtcDAYDHIcxze3F6AnJWaGU8DdNDMehmGGhoZ27CgtkML+\nwqe3tzccDtvt1XwuxnQ3XxQ+Xq/3kUfUr77KOz4QAwOqn/+8iuOI7dsHQdCasWpV+N//nQbI\nsiWG1+tFD2TdLm65hc39Mgya3nMcFwhQ3/ve3KNHTSoVt2PHQEuLBwDMZnO2Xqirq8vtdku3\n5z6mJBwOOxyOd955Z8+ePfxGiqKuuuqqHI8EMyE0TfMhOwRBPP3008K9bW1tbW1tOMoXg8HM\ncnAFR1Gi0Wjq6+ubmpoWLVrUOGZUhlfRMZhZQjgcvnTpks/nK5zC/gLH7/eHw2FQqBSoqBB3\nJcx4OI6z2Wz9/f02Gyl9Cx0/boTLWzP27+8hiGTKU44jrApJXVjEMIxoi1BSueuuiMxzphma\npi0WS2+vet26BUePmmia++Y3B6+4gjl1Sn/xYqXbbZr4FOmBYkqERCKR7373u2+99VYuayhC\noVB3d3ckEkGmlU899ZTFYgGAhQsXYq/TAkRYndHf349sXETgKF8MBjPLwRUcRQxBEBqNBu67\nD+67L99jwWAwOYLjuP7+fuQUIIyx3LBhBNUj5KWwv8BBDeqgUCmwdm0EYHYVdTudTr43RPoW\nKitLPPmk9cCBKo6Dlhb3zp0OmubS7OqPxWKtrezLL497HKZfWHTokHnnzrnhMLl6daitzUbT\n9ZO7uilSVVX1xhsGm00NAIkE8fjj1fyuvXunxeqa4ziv13v06NHHBfcoNzUUdrsdmTiITCuX\nLFni9/uzWLGCyQpVVVU+nw99/peVlaFqo5tvvvk//uM/ampqysvL8z1ADAaDyT+4ggODwWCK\niVAoxJcoT1+M5QxD2LiOEFYK3HFHMB+Dyhssy/LNEbJvoeFh+umnq2iaa221t7YOUBSr1WqF\n5hRKBIPBixcvdndDmoVFfNaD1O3CZOLymATR2SlfvHD11dl/rWQy2dXVZbfb3W63sIZi8eLF\nJEl+73vf+/SnPz1NNR3hcDh2eWkNb1rZ1NTENxBhCge1Wi1rT0vTNEnir/QYDAYDgCs4MBgM\npriIRqPSjVmPsZxhiKoPRJUCGo0lXwPLCwzDSLNC+LeQRsNdvKgVtmYAwMqV1glPm0gkent7\nWZZNv7CovLw8GAwquF2UUFTWHD0z5ZVXcvda/f39kUgEJDUUixYteu2116a1piMmaRzqGLMw\nX7ZsmXQvphAwGo2f//znhQYu2HoDg8FghGCBA4PBYIob4XT9F79w0/S8fI+o4DAajWg+wLKw\nf7+4+cJoNKZ5nvZ2OHgQjh2D3l7weqGmBu68E3bvBnWWzTenF9kkVPQWqq6OOxwqkGvNaGqa\n4LQejwedWbYq5Oab4wDiGhCz2RwIWNetK7HZ1EhSWbQoeuqUXqVSXXNNzSQvr6hgGEYp+LOp\nqcnj8WzdunXFihWtra1DQ0ONjY3ZTZCVnu3s2bMAQJLk4sWLcVptYVI49rQYDAZTmGCBA4PB\nYIoJYd2+dLpeUVGRx7Hlkoy0BpPJpNPphoZi0kqB0tLSNFsh/H5/a6t6cu4SBYVacI9EbyGH\nQ40EDhHS1gzp/b/pJs1XvkKoVBx/jLAP6PbbowAyBgF//WuVzQYwPW4XhS9IIe9bIcIaiquu\nukpY09HU1NTX17do0aKpSw8+n6+1tfXdd9/94IMPUP3Ir371q6uuuurMmTMA0NjYqNVqdTrd\nFF8FMx0ge9p8jwKDwWAKFyxwYDAYTDFhMBjUanUsFpMW9hMEUVZWlu8BTjvJZNLlcj38sOm1\n18adQVNrDQRBJBINd93F9vSohM0XBoPhyisnNmVNJpO9vb2hUKi7eyGfMvvss9YXXiiFIkyZ\nValUer0+HA7L9oZYrdaqiYxqGYZ59FH2lVfGJ8A2Gzz1lDkataJ8WZD0AZGkfB/Q++/Lv8TU\n3S5CodCuXfRLL413bBWgICWtphHWUKAtwpqOWCwWDAZNpsmHucTj8f7+/hMnTuzbt4/fiF4u\nFAr19vYCwLJlywiCwI6VGAwGgylGsCMRBoPBFBMEQcydO7e/XyuNsezrqx0cnOEGHAzDXLhw\nYXh4uLeXyigi9/nnqZ6e8eaL9esb169v/OIXrT/9KTlhpqndbg+FQnB5yuxdd7nQ3hzE1qQf\nvJom1dXVsm+hzs7SYHCCIiC/33/x4sWeHkIpX1bqGGo0smqFqolXXgGOk/nvk5+c/NWxLNvb\n29vd3d3VxfGDXLfOh/YWVI6y9LYIayjQFmFNByi48KRJMpns7u5GRsVCQ9OGhoZIJMKbVjY0\nNGg0mqm8EAaDwWAw+QJXcGAwGEyRodfr33xzvs1GQa5iLAuH/v5+FImSaUSuUqXAsWPM22+n\nWuSPRqN+vx89zn1sDcuyXq/3kUe0r746nusx9UoEnU535EiDzUZDhm+hRCLR39/PcZxSvqxs\nVQhFUekbnUydgYEB9CMTDnLduqHf/KYECixH2WAwUBSVTI4OUlhDwR8jqukIh8Pf/OY333vv\nvffffx/pbsePH7/22mvTebnh4WFkHSoyNO3v71+zZg1/2L59+/bt24dNKzEYDAZTjOAKDgwG\ngyk+PvxQPmBiOmIsC4dwOIz8AiBzrUFaKeDz+U+fPuPzcakrQaQuCSCOrZniZSmSSCS6uroG\nBgZsNpIf5B13eKSDnASnTsmvcKR+C6V2Ev2Xf0lIq0JOndInErU5y7BkGIbPNy38HGWSJKur\nqwEgEAiIgj89Hg/yHxXVdBw7dmzv3r1Hjx5F6kZG0Sp+v19o38A3v8gGpmDTSgwGg8EUI7iC\nA1OIJBKJQCDAMAxBELpIxPyv/wrd3QAAu3fDI4+gY+LxeDgcjsViKpVKH42q/+VfpMdgMDOV\nXMZYFg68uiFkchG5qSsRhIv8/Oo6j9Bd4uc/H6HpuswvJS36+vpQm4BwkOvXD//2t2Uw5UqE\nyb2FpD8CoZNoIqG22dQgVxWyYkW6LzFFZ1A07VcaZAHmKJeWlhIEUV9f7/P5+I0o+POTn/zk\nE088IarpQN0lfLTKggUL0nTJBYB4PC40KOWbX5DD6Pz58w0Gg8JTMRgMBoMpDnAFB6bgcLvd\n58+ft9vtw8PDLqeTXL9+VLkAgGuuAQCO4wYHB8+fP9/X1+d0Ovt7e2O33io6BoOZmbjd0NgI\nBAEEAT/4Qb5Hk2uk2QGHDpnvuacxEKBWrw794hdOOm3RPnUlwhe+wPAbVarxVBGpu0RZmXwp\nzdQJh8P8XL1wKhFEPwLh/d+/v+f06clUhQgJhUKtrdG2NnjvPXC5IB4f7cd5+OHxY1I7ksgK\nUpN7k+SMkZERobrBs2DBAmlNR3Nz85YtW1auXIm6S5YuXSobGiqLqI5G1PxCUdP1ZsZgMBgM\nJmcU3t95zOzG7/cPDAzw/7S2tRmPH2e1WhK5nV1zDQAMDg6OjIykPgaDmTFEo9FgMMgwDE2S\nFRs20LNYy9NoxtfepRG55eUZJMikrkS4/fYwwOhrGY1GgiA4jpN1l5hKnkWmI4QCqETgTTGl\n95+muT/+MVJSMvkIVrvd7vF4hFE1zzxjQeUqfFRNIBBobVWlCOulBQKGdJClpSWTHt70IRv8\nWV5e/stf/vKXv/wl+idf07F//364PFrF6/VWVlam80IGg0GopAibXyiKEv5+YTAYDAZTpOAK\nDkxhMTg4yD82v/FG1XPPBa6/nlm4EACSc+aA1RqLxYSrVaJjuJoasFpzP2wMZjrgOG5gYODi\nxYuDg4Mej4d85BH68GFON5bNOfsEDoPBgKavgQC1bVv9009X0TTX2mpvbR2gaa60tDT9U6Wu\nRKCo8b00TVsslt5etdRd4sKFCo/HnK2rExUmrFxZumePNR4fbygQDvKXvxya1koEpSoJdJNl\n7z9FUbJyT5oRMC6Xy+PxgHJUDcuyPT09Nputuxt4R5I77xy12+AdSZAgpTTI6ROksktXVxe6\nGyL4RhVhtEo8Hk/ztJWVlXyLisjQVLgLg8FgMJjiBVdwYAqIWCzGW51purrmPvwwU1/fv3v3\nkhtuAIBIU5MRIBAI8DMT6THM8uXp9iJjMAWP0+nk5Txey6NdLt3p01BbOwu1PJIka2pqjh1z\nbtlSb7OpkdawaFH01Cm92Ww2mfT19emeSqPRIAdH2UoEUXinzVa1c2fZJDJH0icSiTz6KLzy\nio7f0t9P/fznVRxHbN8+KB1kWVkG5SqZEgwGH32UEg5GUCWhCwSs69aV8PcfaT0AcOWVVbJO\noj/4QeLPf6blTjV+DMdxw8PD6LGwH+edd0b1iJYWGBgYCAaDcLkjyV13uV54oRQEjiQqlaqi\nouL99/1btzaIBqnRaIxGc0kh1nCIEdV0RCKRS5cuCQ8QdpekL0zodLra2tpz587FYrEPP/yQ\nb37hOE7YioXBFCAsy/p8vscee+z999+fRIQQBoOZPWCBA5MjkskkKrMnSVIXiRg+9SmpJyjf\nO00Gg3UPPAAAvW1tqqEhgmEAINzUZARACZFKxzArVmCBY0bCcVwsFmMYhqZpbThMfuxjM95T\nNpFI8K1YUi0vtmLF5DsBihmz2XzkiM5mU8HUtIaSkpLh4WGlTFPhOn8kEtm1izx5Ur56Pyux\nNSMjI4ODgz09C/jWjAMHqpDd6fHjxq98RWaQJdM2TR8YGHC73T09C6WDQX0if/1rlc0GIHf/\nly+/7FSJRKKvr+/CherNmz2yp+JhGEbqncH341x9deTuu+lLl9LNRpkzZ87rr5uU7E6LMUdZ\no9GQJIlcYxDC7hKdTqf8VDGlpaU33XSTsBASNb/gUFhMITM8PDw0NHTy5Ml9+/bxGzOKEMJg\nMLMHLHBgcoHH43E4HKNfzli2YcsWWU/QUYczjpu3Y4fGZuvds4dpbCz705/Q3tiKFRMek8B/\n6mYifr/f4XCMlmGzbMOWLcZZ4EMRCoXQ+qqS3jc7BQ4A+Ogj+aXmjLQGnW7iSgTUIuTxeLq6\nxmf7zz5rRSUDV10FJ09m4XLC4bDD4YDLCxP4PBedjl23boFokEaj0WSalrwLj8eDpr4pwmXe\nf1/+udL7b7PZIpHIhDk1MFFUzZNP9jHMHOkrpnAkOXdO/u4UaY4ySZJlZWW84intLkn/VF1d\nXbKmpDgUFlOwOJ1OZKkrihBatGiRXq/P9+gwGEzBgQUOzLTj8/nsdjv/zxSeoGq1Wq1WG377\nW9ORI/4bbojNnavt7DS8/TbaqzUYwG43lJcDQNmf/iR7jEavB7sdamtzdnUzElQukUgk1MGg\n6tpr81sr4fV6+/v7+X/OHk/Z0SmfgpYXaWrKwHBiZpGtiNwJKxGcTifvCpGiJ2KK8LNW2cKE\nigrVyZNTLVdJH9k+EVGVRJr33+/3I59U2VPdfHMcYFyoEkXVCPtxduwYMBo1wuIFhFABeeaZ\nIZqeL9w783KUrVYrwzAOhyORSJw6dQqpnw0NDWq1On0PDlAwNMVgChaGYfjPpebm5ubm5mAw\niPSOxYsXMwyDzXExGIwILHBgph2n08k/FvkIJOfMoS73EbBardx77wGA+dAh86FDwl2VGzbA\nE0/ovv1tk8lkUDjG2NICTzwB3/72NF7PjIbjOJfLNTw8zLIsqpVQ5bVWgmVZtLiNEL1/uJoa\nYub6UKBiJSUtj1KrsZanRHs7HDwIx45Bby94vVBTA3feCbt3w+XGGhNUIiSTydTSQ7ZSWlPn\nuRDENLbGiGBZlmEY0UbhYO69VwOQbpIo8stQOlVLSxhgvO5ArVZrtdpoNCrbNGQ2m1MoIDt3\nOsrKisM6dCqQJFlfX3/11VcLzUf37du3b98+3F2CmQGEQqFHHnmkvb1d5K/h9/tFkpwwQsjv\n91dlS2zGYDAzBSxwYKYXhmEm9A0VUlJSkjx/XvF011wDAHPnzk2ePZv6GMzksNvtXu9oo3sh\n1EoEg0G+dl36/okuW5ZB63mxgcIglLQ8y913Yy1PCsuyLpdr507D66+Pf7TIulrCRIv8fIuQ\nEH6Kfs010U2bsmP4I6pNEBYmtLXZli+vMxqNSs/NLtLrFQ2GJBelL3CIuk5EpyKICtHx1dXV\nf//7gNA+FvXj0DTd3FxZVUUgEwolBWSyFz0xaeplOaC7u1s2WgV3l2CKmmg06nA4Tpw4sWfP\nHn4j768hLVCaXIQQBoOZPWCBAzNdMAyD1A3K51twxx1qux0AOJVK6CMQWbZM+s2d6uhIJBKB\nQCAajarPnq34X/8L4LL+CIqiqHPnAoFAKBRiGEZ/4ULVZz4jOgYzCYLBIK9uiGolElYrWVWV\n+1hp/ruLrA9FdPnyGSxwUBRVVVWlPXNG8Qis5V1OMpns7u6ORqN9fSVKBpkZnU20RThFP3DA\nSdONWRm2Wq1G3snp5LlMKxRFURSFLlw6GLWaoDNJph31S1K4Ln4vj8FgOHy4PoUz6Jw5c95+\ne1ghG6Ukk4zgDIhGo62t8PLL42KWkl6WA3B3CWbmEY1Gu7q6WJYV+WssWLAAuedKs5kmFyGE\nwWBmD1jgwGSfcDhst9tHS51ZtuG731WPeXAMf/nLUt9QKTRNj4Yg8muskhUqk8k0mnTwt78p\nHYPJCJ/Phx7I1tpAKCSMlsgNo99sFHwo4ldemePx5BiLxTJ49OjIyAia1WjPnVvY0gIAydZW\nateuPA+u8HA6ndFoFFIaZKaPcDIvnaKXlmbN4bOkpCQcDssWJuh0ulwKHABgNps9Ho9ClUSJ\n0lxCtsbhO98xAbhlTwUAsmUpp0/LXyzqxykvLz90SJfLbBS32+1wOLq7ZdJtMtXLMBiMLAMD\nA6iKTeSvsXTpUo/HU15eLrURFUYIYZNRDAYjBQscmCwTiUR6enr4omvU5sCpVEQ8DgDBj31M\n6huaykdg40bYuHGCl0znGEwaoGYi2VqJyLJl1FirUS5BCzgiHwrj0aNor+XLX4YzZ+CJJ3I/\nsJwxZ86c8vJyVKxkPHwYbaQ+/vH8jqoAYVmWrz/KimWGwWBI0RORRbGvvLz8ww9DGzfOERUm\nEARx9dXVEz8/q1it1o6O2Fe+UisaDEVRzc0yOSYcxw0PDz/8sP6118YVn7EaB9NNN5Xee69F\nmlPT2KjTamUafCZ0Bj17Vr5mazocSSKRiMPh4DguK3oZBoOREovFwuGwcIvQX8Pr9ZaXl5tM\nJo1Gw9sDCSOENBrNtLanYTCYIgULHJgsw4vxIGhz0H/wAeX1AsD8++4THlyxfj32ESgcCIJI\nkdlhykchqFarNRqNSj4UAAByHekzDJQuBADwwAPwwAP5Hk6BEovFpEEbQlfLTZsyW+gjSdJi\nsfzzn25pT4RKpTIYyirEJhKThCCIQ4fm2WwEyBUmLF6cnVdJE5qmDx+ut9lI2cGIqiSSyWRP\nT08kEuntXShb40DTtbLXtWcP94lPTGZ4ucxGGR4eRhMtWb3s1ls5AFwbj8FMiZhk4UTor4H2\nEgRRV1fX09PjdruFEUKNjY0Gg8Hn85VOU38aBoMpWnLfU4+ZycTjcT4RQNjmQI31PsiQRx8B\ntxsaG4EggCDgBz/I2zAKBp1Op5TZwZGkYWx5PMfU1tbqxr7xyHDTTTkcC6aYOHTIfM89jYEA\ntXp16KmnbJnYR4xSWVn5xhtzhT0R69c3rl/fePvt8/7852z+9fzgA/mp8nQUJkzIyZPylyYd\nzODgIPrAf+657m3bnEuWRK3W+IYNo9EzVVWK13XNNUUgDaROt1m3Liz3pMkTjUa/+93vfupT\nn0LuwgRBnDhxIrsvgcEUGtKuN1l/DY1Gs3DhwrVr165Zs2bbtm1o4549e2pra9evX5/LAWMw\nmKIAV3BgsgkvxovaHIDjAGBo27b4Qw9pOjsrb7gBID+eoH6/3+/3MwxDAtRs2qTJawZqoVFe\nXh5SqJVo+OpX81Vr43K53H/8I3psfuONuu3bx/fV1sItt+R+SJgCRK1WEwSBVvaklhlGo3zY\n6oR0dsrXfWRXeshlYcKEpDmYdHqCNm/O9uBySOp0G4qqy+ILuVyut9566wlBtx0fIYHBzGC0\nWi3/uY0Q+mugHlWEzWbDEUIYDCZNcAUHJpuILCH7f/hDprGRX36PrVhRW1tbabONHp3bP0sc\nx/X29vb29nq93kgkYvzRjzRvvcXyfeBY4ABQqVTmCxcUd3/nO7kvdYnH4263Gz1GNUEJQWOA\nkkktZhZCkmRJSQkABALUtm31Tz9dRdNca6u9tXWAprlR0+LMeeUV4DiZ/z75yayOvgiJRqMp\nYnSvvjqyaVM+hpU9VCoVesCy8OST1u3b68JhsqXFfeBAj9HI8nunCMuyPT09LpcrHA5v3br1\nqaeeslgsALBgwQJZmxIMZiZBUZSwwUTorwEAFYI/9yhCSMpjjz2W+2FjMJgCBwscmGyi1WpJ\nklRqc1DrdGC3w8aNo1OEG2/M5dicTqff70ePeXMQZuFCAOBqasBqzeVgChbyzBkmGh2w2y+c\nP981VjeRaGgYPyK3SlAoFBod2FhNUPfPf37xD39AG8NNTbkcDKbAmTNnjsNhWLduwdGjJqFl\nxoULFYFAeb5HN/MR9gT97Ge9k+gJKihS6GVarVajmWRNkAgkbQBAc3Pzli1bVq5cyUdIDA0N\nZeUlMJhCprq6Wq/XBwIBj8fT3t6OZNP6+nqVSsXnxAMAy7Jut/sb3/jGmjVrcBsXBoNJTZF/\nAcEUGARBVFRUaBTaHCx3352vNgf0pxE9lmagRpctk7fmn5VoNBqNRuPxeEpPn0Zb6J4eVqsl\no1GAXAscyWQSQDEmNrpsWS4HgylwaJo+cqRBya1zOmJEZzPC/FppT5DJVPTVBxUVFadOhaXp\nNgCQxXQbUdW9MELC4/HMmSOTXIPBzCRIkpw/f35zc7Pwd6Gtra2tre2mm2566aWXAMDlcrlc\nrpMnT+7bt48/BrdxYTAYJbDAgckyFosl0dmpuDtPnSDRaBQ1VMtmoIaWLsUCB08oFHI4HADg\n+dKXkkZj3fbtgeuvp10u3enTbHU1mdtSF5qmQRITy9cEkSrVBDHDmFlGQbl1ziTa2+HgQTh2\nDHp7weuFmhq480560yYTwwRkY3Qn3RNUOKROt1myJAsvkUwmE4mEcIswQgLtpYu9EgaDmYju\n7u4U/hqDg4PDw8MAEIvFtm7dumLFitbW1qGhoUWLFun1mWVjYTCYWQL+w4nJMgRBqDo7eS9P\n7blztWvXAuTHUpRn1C5OOQM1XwMrQNA3CZArdQktWWLkOKnt+fSBKlGVYmLzWBOEKUwKyq1z\nxhCNRltb4eWXx4sybDb4z/+EeHzu5z5nu//+uaIaB51OZzSWlU9/V1B7Ozz/PHfsGPT1EWOy\nC+zeDYLikimRe71MGCExXa+BwRQYyF9DdhfDMCMjo8FMzc3Nzc3NwWAQtXEtXryYYZhsNYth\nMJiZBBY4MNOC2Ww2m80AAG+9Nbopr07XyBBOqRCA1mhwIQAPagiXLXWJLFumYphcWt9RFFVV\nVaU9c0bxCOwOiykG5CogsjkVnz68Xq/dbu/uXrB5s+uGG3ylpYlnnrH87nflAPC3v1FqdYPN\nRkLOe4I4jhseHt6xQ/vGGyZ+I5JdWBYef/yyg99+O/H889zbb1P9/WRGN3+69TKKokRGA8II\nCZqmcfkGZpbj9/tF2oewjcvv91dVVeVpaBgMpnDBfzsx08zGjbBxY74HARqNRqvVKhUCVH/5\ny7gQgIdl2RSlLibBVw2WZRmG4ThOEwpRH/84oMzdbJfqWCyWoWPHLrlc6DuN9ty5hS0tAMDu\n2kW2tmbxhTCYaYJl2d27k3/5y3juhtJUPFM4jjtxIn7wILzzDp3p7D0dGIax2+0cxz33XDef\nBbthwwgSOKqq4ORJeavy6ahx+Oc/2f/+b/b4cQJdqcVSyjDEffe5Pvc5X1lZ8sCBKjSq118f\nv6sMwwwMDDzySMWhQ2b+PNm6+VmhrKyMNxMVRUjMgDYfDGaKCOU/hLCNKxaL5XxEGAymCMAp\nKpgx3G5obASCyH0UaG6orq7mA2tlwIUAY6hUKqVSF44kVUNDAJBMJvv7+8+ePXvp0qWuixcj\nX/ziqLoB03InLRbLFVdcMW/ePIvFUj0wgDaSH/tY1l8Ig8k64XD4/PnzFy8mNxE7L78AACAA\nSURBVG92vfjixUOHzt1++6jh8euvT+nMHo/n3Llzra3Rn/1M/cEHpMsF8fjo7P3hh7MwcgAY\nGRlBwiKvbgDA228b0YMvfSmZsxhdl8u1c2dw/3765ElqeJhIJIiBAdXICJ1MEkuWRK3W+IYN\no3Xs/IJuLBbr6uoKhUJ2uzrrN1+Wf/6T/drXEqtWJauqOLUaGhrgoYcg9RSsqqrKYDBIIyQY\nhslWEi0GU7yQpHieImzjku7FYDAYwALHLCcUCjmdTpvNZu/ri7e0TOscNe8YDIbEBx9cOH/+\n9Ecfnf7oo4u//z3azu7aBRwH11+f4rksy4bDYa/XGwwGky7XzFaCSkpK+FKXhbfdtvC220pf\negntavjqV+kXX0wmk11dXV6vF30Xt7a1GY8fZ/m+FcmbJx6PBwIBr9cbHRiY9K1TqVQlJSUW\ni8Xw9a/nJWYYg5kE8XjcZrMlEonnnuvets2pNBWfBMPDw3a7PZFITOvsPRKJiLZ0dmp/8hML\nAKxaFb7zzlB2XmYiHA6H0+mUvdLjx0fVFl52ueUWFj0YHBxEMUzp3/z2dnjwQWhuBosF0lQo\nEBzHDQ0NCSWYNMUmgiAaGhrWrl27Zs2abdu2oY1tbW3Nzc3//u//PvELTw2fz/eNb3zjuuuu\nw7mbmMJEaiMqbOMyGAz5GBQGgyl0cIvKLIXjOLvd7vV60T+te/eqjhzJVxRozjAYDIsWLWIY\nJhqNao8eRRsnLAQYGRkZGhoazStl2YYtW4wzWgmqrKxMpCx1cblcDMOgf5nfeKPquef4mJXk\nnDmUIGYlmUza7Xa/3w8weutmtoiGwYgYHh5GHx2yFRC33soBTMayN5FIOJ1O9FipeSQrjNoz\nj3HokHnnzrnhMLl6daitzUZRufAtikajyGVQ9krLyhJwuexy110cgIFl2UAggA6WvfktLeIX\n4jhu9+7E5DqJBgYGPB6P3b4QOZUotczIkjpCYpqIxWJ2u/3EiRM4dzP3uN3uxx577L333jt5\n8mQoFAKA48ePX3vttfkeVyFiMpk0Gg3/lUPYxqXRaEwmU8pnYzCYWQoWOGYpTqeTVzdEc1Su\npobIbRRojtFoNBqNBrZsgS1bJjx4eHh4cHCQ/ydfrTCDlSCKoriODpvdjqYHvOdF/JFHVLt3\nA4Dv3Dl0pDRmJbx0qYFlUdUox3Hd3d1RdKNmx63DYEQEg0Hu8uChy6fiADCZmEOh8V6as/fJ\noVar0dSCZWH/fuuBA1UcBy0t7p07HTTNqXNikTqqkCpc6Wc/6xfJLipVAwAkEglpLoPw5n/5\ny1phEWssFuvt7b14ce7mzd5MFYpwOIwUClkJprJyAhkrRYTENJFMJnt6emKxmCh3M/cjmW1E\nIpH+/v533323ra2N34h1pRQQBFFfX9/T0zMyMpJIJD788EP0Fm1sbDQYDD6fr7S0NN9jxGAw\nBQduUZmNJJNJPnZLOEfVXrgAALEVK/I6uvTIiWOIcJkUBEoQs3AhAHA1NTBDlSCapuvr6xcv\nXtzQ0FA7pu+orr0WADiOQ6ZfSjErvCWY2+3m1Q3RrUtYrTP11mEwQpLJpFDdOHTIfM89jYEA\ntXp1aP/+HoJIpnhuCqTGeyCYvTc3M5s2Te7EYkpKSgAgEKC2bat/+ukqmuZaW+2trQNI3dDp\ndNl5mZRIfQSFOoXDodq+vS4cJlta3AcO9JhMHIqNlDbni26+SjX+c+E4zmazRaPRyXUS+Xw+\n9EBWglm7Vtzmk3eGh4fRXW1ubt6yZcvKlStR7mZTU5PD4cj36GYssVisp6eHYRikKz311FMW\niwUAGhsbcxm+XnSo1eqFCxeiNq6vf/3raOOePXtqa2vXr1+f37FhMJjCBAscs5FwOIwkcNk5\namjp0jyPT45kMul0Oi9dutTR0XG+szP6pS/loNkhEAjwy1lSJYhZvnyaXrdAUKlURqNR97Wv\nCT0vUJ82H7PS/8MfMo2NvHtrpKmJ/6LGr7tKb12kqUna24/BzDz4mE+WhSeftAqn4kYjO+kQ\nUOl0SDh7P3hwJFvpoqWlpSMjZevWLTh61ETT3De/OXjFFcypU/pTp/SJRKr+FGS3ec01XKZm\nFlJEUgV/pVdfHdbp2GeeuUx2KSkpoSgKAGia5k06pTe/qkojvIderxcVqsgqFF/8YiL1CKV6\nk1CCueOO4GQuezoRRW8Kczej0ShOppgmnE4naliT6krCpRSMlJ6enty3cWEwmOIFt6jMRkbb\nqpWjQPM5ODni8Xh3dzf/ravsxz/W/v3vOWh24L+2yipB0eXLtamePWPRarXaX/9aNmaFVKlU\nQ0Mwdy6M3T2lQg9tPJ6b5V8MJo+YTKZoNBoIUN/73tyjR00qFbdjx0BLiwcAaJrWaif5ESI0\n3pM2j1gs1dkZPQAAvPZajc1GAEAiQTz++PiZ9+4F2b8VgUBgcHBw505rtpJZ9Xq92+2Gy6/0\n85/3dXToenvVQtlFpVJdc834CCsrKx0Oh+zNr6ysFL4E8kEQIlQobrstClCeYoRSCUbYMqNW\nWzK+5mkmHo8L9R1h7ibam5vmo9kGbwqDEOpKoVCIHevuxEjBzVMYDCYjsMAxG0HLhkpRoJRa\nDXY71ObCPS5N+vv7eXUjl44hJElSPt+CO+5Q2+0A4LvxRqESlJitTbMVFRUwFrNiPnRIuGve\nV74CTzwB3/42AJAkybGskoimn+w3OZZlY7GYSqWifD5obh4t5Nm9Gx55ZCoXhcFMB5WVlR9+\nGLr//rk222VTcQBYvrxiEnXp7e1w8CAcO2bs7l7q///snXt4E9ed989cdJd8t2xjg41swDYh\nJQSSpgSS9E13C+y2TUvSUJImgSZg4iZNmpZubg5ks++GNI1xUtqwD+2z6b7bS9Jns+km6abk\nQiEGUocEbGyDwZZsybIl6y5LM7rMvH8ceRhmJCHbuozl83n4w54ZSUejwZrzPb/f9+vFy8oi\nJMmazXJu9k6SZHqb0j/7LP4gV62Ks9HlclksFgAATDyZrplFXAoKCmQymdPJ8HWK0VH5n/9c\nCOLJLg8/HPu5tLS0vz+ydWux4OQXFRXpdLD5JkbMQ3oKgUIBwGVqiBiVSgU9reI6lUhQySUI\ngm8fy8/dhHtzNrL8JRqNCix7+boSy7KRSATpSggEApEWkMAxH1Gr1SRJahLMUSvvvZebo0oB\niqK45TWxqyWdgTIKn883OTkZCoUwll344x9DdQMAELjmGr4SpFCrpaYEZYeioqLIlM9oHKYK\natRqteq3v40rogGCUDmdQKud1uv6/f7x8fFYbwvDGB58UI0yWRDShiCIDz6oNZkIEG8qfvXV\nl46cUi7A8DBwu8GCBeDOO8HevYA/5aEo6plnsLffVsDnBgCMjckAADgO4Oy9p0dbWVmpUuG1\ntWl7C+++m+qRkUiEc3BIY7YLjuORSN2WLRhfp3jzzeK4Bwtkl3feqTCZ4NgS6iDg8k4isUJx\nxU6ioqIiu93ucrHiUhGVSiXBJEu1Ws35hoDLczcJgoAmJoj0guM4hmH8MgSBroTKNxAIBCJd\nIIFjPoJhWGVlpfLs2YRHSGm6yBlVJmx2SN9rRaNRk8kUCATgrxXt7drOTpYgsGgUAFD1L//C\nP1h3++2SUoKyCdnX5/P5XC4XTdPyvr7ar38dAGEZRWlpaTCBiFa7Y8d0T53b7bZYLNzdYUVH\nh/rYMZTJgpA+p0/HXw/npuIsyzocjqeeUv/5z5caT8Q9HVar1eFwDA5eCiJ96KHa3l4lAPCw\nhLP3rOHz+bg16vRmu/znfypS0SnEnDoVf7tAB9FqtS6XK24zC4Zh2itJsQRBhMO1W7YQglIR\nmUy2alU624XSRXl5OWfDwc/dBACUlZXNOcNLl8v1zDPPdHV1nT59WrKpqxiGqdVqfjMUX1dS\nKBQzduRBIBAIhAD093SeUlRU5Prkk0GrFd6PclGg0bY24plncjy4y4nNaRM4hlDLl6fxtYaH\nhzl1g+uF0Rw/jkUThB0kn1c7nXncQ6HT6WIR9EePxjZdbvelUChkAwMJHz8dSSIajVqtVk7d\nkH6wcaybOq8vAESKJK+AiEQiRqORoiiTqSFJT4fdbofRV/zKCKWSifuccZtHskBcc0rOzGLN\nmtD27TOswE9RpxCTYvlJYWHhqVPe++6rEHcSLVmiTaVx4He/U81MgskJSqWypqamv78/FAqd\nOXMG/mmtra0FAHDOrHOCUChkNptPnDgxJ1JXy8vLOYFDoCvBOBUEAoFApAVUETd/KS4uXrZs\nWW1tbWVl5YKpumLi+usvHZGVKNYrAm8uEzmGyJRKMNVCMksmJyfj9sJgLAsACNx668ULF8b+\n/OfY0Xv3ApYFt9wieBKWZd1ut9VqHTGZQt/6VhaiXnLPtm38mBU++Nmz4VBofGzMZDSa/+d/\nYlsTnLokeL1erklenMkS5hf65xSapkdGRvr7+3t7e/vOng1+4xvz4gJAzAKLxQKL1JIElDIM\nA9MWwOWVEV/9aqzL4Je/jP0XhP/WrcvmO7hE8myX116bmPES9bvvXvYGM/FO//KXGpNJDqYU\niq1bDfDfxx+n1FczXQnG7/c/+uij69ev12q1MJrq+PHjMxz6jCgsLNy4ceP69etbW1vhlo6O\njhUrVsyh3M1oNDo0NBQIBMSpqwK3C4mg1WoXLFjg9/tdLldXVxfUlerq6mQyGXLQRCAQiDSC\nKjjylxRWjwmCiK3Dt7aC1lYAgN/vdw4P0zTNRCKLduxQSWB6plar5XJ5IseQsrvuSlefiN8f\nC/OL2wvDrFpVX18PjhyJHR0vnIymaZPJBJcxK9rb5R99hHooZDJZBSyveP/92Kbp57rBBEeQ\nOM5GCs5sgUDAaDRyN9ZlL72kOnoUXQCIJIRCIS5YIUlPRzAYFE/YuMqIa6+ltm+XRKATPxRG\nbGZRXl6Zw7FdkUReqtdem1K/RupOJZOTk1ar9ZNPPnnppZe4jdkvOhgcHITZNALmUO6m3W6H\nWV2rV69evXq13+/nUletVuuSJUtyPcA4lJSUbNq0iX/m9+/fv3///o0bN7799ts5HBgCgUDk\nE6iCI6/w+XwWi+XixYtDFy/S3/zmdFePx8bGjEaj1+ulabrkxRfh9Gxaz5AJMAyrrq5WTfmN\nxyFNY4vVCEz1wpife442GLjXpa+6CoBk1QoMwxiNRqhucD0UdEMDACBaWQkk1kMBAGBZlqZp\nmqZZhyMbpTqJT90Via0MJ/hopFDBwbKs2WzmZqGCC4BdsECCFwAi58Qccy/ncuUCAFHGB7i8\nMuLVV0ck0ryv0+lgwZ3PR7S21r76ajlJsm1tlra2UYUCT2+2S9rJQpEIAMDv98OOJEHRQUND\nAz/6NwvA3E0xe/bsyeYwZkOS1FWapuM2TOWcPNCVMgrDMDab7fvf//7atWs1Gk1OipsQCEQe\nII3bIsSsgfMrzhe9or1dceTItFaPvV7vxMQE/FngccBUVeE5nZ5pNJrg6dODVis0yOAcQ9Lr\nawB7jxP1wuAyWfLMFJfLBVeTxFEvgaYmRSgknQS4aDQ6NjbmdrtZlgUMU9fSopVAqU4SYM7i\nFdqU0hpnE4lEaJrGcVwxOYlfd90VfTT8fj93Py2+AEIrVqBYAoQYcV0GP6D04MFxkjQAXsYH\niFcZodNJonwDAIBh2MKFC//619EdOxbyzSy6uzWVlZVqNZHGbJe5CMuynFOyoOigsbHR6/UW\nFBTkeoxzCfiFy8FPXYV7pfOdywF1pVyP4hIURbW1tZ08ebKrqyvn/qxwie7UqVOvvPIKt1Gy\njioIBELKIIEjT7DZbJy6MTMLRk7dEE/PJhsbtSybW2d1lUoFG2tpmpZzcn5aFz10Ot34+Hii\nXpjiLVuS98LAm4NEUS/RQEAiN1vRaHRwcJBr+qjo6NB2dkq8k0Kr1cpkskQfTcEdd6Qxzoai\nqNHR0ZjX7JXUn3A4HAwGo9EoNTq69KtfhYnCkaIiIGqiQQIHQgz/b4JYuSgp0cFdKpWKJMlI\nJBI35iPm9SsNVCrVRx8tNplwMEfsNrNJIBAQz8m5ogOPx4MEjmlBEARfIhSkrhJE/PQiBIfH\n4/noo4/2cUFNOVUTAoHA8PAwy7KwuGnFihVtbW02m81gMKAAXQQCMV3QX418AKYMwp/FFowp\n5ozAYulE83OJVHviOK5SqYj7759xs0MSlEplUVHRjNNzo9Fooh6KYHNzJBJJ41Bng81m49SN\nOdFKAwDAcXzhwoVZaFOiKGpwcPBSTvCU+iN+lWg0OjIycu7cueHhYcvIiO6BB+RTZrek2y24\nAEIrVqRleIg8Q61Ww8IxcU8HSbJcTwcM9h4elm/ZUn/0qI4f89HbW+D3l+b0TQj5/PP49xW5\nynaRDuKvUX7RgUS+ZOcQGo2G/ys/dZUgCIVivqvKwWDwhz/8YSIjW7PZPDIy4nK5ctsqxTE+\nPs4VN7W0tKxcuZJzVBkfH8/JkBAIxNwFVXDkAxRFwXWMuPLEZFOTKsUnShDFGmxuLpBSUWXm\nqK6uHv3oI5fLBX/lemHYPXuwp59O/liSJBP1ULA4rpiYAGVlGR18irjdbvhD3FaanJfqJEKt\nVod6eix2++TkZDgc1l68WPv1rwOQ5jal0dFRsY+GuFGLZVmTySTWQWAVjG/9esEFQCoUaW+i\nyRu6usBvfgM+/hgMDwO3GyxYAO68E+zdC6RR8JRZMAxbsGDBsWNjLS21goBStVqt011a0S8q\nKjp8WMGP+eCeRGqVEanbbc43xH9a+UUH0vzDK2XKyso8Hg+cFQtSV8vLy+fz+WQYZmxs7MiR\nIz/72c+4jfzqDIfDAe8ExK1SDoejtDTbminDMFyGHYRf3OT3+1mp3pkgEAhpgio48oHYlCxB\n+UCgqSmVJ5HJZInm54AgZDbbZUdnJ0E26zm10NB06dKlNTU1er2eS8/F1qy54mN1Oh3XQ9Fw\n++0Nt99eNGWKXrdzp+attzI37NSJRqPQsDBhK43IzlA6yOVy+OksX768dqqjKo1tSuFwmNMs\nxJVQweZm7ki3280dKaiCAQDo/vpXwQVQvGUL+O1v0zXOfMLn8z31VKCjA3z6KbDbQTgMTCbw\n/PPgySdzPbJsodPpPvywThxQetttlVMKc4y+vvhKNaqMmCtALyE+/KID8V5EcpRK5cKFCycn\nJ/mpq7W1tRiGkSQJI9t/8IMfrFu3LldZvDmBZVmj0eh0OmmaFqTncr0eXM0vhK8mCHZlB3GJ\nK7+4iWVZaeb+IhAIyYIEjnwguTtmbPX4ShQWFiaan9fu2EH84Q/wdmFsbMw8PBz61remG9GS\nIsFgcHR0dGho6OLAwAyCYFLiSrqJXC4vKirS6/Xq1tbUe2EKCwvVfX2J9uIpSCRZIHkcSbC5\nec4skswikyURXIl4XPUnwBM4vF4v/EGsgyREkuYmuWV0dNRkMplM+I4d9tdfv3D48Lk77ojl\nC7z3Xm6HllW6u2VxtwuUi+zEfCAyh0Kh0Gq13K/8ogMMw0pKSnI3tLlKQUHBxo0b169f39ra\nCrd0dHRcddVVd95558DAwDvvvLN///5jx47B6oB5YlfpdDqh/i7u9RgdHQUARKNRQT+UoFUq\n++scYsMUfnFTKBR6/PHHb7755nklVCEQiNmABI58QC6Xq1SqRPLEgm3bUlk9Li8vTzI/D61Y\nMTAwYDabJyYmFHv3yj/6KBMJshMTEzBEbXJysuD552EQTFpehWEYp9M5Ojo6bDRmSJ3BMAw/\ne9Y4NNTT3d3T3X3hjTfgdv+PfwxYFtxyS7peaDbgOC6XyxNpYYRcToyN5XaEV+TEiUhLS2jl\nykh5OSuXg7o68JOfgLR0rydXf2I5wQAAAMLhMMuycXUQW2vr2Z6enu7ui3/8Y+zovXulcwFI\nB5fLBeMSDx0aam0db2ykKirCd90VWzwsL8/p4NLB8ePhlpbQqlWMXg+SX6hIuZgreDyeH/zg\nBzfeeOOMJ1o1NTUKhcLn8/GLDurq6pRKZdzM4CtCUdSPf/zjeTv3S5S6ajAYQqEQtKvs6OiA\nvhIsy8Lk0fw+RVwXKoRfnREIBEKhkDjGReDPmv2cF4IgBBVM/OKmgYGBffv2HTlyZF4JVQgE\nYjYgD448oaqqipidBSOO40Rvr9lqhU2tnP1EtK0NPPXU4MBAJBQCImOCaGUlkSZbSp/PNzY1\nu55WTm0oFMJxnPR6werVceM8oTs3rIGsaG+H6kwmQkNIkqyrqwsGg8FgkJxK+tDefHMaX2L2\nlJSUkAniSGq+9700xpGkHYZhrFbrU0/pDh++lDUAOxoYBvCc4GeIUqnEcbzwj3+Mq/7IVSrO\nR4MgCAyARIY1NTU1arWaPHky9rxpzfrJG7hCaJ3u0mrhiROx9W0YAz1HoSjKYrE8/XR5hi5U\nRPYJh8MWi6Wzs3P//v3cxhlMtEiSrK+vv/766zmnJwDA/v379+/fv3HjxrenliVSgWEYu91+\n5MiRF154YTZDmtPETV0dGBiARtrQYOLkyZOwogG2ORAEcfXVV2d/qFkjSXUG3KvVagmC4Jdp\nCPxZcxJAo9frTSYT/JkrbmpubgYABINBQa4KishBIBDJQRUceYJarY589tnA+fOC8gHmmWdS\nXz0mCKKmpqapqam+vn6R3R7beP31TqcTqgPigvxAU1O68kHi5tRyryI+PhKJjIyM9Pb2nj9/\nvr+3d/JrX4tblxGJREwmExykwC6BqarKRGiISqUqKSkp+MEPMhH1MntKS0s1/f0Jd0u4kwJa\nvlss8gx1NOA4XlxcnKgSSv/d73KVUGq1OomhrMbtlslk2Pbt0rwApADLshRUGHn09ytfflkP\nALjmmsA990gldWi60DQ9NDQUDAYzd6EisgzDMEaj0e/3w4oAvqnBDIr5jUYjX93gWD0dJZRh\nmKGhIbvdHggE+EOqr69XcmWP8xKKoriYMMj58+fhDzBQefHixVyPYV4iCFUVVGfAvYWFhdwB\nAn/WgoKCnHSq6nS66upqv98vKG5yuVzLli0T9NqMjIxkf4QIBGIOgSo48geNRrNkyRKapimK\nUvz1r3Ajft11030eGMUKdu4EO3fCLZNGI0hsS8kGApe8/mcBXGNJZH8gD4XkvFiFUCg0ODjI\naSsVHR2ajz+OW5cxMTEB70HFoSGTjY3qaHS+LQVgGCbr73e73S6Xi6Io5blzi2+7DYA0x5Gk\nHZ/P5/P5AACHDg1xa/533eX4wx9KAABlZSwAabgnq6ioiCRu1OKuq9LSUn+CKpi6nTulXAUj\nEcSLrocPFzzxRE0ggK9ZM9nRYcLxhpwMbPaMjY3BPzhxL9Q8aL2ZhzgcDn5FABc50dzcbLVa\n4bQ5deIWHUwXm80GW1oEQ2pqapqYmIBix/wkHA4LtnAilN/vBwAsX77c6XSWlZVB87L8Q6VS\n8U8CvzoDwzCof+n1er/f73A4IpHImTNnOH/WycnJysrKXI28uLh406ZN/J6jjo6Ojo6OdevW\nHThwgN9rQ1HU5OSkICcYgUAgOFAFR76hUCgKCwuVu3alcfU4Go0msaVMix8Vy7IsyyZ5FYGH\nttVq5dSN5HUZsGkzkTozs7bnPKCoqGjx4sVNTU2LuZsJaXdScGtucTsaNm1Kz+eI47isv9/l\ndJqMxvPnzo386U+xHZf7aJAkWZDEUlTCVTASAcdxkozJ6wwDXnml4tFHFwUC+ObNzoMHjQUF\nYI7OPaLRKJxEgXxsvZm3CBb8+ROtUCgkrkXKNCzLCmpA+EOKWx4yfxDUL4CpEgYMw7hTxLIs\n9/80/+CHvAqqM4qLi+H5IUnSYDCI/Vm/+MUv3nPPPbkYNQCJHVXg4AW9Ntn/f4dAIOYQqIID\ncWVIkkxSkC+z2UBx8SxfAua66V5/PVlO7aJF8LdIJAIX80G8ugxq+XI175n56ozYLkEh4VTU\nLLFtG9i2LdeDuDLidTl+R8Odd04CoI73uGmDYVhxcXExvKSPHYttFak/+NmzkUjE5XIFg0Gi\np6d60yYApF4FIykKCwsdDofPR+zeXXP0qE4mYx9/fHTzZhcAQKfLTY307IlEIuLFef6Fum2b\nKi2lRohsIvjjI5hohcPhLHeFcGnfcYcUDocZhhHP8+cJKpWK0zIgsIShtLQUtsHCTy1drbUS\nRKPRVFRUXLhwQVCdEQqF+Bfq8PCwwI4UMq1WqfQiKG6CfWHcrzl3QkUgEHMIJHAgroxWqyUS\nF+Qzzz8Pfvzj2b9KYWGhKsGr1O7YwS/7Tx7nOdnUxJ/pymQyze9/n0yd4TWjIiSL4H5d0NGg\nUGSmqjap+kOSZDlsOeAuV2lXwUgKvV7f00Pdf3+1ySQnSfaRR8aWLqXPnFETBLF6dVWuRzdD\nxLoM/0J9+eVhmSyOnRBC4hAEwZ8PxzU1yC2CIc1ncBwvKSnhPIy5EgY4t+dOEVdBlpeUl5cv\nW7aMX8sDez34RrZpaZXKKAqFgi9w8Htt4N6cjQyBQEiefP4Tj0gXxcXFSYwJ8DVr0vIq5eXl\n0WkFwSSoy6CWL+cfpdPpEoWG1O3cye7bB370o3QMH5FZNBoNLBRnGHDgQMXBg+UsCzZvdj7x\nhJUkWZgCmDPmSBWMpCAI4oMPak0mHAAQiWD79l0SNdrbwcMP525ks0Aul5MkCSfD4gu1sDCn\nVylipmg0Gr5vJX+iFbOsyi4kSXKXmXhIMplMCppLDqmsrISpN/wSBjhVhom8AID8tm8YHByc\nvZFtzikqKhILVbAARyaTabXaXA4OgUBIGyRwIK4MjuNYT8/gyAj0AeUSZCd379b867+m61VI\nkmTPnjVaLPBGhHuV8FNPyfbu5R+pUCgwDCv6r/+6YpwnAKCkpCSJOoPNqe/7tBMOh0OhkEwm\nk/l82Jo1cUN2JUJxcbHdbne52HgdDboMlog7nYnihxGz5PPP40/DVq3K8kDSQGdn6LXX2BMn\nCLN5qceDlZVFSJI1m+X8C7WsrCzXw0TMhNLSUpfLBefJgolWaWlpetUEXxjm6AAAIABJREFU\nn8/39NNPf/rpp6dOnYIGUp2dnTfccIPgsKKiIi53TOyzkMbxzEUwDFu0aNHKlSv5k3zYjgFP\naXFxMd+zPP+QfnVGKqhUqrKysqGhIUGvjdvtXrx48RxtY0QgENkBCRyIlJDJZAaDIRAIBINB\n2VQdhOamm9L+KnV1deFwmKIo8siR2EbRvR1BEIWFhZoEdRkV99zD72eB6syQ2QzvbDjdJPhP\n/6T6l39J7/jnED6fb2xsLLYsyTCLd+3SxAvZlQ44jkcidVu24IKOBoVCcc01ae5PYRjG4/EE\ng8FIKFR5331yaZ+Zucu77+Z6BOkgFAqZzea2trLDhy+FSY2NyQAAOA64C7WkpESn0/X3s//+\n78zHHwOzGfd4sAULwJ13gr17QV7PtuY8CoVi0aJFvb29oVCIP9FiGCaN82S/32+1Wv/2t7+1\nt7dzGwmC+MIXviA+WK/XBwKB8fFxsc8CNOilKKqtre3kyZNdXV1JhJJ8JVEJA/zUclzxl1Pm\n1oVRWVkp8M0V99ogEAhEHFjElfjlL38JAPD5fLkeCOISkUiEXrqUBSD+vw8+ED8kEAg4nU5v\ne3vsmHfeyf6wJYLb7e7mYdu+nQUgqlTGzszYWK4HGJ/HH4//abe3p/NVAoFAf3//3DoziFwR\nDofh1dLUFNyxw/b66wOHD/c3NQXiXqjPPef/yle84u0/+lGu3wYiBUpKSsR3UBs3bkzLk3u9\n3p6enu7u7l//+te7du36xS9+AaNely1blughDMMUFRWJh7Rhw4bR0dE33niDv5EgiMnJSZZl\naZq2Wq0PPvjg2rVruTaNzs7OtLwLCZLRT232+Hy+Rx555MYbb8zOZ0HT9J+4aLDLLwypEQwG\nH3vsseuuuy7uzOXpp5/O9QARCAQLV0k//vjjXA8kDqiCAzEnIQgC6+uzTUx4vV6aptUDA4tv\nuw2AeE0EUy0GKgBUcO8c7e9PEwzDjI6Ocr9yIbuk3a7q6WEXLMB4IbuS4tSp+NvT2NEQjUZN\nJhNsbhecmWhlJSHVMzOnOXaMeu019uRJ0mIhvF58bhU12O12GLFx6NAQlwurUsUvDq+psZnN\nVTt22G+91VNcHD14sPwPfygBALz3Hti3L2tDRsyERAGWaTE1YFl2dHSUZVn4hKtXr/b7/Xa7\nHQDQ2Njo8/l0Op34UUNDQ3FTMBYvXuxwOHw+365du1asWNHW1maz2ZYuXapSqaxWq9PpPH36\n9M9//nPu+ERFInlARj+1WRIOh0dHRz/++OOXXnqJ25jRz8Lv9w8PD09MTPAvDIPBILVej2g0\nOj4+fvTo0Z/+9KfcRoIgvF7vfC69QSAQ0wIJHIi5Co7jer1er9ezLIudPBnbuno1ACAcDrvd\nboqimEik8r77FKjFgIfP5+MiBsUhu/RVV2U18HA6ZKGjwel0QnVDfGYCTU2qSCS/vfezTDQa\nHRkZ2bOnhN/cYTKB558HDDM35vzQ+BYAwKkbAICvftVz6pQaAHDgANPSggMAKIq6cOECuFwH\nuesuBxQ4yspYlB0rcTJqajA5OSlOooUv19zc7PF44goccYd08eLFYDAIRELJ0qVLLRYLFERC\noRB/iltfXx/XJ3Vu9TLERbJWFJFIZHBwEHpgCeQG2GGUduAfW4ZhBBdGc3PzyMjIkiVLJCJz\nRKPRwcFBmqaDwaDgKs1yGDMCgZjToJt1xJwHwzB+jIXH47FYLAzDAAAq2tsVR44wSiVOUQCk\nJHCwLBuJREivV+KmmzMmecguJWGBIwtAg9u4Zya4fDkbCBQUFFzhKRApYzKZAoGAxVI5d4sa\n+EkWkP5+5csv6wEA11wTuOsuDAAVAMDj8cC9fB3kxIlYCsDGjQEA8jnTAZEc7m8yR+9UoNjy\n5cv5AS7JgSZZgufhCyVwo2CK29TU5PV6C3lx6SzLut3uY8eO7eP9J8zjQo/sMz4+DiUtsdww\nPj5eU1OT9lf0eDzcwga4/MIIhUI+n08iX23j4+PwghdfpXa7vQIVUSIQiNSY11liiPwjGAya\nzWaobnAtBnRDAwCAqaoCSb8d/X7/xYsXe3t7z/X1TX7tayBP6z5iCzVTIbvm556jDQbV1P10\n+Oqrczm4XMMwTKIzE2xu5t8gImaJz+eDqUyHDg21to43NlIVFeG77oqFApaWMjkdXaoIEjQO\nHy645x6Dz0esWTN54IBRLo/tFc9g+TrIt7/tz85oEdJEnMPS19cHty9btiz1lBYK6vg8+EKJ\noJaBP8WFwi4EFhdYLBaHwyFwA0ENAumCq/yC8D8Lr9ebiaoTsfIFf4DJO4K9uYJlWU6Gg/DP\nTNyGLAQCgYgLEjgQecXExAT8OuS3GCgHBgAAk42NSe4bnE6n0WgMBoMsy1Z0dGg7OxmuHjK/\nBA54k1r85pvJQnbnKyRJJjozLI7LbLbcDi+f8Pl88IfERQ1zAM4akGHAK69UPProokAA37zZ\nefCgsagIVygUcG9yHUQmk0RxOCJXiDtEzp49CwAwGAxKpTJu/0hcxF9wfKFEsIs/xeU3yJjN\nZq7JpaWlZeXKlVyTC1w5QMySaDQq0Mr5nwXDMOK6sNkjuDYEF4ZEGnkikUiSMxMOh9EViEAg\nUgS1qCDyCtgqnKjFQEbTcds4Q6GQ1WqFPwusJSMVFVhZGZHFt5Bp1Gq1SqVKFLJb+O1v80N2\n5xs6nQ5PcGbqdu5k9+0DP/pRjoaWb4jLYfhFDXfeOQmANhfjmh5lZWU+n8/rxXfvrjl6VCeT\nsY8/Prp5swsAUF5ezh2mUqlg0iHDgAMHKg4eLGdZsHmz84knrCTJooXxeY5CodBqtVwZxeTk\n5PDwMABg+fLlGIbFzQGJizizli+UwC00TR84cODMmTOfffYZ3BIKhThroWAwyK/mALwl9Kam\nJpfLVVpaOoM3iOAjdrsQyA2hUOjJJ59Mr/uJ4NoQXBicFJtbrnhmEAgEIkVQBQcir5hZi4HH\n40lU9xFsbuaWmvOGhQsXcqclDvlVsTItiouL1X19ifZiEvDezxsI4jLZUFDUoFDMDVFRrVbT\n9MItW+qPHtWRJPvII2NLl9JnzqiNxkq//9JUsKioiCRJn49oba199dVykmTb2ixtbaMkySqV\nSq4MBDFvqampUSgUPp/P5XJ1dXXB76O6ujqVSgU7uVJBq9XyLSr5Qgm38dy5c7/61a+6urq4\nb8MVK1ZwV6D4tfhL6KmPRCJQFLV79+6bb75Zq9ViGIZh2PHjx3M9KIDjuGChhS83EATxv//7\nv/v27Tty5AhUN9LiflJYWMjJB4ILA8fxuC622YckSYGNN//MyOXy1Nu1EAjEPAf9sUDkFTKZ\nLEmLgdxuj/soaGqVqO4jdY+3uYJcLif7+mzj40ODg/19fSN/+lNsx969gGXBLbdM69lYlp2c\nnHQ6nW63m7ZagcEAMAxgGHj22ZQe73RO+yEZA8Mw/OxZ49BQT3d3T3f3hTfegNsDP/nJDM4M\nIglabaxAQ9zcodUy3F7p88c/FphMcgBAJILt21e1dath61bDP/5j2ZtvXjoGx/FwuFasg/T2\nFrDsIonkFyByCEmS9fX1mzZtWr9+fWtrK9y4f//++vr6rVu3pvgkGIZVV1djGCYQSmpraxmG\ngaUZMLaDiybV6XSFhYVFRUXwV3ELAH8JfW41CAQCgcOHD6ddKUgL/EIYgdwQjUa9Xi/f/WTJ\nkiWpF3kFg8HHHnvspptuEmg6CoVCr9eLLwyXy6VWq6WTDlZcXMz9LDgzqZcyIRAIhFT+qCEQ\naUGn0ykTtxgka76YqvsYfukl2mAonpqdBJub8zJVhCAIePMEAACdnbEfpl+h4PP5RkdHYy3c\nDFPX0pJKKK/X63U6nTDHt66lRS0lP1eSJOvq6iiKCgaD5Pvvw43q9etzO6r8o6CgQKVS2Wwh\ncXNHYWHhHEoEPHUq/vZVqy779Xe/U5lMAEzpINz29nbw8MOZGx1izmA0GmEfk4DV0/mzrNVq\nFy9eXF1dzXdk7Ojo6OjoWLdu3YEDB5YtW1ZfX//JJ5/AXTU1NTiOezweqHGIM0r5S+gZSjDN\nBHa73WazTUxM8KNGGxoaUjc0ySjFxcUURRmNxkgkcubMGb7cQJKkIEBk2bJlPp/vikUWNE2P\njY11dna++OKL3Ea+plNeXr5s2TL+NQYvjI0bN7799tvpf5MAgOmHDZeXlwcCgbGxMcGZCYfD\ngqI/BAKBSAISOBB5RXl5eWT6zRdKpTJJ3Yfa5UoevzLn4YXsTgu4wML5k3HmrMlDeUdHR51O\nZ+wh7e3qY8emleObHZRKpVKpZB96iH3oIczlAqtX52VmcG6JROq+8x3GaJSRJLtli+PECe1r\nr5U5HLJAAK+uBnfeCfbuBSJXAcnx7rspHZaiDoKYtxgMhrR4PY6NjcXNm4DL4Bs2bOAHVfT1\n9TU0NHBTXK1Wi+M4V6khWEKXSJLoFfH5fOPj40AUNdrY2DgxMcE3x8khVVVVy5cvF8sN8Of/\n+I//oGmaCxBxOp3JBQ6KogYHBxmGoWlaoOlw1R+Dg4OzV9BSJxqNTkxMdHZ2TitsGMfxurq6\na6+9NstCDAKByDOQwIHIKwiCiPb0DJrNsFtYee5cw+bNAAD6iScU//zPiR5VVFTkT24t2diY\n4YHPSaxWK3dHLjBnjVZWEvFUIY/Hw6kbKT4k+7AsOzEx4Xa7Q6EQYJjFu3ZJqsYkb3jtNcJo\nJAAAkQj22mtl/F0mE3j+ecAwgHdvPLdJUQdBIGaJWChxu91msxkAYDabBTGcEG6KS5KkXq8f\nGxvz+XyCJfRIJJKJdI9MMDExwf+VHzXqcDjKysqk0BSWSG4AUw1Bv/vd7+Cvy5cvv2KMq8Vi\ngbKUWNPxer1QmUqXgpYKNE0PDQ1FIhGPx8MXXFIJGx4aGsqmEINAIPISJHAg8g25XG4wGCiK\noihK9uGHcKNi7dokDyFJUnf+fKK9eWMt2dUFfvMb8PHHYHgYuN1gwYJZLZKHQiEKll1cbs7a\neOutAIBgc3NcEwVO3RA/JNDUpGGYnLuIMQxjMplgMS0AoGL/fmnWmOQBiYoavvxl3wcf6AAA\n772XPwIHApFzampquru7AQC7d+9+5513cBw/efJkcXFxfX09d0xZWRmO4waDgS+FZHMJ3e12\nP/PMM11dXZ9//vnMMkQEcgDfJzUSiYTDYXHcTPYxGAw0TV+8eDEajXZ1df3tb3/jVADYEMR3\nP0kuTNA0LX7LnKbjcrmyXHrDsuzw8DCUwwSCCwwbTv4tn00hBoFA5CtI4EDkJ7DFAHz/++D7\n30/leKK3NxgM2my2QCAg6+2FdR+Rp58m9+zJ8EizBEVRbW3gnXcuWRvMcpGcW81LZM4aV+CA\n92EJc3xDoZybL0xMTHDqhqDGhKmqwqVRY5IfvPsuOH/+fCgUAgD4fIROF4t1GBpSQIFDGrXk\nCMTcRvxHle+sIY4IdbvdyQs9ZkYkEnE6nf/8z/986tSpuOIFTdNms/nkyZP79+/nHjVdZ1CW\nZQVmqIKoUelYpdrtdphlI1ABYENQ8s+Ij9gHna/pwD+w2cTn8wmGxA8b9ng8fCdRBAKByARI\n4EAgYqhUqtraWgAAO2W6SX7xizkdUdpwOBxjY2NDQ/U7dthvvdVTXBw9eLD8D38oAbNYJI8t\nwiQwZ6Wvuuqyo51OaGPRDIDtwQdVvb1x/Vx1OV+3cTqLr71WbzYDABzf+U7xm28KakzmTLbH\nXCAYDHI335y6AQA4cSJ2mjdvzsGoEIg8Q6lUqlQqbpH/iuEUmVhCh66fp0+ffvnll7mNfPEi\nEonApgaY88IvZ5jWC2EYJpPJYr7XAIDLlQK4Nx1vKA0IEuj5ZReCz4iLuYmLuONGoOlkGXFD\nDV9wCQaDSOBAIBCZBsXEIhBCsO3bAcsClgUbNuR6LGkgEAhAs4xDh4ZaW8cbG6mKivBddzng\n3hkvkisUCpIkE5mzKtRqT2/v8PDwwMDA0MWL9De/CaZsLAiPJ+5DAEEkyvHNKH6/32q1Go3G\nEZMp9K1vycxmuB26sfBrTALNzXDBDZEW+JMQjv5+5csv6wEAq1YFt2/P+piyxcmT0V27wtdc\nEy0vZ+VyUFcHfvITkPWlVsR8oaamhiRJcUooQRBZWOG32+3j4+Msy0LxggtAra+v5xwZbDYb\n19TQ0tKycuVKWM7Q3NxstVqn9XKFhYXczwKlQKPRSCeMQ/BtwqkAixYt4n9G4XA4uWlI8gqd\n7AfHiNWxuRs2jEAg5iioggOByHM4x7W4i+Tf+hYLwEwc1zAMKysrIxOYs5bffffYD3/ovfde\nAEBRe7viyBHOxoKcmIj7kNodO5Ll+GYAlmUtFguXOFDR3i7/6CNunDKbTVxjkrWxzQfEndiH\nDxc88URNIICvWTN58OA4SU5v8XZOwDCM1Wp98knd4cOXGuPzz1QVISkUCkV9fb1erxfHx2ba\nWSMSidhsNvizoBcDNixAPSJJOUMwGIxEIiSZ6v1qeXm51+t1OBwCn1Sv1yuRCBUIQRB831ao\nAgAAWlpauI2pfEYymUyn03EnUKDppF4uwbKs3W5/9tlnP/3009OnT0On9ukaoAAAxBYnfMFF\nCgYoCAQi70EVHAhEngNvU/hwi+TXXBPYupWa8TOXlZVpz51LtBfKAZyNBd3QAABgFyxQDQ4m\nfMbsWniOj49zt/uCcQIAxDUmuExGjI1lc4T5jUql4lYmGQa88krFo48uCgTwzZudBw8a9foc\nu7FkCJPJ5HK5LBb5jh3211+/cPjwuTvuiDnvvvdeboeGyGdGRkbixsdmOpzC7/cLlvT54gU3\nLRfUc/GbGsR7k0MQhMFg2Lhx4/r161tbW+HGjo6OtWvXbptRIHqG0Gg0/F+hChCXK35G1dXV\ncrlcXKFDkmSKp25ycvL8+fOHDx9+5ZVXjh8/Dm8bpmuAAtHpdPySE77ggmEYv74GgUAgMgQS\nOBDSJXZX5HQCgwFgGMAw8OyzuR7U3ENwc3n4cME99xh8PmLNmskDB4w4Pqt6UbKvLzA5OWa1\nmozGsT//GW60tbb2dHdPXncdPypFOTAAAKCvuors6xuzWnvPnu3p7r7wxhvwIdG2NsCy4JZb\nZjOYacEwjMMR69MRjxMAUHD4cMPttzfcfnvR1NLZwvvvB7/9bdZGmPcQBAFXF30+orW19tVX\ny0mSbWuztLWNymSgtLQ01wNMP263G3orprdfDDFvoShq9+7dN998s1arxTAMw7Djx4/HPRI6\na4jZk2EjbXG4LF+84Kbfgs4RgYvEdPtKhoeHc6LmTIvy8nJOCOBUgK9//evd3d0ul2tanxFJ\nkvX19WJNp6mpaevWrVccCU3TJpMpHA4LeogMBsMMOkpkMllFRQUAQCy44Dh+xchbBAKBmD2o\nRQUhOcLhsM1mm5ycDIVCBIbVtbSopuwbUEjnDJDJZLDXl2HAgQMVBw+WsyzYvNn5xBNWkmRn\n77imVqtjfdTvvw+3BJcvB4mjUpQ4XllZWVlZGQqF8Ck/V+L662c5jOkSCATgXVfccSYEXYFp\npbKycmCAveeecpNJTpLsI4+MLV1Kd3dr9Hq9Vquorc31+NKN1+uFP8TtF/vHf6QBSBaXgEDw\nCYfDH3300T5eX9PMltwzilib4IsX3F61Ws3vUuE3Nchksun2NcyJqFGlUrlo0aK+vj6aprlW\nmrq6upk1cZhMphlrOna7HQoZgh6i5ubmsbGxhqmqxtQpKysjCGLx4sW5ChtGIBDzHFTBgZAW\nFEVduHDB5XJB57Oyl15SHT3KcB5aeT+9zEC5ylSHs3CRnCRZlUqVzobYbdsAy7pdLt+NN3Lp\nKubnnqMNBtXUkh3fxkIul5MPPJArP9fYwlSCcY788pf9fX38GhN2z54s15jMB3Acf++9apNJ\nDgCIRLB9+6q2bjV85zuLb71VM+V8kleIy8X5/WJbtgi7yRCIRPj9/gsXLoyNjQlsO3OetC1A\n0IgBLhcvuL1xyxlgf0pBQUGKVSpzDp1Ot2HDBn7Zxf79+5ctW5ZK2YWA2VToJDFAoShKXIOT\nCi6XKxNhwwgEApEKqIIDISFYljWbzZy1OGeLQNrtqp4edsECrKIityPMBNFo1OPxUBTFRCL6\ne+6Rp7tcpbS09MyZwPbtVfxF8jNn1BiGrVpVlZaX4ANLQhKlq5AKBbBYQHV12l93ulxxnI06\nXbSyEpvagq1Zk7Ox5jWnTsXfvmpVdseRFQS+qnxT1Y4Ok1ye+/8XiDlBOBweHh5mGEZs22m1\nWqurqymKevrpp0+ePPnpp5/CrqgZuEWmBblcXlhYyM11XS6XyWQCAAwNDa1YsYIbmFqtrq6u\n7u/vD4fDfGdQhmE6OzslXqUyXSYmJvbs2fPpp59+/vnncVs2sqkCsCybKM8FCkzTcnjlmBNF\nNAgEIl9BAgdCQgQCAYqKeV7ybREab70VABBasSL/qrd9Ph+n6QhSPNIlcOA4/v77i0wmDEwt\nknO72tvBsmWXHdzVBX7zG/Dxx2B4GLjdYMECcOedYO9ekHqdh1qtJklSkyBdRf/d72Y5KiUR\nsPI50Tir7rsPvPAC8dhj4HvfA9/7Xo7GOC94991cjyCLqNVqONuM2y/GRWYiEMlxOBx8cwT+\nkrvL5VIqlR9++OELL7zAHZBbUaC6ujoajVqt1kgk8tZbb8GN8FuPP7CioqJNmzY5nU7ugbCp\n4eqrr961a9eKFSva2tpsNltjY+Pc/Z8ClamTJ0++8sor3EaCIDweDyxmYRhmYmLi2WefXbt2\n7eeffz7jKJPUwTCMIAi+xjFLA5SZ4XQ6n3nmma6urtkEuCAQCAQECRwICUFP2R8ksm/IM4GD\npumRkRF4nyooV2GqqvD0lat89ln8IFjBIrnf729rI99551KF8wzSKzEMW7BggWJqCSgO0ugz\nwjCsqqpKnti4XjzOQCAQDAaj0agyENDdcgtmNAIAwN694KmnMjhQRB5RUlLicDg8Hmz37pqj\nR3UyGfv446ObN7sAAIWFhbM3xEHMEwTZWIIld6vV6vf7+aKAwWDIzjQ1LjiOl5aWrly5Utyz\noFKpOLVicHCQr25wfOlLX2ppaeGqVJYsWRKNRnP4dmYMy7JGo5GmaejlyX06ixcv9vl8Go3G\n6/VaLJbPPvtMIH9kWpyCL839yu8hUigUmf67FI1GR0ZGOjs7X375ZW4jQRCwwAeBQCBmABI4\nEBIiVtA4ZYsw/NJLtMFQPNWLT191VS4HlwE4Zy9xuUqgqUnDsvystdlwxUVyhmFGRkZ8Pt/Q\nUMOOHfZbb/UUF0cPHar87W8LAQDvvTcNgQMAUFBQ4Pv0U5PVCo1UlOfONWzeDABg9+zBnn56\nxu8i7RQUFHi6uoZGR+HiFTdO5pln8LY2/pGhUGhkZCRWS8wwdS0tMXUDSEWvQcwJZDJZJFK3\nZQsp6BdTKBSZ6BdD5CuCbAvBkjuI5xZpsVgMBoPgedxuN1w2//zzzzPXycIwzIkTJ+I6MlRX\nV4fDYTiF5jc1RKPR/v5+fo8DV6XS1NTkdDrL52DmkMvlgqs4gk9n+fLlDodDrVaPjIywLCuQ\nP7IgTun1ep/PB0+vwAClIvN9wSaTKRAIiEUft9ut1Woz/eoIBCIvQQIHQkIoFAqQ2BZBplRK\nxL4hXcAbyrjlKoHmZpKms2YXNzo6Cm3GDh0a4vIdtmyxQYFjBneSOp1Oq9WGQiGaphVHj8KN\nErSxKCws1Ol0wWCQpmnVkSNwI37ddfxjGIYxGo1QrAEAVHR0aDs7Z9NJFIlEJicnaZomCEIV\nDKrXrwfQeAUVg8wDfv97tckEQLx+sYcfztmoEHMLmUzGtXOCy5fc+YfxW1cCgQBN0/BLFgAQ\nDofNZvPx48f379/PHZ+hYgGv11tVVdXd3c1t+eSTT7Zv3w4A+OY3v+l2u8VqxeTkpMDBgV+l\n4vf756LA4ff7+b/yPx2WZcfGxuCvYnHKZrMtXLgwcwNLlOeiUCiSx8RyDTWnTp2aWUONz+eD\njxKLPm63u6ysTGqmuQgEYk6ABA6EhNBoNHK5PJEtQsl3viMR+4Z0EY1GE5WrBJubNZf7fmUO\nmqa5eLm46ZWbN8/kaTEMUygUCoUCtLSAlpZZDzNT4Diu0Wg0Gg148EHw4IPiAxwOB6duCDqJ\nIhUVhF4/rTIbh8MxPj4eu2tkmLqWFoBSkOcT88pUFZEhCgoKuOQLwZI7H0HrCidwRKPRoaGh\nUCgkLhaIZuB7hxZlb/MHJt4Lphw6+PCrVDIxyCwgiCMRfDrctwy3l5M/BCknmQDmufBbhPbv\n379///4kqa4+n89isZw6dWo2DTVJAlwAAH6/HwkcCARiBqCYWISEwDCsurpaJXn7hnRBkmSi\nchUWx2U2W3aGIWjnhnDplatX09u3Z2cgEoVbduN3EikHBgAAwebmuGcvES6Xy2q1cmtiXDFI\nbHd+Xd6IuLz7biwZWfBv3bpcjwwxdygqKlKr1T6fz+VydXV1cZkjLpeLP2MUt65AONF29erV\nLS0tK1eu5IoFrFZrFsafaGAc4tgOfpXKDEI9pIBg2MlPAl/+YBgmeSXF7ElkgJIozyUYDA4P\nD0ciEaiRcUHFBoNBkBWVnCsGuKT+VAgEAsExJ78kEHmMRqOhzpwxjo3BClXOFiEvq/d1Op0q\nQblK3c6dWStXEa+G8dMrDx2aIMnaLAxDssB7rITGtynfgcEiZO7XjNrKIhCIPAbDsNra2muv\nvZYrvgNTmSPr1q07cOAA3CJoXeEWw/mOkuDyZfNYX6EinY7e4kV4/sDiLtFrNBocx7lZvaBK\nRafTpXF4WUOr1Sby8sQwTNCSw5c/cByflmowA6ab6pqkoWZ8fLy2NtV7BoG9iED0maNKFgKB\nyDmoggMhOZRKZV1dXXNz89KlS+u5u7csxsJnjfLycimUq/DvIRgGvPJKxaOPLgoE8M2bnQcP\nGouL555ZfXohCILrJDI/9xxtMHCfWrC5OXX7t0AgwGlJ4mIQSlRioCWIAAAgAElEQVRejkAg\nEIkwmUx8dYODa1QRiAKwAxTuCofD/IcIls0Fe2ePTqfjf8vwB4bjeGFhofghOI7DigBxlUog\nEEiX/XaWKS4u5pQjwadTWloqEHr48gdMkJUODMOIc3w4jczv96eulQhsRAWSHDIZRSAQMwMJ\nHAiJgmGYXC7Htm+PFXBv2JDrEaUfkiRBd/eFgYGe7u6e7u4Lb7wBt1OPPw5YFtxyS3aGodVq\n4f2iz0e0tta++mo5SbJtbZa2tlGSZOfoWlka0Wg0iTqJAEGoXa4Un4ertk1UDJL+oSPSR1cX\nePhhsHo10OuBXA7q6sBPfgIu75pHILIHXHIX09bWJhYF/H4/f5KcfNk87cUCOI7X1NRgGCYY\nGLSxhGbbYsrKyvR6/YYNG9avX9/a2go3dnR0XH/99XfffXd6R5gdYN1NKBQSfDoAAIVCAQUd\nCF/+wDBMr9dTFLV79+6bb74Zfl9jGHb8+PFcvZFIJJLEApZlWX5DDcMwNpvt+9///tq1azUa\njWDwBQUFXE6wQPQpLCxEBhwIBGJmoOovBCKXKBSKhoaGyclJiqLk778PNypvvDGbYyBJsry8\nvKvLvWtXnTi9UqeLu8A2jygtLZ08dQrE6ySq3bEj9U6i2KQiga1saMWKNI8bkVaefTby1luX\nvjFNJvD884BhppegjEBkmsrKyubmZhdPeIWtK3y3SI1Gw7e05C+b4ziuUqnSPiqtVltfX19V\nVcWvOrmijaXf748bLpvIGEL6yOXyr3zlK3y3C/6ns2DBgvPnz4fDYX6UiVqt9nq9n3322T7e\n35oM5d2kiLhzhK+RYRjGaWRer9disXz22WdJjEgXLVp09uxZr9fLveva2tpIJCK1uhUEAjGH\nQAIHApF7YikeDz+cq6BIvV7/3nsak0kOUHqlCJIkdefPJ9ydcieRWq3Gcbzwj3+MWwyi1Gjy\nLAU5bwiHw8PDwwMD1Tt2uG691VNcHD14sPwPfygBALz3HhI4ENJicHDQFa+sjC8KlJWVud1u\nOJkULJuXlZVlqAFkdHQ0bk9NErViusYQ0ie5l2dJScmmTZvEUSbr1q3btm0bP+9m6dKlXOFD\n9oEqWDAY5LbwNTK1Wg0vIb/fPzIywrKsOKyHL5GQJPnlL385keiTxbeFQCDyByRwIOYjDMP4\nfD6KogAAykCg8P/8n1hUZz5amabIuXPxV0tQeiUAAD97lmEYj8dDURTe3V3x938PwLSvFhzH\ny8rK5AlsZYu3bMmzFOT8gGVZo9FI0/ShQ0NciPJddzmgwFFentPBIRAiUhEFFArFwoUL+/r6\nQqEQf9mcZVmZTJbDgeU9yU9CIvlj+fLlAi/PpUuXBoPB5LU2FEW1tbWdPHmyq6sL9gF1dnbe\ncMMNs34TAACg1+tNJhP8WdxQA7cnMSK12Ww1NTXJ3/XcrdNBIBA5BwkciMzgdILVq6WpGvj9\nfrPZHDNEYJi6lpbYOMG8zul8991cj0Da4DheXFwMAO9MTf/2S6/XR86dS7h7Hl9+ksXtdtM0\nDQDg1A0AwIkTMeu7b3wjgr5GEXORgoKCjRs3omVzSSGWP86dO8f3fOV7edrt9kWLFiV6Koqi\nPvzww8x1teh0uurqajg8fkONUqmEA45EInANKe7gufB1gJQvBAKRAZDJKCI9sCzrdrvNZvPF\nixdNQ0Ohb31LmqoBRVEmk4mze6zo6NB2djKckZWUhponOJ3AYAAYBjAMPPtsrkeTDrZtm43x\nLdnXRwWDdpvNPDIywRVx7N2bTVtZROrwb8Qh/f3Kl1/WAwCuuSZwxx2+XAwKgZgtaNlc+oRC\noSR5N4nMWQEA4+PjFy5cGB8f37Vr1y9+8QtYUpH2rpbi4uJNmzbxLWD379/f0NCwdetWwDPV\njjt4sU3pzJCU9yoCgZAOaOkJkQai0ajJZOJiwyra2+UffcQolTjU76WkGthsNu5rteAvfyk/\ndMh3yy2k3a7q6YlUVJAVFbkdXn5A07TD4QgGg9FweOEDD6gkKXXlEKVSGTOHf++92CY0qZAq\nXLIv5PDhgieeqAkE8DVrJjs6TACgHhXEnAQtm0sfwR8fcLmXp3gvxOVywU4QcVdLIBBIo8aR\nXCMTB6jzB08QxCytXliWdTqdR44ckY73KgKBkA6oggORBkZHRzl1g1MN6IYGAACorgZpVw1m\nURTArccqBgdrnnySrq01792rHBgAAASbmxPdMSBSx+PxXLhwwel0BoPB4p/+VHX0aJYLZMLh\nsNvtHh8fdzgcQYtF0vUjsysGQWQBzgyPYcArr1Q8+uiiQADfvNl58KBRq2XEaQIIBAKRFsR+\nKHwvz0R/fKCcwSHoaknj8BIFFe/ZswcOXi6XJxr8LBNSWJY1mUxWq9Xr9fKrVBoaGnLovYpA\nIKQDEjgQsyUUCnFBbmLVgL7qqtm/BMMw4+PjFy9e7O3tPd/fT91228z6XxiGgfHsuN+/6KGH\nAADDHR0ymw2jaQBAcPlycVElhGXZYDA4OTkZsdkkPWHODokFplAoZDab4e2UQOqKVlamX+oS\nYbPZzp8/bzab7Xa71WKJ3nGHNFulEHMFrVYLAPD5iNbW2ldfLSdJtq3N0tY2SpIshmFwLwKB\nQKQdkiT5NqKCvJuCggLxQ8LhMD8AGKTc1ZIJKnjf+ImMSGeGzWaDi1WrV69uaWlZuXIl1G4a\nGxvjRgghEIj5BhI4ELOFq92IqxoEmptn+fzhcPjChQt2uz0YDDIMU/zTnyr/+teZFQXgOI5h\nGGDZhY8/rjCZzM89RxsMqqmv/2Bzs3hJhGEYq9Xa29t78eLFoYsXZ6ytXBlpe1WEw2Gr1To4\nONjf2xv8xjcSnQSHwwHVDbHUFWhqEtx4pR273c5vQUIGK4jZU1hYOD6u27Kl/uhRHUmyjzwy\ntnQpfeaM+swZdSBQnrnICQQiyyA7AwlSWVmJYZjP53O5XF1dXVzejdfrjfvHJ3lXC8Mw2exL\nKiwsrKys9Pv9/MEvXrxYrVYL/EenBWxO4W/hV6k4HI5ZDhuBQOQBSOBAzBZYE5FINaCWL5/l\n81ssFm5iLCgKYBcs4BcFsCxL03QoFEoiFmi12uI339R9+KH31ltDNTXK/n7NiRNwl1ylIsbG\n+AfDMkhu0p72CbPH4zEajefOnUuuGuScycnJCxcuOByOQCBQ+rOfJek6CQaDIHGBDNybIaLR\nKL/+VnCpMFVVV64fkbbGhMgJGIYdPrzQZJIDACIRbN++qq1bDfDfxx8jAw5EPhCNRm022zvv\nvLNv374jR47AdX5kZyAFNBrNokWLNmzYwPfy7OjoWLt27b333is+XrxII+hqmYHzRSAQ+OEP\nf3jTTTelonwJZLLy8vK///u/5w++vb3dYDBAI9LU8Xg8Dz/88Nq1azUaDY7jzc3Np0+f5vby\nq1RomkbmMggEAvUPI2YLXEZIpBqQCgWwWEB19cyePBQKxXXNaLz1VgAAfdVVcJ4diURgNybL\nsjD5VZtALNDr9fSnnwIACg4fLuAyLAAAAFTddx944QXw2GPcFpfLxZV0pt2R1Gw2u91u+HNF\neztUDSRoyxqNRoeHh+G6kPgkEHo9/3aJZVmWYaDUNfzSS7TBUPzmm3BXsLlZBrWwzBAIBJip\n5497qajiPYqiKJfLRVFUNByuuf9+pVQ1JkSm6eoCv/kN+PhjMDwM3G6wYAG4806wdy+Qy8Hn\nn8dfCbj22lmZ5CEQuYVhGJfL9cwzz5w8ebKnpwcK0D/60Y/+/d//3WazpT10AzEz7HY71wXM\nJ27eDUmSarWaq6sVdLXodDq4naKotra2kydPdnV1wZuczs7OG264QfBsNE2Pjo4eP378Zz/7\nGbcxifLFMMyxY8cErp9erzfFwcclGo1aLJZjx451dHRwG2FBCvcrv0oFqRsIBAIggQMxezQa\nDUEQmgSqQfnddwtUg2nBrfkn6n9RAhAOhwcHB7k0Na7OIq5YoFKpFAMDCV/v8oPjeovACXOw\nuVlO0wqFYmbvy+l0cuqGQDVgqqpwKYW5eDweqG7EPQnA7+fumQAAMpms5L//O67UxeK4YmIC\nFBdnaJzcBZCofkQscLhcrtHR0Vh5Tns7bH2SoMaEyDQsy+7dG/nTny6VfJtM4PnnAcOAffvA\nu+/mcGgIREbweDxWq/XUqVOvvPIKtxHH8Y0bN/70pz8FACxdujQUCgl8IhHZZ7p5N5WVlUND\nQ/CL+8yZM1xXi8/ng6YYwWDwL3/5yxXDRyiKGhwcZBgmFArt2rVrxYoVbW1tNputvr6e7wzC\nEQwGh4eHzWYz/+CGhoa+vr7ZBKYMDw9PTk4KxgALUrhj+FUqcrl8lvksCAQiD0AtKojZguN4\nVVWV8uzZhEfMYq4Y+15P2v8yNjbGTW5TaUzAz56NRiIup3PMarX/5S+xF/rJT8DixeDLX+a3\nJ8DWmEQT5tk4SnBtonG9Kmb8tJkArgWl2HVSVFTESV0Nt9/ecPvtRW+/DXfV7dypmqrmyASx\n0twEl0r46qsFxweDQU7dyIkfKkIihMPhixcvXrgQ3bHD/vrrFw4fPnfHHbEGby7GF4HIJzwe\nz8jISCQSgfNGLoTCYDAMDg5ydgZxQ0AREketVtfW1m7cuFHQ1fKlL33p3nvvdbvdMN5V8LmL\nU10tFgssihQYeTY1NXHLMxyRSMRoNIbDYbHr5/j4+Izfi9frhQUmgqddzut9FlSpFBYWzvjl\nEAhE3oAEDkQaKCoqov72t/6+vp7u7p7u7gtvvAG3s3v2AJYFt9wy42eGJRJJ+l+YkRGuADJu\n8mvcpyUIQqPR0DQdmmolpf77v8UWGMkdSXF8hv99GIahaRokLksRhLnQNO31ej0eD221Zt8k\ngmXZJCdBsLJUUFCg6e9P9FRYylWpM0CtVmMYluhSUWo0wGLhHz8xMZHED5XJZDcNQjpAnx2K\nog4dGmptHW9spCoqwnfdFdMfy5HJBiLvYFnWarXCn8XzRr6dwWycIBE5xGazxe1queaaaywW\nC8uygs+9ubl5ZGSE/21O07Rg9YJv5CkWOBwOB9/fVOD6KbY+TRGfz5doDHCvwHuVoihk/IxA\nIABqUUGki6KiosLCQoqiaJpWHT0KN2Jr1szyaVUqlUKhSNT/ov/udyP/9/+y//APIIFYMNnU\nFDdsPRQKXbx4MRqNgttuc912W0V7e/mhQ+L2BJVKpfn97+NOmAFBKB0OMKMsd0FZitirgrvP\noGnaYrHE+mkZpq6lRZF1kwi5XJ5INWBxXOlwCIodyL6+sfFxp9PJMIzy3LmGzZsBAKEnn5Rn\nWJEhSbK0tFSZ4FIp/Pa3Ba1SyStTCIpC/efzAa/XC2dxOt2lW/ATJ2Lhr1/7WggAVKKPyCuC\nwaBAQ+fPG6F3Ixe6kZshImZHoq6W8fFxvhU3/3OnKCoQCGimbmnEBap85Uu8VxBAyz+YZdlA\nIMBvZU0d8YXKPS0AYMOGDXwdp6Ojo6OjY+PGjW9P1Y0iEIh5CxI4EGkDwzCVSqVSqUBLC2hp\nSdfTVldXE1PfamLYVasASCYWxH2U1WrllhSSWGCUlpaGEkyYa3fsmLG3CEEQJEnqXn89rmqA\nkSQ5Pg5qasLh8NDQEPcFn9xbJHMUFhZSCU5C3c6dzPPPg8tPMoZhlZWVFRUVFEXhU1KX/Etf\nysJQKyoqIonrRwRnjGGYJJeNGt3Zzw8E9+UAgP5+5csv6wEA11wT2Lx5EgBUxYHIK7iOTg7+\nvPE///M/wZSdwYxNphDSRFCSI9ALKIriBA6xjQXfyFO8V1CjwT9YvDd1BI0z/Kc1m82pe68i\nEIj5xhxoUfnTn/4Eg6mefPJJ8d6LFy9u3bq1srJSqVQuWbLkySef5Oyj5w+RSMTn87ndbmp0\nlGth8O/e7XA4ghbLXE++VKvV7JkzQ4ODcftfyK98BcfxJMmvgsYEAEA0GuXqHpN3tajVat35\n8wlHNguJobCwMJFXxaIHHsB+9zsAgM1m49SNHJpEKJVK7blzifbiCYp0oNqlaGkBLAtYFmzY\nkLEBXvaisv7+YCBgt9nMIyMTnByzd6+4VSp5ZYpiYiILA0bkHMGd9+HDBffcY/D5iDVrJg8c\nMGLYDO/LEQjJInZb4OaNNTU1yM4gjxGUdQhkCP5epVIpUDH4Rp5ik1FBYwj/YBAvvDZFNJcX\nyfKftqamJhgMsiL27Nkzs9dCIBD5hNQrOOx2+/3336/VarmsUD49PT3r1q3zeDz/8A//YDAY\njh49+txzz73//vsffPBBXJPn/INhmNHR0Vg/JMPUtbRwRhKOujqfxcLfMneDIZRK5eLFi6Gb\nt2zKNQP2v2AYVlxcrEo5+RXwai9T6WohenuDwaDD4aAoijx7tu4b3wAAsHv2YE8/PZt3pNfr\no319CXdfey0AIK63CIwvCTQ1aRhmxiYg04Xs63M4HDabLRqNcl0n1OOPK597LjsDmBaxMiLA\ns4jklnScTrB6NfwfUbl7d7i/HySoTJlN9A9iDsHdeTMMOHCg4uDBcpYFmzc7n3jCSpLsjO/L\nEQjJolKpMAzjz2bhvLG2tranp4ezM2BZVtAggJjrKBQK/r20QIbgF+yQJFlYWMh5bQiMPEtK\nSgTPzL9LFxxMEMSM+z2Liorsdju8ZxM8bUFBAT9IBYFAIPhIvYLjgQcewHH8kUceibt3+/bt\nbrf7V7/61VtvvdXe3v63v/1ty5YtJ06cePHFF7M8zpzAsqzRaOS+gbgWBvhrsLlZsGXuChwQ\nHMeVSiVx//2CooCKigr1lcQCPrF1iQTGmfRVVwmOV6lUNTU1DQ0NdVPRJ7P3FiEIgujtNY+M\nnO3p4ZelMM88A2sNotEoXFtOZBKR5VvP0tLSxsbGJUuWLLTZ4BbljTdmcwAzYds2wLLRSGRi\nzZqRkZHBCxfob36T0/vU69ZN67JB5CWwM9znI1pba199tZwk2bY2S1vbKEmy3F4EIp8gCII/\nQeXmjUNDQ/zQjauvvnrr1q25GSIiMxQVFXE/C/QCkiQF5RJVVVVKpVJs5CmTycRdTiUlJQqF\nQnywy+VSKpWCxRiKonbv3n3zzTdrtVpYoH18auFKAIZhdXV1NE0LnjYcDmu12tmfEAQCka9I\nWuD49a9//eabb/7bv/2bWC0GAJw6deqTTz5ZuXLlvffeC7fgOP7CCy/gOP7qq69OKzZ8juLx\neLh+HEELQ1ivV586dcXAVGkSDAadTqfdbveZTFfsr4EftKyzM7poEdzi2rIlti9eYwIAQKFQ\nJOlqkSmV4q6WGNu2pbHhgiCImpqa5ubmhoaG2qmGCPy662I/wBuCDGS4zBgMwxQKRZa7TmZJ\nIBAYGBgYGxvzeDy6f/1XxZEjnN6HrV5N9PaODA8LWp9CTz45y+gfxBxCq9U6HMVbttQfPaoj\nSfaRR8aWLqXPnFGfOaMOBMqRBwEiL6msrCwoKBBMR8WsXr069bkoQiIEg8HHHnvspptuEn9k\nKpWqrKxMLEO43W6tViu4qSAIor6+ftOmTYK42cbGRrHyheN4XV2dOJuW/ysAgGVZj8fz/vvv\n79u378iRI9ACiSCIL3zhC4nejlwu/7u/+zvB065ateruu++ezVlCIBD5jXTrb41G48MPP3zf\nffdt2rSpvb1dfMAHH3wAANhw+Syrurr66quv/vzzz8+fPw+7CvMYzmBJ3MIQqqsTbKGvukr6\nTTvhcHhkZIQfGpKov4ZlWYfD4XK5QqEQG40aHnxQPTwMdxXr9bGDEnhNJe9qqbz33my2J2AY\nplQqwY4dYMcOwXa1Wq34f/8vUT4uOT4OqquzM8g5SjQaNZlMsBBGYCUbrawkKioIABYuXFhV\nVUVRFHHkCHxUdvxQEdLhf/93gcmEAQAiEWzfvipu+0svsVN6IwKRV2AYtmjRopUrV7pcLsEu\nfggFwzAffPDBvn37uL3J56KI3ELT9NjYWGdnJ7+KWfCRVVZWNjc38z/3JOEjQ0ND4isEJDDy\nHBkZEcfH8g+ORCImkykYDNrt9l27dq1YsaKtrc1mszU2NibpYRkcHHQ6nSmOAYFAICASFTgY\nhrnnnnuKiopeeumlRMecO3cOACBWMZYuXTpPBA5YJRi3hUFx/rxgCyV5gYNhGKPRSNM0/DVJ\naAjLssPDw5xRaEVHh/rYsUtH/tM/gXiKGJ+KioqI5NsTysrKmAQqTPX27cgk4oo4nU6obsQ1\nMZHTNFyfJ0lSq9WCBx8EDz6Y4xEjcsFnnwkTASDXXht/OwKRBwwODiafuwYCgeHhYbPZzJ+L\nLlmyBOVnS5NgMDg0NMQwDE3T/I+soaGB/5Fd8XPnkyhuNi5XPHhkZCQYDMLXWr16td/vh5m1\nS5cuZVlWnMwygzEgEAgERKICx4svvvjXv/71vffeS2LiDesXxAfAJsO4QrKY0dHR22+/nZtU\nxwX+CZbgX1iCIBLlXJJut2BL+OqrczfSlHA6ndwHIVhvj1RU4OXlOO9ITt2IuzJ/xdfCcVzW\n3+90uTweD03TqvPna7/+dQAA2LsXPPVUBt7cTCgoKIgkji+RiAojZWApUEITk0AANSAgAADv\nvpvrESAQWSf5vDEcDhuNRoZhBHPRZcuWORyO0tLSLI4UkRIWi4VhGCCSDxobG30+H2cnlCu9\nYHJyUpDJ3dvbC0fS2Njo9XpRZA8CgUgjuRQ4GIZ56KGH+FseffRRg8HQ3d391FNP7dy58ytf\n+coMnhb+xUwkBgsoLi7evHkzF6sRl5MnTw4PD6f4hNlErVbL/+M/4rYwBK65hlGr+VuUGg2w\nWKTc1BA3uhWutwebm7FAgPOU4tYf4q7MaxMvBfDBMKykpCRm79LZGdsqsaJHsq8vGAy63W6a\npmW9vdWbNgEgLRVGyjAMk0gBDDY341GUAIpAIBBxsNvtcLYM4eaizc3NNputpKREgndE8xmK\noihYwToF/yNzu90590sWqBsAgN4pT7Hu7u4NGzacOXMGHtPZ2XnDDTdke3wIBCK/yLHA8fOf\n/5y/5c4771y8ePHdd9+9YMGCF154IfnDodzL+VBwJKrsiItKpUoU0cLx6quv/td//Vcqz5Zl\nSkpKJhO0MKg/+6zuck+HgjvukHhTA4wFSbTeLueFhsBCj4RHhkLTXpnftg1s25a2d5JWLoWe\nfvBBbJPEVBjJIpPJElnJsjiumJgAZWW5HSECgUBIEPFiO/xh+fLl0WiUoqjYtxJCGojLkPkf\nWfIi5ezA18sgfX19AAAcx//nf/6H24hMXhAIRFrIZYoKSZLs5dx4443RaPT06dNDQ0M6nQ6b\nAmoQzz33HIZh3/ve9+DDocXGOVEN/8DAAABg6dKl2X03OUAmkxUMDEzjATNoanA6r5hjki74\nHTfi0BCCIODPserKxEdmdJAZgqZpj8fj9/vD4+MJT3haM1wkQYavroKCAs2UAthw++0Nt99e\nNGWiVrdzp+att9L+iggEApEHRC8vcOPmovC+K8sh5YgZwH1kErkZJknheurZs2cBAFVVVbt2\n7frFL36h1+sBAMjkBYFApAXJxcTiOL5dxBe/+EUAwMqVK7dv375u3Tp45Je//GUAwJ///Gf+\nw0dHR0+fPl1dXS2Rv+mZBj97NhIO28bHTUajeUoFDz/1lN1mM4+MTHBlHQkCU+Pi8/lGRkYG\nBgbO9/dTt92WKMck7Wg0mkTr7YAg1FNtKTCsNNGRGEnKbDbBMzMMk02lZlrAHNOBgYGRkRHj\n4CD9zW9m7YTnBJZl3W736OioaWgo02+2oKBAndhKFl+zJu2viMgOXV3g4YfBtdeyej2Qy0Fd\nHfj/7N15eBv1nTDw7xy6Jd+WbMeOHee0k0ABB8ouZKHts9umB21xKBQKJRRykHK0dKHhCKRn\n0meLMX3Sp7ybfd8uu2Vb2MK+XZa3kKbQkCyUlHYTx0cOW7Il27plnaNjZt4/fvJEkTSKZMmH\n7O/n4Q95Rhr9ZDlI853v8dhjkLPQECFUgLTTUXIu2t7erlarM/eieaeeHn8ukd6yC0mgpUBm\n0F5//fWFjg1Oq5EJhUKjo6MA0NXVtXPnzo985COpTV5mvDycaowQIhbcpxRN0//4j/+YtrGn\np+e999779Kc//d3vflfaeOWVV1599dV//OMf//mf//nOO+8EAEEQ/v7v/14QhB07diydAlGW\nZUnkG373O7JFce219fX1AABvvpm8U95FDRMTE9Kni6mnR/2HP2SdYzIbamtrQx9+CNkqblq3\nb0+tr6mqqmJlanOW33efdM9IJOJwOMLhMB+Pr9i1S7fwAgeRSIQ0ciM/5hgcszjE43GLxUJK\nhU09Pap33pntF8sODNgmJnw+nyiK6qGhVd3dABD59rc13//+bDwdmgPBYPCJJ6jf/lYHkPyf\nvMUC+/eDIEDKOMsCnDgBL74Ix47B6Cj4fNDUBLfeCvv2gVJZymUjVEb0er3U00E6F12/fj0A\nsCybeTqN5pdKpdLpdFJhUdpbVl1dXfxTiKLocDh+//vf55hBCwAcx+3du/f9998/ceJEak8N\nlUpVW1vrdrsDgUAikTh58iTJxm1tbfV6vf/zP/8jdQxxOp0z6GIriqLb7f7DH/6AU40RQrAA\nMzgKcujQocrKyrvvvvvzn//8ww8/fPXVV//rv/7rNddc881vfnO+lzYfMksYCixq8Pl8UnRD\nmk4SXbUKAITGRshjOkkxWJY1nDkjuzvlBLi2tjbHlXlyT7/fPzw8HAgEeJ439fbqjh0TpO9k\nBZ5L8zwfDAbdbrff789VQlK48fFxKbox97/wOSaKohTdSHuxfEPDLL1YmqaXLVu2du3a1tbW\npokJslEznQWGyo7H4zGbzaOjzPbtzpdfPnf48NAtt3jILimcW6jvfCfR2wt/+hM4nRCPJ8Ml\nTzxRsjUjVHbq6upYlg0EAl6v98SJE6nnohqNZulcQCojzc3NCoUi8y1TKBS5++jnaWxszOl0\nchyXWlGSNpMlFov97ne/O3DgwDvvvEOiG6khhoaGhrq6ui7jb8QAACAASURBVE996lObN2/e\nvXs32djb27t58+Yf//jH5Mf169cnEolCF0y+XUxOTvr9/tTlrVq1Knf2CmZ8ILRYlXeAY8OG\nDX/605++9KUvHT9+/ODBg16vd8+ePb/73e+w/dXMuFwuciN1Oon67FkACHd0zMEC6NOnBZ73\nejzjNtukVHyUUV9DUZRicNDpcJwZGuo7dercK6+Q7eIzz5B7JhIJq9VKPnfTzqUTJpNA0lvI\nQ0QxEAi4XC6n0xkcHc0MXjidzqGhIbPZPDExMWo2l7CqIh6Pk5nwkO0XXqbNRHIIBAIkupH1\nryuzxXoJsSxrMBi0u3cvtiYmS0wsFpuYmACAQ4dGdu+2r1vHuVxsJJL8FDt5suBalUQiMTw8\nfPZsooThEoQWAZZl29rasp6L7tq1a37XhrJSKBSrVq3asmVL2lu2bt2622+/vciD+/1+v98P\nGRUlnZ2dNpuN3CcUCp07d87pdKZFQKRwGEVR4XA4czIATBc9SU1e+AJnnDmdzmAwmLm8devW\nWa3Whx9++LrrrssMYQSDwbfeeksuHIMQKmsLrkQlq4ceeuihhx7KumvlypW/+MUv5ng9i5Io\niuT8M+t0knBnpyoeVygUs70Mmqarq6urq6tBCnBkq6+hKKq+vr6+vl4QBJAacEx3VZiamiKZ\nEVknzvJ+f1VVFQCEw+GxsbF4PA4AIAhtO3emBS+cTqfdbpeetLQlJMnnlf+F64o5+sJDvkDI\nzb6BUEinK/Ur9nigqyv5nuJs3fJHSo0AwGBIfv39x3+sP3y4gtwWxcJqVchFv0gkcujQiHTA\nO+5w/+pXNQCQEgVFaCkaHx/Pei7ahWO8FiqLxeLz+TK3F/+WpR02dQZtLBYLh8MqlWpsbEwQ\nhK6urq6urmAwKEVARkdHV69eTdM0pGR8DA0NSV+BAOAzn/kMpDR5KfSrpsfjkVve0aNHe3p6\npF1SCIOUY7vd7l27dm3cuHHv3r0OhwNbnCK0aJR3BgcqoUtOJ0lNRJwL+dXX0DRNf+1raffM\nEamJrF9P9kajUbPZLH3ESsGL5HGvuiqRSDhS+pWmZYKITU1FVlWQz3u5X3h0w4bMh4iiGAqF\nyKdy2GpdmG1T5fA8n+Ovq9ArNnI4jnM6nVardXJ8PLF16+Lu2LrUZA47PH9epVSKALBhQ2Tr\n1mQf4jyTLwKBAEmhkqIbAPDee3py46ab4tkfhtDSQM5FMz3zzDPzvTSUXaneMo7jHnnkkc2b\nN0tZD++//37qHVJn0AJALBabmppKna2TGmKIx+OBQCDtKSoqKqTbaR1DtFptQV1sE4lE2lif\n1OXFYrHUjJIVK1YEg0Gv10vKsTNbnGaNECGEyk55ZHCgOUDTNMuyhpdflptjwtrt0NIyr2ss\nxPS59Oizz0bb26tfe41sjnR2qgAAwG63Z/a/YJ1OTV8f39DAmEwBr1eK6WRmgkQ3bCiyzZpK\npWIYpuKVV7L+wpUaDdhssGyZdP9wOGy1WpO1qdnyTWZVPB5nWZbyemecE8GyrNzsG5GmVS4X\nNDYWs0JRFCcmJqQrOaaeHvbIEVGjoUgdEAY4Fp3DhysmJxWxGLVpU6i31+J0Kl5+uRryTr4g\nKc2pBgfVzz9vBIArrgjffHMIALM4EEJLCM/zdrv9D3/4Q1on0Y6Li5TTxgbTNB0Oh1PvkBYB\n4TiusrIy9Q719fV+v9/j8aQ1HJ2amqqrqyvyVaQuT61Wp2aUrF+/3ul0pmWIpIZjXC4XyfBF\nCJU1DHCgCyorKzUy00nS5pgscDnmyIo0rfF4oKFBOr3JDF6EOzp0giBdE8iaCcIVHeCgKKq2\ntlYp8ws33nln6i+c47h5mbcSiUTsdns4HBYEgRLF9vvv18w0qlJRURGTebFtO3YkfvAD2Lix\nmKU6HA4pupEWsYJlyxZZx9alSa1Wk5x5QYCDB00vvFAvitDd7Xn88QmWFX/zm2TyRXd3XkdL\nSxo6fLji8cebw2GahEsoqqbUy0cIoYVLEISRkRGO40gnUalwo729vaqqKjW1IW1ssFqtTsvR\nSIuASF9dJCzLrlix4qMf/WjqYXt7e3t7e7ds2fL666/nv2yGYRiGSf3/edry4OIQhiiKaU1M\nU8MxHMcJgpBMsEUIlS0McKALjEZjYvp/9FmUzzXwysrKkPy5tLB/P792LfnElStjUSUSaSUk\naZkg8csuK36d9fX1icFB2d0pv/BL5psUv5hMgUBgdHRUSmMxPvec5ujRGUdVtFqtQv7Fstdc\nU8RKgef5rC1yScQqtnFjOU78JJm3qlCI2rQJO4kAQFVVlcPh8PvpRx9tPnrUoFCIe/aMd3d7\n4eLki3vuuXQR9YkT8NOfVr/3Xt3EhCIQYDQaIRBgAC6ESwrKkUYIoXJHhqQAQGYfDUEQKIoi\nXwbSKkoMBoNSqVRePFU7LcSgUqmkXdIc2Q8++CAt74MotGMIRVFVVVXSBMC05RFpGSVpMsMx\nGOBAqNzhdzh0AcMwYn//2MQEuUyqHhpa1d0NAPzevczTT8/z4gqhUChyTJylN20ChqEoShQE\nuTKWWoYhPS/lMkHUOl1aCckMkHEwgUDA7/dzHKceGlr26U8DpJ/HiqKYO99EL4oln9snCILN\nZpOiG2lRFbGpiSo8qsL099smJrxeL6T8dQW+9S39/v1Frj8UCpGlZo1YhTo6LhHgWGDtSN1u\nt8vlisfjpBZJj51EAABAoVBwXPNtt2ksFiXLig8/PLlmTfTkSe0HH+h+9rP6SCSZfMEwHQCy\nf07hcNjhcHz72zVSd1IAINGNv/qr4N6942SLXq+f7ZeDEEILB/lolqRmPQSDwcbGxjNnzsTj\n8dSKknA43NDQAACVlZUOhyNrBISmaanjhiAI77777oGULtAMw/j9/iJbexqNxlAo5HQ60wpe\nvF4vmaGWFsKQgjVEajiGFGsXsxiE0EKA/4zRRViWbWlpWbZsGcdxzNGjZCNT3NX1ecH09weD\nQYfDEYlEVIOD5Fw6/uSTin37yB10Op3ixRezBi8UajUzOcksW2YwGHQymSCGrVtLVbNjMBgM\nBgMAwDvvJDddfAWD5/kcZ++R9es1PF+aj+SU8/zYnj2J224jmzOjKvHLLptBTgRN08uWLauv\nr49EIsyRI2Sj4cYboejoTDI9Vb7xSnXGQwRBmJqa4jiOj8eNd92lXDBBBJvN5vV6RVGkKGrO\napHKxa9/XWmxAAAkEtSBAxc1bSHJF2o1kyNYNjU1RQZI22wN27c7N2wI79/fZLUm67GtVuXJ\nk1oAWLlSK+U2I4TQoieklOUSqVkPgiDo9fotW7akBkFSK0qUSqXRaDx37lxmiKG5uZl8P+E4\nbnR01Gq1pta/rFq1SqPRFLl4hmHa29s3bdqUWfBy/fXXHzx4MC2jRKfTSReN0sIxab1CEEJl\nCgMcKAuaprVaLezcCTt3FvRAMuaD4zhRFNXhsOHGG+fxqrher9fr9aIoCsePky2Ka6+V9hqN\nRrmWEE3btpHgRXNzMz8wIPsEJT/b3LYNtm3L3Jy7WCbS2VlMOqUoin6/PxKJJGKx1PN8bjqT\nU64FSVqAg8yKi8fjymCw4mMfo8xmgOzvezKd9YEH4IEHZrzsNOT7k1y6DaNUpqXbRCKR0dFR\nMkPH1NOjfPvtBRJEIA3eAYCiqLSsmYTJxC75TiIffph9+913u77xjUm4uDl/Gp7nx8fHyTdv\nMhq2t9ckRTcAYHRUefvt7QDw7LNiGQZ1EUJohjLjwmlZD2azOS3Fg5AqSurr69euXSsXAREE\nwWKxxOPxtPqXdevWTUxMNDU1Fbl+s9mcdQDK+vXrM6e0LFu27Pz5816vNy0cEwgETEv+Qxah\nxQEDHKhkIpHI2NjYfI35kENRFHPvvXDvvWnbtVqtSr6MhSyYYRh6cHDK7w+FQrFYTHPmjOnv\n/g5gruM1NE1rNBr1L34hN2+FnpiYWbFMNBodHR0l0zfTzvOTTUZkoiqxlIagoiiOj48nv9YI\nQtvOncnoBszd+67T6Wialku3Md11V2q6Dc/zZrOZJH3MWUOTPElf0TKzZiKdnUw4XGQqb7l7\n4w0AgHA4/M47tp07W0mtyje+MXn55ZGTJ7UMw3R1GeUeOzU1JTWiI6NhBwayXzm86qoSF3wh\nhNBCRlGUSqVKHcWdmvXAsuyaNWtSyzoyDQ8P54iAeDweckWBSK1/8Xg89fX1aZNNCkVG5KZu\n4Tju1KlTHMedOHFCCmFEIhGTyaRQKNrb200mU/EtThFCCxMGOJaqUjcdiMVi0kkjzOGYj2Iw\n/f2JRILUKSj6+41/+7cA6b8NiqIqKyuTWYvTVRVQYBOs4hmNRl7m7L3x7rtnVixDrqiQgFTm\neb6ypQXGxuRyIlKn2NpsNulbwny97zRNG41G9enTsvdIWYnb7SZ/qFkbmmh5nmGY2V9yduT7\npVwtkiIaXeIBDkKr1R450mqxKCGjVqWnBx58MPujUr+7Ez/9qXlwUL1t24pAgNm0KXb8uBKL\nrxFCS1NNTc3ExAS5nZb1UFNz6alSmSGGVKFQKPXHtK6foVCo5MNZ1Wr1Jz/5SWm2GlwcwrBa\nrVkzPgptcYoQWpjw29wS4vf7p6amotEoJYrN996rKml6hcPhkKIbZZRaz7JsbW0tAMBvf5vc\nlOPjTaaEZA4YDAb+UvkmhfJ6vSS6kfU8X6VSsSyrPXECskVVKm65hURVIpGI9C0h7X2PG42J\niopii2vzVldX5zh27LzTSb5j5WiRS4pv5YIIEA4nW6LMB4qictQiKUvdSrZ89fVlbwJz5ZUF\nHCR1NOyLL/pZtvHSj0EIocWopqYmHA5brda0wo14PF5kegVkjOVOq39J21sSw8PDqdENCQlh\n5A7HIITKHQY4lgRRFK1WK5mNAgCmnh7VO++U9jJ7jjEfkc5OTSKx0BtTz1/wIk9Mf38sFpPy\nTRo++UmAorJvyBUVufP8RCTS3NysuFQLkkAgQH7KfN+59esjgUDx/cPyZzQaq6urQ6FQNBrV\nTafbZLbI5Xk+VxChFN+04vF4NBpVKBTKYLCgIa9qtVr1r/+aNWtGpGmNxwOlvsxVpkitSkFS\nRxUKAhw8aHrhhXpRTHYnra/H6AZCaOmiKKqlpeXyyy+X66NRzMHTvgGmdf0sPoCSCUMYCC1l\nOOp5SXA6nVJ0Q7rMHl21CgDEpia4ZHqFxwPt7UBRQFHwne9k7hdFkfTfljtbTq29RDOmVCrr\n6+tbWloabLbkpiLSKVPP863f+160vV0znTUa6ezkeV6v14snT45aLAP9/X2nTo28+mrykfv2\ngSjCjTcCAHlnZaMkF3dlnwMKhaKqqspkMukfeABEEUQRPvWpzPvIld6INK1wOIpZQCAQOHPm\nzNDQkNlsPjs0FL7ppoI60dTU1EidRFZt3bpq69aq6a+VbTt2qH7962LWtsRVVlaS4qNAgNm9\nu/VnP6tnWXHvXtveveMqFY3N8xFCS1zuPhqXFIlEHnnkkeuvv16v11MURVHUf//3f5NdqXmR\nmXNkdTpdCVaPEELTFvZFdVQKoii63W5yO2t6Rdaa/kAg4Ha7I5GIkEis2LVLm/MkjaIohmH4\nREJ2SOcCT98oO6XIN2FZNsd5vtLphLo6tVq9fPlyABAEgf7jH5OPTPmuwzBMjmwI9fw1s8hB\nr9ezMg1N2nbsEA8cgG99a2ZHnpqaGhsbk3409fbqjh0rKFVKrVYrSl2LhAiGYZYtW3bsmF3q\nTvrww5Nr1kRPntSaTCatlmltne8lIoTQ/Jlx1oMoii6X68iRI//wD/8gbWQY5vLLLye3q6qq\n3G630+nMnCPb2Ng4j32vEEKLEp52Lh48z5PkfJZltRyn+uu/JpeO+aee4rduBZnL7KGOjswA\nh91uJxO8AMD03HPad9+95EmaTqdjfv7zrGfLrEqlcDhmNuYDlRb5VkF5vdDV1TIdtMp6ni/s\n3w8dHdIWmqazRlV0Oh3/v/6XXJREPzV16fygOVdTU5OQL72hZpoUIwjC+Pi49GNaRxKxqYnK\n71fB9PeHQiGXy8VxHHv69MqbbwYA4emn6b17Z7YwJKmoqHj7ba3FwkIh3UkRQgjlQHqNRyKR\nXbt2bdy4ce/evQ6Ho729XWquQVFUW1vbpk2bcHAJQmgOYIBjkXC5XA6HQxAEgOSoTqmHqHDF\nFQCy8z7DKSexhDSfHAoZomk0GjmZq+LL7rlnZmM+UKmQqys+ny8Wi4EgXJSSI4PetCmfIxsM\nBvjLX0AmSrIw33eapqm+vuGxsXA4DCntSMOPPab9wQ9mfNhgMCh9mctMlYpu2KDO+1A6nS6Z\nsnvsWHLNV18944WhVCdPZv/UK6g7KUIIISIQCJCwRVdXV1dXl/QdsrOzc3x8fM2aNRRFAcDo\n6CgOLkEIzQ0McCwGbrd7cnJS+jFtVCe9aRN4vXLFCKxKJc37JKS+01mHa6hl+mmr1Wrl2bOy\nS8TU+vkjCILZbCYn85CRkjP2wQdTajWknOcH//7v9fv35398/dCQ7L6F+r4rFIr29vZwOMxx\nHDsdl9Fu3lzMMclIGpBJleIKCXBcsOB735adGXQnRQghJEdq8Ub09/eTXNHOzs54PB4KhfR6\nPWDXT4TQHMImo2VPEAS73S79mNZDVGhsZJct0+l0cp0Ll91zD7z0UuoBORIWkWkbGY1GsyzC\n44H2drq/HwAi3/72uM3mePPN5K6UhpRoXjgcDim6kfnn0dLVtXLlyqamJuN08wj9DTcUdHyq\nr08UBP/U1OTExOT/+39ko/jMMwv/fddqtTU1NRUPPSTXjrQg5CKVXN/W2MaNxS8YIYQQWlCk\n4D7RP/2pR3qIpu1FCKE5gAGOshcOh5OVKRfnXKjPngUAbv16AGhsbJROtLLIvMwuP1xDCsCL\noujz+SYmJsYsltjNN0ujIjTXXZd6tlzMmA9UPFEUpabomX8ekc5OANBoNEWe51MUVVFR0dDQ\nII13ofKrcFlMtFotAMilSik1GpBm3yCEEEKLQjK4P21gYAAAaJpeu3ZtNBp94oknrrvuusy5\nKgghNHuwRKVciaIYi8UEQYjb7Ws++UmlzQYAfFUVZCTGawHUajV38uR5my0SiUBKMULWzoUq\nlUr3y1/KtY1Uu91gMHAcNzo6SgLzpp4e5dtvp3chxdT6hSEej5PGELItZkUx7dtJUZbw+67R\naLRarU6mE03VrbcuzI4kCCGE0IxpNJpQKCT9ePr0aQBob29Xq9UnT5786U9/Ku1KnauCEEKz\nBzM4yg8Z1jAwMHD27NnzZ88q7rxTOX1lmPH50nIu4pddRm6o1eqVK1euWbOmtbW1xeEgG7N2\nLqyqqpKrZ2nbsUPxyiuCIFgsFhLdSCt54BsaFuDUjDIiiiKp9wGKAoqC73ynVMeVS8kpzfER\nAAC0tLQUliqFEEIIzTnSevyBBx4oPr2ipqZGukwSCoVGR0chpT5l165dP/3pT41GIwCsXLlS\no9GU7kUghFB2mMFRZgRBGBkZIYkYkNFPNLB5c1rOhUqrTe0hqlQqlUol7NwJO3fKPUVlZWV8\ncFB2BVdd5fF44vE4FNiFFOUQiUQcDkckEknEYit27dJJI06KPiVWKBQ0TVf++7/LtZilxsdx\ngm+pKBQKYXDQ6XYHg8FYLKY9e7bls58FANi3D558cr5XhxBCCEEgELDZbB9++OHzzz8vbZxx\neoVSqVy2bNng4GA8Hj958iQpZG5tbfV6vWvXrk2dq9LR0eHz+aqrq0v1QhBCKCvM4Cgzbrdb\nim6kZU8AgOEPf0jLuai69da0HqL5YAcGXE7n4MBA36lT5155hWzk9+4lbSNDoZAoinJdSKV+\nlihPU1NTw8PDgUAgkUiYent1x44J6umBG5cKcAiCIAhCjqQPiqJypORktphFRaJpur6+fsWK\nFWvXrpVSpUrfiWY20nzK04kT8OCD0NUFRiMoldDWBo89BtjVDiGEsgoGg6Ojo4lEIi29or29\nXalUzuyYVVVVW7Zs2bx58+7du8mW3t7ezZs3P/roo3DxXJVAIFCi14EQQrIwg6PMSFPEM7Mn\nZBWeBUBRVF1dXV1dXSKRgOPHyUbmmmvIDZ7nKQBS8jD67LPR9vbq114juyKdnSzPF/p0i5bH\nA11dyfarMtfwE4mEzWYjn/1SxIp1OjV9fQmTia6vzxqDFEXR6XT6fL5YLAaC0H7//Vr5pA+T\nycQPDMgucmnUTYiiGAqFIpGIIAjqcLjiYx+jzGaAWc6tKGlHkmAwGAgEOI5jKKrh7ruVpUvz\nKV+iKO7bl/jNby6kjFkssH8/CAIcODCP60IIoQVqcnKSfOXo6upKTa/o7Ox0Op2NjY0zOObw\n8LDUzjwVKVRJnatC8n8RQmhWYQZHmSGdL7JmTzh277ZZraMWi/Ott5L3LnpEK8uy7H33pQ3X\nYFlWblSESNMK6ar1khSLxVwul81mG7da493dcKmzUJ/PR4bgZB1xkvVaBylTcjgcySavvb3a\nd9/NkfTBMAzT3z8xPj7Q35+aklMWk1xLIhqNnjt3zmw22+12p93O3HFHMroB8x8dCIfDHo/H\n4XD4zWZxxYqsSRmiKFqtVrPZ7Ha7Q6GQ5rvfJW19k7vn+yXMl1gsdu7cuXPn+O3bnS+/fO7w\n4aFbbvGQXdKUaoQQQpJYLMaRfvDTsqZXcBz36KOP3nDDDXm252hvbxen+Xy+U9Puv/9+uHiu\nCk3jeQdCaNZhBkeZoShKFAS57ImGujqVSgXS+IbZGdFqMBgomVERbTt2CPv3w7p1s/G82eWR\nJTFnXC6X3W4n3xVMPT2K3/8+fbhMBvJVQ67eR+S4ysrKtIc4nU6pDigt6UNobKSzNXllGKax\nsbGxsTGRSNDT0aglMsmV5/mRkZFEIkF+TGtbM4/RgXg8PjY2lnwrBaFt5065sIvD4ZBSt9Le\ncbGpiVqSbX1FUbRYLNFo9NChEYMhmTV2xx3uX/2qBgDq6+d1cQghtCBJH4WSzPSKeDx+5MiR\nAylZcAW15yAT01OlzlXR6XQzWzlCCOUPI6llRq1WV736atbsCYpllU4nAMC2bWk5F6VVVVWl\nlS95oGf/tDkSiZAsCfvERGLr1ktmScwNn88nZX6mtUcRm5pyDZcpcMSJlAiamfQR7ujIvUiW\nZemvfW1W/zwWGpfLJX2lS3tfEiaTaDTO2UoEQUidkqNQKvXPPkt2SWGX5F1T/pIFQXC73eR2\n5jse3bBhzta/oPh8vmg0CgBSdAMA3ntPT27cdBNmQSOEUDqGYdK2pKZXMAwTDAbPnj3rcDjS\npp+oVKo8n0KhUKS2EU2dq8IwTE1NTWleCUIIycMMjjJTW1srfvghZMueWH7fffCjH8Ejj8z2\nGiiKok+fHhkbI5PP1UNDq7q7ASD06KO6H/4wxwNFUYxGo9FoVKFQqMNh+uqrC828EEVxfHxc\nOsM39fSwR46IGg1FGq/Oa4DDMV2bk9kehVu/Xm4wmkqlylHvo/F40iIjPM+T0/WsSR/hzk4N\nz2d+g1nKgsEguZH5vkQ6O5lIJPNyU2kJguBwOPx+fywWo0Rxxa5dUsMUEsPK3XuF9A0BmXc8\n1NGhzvakix75n0+qwUH1888bAeCKK8Ld3WGAuvlYF0IILVxKpZJl2dQ8jtT0Cq1WOzo6KghC\nWnuOjo4Oq9Xa2tqa57OQdNHx8fFEIiHNVVmxYoVerw8Gg1VVVbPwyhBC6AIMcJSZysrKxNCQ\n7O65OsNnWXbFihWRSCQSibDTcRbd3/xNjod4vV673Z78WBWEtp079YVnXkxOTkrRjbTTQli2\nLFeWxCyLxWI52qME162TC3BUVlaGc9f7ZE3KmE76yCxTKt1rWiRyxIMi69erM/J1S4sUyEg1\nz8bnniMNU0iBTKSzM2vYBUIhg8FAHkKiG/iOp0n+WqYdPlzx+OPN4TC9aVOot9dC0xjdQAih\ndBRF1dfXT0xMkB9T0ysoimJZNvV/rWntOaLRaJ55HDRNt7a2XnHFFamdR3t6enp6erZs2fL6\n9Bw3hBCaJViiUn7YgQH/1JTFbB4aHBx59VWycV4aRmo0mpqamoqHHrpkyYPb7bbZbJl9EJK7\n8wtwxONxjyfZRDAzVz+2cWMxr6VIPJkdI1NskqNyRKlUGs6ckdubWe/DMEyOJq+0QsFMThb5\nWhYZhmFyFAHNdrbL5OSkFN1IK5CJG42CSpU17JJ6eU2hUACA3DvOKJVgs83qS1iYWDYZnRcE\n+MlPTN/4xvJwmO7u9rzwglmvF6S9CCGEUtXW1tbV1QUCAa/Xe+LECRLCaGtr02g0Lpcr9Z6p\n7TkAQGr+lQ+5uSpds9MbLlM4HP7mN7/5N3/zN3n2SUUILSb4LbAsVVRUVFRUAAAcO0a2LOSG\nkYlEwm63Sz/OuEtiKBQin8RyV+NnOMB9miiKlNc7s5al5IQqR9wBbDZYtizrY5n+/kAg4HA4\nOI5TDQ6Sep/4k08q9u3Lev/q6mqVTNJHy733zk2ZUhnR6/Wal16Sa1uj9Xph1nqeCYIwNTVF\nbmctXJJLylCnhF3UarVSqdTJvOONd9+9NN9xg8Hg8XgCAebRR5uPHjUoFOKePePd3V4AoChK\nr9fP9wIRQmiBamho6OzsTA1APPfcc88999wNN9zw/PPPSxtT23NARt5cbmSuSqEL4zjuqaee\n+uMf/3jixAlSh3j8+PFrr722oIOEw+HJycn33nvvxz/+sbSxoD6pCKFyhwGOMrdtG2zbNt+L\nuIRAICB9Lmae5kU3bMiziUBalkTmaWH6uJH8eDwer9cbjUZFnk/tj1BQvY9CoVCr1XJnoZeM\nOxgMBoPBIIoiPx2xUsh/otfX1yfkm7zKLbuY8E1Zq6urC81T25p4PJ6jfYZIURVHjmSGXYBh\ndD4fkAgmAAA0NjYqp6+kZbEkx8QaDAa3u/quu+otFiXLig8/PLlmTfTkSS0ArFljUCqLjHYi\nhNCiJZde8ZGPfCT1x9T2HAAwq/9fFUXR5XK9/fbb6eqB+QAAIABJREFUP/rRj6SNM4hKBAKB\n0dFRURRjsdiuXbs2bty4d+9eh8OxevXq2e63hRBaODDAgS4gTUATiYQyGFT+1V+V6kyYDB4D\nmdM8Lu8ARzFZElmJojg2Nub3+8mPpov7IxR63mgymYo8C6Uoir3vPrjvvtx3o2laMTjocDp9\nPl8sFpOavIrPPEM99VTanf1+v9vt5jhOSCRmHL4payzL5igCmovfg0xIjg6HIVvYpXX79rSw\ni8Fg8J84MTI+TkpXpHdcePppeu/eWV//QvXb3zZZLBQAJBLUgQON0vaenlkakI0QQouBXHqF\n3+8nLTng4vYcAMAwzOxNeBVF0Ww2h0KhUCiUGpVYuXKlWl1AH21RFG02G3lpaX1S165dGw6H\nMcaB0BKBAY5FKBqNchwXj8fV4bD+hhvyiVOIouh0Ol0ulyAIpAOosnRnwjRNk+fIepoXv+yy\nPI+j0+koipLLkjDeeWehV+O9Xq8U3UgrnOEbGpgCW5YaDIapDz4YHh8nmSbSWWiOYpMZoyjK\naDQajUZBEKj3309uzChTstvt5KMdig7flDX69GlRFP1+fyQSoU+dMv7t3wLMRQ6LQqGgabry\n3/89a0hONTYm+8iMd6eiosJgMEQikWg0qn7nHbKRvvrq2Vl4efjzn6ms26+8co4XghBCiwH5\noEmbftLa2ur1epcvX578LjcLXC4XKUjJnN5it9sbGxsvdYCkYDCYuLhxeGqfVJ/PhwEOhJYI\nDHAsKjzP22y25Em7ILTt3An5xSlsNpvP5yO3pQ6gpToTJp8ocpkXKq02z8wLlmXr6urUp0/L\n3qPApWZtWUoKZ8IdHXpRpKjsZ1ByKisrDQZDMBjkOE575AjZmKPYpHg0TcM998A992TuCoVC\nUnSj+PBNufJ4SFUOBVC5b1/lk0/Cf/1XctfsX+WnabqyslIuJKew2cQDB6buvZeEXUx/93cA\nucIuFEVptVqtVgv33w/33z/bi1/43nhjvleAEEKLy/Llyz/ykY9IXwgBoLe3t7e3d1ann6TV\ny6RFJRoaGvL8MhaNRtO2pPZJJaPuEEJLAU5RWTxEUbRYLBcKLvKeVBIMBqUPs7RBDwmTSaiv\nL3Jh5KxMOs1btXXrqq1bq6Y/KStuuQVeeinPQ5lMJt/Ro6f7+vpOneo7dercK6+Q7fzevTMY\nIkM+C+Vals7ss5Cm6YqKCqPRqPv61yPhsM/rDVx3Xdxuh/Z2oCigKPjOd2Zw2BnIMXEm3NEh\n1Q0tPj6fb2RkZGBgoL+vL/L5z6fH+LZtu+TQn1Qk9cPhcNjt9qmRkULfx4aGBq18wxSqq6uq\nqqqxsdFktSY3YXHFjAiCcPRoZMeO6JVXCkYjKJXQ1gaPPQb4hRYhhPI3MjKSGt2QzN70E9Iv\nI3VLalSC5/n8v65kxkFS+6QWeskKIVS+MINj8ZiampKGeKVdsU+YTFRdndw8zByDHiKdnWIw\nWJHS73BmWlpaxLz7Ysbj8VAoFIvFWJbVcpz6uutSq2waGhpqampCoVA0GtVPZ0kw11xT6JJE\nURRFseQtS4lAIDA+Pp78VBaEtp07FXPe/IJMJ5UL3wDHkfmji4zVar2Qi9TTozl6tJhcpHA4\nPDY2lvo+5pkSJWEYhhoYmLDbp6amEomEbMOUcugWPGdOnIAXX4Rjx2B0FHw+aGqCW2+FfftA\nrsOdy+VyOBz79jUfPnzh/1QWC+zfD4IABw7M0bIRQqjczWz6SWmlTW/JPzCh0Wik29Fo9ODB\ng0eOHAEAQRA2bdr0n//5n62trbOwXoTQgoMZHItHIBAgNzKv2Ec6O0l9Y1Ykdl7aRIY0CoWC\nHRhwu1yjFsvZM2fGpTKBffvSMi/sdvuZM2esVqvD4Ri3WhNbt2aeUiqVyurq6oaGBv0DD+Rz\nNZ5cHxBFETwe6Qo89d3vqlQqucIZimUVDsfMXixp4i1dc8g/laa0UsM31u99L9rerpm+KhLp\n7Jz3bzCzwev1yuUiCY2NUGBVTiwWM5vNxb+PNE03NjauW7euo6Nj5fTyFvJc53kkiqLb7X7i\niVBvL/zpT+B0QjyeDFU88UT2hzgcjsnJSUEQbDbl9u3Ol18+d/jw0C23JNOX3nxz7haPEEKo\nUBRFpc1nSZ3ewjAMaTCfD61WKzUlHRoa+qd/+idypQcAaJr+67/+69KtGiG0oGGAY/EgZ2Jy\ncYocOX40Tec4Ey5VWymapmtra5cvX7569eqmiQmyMbhuXdhqlYIOocceczqd0rl38aEBjuPM\nZnN/f/+ZM2f6+/rCN92UGi6pqqqSK5xZft999C9/ObNXOjExIb2E4k+zZyxH+EakabXbPTfL\nmEvu6ReVGeMLrVtXaEzH4XBI440zS7dm8D4yDEPdc09BBTJLCimym5iYGB1l8gxVxONxqdHM\noUMju3fb163jTKb4HXck/xKKLrBDCCE0u6qrq6XbadNbqqqqCiotaWlpoWk6EAh4PJ4tW7ZI\n29va2kgDeITQUoAlKosHwzCiIMgWXDByFSqg0WjYf/5nuTNhrdcLNTUlXKcoivYtW9x9faIo\ngiC0felLUtDBlZI9mFZlIzQ20gWeUoZCIbPZLJ3WGjPGiNTV1cXzLpzJUyQSkXJeMkt+uPXr\n56yFd3V1tSDT3rJtx460iTOCIESj0Wg0qlAo1OEwc801pRoSPGfIkGOQj/Gp4nGlXJFDNsFg\nkNzIWrqljscXZY3PPHK5XOR3fujQiMGQ/CZ6xx3uX/2qBmRCFYFAQPoHLj0EAN57T09udHfP\n5ooRQggVra6uLhQKTUxMpE1v4ThOpVIVdKhoNCoIwqc+9Smp+JoYHh6+5ZZbfve735Vy3Qih\nhQozOBYPnU5X8x//IRen0GXrGkXU1NTIJTK07dihfvXV0q7TZrO5XC7yAZaWoxHp7CQ3Mq/A\nRzdsKOhZRFG0Wq1ymRRiUxOYTBRFKQYH7ZOTgwMDqS1LhaefnkHLUkLKlMl6mh2efoFzoKKi\nQjc4KLs7JXzjdruHhobOnz9vtVpHzp/P0pizTJS2KodMm8uVEpVS8TRnvWMXMaktbv6hiqyJ\naYOD6uefNwJAV1c023whhBBCCwhFUa2trZ/+9Kc3b968e/dusrG3t3fTpk1f+cpXCjqUx+Ox\nWq1p0Q1i7dq1mMSB0BKBAY7Fo7q6Wv/hhyATp1BMn71nYlm24uxZ2eOW9BQ3HA7LdUmIG42J\n2lqQOaXkCgxwBINB6eQnM1wS27iR7KIoymQykf4IK6bPr+irr57xC0xW9MicZkvPOzcUg4Nu\nl+vM0FCOiTNOp3NiYkL61J+vjiHFI3W8clU5wDAXNVXJIzbBsmyOcMnI+fNpFU+z9sqWhKyt\n8qVQxRVXhL/61SxfTDML6A4frrjrrvZAgNm0KfTii+68a7cRQgjNm5GRkbRhsUSh01s4jmtu\nbj417Zvf/CbZ/i//8i+7du3KnCOLEFqU8Nvf4sEwjOHMGdndOU/A6NOnY7GY2+2ORCJMX1/r\nTTdB5qCHUpCm2GYt3wAAubEmicsvL+iJpI8x2YKFi+/PMAzcey/ce+/MXxsAAGg0Goqiql59\nNetptkqrBZsNli0r8lnyV1tbW1tby/O8ePw42ZI6cSaRSDhSTvvTyoLEpiZqrjqGlERVVZVS\npiqndfv2+Pe/P/6Vr0QikUQs1rZzp+ZSsQmdTkf/n/8jlxLV+IMfpFU8zdrLWhIy82sOH654\n/PHmcJjetCnU22thmDWZj9JqL5R8CQIcPGh64YV6UYTubs/jj0+YTE2zu2iEEEKlUKrpLWkH\nSRvIghBaIjDAsajQp08LguDz+SKRCH3qVCNpZJhfGwWlUtnY2AgA8PbbZMtsDHrI3QkVAOSu\nwM8wNCATLpm9TAqGYWpqajQyp9m1t9+e1vxibjAMA/fdB/fdl7bd7/dL3wYyQ06xjRsLK36d\nb3V1dQn5piq2hoagzwd5j481Go2RHE1MAMo3ErQAsSzLsiwpC8oMVajV2Rvp63Q6rVYbDocD\nAebRR5uPHjUoFOKePePd3V6lUllZWcysZ4QQQmVGpVKFw2Hpx9SBLABQUB8uhFD5whKVxYam\n6ZqammXLljWOjyc35Ujwy5qov23b7A16yD2xRaPRyHUDqbjlFnjppfyfiHyYyYVLFGo12Gyl\nfGEpGhoa8mx+Me9ydwyRyoJOnIAHH4SuLjAaQamEtjZ47DEoxfjgEqNpmh0YmBgf7z99OrUq\nJ/7kk6f7+oKbNkFGYRTf0CA3D0WlUuUo3YqbTJkVT6gYVVVVABAIMLt3t/7sZ/UsK+7da9u7\nd5xlRbIrq5aWFrvdcNttK48eNbCs+PDDk2vWRPv7KyYn28bG8AMOIYSWkNQPi7SBLAaDIf+J\nswihsob/1Bevbdtg27bMzYIgBAIBjuP4eLz+zjsVc9tEQKvVwj/9k1zafwPPK0sUGtDpdCqV\nSidzBb76tttmL5OC9C71+/2BQCAajWrOnCkolWYupXUMSctziV92GQDEYrGnnxZff/1CMofF\nAvv3gyDAgQPzseicaJpubGxsbGyMxWL0dFVOYO1akqiSmaUS7uhQRqNyfdpJStTU1JTX6xX+\n/OdV3d0AwFdVUbGY+YUXUiNB5ZXqsnB88IH4858Lx49TVivt8zXU1NRHIuD3M1Ko4uRJrVKp\nvOoqo9wRFArFkSOtFgsAQCJBHTjQKO3q6YEHH5yDF4EQQmiGOI7bu3fv+++/f+LEiVAoBADH\njx+/9tprZ3a06upqv9+fOZAlEAgYjbKfIwihRQYDHEtLJBIZHR0l1+1NPT2K3/9+jpsIVFZW\n0tOdULPPLh0cDIfDwWAwFouph4bqPvEJgJmEBiiKam5upqfTQ7K4+PWKohiLxXieV4VCJRmS\nWlFRUVFRAXCh5CdXKs08IS0McpQF+U6ftgEMD6/cvt35iU9MVVfzL7xQT8Z2vvnmQgxwSJRK\npVSV4xsehnBYrjCKj0RyDKKjabq6ujoej8en/5YYn2/OKp7K14kT8POfC8eOgdVK+3zQ1AS3\n3gr79oGUIMzz/OTk5J49hsOHK6RH2e3JadYFhSo+/DD79iuvLPZVIIQQmiWiKPp8vnffffdA\nypcJhmFefvnlb3/72zOLd5CBLFdccYUvZXRgb29vb2/vli1bXp9OCkYILW4Y4FhC4vG42Wwm\n8zLS2kkKjY30nDQRoGn6khNbtFptsnfgm28mt88oNKDRaGJ9fVaHIxQKxeNx3blzK77wBYD0\nsIUoig6Hw+12C4IAgtC2c6e+tFktMqk0+fJ4oKur+IBLVqSFgVyei2Hr1slvflP86lcPHRqR\nJnfecYebBDjq60u4kNmVOj42LTYR6exU5NHYTK1WO77wBQBY9tRTmZEgpUYzx71jFziPx7Nn\nj+KttwzSlrSsH57nh4eHo9GozVabGTvLlDtU8cYbpVw8Qgih2ZZIJMxmM8dxbrd7165dGzdu\n3Lt3r8PhWLZs2bPPPivdjWGYywvsMT8yMpIa3ZAUOpAFIVS+MMCxhLhcLhLdyEzUD61bpxdF\niqLmYBlUXx/P816vNxKJUCdPNn/mMyA3saXI0ACAUqlsbm4GAEEQ6D/+Mbn14g+50dHRQCBA\nbktDUud3NIbP5/P7/dFolAZovvde1WyWEbW0tIjyjTkjnZ0AIEU3AOC99/TkRnd3ydcyW1Qq\nlfoXv5ArjFK5XFBdnfsIer1eoVDIRYIqv/SleekduzBNTEy43W6rdVWOrB+n00nmHGWNnX3i\nE/DWW/O2foQQQrNtdHSU4zgA6Orq6urqCgaDTqcTAFpaWj7zmc9I8Y5Vq1alTsvKR6kGsiCE\nyhcGOJaQYDAI8hNMWI7TaDRzsxKGYerq6gBAOo+ZjYktqWiazhoumZqakqIbaVktCZOJnfPR\nGKIoXhRw6elRvfPOrAZcFAqFMDDg8flCoVA0GtWdO0c6hojPPHP6i19Mu/PgoPr5540AcMUV\n4bvvVpdLl+LKykpefh6KeOAAfOtbuY9A03RzczObd8XTkhUKhdxuN8hELqSsn6mpKXIja+zs\n5ptFgLkItiKEEJp7oVAoddYJAPT395OoxObNm7/85S9L8Y5169Z5PJ6amuzJfQghlFV5nJ+g\nkuB5PscEEzKgca7N5sSWfEh5jKlZLWQ0RqSzMxKJzPF67HZ7ZsCFzPsQm5rk5n0UiUzeaWlp\nWbVqlTR8R8ioCjh8uOKuu9oDAWbTptDBg2aK4jOOtEAZDIYcc22o/NJWdTodnDo1Njo6ODDQ\nd+rUyKuvJnfs2weiCDfeWJKlljvpH1TWyMVnPxsFAFEUpfE9ktTY2Ve+svAm9CCEECqRtOgG\nAPRPfx0lE0+keEdnZycJmiOEUP4wwLGEsCwr105SpGmFwzG/y5uxWCwWjUZFtzvLyNs8Hgvy\nWS2xuR2FKgiCx+MhtzMDLtz69XOxiOmQE/3pT0slS4IAP/mJ6RvfWB4O093dnhdeMBsMYnmN\nW2MHBpwOx0B/f9r42IJiEyqVqqWlZd26dZ2dnSum36kF2Dt2HmX+k0mNXNx6ayjro9JiZ0ol\nfjAhhNCiJQhC2paBgQEAoGl67dq1cHG8IxqNZt4fIYRyKKdTFFQkvV6vLi5Rf0EhzUE9Hg/P\n8zNuDkpRlCgIcu0n1XPSlETCcRz5FM8acAl1dMxRBREAAFAUZTAY/H5/IMA8+mjz0aMGhULc\ns2e8u9sLAHq9YW46tpQKRVH19fX19fWxWAzefZdsVMx0EJ1cxRNKc/hwxeOPN4fD9KZNod5e\ni0LRCAAURWk0GpIeJQhw8KDphRfqRRG6uz2PPz6hVjPlFTtDCCFUEIVCkbbl9OnTANDe3q5W\nqyEj3oE9NRBCBcELZUtIXV2dRr6JQJ6J+guEKIpms9npdJK2qVJz0OTuvAMcGo2m5j/+Qy6r\nRev1zsLa08Xj8UQiAR6PZv36DRs3bti4ceWXv5y1jCjrw0nC/8wSWHIzmUxWq/q221YePWpg\nWfHhhyfXrImePKk9dUoXjTaU5CnmnlKpVO7YMb+FUYuY1McnM+tHrxfU0/9CSUF1IMDs3t36\ns5/Vs6y4d69t795xlhVramrKK3aGEEKoIAbDRddIQqHQ6OgoTNenwMXxDpZlGYaZl3UihMoU\nXihbQliWTZw8eWZ0lKSRq4eGVnV3A0D4sce0P/jBfK+uMB6PhwxIh4zmoHxDA5N3r4ra2lpO\nPqtFGo2RSCQCgUA0GqVpWhOJGG68sfi5rTzPT05OTk1NkfG0K3bt0pnNZJdqZCQz4MKqVGmz\nSEOhkN1uj0QiIs+XfrotgEqlOnKk1WJRAEAiQR040Cjt6umBNWtK8iRoUamurna73X4/nZn1\no9FopPBHdXX1wED8y1+usliUqbEzrVar15fP/GGEEEKFUygU9fX1DocjEAgkEomTJ0+SHI3W\n1lav1xuLxVLjHZWVlfO8XIRQucEAx9KiVqtXr14dDAY5jlNOTzDRbt48v6uagazNQcnI23BH\nhyaRyMxyj0ajkUgkkUioQiH9DTdQZjMAqPftU5w5I/s0V10FAG63e3JyMpkhKQhtO3dC0aGE\nRCIxPDwsNSww9fbqjh27MC0lW8Cl8e67U2eR+nw+m81GVjV7021PnUrPIyUyOpCWJVEUBUFg\npqagq6v4iBUCAJVKxXHNt92mSYtcMAxz1VVNqfd8/XWjxQKQLXb24INzvGqEEEJzymg0UhR1\n3XXXSUO1AKC3t7e3t3f9+vVSvCMcDjc2NsofBiGEssASlSWH9Faor6+vfPjh8k3Uj0ajkHdz\nUEEQxsbGzp49a7VaJ8fHqS9/mZrOlYCrrmL6+yPhsHVs7OyZM9JoDPGZZ0j7SZ/PNzExIdV/\n5qiF4Xk+EAi4XC6v1xudmMhdMGK326VFpk9LydGAYPrpEonE+Pg4WVXawxMmE09G8JbCG28k\n/0bS/rv++lI9w/zw+/3Dw8P9/f0Dp0+HPve54iNWSPLrX1daLEqYjlzcfnv77be333pr6+uv\nXxQs+/DD7A9fHLEzhBBCuQUCgdTohoTUpwBAb2/vNddcc+edd87tuhBCZQ8DHKgsURSVY+Rt\nWg3/6Oio9CGaNUKh0Wiam5tXr14tjcagNm0iN+x2u3SctFAC39AgzW11u91DQ0MWi2VyctI2\nNhbv7s5xziyKorSezGkpoc2bz50923fqVOq8D+Hpp1PnfSQLW2Sm22b9xoAkDodjdHQ0HA6L\noijlziT3YYCjaHlGLhZr7AwhhFA+2tvbxRTnz5/PereusuoQhxBaCLBEBZUljUajePHFrM1B\ngWFULhe0tJCfAoFAMBgkt9O6dcSNRqipueiy8sWjMTiOi8fj5HZmLUyks1MPAAAej2diYkJ6\n1CULRuLxeI5pKeHOzpUrV8bj8Wg0qjx6lDyEvvrq1CPkTmDho9HCf6NLRSQScUxPRE77e0iY\nTGze3VuQnDfemO8VIIQQKjck3jHfq0AILQaYwYHKUk1NjW66OeiqrVtXbd1a9frrZFfr9u30\nL38p3dPv95MbmckO3Pr1Uuwjq0QiQW7IhRIAQBCEHFkeQmMjyJ0zy2egAIBSqTQYDKqdO2XL\niHI+HMnxTk/GyZr8Eg6H53V1CCGEEEIIoZnDAAcqSxUVFfqhIdndKUkTJEghF6GQEjSySnYq\nlQklxDZuBIBwOExG1YJMDCXzsAqFgmGY6tdey5qBolCrqfHx3C9frVbLPXzOptuWqdzJL1FM\nfkEIIYQQQqhsYYkKKlfswIDf7/d4PBzHKQcG2r/4RQAQn3mGeuqp1LsxDCNFKEaffTba3l79\n2mtkV6SzU59zuDoZwG54+eWsoQS1Tgc2W0KnIz9mPWfmNmzQZhyWoqjq6mq1zHjapm3bUqel\nZFVZWcl8+GHWh7ft2CHs3w+Yx5GD/N9D9pkxCCGEEEIIoXKAAQ5UxioqKioqKgAAjh8nW6Tm\noBKdTkf97/8tl+ygn5qC2tocT2EymSiZSETt7bfDj35E33cfgOw5c/yyy7Ie1mg0JgYGZJ/1\nUq0uGYYxyE+3pTN+CUiiUqmU//Ivcn8Parcbqqvnd4UIIYQQQgihmcEAB1oULm4OmqqyspL5\n859BJtnhkrkS1dXVvHwoAa66SqfTURRV9eqrWc+ZlRoN2GywbFna42iaVgwOutxun88XjUY1\nZ86QDBTYtw+efPKSLxcA6NOnSb/McDis6O9f1d0NAImnnmKfeSafhy9Z1dXVUZmIVT5/Dwgh\nhBBCCKEFCwMcaJGjaTpHskM+Y0GZ/v54PB4IBKLRqHJgoPbjHwe4EIlgAGpqajQy58zVt90m\nd85MUVRdXV1dXZ0oitT77ye3FjIOTaPRtLa2AoA4ncDCfvSj+T98adJoNIr8urcghBBCCCGE\nygs2GUWLH9XXJ/C81+MZt9kmpCGW+/aBKMKNN+ZzBIVCUVNT09jYWGs2JzelRCIaGhp0g4Oy\nD77UOTNFUbBtm+y0lDxQ99xTzMOXGnZgwD81NXz+/Om+vnOvvEI28nv35v/3gBBCCCGEEFqA\nMMCBlgSapqurq5uamhqlASWF5EpckC0SQVGUYnAwFAzaJydHLRbHm28mdxQSQ0FzqaKior29\nvbOzs3164gxzzTXzuySEEEIIIYRQkTDAgZaY4nIlctDpdCaTafny5caxseSmmcVQ0FyhKIr+\n2tcw+QUhhBBCCKHFAQMcaKY8HmhvB4oCioLvfGe+V7OQzFoMBSGEEEIIIYSQHGwyigoQjUan\npqY4jgNBaLj7buXISHIHtmZECCGEEEIIITSvMIMD5cvlcp07d87hcPj9fs13v6t8+21BrU7u\nwwDH0oGZOwghhBBCCKEFCQMcKC9+v39yclIURQCoeOut+kOHAjfeGF21CgDEpiYwmeZ7gWh2\nBYNBh8NhHR2N3XwzYOZOcRKJBM/zGCpCCCGEEEKotLBEBeXF6XSKokhRlGp4uPmJJ6KtrdZ9\n+9Z94hMAEO7o0M338tDsSSQSY2NjoVAIAEw9PSRzh+Y4AAxwFCaRSNjtdr/fz/M8CEL7/fdr\nMVSEEEIIIYRQ6WAGB7o0QRAikQhFUXQwuPyBBwBgtLdX4XBQ0SgAhDo6BEGY7zWi2SJFN9Iy\nd/iGBszcyV88Hj9//rzX6+V5HgBMvb3ad9/FIi+EEEIIIYRKCAMc6NKS8QtRbNmzR2WxWL/3\nvWh7u6a/n+yNdHaWe4CDRHACgUBschKrBlIFg0ES3UjN3FGfPQsA4Y4OsgvlY2JiIh6Pk9tp\noSKhsRFDRQghhBBCCBUPS1TQpTEMQ1FU1auvGn7/e/8nPhFrblYPDuree+/C7slJaG6e1zXO\nkCAIdrvd4/GIogiC0LZzJ46GSRUMBgEga+ZOZP16CIV0OqxPurREIhEIBMjtzCIvbv167bwu\nDyGEEEIIocUBMzjQpVEUpdfrdX/6EwBUHD68auvWVVu3Vr3+Otnbun079W//Nq8LLFBKc0f/\nt77ldrtJ81RTb6/++HGsGkjF83yOzJ1EIjF7Ty2KYiAQcDqdExMTvuFhccWK8s2sicVi5G9M\nrsgrkUhgz1GEEEIIIYSKhAEOlJeGhgbpzDaLBR8L4DjObrdbLBbz8HD0i1+U5oBMrVpFbqRV\nDSRMJtFoLPJJRVEs9xNXlmWrX3sta+aOSNNKp3OWnjcajZ4/f95isdjtdrfTyX7lK5TZnNy3\n4P/YZMmEisIdHYP9/aHPfa748TQnTsCDD0JXFxiNoFRCWxs89hjEYiVZ/eweHCGEEEIIoeJh\niQrKi0qlCv/lL2es1lgsBgDqoaFV3d0AEH/yScW+ffO9uktwuVx2uz2ZptHTo3rnHWkOSKSz\nE7JVDUQ6O5lIRKudYelAKBRyOBzhcFjk+badO/VlW/ai1+vj05k7FYcPp+5q27Ej/v3vw+WX\nl/xJBUEwm81Sxwops6YcR7eQ4hSO4yiKqvnFLwy//z0ALH/4Yc+tt9LTRSsiTTf+8Ie6Y8eK\nfI0ej+eJJ1S//e2FoiGLBfbvB0GAAweKfSHtnDsGAAAgAElEQVSiKO7bl/jNbxSzcXCEEEII\nIYRKAjM4UL60Wu3q1avb29ubmpoax8fJRsW1187vqi7J7/dPTk6S6EZmmkaitlauwURsptem\nvV6v2WwOhUKiKJZ72YtOp9MNDsrtVXz0o7PxpG63W64fZ8JkEurrZ+NJZ4PX6z1z5ozNZnO7\n3SLP1x86JO2q+bd/k4q82nbsqH3ppWJ6joqiaLFYxsfHR0eZ7dudL7987vDhoVtu8ZC9b75Z\n7AuJxWLnzp07d46fjYMjhBBCCCFUKhjgQAWgKEqr1dbU1Oi+/nUQRRBF+NSn5ntRl+CcLqPI\nnAMS6ezM0WCCpmfyryMej09MTMjFU/i6utK8qtLKWUTD9PePjY72nTrVd+rUuVdeIRv9jzwi\n8DzceONsLId0NgWZtywcDs/Gk5ac3++32WzSgCFTby/rcMjdOW4yXfRnWSCXy0WamB46NLJ7\nt33dOs5kit9xh5vsLTIiRBJqotHobBwcIYQQQgihEsIAB1rMRFGMRCIgPwckR4MJrdc7g2ec\nmpoi57RZT86lURrzLh6PT05ODg8PDw0MRD7/+RzdH2iabmlpWbVqVVNTk3FsjGys+NjHZhYA\nygfpXSr3ls1qZ9MSstvt0m0p1BXZsAEA4kajFCpKVFUJWq35hRek1xguPMDh8STzKQwGXtr4\n3nt6cqO7e6avAQAAvF4vyWbKevAvfIHP/jCEEEIIIYTmHAY40GKWvH4un6YhNxqmbccO9uWX\nZ/CM0WgU5E/OOdJhYb6FQqFz5865XK5wOFzzD/+gOXr0kkU0arW6pqam4qGH5iBzh2XZHG8Z\ny5ZB56BYLEb+EiBbqItbv156RazPl/YaufXrC3ounuelih7J4KD6+eeNAHDFFeGvfrWoGEQo\nFMpx8FtuWSgxO4QQQgghhDDAgRYzmqZpmpZL0wCGMeQcDROLxUKhUNxuL2wMivzJefGvqHg8\nz4+NjfE8D7MzO6Z4Op1O7i2jWHZmmTVzjPx6QT7U5fviF2379gFA5mtUqNVgs+X/XKQeKtXh\nwxV33dUeCDCbNoUOHjQzTPodZvZash6cojCDAyGEEEIILRRlcC0UoRmjKMpgMOhk5oC0bt8O\nP/pR9Otfd7vdHMcxfX2tN90EAOIzz/i+/nW73Z44cwYEoW3nTkXeY1BUKtUlyl4aGkr+Mgsy\nNTVFqjyyzo6hQiG9Xj+/K6ytrQ1++CFke8uW33cf/OhH8Mgj87S0fDEMA3Ah1DX67LPR9vbq\n114je2MbN1ZVVcn9WTZ89asFvUaWZRmGIWEIQYCDB00vvFAvitDd7Xn88QmVii4y5yX5WrId\nnGXFskioQQghhBBCSwR+N0WLnMlkEnOmaahUqqamJgCAt98m26ZWr7ZNX0IvdEZpVVVVUH6u\nqnjgAHR0zPCVlAhp0imXWUBFIvMe4GAYpuLsWdnd5TCJRqlUKpVK3S9/mTXUpdbpqnmeHxiQ\nfXyBr7GqqsrtdgcCzKOPNh89alAoxD17xru7vWRXEa8DAMBgMPj9/qwHpyhKp9Nd8ggIIYQQ\nQgjNDSxRQYucUqkUT548d/Zs2hyQ+JNPgiheNAdk2zYQxSjH2S67jGzIOlY299OxLGs4c0Zu\nL9XVVeTLKZ4oijmKaKSpHwUdMBAIOJ1Op9MZsFgKK+eRQfX1iYLgn5qyT04633oruXXfvvS3\nbAEzmUxyHV7q7riD+dWv2IEBh91+7uzZ0319I6++mnzYjF6j0WicnNTfdtvKo0cNLCs+/PDk\nmjXRkye1AwOVkUixNUdVVVV2uyHz4CdPajnOhBkcCCGEEEJo4cDvpmjxU6vVq1at4jiO4zjl\ndJqG4tprs955amqKNDXIWsER9/trampyPx3T3x8Ohx0ORzgcVg4MrOruBoDEU0+xzzxTwhc1\nY0qlMkcRjdrtBpPpogd4PNDVlRyzsm8fPPlk6s5gMGi1WpOTTQShbefOHANZCkJRVEVFRUVF\nBUzHBUAKD+Vc0gJRWVnJy4e64KqraJo2Go1Go1EURer995PbZxQCYxjmyJFWi4UCgESCOnCg\nUdrV0wMPPjiDQ15AUdThwy0WC5314OWQT4MQQgghhJYKDHCgpUKtVqvVati9G3bvznE3MhFT\nroJDiMXyeS6tVtvW1gYAwvHjZAv70Y8Wt/ySqays5OSLaIT9+6GzUxTFqakpjuMSsZjxrruU\nMjELjuMsFovU5LLQcp58bdsG27aFQqFQKBSzWhUMU3vHHWyJwiiziunvTyQSfr8/Go0qBwZq\nP/5xgCwRGYqiyGss5rn+/Gcq6/YrryzmqEl/+Uv2XL+SHBwhhBBCCKFSwRIVtNBJFRB2u91v\nNl9UAeHxlKQgIhVFUTkqOCgq+2mkHPprX5uDuaoFUavV+qEhub30pk0cx509e9ZqtbpcLtW+\nfcq335YbImu326XoRlo5D9/QkJ4JMlOCIFgslpGREYfD4fP56CefZI8cETWarEtaaFiWramp\naWxsrDWbk5tmp0zpjTeSf2iiCB98AA88AFddBfX18PGPQ1sbPPYY5Beau/TBU/+7/vrSvQCE\nEEIIIYSKhhkcaEELh8NjY2PxeBwgvQJivKGh+vOf15T6Sr5Go4HcY1BKdN4+j9iBAZfL5XQ6\neZ5XDw2RIhpuzx71977H87z57FlSciLFLFinU9PXxzc0MCmvXRTFYDBIbmeW84Q7OnSCQNMl\nCKFardZAIEBupy0Jli0rm7ej6ByNPHEct3cv/Nd/qaUtFgvs3w+CAAcOzMHzI4QQQgghNG8w\ngwMtXLFYzGw2J6MbKRUQ5EfV229rjh6VSy6YscrKSv30jNK03pBtO3bof/ObkjzLvKurq1u3\nbt3q1aub7XayRX3ddQDg8Xgyh8iqz54FgHBHB5lFSvA8T9I35Mp5Uu88Y5FIxO/3k9uZS0pc\nfnnxT7GYeDye8+fPj4zA9u3Ol18+d/jw0C23eMiuN9+c36UhhBBCCCE06zDAgRYuh8MhDfVI\nr4CorKz9xS9S55uU6ko+wzALfAxKJBLx+XxTU1PRiYliKnQoilKpVOpdu1KLaEhShlzMIhQK\nSQ9nGCZ3OQ/DMMW/WClJRG5JxT/FohEKhcbHx0VRPHRoZPdu+7p1nMkUv+MON9lbXz+/q0MI\nIYQQQmjWYYADLVxZKyDIpXs6GEz9MdLZmXruXST69Ol4LDYxPn7+3Dlpfqfw9NPzPqM0HA6f\nPXv2/PnzVqt1zGKJd3eXamSJhOf5HDGL1KQMiqK0Wq3cQBalRkNPTJRmPQByS+IwwJHC5XKR\nGwbDhbfpvff05MbNN4vzsCaEEEIIIYTmEAY40MJFaiWyXroHmk67kh+JRPI/bCwWy92gVKFQ\nNDY2rly5coUnmeFPX311iV7WDEUiEbPZHCUvP6Ngp1QBDpZlcwyRVTgcqXc2Go266YEsaeU8\njXffDS+9VJL1AIDckhilEmy24p9lcQiHw2lbBgfVzz9vBIArrgh/5StFdBlFCCGEEEKoHGCA\nA+USi8USicRsDCvJB8Mw6ZfuT58mu1x3352WXCAVs8gRRdFutw8ODg4ODp4ZHAx+9rN5pT9s\n27ZAxqBMTk7KFuyUbmSJwWCQi1m07dih+7//N/XOOp0uRzlPSWIuBoMBAOSWVPPlL5ckjLI4\npP0TOHy44q672gMBZtOm0MGDZpq+xD8QhBBCCCGEyh0GOFAWPM/bbLb+/v4zZ84M9veHPve5\nkpdC5EOv16deulcNDFS/8grZFa+v1x8/nppcoJrOz89KFEWz2ex0OklWyCylP8wenuelGpzM\ngp1IZ2epnqi6ulo7MCC3N7MFCdPfn4jH3S7XuM3mkPpY7ttXqnIelUpVXV2tng5sZbHg37s5\no1AoyA1BgJ/8xPSNbywPh+nubs8LL5j1ekHaixBCCCGE0GKFY2JRukQiMTw8HIslE9pNvb26\nY8cEtZrmOIA5PZ+sr6/npi/dVxw+nLqr6XvfS/2xbccOYf9+kD/Pd7vdUoAg9/TThUkaJZO1\nYCfc2akTRYqiin8imqapvr7hsTFS7yANkQ09+qjuhz/M+hCWZWtrawEA/n979x4dVX3vffy7\n55aZ3BPIjSCXgAjxVmnAK6tica1qL9oaKog+WLGCClit1gpYCh5OLXqWFJVVOOV4Wp9Wqx7l\nsVo9EMEapVQBESK5cAkJuZCE3JgwmZnM7P38scMYQiYGmGTPTt6v5R8zv/3L3t/onnHmk9/l\n/fc7myK6FOuIESNqPvywublZfxoqSf31ry3Ll0fwQmaXlJTU0NDgdlsff3xkYWGC3a4tWVKT\nn98sIrGxsfpkHwAAAGAQYwQHuqurqwulG92mQqhZWZGaCtEXTqcz8cCBPna2TJnSy9GWlhb9\nQY+7n37t9BbDWSwWkd7W2oxIuqGz2+05OTk5OTlZWVnpR4/qjXHf+tbX/2T/TOdRFCU7O/vC\nCy/Mzs5OS0vLqqnR2w1fFSXaDB8+vLY2bvbscYWFCTab9vDDxyZM8O3dG7tvX1xHxwijqwMA\nAAD6HQEHTqOqamtrq/64xyxggOuxfPllMBBobmqqralp2LJFb2xYtKho376Dp6areJcs+doJ\nEfranOG2Gg0FOlHL4XDY7fZwa23GxMZGfK3N2NjYYcOGJf7sZ1GyBIk+VyUjIyNu0aIoKSna\nWK3WrVtHV1Q4RCQQUFavzpozJ2fOnJw77hj7/vvOr/1xAAAAwOwIOHCajo4OfThDuKkQmjbQ\nm01ardaUlJSsrKy0ykq9Zfh3vjNu3LjsY8f0p87rruvTicLvftoPVUdeWlpauLU2M+bOHdxr\nbQYCAVVVjVrs1kT27On5LX3y5AEuBAAAADAAAQdO0znTITqzgFMzIJSbb3a5XK4HH+z7X/Kd\nTme44Q+KzeZoaOjn0iMgNTU1vrQ07OHBuNZmIBCorq4uLi4uKSnZX1TkueWW81zs1u/3e71e\n9fjxwRqUvPde52ui2z/TphldGQAAAND/WHYOp/H7/YqiJL/1VsK2barLNerhh0XEO2GCftTu\ndCo1NZKdbWiN5yI1NVUJs17pqPvuk2eekUcfNai0s2ArLvZ4PK2trT6fz1FcPOLmm0VEVq6U\nJ580urTI6+joOHz4cGh11Yy1a2M//vjcFrvVNK2+vr6pqSkYDIqqjrn//ngjdgUCAAAA0K8Y\nwYFOgUCgvLz8yJEjmqbF7dwpIpb2dv2Qs6xMfzDinntMOhUiJSUlrqQk7GHzfMuNjY3Nysoa\nM2bMiNrazqZIbFmiaZrf73e73R6PJ0oGONTU1ITSjfNZ7FbTtIqKioaGhmAwKCbcIRgAAABA\nHxFwQOTUl8DQRqrxhYVhu5r2C6G9pKS5qenwoUP7v/zy0P/8j96orVjRuUCp6dZ3iNyWJW63\n+8CBA2VlZRUVFYcPHjz/mSDnLxAIuN1u/fGZi916L76476dqbm5ua2vTH3cLSoKZmQO5KxAA\nAACAfkXAARGR1tbW9lPjNRK3bLE1N7unT2+/5BIRCaSmdnZaufJrNyuJcikpKTk5Obm5uTnN\nzXqLJze3tra2qrLSf9tthn+rN8SJEycqKytD+8hEyQAHfdcbCb/Ybd9P1fsOwaFBIgAAAADM\njoADIiKhP3Gf+SUwkJ7e2SkSUyGihDJvns/rLSstLZ84sbGxMWblSseHHxr+rX7gaZpWU1MT\n2hnnfGaC9FN9PS52e1YjOPTsJtwOwZ1JSjSN3/n0U/XBBwOTJ6vp6eJwyJgx8stfStRvZAwA\nAAAYj0VGISISCAQkzJfAEzNmuD/6KC0tzeASI0pV1SNHjuh/vQ99q7c1NLiKioKZmdYhM23B\n4/Ho/+nl9Gxr4owZIuK75BKXQYU5nc7QYrdnbnxjdzqlurqPi90qihIKSiqfe86Xk5OyaZN+\nqD03t7W2VlT1gvvuc0bB+B1VVevq6pYujSsoSAw1VlTIb38rqiqrVxtVFwAAAGAOjOCAiIjV\nau1la1ibbbAFYU1NTXq6McSnLYRmpoQb4GBUYVarNTExMe7UxjfjZ84cP3Nm8rvv6kcz5s7t\n+2K3vewQrFks6tGjyc884/zoo2gYv1NZWdnY2Fhd7Zg/v+H11w8WFJT++MdN+qHNm40qCgAA\nADCNwfbFFecmPj7e8t//He5LYHxrq6SkGFthZOnLqYb7Vh88eTI5OdngEgeExWIRkXADHPyX\nXmpgbVlZWWpxcdjDfY4hhg0bFgizQ/CYBQtafvCD5LffjobxOy0tLfpMsY0byxMSgnrjnXc2\nvvZaqogMrhFUAAAAQL9gBAdERJKTkxM+/1x6+mv5mAUL7G+8YWh1kRcMBnsZsaLvJzoUuFwu\nEQk3wCEmNlaqq42qzWazWffvP1ZbW1ZaWrRvX2jjm7Nd7DY+Pj6+tDTc0cT//d9u43eM+q/f\n2tqqPwilGyKyY0e8/uCWW74aVfSvfwUfeKCDRToAAACAbhjBARERRVESysrCHh50i27abLZe\npi04Ghpk2DBjKxwYDoej60yQbgMcUmbPlmeekUcfNag6sVqtmZmZmZmZmqbJv/7V2Xr2i93a\niovb2toaGxu9Xq+juHjsD38oIg333Zf4v/9rb2joNn5HaW+Pj4+P6O/RJ/4zIoqSEufzz6eL\nyBVXeGbP7hBJ6ujoqK6uXrYslUU6AAAAgDMxggOdlKIi0TT3iRP1dXX1oRn/TzwhY8fKDTdE\nw+4SEZSQkBBufYcxCxbEvf22odUNqOzs7NhIzATpV4qiKPPmiaaJpslNN53DGeLj40ePHn3R\nRReNbepc1SJ+x44ex++E9pTpWb/tt9I5XeiUgoLEuXNz3G7rlCkn1607YrcrgUDg8OHDbW1t\nLNIBAAAA9IgRHDhNQkKC0+n0FBXpT31vvx0TBbtLRFxycnJH+G/1lilTBrIYY1mtVktJSVNz\nc1tbm8/niz1wIPu73xURWblSnnzS6Or6wT33lE+f7vi//zf7V78KO34nISHUXdM0fXUMn89n\nVZQR8+b10yvC5XK1t7eLiKrKunUZGzakaZrk5zctXVprs2kul6uurk5f/pZFOgAAAIAeEXDg\nNB6Pp6KiInjjjUf37ctYsyZt40bV6bR4vSKDKuBQFMXy5ZdHqqr0ZR2dpaXj8/NF5OTjj8c9\n/bTR1X1FVVX9q7WIuNrb46+/XvRv1xFNHxRFSU1NTU1NFREpLOxsPfuZIGaRlJRkCb/saNdZ\nOaqqVlRU6EvSikjGmjUx//hHP70iUlNTm5ubT5ywPP74yMLCBLtdW7KkJj+/WS/YbrefOHFC\n79njIh233aaJKBGsBwAAADAdpqjgK6qqVlZW6ossJm7ZkrZxo3v6dN/48SISyMhQB9ffiG02\n25gxY8aNGzdixIj0o0f1xrhvfcvYqrpyu91lZWWVlZV1dXV1tbUye7b082gaTdPk1ltl7FgR\nkZtvHkyTkrpKSUnp46ycmpqaULrR7RWhjRghEd1vxel0er0jZ88eV1iYYLNpDz98bMIE3969\nsaWlKR0dI4LB4Jmrn3ZdpGPu3EAEiwEAAADMiBEc+Epra2sgEBCRmMOHRy5bpu8uMXHGDBFp\nz81V3e6kpCSja4wwl8vlcrnkZz+Tn/3M6FpO097eXllZGVoPImPt2vjt2/tp7IDH42loaPB4\nPMGOjpwHH4wdjJOSulIUxVZcXF1b29LSomlaaPxOx5NP2leuDHXr6OhoaWnRH5/5ivBdcokz\n0oW9+WZSRYWISCCgrF6dFWpfs0YWL+6+MkhBQeLSpSM9HsuUKSfXrq2w2ydEuhwAAADAZAg4\n8BV9CQBLW9uoxYtFpNvuEmp7++ALOKLWsWPHQulGaOyAraHBVVQUzMy0Rm7sQHNzc01NjX6t\njLVrYz/+eFBOSurGYrFkZ2dnZWV5vV7LRx/pjfarr+7aR385SJhXhCc3N+IBx+7dPbdPniyK\nojidTq/XKz0t0hEba7fZeDMHAADAUMcUFXxF0zTRtAuWLDmX3SUQOaqqejwe/XHXsQPOAwdE\nxDNpUqT+W/j9/lC6ceakJC09PSJXiVoWiyU2Ntb5wAM97s+iqqqIhHtFeC+++DyvvnOnPPSQ\n5OVJero4HDJmjFx+ufh8nbV0/WfaNBGR4cOHi4jbbV24cPT69Wk2m7Z8efXy5TU2m6YfAgAA\nAIY4/uiHrzgcjpRNmxK2betxdwlnY6NkZfV+BkREMBjUQ4dwo2lcwWBE/mLf3NysX6jHSUnK\nyZPx8fHnfxWTstvtIhLuFWGLiZHqasnOPreT+3y+FSuUd95xhFoqKuS3vxVVldWre/6R5OTk\n4uKO2bOTKiocXRfpSExMTEjoXCIWAAAAGMoIOPCVpKQkT/jdJYJPPy2XXGJQaUOL1WoV+Wrs\nQOVzz/lyclI2bdKPtufmdnY4b16vV9M068mTPcYoFq93KAccsbGxdrs9LswrIv3//J+u+62c\nldra2sbGxkOHxs+f3zBjRmtKSnDjxsxXXkkSkc2bwwYcIvLOO2nhFul46KFzKAQAAAAYVAg4\n8BWHw2EpLQ131Dp16kAWM5RZLBaXy+X8y196HDsQExur1NSc89iBbhSRcDFKXEQuYFqKoowY\nMcJxak5KD85pjZK6urrGxkYR2bixPLTh6+zZ9XrA0ftWRb0s0gEAAACAgAOnsRUXt7S01NXV\ndXR0hHaX8C5Z4ly1yujShpaMjIxAmLEDmXfffc5jB7qJiYmx9T4paWgv7pCQkODetauittbv\n94tI6BWhrVih/OpX53DCYDB4/PjxUyf/atvXHTs6R8rk5/f24++9dw7XBAAAAIYKAg50l5yc\nnJyc3NHRoRYW6i3O664ztqQhKD4+PlhWFvZwhPY3SU5O9oaflKStXi2PPRaRC0UDTdOU5mbJ\nyxN9H9yVK+XJJ7/2pxISEuLj4/1+v8/nizn1ilCmTDm3Gk6ePHnmArElJc7nn08Xkbw837x5\nMed2ZgAAAADsooKe2e32mPvv73F3CQwM6/79HX5/4/Hj1VVVDVu2dLauXCmaJtOnR+QSTqcz\nPvykJCUvLyJXMVZzc/OhQ4f279+/v6jIc8stnemGnEVIpChKTExMYmLi+b8igsFgt5aCgsS5\nc3PcbuuUKSf/8z+r2ewVAAAAOGcEHED0stvtw4YNy87OTqus7GyKdOhgKy5ubWk5eOBA0b59\nB994Q2/sePLJCMYoBqqqqqqurm5vb1dVNf13v4v9+GPV6ew8FqFRMGel6943qiovvJDxyCOj\nPB5Lfn7Thg1HkpPP5Q15505ZtEidPFkNbTf7y1+K3x+5ogEAAACT4M+FgBncc4/cc89pLU1N\nZzvbIpykpKSkpCRVVdXt2/UW+9VXn3upUaO5ubmlpUV/nLhlS9rGje7p020NDa6iomBmpjUj\nY+BLiouLs1gsqqq63dbHHx9ZWJhgt2tLltTk5zeLyDnsWdPU1LRkiX3LloRQy9duNwsAAAAM\nVozgAMxB07Tjx4/rsy2Kv/yy/dZbz2G2RS8sFovtvvsG06SkpqYm/UHM4cMjly3zjR5dtXKl\n88ABEfFMmnTmWhgDwGKxpKWlVVY6Zs8eV1iYYLNpDz98bMIE3969sfv3J7rdqWd1tmPHjtXU\n1FRV2efPb3j99YMFBaU//nHnr7x5cz9UDwAAAEQ3RnAAJqCq6pEjRzwej/40Y80aV2Gh6nRa\nvF4RY2ZbRD+v1ysilra2UYsXi0jl2rX2+nrF5xOR9osvdvj9MTEGrOiZlpa2eXNsRYVDRAIB\nZfXqrNChNWvkoYf6ep729nZ9Q5au283eeWfja6+lisjw4ZqIEtnKAQAAgCjHCA7ABOrr60Pp\nRmi2hW/8eBFRs7LEiNkW5qBpFyxZElNRUbVqlS8nx7V/v97cnptryAgOXWlpXI/tkyefxUlC\ns2963G72+9/3nWNxAAAAgGkRcADRTtO05uZm/fGZsy3ac3MNrS56xcTEpGzalLBt24kZM/wj\nRzpLSuJ27Og8ZrU6GhqMKuy99zpnAnX7Z9q0sziJ/4x1REPbzV5xhefHP3ZHsGAAAADAFAg4\ngGjn9/v17UV7nG1xctIkg+uLVsnJyXG7dolIYkHB+Jkzx8+cmfzuu/qh0fPnW/761/679M6d\n8tBDkpcn/beziaKcNgOl63az69YdsduZnwIAAIAhhzU4AJM4Ndui8rnnfDk5KZs26c36bItu\nX3chIsOGDesoLg57uH8WLlFVtaGhYenSuM2bv9oSpT92NnG5XCdOnBARVZV16zI2bEjTNMnP\nb1q6tNZm01wuV8SuBAAAAJgEIziAaGe32xVFCTfbwhYTo9TUGFthdFIUxV5SUl9XV1JcXLRv\n38E33tDb1V//WjRNpk+P+BWDweDhw4cbGhqOHrX1984mycnJFovF7bYuXDh6/fo0m01bvrx6\n+fIam01zOp2xsbGRvBgAAABgBozgAKKdxWJJSkoKzbZILCjoejR73jx55hl59FGDqotqiqKk\np6enp6cHAgHln//UGy1Tp57XSZuaJC+vc4/elSvlySdDR44dO6Zv3dLjziZpaed12W7sdrvf\nP2r2bEdFhaPrdrNWq3Xy5CxG9AAAAGAIYgQHYAKZmZmhHUB6EGXbxAYCgdbW1vr6+qamJm9N\njeTkiKKIoshTTxlVks1ms/70p52Led50U29dm5rOLDgQCNTX11dUVBw6cMD7wx92phty2r95\nVVV739kkPz9iv47u9dfju243O2dOzpw5ObNmjf773x0RvhIAAABgBozgAEzAZrPJ/v1Vx461\ntrZqmuYsLR2fny8i2ooVyq9+ZXR1p2loaKivr+/chFVVx9x/f49xQFRpaWk5ceKEz+dTNG3k\nT3/qPL3gtra2o0eP6uu8ZqxZ4/zoI9XptHi9oQ46n8935tazXXc2mTcvwtNGdu/uuf2stpsF\nAAAABg1GcADmYLPZRo4cmZubO2HChLFNncs6KFOmGFtVN8ePH6+rqwt9z89YuzZ++3bV6ew8\nPGABR0+jMHqkaVpFRUVVVZUecCStXpPAMScAAB8DSURBVK3nF52Hv/nNQCBQWVmppxuJW7ak\nbdzonj7dN368iAQzMyUjo5eTd93Z5Pe/r7RFOk+OyHazAAAAwKBBwAGYiaIoDoejr7MtBlYw\nGKyvrw897RYHqFlZvccBZ0vTNE3TumYZnieeqKqqqigv9/3oR30cNlJfX+92u3ssWBsxQjIy\nmpqaVFUVkZjDh0cuW+YbPbpq5UrngQMi4pk0yefzhU7lcDhCK1+oqrzwQsYjj4zyeCz5+U0b\nNhwZNswewd8dAAAAwJmYogIgMk6ePKlnAXJ6HDBxxgwR8V1ySUR2LlVV9fjx4y0tLR0dHaKq\nYx94IPZUltEwapS7pSVjzZqYf/yjx1kk3Wia1tjYGK7g9tzcWBGPxyMilra2UYsXi0jl2rX2\n+nrF5xOR9osvDra3x8TE6GewWq2JiYmtra1ut/Xxx0cWFibY7dqSJTX5+c0ikpKSEonfHgAA\nAEBYjOAAEBmBQEB/EC4OOP9LqKpaXl5eX1/v9/s1TUv/3e9iP/44NKOkPTf3rGaR+Hw+PZHp\nseCTkybpVxRNu2DJkpiKiqpVq3w5OaHVXttzc0OBji4rK6u2Nm727HGFhQlddzY5eHD4iRME\nHAAAAED/YgQHgMiwWq0iEooDKp97zpeTk7Jpk36047LLzv8SdXV17e3t+uNQlmFraHAVFXWk\np1tbW7uNwvBMmhQbDHYWdobOeCJMwZ5Jk0TEbrenbNqUsG3biRkz/CNHOktK4nbs0DtoFouj\noUFSU/WnO3fKyy/bXntt7LFjIqd2Nglda80aeeih8/8XAAAAACAsAg4AkREXF6coSvJbb/UY\nBzjj4qS6WrKzz/n8mqaF9mHtYQrMRRf1OGxEaW+Pj4/v8YQOh0NEwuUXtpgYqa5OSkpSd+0S\nkcSCgsSCgq4/PmbBAm31annsMRFRVXXlyuDf/hZ2oQ12NgEAAAD6GwEHgMiw2WwpKSmxYeKA\npNtvl2eekUcfPefzd3R06LuZ9DijxF5VdeYojPbc3JhgsJeC4+Li4sIUnD1vnjzzTOKjj/qL\ni8OdQcnLExGfz1dRUXHw4Kj581tmzGhNSQlu2JD22mupInL55bJnzzn/xgAAAADOAgEHgIjJ\nysoKlJSEPRyRbWLDzCiJKS8PO4skKamXgpVTa2qEK9hWXFxz7Fhzc7Omac7S0vH5+SLiXbLE\nuWqViKiqWlFR4ff7N24sT0joDFPuvLNRDzjS0iLwGwMAAADoCwIOABGjKIq9pMTj8bS1tfl8\nPldZ2fAZM0REVq6UJ588z5Pb7XaLxZL0P//T44wS+bpZJD1yOp3evXsP19Tou6WE8gv117+2\nLF+u97FYLCNGjMjIyGhvb7d8+GHnD153nf6gpaXF7/eLSCjdEJEdOzonxdx6a4C3WQAAAGBg\n8MkbQITFxsbGxsaKiGzZ0tmUl3f+p1UUJTk5OdwUmLA/9XWXdjqdOTk5gUDA6/U6Pv5Yb7RM\nndqtm9VqjY+Pl4ULZeHCru0nT57s1rOkxPn88+kicsUVnpkzvSKpfakTAAAAwHlim1gA/eae\ne0TTRNPkppsicr6MjIzY8CtilG/cWLRv38E33tCftj/xhGiaTJ/elzPbbLb4+HjHggVnW3Dw\n9DU+CgoS587NcbutU6acXLfuiKKEXQEEAAAAQGQxggOAaVitVqW4+Fh9fUtLSyAQCM0o0Vas\nUH71qyyv1+v12rZu1Tu7pk0bgJLs9s6dU1RV1q3L2LAhTdMkP79p6dJam00LHQUAAADQ3xjB\nAeDrNDVJTo4oiiiKPPWUsbVYLJbMzMyJEydOmjQpp7lZb1SmTBERp9OZnJwcv3hxZIeN9C4h\nIUFE3G7rwoWj169Ps9m05curly+vsdk0RVHC7VALAAAAIOIYwQGgB16vt6mpqb29PdjRccF9\n97nKyzsPRGQnlEiwWq1y771y773GlpGYmHj8ePLdd6dXVDhsNu3hh49NmODbuzdWRCZOTLLZ\neI8FAAAABggfvgF019zcXFNTo2maiGSsWeMqLFSdTovXKxJFAUf02Lw5u6JCEZFAQFm9OivU\nvmaNTJ5sXFkAAADAEMMUFWAwOo9JJV6vN5RuJG7ZkrZxo3v6dN/48SISzMyUjIx+KdjMPv9c\n6bGddAMAAAAYSIzgAAaJ1tbWpqYmr9erBYNj7r8/9lwnlTQ2NurpRszhwyOXLfONHl21cuXE\nGTNExDNpkisQYNpFN++9Z3QFAAAAAAg4gMGhurq6+dSKmxlr1sR+/PE5Typpb28XEUtb26jF\ni0Wkcu1ae3294vOJSPvFF0t7u76sJgAAAABEFaaoAKbX0tISSjfOf1KJqqqiaRcsWRJTUVG1\napUvJ8e1f79+qD03Vx/cMfhF08YxAAAAAPqCgAMwvcbGRv1B10klzgMHRMQzadLZRhIOhyNl\n06aEbdtOzJjhHznSWVISt2OHfkizWBwNDZEtPnr4fL7q6uqDBw8Wf/ml55ZbJPo2jgEAAADQ\nC6aoAKbn9Xol/KQSh98fExPT97MlJSUpu3aJSGJBQWJBQddDYxYskGeekUcfjWT10cHtdh89\nelRVVTnvOT4AAAAADMEIDmBQCDOpJPWVV2KczrOaZ5GcnBxbXBz28GD8th8IBELpRrc5PoGM\nDC09/fwvsXOnLF6sffObWnq6OBwyZoz88pfi95//iQEAAAB0YgQHYHoOhyP21Vd7nFRiOzV7\npe/BhKIotuLi2rq6pqYmTdOcpaXj8/NFxL9smWOQrkbR0tKipxtnbhzTnpurnDwZHx9/Pud3\nu91LlyqbN391kooK+e1vRVVl9erzrB0AAABAJwIOwPSSk5PtYSaVfOVsRl5YLJasrKyMjAyf\nz2cpLNQbHddcc96VRqneN45R2tvPJ+Boamqqqak5enT8/PkNM2a0pqQEN2xIe+21VBHZvJmA\nAwAAAIgYpqgApjds2LDeJpWc6zwLi8Xicrli7r9fNE00TW666TxqjGqapp3txjE7d8pDD0le\nnvQ+5aSjo6O2tlZENm4sX7iwbuJEb0ZGx513dg6rGT58aGxJAwAAAAwIRnAApmexWGzFxYfK\ny/WRCIkffDDqZz8TkeP33jvs5ZcVn689N1fa2hISEoyuNEr1vnGMs7FRTo+H3G738uX2v//d\nGWoJN+WktbVVz0cSEoKhxh07OseD3HyzRySuf34nAAAAYMhhBAcwGFgsFpvNJiKWtrYRp1bK\n6MjMDM2z8Pl8RtYX3ZKTk+NOzfEZP3Pm+Jkzk999Vz80ZsGC+L/9LdRTVdUjR45UVFSUl8v8\n+Q2vv36woKB01qwW/ejmzd3PfOa/9pIS5/PPp4vIFVd4Zs480T+/EAAAADAUEXAAg4emqhcs\nWWJrbKx87rmiffvUU7vDtufmGltYlHM6nfGlpeGOWqZMCT2urq5ua2uT06ec3HFHg340La37\nzyqK0vVpQUHi3Lk5brd1ypST69YdsduV7j8AAAAA4FwRcACDhNPpTP1//6+3eRYIz1Zc3NTY\nWFJcXLRv38E33tAbfUuXiqbJ9OmdT32+1tZW/XGPU07y87uf1unsnMaiqvLCCxmPPDLK47Hk\n5zdt2HAkPl4NHQUAAABw/liDAxgkUlJSPGH2UhmzYIG2erU89phBpZlDampqampqR0eH9vHH\nekvMtdd27XDy5Mkzfyo05SQvzzdvXky3o0lJSXV1dS0t8vjjIwsLE+x2bcmSmvz8ZhGx2WyJ\niYn98psAAAAAQxIjOIBBwuFwJJSVhTuq5OUNZDHmZbfbHQsW9LhxjKqq3Tp3nXLy0kt1tjMS\nY6vV6vePmj17XGFhgs2mPfzwsQkTfHv3xu7bF6dpoywW3oEBAACAiInqj9cffPDBrbfempGR\nERMTc8EFF9xyyy0ffvhhtz6HDh2aM2dOZmam0+m88MILly1b5vF4jCgWMJ51//42t7v88OH9\nX34ZmmcRXL686zwLnDNblwDjzCknyck9v52+9lpcRYVDRAIBZfXqrDlzcubMybnjjrGbN8cO\nUN0AAADA0BC9U1SeeOKJp59+OiYm5qqrrsrIyGhoaPjkk08uvfTS66+/PtSnqKho2rRpra2t\n3/ve93JycgoLC1etWvXBBx9s3brV5XIZVztgmPj4+Pj4eBFRt2/XW6xXXmloRYNHfHy8oiia\nprnd1jOnnISbb7J7d89nmzy5/yoFAAAAhqIoDTheeumlp59++uqrr3799dezs7P1RlVVm5ub\nu3abN29eS0vLSy+9dPfdd+sd7rzzzldeeeU//uM/li1bNvBlA9HDcu+9cu+9RlcxqNhstvT0\n9M8+a37ggTEVFY6uU06cTmdCQs8Rx3vvDXihAAAAwJCkaJpmdA3d+f3+0aNHu93uQ4cOZWRk\nhOu2e/fub37zm9/4xjc+//zzUGN1dfWoUaNGjBhRWVnZbYPGc7Z+/foFCxa43W79D+MAhrKH\nH/asWdPD7JI1a+Shhwa+HAAAAGBA+f3+mJiYTz755JprrjG6lu6icQTH1q1bjx07NmfOnKSk\npL/+9a9FRUUul+vKK6+84YYbumYWW7duFZGbTl8FMDs7+7LLLtuzZ09ZWdlFF1000KUDGOxK\nSnpeO4MpJwAAAICxojHg+Oyzz0Rk2LBhl1122YEDB0LtV1999VtvvRUa01FaWioiZ6YYEyZM\nIOAA0E+eekomTJBPPpHKSmlpkREjZNYsWblSHA6jKwMAAACGtmgMOOrr60XkxRdfHD9+/LZt\n2/Ly8srLy3/+859v2bJl1qxZ27Zt07u1traKSFJSUrcfT05OFpGWlpY+Xm7v3r0dHR29dKis\nrDzbXwHAoNTR0bFihfbOO1+FGRUV8tvfiqrK6tUG1gUAAADA0IBDVdXFixd3bXnkkUdycnKC\nwaCIKIqyadOmiRMnisill1761ltvTZgw4cMPP9y5c2deXl4vp9VXFenjAhyHDh2aPHmyfsXe\nWa3WvpwQwGDV1NRUW1t76NC4+fMbZsxoTUkJ/ud/pv/1rykisnkzAQcAAABgMIMDjhdffLFr\ny6xZs3JyclJSUkRk4sSJerqhi4uLu/HGG//4xz+GAg597IY+jqOrcCM7ejRu3LgTJ074fL5e\n+nz66aff+c53CDiAoezEiRM1NTUisnFjeUJCZyQ6Z85xPeBISzOyNgAAAABibMBhs9l63MNF\nXztDn2nSld7i9Xq7dtNX4uhKX7ZjwoQJfSwjNjY2NrbnVQN1CQkJfTwVgMGqrq5OfxBKN0Rk\nx47OnZXy8w0oCQAAAEBXFqML6MG3v/1tRVFKSkq6LY2xb98+ERk7dqz+9IYbbhCR999/v2uf\nmpqaL774Ijs7u+8BBwD0LhAInDnOq6TE+fzz6SJyxRWeefOMKAsAAABAF9EYcGRnZ//whz88\nfvz4qlWrQo3vvPPO1q1bhw8fPmPGDL1l8uTJU6dO/fzzz//0pz/pLaqq/uIXv1BVdcGCBX1c\ngwMAvtaZy/QUFCTOnZvjdlunTDm5bt0Ri0U1pDAAAAAAIdG4i4qIPP/887t3716xYsXmzZsn\nT55cUVHx97//3W63/+EPf4iLiwt127hx43XXXfeTn/zkzTffHDt2bGFh4a5du6688sqf//zn\nBhYPYJCx2b56q1RVWbcuY8OGNE2T/PympUtrY2IsFks0hsUAAADAkBKlH8pHjBjx2WefLVq0\nqKamZsOGDf/85z9vueWW7du333LLLV27XXLJJbt27br99tu3b9++bt265ubmJUuWfPDBBy6X\ny6jKAQw+VqtVf1dxu60LF45evz7NZtOWL69evrzGZtPi4+ONLhAAAACAKD0u84mutm/ffu21\n1/p8PofDYXQtAIxx8uTJjz6quf/+0RUVDptNe+SRY5df3i4iiqLk5WVfeCFvDgAAABgS/H5/\nTEzMJ598cs011xhdS3dROkUFAKJKXFzcBx+MqqhwiEggoKxenRU6tGaNPPSQcZUBAAAAEJGo\nnaICANHmyy9jemyfPHmACwEAAADQA0ZwAECfvPee0RUAAAAACI8RHAAAAAAAwPQIOAAAAAAA\ngOkRcAAAAAAAANMj4AAAAAAAAKZHwAEAAAAAAEyPgAMAAAAAAJgeAQcAAAAAADA9Ag4AAAAA\nAGB6BBwAAAAAAMD0CDgAAAAAAIDpEXAAAAAAAADTI+AAAAAAAACmR8ABAAAAAABMj4ADAAAA\nAACYHgEHAAAAAAAwPQIOAAAAAABgegQcAAAAAADA9Ag4AAAAAACA6RFwAAAAAAAA0yPgAAAA\nAAAApkfAAQAAAAAATI+AAwAAAAAAmB4BBwAAAAAAMD0CDgAAAAAAYHoEHAAAAAAAwPQIOAAA\nAAAAgOkRcAAAAAAAANMj4AAAAAAAAKZHwAEAAAAAAEyPgAMAAAAAAJgeAQcAAAAAADA9Ag4A\nAAAAAGB6BBwAAAAAAMD0CDgAAAAAAIDpEXAAAAAAAADTI+AAAAAAAACmR8ABAAAAAABMj4AD\nAAAAAACYns3oAkzA4XCISExMjNGFAAAAAABgPP1rcrRRNE0zugYT+OKLLwKBgNFVRLunnnqq\nrq7uwQcfNLoQwADvv//+xx9//G//9m9GFwIYYM+ePS+88MIf/vAHowsBDHDs2LHHHnts7dq1\nKSkpRtcCGOCuu+76/e9/n5eXZ3QhGFA2m+3yyy83uooeEHAgYh544IGmpqZXX33V6EIAAzz3\n3HMvv/zy7t27jS4EMMC77757++23t7W1GV0IYIADBw5MmDChqqoqOzvb6FoAAyiKsm3btuuv\nv97oQgAR1uAAAAAAAACDAAEHAAAAAAAwPQIOAAAAAABgegQcAAAAAADA9Ag4AAAAAACA6RFw\nAAAAAAAA0yPgAAAAAAAApkfAAQAAAAAATI+AAwAAAAAAmB4BByLG4XA4HA6jqwCMwf2PoYz7\nH0OZfvPzEsCQxf8CEFUUTdOMrgGDRGtrayAQGDZsmNGFAAbwer1NTU0jRowwuhDAAKqqVlZW\njhkzxuhCAGMcPnw4JyfH6CoAY5SXl48ZM0ZRFKMLAUQIOAAAAAAAwCDAFBUAAAAAAGB6BBwA\nAAAAAMD0CDgAAAAAAIDpEXAAAAAAAADTI+AAAAAAAACmR8ABAAAAAABMj4ADAAAAAACYHgEH\nAAAAAAAwPQIOAAAAAABgegQcAAAAAADA9Ag4AAAAAACA6RFwAAAAAAAA0yPgAAAAAAAApkfA\nAQAAAAAATI+AAwAAAAAAmB4BB77em2++uWjRomuvvTY+Pl5RlFmzZoXreejQoTlz5mRmZjqd\nzgsvvHDZsmUej+ecuwFRa+LEicoZMjMzz+zJ3Y7BjTscQwHv+Rg6+NgPs1M0TTO6BkS7vLy8\nXbt2JSYmZmZmlpWV3X777a+++uqZ3YqKiqZNm9ba2vq9730vJyensLBw9+7dV1111datW10u\n19l2A6LZxIkTDxw4cNddd3VtTEpK+t3vfte1hbsdgxt3OIYI3vMxdPCxH6anAV9n27ZtBw4c\nUFX1b3/7m4jcfvvtPXabOnWqiLz00kv602AwOHv2bBF56qmnzqEbEM0uuuiimJiYr+3G3Y7B\njTscQwTv+Rg6+NgPsyPgwFno5Z1u165dIvKNb3yja2NVVZXFYhk5cqSqqmfVDYhyffmwy92O\nwY07HEMH7/kYgvjYD5NiDQ5ExtatW0Xkpptu6tqYnZ192WWXVVVVlZWVnVU3IPqpqvrv//7v\n8+bNW7hw4YYNG5qamrp14G7H4MYdjiGF93wghI/9iGYEHIiM0tJSEbnooou6tU+YMEFEQm9h\nfewGRL+Ojo6lS5f+13/914svvjh//vzRo0e/8sorXTtwt2Nw4w7HkMJ7PhDCx35EMwIOREZr\na6uIJCUldWtPTk4WkZaWlrPqBkS5uXPnbtmypba21uPxFBUVLVy40OPx3HXXXYWFhaE+3O0Y\n3LjDMXTwng90xcd+RDOb0QUgWqiqunjx4q4tjzzySE5OznmeVtM0EVEUJSLdgIHUy4viiSee\nCDVefPHFzz//fFJS0qpVq37zm99Mmzat99Nyt2Nw4w7H4MN7PtAXfOxHNCDgQCdVVV988cWu\nLbNmzep7wKGns3pS21W37LaP3YBocFYvinnz5q1aterTTz8NtXC3Y3DjDsdQxns+hjI+9iOa\nEXCgk81m0/PUc6PPr9Pn2nV14MABOTXXru/dgGhwVi8Kfbylz+cLtXC3Y3DjDsdQxns+hjI+\n9iOasQYHIuOGG24Qkffff79rY01NzRdffJGdnR16C+tjN8B0/vGPf4jIuHHjQi3c7RjcuMMx\nlPGej6GMj/2IZgQciIzJkydPnTr1888//9Of/qS3qKr6i1/8QlXVBQsWhGbZ9bEbEM0+++yz\nvXv3dm3ZuXPngw8+KCJ33XVXqJG7HYMbdziGCN7zgW742I9oppzPrAQMEW+++ebbb78tIlVV\nVR988MGYMWO+9a1vicjw4cOfffbZULeioqLrrrvO7XZ///vfHzt2bGFh4a5du6688spt27a5\nXK6z7QZErWefffaxxx4bN27c2LFjExMTy8vL9+zZo2naD37wgzfeeMNut4d6crdjcOMOx1DA\nez6GFD72w/Q04OssXbq0x5tn9OjR3XoePHhw9uzZaWlpDocjJydnyZIlbW1tZ56wj92A6LR7\n9+6f/vSnl156aWpqqs1mGz58+I033vjyyy+rqnpmZ+52DG7c4Rj0eM/HkMLHfpgdIzgAAAAA\nAIDpsQYHAAAAAAAwPQIOAAAAAABgegQcAAAAAADA9Ag4AAAAAACA6RFwAAAAAAAA0yPgAAAA\nAAAApkfAAQAAAAAATI+AAwAAAAAAmB4BBwAAAAAAMD0CDgAAAAAAYHoEHAAAAAAAwPQIOAAA\nAAAAgOkRcAAAAAAAANMj4AAAAAAAAKZHwAEAAAAAAEyPgAMAAAAAAJgeAQcAAAAAADA9Ag4A\nAAAAAGB6BBwAAAAAAMD0CDgAAAAAAIDpEXAAAAAAAADTI+AAAAAAAACmR8ABAAAAAABMj4AD\nAAAAAACYHgEHAAAAAAAwPQIOAAAAAABgegQcAAAAAADA9Ag4AAAAAACA6RFwAAAAAAAA0yPg\nAAAAAAAApkfAAQAAAAAATI+AAwAAAAAAmB4BBwAAMF5VVZWiKLfeeqvRhQAAALMi4AAAAP3C\n6/UqXcTExKSlpeXl5d13332bN29WVXUgi3nzzTcXLVp07bXXxsfHK4oya9asgbw6AAAYAIqm\naUbXAAAABiGv1+tyuRwOx09+8hMRCQaDLS0t+/fv379/v4hcddVVf/7zn3NycvTOfr//008/\nHTZs2KRJk/qjmLy8vF27diUmJmZmZpaVld1+++2vvvpqf1wIAAAYhYADAAD0Cz3gSEpKamlp\n6dpeWlq6ePHizZs3jx07dufOnampqQNQzIcffjhy5Mhx48a9++673//+9wk4AAAYfJiiAgAA\nBtRFF1307rvvXnvtteXl5atXr9Ybz1yDY8+ePYqi3H333QcPHvzRj36UmpqamJh48803l5WV\niUhtbe3dd9+dkZHhcrmuu+66Xbt29X7R66+/fvz48Yqi9N/vBQAAjEXAAQAABprNZluxYoWI\n/OUvf+m9Z2Vl5dVXX11bW3vHHXdMnTr1vffeu/766w8ePDh16tQvvvjitttu+/a3v/3JJ5/c\neOON3caJAACAoYaAAwAAGGDatGkOh+Po0aO1tbW9dNu2bduiRYv++c9/vvDCCwUFBffee29t\nbe3UqVNvu+223bt3r1u37p133lm2bFlzc/P69esHrHgAABCFCDgAAIABHA5HWlqaiDQ0NPTS\nbfTo0UuXLg09vfvuu/UHv/nNb0LzTfTGPXv29E+lAADAHAg4AACAMfSVzntfF+OKK66wWq2h\np9nZ2SJy8cUXu1yubo1VVVX9VSgAADADAg4AAGAAn893/PhxEdHHcYSTlJTU9anNZgvX2NHR\nEfkqAQCAeRBwAAAAAxQWFvr9/lGjRmVmZhpdCwAAGAwIOAAAwEALBALLly8XkTlz5hhdCwAA\nGCQIOAAAwIAqKyv77ne/u3379nHjxj322GNGlwMAAAYJm9EFAACAway9vX3BggUiEgwGT5w4\n8eWXX+7fv1/TtGuuuebPf/5zSkrKwJTx5ptvvv3223JqLdJ//etf+t4rw4cPf/bZZwemBgAA\n0K8IOAAAQD/y+/3r168XEYfDkZiYOGrUqHvvvXfmzJkzZszoff+UyNq9e/cf//jH0NMjR44c\nOXJEREaPHk3AAQDA4KDoO7QBAAAAAACYF2twAAAAAAAA0yPgAAAAAAAApkfAAQAAAAAATI+A\nAwAAAAAAmB4BBwAAAAAAMD0CDgAAAAAAYHoEHAAAAAAAwPQIOAAAAAAAgOkRcAAAAAAAANMj\n4AAAAAAAAKZHwAEAAAAAAEyPgAMAAAAAAJgeAQcAAAAAADA9Ag4AAAAAAGB6BBwAAAAAAMD0\nCDgAAAAAAIDpEXAAAAAAAADTI+AAAAAAAACmR8ABAAAAAABMj4ADAAAAAACYHgEHAAAAAAAw\nPQIOAAAAAABgegQcAAAAAADA9Ag4AAAAAACA6RFwAAAAAAAA0yPgAAAAAAAApkfAAQAAAAAA\nTI+AAwAAAAAAmB4BBwAAAAAAMD0CDgAAAAAAYHr/Hzge4h39Kq5bAAAAAElFTkSuQmCC"},"metadata":{"image/png":{"width":720,"height":720}}}]},{"metadata":{"trusted":false},"cell_type":"code","source":"FG<-pseudoTimeOrdering(FG,quiet = TRUE, export = FALSE)\nplotOrderTsne(FG)","execution_count":6,"outputs":[{"output_type":"display_data","data":{"text/plain":"Plot with title “Pseudo-time ordering of k-means clustering”","image/png":"iVBORw0KGgoAAAANSUhEUgAABaAAAAWgCAIAAAAnwnOfAAAACXBIWXMAABJ0AAASdAHeZh94\nAAAgAElEQVR4nOzdd3xb1d0/8HO1JUuyhuW9V+zYWc5OyCRkUSCDQJhhFlrKaBlllgKheWh/\nFMrTp3l4KKOFQoDghJ0EAkkgAzKcxHaGHTveW7K1t+7vj5soinRly0uynM/7D17Suet7r2+E\n7lfnfA9F0zQBAAAAAAAAAIhmnEgHAAAAAAAAAAAwWEhwAAAAAAAAAEDUQ4IDAAAAAAAAAKIe\nEhwAAAAAAAAAEPWQ4AAAAAAAAACAqIcEBwAAAAAAAABEPSQ4AAAAAAAAACDqIcEBAAAAAAAA\nAFEPCQ4AAAAAAAAAiHpIcAAAAAAAAABA1EOCAwAAAAAAAACiHhIcAAAAAAAAABD1kOAAAAAA\nAAAAgKiHBAcAAAAAAAAARD0kOAAAAAAAAAAg6iHBAQAAAAAAAABRDwkOAIB+iIuLoyiKoqh7\n7703PEfcvHkzdd6pU6fCc9DIGr6LHF0X8+9///ukSZMkEgkTcEFBQbA1o+u8osWouaqj5kQA\nAAD6hAQHAIRbRUUFFUAoFKalpa1aterLL7+MdIAR4L0O69evj3QsMCK88cYb999//9GjR61W\na6RjgWGEf/sAAABDiBfpAAAACCHE4XA0NTU1NTVt2bLlzjvv/Oc//xnpiEYKlUo1efJk5rVY\nLI5sMNEuii7mu+++y7zIycn561//KpfLJRJJZEOCKBVFtz0AAMAgIcEBAJGUk5MzceJEj8dT\nV1dXVlbGNL755ptLlixZs2ZNZGMbIRYuXHjo0KFIRzHEjEajTCYL/xGj6GLW1dUxLy6//PKr\nr746orFAtIq62x4AAGCQMEQFACJp6dKlmzdvLi0tPXLkyGuvveZt//jjj72vP/nkk6VLlyYk\nJPD5fKlUmp6evmjRoscee6yrq8t3V/v27bv55pszMzNFIpFUKp04ceIf//jHnp4e33VeffVV\nb4dw30UikYhpfPrpp72Ndrv9+eefz8nJEQqFOTk5zz33nN1uD3Yiu3btWrt2bUZGBnP04uLi\n3/72t/X19X1egUWLFlEU5X37zDPP+EXIOn5+06ZN3sYTJ068/PLLeXl5YrG4uLj4X//6FxP8\nM888k5mZKRQKCwoK/vGPfwQeOpQr1otQTtk3+BMnTvztb38rLi4WiURXXXUVs0K/LnIoAfd+\nRNaL6df4/vvvT5s2TSKRqNXqG264obm52Xf/DofjhRdeyM3NFQqF2dnZzz77rN1u71fRkD6v\n22233UZRVGNjI/P2//7v/5idP/TQQ33u3Nf777/P4/GYbdeuXetyuYKtGbbbqaOj4/777587\nd25aWppMJuPz+Wq1evbs2X/5y198R+L06y8S4ucDq7KysrvvvnvMmDFSqVQikWRnZ19//fUH\nDhzoZZN+fYb0Hluf//ZDv7Aj/7YHAAAIExoAILzKy8u9H0H33Xeft/3s2bPe9rlz5zKNr776\narCPr7KyMu+2zLNB4Drp6elVVVXe1V555RXvop6eHm+7UChkGp966immxe12L1myxG9vixYt\nUigUzOt77rnHu/nDDz/MGqFUKv3qq696vxqXX355sBPs7u6mado313Py5Elmqw8++MDbOH/+\nfL8N//a3vwU2vvrqq77HDfGKBRPiKfsG73umzN+3Xxc5xIB7PyLrxfRtXL58ud/+CwoKbDab\n965YunSp3wqXX345a8ADvm7r1q1jXefBBx8MttvA8/rggw+4XC7Tcscdd7jd7l6iCtvt5O2l\nFaikpMRkMvX3LxLi5wOr9evXczgsP/Ns2LAh2FWl+/MZ0mdsff7bD/3CjvDbHgAAIGwwRAUA\nRorPPvvM+zoxMZF58be//Y15ccMNN6xevZqm6cbGxkOHDn399dfelTdt2vTCCy8wr2+//fbl\ny5fr9fq//OUvp0+fbmhoWLVq1dGjR70Pe16szzZeGzdu3L59O/M6PT19xYoVHR0dH330kcfj\n8VvzX//618svv8y8zsnJWbNmjcFgePvtt61Wq8lkuu66606fPp2cnBzsQHfdddeiRYueeOIJ\n5u3ixYsXLFjAvA5xtPyuXbtKSkry8vK2bNnicDgIIQ8++CAhZNKkSfn5+aWlpU6nkxDy0ksv\nMe1koFdskKe8c+fOtLS0ZcuW8Xg8m81G+nORBxZw4BH79NVXX+Xm5k6dOvX7779va2sjhJw6\ndWrr1q3XX389E/C2bduYNTMyMlavXt3W1rZp06bAgAdz3dasWVNQULBhwwaDwUAImT59+ooV\nKwghU6dODeUohJCPPvro5ptvdrvdhJAHH3zwlVdeYX1IZjXct1N+fv6KFStycnISEhJsNtvu\n3bv/93//l6bpI0eObNy48ZFHHvGLp/e/SCifD8EukberhUAguP7663Nzc5uamrw3ZCh6/wzp\nM7Y+/+2PjtseAAAgrCKbXwGAS5BvD46cnJzVq1evWrWqpKTE96Ppo48+YlbWaDRMy44dO3x3\n4nQ67XY783rChAnMOmvXrvWuUFlZ6d3btm3bmEbfX1/NZrN35cBfX4uLi5kWpVLZ2dnJNHqf\nWIjPj5beNdVqtfd3V99kzTPPPNPnNfGu/MILL/gt6rMHx/Lly5nf55966ilv4+LFi5nGZ599\n1tvY0NDQ3yvGKvRT9g1+3Lhxvr949+sihx5w70fs86fsWbNmWa1Wmqbr6uq8SYHf/e53gQF3\ndXUxjX/9618DAx7kdaNpOiUlJZR9Bp7C888/z+Od+/XiySef7HNbOoy3k29HEpfLZTKZjEbj\n4sWLmdUWLlwYeDq9/0VC+XxgNXHiRGZDgUBw5MgRb7vD4aivrw8Mg7UHR++fISHG5t1b4L/9\n0XHbAwAAhBNqcABAJNXU1HzyySdMDQ5v42233eatMDp9+nTmxeLFi7OyspYtW/bb3/528+bN\nDodDIBAQQoxG47Fjx5h1fEsJFBUVeXfIWmCvl19frVar9ynimmuuiYuLY17feeedfmsaDIaK\nigrm9cqVK719tq+66irvVvv27WNeLF26dMbFqqure7s6obn33nuZcykoKPA2/vKXv2Qax48f\n723s7u4mg7hijH6dsq9HHnkkNjbW+zb0izzggP2OGIrf/va3IpGIEJKRkeF9QGWum1/AarWa\neX333XeHsucBX7f++sMf/sCU29iwYcOLL77ouyiUO3BYbycOh/Pxxx8vXbpUo9HweDypVCqT\nyXbs2MEsbWpqCjydXv4iJITPB1ZGo/Ho0aPM69WrV0+aNMm7iM/np6enB9vQT+89OAYWm2+Q\no+C2BwAACDMMUQGAEUEgEGg0milTptxxxx2+c0b8/e9/r62tPXHiBCGkrq6urq5u27Ztr776\nakJCwu7du8eMGaPT6frcOWvhTPr8b6dut9uv/qJer/cu9Q6WIYTExMRIpVKTyeRt8T5o+a3J\nvGVKCXojPHTokFar9V3HbDb3GXyfMjIymBfMIwojKyuLeeHbg53pUj7gK8bo1yn7Gjt2rO/b\n0C/ygAP2O2Io8vLyvK+9v8kz1y1YwFKp1C9gVgO+bgPD4XCys7P9GkO5A4f1dnr++ed9+4D4\nYa0v28tfhITw+cB6IN+YMzMz+4w/mF4+QwYcG2uQwYz82x4AACDMkOAAgEi67777/v73v/ey\nQkZGRkVFxYEDB/bt21dVVXXy5Ml9+/a53e729vZHHnnk888/V6lU3pV/85vfsBZx9P6e6fuL\nq9VqZQa6nz17lqlW4Ls+RVHM13pmRDrDbDb7faFXKpXe175r+r71jXA4sP4a7G2kffrAB8bT\n5xULNOBTlkqlfocI8SIPOGC/I4bC+3RHAn6f9w24vb3d2240GkN5zAvbrVJYWHjy5EmPx3PT\nTTeJRKL+TjE7fLeT3W5/6aWXmJY5c+b885//zMnJ4XK5q1evLi0tDRZPL38REsLnA+s+fWP2\nrW0cihA/QwYcG2uQ0XvbAwAAhBmGqADAiNbV1UVR1MyZMx9++OHXX399z549zz33HLOIqeUh\nk8m83ea/+eabhISETB8pKSk7duzwfuP3jgsghOzfv58QQtP0n/70J7+DisVibz/wTz/91Dvf\n5Jtvvum3plwu9w5Q37Jli/cH1c8//9y71axZs7zn4jdK0FsIwFs0wXeyzGHSrysWqF+n3IvQ\nL/IgAx4qzLSpzOvPPvvMe+Kvv/56KJsP1XXr07///e9p06YRQlwu13XXXedbNbOXO3DAQv/r\ntLe3WywWZs1rr702Pz+fy+Xq9fq9e/cO+Oh9fj4Ei9l74qWlpb5zu7jdbtaRMl4hfoaEHluw\nf/uj47YHAAAIM/TgAIARbdmyZcx/MzIy4uPjOzs73333XWZRfHw88+L3v//9TTfdRAg5ffr0\nzJkz77nnnpSUFK1W+9NPP5WWlnZ2dt566618Pp8Qwjz4MW644YYlS5bU1NSwTl35y1/+8oEH\nHiCEdHd3T548eeXKle3t7R999FHgmo888shtt91GCNFqtVOmTLnuuuv0ev3bb7/NLJVKpffe\ne2+fp5mamlpXV0cIeeutt/h8vlQqTU1NXbt2baiXqZ9Cv2KshuSUSX8u8iADHir33HPPb37z\nG0KIVqudNGnSNddc09ra6lussXdDdd16x8w4O3fu3BMnTtjt9pUrV3711VeB87wOoRD/OhqN\nRiQSMfN6vPLKK0zXgFdffdW3X0B/hfL5wOqJJ55gpghxOBwzZsy4/vrr8/Ly2travvnmmzvu\nuOPxxx8PtmHonyEhxtbLv/3RcdsDAACE1fDVLwUAYOX76+V9993X+8qTJ09m/ezicrlbt271\nrvb000/3MhEmM0EA45prrvFbesUVV3gfErwzILhcrkWLFvmtOX36dLlczrz2nTjgd7/7Hetx\nmUfNUK7Jo48+6rft7NmzmUV9zqLCOi1CeXk507hlyxZvY1lZ2QCuGKsQT5k1eK9+XeQQA+79\niH1OJ+G7ibcaxbp167wBL1261O/Q8+fP944U+NWvfjUk140exCwqzCk0NTV545dKpfv27etl\n87DdTr///e/9FqnV6hkzZjCvMzIygp0OI/AvEuLnA6v169ezVgndsGFD72GE+BkSYmy9/NsP\n/cKO8NseAAAgbDBEBQBGtMcff/yuu+4qKSlJTEwUCARCoTAzM/PGG2/ct2+f72PGCy+8sH//\n/nXr1uXk5IjFYolEkpWVNXfu3GefffbAgQO+5RLff//9Bx54ICkpSSAQ5Ofnb9iw4csvvwx8\nzuFyuZ9//vmzzz6blZXFTKzw2GOP7dy5k/X30pdffvm777677rrrUlNTBQKBRCIZO3bsgw8+\nWF5ezvyK26cXXnjh0UcfzcjI8PZXH26hXzFWgz9l0s+LPMiAhwTzaPrcc89lZWUJBIKMjIwn\nn3xyy5YtRqORWcG30AarIbluoUhJSfnmm2+YngImk2nZsmWHDx8ewv37CfGv86c//en1118f\nP368WCxWq9XXXnvtgQMHcnJyBnzcED8fWD311FMHDx6888478/LyxGKxUChMS0tbvXp1n71d\nQvwMCf2zq5d/+6PjtgcAAAibc4WjAAAAoE/eupJe//nPf26++Wbm9datW/t8rgaIOrjtAQAg\nWiDBAQAAEKqFCxcqFIqFCxemp6ebzeb9+/e/8cYbTF2JMWPGlJeXh6EmAkCY4bYHAIBogQQH\nAABAqC677DLWWT+ysrK2bduWn58f/pAAhhtuewAAiBbcP/7xj5GOAQAAIDoIBAKXy2W1Wl0u\nFyEkLi5u+vTpDz300BtvvJGUlBTp6ACGBW57AACIFujBAQAAAAAAAABRD7OoAAAAAAAAAEDU\nQ4IDAAAAAAAAAKIeEhwAAAAAAAAAEPWQ4AAAAAAAAACAqIcEBwAAAAAAAABEPSQ4AAAAAAAA\nACDqIcEBAAAAAAAAAFEPCQ4AAAAAAAAAiHpIcAAAAAAAAABA1EOCAwAAAAAAAACiHhIcAAAA\nAAAAABD1kOAAAAAAAAAAgKjHi3QA0YGm6SVLluh0ukgHAgAAAAAAo9CCBQv+8pe/RDqKEeH+\n++/fv39/pKOAQYnU/UzRNB3+o0Ydh8MhFAofe+yxnJycSMcCAAAAAACjyvbt25ubmw8cOBDp\nQEaE8ePHf8R1RDoKGJTbhIqI3M/owdEP11xzzaxZsyIdBQAAAAAAjCrd3d1btmyJdBQAUQ81\nOAAAAAAAAAAg6iHBAQAAAAAAAABRDwkOAAAAAAAAAIh6SHAAAAAAAAAAQNRDggMAAAAAAAAA\noh4SHAAAAAAAAAAQ9ZDgAAAAAAAAAICohwQHAAAAAAAAAEQ9JDgAAAAAAAAAIOrxIh0AAAAA\nAAAAwAUcPjfSIUBUQg8OAAAAAAAAAIh6SHAAAAAAAAAAQNRDggMAAAAAAAAAoh4SHAAAAAAA\nAAAQ9ZDgAAAAAAAAAICohwQHAAAAAAAAAEQ9JDgAAAAAAAAAIOohwQEAAAAAAAAAUQ8JDgAA\nAAAAAACIekhwAAAAAAAAAEDU40U6AAAAAAAAAIALOAJupEOAwXFF5rDowQEAAAAAAAAAUQ8J\nDgAAAAAAAACIekhwAAAAAAAAAEDUQ4IDAAAAAAAAAKIeEhwAAAAAAAAAEPWQ4AAAAAAAAACA\nqIcEBwAAAAAAAABEPSQ4AAAAAAAAACDqIcEBAAAAAAAAAFEPCQ4AAAAAAAAAiHq8SAcAAAAA\nAAAAcAGHz410CDA4rsgcFj04AAAAAAAAACDqIcEBAAAAAEAIIQaL02yP0M+OAENNq9U+/PDD\n8+bNk8vlFEW99957vay8fv16iqISExP92vV6/a9//evExESRSFRSUlJaWtrfFQDCCQkOAAAA\nALik0TTZX9X53EfHnt987NkPj/6ptPxYfXekgwIYrNbW1nfeeUcgEFxxxRW9r3ny5MkXX3wx\nISHBr93j8Vx55ZXvvffeU089VVpampWVde21127dujX0FQDCDDU4AAAAAOCS9sXhxt0n2r1v\ndSb7u7trTNPTZ4+Jj2BUAIM0duxYrVZLCPn222976Vjh8XjuvPPO2267rbq6uqKiwndRaWnp\n3r1733nnnXXr1hFClixZUlJS8sgjj6xYsSLEFQDCDD04AAAAAODS1WN2/HCyI7D96yPNTrcn\n/PEADBUOJ6Rnvddee62+vv6//uu/Ahdt3bpVJBKtXbuWecvlcm+55Zaamprjx4+HuAJAmCHB\nAQAAAACXKJomW39u8NB04CKb092stYQ/JIBwqq2tfeqpp/77v/87NjY2cGllZWVeXp5QKPS2\njBs3jhDi7ejR5woAYYYhKgAAAABwifquorWisSfYUvTggFHv7rvvXrx48apVq1iXarXa7Oxs\n3xaVSsW0h7hCMB9++GFZWVmwpXV1dUSdE0L4AP6Q4AAAAACAS5GHpn1Lb/ihCEmIFYczHoAw\ne+ONNw4dOnTixIn+bkhR1CBXaG1tra2tDbbUYkHnKRggJDgAAAAA4FJksDgtwSeFHZ+hlEv4\n4YwHIJy6uroeffTRxx9/PCYmpqenhxDicrlomu7p6REIBBKJhBCiVqt1Op3vVsxbpptGKCsE\n89BDDz300EPBlorFyC3CACHBAQAAAACXIk7wH5k1MtGamZlhjAUg3JqamvR6/ZNPPvnkk0/6\ntiuVynXr1r3zzjuEkKKios2bN9tsNpFIxCxlqocWFxczb/tcYcA4Au4g9wARZo3MYVFkFAAA\nAAAuRXIJXyUVsi5aOT1dNMzPVzUdxp0n2r4+3nLorM7hQrEPCLfc3NzvL1ZSUqJUKr///vvH\nH3+cWWflypV2u/2DDz5g3rrd7nfffTcnJ2f8+PEhrgAQZujBAQAAAACXqGWTUv7zg38hgNxE\nWV6yfPgO6nB5Pj7YcKbd6G3Zc7rj2qlp6eqY4TsoXJo+++wzh8NRXl5OCDl48CDTz2LVqlUc\nDkcqlc6fP993ZaVS2dzc7Nu4cuXKWbNmPfDAA3q9Pjs7+6233qqoqCgtLQ19BYAwQ4IDAAAA\nAC5Rk7JUHA712cFGvcVBCOFQ1Mx8zbJJKX0USByc7RWtvtkNQojR5vzw54YHFuUL+eiWD0Pp\n1ltv1ev1zOvXXnvttddeI4RYrVbviJLecTicL7/88oknntiwYYNery8sLNy8efOKFStCXwEg\nzJDgAAAAAIBL14QM5bh0hdZot9jdCQqRaJhTDC635zjbxLQWu+tkq2FiunJYjw6XGqZ6aIi+\n/fbbwEaFQrFx48aNGzcG26rPFQDCCQkOAAAAALikcShKIw/pB+3BM9hcLjd7xQ2dyRGeGAAA\nRisUGQUAAAAACBMhL+jXbyEf38wBAAYFH6MAAAAAAGESI+QlxrL3FsmJl4U5GACAUQYJDgAA\nAACAfjM7XDanewAbLh2XzOX4lzGdkqUKlvgAAIAQoQYHAAAAAECoaEIqW/U/1+ksDjchRC7i\nz8pW52qkoe8hIy7m7vm531S0NWjNLo9HFSOcnadBeVEAgMFDggMAAAAAIFT7arvKfKZBMdic\n2060zcvTjEuODX0nCXLRzbMyaZq4PR4eF12qAQCGBhIcAAAAAAAhMdldR5tY5t3cX6stTJTz\nAgae9I6iCLIbAKy4guGdsBlGK3ykAgAAAACEpEVvpWmWdofb02m0hz0cAAC4CBIcAAAAAAAh\ncXnY0hvnFnnCGQkAAATCEBUAAAAAgJCoJIIBLPLloekug73b7IiTCVUyYf/GtAAAQK+Q4AAA\nAAAACEmCXBQvE3YEjEbJ0UhjhH1/r65tN328v65FZzm3VYLsulmZiUpx6AF09FjrOkwUIVkJ\nsjhMKwsAcDEkOAAAAAAAQkIRsrwoafuJtlaDzduYqY5ZmB/f57YtOss/tp1yui+MZKlpN772\n9cnHV4yTS/h9bm5zuN/9rvqHyjamCAiHouaPT7ppQa6AhyHnAADnIMEBAAAAABAqqZC3elJq\ng87SabJTFEmSi5NC60nxzfFW3+wGw2xz7T7RdtWUtD43f/3rU4eqO71vPTT93bEWp8vzy2UF\n/YofAGAUi9aM7969e5cvX65UKiUSydixY19++WXfpXq9/te//nViYqJIJCopKSktLY1UnAAA\nAAAw+qSrJJPTlSVpyhCzG4SQ+k4Ta3tdp1lvdpTX6U419ljsLtZ12rotvtkNrx8r27pNmL0F\nAOCcqOzB8eGHH950000zZ8588cUXZTJZbW1tR0eHd6nH47nyyiuPHz/+4osv5uTkvPnmm9de\ne21paemKFSsiGDMAAAAAXMpY55clhLTqLL/6770emiaECPnclbMzr5mRTlEXlR+t72BPjtCE\n1HeYlFLhUAcLABCVoi/B0dbWdtddd61YseKjjz7icFh6oJSWlu7du/edd95Zt24dIWTJkiUl\nJSWPPPIIEhwAAAAAECmpaomOrbdFp87qOZ/8sDvdm3bVOJzu6+Zm+67jl+84jyaECrIIAOBS\nFH1DVN555x2TybRhwwYOh+Nhm29869atIpFo7dq1zFsul3vLLbfU1NQcP348vJECAAAAAJxz\n+bgkLsc/GUHTxGJ1+DV+8VOD3en2bclJlLHlMSguh8pOlA1tnAAA0Sv6enDs2bMnNTX16NGj\nV111VVVVlVKpXL169Z///GeFQsGsUFlZmZeXJxRe6Ko3btw4QkhFRcX48eN72bNOp+vp6WFd\n5HQ6h+4MAAAAAOCSkxkvvWNh3sf763rM5zIaUhG/ocXgdvuPXXG4PA0d5rwUubdFLRfNH5f8\n/fEWvzWXlKTKxH3PwAIQdTh8bqRDgEFi6YsQBtGX4Ghpaenp6bn99tufeeaZyZMnHzhwYP36\n9eXl5Xv37mVGrGi12uzsizr1qVQqpr33PY8fP765ubmXFfbv3z9r1qxBnwEAAAAAXIqK0xVj\nUsbXdZh6zI44uai6Sf9WfTfrmnRAxY5bL8+VS/hfHWp0ujyEEAGfe9W09KtnpA970AAA0SP6\nEhwej8dkMr3yyisPPfQQIWTRokUURT399NM7duxYunRpLxv2OUCxrKzMaDSyLrJYLOPGjZNK\npQMOGwAAAACAz+XkJZ3rmkEH9N3wrpMWf+5rZ7PWXFaj0xptmljxvHFJy6emNXWZORSVEicR\nC6LvmzwAwLCKvo9FtVpNCPHNZSxfvvzpp58+cuQI06hWq3U6ne8mzFumH0cvNBqNRqNhXWQy\nsVeuBgAAAAAYmOwk2YRs9bFa/17Gy6amiQVcQkjpvrovDzV5POfyIFsP1K+ZnXnFpJRwBwoA\nECWir8goU0fDt7wo89o7o0pRUVFVVZXNZvOuwJQXLS4uDmugAAAAAAC9euCaonnjkrxv+VzO\nilmZa+dnE0J+Ot35+c+N3uwGIcTp8ry/u/Z0sz4CgQIARIPoS3CsXr2aEPLFF194Wz777DNC\nyIwZM5i3K1eutNvtH3zwAfPW7Xa/++67OTk5vVcYBQAAAAAIsxgR71e/KPzvX896ePW4x6+b\n8Pf7Zq2dl82hKELInoo21k2CtQMAQPQNUZk7d+611177zDPPGI3GKVOm7N+//+WXX166dOn8\n+fOZFVauXDlr1qwHHnhAr9dnZ2e/9dZbFRUVpaWlEY0aAAAAAICdJlakiRX5Nbbrrawrt3Wz\ntwMAQPQlOAgh77333gsvvPDvf//7pZdeSkpKevjhh//4xz96l3I4nC+//PKJJ57YsGGDXq8v\nLCzcvHnzihUrIhcvAAAAAED/iPg8QuyB7RJhVH6BBwAIg6j8fBQKhevXr1+/fn2wFRQKxcaN\nGzdu3BjOqAAAAAAAhsq4TGWz1hzYXpyhDH8wAABRIfpqcAAAAAAAjHrLp6TGyf3HraRpYhaM\nT2JdHwAAkOAAAAAAAAgfuu9VCCFEJuY/e+PEBeOTZGI+IUQRI1g+JfXJNRMEPHyBBwBgF5VD\nVAAAAAAAos7PVZ1fHWpq1lqEfE5+Suya2ZlJKkkv60tF/FsX5t66MNfh8iCvAZcUjoAb6RBg\nkDwROSoSHAAAAAAAw27TntrtR5qZ1y63p6xGW1Hf/eiqcXnJ8j63RXYDACAU+Hln6yMAACAA\nSURBVKwEAAAAABhezVrLjrJmv0any/Pe92ciEg8AwKiEHhwAAAAAAEHZne6vDjZWNnQbrc4U\ndcySktQxqbH93UlFfTfNVnujodPcY3YoYgRDECgAwCUPCQ4AAAAAAHY6o339prJOvY1526K1\nHKzqXDUrc+WszH7tx2p3BVtksbuQ4AAAGBIYogIAAAAAwO6D3TXe7IbXlv11jZ2mfu1HEyti\nnT6Fx6XUMuHA4xsQu9NNs/YnAQCIcujBAQAAAADAwu2hD5/pCmynaXKwuitNIw19V5Ny1FKR\nwGRz+rVPy9cI+WGaLcLtobcdavz8QEOn3ibgcYozlesW5adqYsJzdACAMEAPDgAAAAAYuWhC\ndGZHnc7SYbS7PWHtd2C1u5wu9pkO9WZHv3YlEfJ+fWWBXML3bRyTEnvT/JyBx9dPr26peGt7\nFdMhxeHyHDmjffSfP1U368MWAADAcEMPDgAAAAAYoXQWx4812k6TnXkbI+DNylalKyXhObpE\nxBPwuQ6nO3CRUtrvcSWFaYoN66b8UNne1GUWC7j5qbGTc+MoQjw0fayuu77T5PLQKSrJlBw1\nnzv0v0FW1nfvO9Hu1+hwed75pvrF26YM+eEAACICCQ4AAAAAGImsTvdXlW12nz4UZodr5+mO\nZWMTE+WiMATAoahp+ZofK9v82rkcatoYzQB2KBHylpSk+LboLY63vjvTrLN4W76vaFs3PydF\nNcRJnGO1Otb20409dqc7bMNkAACGFYaoAAAAAMBIdLLNaA8YIeKhybEwjqq4YV52SpxvlQqa\noqhJOeqaVkN7j3Xw+9+0t843u0EI0Zns/95dM+SDcWwO9mlcaEKsDpYuKgAA0Qg9OAAAAABg\nJNIGqXPhHbESBnKJYP0tk7892lJZ360z2nUmu8nqPFTddai6i8uhFpekrp2XzaGoge28x+yo\nbjUEtmuN9po2Y36yfHCxXyQpSJcQqZjvVxkEYCTgCNCrKNr511QOD/TgAAAAAAAIisflLJ2c\nev/VRRaHy2S98JXd7aG/PtT46f56b4vd5TnRathbqz3a1NMVQhZGawy6TlfwRQMzfYxGJhEQ\n4p+LuWJSyoATNAAAIw16cAAAAADASBQnFTR0Wwih/R7LNf0v8Dl4h890deltge07jjRfPSOD\ny6HqtObd1V0217nhHj9TZGyifHZOXC/JA1Hw36jFQ/fzdYvO8unBxtoOY5xGoqaJwWjr1ttp\nD00Iuawo4fr52UN1IACAiEOCAwAAAABGosIE2Yk2o+3iSUw4FDUxVRH+YJo6zaztJpuz22QX\nCLjfnu7wLZxB06Sy1SAX8cenxAbbZ5JSLJfwDRb/jtw8LpWbJBuasLWW/9l2yuk+V8qEokis\nXJSolhSnKMZnqgrTI3AlAQCGD4aoAAAAAECEWZzun+p1X1S2fVbR+mOtVm91EkJEfO7ysYkJ\nsgv9NWRC3qIx8b4tYcPjBu2KweNyTrebWMuCnmhjKbHhxaGoVdMzAkeILJ2YIhMNTV2Mr8ua\nvNkNL6vTk5+mQHYDAEYf9OAAAAAAgEjqNNm/rer0Pof3WJ11OsvsLHWWWqKU8H9RnNRjdRps\nTomAp5LwI1UwIj+VrSMGTRJVYkWMwNDUw7qVwer00HQvMRenKe5fVvBVWXNDp8lN08lKyRUT\nkguDd/roF5qQmnYT66IzbYZJWaohOQoAwMiBBAcAAAAARNK+Op1fLwMPTf9Ur0tRiARcDiFE\nIeYrxBGe6WNsunJcpqq8TndRK0Wun5tDgvfv4HKpPjMycXJRUbpCJReJBZzseNlQZTcIITRN\nuz3+3TcYjoD5dwEARgEkOAAAAAAgYvRWp97KMpugw+1pNdgylOyTm4YfRciD1xSV7qvbcaTZ\n5fYQQuIV4psX5E7KURNCUhXiU23GwK1SFX3EX91m3LSvzmx3MW9/PNWZmyC7ZU6WkD8ERUY5\nFBUnE3UaWGqjJirEg98/AMBIgwQHAAAAAESM1eUOtsivvGjECfncG+blrLksq63bKhZw1XKR\nd1G2Wpoca2zRW33XF/A40zJ6GwZisbve33vW6rjoNM+0G78oa149LX1IYr6sIH7Lzw1+jUI+\nd2pO3JDsHwBgREGRUQAAAACIALPNtauibVd5W7AVJIKR+FMcj8tJjYvxzW4QQiiKLCtKnJKu\nFPO5hBAeh8pUx6yamKKU9DayprJJ75fdYByt6w6sDDowswriFxYncjkXhskoYgS3L8iV9xoY\nAECUGon/2wAAAACA0e3oWd1/dtcwQzPGFcTHXPy8TRMi5nOTLk4ijHA8DjU5XTk5Xelwefg8\nTiilULUmO2u70+0xWJzqoZgshiJkeUnq1Ny4M21Gg8WZoBCNTVUIePiNEwBGJyQ4AAAAACCs\ntEb72zurvZ0Uauq7C3LVAp+qE3wONTtLHVqWoH9ompQ395xqMxpsTrmIX5goK05RDO3ELKGn\nD0TBC22IBBcW9Zgd7T3WGCEvUSnpZbbaXmjkIk1UZYsAuENRhgYuQUhwAAAAAEBYHfCZFJYQ\nYrE6j51oT4qXqmJF2YlylZg/NlEuEfg/3rg99MGz2toOk8XhVksF07LVKf0sQer20KVHmuq0\nZuZtt9lRrzWfajOunpwakdln85Pk2461BLanqSUxQh4hxGBxfPjj2YPVXUy7IkZw/WVZk3NR\nPgMAgB0SHAAAAAAQVp16/3k93G66qdXY1Gq867JsHpelB4TF4frXj2c7zk8I0txtKW/SLyiM\nn5MfH/pxjzX1eLMbXnVa87HGnknpyv6cwdBIVopn5mn2V3f6NvK5nKtKUgkhLjf9ymeVzVqL\nd1GP2fH69tP3cqiSbHW4YwUAiAYYgAcAAAAAYSXks38F5XM5XLbsBiHk28q2jounO6Vp+vuT\n7e0BuZJeVLWzzOTaS3sYXD0l9fqZGYkKMZdDiQXcotTYB5cVpMfFEEKO1HT5Zje8Pv/Jf1YU\nAABgoAcHAAAAAIRVYarihxPtLO1psawDRWhCTjTrWdppUtmiT4gNtbqEhW3KEkKIOUh7GFCE\nTMpUTcpUeWjab5hMbZC0S7POYne67S7Pt0dbGrrMhJB0TcwVE5Jl4mGZGKWuw1TR2NNjdqik\ngklZ6iSleDiOAgAwJJDgAAAAAICwmpCpKkxVnGzq8W0UC7grpmWwru90eewu9mlTjVZn6MeV\nCnmsE5fIhJH/ShxYBISmg65c3Wp8e2eVd4rZqhb93pMdv15WkJMoG8KQaJqU/lS/v+rCCJpd\nle2LJyQvGp80hEcBABhCkf80BwAAAIBLCkWRXy0ds7O8dXdFW4/ZIeRzC1NjV83IiAsy0wef\nxxHwOA62HIdUxCOE0ISc7TJ3mOw0TatjhLmaGA5F1bWbys/qjFZnkkoyozBeLOAWJsrqA2pw\nEEIKk+RDe4K+rA73zoq2Uy16o80ZJxNNz42bmq0OpaRpuiaGtT0+VrTph1rrxb1OrA7XO99V\nP7d2Emfopp75+UyXb3aDEOKh6W1HmzM0MXnDecUAAAYMCQ4AAAAACDcel7NkYsqSiSl2p1vA\n72PuU4qQwuTYYw3d/u0UGZsca3a4tp9o7/TpmnG0iW/QWr873Ow53wviwz019/1ibHGmqk5r\nOdVm8N1JYZK8KDl2SE4qkMHq/J8dVXqLg3nbrLOU/txwusVw85ysPvMQU/M0Xx5q6jL4FxmZ\nnBv3DdvcK1qj/WyHaQg7cRyq6WJtP1ij9UtwaI32IzVdWqNdKRVOylLFKzCMBQAiAwkOAAAA\nAIgYId9/OlhWVxQlNuksfgNM5uTHJynEn5e3dl7c3m1xmmiP7xCPHpPjr6Xlf71nxlUTkguT\n5CdbDQabUy7ij02W52ikQ3Ee7HYcb/VmN7wqm3pONOmLUvvIqgh4nIeuLvrXd9XVLecyMiIB\nd8X0jNgYQbBNAo81GDoT+960xouu9nfHWz/ZX+c837/m05/qfzE1ffnk1CGMBAAgREhwAAAA\nAMBIFyPk3bsg90CNtqbDaLa7NDLRtBx1hjpGb3O26K2B60slArlcoDdceBS3Otw/VLRdPSMj\nN16aGz+MSQ1fp1pYaqMSQk42953gIITEx4oeWTmusdPUorPIxPzMeFmMiFfdagi2vnxI64yK\nBVw9yywuRCK4kJM63azf9EOt71KXm956oD5ZJZmYpRrCYAAAQoEEBwAAAABEAR6Xc1m+5rJ8\njW+jIXiRUYmY75vgIIS0sM26Oqwsdvb5WSwOV4h7oAhJ10jTfbqZZCXIYiWCwM4aSqkgK2Eo\ni4wWpsS29bAkjwpTFd7XeyrbWLfdU9mGBAcAhB8SHAAAAAAQJnaXR8jjDOEO+dyge3MFFCUN\ncTjMEFJKBX4DOhiqGOGA98njULcuyHl9+2nfqqsCHufW+bncoaswSghZOC6porGn8+IiIJnx\n0ul5cd63HT3+JUIY7WyZEYDQcQTh/tcKowMSHAAAAAAwvBxuz+GG7tPtRrvLw+dyMlSSGVmq\nGMEQfBHVSIVCHidwElmPh+7R+z97jwt7n4KSTNU35a1+jRRFSgYXSWGq4g/XT9xe1lzfaSKE\nZGikSyelKqVBa3MMjFjAfejKwm+Ptx5v6O4xO+Jkwik5cXPHJvimUYLljETn210eullr7jY5\n1DJhiloSOBsuAMAQQoIDAAAAAIaRy0N/drxFaz43pMLp9pzpNDX3WFdPTIkRDva7KJdDzcpW\nf3/xbKaEkIYmg7fsJaEJocj4LNVkn64H4TF/bEJLt7WyqcfbwuVQV05KSVYOdp4RlVR4w5zs\nQe6kT0I+98rJqVcGrxg6Nk1RxVZnZGy6ghByqln/wQ+1neczTSlqyU1zczLDVQAFAC5BSHAA\nAAAAwDA61WbwZje8rE734caeublDkHHIj5dJhbyf63SdJgchRCnhT8lQccckvLuzurK+2+2h\nZRL+kimpV8/ICH/nAS6HumVO1ukWw8kWvdHqjJOLpmar42QDH58y0iwcn3SgqqOt+6IBKWqZ\ncOmk1MYu8z++OunyXJjMpllree2LE0+tmaAeRVcAAEYUJDgAAAAAYBg1BynH0NQzZCU/k2PF\nKyakMBPDegdBPLl2ottDm6zOXuZVDY8xyfIxyfLIxjAczDbXvpPt6eoYIZfTbrDZHG6JkDc5\nR71iRkaMiPfh3rO+2Q2Gzen+7njrmtmZkYgXAEY/JDgAAAAAYBg5A55yz7W72dsHLLC+A5dD\n9ZndsDrdP5zurGo3Gm1OhUQwIU0xPVs9tNU6R6Vjtdr/+fykwWcyl1mFCb++qpB3vuwrUx8k\nULB2AIDBG8oq1gAAAAAAfmJFfNZ2hZi9PZzMdtdbe2oP1ekMVidNk26zY9epjv8cqGc6g0Aw\nPSbHK1sqDBdPVbvvZPsnP9Z53wZLEaHMKAAMH/TgAAAAAIBhVJgoO9luCMwYFCVFftTG3uou\nvdXp19iksxxr7JmUruwy2LYcqD/VpLc6XElKyRUTk6ePicfjOSFk74k2m8Md2L7zaMt187KZ\nS5SukXYETGRDCMlAkVEAGDbowQEAAAAAwyhOKpyfpxFwL3zt5FBUSZoyVxP5B92aIMMlajpM\nDZ3m5zYdPVjdZbQ6XW66scv81rfV/9lVE57ArHbX6caekw09ZpsrPEfslzYde10Vg8VhOR/w\n4onJfK7/s4ZYwF04Lml4gwOASxh6cAAAAADA8MqPl6UqJLVas97qlAp56UqJweY80tgt4HKS\nYsXqsBQBrek01WnNZodbJREUJ8sVEgEhxO5k6YZACLE7PR/+WBu4dE9l2+zChKyEYUzNuD30\nR7tqPtp97ugCHmfVnKwbF+byApIFIarvNJ9q1hutTo1cWJKjlgUZMdQvQgGXtZ1DUQL+uThT\n1TH3LS/c9ENt2/kqs+mamBvn5KikmEIFAIYLEhwAAAAAMOwkAm5xkpwQorM4tp9s6zLZmXaK\nImMT5fPyNIElQoeKy0N/cbzlrNbMvK0h5Ehj97w8zYRUhUIisDhYOiPIxbw9rQbWvVXU64Y1\nwfHm16e27q3zvnW4PJu+rzGYHb9ZUdzfXXlo+uN99ftOd3hbvi5rXjs7a2KWapBBjs9SffFT\nQ2B7YbrCt9dGfrL86TUTmnWWbpMjTi5MUkpQgAMAhhUSHAAAAAAQJm4P/UV5q8F2oewFTZPK\nVoOQx52VrR6mg/50VuvNbnjD+L6qI0UhnpCuaGGbxTY/XhaszKiFrfbEUNGbHZ/vrw9s//pg\n0/ULcjWxon7tbVdFm292gxBitbv/vbsmVS2Jk/dvV37GZ6mm5msOVnX6Ngr53Fsuz/Vbk8Oh\n0uJi0uJiBnM4uARx+Oy9hAB6hxocAAAAABAmZ7Vm3+yGV0WL3h1kNtnBO9FqIMR/5zRNTrUZ\nJ6YpJ2de1J2By6GuKErMTZQJgzxfaS5ODdA0aey2Hm/RV7QaOs53Sxmw6mb260DT9OnGnv7u\nbf/FCQhCCKGI20P/VN01sPB8PbSy+JbLc9VyESFEwONMylVvuH1qZoJs8HsGABgw9OAAAAAA\ngDDRmR2s7Q63x2h3DcfEsTRNTHYX66SlepuTosiS4sRxqbFVbUaDzamSCIpSYpUxAkLIjDGa\n3RVtfpuI+NwpuRd6mhhszt012m6f2VLTlOLLstSBxTVD1EuSh+7nzLU0TboM7AmXDgPL5Cb9\nxeVQV05Lv3Jaus3hFvI5FAafAMAIgAQHAAAAAIQJlxP0MbiXRV4emm7R2/RWp4DHSZCJ5KK+\nv8pSFBHyOHaXJ3CR+HwfjWSFOFkh9lu6elZmp952wqffhETIu2NRnlwi8AbzXXWXX4eUxm7r\nz9zu2VkDHG6TnSijKMKayshJju3XriiKCHgcG1sVVdGQdv4XBSk4CgAQfkhwAAAAAECYBOYR\nGLFivkzYx/dSndmx+0yX7nx3CQ5FFSfLp6Qr+8yLZMdJT7axVAzN7rUwhIjPfejqomNndaea\n9RabK1klmT02XuozBUmrwcY63KZWa56aphTwWDpxuD10h8FmcbjUUqFCwjJ3TFysaOHElJ1l\nzX7tc8YlJqslvUTLqiAl9midLrC9MKV/uRIAgGiBBAcAAAAAhEmSXJQTJ63pMvm1z86O631D\np9uz/VS7b4FPD00fb9YLeZzxfXVtmJ0T19htMdpdvqmQgkRZprrvypcTslQTgsw5ore5WNtp\nmhjsrjief/6islm/vbzVeD4nkp8ov3JCsjxgVM79K4qEfM7XB5uYMSkURS0qSbn3qrF9hhro\nysmpVa0Gi/2iOPOT5RMyBzuLCgDAyIQEBwAAAACEz+LChMONgrLGHqfbQwhRSgRzcuLSVX10\nT6jVmlmnL6lsNfSZ4JCJeLdMzzhwVndWa7Y4XCqJYEKqYmySfMCnwOAFH1MTuOh0q2HzwXPz\nqtKEUIRUtRm0Jvu9C3J5FxfsEPC5v1lRfO28nDPNepqmc1Nik/q6OMHEx4oevabo04ONJ5v0\ndqc7ViKYUxi/oDgJ5TIAYLRCggMAAAAAwofLoaZlqKamqww2p4DHEYdWD6LbwjIYhBBicbht\nTnefRSVEfO78fM18oul3uMElyoSs7WI+N1bk3y9j16kL07V60wtak728ST8pQ8myc6U4Uck+\nnKdfVFLh7QtyCSF2pzvYvDAAAKMGpokFAAAAgHCjKBIr5oeY3SCEcIP3OuCEUJ10OMhF/DHx\n0sD2KWlKv2DdHrpNb2XdSVO3ZThiC4TsBgBcCtCDAwAAAABGugS5kLSwtKtjBIKBzsk6eNPS\nVQox/3iLwep0E0KUYv7kNGVyrCj0PfR38lcAAOgFEhwAAAAAMNKlKSWJclGbwebbyKHIlHSW\n8R1hQ1FkTLxsTLzM5nRzOFSwVAuXQ8XLRR0XB89IVvSvvobd6W7ttlKEJCrFg++UQRNisjhl\nEv8BNQAAUQoJDgAAAAAYcbRmx5kus8nukgi4aQpxqkK8uCD+cGPPyTajh6YJIQoxf2aWKjl2\nCApVDF6fRUDm5Gs+OdTo1xgr4Y9PU4R4CLeH/upw07YjTU6XhxAi5HOXT05dUpLCGVDJUL3Z\n8eEPtQdPd9qcbpGAO7MgYc2cLFnAlC4AkcIRYFAVDAQSHAAAAAAwshxt1pe36mn63IQj1Z2m\nDJVkTrZ6RqZqWoZSb3UKeVxJVD3/FKcqnG76m8pW6/m5YDLiYq6emCLghTq+ZtMPtbsr2rxv\n7U73lgP1Zrvr2lmZ/Q3GaHE++95hrdHOXF6bw/398ZYTDd3P3zJZIsTTAQBEMXyEAQAAAMAI\n0mqwHW/RM6+9nRPqdRaNVDg2QcahKKVEEKnYBmNShnJsSmxLt8Vsd2lkooT+lOroNjn2VLb5\nNdKEfHusZWlJijRg0pbefXmwQWu0E5/LSwhp77FuP9y0sv/pEgCAkQOzqAAAAACMZjqjfdex\n1k9+OLuvst12vvvASFarNbO3d7G3RxEhj5OlkRanKvqV3SCEnO0wBlYjpQjxeOi6dlN/w6hs\n6GFtr6jv7u+uAABGFPTgAAAAABi1Pt9f/86OKqvdxbzVxIoeXDWuJC8uslH1zhwkC2N2uMIc\nycjh8QSdbMXd/3lYguW57M4oyH8BAPQCPTgAAAAARqe9le0bPz9htV3IC3Tqbc+/d6RVZ4lg\nVH0SBilLIeRFU9GNoZUWF8PaTvksqm4xvLfzzKtbKt7/vqaho7duHUkq9sqsSar+TekCADDS\noAcHAAAAwOj06b46Qi4utECIw+n+6qeGO5cVRCKikKQpxPVsKZg05YiYMCVEp9uNDTqL1elW\nxQjGJcfGDm6CkgSFeFK2uqxW69c+fYxGJRXShLy9verrg42e8705tu6rWzM3e82cLNa9XT4h\nuazGf1dM+2CCBACIOCQ4AAAAAEanhnYja3tdm5EQ4nJ7jtXqmrvMKpmwKEOplAnDG11QWeqY\nOp2lqcfq26gQ88clySMVUr843J7PjrU0dp/L0dR0krKG7oVj4ouSYwez29sX5Ql3cw+c7mDe\nUhSZXZiwdk42IWTP8dYvf27wXdntoTftqslLlk/MUQfuakK2+sb5OR//eJaZcZYQIuRzb5iX\nUxDynLUAACMTEhwAAAAAoxM/yFgPPp97qqHnb1sqWs6X8xQJuDcuzF08OfXT/fXldd0mmzNF\nHXPltLSiDGUY4z2HImRBnuZMp+lMl9loc0oEvDSluDhRzuVQfW88Auyr6fJmNxguD73zdEeK\nUqIYRD8OEZ97x6K85VNSGzpNFKEyNDHxinNdWr471sK6yXfHWlgTHISQZVPSpuRpjtR0delt\nmljx5Lw4dUCGi6ZJu96qNdpjJYIkpTharj8AXMqQ4AAAAAAYnYoyVXuOtwa25ybJnnvvsMWn\nNofN4X57e9VnPzWYmUaatOusR850rZqded3c7LAF7GV3ecR8br5GKhfxNNKR0rUkRCfbWDrO\nuD306TbD9Cz2dIOfHrOjqsVgsDri5aLCVIVvoipRIU5U+A/V6eixse6nvdvK2s7QxIqWlKQG\nW9rSbfl4f33j+Zlr4mTCVdMz8pOjoxMNAFyykOAAAAAAGJ1uXJh78HSndwoVRrI6hqYo3+wG\ng8/nmL2N53+q37KvfnpBfEa8dNhj9XGy3Xi0We90nxs9ERcjmJ2lHmQNi7BxuDy2IHOR6AOu\nOatvj7d8cbDR4fQwfwW1THjL/Jz8Xoe3xIjYv9LHiAZ40QwW58btp60+k610Ge1vflf9m6UF\nwcqdAgCMBEhwAAAAAIxO6fHS/3fPjI2fn6g4qyOEUBQ1f0LSncsK3vz6VODKXC7LeBaapg9W\ndYYzwXGm03Swodu3pcvs+Kaq45riJD5bhCMNn8vhcig326yuYn7fs8D8XN1Vur+ekAs5Jq3R\nvnHb6T9cN1EpFQTbqiQ37ixbt5HJA50P+MfTHdaAqWTdHnpnRdtt83MGtk+AfuEKLt1Zk2Aw\nkOAAAAAAGLWyEmV/vnu6yerUGmyJKomQzyWEBGYKKCpoeQW92TG8IV6sos0Q2GhxuM9qLfnh\n7UgyMBRFsuJizrDN0poTQt+H78tZhhTZne69J9t/MTUt2FbXzMw4cKqj+fxwEkZesnzx5JRg\nm5htrq0H6o/XdevNjiSleMGEpDljE713QdPFu/JqDNIOADBCREEiHAAAAAAGQyrmZyTIhOd7\nEBSm+0+WQdMsPQ4YarloGCO7mMtDG4KM49BZwppnGYy5uRpJwI/P41NikwNqZwRq6WaZH5cQ\n0sw2b65XjIj30p3TrpmZkaAUczlUkkqydn7OC+umBOvzojPan3nv8LdHWzp6rHanu67D9PY3\n1f/46qT3Jgh6NwwRo82pNdqD33QAAAOEHhwAAAAAl5YFk1I+P9DQ2HlRLwPaQ1N+02TQhMul\nZhTEhy2w4P1Iels0MC4Pfayp56zWbLS75CJ+nkZanCznDMVhYsX8W2dkHqjV1uksNodbFSOY\nlKbIT5CFsi2PQzlZ27l9BCYWcG9dlHfrorxQjvLJvrpuk3/C6FB117Fa3cRsFSEkTS2pbmXp\nSpOmloSy/15UNvZ8dqhRa7QTQoR87sLixPlFiZifBQCGChIcAAAAAJcWAY/z4h1T395+etex\nFuZX9KxE2a1X5H+4p7beZ2wFl0vddkV+orLvfgdDhUtRKomAtbNG6HOp2J3ubrMjViIQBx/D\n73B7Pj3eojs/+qbLZO8y2et05l8UJw1JjkPA5YxPiZ2WpYoR9O/Ldm6SvLy+O7A9r9cio4Fs\nTvf2w01VzXqny5OmkS6bkhrn0xPn2Fkd61bHzmqZBMfsgvj9VZ1+ZTi4HGrhuKR+heHnUI12\n096z3rd2p/vrsuYOve2Gy7IGs1sAAC8kOAAAAAAuObExgodWjbvnysJWnUUpFSplQkLIhBz1\nzqMtFXU6g8WZpolZMjk1NexTZkxMif2uutOvUSkRZKr67jugMzm+ONJ0olnPvM1LlF09OVXD\nNsTmWJNed3FtEZqQVr3tVJtxbNKgZkJ1uDx7a7rKGrqZOqNqqfDygvgMfe3U1AAAIABJREFU\ndaiX8RdT0k41650uj29jklI8M1/T+4YmqzNGxGNqqbTqLP/18TGd0c4sOtnYs6u89VfLC6fk\nxRFCaEIsdvZxQCbrufZYieDexWM+3l/XpD03NEYlFa6anp4+iPuBpslXR5oC2w/XaucVJSQr\nB9s3BACAIMEBAAAAcImgaXrHoabdx1s7eqzxCvHc8UlLpqRm+zzPcznU4pKUxSVBK1OGQapC\nvDBP83NDt+n8Q3iWOmZqmqLPjhUmm2vjN1VG24URHtVtxo3fVN2/tEAZ4z//SL3Ov1gms/c6\nnWUwCQ6aJqVlTY0+9TK0Jvvmw02rSlKzQksNpMXF/O7qog9/PFvXYSKEUBSZnq9ZOT2Dz2Ov\npmF1uD/eU7vzaIvJ6hTyuVPy4269PO+f2097sxsMh9P9xrZTY9NnSIQ8ihC1TNRlsAXuTRN7\nIRmUopI8uHxsa7ely2hXxAiSlWLe4Gax6TTYDFbW8Tekts2EBAcADAkkOAAAAABGP5fb8+y/\nDped6SKEEJq0aC1Ha7Q/lLc9t25Kn/UdwixVIU6JFRtsTpvLEyvmi4I82/v58XSHb3aDYXG4\nd59sXzHl3PwjNE2q2wxtelurzkIoShAwhsXm9J8btV9qu0yNAdVAPTS9p6ojKy7UURgZGulj\nK8eZbS69xaGRi5jURrfJcaCqs8NgixHxClNii9IUhBCn2/OHfx+qbT03Qazd6d5b2X6sVuei\nPYH5IIvddfysjqmoMnts/KcHGvxW4FDUzMKL6q1QFElWSZJD6DsTCpfHE2yR0x10EQBAvyDB\nAQAAADD6ff1z47nsBjnfXYGQsjNdX//ccNXMDOatweyIEfNHQsVHiiKxYn6/yk6c7WSZmZUQ\ncvZ8VZFus+PDA/W+05GIRTyVQuybCpCLBvXduKnbytreYbTbXR5haJkaRoyIF3M+mJ+quzbv\nr3OcH7fyw4n2ojTF7Qtzvzva4s1ueJmsTh6XIxCw/BG93Tp+MTWtodNcVqP1LuJxOTfOy04L\neQSK1eH20HSMsB+XSy0T8biUy+0/dQpFEa3ZsfmnBiGfk6WRFqUpIn//AUDUQoIDAAAAYPTb\nW9kerH3xlNSPdtd+eaDeYHHyuJySXPVdVxaGv/rGILkDnpwZLg9NCPHQ9H/2nm3XXzQuw2pz\ndettKsWFcRn58efmOrE53c06i8XhjpeLEmJDnSjXHbyTAlOSYwA69LYP957127yysWd7WcvJ\nOvZaoW6PhxC/ZApNCBV7fqgOj8t54Kqxx87qjp3VGSzORKV4blFCfAiz2BJCKhp7th1r6TLa\nCSFyMf+KcUlTctShpCSEPM60XM2+0x2+jRwORyTkHms4V1f15xptxpmumy/LEvGDFogFAOgF\nEhwAAAAAo5/ebGdt7zban3n7YEXduSdMl9vz8+nO8rruv947IyO0mU1HiESFuClgeAghJEkh\nJoTUdZr9shsMq9XlkdMcDkVRZGKKIl0lIYTsr+7acbzFO4dIXqJs1bT0wEIegeKCTPUSI+SJ\nB/rEfrhGy5oc+elMpyBIOoU61ymFvtBXh1BCPnd8psp3tQlZqglZKtIf+6s7Pz10oVCower8\n5Od6ncm+ZEJyKJtfPSXV4XIfOt9zhKKISMj1O7f6LvNXR1tWTU3rV2Aw+nCQ5IIBGVStIAAA\nAACICmq2yUQIIVwu5c1ueFntrn/tqBr+oIbSzPy4wME1HIqaPUZDCOlgq6lJCKEJrY7hj0uO\nXTkhZVqmihByqFb76aFG3xlSq9uMb35/xhVCnYiCRDnrqI3JGcoBTz6rNbFnppieF6yLUtWS\nGBHPm92gCeFyqHWX58kk/AEGQQghxOn2bDvWGtBM7T7ZEax6qB8el7N2dtbvrhq7anrGlSWp\n84qSWLu1lDd2oyoHAAwMEhwAAAAAo9+88Ums7VIx+0Nv2Rkta/uIlaKU3Dg7U+pTREMi4K6Z\nkZGlkRJC+OwzgNCEkDk5mlnZas35zhffs43l6TLajzf29BmDgMe5dnKaRnahHweHoqZlqadl\nqvt1Lr4kQepcCHicK0pSWQumXD0z4893TFtckpKVIEtRx8wqiH/hlslzihMHHAOjRWe1sxVh\n9dC0t9BJKJKVklljNAuKEwU8DmuCw+WmTTb2iWwBAHqHISoAAAAAo9/lk1LKzmh3HWvxbZw/\nIZl5yA/kcHncHnokFBwNXVGqIidBdqbN2G12KCSCnASpNzuQqYmhWE6VihHyfEtsWB3uYD0m\nmnWWkvNDPJxuT5CMCYmXCW+dmVmvtXSZ7CI+N00pUQyu30RRauzuyrbA9rFpiswE6W+uLnrj\n61OW81PqcjnUNTMzFkxIJoTcsjBvMMcN5Ajeq2JgHS5EAvb5eyhCUIMDAAYGCQ4AAACA0Y/D\noR67fsK8CUm7j7W0d1sTlOJ5E5KnF8T/Z+cZ1vWT1ZLoym4wRHxucZoisF0tFU7LjfvJO4/M\neUvGJ/nOqNrLQBKKUDRNf3e09dMD9a1ai1DALcpQ3np5bkpANVYORWXFxWQNUZXWMSmxk7PV\nh2sv6lAjE/GvnpJGCJk7LnF8tmr/yfZWrUUtF03OixtMdViL3SXiczlB/u7xciFbkohZFGod\nVl/5ibJvygPHvJC0uBhxwAy+AAChQIIDAAAA4FIxvSB+ekG8b8uikpSP99Q6AoYe/GJGehjj\nCoflE5PjZMJdJ9rNdhchJE4mXDw+qTD5orloRXyuRi7sNLB04khTS/7xxf9n777D4yjPheHf\nz8z2vlqteu/VvYOxMRiDaTYQQgg1hJBy3uQkIflCcpKPkxfSTirJCSeH5oQEEmIEAZtiTLOx\njbssybJlWb2X7b3MPO8fK69Xu7PSqkv2/bsursv7zOzsI9lIO/fe5cz7tb2hu3+PL3js3FBd\nq+k/71tekjmhgbYTdu+GwpIMzf4zAwNWr1ouLsvUbl2Wqb5QW6RTSm5YMaWWnEGO33uqd++p\nPrvbL2KZ0kztZ67Iy0hSRJ2mVUjKs7SN3bao9WyDIvH5spHSdPJ1xcaDzUORiwyBAYvnR/+o\nzdDLN1Wnl6RrJnFlhNBlCwMcCCGEEEKXr1S9/PufW/Lbmnqr0x9aYRhy85qcW9bmJvL01iHn\nwebhIYdXJmZzk5UbS1Pi9YyYcwwha4qS1xQl29wBsYgoJML7vLYq/eWD7VGLaTq5jGU+uBDd\nCPMH+effPfezL6yckR1fQAisKTGuKTFOy9XcvuBHdX3dwy6JmCnL0q0qNT799tn6jotjdE53\nWpp7bd/ZXp2boop67mdW577MtZ/rs4dXcpKVd1+RN+lUnxuWZOQkKz9pGhywecUixukJ+Pyc\ni1IAON/vON/vuGFp5jXVwu1jEEIo1jz9DYQQQgghhMI4nnIcL5mZxgSrylKe+dZVn9T3dw+7\ndCrp8uLkvLSEBsR+dHbg47ODoT/bPYFBu/d0t+3B9QXJauFpqfOEdsymGItz9Rylb53sCfe5\nrMrW3bI8692I8aiRzvfYnN6ASjalRhuzprHT8oddjQ73yNCT92t7X/9UZY4ZIewP8jsPtn97\nW1XUulzCfmFjYeugs2vYxVGaqVeUZGimWMhUmaWtzNICwK93NZp80b1F363tXZqflBRn/i5C\nCEXBAAdCCCGE0PzV1G177u2zZzutHM+nJyk+u7Hw2mWZZNJDR+NQysRbVk6szGHI4dvXNBi1\n6PYH367rvfeK/HGfbnH6DzYN9ls9SpmoNEO7JD9p/jT8WJaXVJ2tG7B6Xb5gilamV0oAwBNz\n7x1CATw+broCHG5f8N3j3S39Dp6n+anq61dkqeOMuZncxf/wZqNj9EjXQZtbJBIInDX32oMc\nFQm1AS1IURXEJHdMkdXl7zW7Y9d5Ss/22NaVpsQeQgihWBjgQAghhBCap442Df34xeMcP9LY\nsdfk/s2r9S299i/fXDG3GwOAc/12KtRwsm3Y6QvyUpHwkJGQYy2mfxxo8wdHRm8cahoqOqN+\neHPJ/JmdIWaZLMOoJhRpMT0pQmQSVq+STMuLtg84/uvVert7pFaovt38/qmeb26rLs1KqMcH\nT+mQzWt1+tOT5BqFwJZqW01R0Q0AiBcs4yn1BzkRO0s3C25/3LmwLp/AbFqEEBKEAQ6EEEII\noXnqT7saw9GNsDc/7dy6OicnRdVr8XSZXAGOT9cpClJVk8iAoHSsuSFjc8fLaKDg8Qelorj3\n/Gan7+VPWoPcqK/rfL9j17HuOxJr/DEJQzbvvz7taB9wAEBeqvrWNblG7cQGf6wpS/nr+80u\nb/RXvXFRuijOyNgJoQBPv3UmHN0IcXmDT7915pcPrRr3JU53Wv6ytzmcBLGi2HjvpqKk0bVC\nQzavwOvyFITCShqFZDbbqegUEoYQXihmljRN8SOE0OUAAxwIIYQQQvNRv8XTaxJI2qeUHjs3\nfKhl+GS7JbyYZ1R9dm1uqJhiXAGOP9A8VNdltbr9Gpm4PEO7oSxlotkTWoUEAChAVISEZYhq\nzBvjE63mqOhGyLGW4dvX5E538Q0AQF2b+Xf/aggnjHQMOg+dGfjGrVWL8pMSv4hGIX70jkW/\ne63B6roYg1henHzftcXTssnOQafgX7fJ7j3XY6vI0Y/x3KZu23/trIuMhR1rHuoacv7k/hWR\nfVsEAxZBjheJ2Oi/RYANVWkT2v8UKaSi8izt6S5r7HpFlsDcX3TJY3BUMJqUaYg3I4QQQgih\naRev6QMAnGwzRUY3AKB9yPni/lbBD8CjBDn+hf2tH58dtLj8lILNE/i0Zfh/Pzrv8U+sEKA8\nQyNimdhwREWGdux0A4tTYAgrAHj8XLyskKkIcvyz7zaFoxsh/iD/7LtNQY6P9yxBi/KTfv+1\ndQ9dX7p5Web2dXn/cffS79+1RDpNZTVmh/C3BQBM8Q+F1Bxsj830GbB69jX0R65U5yUxMQEk\nSkEjF0WVtFxRnnLj8qzxNz2tbl+Tm66XR67IJezn1xfI8UYXIZQwzOBACCGEEJqPUvVyEUsE\nkx1M7oBGEz1XotfiaRt0FqaOMwDlWLu5z+qJWrS4/J80D22unMCH9mqZ+JYlmW/U9kSGCVK1\nsuvHG+oZr/CBZYhsBm5lz/fZBUMqFqfvfK+9LHti2QEKqWjrBLuxJkijEAslxIQOjZOYcz5i\nbmuk5l77tUszww/T9PKbV+f869OOyHPEIuZL15flGFVHzw/3md1qubgsS5efOs09RBOhkYu/\neVPFsRZT64DDH+TT9fK1JSkqGd6tIIQmAH9kIIQQQgjNRwqpaMOijPdP9kStJ6mlqjhdCfpt\n3nEDHOcHnHHWHRMKcABAdbYu26A40moacvikIibfqFqaq4/NEYhSma3bc6o3dr0sUyuUETJV\ndld0W80w2+iGF3MrL1VtUMtikzXUcnH5mE1GKQAfk74REpvWcceV+flp6jcPd3YNOaVitjRL\nd+f6/PQkBQCsr0idwvanB0PIqqLkVUXJc70RhNBChQEOhBBCCKF56iu3VFidvuPNw+GVNL38\nizdVvFEbHfUIESUQIPAFhUtRvIHJzKrQKSTXVY2TshElL0V1ZXnKJ2dGjZhVykTbV+dMYgPj\n0qvjpj9E9eCcWyxDHr6+7Fev1QciqmlYhnzhulLJmFUwBCDHqGztd8QeyhUa5rq8KHk5RhAQ\nQpcoDHAghBBCCM1TCqno/z648kTzcEO72efn8tLUGxZnsAzZ29jvFmqZkS90QxtFr5R0mwWa\nWSYl1qAUACiFum5rfbfV4gpo5KKydM2KvKQJJV98Zm1ecZrmw9P9A1aPUioqzdTesDRTLReP\n+0SnN/h+Q3/7kNPj51J1sg3lqQXjfckFaRqjVhY7QCRFKy9I0yS+51lQmav/+YOrXjvU3trn\n4CnNT1XfujY3I8542kg3rsr5/RunoxbVcvHGRROLPSGE0EKHAQ6EEEIIoXltWXHysuJRH7lv\nWZzx2tGuqNNWFhpSNOOPPl2ao6+PmVUBAEtzExopQim8crSzZXCkzsXhDfRYPI299s+vyZWI\nJtC9fkl+0pKJDDEBgAGb9097z4UjO1a3v6nXvmVxxtVj1lawDPnKjeW/qqmPnPCqlIm+fGPZ\nTFTETJFRK/vS9WUTfdaqEuMXriv5+8et4S6t2UbVl64vTSRm1DXk6ja5pGK2IE2tSzjIhRBC\n8xMGOBBCCCGEFpjVRclyCbv7ZG+oi4RUzG6qSL2yLCWR5+YbVZsr0z44MxBu0EAIWVtoqE6s\n3WZ9tzUc3Qjrs3oOt5rWlxgn8kVM2L+OdcXmrbxX17coW2cYs9ikOEP7Xw+tfutoV1u/AwDy\n09RbV2SrFePf/C8gVy/KWFlsbO6129z+NL28JFM7bjMUi9O/Y++5ugvjeEQsc8PyzO1r82Zi\nUi9CCM0ODHAghBBCCM0LLl+wpc9hc/tTtLKidM3Y+QWLcvSLcvQ2tz/A0SSVZNy72Ujrio2l\n6ZqGbpvZ5dMqJBUZmjStfPynAQBAk1Cvh9D6jAY43H6uLSawAgA8pWd6bVeWjhPcUcvFn72q\nYGa2Nl+o5OKlhYYET+Yp/d0bpzsivqVBjn/zSBfLMLeuiW6GwvG0a8hlc/nTk+Qpuov/VJze\nYOew0+UNJmtkeUZV7L/BIMfvOdbd3GMLcrQoU7NlRfZMDMpBCKEwDHAghBBCCM29A2cGXjvc\nGS4xSNPJ79lYWDDeSBTteANE4zGopBsSy/iI4hHq/QEA4Z3PELcvKDwpBCCy9gQlqKHD2iEU\nMHr3ZM+NK7NF7MVYxfHm4R3vnQs3MVlckPTQltIUnfyTM4Pv1PaEe9NmG5R3rstL118Mf/Sa\n3P//n4/1DLsAACi8f7KnZn/bj+5dXpgxv1qfIIQuJRMolUQIIYQQQjPhVLv5b/taI2ME/VbP\nf791xuaaR3NMQ9Ry4Y/HNAm0e5jS68pE8VJasHPEJHQNCU8L9viCgzZP+GF9u/nXNfWRLVpP\ntZp//NLJg01Drx/tjJy802VyPbP3nDci/vVf/6gdiW4AAAEAGLJ5f/pybezwWoQQmi6YwYEQ\nQgghNBuGbd5X97e19tqlErYiV7/9yjzphfGfe0/1xp7v8XOfnBm4cUX27GyP42mn2W12+ZRS\nUZZeoZIKv0uszNCe6bXHrldlaWd0e1IxW56pbYhpjyoRMRUz/NKXJBK/pimy3OnVT9pj4xHD\nNu+7JwUGFds9gWOtplAvmK5BZ1O3LfacXpOrscNSPcH+sugyxGA1E5oUDHAghBBCCM24g6cH\nfvbyyfDn2582Duw61PGLR1ZnGJQA0GMSmNsKAF1x1qddr9XzdkOf+ULCiJhlrihKXpkncBda\nkqZemZ90tM0cuVierlmWq5/pTd66PGvI7h2IyCYQs8wdq3PUskuqXej08viD53vtZofPqJWX\nZGpE7Ej6dmG6cPWTRiFO0V2cxdPSJxDMIoS4/cJlQT0XJhAPWD2CJwBAv9mNAQ6E0AzBAAdC\nCCGE0MxyegK/fOWUd3T3ikGr59f/rP/ll9cAABOn+GJ25pi6/MGdx7t9wYvbC3D8R02DCglb\nmSGQHLG5Mq0sXVPfbbW4Ahq5uCxdXTxer5BpoZaLv3592eHzw21DTq+fS9PJ15UY9ZdZfYov\nwIlYJsF/GEeahv7yQXO40ClNL3/outKybB0AlGZqK3P0pzstUU/ZvjY3nMFBR/6LReMVmYSf\nq4pfspTI8FqEEJocDHAghBBCCM2sY01DTk8gdr2u1TRs8yZrZQWp6oaYW00AKEybjcBBfbct\nMroRdrTdLBjgAIDsJEV2kmKG9yWAZci6EuO6GZ5HOw9RCsfbTB+eHjA5fQwhmUmKrUsz8o2q\nMZ5ytsv6x92NkQUm/RbPL2vqf/LAihStHAC+dlP5zgNtH9X185QCgFouvuPKvKsq08LnE4C8\nNFVzT3QSB6WgkYvsHoEkjrwUZegPRRlavVpqcfiiTpBLRdUFiY56QQihicImowghhBBCMyuy\nR2OUYZsXAG5ckRWuHQhL1siumNSgk4kadkbfhYYMOX1v1va+Wdt7rN0c5Ka5MaQ/yPdaPGan\nP24yAIqw62T3zsMdJqcPAHhKu0yuZ94/f7o7uiNJpLeOdcW2z/AFuPdOjLTPkEvYe68ueuqR\nNd+/c/Hjdy/99RdXRUY3QratzYu9sk4p2bYylyEkKr8jTSdfmjcSvBCx5Gu3VI7KNKEAAF+6\nsVwpw09YEUIzBX++IIQQQgjNoH6Lp2vQyTAEgPJ89FGdSgIAuUbVN26q+PsnreFmHEvykz6z\nLj/chXRGxa13oFDfbQWA+m443Gq6c2WOUS0d92omp6992OX2B41qWXGqOvbiviD/fkPf4fOm\nUOKAViG+YXFmJTYKjW/Y4Tt4bmhkEskFPKVvHu+pyNTF6xbaKTQFFgCipsMqZaLi+HNblxcn\nf+3mihffP293j9S5FGdqHtlanpWsfFBc9PqRTpPDBwAEYHFe0q2j58uuq0z93dfW/eW95rNd\nVp6nxZmaz19TXDHzvVoQQpczDHAghBBCCM2Ulz84//ePWgJBnmEIAGEIcDylF5IWijK1aRcK\nPQrT1N+/Y7HZ4bO6/Kk6uWoWP+XO0skbegQGXtCIcIzNHXj9RPcXryqMP3wDKKV7GvqPtI5E\nLgDAoJLetjw7Qy+/eA7ASwfaWiPusW3uwN8Ptd+5Jrc6WzflL+XS1DLgEMxzsbr9ww6vUSOL\nXOwZdjV0WKxOv4+LCacBwOghKYlYX5W2ssTY0me3Ov0ZBkVemjr0/PJMbem2KpPD5/AEUrTC\n/2IL0jWP37d8Qi+HEEJTgQEOhBBCCKEZ8d6Jnhf3No9aIsCyhOMopaCUib51R/Xog2BQSw0J\nZElMr4oM7fFOy1BMuwRudMLJkMPXb/Ok6+QQx4Hm4U9bhiNXTE7fS5+2/9u1JbILqSitA45W\nocyC9+r7qrJ1s9FSde7wlH5c3//+qd5+i0ctF1fl6W9fl6dLoEmqPygcqgAAX+DiIQrw4t7m\nXYc7Q5UpUjHDigSq0Ysz4+ZrxCOTsJVCmRcMIUaNLCrCghBCcwh7cCCEEEIIzYg3D3UIruvV\n0pvX5j736MaizHlRl8Ey5M4VOVURm6EUghyNzRqwuAVapY48BeBw63DsussXbIhoFdEZZ/Ct\nxeV3CPVhvZT8cffZv3xwvsfk5nhqdfk/OT3ww7+eGIzfnyUsOU7MiyEkMhy2+3Dnvw51hPtu\nBLjIv8CRP2qVkuuWZk36S0AIoXkOMzgQQgghhGZEV5wmCOU5+v+zvWqWNzM2hYS9oSr92vJU\ni9tvdvprjncLniaP3xPE6+ecXoGxGgAwGJEbwse0vQyL7Yh5KalrMx8/Hx0AcnoC//yk7Ws3\nlo/93OJ0TZJKYnb6o9YX5+rkkot/I28f7Yo8yvPU5+MkYiY8hLgyR//A5mK1Aqe0IoQuWZjB\ngRBCCCE0I6Ri4TdaUslstA6dBDHLpKhlhSkqmVAgQyZms5Li1qdEdpeMPhTRZzRVJ1zOIJew\n2kv6xruuXWAMMADUtZnHjeuIGHLv+oKoPI6SdM2tK7LDD4Mc32/xRD2R56nXxy3OT/rBZ5f+\n9pG137tzcZp+Dob7orliMpm+/e1vb9iwQaPREEL++te/Rp2wf//+Rx55pLy8XKlUZmVlbd++\nvba2Nuocm8321a9+NS0tTSaTLVu2rKamZqInIDSbMIMDIYQQQmhGVBcYDjT0x64vKjDM/mYS\nJ2aZLVVpb9T2RFY4EAJbqtLEMbNsI5+VoZf3xtxjA0Besir857IMrUElNcUMpl1XYpxo88uF\nxe0TTm/xB3mO42OHBEdJ18m/ubW8rtPaZ3VLWCbXqCpOU0eewDKEZYhgFoxGLinDBq6Xpb6+\nvh07dixbtmzz5s2CcYef//znnZ2dd955Z0lJSU9Pz1NPPbV69er333//yiuvDJ3A8/yNN95Y\nV1f35JNPFhYWPvfcc3fccUdNTc22bdsSPGHSmFmZIYUuPRjgQAghhBCaEfdeW3yiedgz+s62\nIF2zeVlmx5DrVLvZ5PQZVNJlBUlZBmW8i/CUzv6df2WmNkkp+bhpqM/qAYB0nXxDqXGM9qIh\nmyvTXzzYFlWEkj/6VlzEkPvW5796tKtz2BVaYRmyrsS4oSx1ur+I+SVFK5y6oldJx41uhLAM\nWZqnXwrCY1YJIRW5+vo2c+yhqjyczHqZqqioMJlMALB3717BAMdvf/vboqKi8MM77rijrKzs\nF7/4RTjAUVNTc+DAgR07dtx///0AsGXLlmXLlj366KPh+MW4JyA0yzDAgRBCCCE0VRQcPDUD\n+ACkDDEQUAFATorqN19Z+6ddjbUtZkqpRMRctyL73s3Fu45372scCM9S/bChf1N12s0R5QYA\nwFFa121r6LPZPQG5hM1LUq7JNyhmsbYlXSe/a3XOhJ6Sl6y8/4r8d+r7QmEREcusLjBcVZoS\nFZ5JUkm/eHVR57BrwOaVidmcZIVOMf4kkYVuTVnKrqNdsRkWV1VOW2Tnnk1F//HnY4HRI1fK\ns3VrylOm6yXQwsIw48TOIqMbAFBQUJCXl9fb2xteef3112Uy2V133RV6yLLsvffe+53vfKeu\nrm7RokWJnIDQLMMAB0IIIYTQlHC0jacD4Yc87WdIOktyACAnRfXkF1b5A5zZ6U/RyhiGHG81\nfXR6VN0KT+neur48o6r6wiROSuHN+t7uC+Uebj/X2G9vN7s+szRbLZvXb95yDMovbSzyBji3\nL6hTSuLlnhCA3GRlbnLcvJVLT5pe/tB1JX9+/7wvwIUXVxQl37RqYlGkMRRlaH7ywMrn9zSd\n7bRSAKmYvX5F1meuKri0a3/QNOrp6Wlvb7/33nvDK6dPny4uLpZKL/Z/qa6uBoCGhoZQ/GLc\nExCaZfP6dyRCCCGE0DzH0+HI6AYAAFCe9hJQM2QkYCERs2n6kfqOI80Cs1QB4HDzcDjAcX7I\n2R3TzMLt5z5tN21eCKUcMjEr2Kb0Mre2LKUsS/tJ40Cf2aNRiKteqHSZAAAgAElEQVRy9VW5\n01w8UpCufuL+Fb4AZ3P5jVoZWSChjTmpw0JReJ5/+OGHJRLJY489Fl40mUwFBQWRpyUlJYXW\nEzwhnlOnTp07dy7eUY7j4h1CaGwY4EAIIYQQmjwehgTXKQyBULsEc0x/zZDIvpsdZpfgOR1m\n98Q3iOYRvUp68/SlbMQjFbMp4zVMmYTWPserB9ra+x1iEVOapbvzqgKDRjr+0+KzuwM1B9tP\ntZltbr9RK99YnbZ5SeYY43jQzKGUfvWrX92zZ88//vGPqLoVQePGzsY94X/+53/27NkT72gw\nKNyUF6FxYYADIYQQQmgKqHDAglIfCL3Dl0uE3n1RUEgvrvs5XuAcAH9QeH2hoBQo4Gf1C9Le\nkz1/2n023Dima8j1yen+H31+WWmWdnIXNDl8T/y91uryhx4OWj2v7G+rb7c8elsV/guZZZTS\nr3zlK88+++xf/vKX22+/PfKQwWAwm0d1rg09DKVpJHJCPE8//fQYR+Xy6Y/QoctEQk2bEUII\nIYRQHHE+LiLC65WCAzsJVEbcKGplYsHnauViAKAULC6/x7+QUrj77d6dJ7r/8HHz7z88/9cj\nHeeHnHO9o8sdBWgbcBw8M9jQYXHFGWEbZnf7n3/3HE9HdUj1+rk/7mqc9AZeP9QRjm6Enemy\nHm4STolCM4RS+qUvfemZZ57ZsWPH3XffHXW0srLy3LlzXq83vFJXVwcAVVVVCZ6A0CzDDA6E\n0MLHB2nDXtp6lDrNRJVMiteQ8qthvM7hCCE0LRii56jA7ToTZ5znxsrU2nZz7+hik5xk5fqK\ni801ytI0tT1WGj1wA8pS1R+c7j9wbiiUypGqlW1dklmQoprilzDTzg85d9f3hW+Phxy+N+t6\n1xYY1uQb5nZjl60ek/v598619jtCD5VS0WfW52+oSot3/qlWc2Rv1LDuIVevyZ1hUIz7irFd\nNuo7LIJn1rdb1pbh2JdZQin94he/uGPHjhdeeOGee+6JPWH79u1/+9vfXn755QcffBAAOI57\n8cUXCwsLww1Exz0BoVmGAQ6E0AIX9PO7fkEHW0OPqNtGB1tI23Fm67eAwR9xCKEZx5A0npoo\njApYEFAzRPgmTSpmv3lTxft1fSfaTGanz6CSLi9M3lSdJmYvhmUNSsmmkpR954cDEbUqFWma\nM53Wpj57eGXA5v3zvta7r8grTddM95c1bSiFD5sG+ZhozeE2c1WGViXFH9Szze0L/rKmPjJ7\nwuUL7tjbLJewq0qMgk+xu6NTLcJsLv8YAQ5/gNt1pOuTxv5hm1evlq4sNt52RV6oGssTJ23E\nPV46CZqQN954w+/319fXA8DRo0dlMhkA3HbbbaEJst/61reef/752267TaFQ7Ny5M/QUhUKx\ndevW0J+3b9++bt26r3/96zabraCg4Pnnn29oaKipqQlff9wTEJpl+EsFIbSw0Yb3wtGNi4u9\nZ+nZfaRi05xsCSF0mWFFTBVPe3lqouAjIGNIMkPSxigEloiYG5Zl3rAsc4yLlqdpsvWKpgGH\n1RNQSNg8g9LrC35Y1xd1Gk/pnrq++RzgMLl8TqFbVp7STrO7Yh7v/FJ1oHEgtjYEAHYf7YoX\n4DBoZPGuNkafUX+Qf+LvtW0DI3kiZofv3RPdJ1tN//n5ZSq52KiV95gEmummzkB71MvZfffd\nZ7PZQn9+6qmnnnrqKQDweDyhSMehQ4cAoKamJjIkkZmZ2d3dHfozwzC7d+9+7LHHfvrTn9ps\ntvLy8p07d27bti188rgnIDTLMMCBEFrYaHttnPWTGOBACM0WhiFZDMma3ouqpKLlORfrXD5s\nHBA8bdDudfmCyvmaCuGL3xh1ofdMXRA4nr5f21vfbnZ4Aml6+bVLMzuGhGf0dA27eJ4yjECD\nz8UFBo1CbHcHotbLsnVjjGv54FRvOLoRNmj17DrSedeGwquqUl/+OPrzCYaQKysXwCDkBcRq\ntY5x9NNPPx33Cjqd7umnnx6jJ+i4J0wSjppGkzJPfxcihFCCqDf6zdMIT5x1hBBamIJxRqsA\nQCD+oTmnkws3TAUAnSLuoUuJ0xvYc6KnfcDBMkxBmnrzskzZbN25Ob2Bn71yqmNwpEdMS5/9\n0JnBnDhNWwghgnN/AEAuYb++repXr9ZH1pUka2X/dkvFGK9e12YWXD/VZr5rQ+G1SzJ6TO59\nDf3hdYmI+fzVhdnJyjG/JoQQGgsGOBBCCxtRJVH7oMABNfauQwhdUowaGQCFmHtQuYTVxA8i\nzDmlVJSfrGwbjs4a0MnF2frxm1MudOd6bL95vcHpGcl9ONY89N7Jnu/esShrVm7j/7m/LRzd\nCOEpbR90sAxDYkaxFqSqx5jPurTQ8Puvrt19uKu13yEVM6VZuhtWZknHjNSM3WWDIeTBa4vX\nV6bWtpqtTl+aXrG2PMWgjlvwghBCicAAB0JoYSNFa2nvWcH12d8MQgjNnIpM7Xtyid0TXSaw\nqjB5jPvS+eC68rQ36nr6bBcHSWrl4puqM1ihaohLSZDj/7ir0Tn6r8zi9P3PW2eeuG/FLGzg\niODIVQpyqcg7eswww5Bta3PGvppeJb3nmqLEXz1FJz8f0RM3LLLLRlG6pgj7sCCEpg8GOBBC\nCxspvZL0n6PnDoxarLyGFCyfqy0hhNBMkIiYe9cX/PPTjkH7SKSAIWRFQdKmed+zQCFh71qR\n0zzo6LN5gzxNUUvL0jSiSz26AQBN3TaTwxe73jHo7B52zXQSR5Djnd7ocFhIWaaGYZgTLabQ\nw2SN7PMbCytzhAcbT9qG6rSDZwQax2yoTp/eF0IIoTAMcCCEFjhCmI0P0aI1tPUYOIdBnUyK\n1pL0krneFkIITTObNzDs9q8sM3p9XDDIaaTi3GRlqjbueIv5pjhFXZyinutdzCrB6MbIIbt3\npgMcIpZRykQur0CdSIpO/rkNhRanr8/i0cjF6UmKmcimqcjR37WhcOcnbeH2MQwh16/IKkjX\n/O3jll6zWy4RlWZqNlanR85IRgihqcAAB0LoUkCyKklW5VzvAiGEZkp9n+10v53SkYcMIWl6\nxQKKblye1PF7o6gVklnYwMpi40f10aOFAWhoHKxeJdWrprnnhc3l//tHLafazDaXP9Og3Loy\n+8n7VxxuGhyweJI1spUlxq5h14//fpLjR/4p17Wb950e+Pa2Kp1yNr4hCKFLHgY4EEIIIYTm\ntW6rp2F0LwOe0vo+W5JCnKGNO6RzPus0u9tNLref0yskFekatWy235E2DTqaB50Ob0AtExen\nqEoTSC1x+oNOb1AmZjUyUYJNT8qzdQqpyB3Ta9OgkeWlCo8ymV53ri9o7rX3mEZ1eL15dW7h\nzLS96Ld4vv/CUZvLH3rY0mf//Runr6pO/8a2kU8gzA7fS/taw9GNkAGr5+/7Wr98Q9lMbAkh\ndLnBAAdCaOHzO6mzn7ASUKUCiw3YEUKXmlZT9AiSkBaTa8EFODievtvY3zRwcZL3kQ7TptLU\nyhnuNOkP8ianj6egV4o/ODfUfuFbanL5202u84POGyrT4oUtnL7gsS5L34XWJyqJaHm2PiOB\n9BmZhH1gc8mf3joTeUsvYpmHt5TOTl9YtUL8f+9d/u6J7vp2i9XlyzQor12SWZGjm6GXe/H9\n5nB0I2xffd+mxenV+UkAcLLVJDTtmJ5qN/sC3NgzWRBCKBEY4EAILWScjza/TXuPAuUpALBS\nUnANybkydowiQggtXM444zYdcdbns2Md5qYBR+S02yBH954ZSNfIkmamSIFSONxq+qR5yB/k\nAUAmYSXS6BvpNpOrsc9elaGNfXqA499vHnRHzBxx+oP7Woc2FaWkJDDTdG1ZSpZB8fqhjrYB\nB0NIUYZm+7q8yDEiM00sYm5alXPTqnEmpEwdBTjRPCx46FjzcCjAYY0JfwAAAOF4avcEjBjg\nQAhNGQY4EJovaPcZ/vg71NQNcg2TW8WsvAnEmIwwDlr/MjU1XXzM+WjzW8AHSd7Vc7cphBCa\nZmJWOGgrWYCtGU/32SEmCM1TeqbffkVh8ky84r5zgwcibryZON/M80NOwQBHq8nlHj1RFQAo\nhYZ+2yZ1SiIbyDaq/s8tl3KXqCDHi1jGH+BCIaRYjguDcjUK4aYkDCFqWdx+JQghlDgMcCA0\nL3CfvMIfqrnwqIfrPsOf3sd+7nGimuaZbZcUe9eo6MYFtP1jkrMeGPz5hhC6RKRr5MNCH32n\naxZYk1FKwR5ncKnVI7w+Rb4g/+mFYagh8WpDXDFRjBCTcNIBCP6NXFY6Bpwvvt98pssaCPLp\nBsVt6/K0SklsiQoAhDNWFuclvXqogx/dgwMAyrN1Mgmmb6DRxPhGDk3Gwgv8I3TpoYPt/Kev\nRS9aB/iP/jon+1koqL1b+ADnA9fg7O4FIYRmUGmKShPz+bZWJk6kNea8QkjcrBOpaEbelA7Y\nvFEtLXmIvrsOUcbUrYQIn33Za2i3fPe5IydbTF4/x/G0e8j11L9OG4RqdliGrK9KC/05RSff\nviY36gStQnL3VQUzvmOE0OUBA2MIzT3+3BGgAu+g+OajLOWBYCAyDqFv2oVDwlmyCCG0EIlZ\n5rrSlIZ+e6fF7fZzCgmbq1dUpmlFzMLrN5RnUEZ2GA3LT1bOxMvRmABFIMiLhIIsxUbhaJFe\nIe60CKwnzcqc13nruXebYtuFtg04y3N0Zzqt4RWJiPnS1rIMgyK8smVpZnG65r3anh6zWy4R\nlWZqr1+WqZDiLQlCaHrgTxOE5gGXTXg96AefG2SzMUluISLqTOEIBysGZeosbwYhhGaUmGWW\nZuqWZup4SmdnAMcMuaIwucvijupqUWhUFSbPyC87o1pGyKh4eMDPi1hePDphpDBZVZEmPMal\n0KBqGnB4Y7pLVKTO7NiX+czs8HUOOmPXKaWrS1NuWZNb22Kyu/2Zycprl2YaY8bNFKSpH7ke\nh8IihGYEBjgQmntEHafRhkQG0hn5RGuh42wWRiYnuhzQF4ClNeooyb4SWOxVhhC6NC3o6AYA\naOXie1bnHmwxtZtcbj+nU4gXZ+kWZQp095wWCgm7JEd/smNUDobHEzQYFMkaqd0b1MhEJSnq\nImPc8IpUxFxdnHK4w2x2j3SXkImYpVkJjYm9VLnjj+9x+YKrSo2rSo2zuR+EEArDAAdCc4+U\nrIGDNbFVFUzpWlhAb2R9Hn6wA/xeYswmGsNMvAINBp1v/sP+6ou8zQKEkRSX6+5/RJKqpgOn\nRs5gRCT3KpJ/zUy8OkIITSvK00EeLED9hMgYMBJyuXSVVkpEm8tnL8/uuso0lpATHRb+QiJH\nZaZ2S1WaLOGhpDq5+LqyVIvbb/cG5WLGoJQuxOKgqRi0elr7HQBQmK4xamXJGpmIZWJLVAAg\nI2n2JuAihFAsDHAgNPdIcha74W7u45ciYxzEmMNuuHsOdzURlDu8O7j/FfB5AACAsIs2iDY/\nALJpTj8x/+Y/3fv2XHhN3n/u9OCPvmn84a9k6zZTRx+wYqLOBAlW9CCE5j8uyJ+lMNKKglI3\nD2YGUlhyGbVapBTO9tv7bF5KaapGVp6uYWcmasAy5LqqtNWFhj6rh6eQqpEZVBNun0EAkhSS\ny7Dvhi/AvbDn3N6TvZRSAGAI2bws84HrSq6sTP2ori/qZJ1SsqIEczcQQnMJAxwIzQvMyptI\nbhV/4l1q6gG5ismtZpZeB8zCGJnGHXoj+EHkwBfK1X1EbcPie34EMG3vVv1Npy9GNy6+Nmd9\n7rdpf/wHkc9IzghCCM0EjvaGoxthPB0koGcujzwOuzfw2onuQYcvvHK4zbR9aVaScqYiCFq5\nWCvH6sUJ++83Gw+cHgg/5Cl993i3N8B98fpSk8NX32YOH0pSS791ezW2C0UIzS38GYTQfEFS\n8tjrH5nrXUwczwUPRs+4BQC+o4HvPMPkVEzX63jrjgEVCJgEuto58zCblBy1zpkHfUf3cYO9\njCFFuvQKUXr2dO0EIYSmiFKz8DqYAC6LAMfuut7I6AYAmF3+N0713L82fwGVZl7yBiyeyOhG\n2L66vrs3Fj5+z7Jj54YaO60eXzA3Vb1xUbpMsjA+mEEIXcIwwIEQmhJq7gOvS2Cdp4HaT0QK\nA5s8PYXW1OeNlw5CvZ6oFc8Hbzhe/iP1eUMPnf98VnXLPcrtD0zLThBCaIooBCa0fokxu/zd\nluif2wAw5PD12jyZOmziMHuONg3tPtzRPeTSKCVLi5LvWJ8vj0jBaOsXGOgLABSgtd+RrJWt\nKDEmWJNidfklIgbzOxBCMw1/yiCEpoZGj2qlHO8ZdPodPmh+CXa+JMrMU9/375LyJVN8HXFW\nruA6kclZ46gYSqC5wb7j16NO4oLO13awGbmy1VdPcRsIITR1BMQUBOZQELgsWjxYL4wjETyE\nAY5Z88xbZ3Z92hn685DN29Jr31fX97MvrjJoRgbEjJFNk2C/FI6n753s2XW0y+UNAkCmQXnX\nVflVuZdFmhJCaE5ggAMhNCUkKR2kCvC5Qw8pT52dVs7PhatJgj3tll88mvTYb8Ql1VN5Ifma\nDazByJmGotZVW24l4lG3BJ4PdwlewfPhmxjgQAjNB4QYKO2OXWdgQbYTOtg4sPtIV/ewSykT\nVebq79pQEL5DFiQRMfEOSUVY4zBLznRaw9GNsEGrZ8eec9++Y1HoYVGGlhBCYz7JYAgpykho\nsu8Le5sPNF4scukxuX71WsNXbyxfWRxdWIpQtITnHCEUKe4vGIQQSggrEq29Nfwo4PByfg6i\nemVwQWfNC1N8HSKTJ//wV6LMnMhFxYYt2vu+FnVmcEDgtgEAuP6uKe4BIYQmgaMmP9/o4477\nudoA30LBx5IMAuqo0xiSSohuTnY4Fc+92/TUv0639Nl9Ac7s8O1v6H/02SOdg84xnpKmlQuO\naBWzjFEpOddnP95q6hp2xdxWo+l0+IxAcw0AOHJ2MPydN2ikm5dmxJ5zw8osXQKTaDqHXJHR\njbC/72ud0N8tBbC6/AGhqbQIIRQFMzgQmke4wd5A21kIBkU5haLswrneTqLYK7YDQPBADQR8\nAY9A0jUABM7VA6VjZbsmQFJYmvaHlzwHPwp0tDAKpbRqmaS0MvY0IhVObyYyxVReHSGEJiHI\nd3J0MPRnCkFKLX7OJmFKRUwlTwcpWCj1EyJjIIWQhD4Sn1da+uzvneiJWvT4gn/e2/zDu5fG\ne5aIIZvKUt6qjx4ymqtX/GpXo9s38nskJ1l557q8NKxYmRl2t3DDF6+f8/qD4U4cD11fqlFK\n/nWoIxDkAUAiYraty7tjfX4iL9HUbRVcNzt8g1ZPagJ/s74A905t7ydnBvxBniGkME192+qc\njCT8bY4QigsDHAjNCzQYcP7tv90fvgH8yAcU0uXrNV94lFEviPe7hL3iNmbZZtrfxrzyPNhq\nY8+glAfBISgTfSWRWHHVZoDNY5wjrVrhbzgauy6pXjnFV0cIXa74ySW9UuoORzcirxakXWJS\nxpAUgJTpm6Y9B062mATXGzutvgAnjZ9hXpmh1crF+5uH+m1eCpCilmZo5G8dG5Vn1zns+tN7\n5757a5V8gczmCPLU5gkoJaxgfsp8E6+MSCUXR/YZFbHM5zYW3ro2t3PQSQjJNioTbxTqD8bN\nufAHxk/H4Hn6x3eb2i9kA/GUNvfZf7Or8d9vqsjEGAdCKA4McCA0Lzhe/L3nwzciV3zH99vc\nDv33fjPFrIdZQ+Rqkr9IXLnKd1ogwCHKKgAySzVx8mtu9XzyTrC7LXKRTUpR3vT52dkAQuhS\nQSnt52kvgBeAJUTHkDyAsbpLROHBJrxOnQAcwAK4DR6b2yuctUcpdXmDYwQ4ACBLr/jcqlxK\ngQJlCPnNrsbYcxyewJHzwxsqpmca18zxBLgDLcNNA45Q5UWqRrahyJiils7xtsZ0ZXXazv2t\nPB9dLLJhUXrsyQqpqCx7wvVT8VItRCyTohv//6MTbeb2mFonf5DfdazrketKJ7oZhNBlAntw\nIDT3eJfds2937Lr/TG2g9ezs72cq5FfdwCg1sevKrXfN2h6IVJb0H39QXP8ZRqUBAKJQyq+6\nIek//8RosW07QmgCeHqOp60AoYHTHKUmjq+lIDAYOx5KubiHYqaoBDh+f/PQM/tafrnn7P/u\na9l3bsg/75sOpMSpMpBJWK0yoYkwhABDCKXQJzQ4FgB6zO7J729WBHlaU9tz9kJ0AwAG7N6a\n2u4hp28utzWe3BTVF7aUsqOnoZRm6+65tngql42Ml1Tn6VO0AoGM9ZWpYwe/Qpr77HHWHdie\nBSEUz8LO4HjiiSd++MMfpqam9vf3R67bbLbHHnuspqbGarVWVFT8x3/8x2233TZXm0RoXMHu\nduCE3wQHO5vFheWzvJ+pYDR63Xd+YX/mZ8Ge9tAKkSlUdzwkW3vNbG6DKJTqu7+mvvtr1O0i\nCuVsvjRC6NJAqY3S4ZhljtI2QqoSvAghUhC+FWOiJsL6g/xLRzqGHCO3xDZP4HCbqWXI+fnV\nuWPMHJlza8pSXv6oxReI/hW2vjItdOcc4Pjz/Q6T06dVSApSVMp41Q0ECAHB79X8T2I802+3\nxAy+DfL0SLv5xiqBbIixufxBizsQ5KlWLtbLxdO0R2E3r81dVGh450hX97BTo5AsK07etCSD\njJc36g9we453n++xA0BRpmbz8iypmKUAB08PvHm4o3vYLZewFTm6z20sTEtSfOPWyv9562zX\n8MWw4OpS411XFSSyvXhdRYM8T3lKEhxUixC6zCzgAMeZM2eefPLJ1NTorEWe52+88ca6uron\nn3yysLDwueeeu+OOO2pqarZt2zYn+0RoXISN/zkGs/ASmMUFZYYnnvOfPRXs62C1BnFpNaOZ\ns9QJjG4ghCaHglWwcxCldiCJtuRgiB6gGyD6/p8lhqgrn+i0hKMbYcNO3/EOy9rC+Ts7VqeS\n/NstFU/vOhPuDAoAlbn6uzcVAUBzv+PVw53WCzf/cgm7dUnmSqEvhwBkJytj6xEAINeompm9\nT5tem3DuSbz1eHhKG/rs7RZ3eIhJkkKyMls3ox09clNUj9w0gc9ROgadT/z1xKB15Ev78FTv\n6wfaf3jv8g9qe989PjLCzOHhDzcN1baaf3j30qIMzeOfX3qq1dw17JKK2ZJMTX5q9PygeFK1\nQvlBFFK0MgajGwihOBZqgIPn+YceeuiBBx5obm5uaGiIPFRTU3PgwIEdO3bcf//9ALBly5Zl\ny5Y9+uijGOBA85Yop5BIZdTnjT0kLk70c8L5hWUllcsklcvmeh8IITRpwTjZAxSASzDAQUAk\nZgqCfFtkQQpDNCImK+rM1iHhuaqtw875HOAAgJUlxpJHtB+c6u0ecillosq8pFWlRgIw7PD9\neV9rMOJDeI+fe/VIp1YhLkkXqGS8bnHGs3ub+dGzYZPV0qiACE8ppcDOp/tbPk4hUTCmvcXY\nTvc72kbX45jd/vfODS3J0Gbr58UoGUrpL185FY5uhAzZvD996aTVEz2TxRfgdrx37on7VzCE\nLC00LJ34P+PVxcl763qjO5USWD/ve7IghObQQg1wPPXUUx0dHW+//fbtt98edej111+XyWR3\n3TVS8M+y7L333vud73ynrq5u0aJFs75ThMZHJDLlzfc4dz4btS67cosoI3dOtoQQQpc5AvI4\nt6cigAkUDjBEK2GrODpMqQdAxBA1QwSaNfrizJuIrf6Yh7RKyfZ1eVGLh5qHgkIlBvvPDgoG\nOErSNQ9cXfj6kS7zhdYVldm67atyxBcqdDqGXe/V9/WY3TyFFI306sq0isx5MWgsSSlpiS1m\nAkhWTqDJaJCn7ULdRnhKT/RYg5Tmz4O5Ied77Z1CWTZ9ZrdMKoqtGTnfa3d4AurJFtrolJIv\nXlP8t/1ttgtJQAxDNlWlXVmOAQ6EUFwLMsDR2tr6gx/84MUXX9RqBX6xnT59uri4WCq9+Eul\nuroaABoaGjDAgeYt5c2fJ3Klq+Z53uUAACKWKG74rHLbfXO9r0sSBc4MnAMYKbAGIAm1wUMI\nXW4ISQbaARB9i05I2sQvJmJJ2tjNJLRy8bBQT0qdYqH+jBqwCqQlAkB/nHUAqMjSlWZoB21e\npzdg1Mh0EW1K67usr3zacfEiNu/LB9uvrUrbMA/udSvTNbXd1tiGEUuyJjB2xOULRmWvRDo7\n4MjVy5m5nqo2FP/vjqfCM4Hc3uCkAxwAUJqp/cHt1fWd1kGbRy0Xl2RoU4W6liKEUNiCDHA8\n/PDD1113Xby+oSaTqaBgVO+ipKSk0PrYl73lllt6e3sFD/E8DwBWq3Uy20UoEYQoNm+XX30T\n19tJgwFRRi6RzYt81FnjOXHYW3uUdzrEOfmqa7eGBqBMP85GPUeBM488JGIiWwSSwhl5LYTQ\ngsZ7SMBORYrIEdeE9zMQmIl3T9VZ2hahKpXqrHmRpDAJ8apIxq4uYRmSrpcDjPoNyFP6Vm1P\n7MkfNg4sy09Sy2a2E+e4VFLRTVXp7zcN2r0jZRpillmdl1ScMoHuIWO39vRzvMMX1M7YV+rw\nBCwOX3qSQjxmR1ulPO4/fcHdS0RM0pRn5UrF7Ir5XaWFZspMdp9Bl7CFF+B45plnjh071tgo\nMCx9bOM2hX7wwQeHhoYED3m93pMnT6pU873NFVroiEgsyrnsbrapzzv00x+4D30cXrG99Fzy\nd38sX7E25tQA+M7ToBkIS0QGkBZObNY19VPXx0AjPoCiAeo5TogYxDlT+RIQQpceynUT3k8C\nAcpIgbAAPHBBQgOUdBNR0bS/XHGKem2h4dNWU/hTfEJgTYGhOCXRjowzyuLyf3h2oMvkDvJ8\nqka2vjQl1zBOC+c8o6pJaMxnfsqEez/3W71Ob/RUXQDgeNo26FyUM/cjwDN18rtX5nSZ3VZP\nQCFls7TyuPNi4lBJRRKWGWMwcLxOH1PU3GN79p2m8712AGAI2bQk455riuLlXJRn61RysTOm\n3YZSJpbJWLcvupzqisq0sSMmCCE07RZYgGN4ePg73/nO906gCuYAACAASURBVL73PaVSGcqn\nCAaDlFKr1SqRSBQKBQAYDAaz2Rz5rNDDUB7HGLZv3x7vkNPp/MY3viESLbBvF7qEBfu63Hvf\nDPR2stok2dI1stVXzfWOJs/ywn9HRjcAgLNZhp78XuYLr7G6iP9tg8PU+THwI73NqO88eM8Q\n9SZgEo48+ttHRTcuoL6zBAMcCKEo1A0AQCnhRv/coC7B06fuyiJjaarmbL/d7glo5OKyNI1x\nyp9+T4tOk+vFg+3hhhpOr7Nl0LmlOn1NYfIYz1pTnHz4/LB19PxUqZjZVDnhGp8xGpH4AjNz\n3z9xIobkJ09+bhdDoCxVXddri3dUJZ3+T7Obe2w//PPxcHENT+nekz3NvbafP7RKzAoEJiRi\n9pGbyn/zaj0f0T+VYchXbi7XqqS//9dpR0TsozJXf9810x8KRAihsS2wO/bu7m6bzfb973//\n+9//fuS6Xq+///77d+zYAQCVlZU7d+70er0y2UiRXl1dHQBUVS3MaRQIxXC9U2N74Xc0MPI2\nwrXnddmytUnf/QmRzIu3whNCOc65583Ydd7tcu3bq7nlzgsLHHXuD0c3LqzZqfMg0VyX6Gvx\ncarMOJvgMEiE0OUtznskMoMFEUa11Kg2ztz1J+fN2p7YdqF7T/dXZmrHKA+RS9ivbC5+80RP\nQ9fIz95co/LW5dlGzYR7KCSpBH+7UQBimB8xoGmRn6RggJzqs8b24sjRKwQjDlP08ketsa1D\nOgac++r6r1maIfiUq6rTM5KUL314vrnbBgDFWdq7ry4qytQAwG+/vPbDU71dwy6FVFSRo1tR\nYsRfqwih2bfAAhxFRUUffvhh5Mq3v/3ttra2mpqatLSRDwS2b9/+t7/97eWXX37wwQcBgOO4\nF198sbCwEDuMoktDoKPF+uyvo3JVvScOOXb+WXP3l+ZqV5PG2628S3g4YrCn8+KDwADwQp+a\nBgeBdyacxDHp91o8UB7IAvuBiRCaCsKmUq5b4AAz910tE8FTen7IaXL5CSEpKml+snJyPwHN\nLv+wI7r7KQXgeNoy6FwyZnmIViG558p8X4AzOf1ahXiiVRsR1xEXp6mb+x2jl4lRLc0zTj5p\nYh7KTZIbVZJjXRZLRCpEjl5eLTR3ZurOdFoE1xs7LfECHABQlKn50T0CY+CVMtFNqzEdEiE0\nxxbY+3WVSrVx48bIFb1e39PTE7m4ffv2devWff3rX7fZbAUFBc8//3xDQ0NNTc0sbxWhMXDD\ng/Z/7vCfawTKiwvLNHc+IEqN+04iimffu4KVuO4P31qIAQ4ikwNhgAp8RYwyImwhGN0I4RwJ\nBjgIa6DQJnCANcSNfQQHqPskBE0AFFg1kVWDtED4TITQJYZNB9YI3OjmXERGxKVztKEJsLgD\ne870WyNuklPU0i3laQrJhMscPH6B5hehn5hun8ChWFIxm6Gfatvs21bmvHSwrct0cZCqQSW9\na13e7AwWCXD8TCRQCFJI2KsKk00uv80bYBmil0s0shl5ux6KUgkeik3rQAihhWKBBTgSwTDM\n7t27H3vssZ/+9Kc2m628vHznzp3btm2b630hNMJXf2Lox9+i3pFqC39Lk/ujtw3f+5l85RWJ\nPD041C+4zpkGKRck7AL7n5qRK2SVi70NJ2MPyVesu/hgjGGuJOH8ZHEu+JqAj/4MkMgqhc/3\nd1Dn/osPOQd1HQTORhRLE31FhNACRohkNQTbaLADqAuIDNgUIiqb/7OlKYX3zg5YR3eCHHT4\nPjw3eGNV+kSvppXH/XqncYQtx9Nhh48hkKSSCo5ZUclED28qbuy2dZtdQY5m6OWLcvRjD2SZ\nOrsnsPt4d32HxeUL6pSSNcXGaxalS0QMAPiDfKfV7fAG5RI2QyOf9hiEQSkxKGf2XxoByDaq\n2gccsYdyJzL/BSGE5pUFdi8Ua+/evbGLOp3u6aeffvrpp2d/PwiNg+fNv/1xOLoRQv1+y1NP\nyJ57PZEmGvHmpzIK1YKLboToH/lm/6Nfor5RbfxU194orYgoKxOnAWGBxvSZYxQgSrh/PmGJ\nciP1noTAhbRzRknky0AkmHBOqfuYwLK3EWTFE+hsOuqSfggOAfUCowKRcWIjYBBCc4CAqICI\nFljeVr/daxnd2jOk2+pxeIPqCd6Kq2SighRV62B0LaFSKipKnYYJLzylB5qGPj4z4AvyACCX\nsJsq01YXJceGLghAZZa2clbm5vKUmh3+373VaHePxImsLv87tT1ne21f31reY/McajeHNgwA\nDLFWpWuWZC68gb43rc7+wxvRcwkVUtE1SzKncllKYX9D3+uftHcNOZUycVW+/r7NJSm6qWbx\nIIRQIhbk7RBCC5e/+UxwsC92nbOafadrZUtXj3sF2YorXO8IlFzJEksAmYekJRUZf3zJ8uzv\nvKeO8R6PODNLc9s96q2js66IlMiXUffR0U8lRLl6Yp01GDlRrAPqBc4BjBQYddync9bonqYj\nKAT6QTrxzvD+Vuo9BfTCZ6qMmihWAjvWDAKEEJoEmzd6imfkoYkGOADglqVZfz3YFtmJQyZm\nb1+RLZmOCaBv1/Z+en4IgISahnr83O6TPW5fcBLDVqZFt9m9p66vy+RyuoP+QHQNTvug8+PG\ngV5vgI9oBMpTWtdrU0tFhVOYojInrl6cYXX5X/m41X8hWJOml//brZU61ZSSR/5395m3jox0\n0fI7vfvr+482Df30oVUFM9NJBCGEImGAA6FZxVnNcQ9ZTIlcQbZsrXz9Zs/+9yIX2SSj5p6v\nTHVzc0eclZPy+K8AgPr9RBLnfZWslIh01FMHQQsAA+JkIl8CrG4yr0dkIBqvjT+NX1s+xqF4\nAj3UMzofhHdQ136iuh4Y/FALITSdJPG7RUyukYRWLv7y1cUnO8ydJneA49O08pUFSQrJNLyH\ntHsCR1pMFwLNF8PN+88OrisxysTTPxh1bE199pcOtIeCF0FOeDbtyQ6LMUUgkNE06FhwAQ4A\n2L4ub31VWmOHxeL0ZxgUSwoNU+w20tJrD0c3ACD01+r1c8++ffYnX1g1tc0ihND4MMCB0Kxi\nk5IhzkhS1pDoaMCkf3/cvWS1652aQE8Hq02SLV+rvvML8UpXFpa40Y0QUSpRb56lrYwkdwg1\nYGMn/K2m/iah1QD1txAZTrBGCE2ndK2MZUhs/0iZmDVO9pN5liEr8g0r8g1T3t0oXSY3HzsT\nFSDI0x6zu3A6SmASRwF2n+wJ70doXwAAHp9wgkxU05MFJFkju6p6ws1ZwpyewLHzpkGrR6eS\nLMpLOt48JHhaY4fF7QsqJjtJByGEEoQ/ZRCaVZKiUlFGdrC3K2qdNRilFYsTvQohiqu3Kq7e\nOs2bQ5EYGUiywd8Zvc6qQTzxxGnOJrzOWyd8KYQQGpNczK7I0R9uj04YvKLAMDszRxIXb4oH\nAATjH5ohJofP4rrYu4RhQDCHQx2ntapotsaszCtHzg395f3zrgvzdF7Z35aiFu4mRim4vBjg\nQBMhxn8taDLw3w1Cs4swhm89PvT4v/POC33LKRCFIulbjxPxfO/Mf7khyjWU+iEQMbaG1RDV\nVZNqDhrvpuJyfEOMEJppS7J0Orn4WKfF4g4AgFElXZ2XlK4drzRv1qXG2RIBSJ+mnpRBnjYP\nOEwuv1zMZCcpUtRxvwnhPhQhErHIw0UnZTCErCg0tFgFOjSla+bdt3em9Zjcf3qniY8IRXE8\nbR+I7kcbIpOw+tEJRJTSA40DpzutTk8gI0mxaUlGKjYiRQhNGQY4EJptktKq9D/ttL/6ov/8\nGeA4SVGZ+rZ7Q6UraH4hEqK+FgK9NDgElCMiPUhyJxmSYJMh2CvwCthkFCE0M/IMyjyDkuMp\nITDfEjfCUrWy4jR1c3/0pNLqHJ1GLo5c8QU46cRbcvRYPbvrem0XikcIgUWZumsrUgW/IXql\nhCEkXKIiFjMcz/r9F7M4JCLmtjW5q4qSLY0D5tFzaiQiZiFOUZmifQ39fEyijVjCsj6G4/io\n9Q2L0iOTXNy+4C9frW/uHUlvrG017TnZc/+1xRunUCyDEEKAAQ6E5gSj0eke/D9zvQuUGHEG\nEWdM8RpEVkGd/QCj3/AxSpAssPGTCKGFhWXmaWgj7DOrc18/1tXYc7GOb3GO/pblWaE/2z2B\nd+v6mnptviCvkIqW5ydtLE9NcHqLN8C9dqLbE7gYoaAUTnVblVLRFUUCwWW5hK3M0tZ3Xawc\nlElFEjHDElKWoU3VypYXGHRKCQBcV5ZyqsfWPOQM8pQhJEMrW5GtV19+xRf9FnfsIsMQnUbq\n8wadEU1JqvOTHtxSGnnaK/tbw9GNkCDH73jvXFmWLk2PeRwIocm77H4WIzRLKHW8/6772BHe\n6ZAWFmm3fYbVJ030Gq6Dn7hrj1OvV1pUorl+K5EI17UudNTrDtYd4Id6iEorKlnKpOdN26U5\nG/WdBs4MAMDqibRyklNXpo5NIqqN1HMCuAtvncVZRLYECP4QRghd1uQS9nPr8vqsnl6LhwBk\nGRQpF2o9bG7/03ubw/0d3L7g/rODLQOOhzcVixII3Jztd0RGN8JquyzrCpMFk1puXpbl9AXb\nBi8WWRjUsrvW5mWMvuWWsMzKHP3KHL3bz8nEzLxNkJm6s13W3Ue6uoddSpmoMld/69rcyCYa\nkjg5NVqV9PEvrdl7oqdz0KmQiRblJ60pT438JlFKD50ZjH0ix9PDTYO3rsmd7q8DIXQZwffW\nCE0/zmHv/fbXPLUnQg8dAOa/vpD++M+U6zcmeAXe6ej57r+7Dh0Irww/83TmL34rK6+Y9t1O\nWqC7w31wX3CwX5SRpbzqGlFyyiQuEjx7zPOP31GHZeQxw0jW3SS79WGY+vvFQDd1H7o4BoV3\n00AvUawBcfZUrzw5bDJRXQe8G6gbGA0QbLmCEEIj0nXy2KYbHzYOhKMbYb0Wz4k286rC8ee5\nmJw+wXW3n/MEOIVE4OZcLmG/sKGwqc/eOewKcHyqVr44Ry9i4/4+ErzIJaPmQPsr+1rDD8/3\n2vc39D9+z7KUC39TFdm6Y83DsU+szNFplZLb1+fHu7Lbx7lj/mZDhm3eqe0aIXS5w/52CE2/\nod/8PBzdCOEdjr4ffpezRPe0j6f/J/8ZGd0AgEB3Z8+3vkZ98+UXv+Uv/9v94GdMT//a9upL\npt//ovu+7Y7dr030Irx12PPnn16MbgAAz/s/ecO/7/Upb5CnnuMxQ14p9RwHKtQWf9YwCmCT\nMbqBEELjOh+nXWXLQHTPDkHi+GNNxk4AKU3XbK5O37okc3l+0hjRjUtb97Br5/62qEWzw/fn\nvc3hh+srU3NTVFHnqGTiW9eOk4Ihk7Dxhs6oFWLBdYQQShAGOBCaZtTvc7z3Tuw673Y5PtiT\nyBU4h92x5+3Y9UB/n/PA/qnubzq4PtpjeeFpGrxYXst73EO/fsLXWD+h6wSO7aUBgU/Y/Aff\nmuoWg8NAhT67o37gBD5uQgghNN/4g8LxaK9Q4Ums7CSF4Hq6VpZgF4/L2bFzQ+F+q5FqW0y+\nC99/Ecv8f3csun55llwqCj1cUZT8o7uXGMcbKMMyZEmBcN3ucqH2KABAKfRY3HWdlrZBZ9S8\nG4QQioQlKghNs6DJFC/PItDTncgVAj3dlBN+9+bviP44ZU7Y33hVYJXn7bteNVZUJ34dfqhH\neN3cD1wQ2Cn8gKL++IeEk5YRQgjNK8lqaZdJoI1lcvxRr5Hyk5UFRlXr0Kg0EJYhG0snU1B5\nubG5o0fkhnA8dXmD4Yk2cgn72fX5n12fb3f7VXJx4u1IPrexsLnXbnON+mV93bKswnRN7Ml9\nVs/rx7p6LSPTeVUy0dYlmdXZc9RUCyE0v2GAA6FpxqrUQAgIfe7BahKaIccooxM+Lx5SKCe/\ns+kT6O4QXu9sn9B1iCTOm1RWBMzUCpsZ4Q/uAACYefE9RAghNLYVBYbYAAdDyIo4H/7HunVJ\n5pE207F2iy/IEQIZWvnVZanp2oTiI5c5g1q4r7mYZdRygSoSjWJipZcpWvnPHlz52sGO0x0W\npyeQYVBsWZ4lmL7h8gV3fNzijpjX6/QGX/m0Qy5mi9LUE3pRhNDlAAMcCE0zRq2WL14a1YMD\nAIBhlFduSOQKkqxscWaWQLoHwyjXXDHu0zmrmVFrCTuDnc+IXHiEG5HHDysIYYsWwyGBYhxR\n0eKpNhll9cBogLdHrzNqYCc8zmaqeAf1nQXOCkQEbDKRlgHBGmOE0ILB8dTpD6okolkeOrs0\nL2nY4TvQdLFWQipiblqWFduONB4RQ9YVJq8rTHZ6g1IxM0ZXDhRlVanx7x+3BrnoYpBVpUbx\nNBX4qGT/j73zDoyrutL4ua9NH81oZqRR75It2Zbl3rENGNNLqCEsBNhACiGQDSkQ0jaBsMkm\nkGySZZMAIaFXY7oBG2Nw73KTZPVeprfX7v6h0Wg088bq/f7+8juvXcmjee9+95zvsLdsLBz0\nsH013dHqRoQdpzqIwEEgEOIhAgeBMPakfPdHjXfdKvt90UHzjbeoCouHdD5CqQ881Hz/N2MK\nVZK//G9sRgZ/8rDU1UYn29jCMqTqX4aS/T7ns391b31V9noQy2mXrUr++v1sxhh1DMFYaG0W\n21oZexqblqFZvEIxWUO7ZMWwrsrOX8UXzJNqjkcHEadWXXrbKMYavgzSrsD+nSAHomJqpF0B\nMLGOcXwdDuwH6HtHFDsxX4v064FSyMIlEAiEKYU3JO5rdDQ5AhgAAWSZtUuyTHrVBL09IoBN\n89PKs81nWt2ugGDRq+ZlJRnUIxGI9Wryxjs8Ukya2y4seuqDM5Lcn5GaadX92wVFEzySFmdA\nMd7qUChfIswoEvQhJhDODfm6JxDGHlXJ3Jzn3+j+8+P+A/skj0tVWGy++auG8zcN/Qr6deuz\nn3qu43ePBY8fxaLIZWVb7rhbU5jT/cPbpPawbwVlthpvvU+1aDUAYFFs/e5doVOVvbuwwPt2\nbQ8cOZDx52fZzNH2kw9VnW5/9OfBY4d7N9Xzym1f/7bv04+k7s7ow7i8QuNV1w/v0hSlvfNn\n/Ecv87u24oAXaIYpXKC+4k4qNXuUYwYAoE1IfzHw1VjqAQBEJwNXCGhiv/QkNw4c6Fc3esFB\nHNiPdBsndCQEAoEwTAKC9O7J9kCfoyQGaHD4O72hy8vs6gmceKQmqVNJUclkcEFFxtxs03v7\nmhq7fHo1U5ZjvnBRRqLuJ+NHwkWJ0beTJxAIMxEicBAI4wKblm7/+a9HcwXN/PKcv/8LiyIW\nBEqjkdoaux+6E/P9Bpmyo8v5h4eTH/wDW1jq/XBrRN0Ig0H2ehx//3PKw4+OZhhCa3PjXbfK\n3v6efMHjR5q//53MPzzp3vKSf+cncsBP6Q2Giy4333Z3dEbJEEGsSrX5K6rNX8EeB9IaRmUs\nqnB1BlRzxvoNCAOWBhdK+DrsPwSyDwAAASAaUNR8QOwCOQDUULOsCQQCYeKpbHMH4vqVBASp\nss2zOMrf0R8SW3sCBg1jTVIP3WOSMC3IsOju2FwyuWPItGhPNLvi45EuOZKM91d3NXf7aRrl\npRjm55rJp5BAmM0QgYNAmED83djbjjg9GNKAHlKSLWIYxDAA4P/gNZkPxT6zJcn37gume34e\nOLA37kwAgMDBPaMcsuOfT0WrG73IXo/rrTdSf/if8AMsedxDNE89N8hgHv1FxhfJif0HQWwH\nLAGlQ+q5oC5RXlsKnsT+A/2bGABLgGCAcyoOABCBg0AgTCHaPMEmZyAgSAYVk5esa/co95xq\n94Q7hXmDwks7az870d5rkWEzqm9an1+Rb5mwAU9BvCFx5+nOhh5fSJRtBtXKAmuulThbj4ol\neZbdVV3uwICuLjSF1pemAkCrI/B/H5xuiypjKbAb7rqoRD+iUiYCgTADIAIHgTAhBJ248nXc\neRIAMABwejTnMpS+aOgXEOqrFFckxPpqAMBB5QpVOTDaCtXgsaMJ4ocBABAaE3VjWIgt9fzJ\nI7LPw2TkqhcuH+Okj4R37cSebYD7FjNlH/bvB7EL6dfEHolFHDiicAUsAUQJHIioGwQCYaqA\nMXxR313f0//IONXhYSnlYoReUwZZxr99/Xhde38T1k538IktJ+69omzhkLucTDwhUZZkrOXG\npcSmwx385xf1wb60F29QrO30nVeSsrpIoTkIYSgcreupbHDqGQrUjDso9gaT9arLKzJyrDpJ\nxk9+cLp9oElHTZvn2e01X988ZzLGSyAQJh8icBAI448kyHufBH9Xf4T34qMvAMUi+/whXgMl\naptKUQDAZuUq7uRy8oczUAWwrGBdDgAgxzqrTwSy7H72j74PXoM++1UmM9f0rYfZ3HH3PMP+\nA/3qRgS+DsRiYFIGBKVuwGKCq8iAKAAAxkbqUwiEaQ0G2H6k5a09Dc1dfr2amZ+X/JWNhdZp\naxVxptMTrW4AAMbAS7Ji03OThgWAgzXd0epGhFc/r5uaAkeTM3Cg0ekKCgCgYqj5aUlzUw1j\nW1Lz/vG2YFxRz6dnOuamG5N1w+uiOuaIEt7yRd0nh1vaevwpJs2aefYvrc3jprCJoyDKf3nv\n1LF6RySiUTHnl6cvK7alGFW9xVBVre52JQvS4/UOp483TfbvnEAgTAqkXRaBMO7gtiMD1I1I\nvPrDoV+ELSxNEC8DAMOlVyFGqS/9FdcN/RaKqIqV10BUJXNHeeUR4H39H753X4ao5jJiU13P\no9/DAd85zhoDsACiwv8gAGChNS6UQBKKgNRIs2RARHJA6AQOHAS+GjA/4mESCIQJ449bKv+4\n5UR9u1eUZKeP33m87b7/3d3QoTDhnxbUdit/i8a3hUUI5qQaAKCqNa4PNwAANHX5gkpNPSeX\nmi7fx1WdveoGAIREeX+jY3d9zxjeIsBLjT1+iNODMIbqyf5gCKL8w7/t+es7p2pa3L6gWNvm\neXZb1f1/+SIw9f6nIry1rzFa3QCAQEh8d38jwjhi9dLpCiqeiwE6EuwiEAgzHiJwEAjjj6tR\nOe5tA0nAPB86cypUdRrz55rZajd9iTLEFoMglVp3+c0AwGbmpDz8SHS1CKJp001fNVx69SjH\nbv7yrYiNlU4Qy5pvvm2UVx42WPa9/2p8WHZ2B774ZJxvnSAjAwCwEBtBXEJrd9oMqrnIcDFQ\nhsj5OLAfe97HgSMQOo39+7B7KwjNYzBmAoEwbpxsdG4/Eitu+kPi0x9WTcp4Ro8vwUTXpOE4\npv9dkWOo1XkWm14FALIcN5XvQ4rP+phUMIYDTc74MVV1ep2BuO/wkRI2ZFX6+veHEj9EJoT3\n9jVW1jligmdbPa/trJ2Au2OM2xyB1h7/OT4z8Xx+qiM+KMl4z5n+Dm4qNuFEZiIb/RAIhCkF\nKVEhECYNLOOef/y955m/yn4/AFA6veWOu81fvhWUyp4ps9X8o8c9T/+OPx32d2Bziw233cek\nhTuq6taer1m4xLtjm9BYz1hsmuWrR1+fAgCqwuL03/5PxyM/E1rDs242LT3l+w+risbfVr23\nCqbvtyG7nbLbqXig2DTOr2iUGhCnmFuBaGP/htCAg0dADgDFAgBgCaILfBgbMlwUe37oFIQG\nzohwCPt3IcOlQBFfOgJhinKoulsxfqy2RxBllpl+q0ccQ/GSQuGhScNeUJzS7Ap4Q6JexdgN\nal6URRkzFMqyKX9HWYwqnWpqvV66gkJQkBSF53ZPsLfiZvToVQyFkKwk7iRpJ9nwco+SWNAb\nv/n8wlFe3B8SO13BJB0XXxKCMXywv/GfH1W7/TwA6NTMTRsKL1uRjQYrDRJEufeUeLrc/d63\nxelJNIWkON3EqGUzLdph/yQEAmFGMLWeQATCzMSYqRju+qjOtfOdyKbs83Y+8RvJ6bB+637F\n45mMXPODj0tdbVJnK21JpW32sKFDH5QhyXjZl8Zw4L3oVqzOfWlL4NgRoaWZTUvXLFg4gnaw\nw0KuOSx8/ILcVgsAVHohe8HNVE4pMAmLaRE73nW2CFQFEDwZF+aAywn/m6/BgQMD99JAIZBF\nAAAmBenXxl8Xh6oV7oYl4GtBPW/UwyYQCOOCL6i87C9jHOBFNvGX1dQBAwaMUd9DJCNJc7oj\ntmFWb1zFUPkWnS8kbj/d8Xpbk4wxQpBv1a8psFoMqu64TiuXLM4a99EPk/gJcARxODkF54Zj\nqGK74VRc5Q7HUMWpBsVTJgyPX/kT6/aNqijS4eVf+uzsgZqw3pdj03/5vPy8qB/2xe01z3/S\n/5jzBcW/vnuq2xO6bVPxua/MMBTLUIKoILppVf2pGSYdt7ki4+0DTTHHXLcqj4ovryIQCLOD\n6bfIQCBMO1DaQtDGts0T3UHXrlPxBzuee0ZyxuaRRkNb7dzcCjolPUbdGFeQSq1dsjzpimu0\nS1eMt7oh7nkn9I+fy01nQBRAFOSGk6G/PyQd/oTS6dkc5YUmrrRiXIcEAEizsF/L6IXSIP15\ngFQAACDj4DGl0yiknosMFyDjJiVXUQyycmE2lpWL2wkEwlQg1axsEqxXs4YxSgcYPwQ55OQ7\nuoPN3aGWnlBrUPICQJndaIhLu0gzqnOStQAQFKR/7ak/0eruTU/AGGo6vS8fbLzrkjn59v7Z\nLMdQ167O3VieNprhSTI+1OB4+1jLliPNu892x3t2jgCDmqESpAwMN33DExRanIFE1hUXzbOn\nGMOPSAwAGFiaunxh+qSntKQk+MTak0dudx0UpP96/VhE3QCA+k7vb9443tgV9nPxBcVXdp6N\nP3HL53WOBB2IIyCA+TnKzeMX5A6wsL10SdYdFxTbzRqEgKZQXqrhvivKFhfM6l7FBMIsh2Rw\nEAjjD81SS7+Gj7+Cu/uKEThdiCkA+aP4Y7EoBvbvDZ45HTh2GPOCqrg4+dY7Wfuw3xdxKOj/\nZKtw9jQgxBXM0Wy4bPzTHMYAHPQJHz4bHxfee4ouW2348td7Hnsg2mQUAFQLlqkWLB33kSEa\n6deCWIKFNsA8opOAywXU92YsuROagzImYO2JLgqIJww+LwAAIABJREFUTmBKOtXnSATCbGZ1\nqf2F7WdDcXPv9eVpg+beTy68HHDz/TNSGUtewSnKgp41XzTHfqLd3eQM+HnRoGbzLboiq773\nhznY4HDFeVX4eanO4X/ohoXVre7mbr9ewxSmGUfZt8IZEF450OjoSys43eY5UN9z5cKMTPOo\nyg04msq36qo7YwXlJA2bZhyqZN/iDLx3rLWtz7qy2G64sMyeNFAf0amY29fmHWl0NnT7Q6Js\nM6iW5JoN6sn/Pj9/Yfqu420K8YqMEV9zZ2VblzvWyFMQ5bf2NX7j4jkAUNXsUkzBkGR8qtG5\nsjT13Nf/0srcqha3Z+AHb1mxrTTLFHPk4gLL4gKLIMkUQjRJ3JhJECMVwoggAgeBMCFozGjp\nvyNvO/Z1IlYLxgzYsV35SAytP/+x5Akv4PsP7nO98Wr6o/+tP2/j0O8mNNQ4Hvu+1NXeuxnY\n8a5364vJP3iMSR+Yg8AHxc9elU9+jl1dKMlGla1mVl8DrGr4P96YIdefAEFhYQcHvHJzlap8\nmeXB37mfflxoqAEAxKl0F1+nv+bWhKaeg4J5ELtA8gNtANY2eFIbk4JimsL2DTzxLc65/MjY\nFS1FUUJNhEAgTD4Wo+req8r+uOVEtHnkokLLlzcUTOKohoJPcMUHg5JPzehZmi1PTypPj3Wz\nBoDGgR1kIzT0+BGConRjUbpR8YDh8u6xVsfAogk/L711tOXf1xQw9KgmrsuyzaIk10X9IBYd\nt67AmiizI4Z2d/CfX9SJUn89y5k2T7sreOd5BaqBlisUQhXZ5ops5eyDyWJFaeq16/Jf+6w2\n2ubz4mVZFywaucBR3apQ0wQANX1FOorqRnjXQMOXY7U9x+ocgZCYadOtm5+m4WgAsCWpf3Jj\nxZt76k80Ot1+IS1Zu2F+2qo5io9gAACWJmnpBAIBgAgcBMKEok9F+vCShapQuQBVlmTwDChP\nkIPB1p/8sODtjyidfkh3kWXn738SUTd6kTpanE/8zPrI3/q1gFCAf+ZB3Bnu8IKd7dKu1+Sq\nA9xtv5xMjSOk0NC+FxzyAwBXWmF97GnZ45I9LiY1A+hRqPuhWuzbB3LfAhSdhAyrgLGN5FKU\nAYBSljno2LWmaJC6HIsdsa1YmFRglX1bCATCFGH5nJSSLNMnh1saOn0GDbsgP3lJkXWyBzUI\nEhalBD2hBCnEKPUa7yWRUcUYGlgAgCsgNDkUlBRvUKzv8RXYhvYETABDoXUF1lI73+kNiTJO\n1rLpSZqhSyafnemKVjciAz5Y37OyYKr/v/dy++aS1fPs2w+3tPX4bSbNuvn2eXnJg5+WmETO\nJmKfeJGTmvC/LLfPpyMkSL977fi+qMYoL39ae98188pyzABg1LK3bBitByqBQJhtEIGDQJgc\nuJw8/boN3k9j+ptiUDJgl1wu3+7PDedvGsqV+eoTYkt9fFyoqxLqq9ncovA1974dUTf6b99R\nL+17l1511VBuNHpkR2dw+xaptQ6ptWzRAtXqzSg5YeYCldxfp0MZkuKb5g4PoRV7dgLg/p5+\nkgu7tiHzlUANPxcascDlAV8TG6fNgygmdBIyXIQDh0BoBZABqUBVjFRzlTsNEgiEqYRJx129\nOneyRzEsEuoR+BxpaAAWvarFqaA+W/VjqYa7E3dsjS+QGRlWHWcdURGNovICAE09AZjqWTv9\nlGQmlWQO79EpSrihw+Py8Vk2vTVpQDlPpkV7tK4n/pRMa7i9TopJs2Ju6u6T7TEHVBRaslPC\n2sez26qj1Q0AcHhDj7189H++uUo/5e1sCATC1IQIHATCpGH/6SPtj/7c80F/IxXtstXeXTsV\nD/Z98ZnQ1sraUjSLlzGWc7lnSZ0KdbZ9u1ojAodcc0jxGLn64MQIHPzBT33/+A0OhRMo+H2f\nBHe8ZbjnV1RqjtweK9BQ2XORbSyTGrD/OADE6ghYwIHTSDcSy1KkWYhBBj6qYS1jQ5rlg0sV\nlAHp1gFgwAKgaeCTQiDMHHAQsBcQC0gPMPOLvSnEIEBYSeag0bkmkxVZpspmV3wD1EVRhRiS\njI+2uGq7/d6QaFAzBRbdvHTjEAtAeuktTFDeNdml+IrNX+Gc/VlmALuOt/1py4nOPm1r9Tz7\nN68si8gc68rsHx1tjXei2RTl63Hv1fMoBJ+f6Nc4lhTb7r0m3CNMlORPjrbE39cbEL442XHh\nKMpnCATCbIYIHATCpEHpDWn/+V+W2+8KnqwEhNSl8wBR3l2bFQ92vvpS+CydznbP/cbzL/B9\n+KbYWEfpjar5izVrLojUnlD6hO3oKF1/mTQO+hSPwSHlOACApxu7OpDeDEkpo+zhIrt7otWN\nXqTms/6X/6y74Xuh5x7BXf3OFFRqDnftfaO5nQJSglY1ksJ61NCgkWYpcMUgdQNIQJmGWe2C\niLpBIEwcmMfiCZD7JleIQ3QJ0FOuv+nYggCpaG1Qiv2SpxDN0efy2kw1qi9dkLbtRHugbzbL\n0dR5JSl5fWv1giS/XdnW3Wef0ePje3x8vcN/Sal96KaPVr3KpGGdcckaDI1yLLohXmT0YIAT\nza7aTm+Al6wG1eLcZKOGTTWq67oUHo6pQ/YonXbsOdnxs38M6H2+63hbfbvnL/et4xgKAJIN\nqm9fVvrMx1UdfcarWhVzzcqc8qguJ1o18/0bF1Y3u6tbXDLGBWnGkiiLUKeXDyboR9OawPaF\nQCAQBoUIHATCJMPlF3L5/SWmqqLiUNWZcxwv+3ztj/7C/eyfGC68cOTbtoV77zXLg/9FafUA\nwJUsQFod9se9whpNbFFZZBOZU3G3kr2lWalIxNkmf/w0bgh3QkWWTLTxqyhjTvyBgUN7Qycr\nQZa4gmLtirWJ7D+Fw5/HqBu98Id36W65X/2N30vHPpVbagAhKqOYnrcaqDE3D0v0zj262hA6\nCejR1c4QCIRxB2NhH+Aou00cwuIxBAjoIWWKyTJ2+vkkLTftWjboWJMMMi/115vQiDFwFjTY\nV98cuzHXoqvp9Dr9glHD5lt10a1PK9s83b7YTlIdntDJds+8tGH4j24qs796sCkmLWJ9cYo2\ncXLH2BIS5ed319VHaRlfVHddUZGxPN8SL3BwDLU4d2qZiY4hz36o8CrS1On7+GDz5mVhKbAo\n3fjTmypON7vbnQGTjivOMOqVWsYUZhgLMxQ+BmpV4pydwRrrdrmC/pBoT9ZyUSavvCjvq+5q\n7fFzLFWUZpybeS4PLAKBMFMhAgeBMLVI++mvGu++XRroMxoP3+1iot4a+ZNH3P/4k+nuBwAA\nqTVJt33H+adfDjiBopLu+C5i+9886PINcvXB+CvT5XHtWoJe6eVfgM8ZCeDuJvzao9QNP0Ep\neZGg5HZ1/OIHgf27IxHV3PmpP32MSVFQTGRHZ3wQAEASZVcPnZJBL9xAL9ygfMyYwNiAb4gP\no5GZjBIIhGmE3DFA3QDoVTaxVIUGEzicPv7Vz+sO1HSJEqYptKjAcu2qPLN+2qRfIUBG1iLQ\nIVHmZZAZxKpozRCFXTVLlyn1WAGAxgQWFY0O/7AEjhyL7qur8nZWd7Y4g6Is243qlQXWDJNm\n6FcYJdsqW6PVDQwgSPKbh5q/dUHxZeXp2060B/tyWMw67tLydOMM9YkQJVzVrNBwBwBONjjW\nzLe7/UKqWUNTiKGpsmxTWfZIpAS9mi1MN1a3KLzwLCxIWIp7qLr76W1n2h0BAKApdOGijBvW\n5WtUTG2752/bqnq84UZs70FzWZbpjguL1ZNd30QgECYYInAQCFMLdem8vNff7f7bXwJHDmFB\nkNwusV3BU0OOy+r0f/q+6d+/29tVRLNuM5Oe43n5r8LZ0wDAFpYarr+TzRvQt4Was4JedbX0\nxZuA+7zlKJpefQ1VtDjmyvjotmh1I4wk4D1voMv7K0c6H304Wt0AgNDJY+0/+Y+M//lHfP4F\n0iV45UWISrRrTEHa+Vhoju3hSmlBUzIBdycQCJMIluO+0MI7AoCDgBIWHXiDwqOvHI3MoCQZ\n76vqqmpx//iGhYZpNdFlKRVLjaU/aEhQ9igNJm4UmgizjruifHLMFzDGRxsHfDYQAAYQJbmy\n2bWq0FpkNzR2+91BwaLjsi26MczfEWUsy5hjpk6jU2VvEYpC+850bj/aCgAcQ122IueG9QXq\nUeTXfHVT8U//eTCmoezGhemJGg/vO9P521ePRTYlGb+3v6mx0/e96xb87wen3f4B9U2Vjc5X\nPq/7ynnTxwaWQCCMBUTgIBCmHIzFkvrAg73/bv7evR4lgSMeHAxIHidtCi96sIVzk3/420Fu\ntOHLdOkq6eQX4OoCk40uXYVs2QpXbq1WvmNrVeTfYkeb/4tP448JnawMnapUlc6PibPzl8Pr\n/wdy7LsvUzgf6RJ6iIwljAUZL8S+PSD2mXFw6Ui3fIARhtCA+UbAfqB0iM0DNk3xSgQCYZaw\n7XBLRN2I4PTx7x9qvnZV7mSMaKqgVzGuoEKjE8NghQZTCj8v8XGKTK+G4fTxAKBh6WJ7+Anl\nCgidnhBNoVSjejQVNM3OwL4GR4+PxwAGFVORZSoaXUPcMYGhqZxUQ12bJzpIUUjF0b5guNMw\nL8qvfVZb0+L+2a1LhmMmO4A5WabH7lj27EdVlfVOXpBSzZorV+VeUJGe6PgXtp+ND1bWO7bs\nbohRN3rZW9V1/eq8qaQcEQiEcWc6PXgIhFmItmKxZ9v78XEmvsyVpns9OIYFSs1lUnMHOQgn\nWIKLigsNtcrHIODrz8YLHHRqpubSWwJvPTPgWK1ed+O3BhnMOfC7cFcDIApZs0EzBJWETUGm\ny0DyguwD2jiwO6yMfZ+BEPEo6cJ8PXD5SLt85MMjEAhTA0QlYUVnQ6QGdK68hlMJkvZPNynH\nZw+FNl2zS6GP7FSYqw8dFUNTCCk2TInu8BIUpA8r2481hXM9GBqtLLCuKbKNYJJf3endUd0Z\nKRHyhMRPq7ucAWFp9uRbe9y4ofDR5wd0W2OVZIIjZ7sPVHUuKR55dWd2iv7BmyowxoI0SA6L\nJyA0dyv7oFcp1bkAgCjJ3Z5QmnniqpwIYwhiyUSVMBLI54ZAmNIkXXWt48V/8Q0D26YixJlj\nk6jVC5YibixTjvux5UDdkfgwSsnt/7dKDVi5jhuplfO9NZfczOSXBj94SWqpQxotU7RAc9kt\nlDFZ8eBBEHl514v4yIcgSwAANIsWXUytvBaoQVfVENAGoOPUkFB1lLrRB38W2DRgFZJcCATC\ndIJKBaQH7I0JI7rg3G4UQoKCi5Co3Ali9lBo03d6+RNt/ZNMhGBeWlJOsvYcZ001GBrlp+ir\n2z3xu0rs/RUTrx9oqo3y6RAlvPNMJ8awrmR4k3wZ4z31jviP3LEW19xUg36yk182VqQHQuLf\n3j3lDQgAgABRCUpyjtc5RiNw9IIQ4phBJCJJStiU9xxnEg8OAmG2QQQOAmFKQ2m12X/7Z8dv\nH3W//w5gDADqsvn6ssLQvh0DDtMbk26/d7zGsOAC6ciHwA9cnaNotOTyyJaquJTS6eS41i2I\nYTQLYk09IrBzKtg5FaMfobzt//Cpz/u3JQHv2yLzQWrDrSO7IBbqleN8PSICB4Ew7UGIXYbF\nYyBH3I4ZxBQBnXPu09LM2kalXqHp5uk0jR8nVuYl51t1td0+T1A0qJkCq86mH7bm3tDjb3YG\nBEm26FUlqQZmwpvUXDQvrdnhDwx0uVqab0nvSwFodgRqlT4De852ryy0sPQwSiF6/ELEsjQa\njKHVHZwKyS+Xrsg+rzztdKPT4eVtRvXD/9iveJgwUQJfko41ajm3P7ZfDwDMyzU3HVZwuk0z\na6aRBzCBQBgTiMBBIEx1GKst/ZHf2h/6Gd/QwKSkMBYrAAS+2O5963mxsZbSG1ULlhhu+veI\n+8bYY7BQV/9A/vBJ6OlLatAmUev/DWVFNZ1VqZK/9u2u3z8Sc6rppq/SFuvQbyXXVQqfvCC3\n1iKKQumF7AU3U+mD2YM52weoG33go9tg+dWgHZFlqayQaw0AICt3CiAQCNMMpEbsUsBewB4A\nFqgkgMFdQs+bZ99bpdABasMCYtADAJBqUKUaRphIyIvyO8dba6MKEPbUdl86Ly3VmNDzdTyw\nGlTf2Fj8ycn2s51ePy+mGNUrC6ylGf29Y9qUKnEAQJDkLk8obTgNX0QpoQOrmDhVYYLRa9jF\nxTYAwBjMepUjzoMGALJTJsQ5CwAhdMmyrBe218TEbUnqixZl+AXp08oBnmU0ha5fnQcEAmGW\nQQQOAmF6QOn06rmlkU3NyvWalesn7O4orZD+yiO4tRqcbWBIRvYi4GJfOo1XXk8nW3uefEJo\nqgcAJsVuvu1uw+Yrhn4Xcd/7wtb/7f03BsA1h0O1R7nrvkuXrgwfgUOAQ0AZotNRcbuC5RgA\nAJZxRy3KLR/6APpBKgClQl+K1PESCDMIpAc0jHXyonTjrRsLX/ysNti3wq9i6etW55ZkKDdP\nJQydT8501A60V3AFhC1HW25bmTustIjRo1czl1dMRBuXJA2LEnQrSZp6TXkQgktXZP9zW1VM\n3KxXrZ2n0Ax+nLhyRXYwJG7d2xiRh/LTDN+8vFTF0jeszsu26j443NzpDjIUlW83XLMiJ8uq\nm7CxEQiEKQIROAgEAkgup2/HNqGpgbHatMvXsDlKKx4UjTJKIONcXVR1azfq1m6UPW4sy3SS\naVhjwAGv8MEzsVFZFrY+SZcsBdyFAwdB6nXyo0FdglTzAI1XYS3isnCgRyHOZo7THQkEwrRg\n9dzU+Tnmo3WObk8o2aCan2M26UgC/GjhJfm0kvOFNySe7fKVpE5QgsBQSJSjwdKUzTC8ZBMN\nS+dYtHXdsYmBZi1nN46Po9bouHZtnscvbN1dL8lhWSbTqrv/2gVa9cTNJhBCN64v2Lgw/XST\nyxcUs2y60mwTQggAEIJVc1JWzUkRRJmmETXizi4EAmGaQwQOAmG24/3k/a7f/Ur2hM3hep58\nPOmGf0u+8x4Y2suB5HIGjx+RerrZrGz1/ApE05QhripElvhdbwuHtss9HZTZxpav4dZeCfSA\n7x+5/gTwwfjrY59Lrv8CJTdHLXRJEDyBRQfSrwcAlJogARVR0Taow4MrAaEVxI4BQTYTOJLs\nSiDMdoxabk1p6mSPYkbhDgiROXMMDiXDhUkk3aTJt+nPdsY61K4ssDD0sGfUa/KtgtTZ7Owv\ne0nWcueX2Kbm5BwhdPvmkouWZB6vc3j8fFaKfkmRjaYRAIQEqbUnoNcw1nEoKfKHRJpCqiiv\n0BSTJiVxNZBitxcCgTB7IAIHgTCr4evPdv7yQSz1O4RhSXI+9xSbkW245KpBT3e98VL3X34v\ne8OvelxBUeqPfqEqKR1wkCj4/vchqbayd0vyOqXGKuHoLu3XH0Fs1CJVULn3GwDInko6OS4D\nXGwFsQ0YO5jsqHgFPrM7Zj9acD5oR5o3jmik3wihaiw0guwHSoe4POByR3g1AoFAmE3wotzp\nDYmSbDWodNzgr5qJmoNiAG5i61OGwtWLMz860Xak0dnbT5alqdVF1pUFw3CbiqBiqM1zU1tc\ngQ5PSJSxVafKSdZOSXGjnwyrLiOq7sMbEJ7fXvPxkVaMMQCkW7Rf3VQ8P1e5IZrTx9e3e2QM\nOSn65MHsWjDA5yfaX/u8rsMZQACZNv0N6/LL80bUai1+JAGhodvnC4lmHVdg009wGRSBQBhX\niMBBIEwOWBS9H2wJnTgqB/xcfrHhsmuHW9MxJnjefj1a3Yjg3vLyoAKH9+P3O3/zn9ERvqaq\n5f67s//1Jm0y9wd3vxtRNyJIDWf4nVtUG6+LRJA54YooZVB+3cNiB2LsAEBtukvWGPDRjwDL\nAAA0gxZeRK26/tzjHwwEqiKkKhrdRQgEAmEWgTHsreveXdPNSzIAIATlmabzilMSSRi9GNWs\nWcvFJ2sggOwp02i2sdu/7XhrqyNAUyjTor1pWa4MMk2hVKN6lI1I05M06UnT0uBJkvGvXjx8\ntrW/vKil2//oi0d+cH35/IFKhCjJz2+v2bon7J1BIXTR4sxbLihUJf7VvfpZ7Zu7wx3NMEBj\np/c3rx6986KS8+aP1tN3V3XXntruSNKQQc1cPC8tx0LcOgiEGQIROAiESUDs6uh48B6+LuwE\n7v/sY/drz9kefESzaPkEj0RoVO6HKjTUDXqu47mn44OSy+l+61XzLXdGImLlHsXTxco90QIH\nlVWCLOm4uyXmMCqrCCUn6ISC+6QZhqM23AZLr8Cd9YAoZMsB3SSoRQQCgTDL+bym6/Oarsgm\nxnC40ekNiVdXDGJgtL7Y9uaRFhkPKFQpzzRZh99rdjzYV9P9+r7GyPAcPv5Ek+vWdfn5aSNq\n1JUYSca7T3fWtnkAIC9Vv2JOCj3hvXKHzt7THdHqRi+SjF/YcTZG4HjqgzPv7W+KbMoYv7u/\n0RMU7rt6nuKVHd7Q1r0NONpRHAAAnt9Rs7o0lRlFwsXRJmf0RxQAPEHx9UPNd6zJM6innLcr\ngUAYAUTgIBAmge7HfxlRN3qRve6uRx/MeOoNSjcMS//Rg9TKq0ZIM9hqEsZ8zRnFPaGq0wMO\n9ClYxwEA9roGbFM0d8P3+Od+hZ39XRiRNYP70n2APges0JoO0QNVDH0y0o9N8iqBQCAQhosg\nyfvqFOyZqzu87e7guRu+5lp0Ny7J2lHV2eYOSjI2adilucllaVOiPU2Al9462BQjvkgyfm1v\n4wNXlI6hX0aHK/j4m5UtPf22o+/sb7r3itJz+E1MJE5vqMfDpyVrNKrw9OFUo0vxyLOtbkGU\nI14YLh//wYHm+MM+O95243n5aclaUZI/OtJa2ehw+wW7WbNhfprDE5JkHP+b9QXFunZvYfqQ\ndCVelD8+3lrd6vGHRLtJs36ePduqO9jgiD9SkOSjTa7VhSOpMyIQCFMNInAQCBON5HIE9n2h\nFHcG9u7SbbhoIgejXbzct/2D+LhmyYpBzkQIKOUlFMQM+GJBZhu0KHRypZJTYiOpOepv/UE8\nuE1uq0WIQhmFzMKNQNM4WAzBY3Hna4HNGmSQBAKBQJgoury80Ne8M4ZW1yACBwCkGtXXL86S\nMZZkPKU8EWraPbyo8HM5/XyrM5BhHpsiGgzw57dPRqsbANDS4//TO6d+8uWKyc3iON3o/J8t\nJ6qbXQCAEKwvT7/z4jlmgyqRNSwGEGUcSYeoa/fEyEMRalrceg37mzeOt/b94C09/oM13XMT\nt14OCgp1tfE4vPwT75zs8YZXR5p7/Idqe65YmtXtVbat7fYprKMQJpnRFX8RZi1E4CAQJhqp\noy1sFRGH2B5boDHe6Ddf7t76auj0ieggbUwy33b3oOeq55UHDuxViM9fGL3JLlqvWKXCLtqg\ncFGWY5ZfEhND6jKMeQid6W+kQpuQdiUg8g1GIBAIUwflSSwA4ATz23gohKjhtyMZVwJ8whm1\nPzSkyfZQaOz01XXENmcBgPoO7/G6ns+Pt59udMoYF6YbrzuvQKdmDpzpbOn220yaRUXWQQ07\nR0N1s+v7/7cnIvFgDJ8cbqlqcv3hntXZNmXfihSTRsMNdWr6xu761p6YXrm4stGpeDACSDaq\ntuxtqGx09qZ7rCtNrci3xB/52p76iLrRi4zxlv2NGXZDbN0LAAAwCdZsCATCtINMDwiEiYYy\nJlyXOMeucQIxbNp/P+l89q/ut1+TPW6kUmlXrLXc9R0mLWPQc5Nv/0bLkYNYFKODbFaO8eIr\nB0QWrJFWV/K7tvZu9pbUsssvYpecP/RhIs0iUBWB2AGYByoJ2LS4ytwZh9CIxXbAIaCMiMsH\nivifEQiEKY1Fr2IoJCqt6tunp4lmLyYdBwB9j68BmMO7xoAOVyDBHvzL5w65+/IOzjS53tvf\nzNLgC4Qfvlo18++XzL10RfZYjSSGf35UHZ/A0tTl+2B/08aKjFd31bl8sTkRly0fMJg8u4FC\nSDGJozDd+MrueC8wRFGQpOPirzwvN/nP757q9oSVC5efP93sWj0n5ebzCqIPEyRZUSKRZcwC\nKKZwZE0ZO1sCgTBKiMBBIEw0TGo6l1sQ48EBAIhhNUtWTfx4KK0u+a57k++6V3I6aGNSosKT\neDTli9J/+5fO//4lX1/bG9FvuND67e8j9cA8ZITUV9/NLlwrHNwuOzook40pX8sUlQ9/oAbg\nDMM+azqCRez/DMSOvu1mzJ9B6sWkTy2BQJjKcDS1OCd5T213TDzXoktLGqQ+ZSqTl6I3aTln\nXJOXbKvOOnapE9qE/XRRKCqFBGPgeTF6KP6g+Phrx1LMmqUltrEaTDSVdQqmFQBQWe+4fGXO\nD29Y+KetJxr6ck84hrpqVe6mRQPWSIxa7qIlme/ua4y5wrr5dmuSxh8SQYmSbFNHT6Cuvd/G\nqzTbbDGp62piP2C7TnUsKbSWRFW1+ENiovIZq5br5MWYWqpUo7psaL4eBAJh6kMEDgJhErB8\n+0dtP/wmDgUjEQxgvu3rTIp9EkcV3dt1iGgWL8t+9nWhtVns6uRy8s5xBTqvjM4rG90AZws4\neCxK3egNSTiwHzFWoAZ60GIeJDdQHFCGmZ/SQiAQpjxri2wIwb66nsj0sjTdeP6chF3ApwUM\nhW5anfvszrPeYP9UPFnPXb8iZwzvUpBm0HB0fDkMxlgQogUO5Xn7G7tqx0ngEBP4qgiiDAC5\nqfpHb196ot7Z1OVL0rElmSbFepmvbirWcPRbuxt6lQWaQpuXZH7l/CKGRjo14wsqaBzpZu03\nL5l7sLq7rt1D06gwzTg/N/k/nt6nOJgjdT3RAodWxdAUUtQ4Uk2aC/OSPz7V3tjj7x1JeaZp\ndaF1DM1iCQTC5EIEDgJhElCVlWc8+ZLjmT+HKo/goJ/LL0666XZ1+ZLJHteIoCg2I4vNIH6f\nY4eg2LtXBqEBVKXhLczjwGHgz4aL3ikt0iwirqsEAmFyQQjWFtkW5ZibHYGqDk9IkGmETrd7\n5mckTesJZI5V991LS3dXdTU7/BRCOVbdskLrmdWnAAAgAElEQVTL2Dqhqlj6xvPyn/qwKibu\n8fLR8/REAkd9m4J/x5iQazecVir3yE8L51RSCM3LNc/LPdcaCU2hmzcWXr4ip77dI2Ock2Iw\n6cPVPUsKrTuOt8UcjxAsKbRSCC0psi4pCjc3kWWcKN3DExCiN1mamp9tPhzX04emUHmu2WZQ\n3bg0m5dkX0hM0rDT+pNJIBDiIQIHgTA5MPZ02/d/MdmjIEw9sABY2eMdy76+tzCMvdtB6o7a\n5QffZ0i3hmgcBAJh0nH4+A9PtPn7khGONbsO1DuuW5xpULPnPnEqo+HoDWVjnIqCARo7fR2u\ngEHD5qTo15XZU5I0r31e1+s2mpuiX15s++1LRwaehBTNXDl2vDwyr1mT+8jzh2OCWhWzeemw\nHzdGLTs/L7ab+1XLc2rbPQ2dvujgZUuzc1L0MUdSFErScq64WiEASNbHpo1csyK7qcff5e5P\nlaUQunJZtq2vmw9HU5x2zCxUCATC1IEIHAQCgTCVQAwABaCUEoz6XuCE5mh1A/qqU3DwKCIC\nB4FAmFQESd5ypMU/sNSi2xt673jbdUvIF1Q/LT3+f+6oqevw9rqX6tXsl1bmLC+2/ej68l4/\nTgohjPELH1c3d/VP/hECxRyOBUqdRMaEtfPTHF7+mfdPR8pn0pK19127wDJY398holMzP7y2\nfMfxtsoGhzsg2E2aDfPT8u3KllvLiqwfHontN4cQLO3L8oiQpOV+cPW87cfbqlrdfl6ymzTr\ny+yZFuIkSiDMfIjAQSAQJhoc8AHLIWYaL+XFgEGWZAEQohGLRuuFgYBNA6FZYQeTHr6d2KV8\nquQGzAMiS1KE6YnYiKV2wEFAesTmAmWa7AERRkJdt8+nVEdQ1+3zBkW9mrx5AgD4QuITW0+4\nA0KkN4s3KDzzSbWGoxfkJkeKJhBC/3Hdgoee2u8LChgwAkRRCOPYOhWjlrtpY+H4jfaKlTlr\n59mP1fZ0e0IZVl1FgYVlxjJhhKbQxgVpGxekDXrkpUuy6ju9Z1rckQhFoauX52RaFBqNsTR1\nYXn6heXpYzhUAoEw9SGPGQJhOoFDQc9rzwb37pS62+nUDN2GS3QXXQ30ULvNTzJY9n+81fvG\ns1JnG9AMVzjXcPM3uOJ5kz2sfjAWg3KPJAcAMIVUatpCDSYWYMAB0RmU3L05wwgoDWNS06My\nY0fqcix2AQ4NiHL5wERWqJRrsAFAOfWDQJjqSDj4BUhdEG7F2YXFesSVADtnsgdGGDbugLJL\nAgC4ggIROHr54lSHu9c2YqAk/t6h5gW5A4o4SrJMf/3uupd3nD3T5BQlXJSZdP6ijFd3nN1x\ntFWWMUKwtCTl61eUpprHtxGv2aBaNwQBYrzhGOrey8v2VXWdaHS4/UKqSbO2NDWddHglEAhR\nkMcMgTBtkL2ezgfvFhtr+zZPOWtOBfbutP74v6eFxuH6++/8H74R3pBE/vSx7p99K/k/HlFV\nrJzUcYWRcNAnNOI+gUDCIUH2aBk7S51LrfAJXbzcnzyMQfaLPRhkDT2KxWdKjwybcbASxDbA\nIaCMSFUcba6BaLOywkFpAE3jXoyEWQvmT/eqG9A/3cOYP4XoFKBiK/YJUxx1YjMIDTsNHlXj\niiRhmkYA0NDlUzygscuHMcS4XibpuDsvGSD2/fDLFd+9vrytx59i0qi52fVbRQDLiqzL4mpS\nCAQCoRcicBAI0wbP689G1I0IoaP7/Nvf1Z5/2aQMaeiIDWf71Y0IkuR+5gnb1BA4AmIbjk1/\nwAGxneF0CJRfHyUsRKsbfSdBUHSp6aRR1aogFdIsSriXzQLqGMixnvko0mOFQJheiE2KYSw2\nIS5W4MBYRmi06fEYRBl3yjiAgEZIT6Nk0mh5rMi16BQ7dJq1XLJulhbQYYw/OtTy+q7a1m6/\nmqPn5yWbTAnF6OpWd6czkGLS5KbqmcSNWjiGyo6z4SQQZhSzXhIljAwicBAI04bg/l2K8cD+\nz6a+wBGqPKAYF9uapM422maf4PHEIGNeiikJAQAADLIo+1lK2e1MlBVOAQQYsCTzDBVr6j4k\nsAB8PZZciFIDYwdGyTcO0Ui/Hvv3gNjZH1GVgap4JHckECYdHFCOy/1xCYsewS1IPAZMIUrD\n6LSMbmQyoozdgnwWoK+SAndKqJ2lihF5KRoLdCpmXbHtk1Md0UGaQpvKJvl7fhL545uV2w6G\nnZV8QXH3yQ6TUa0zxMo9giD7Q9KPn9nfu5lu0d65eU5pNjGjIRAIhGFAnuWEyUHs7HC++iJf\nU0Xp9ZqKJUmXXQkUkWkHQfa4lONu5fiUAgcTTGAAcMA/kSNRRAYp0S6ME9aTn9ML4xy7EiO0\nYN8XvZO68PmqQqRbrrC2TBmQ/gIQu0B2AeKAtgI1vgXYBMI4glSAg8pxAAAQZcER6sZ9fxYy\nln2CR5B4k2oEBSzSAHUDAAAw9otyPUsVDP9qBAWW5CSnGNRf1HR1eEIMhTJMmjVFtjFM3wiJ\nsickalhKx02D99gzTa6IuhHB5QkaDFx00qAkYY83FP3oaOn2P/ri4UdvX5Y+y3p/iJL8/sHm\nz092dLqDZr1qaZH18mVZKrKYTyAQhsY0eDAQZh7e7dvafvoj2R/O7Xe//abz5ecyn3iSNpNa\n63PB2Oy8s0chnjIOvl8Yh6pOCXVnabOZKymjjUmjvB6TptwdELEsPR7jHyZU4i9DhBLuoqlE\n7+so8a7EyAHs/RRi9JRQNVA60MxXPoWxApA6ZML0h0kDIbb+DqI6B3lFD44TDXk5FJKCKnp4\nvjMSdsaoG73I2AkgQYJ6NMJwyU7WZidnj/llfbx4oNHZ7Aor5mYttyTLbJ3alS+HqhX6XmEM\nre3e9YszTzY5wxFJjhfGeVHeuqfha1EGHL39U9DMLagSJfnXrxyrbg33SelwBt7e13iwpvuh\nG8q1KjJtIRAIg0O+KQgTjeToiVY3egmdPtnxX79M+9VvJ2tU0wLteZv5qhMK8Q0Xj+2NhKb6\nzsd+Fjx2qHeT0uqS7/yW8eobR3NNVcVKOtkm9XTGxDVrLkLqyU89oBBLI7UUt4aMgGYoheZz\nvTAgs4gR4lI81LQBwfA9AvjaWHUDAABw6AxKJHAQCDMCxM7FUhfIngFRJgfolN5/ChKveCIv\nh4YrcAAoXwoAY8wjNPlfR4RE8JK87UyHn5f6OquCw89/XNVxYUmqWTN1+477gsppgKIo37Qm\nD1Go3RkwatnH36h0exU+nGfbwn8XR8/2PP9J9dlWNwAUZiR95fyiuTOxemXH8bbqVjdE2ucC\nAEBrj/+d/U3Xrs6NPjLASztOt1e1ebwh0aJXLclLrshOnsHSD4FAGCJE4CBMNN7tH8WoG33x\nbbLfT2lnVx7mUJD9PteLzwQOH5C9Hjo5Ve5p799HUYZrb1MtWDqGt8PBQOt37xY72vq2Qfb7\nup74NdJoDZuvGPFlkUpt/t6jjt8/LLX3Z+qqFq0y3vrtoV9Eam3wvfY3seo45kN0Zp72spu5\nBStGPKQYNIzdJzZFF6QgoLSMXVmqkH04dBjENh1i/HQ2T5kje9S0UcuYFU4ZDCy5lXfIAcAC\noKn7+k4gjBbEIc16EKpxuHOQHjG50Je+gQHHp2+Ed+ER1IIlztFAJH1jSlPd5fXzEgys2ZNk\nXNnqWpM/dXPZ7MnKqplGxZj1KppGFoMKIKEo3jtjf3dv41+29q9wHK/t+cFf99x/7YLzpkDr\n1rHlaG1vpmqsUHGktida4HAHhL99WhNutQvQ6gy8dai5ut1z3bKc6DMbHf49tT2dnhBNo7Qk\n9ep866x1uiUQZg9E4CCMMxj7dm0PVR7FfIgrLNaff4nQ1qp8oCiKne1cTt4ED3CKI7Y2t3zn\nzn65AYDiaO3CRXRSEmPP1K7fzOaXjO0dvdvejb5d5B3D+a+/6zdcyO/5UGquRSo1k1/Glq8e\nVposm1ds+82zwd0fC/U1lFrDzS3nyhI3ColDOHnI9fsfgBgWIMSaE+7HH9RedZv28luGfpFz\nQCOVgc0LST0SDgLGFKVSUcmUYn0KFrB/R68tIsKiTjyrRmqJ0iGulKETnDIUEp6IyLyLMAug\ngS1BrMIXGgJEI1rCCkY5DDXsPzcKJQGgeJcchLQIyMxnStOllOAAAJ2+RFk5U4LVZfZnt1X5\n4/I4Ni5M720Z20thurG+I7Y3Vm/cHxSf/uB0/K4n3z65sjSVY0bbVGhK4QspJ7zEJMJ8fLI9\nom5EONniPtPmLrGHm7sfaHB8WtUZTgYRobrDW9vlu6o8IzuZrKURCDMZInAQxhHJ5Wz/8f2R\nSgcAcD77N/WKdcpHI0QnzcBky1HS9cSvB8gNADIv+Q4eynz6NTZD2dVilISqTynGhaZ698/u\nwF5H+LDtbzB5pbqv/QTpjEO/OGJZzdqLNGtHMjDvPx+PqBsR/FueVa28kLaOjTk/AkpND2EZ\nUDgb0/SBxkFaCoJQjZhVI787m4aDSr98NjXx2h6BMCtQ0xqfqDD3G359CiBQ0ShNwi0DwxSD\nckY6OsKocAWEZncgwEt6FZNt1moSe0nKCRJ2EsWnCEk67oHry3/36jFXlBCzuNh266YBfa8u\nW579WWV7SBgg5Gk4+rJl2ZX1jiCvIPB5A8KZJue83BnlX2Y1qmtaPfHxlKQBf+xVbQrHAMCZ\nNk+vwOELibtqet1P+lUkScYfnWr/6iqylkYgzGTISzNhHOn6zc+j1Q0AEFqb/J9uU2yYolmw\nkDaNJLF/BiMH/IF9n8fHsSj6d20fm3vwAdx+FruirDFQwq8F7B5gcSrWnvC/9MexGcZgSB0t\nUlujwg5ZEir3T8wYImBJweoVACBRfIiwGcCmxwYRg7TDyHMhEGYkMsiKcT6BN8e5Yah0liqi\nkB6ARsBRKJmjyyiU0G2HMH4caXG9e6rtSLPrTKf3YJNza2Xr2W6FItZezFrlFBuzZqqn3lQU\nWv/07TV3XDznwkWZV6/Je/iWxQ9/ZVFMW5C0ZO1DN1XkpOgjkdxUw4M3VaSaNf4ESQ2Q2OBj\n+rKmNFU5XtYfxwABQbn3WYAP/0IaHH5JVlC+nAGhe2qn/BAIhFFCMjgI44Xk6Pbt2qEQ7+4w\nbrrI/d470UFKp0/53kMTNbRpg+xyYkn5ES52x7p1DpuAR9rxnHx8e68nO0pOp8+/DeUuUJfO\nd7/xYvzhtIbFVGxFinBkF/Z7kVYff/zYggMKi7fhXT6lZRyMQQwBO+zV3SEOZ5jxoYIM50Hg\nJA6dAjkIQAFrR9rFQI+2hQ2BMJ3AQSzXA/YCohEyA5UFQPFSSPFYXg5pYCTZ5hRKohD5y5pk\nznb7TrQN8B4SZby3ocekYZOVtIxCq+5Mh0eMm7XOTTWM4yjHCL2GvWLlIFlCRRnGR25f2tzl\n73QFUkyaDIsWIQQAqeaE3rf2GVdtMS/HfNXKnC17GuSo/+jzy9NXRwkfCMCkZR1KOkVEBQsK\nyqooAAQTiCMEAmFmQAQOwnghtDQDVn66aOaXGy65yvHMX0M1VZRer1201HLXtxibsmY/m6FN\nZsQwOK4uAwAYa8oIL+pqwx01IASkL97GPf1+KLinRXz118yXfqDbsIl7/mm+tjrmPG2qXsFv\nQ5bl7jZaWzjCwQwZypIKCIFSHjKVMjDrwdspH34dt50CiQe1gSpch+ZcABQCyQ0gAZ0EaNRr\nfVQSgJKVDDX6+RINmnlIMw8wD4iNd1kjEGY4cjuWjgJIAAAYMLSB3IDopYlNRhPOYQhTn5ou\nBeUaY6jp8iVnK3xR6zhmfaFtT32Ppy+jgWOoigxTmnGctOyJQJLxkbM9zd2+JC07N9tsS1Jn\n2XRZtgH5RCWZSdkp+oY4h47CDOPhup7ndp71BIRUk2b9PHv5jChXuXJ59qICy+5THZ2uoMWo\nXlxoKUyLLYYtzzJvP9UeE0QILcgO5wIbNQnnOElTuOcOgUAYPUTgIIwXlC5hui+l0+tWrNat\nWD3KW+BgwP/FdrG5gTKa1OVL2JyCUV5wqoHUGu2Ktb7PPomNM6xu7cZhX04W5T0v4tOfApZx\nSMCe2JaoIEvSzheYW36Z9pu/dP3hMd/2D3rDTIrdeP4GOB47jPBgxj99AwAofRJXvpI/HFuw\nQyUlc/OXRTaxq1X+6Lcg9P1oQY98/B1KboRME+DepR4E6hKkWZjY0XNwEJePhRrAsfZmiBs7\nw9fRqzAEwvSDx9KxsLoRAfuwfIJG2bKSlkEP32SUMHXwJKi88IRiv10j2PSqS0rtHd6QJyhq\nOdqmV3H0NK62rml1P/FGZVNXuCqHpalr1uRety4/RttGCH3/hoX/+a+DrT3+SP/UdIuOVjPv\nHwo3JnP6+NPNrnWlqTeuzZ/Qn2F8yLLqstacyyljTbGtzRU41dqfAURT6KL5aal9aleOWadX\nMd64z1hOslavIt8b0wSW/E8RRgL53BDGCy47j0mxxxhkAgCiac3i5f3bkiT1dFBJZsQNbwUm\neGRfz+9/LnV1hLcpynDZdaY7v3MOC4npiOWeB/iaKqG1KRJBNG255wHGHmfWMBjy/tfwqe3h\njQT5mbjtLEgCnWxJ/cmvpW/9B99QSxtNbE4ednS6T+wAOXaCQadmUZaxMfgcFP2t97sdXWL9\nmUiEMpoNdz+MVP25u/jYln51AwAAUK4dMrR96gYAYAiewrIX6dePfChIgzRrcPAAyH2vVkiF\nVAuAIVlIBMIokDsAlGa8cqeGLhRkhUmvmp5p+fmzCpaiQkruKix1ruc4hZDdoLZPg6qUQfAG\nhP987pDb3//BFiT5xR1nDRr24qWxJuKZNt0fvrX6kyMt1c0uhFBxZlJjT2BvdWyx6qcn2pcU\nWuPzHWYeNIVuWJ5T1e6pavN4Q4JFr6rINifrVZEDGBpdMi9t67EWf5Q/q0XHbSqdoJcWAoEw\nWRCBgzBuUJTlngc6fvq9GBcJ05dvZ1LTAED2ub2v/C244x0sCoAornSh4SvfZjJyh3Jtqaer\n65cP4IC/PyTLni0v0pYUwzVfGcufYrJhUuyZT73seuW5wOF9stfL5RUkXXszl18UOYCvrw0e\nO4SDQS6/UFOxNGHfVknApxUsUeLAIElAswBAW2wai603imzp6guuD3zwwoCr04zmum+O8Acb\nPpTRbHrof0J7PhKqjmM+xGQVqNddgjQDEoVw28BGehRCeZkK1+KbQOwGxjLy0dDJSHc+SN0g\n+wBpgE4GRFJeCYRREpdWFgaraUrEOr/Ybz+JAOlZI0uRv7tpjN2orlaoUsH26VxyMnR2HGuN\nVjcivLWnIV7gAACWoTYtzty0OPxQu/+pvYqXPVzXMxsEjl6KUg1FiR1YMkya21bmHWt2dnhC\nDIXSkjRl6cY4MzECgTDTIAIHYRzRrdmQ/sRT3U8+HjpxDEsil51nvvVruvWbAAALvONX3xEb\nz4YPxTJfebDnZ99I/smfhqJx+D5+Z4C60Yf37VdmmMABAEilNt18u+nm22PiWBC6Hv+1a8sr\nkcQK9fyFqT/+FZuuMKXHnk4Qo+y4GBpA4b0KmVIhQSqN+rJb6cyC4IcvSi11iOWY/DL1FbfT\n6bkj+plGCkWpVl6oWnmh8l4sgzTgh0J6DTAJOg6KXaMSOAAAKKBtQNtGdxECgRAhcWUW4vQs\np6Y1ISkoYYmhGBWtoVHCfqKEaUFZmrHJFYhxfEzWqgqss6KjTWOncr+YdkcgJEiqxO1yAUCU\nZMXGsQDgDSQs8JmFqBhqSc5M8CUhEAhDhwgchPFFVTo//fd/BVnGkojY/pfX4M73+tWNPnDQ\n733t76Z7fj7oZcWmeuV4RysOBZFqViz+dP/5d643XoqOBI8dbn3gnqynX0ZM7J82GlipjlQM\n9iOI86Knll1+jjuyC9ewC9eALCk2+p18EAV6K3ijUnbxOfKcR9vxhEAgjDHIBoAU/jaRqVf7\nYCiWGbuUDQwSxgEAjJAGkdehyUDL0pvnpB5udjW7AoIkqxiqwKIvS5sta+xMgkochBBNDfIb\nYGjKoGE9SlqGOapMg0AgEGYh5IlOmBAoClEDlub4k4cVDxQSxGNIKGHQtOxy0DZ7wkqNmQIO\nBl1bXomP83U1/r27dKvOi91hsIHWBH5neBMhyqiVvQEQ++qfGY5eeQ1VfsHg956a6gYAAFAF\na+Qjr0c2sd+PZBkUXyKZmCUdDFIPSG6geutNiMcnYejgoOQX5BAGzCBWTesoklkwMpAG0cVY\nGlhoBiyiy8b6TliU2wW5HcIGEIihrCyVDjCjLJymBRqWXpmbDACCJLPT2S50BMzNNr2zvzH+\nZaU4w8gM4VextND68bHWPsvRMBRCSwutYznKGQ3G0OULeYKiXsVY9Bw9018dCYRZAhE4CJMD\nFkLKcV6hq3k86vIl3ncUpvcgSS13XEVpdYarbzZccwviZuw0VWhpwkHlenW+pkpB4ECIWnyN\nvPPv/RGGokw6ZCmAzArQGVHGHKQ3j9t4JwhUsgH5unDNrnCLYknGzd0oK66KhEkBJqrPruTE\n/j0g9fRdhUOahcDNtKY8hPFAxpKL75L6WurwEAiIXgOXzFGzIo9s7KHyEDJh6SyAB4ABZEZ0\nIcAYr0gLcosod0QFsCh1YixydO7Y3ogwdGabugEAK+amFKcbq1rc0UGaQjdvHFLn9cuWZjV0\n+aqj2ohQFLpmRU568qx23nUHhfoun4+XzFouz6Y7R5Oddk/wkzOdXd7w66hJw64vSsk0axId\nTyAQpgtE4CBMDkx6TujgLoV4Zu5QTtesPE81b1Ho+EHFvbLf5/rXk3zNaeuDj41mkFMZxCbM\n044uBRoQL1xJMSp574vg6wEAoFk0bxNVfmmvpegMAVHU4htw/kpoO4UDLmS0o+xFWDwNwVMQ\n8ernspGuv7MsYB57PwEcHBDx70WIA1bB5o1AiMYrOCQsRC+iYpA9Qk8yZ0czq6PTxIHMiFk8\nfpfHIIlybO8JQCBhB8Z2hGapMlXv8Dc7gwFBMqiZIqverJ1Bz4WpCk2hh26ueO7jmg8PNssY\nA0BOiv6OzSVlOUNabFCz9H2Xl+2t6qxsdHoDQqpJs7Y0dZarG7vPdn9R0yX2ld/q1czmsrQ8\nJUsXd1B480gLL/U38XEGhLeOt1xXkWklNT4EwjSHCByEyUGz/jL/+69gITZfQ3vhNUM6H1G2\nn/y3++WnvVtflv3KNl2B3TtCxw+q5i0a5VCnJmxGFmOxid1xr+kAmoUJ5wYodxGduwi83VgM\nIWMKUDPzGwCZs8GcHck0RdwiUM8BsQtABtoEtGnA0fzZAepGHzh4AhGBg3BOZCzzchBgQIo4\nAGAs83JQRTqYTkl6fTcUd8ngp2HWCRySjLdXdzW7AuFtF5zu8FRkmObNmk4ck4hezX7tkjlf\n3VTc0uM3atnh2mcgBMuLbcuLidc1AEBli2tn1YCXIm9QfONw0+2r85M0sYLdkSZXtLrRiyTj\ng43OTXNJx3cCYXpD1pcIkwOdkp507y/+n73zDoyjuvb/uXdmdmb7qvdq2bjjbmxjbGNC7y0m\nBEKSl0dCCr9ASCDJCy95kB6SQPIgkPAIkFBCJ6abahs33Hsv6tL2Ojsz9/7+WHm12p1VXWlX\n0v38pT3TrqTd2bnfe873YEd3GwvEC5ZrvyotOb+fZ0CS0X7TNyqee7/kwafT7SPv0k/xGAtg\nXPD121PDlnMvECf3Va9uKUCO8rGqbuiDTWCoBkNtsroBQDW3/iGamxmRMnqHUA1A/22iUXWE\nB8PIAHQ8fuR3t/i61Q0AAKAUtjZ6OgL6laSMjCPwuKbYwsxBh8jWEzrf5qpGdzZ6UuPtAf0i\n3zZ/umbVDAZj1DCeZjiMHEOcubDw10/J29arrY2co8AwfR5XVDaI82Bz2hboRB7LX1TWiy5H\nRlPnQ79R21oAAIli3g235N38H9ke16gjnakYSr+JwQAAwLEiFL23CfMZzVkQMur3agFAaDwm\n3Rx16mdBHmgPbG/0tPoiGqEFZsO8mvwy27hLb2GMIpxBfRO3zoFIdexbn8EYAzCBg5FNkGSS\nFvWjbUevcPmF2GwhwUDqJqG6fognz3Esy8+zLD9PbW0mkbBQWZPaHZbRJ4groHBMZwNfoBNk\nMBLAiOOxoJLUTo2ImYzmLAg4Hhem2nBwyI7RePQXDEY13fihDn8w0pWIFJDVE67QsolFM8rt\nIzg0BmMAcBgpeu9l3Z67xVapxauzBlZiZbfuXEJgSwWMwcBKVBijHsTzlot0nDu4giLT4hUD\nPZvm9Tj/9kjLPd9tvfduz4vPUkWnyXyuwZeWG2onMHVjkBjqAFtSoghJM7IwGMZow8LnoZQ1\nPzNvYxkcuYyAK3hckrhYy+ECA1eTxSFlEYnXfxRMMSiAdUc6Q2nUEAYj61SlMVitztcxGZ1V\n4TCkvPM5jOZUj/p2cgwGg82IGGMB2423aj5v8J1X4xGhsrbgrv9B0sCW40JbNrbc/V3N5429\n9L35uue5pysffIwvHUztDGN0gHhkWUnDn4HS2BXBFmScCzyzGWP0DY8NeWJpUPWqJEqBckgw\n8VYBs1r6HAcJuJzHRYSGAShGRgRjtqd4n1TmGQ+266RAaikKh0roSXdocol+WSil0OQNu0NR\no8CV2SWzgT1hMkaUJRMKT3QGk6xDi6zi9AqdtCOrxF85s+KDg+1xr5k8k2H5xKIC8/i9FTAY\nYwb29cMYCUjQr3W0c8Wl2KSjow8dxPP53/6h9dLrIru30mBAqK6XFiwdaEYDCYVafvS9uLoR\nI3r8WOv9P6l86LGMjpeRY2ATMi8FEgLiByQBZ2N1uIz+gxFnFfKzPQoGAADQMBAvAAB2QF8N\nXxEIHGLNUGFWub3FG/HLPWxxFZUoqSkcABHdGgCAzoD83oH2uNkBj9H8mvy5bDGcMYIUWcUv\nLKxZs7/tlCsEABihGRX2pZOKeL0SFQAotorXz61yBaO+sGKR+AKzASP21c9gjAWYwMEYXpRT\nxz1/+W1kx+bYS+NZyxxf+y5fPCwJEb5CY4QAACAASURBVELdRKFu4qAPD21Yp7ldOvFNn6od\n7XxR8RCGxhgNYBPg8WgxyGCMCTQa3Q3K8dPuoQiEemSYCsBqhfpAErjLppftavY2eSMhRbOJ\nfHW+8f397bo7W0Wd50ZZJa/tak6sXlEJ/fSYUxTw9DLm2cEYOYqs4qr51VGVBGXVbhL6FCwQ\nQIHZwLI2GIwxBhM4GEOAkOjmNerRvVQOceV14pKLkdmWuF1tbWr//n+QgB9o14p4eMNH0YN7\nSh/6B7Yld+vMOkpri/4GStXW5gEJHFSWQ5vXK40nufwC46x5fHFpZobIYDAGi0YVQjUO8Rix\nL74xCJW3g3oqMQDKEUpVJM7O2phGDzxGsysdsyu7Iwda/U2ecNJuJgNXrWdzcKDNr+vNse2U\nhwkcjJHHwGMDzzQLBmP8wp7zGIOE+NzBh3+sNR2NvVS2fiy//5L5y/fwZ3Q/TfpeeJIE/AA9\n8v01V6f/1WftN319RIcbgxDAaY11OXva5zBsH4AcE96+peNX96qtzbGXSBTzbr7V8YUv9/8M\nDAYjg6gkElSdGu0yDBawZOILWG3CmIKGeqobp1FPgGFKn7UqjFRWnlH82s5mT7jbZlvk8fmT\nSwVO5zvUGdRvw+kNK4pGdA9hMAZEVCVrdrXsb/IGImqxTTpnasmUSqadMRgMfZjAwRgk4ece\niqsbMWjIH3zil7afPI6MXUYb0b3bdY+V08SHCRqNuv7xhPflF6JNTXxhoeWcFUXfvJ1zJNcG\nmxYuRgYDjSb3UTfU1huqa/t5LbWjre2Ht5NwqPvqsux67EGusMh6/qVD+CUYDEa/oQrQACAR\nkEmlUZ/SdrpsAQBAIRG/0moXytFAGp1oVNFoFAHmsYhYA7JcQ/Ok3UQ8wLEcurR4wspxVygY\nVY0CV2aTymxdYpBNEm6YV727xdvqjaiUFprFmRV2Y5qWjb0UAoxnUwMKsP2o80iLnxBaXWSe\nP6lIt18po098IeWPq/d2+uVYOnCnL7K30bN8WulVC6uzPTQGg5GLMIGDMRhoyK/s2agTD/qU\nPZsM87qas1JVTd0HAEay9yrVtFPf+Gpoa5cJiNre5nnh2cAnH9Y+9XxS1QlfWFT4rTs6Hvhl\nYhAZxJIf/nf/L+d/45VEdSOO74V/MIGDwRh2aIQqe0Fr6nqJrRHUkKhuxCBUixC/ketXZhah\nalB1KqQrXR8BNvIOibP1fhSDkfvsbfPva/PR05+Po85ghd24sCYvpkpwGJ1Z4Tizou/zlNml\nXc3e1HiJTRq3U3pfKPrQ6/sOt/gAIDYtf33TqW9fNrU0b2DN3RgA8NqWU51+GaBHOvCHe1pn\n1eXXFad2eR8qGqXcOBbmGIwxAFuGYgwG4u4AouOvDgDE1Rb/Waiu191HqG0YlmHp4Vv9Wlzd\niKO2tXb+5U+pO+etuqnqL383LVzMOfKEsnLr+RfXPvuqcdbc/l8uevyIfvzYYaDJsywGg5FR\nNCp/2q1uAADxqzTZRyCGSvST6lOgfqUtrm4AAAUSUl1h1aPRCAX92yBjpOHSdetAgEUAfal9\nnNMRkPe2+pK+l5q84YMdwYCshtN0S9GlochSZElujYwRWlRbMPRxjlIee/tgl7oBXdPyZlfo\nT//eS8jIPQlQCu2+yKE2f7tfHtUPIDtPuHXjO47rGMMPGpXQzSdcf994/OGPj/xt/bEPDrYP\n6FPAYDByB5bBwRgMyJRWMkcma/xn6xU3hDd+nLwDL1gvvX64RpZCYF3yAGIE132iGzfOnlf5\n0LxBXw4J+r5WSDAAWxBgMIYVtRFoICU6pM9dlITi5h2JhDV3lEQBkMg5RK6QFa1kBQoUABAg\nQEbga0A9kbyHIFLYCRQALAg1ALCi/W5OuvW1v90t3m2NbgAwG/hZFfbqvL57S2GErphZvv6o\nc1+rLzaPzjMZzmkorByv2QpOn7xbb07e5AwdavadMSLmEadcobd2tXT4u5TcEpt00cyycsfo\n+48oGpHTCA3+cMbSgTVKX9nR1OqLxF6GFW1Pi++4K3T9nEqzgc2VGIxRBvvQMgYDzivmymu1\n5uPJGzhemNqtDogz5uTf8d+exx4gfl8sRZPLL8y77W6hdkJ8H+XE4cAr/1ROHsEmi2HqmdYr\nbkQmcwaH2uVymoLm9+nGh4hx5pzAu6tT49KZA0gDYTAYg4ASndU8joYJsurEUb889lWSbMpz\nGkQBIaCy5tZo1MxXptmNMUS8lB4HiOlWNoRqAawAIGuRoOJXqQoAPBYsvNUgnkmRAZQj0JVW\ng0AQQYjbiwYo3YHQTICca+CVLUJKmhrS00v9wai67pgzqpGGwr6rACSBO/eM4rMbCj0hRRKw\nTRrXJr5tKQ1o4rS4wxkUOAihu0+4m10ho8ifUWGP1790+uVnNp5Qte60jTZf5J8bTnz1nPo8\n0yhrLyJw2CTyIVnn7ZpnTs4bGjT7W/1xdSNOUFY3H3cvn1SUqaswBkwa3x8Go3eYwMEYGJrP\nx1ksgLHx+m8H/3wPVXo8/UsX3YjzSxIj5hUXGectjuzYonW08iXl0uyFyNi9HBR8+2XPXx8A\nrUubl/dsC635d+H//Jkvq8rUgA2V1UHdeFVNpi6RiOWCS70vPxs9eigxiEQx7yu3DcflGAxG\nAjpJ2BLtUJAlKY8DAZY4HdUjlf6kf6gkqNIQj/pe6GYMkBZKDya8dFHqRmhKSDUGlG7lWiWK\nJ+qyCnajUEdpBDQXgAaYAC/2/AdSSo8hxLrGdmHoX3OTnc3e+gJzP71CDRwutmZszjl6kQxp\nZ2XG9JsSOdLmf3tbU6MzxHGotshyydzK0pTki1OdwUffOtDY2fWMgxFaeWbZqmX1GKENR52J\n6kaMqEo2HXVeML1sIL9KTjC3vuCTfW1JQYRgdn1+pi5x0q3jngYAJ9xBACZwMBijDJZVy+gX\nJBzu+PMfDq1YdGjZggOL5zR979vUYLXe84gw5xxsL0BGMz9huuUb90nnr0o9FlvtprNXWq+6\n0bh4RaK6oXW2eR//Q1zd6Aq6Oj1/+W0GR26//Crd1rD2K6/J4FXiIMFQ9sCj1ku6LypOnVH+\nx8fFiZOH43IMBiMOwjrGnzwEzeDECQ1TOCRYhRKM+qXvczjdbI2iBD1FI2kXbBmDRaU01dKI\nUnooqKQWIkFUOUlDa0A9BTQINAJaFMI+0JIy2H3AbFNOU27Xr1agPYVCWSXezBUCjBOqiywW\no04OC8+h/qRvfLKv7cHV+w40+4Ky6gspO0+4f/3K7n2NPWxcZUX7/Su74+oGABBK393e/NrG\nkwDQkiaFpNmdnKQwKrhkbmVNUY/cXozQ5fOqKvIzJitHVf07Q7o4g8HIZVgGBwMAILzxo/DH\nb6utzVxxqXHRCtPS8xMNI6imnfr6l8M7u3q7UjniX/NuaPPGmiefN99yz6AvGtmyTredirzr\nM+L3YmtmcjilaTNKfvDj9t/9ika7PQUd134+77obMnL+VDi7o+h7Pym8/W6l8SRfWJSpX4TB\nYPQBVwXqYUixzBD5cgNXphKZUJVDAo8N/TfmMGATj0SVJjuSIuZbOex4AXQL7xUehxTSY3KO\ngFrIkZT9KURDYLQnBxkAAFDpMJ50Sy090/IppVqKHWVKKgCjD3gO3XBO/WNvH0iKX76w2mHu\no0IkGFFf23wqKagR+tz6Y/deNyv+aLblUKc7oFNAt2Z7y+VpmqfSFPVqtGA0cP/v0qkbDnbs\nb/L5w9ESh3HplJIMqhsAYDcKp/ScTO16QhWDwchxmMAx7qHE9fv/Dq99L/ZKOXYwsvHj8Mfv\n5N/zK8R1vT18b7weVzfiaD5fx//+seJXvx/0lTW3M92QNI8rg7pA3vVfsCxe6n3zdeXECb64\nxLL8XOPMWZk6eTqQYDDUjVyzGAaDAUhEhrOosh1IvH6BR8Jk4MoRgICl3o5Nj1UoDqquKOla\nKUUACFSEeizrYcTS8jNOWgkJoWThg6cBDHpZBpSApgIXf9SRAFhFdxcIYHFdwXFn8JgrFJBV\ngcN+WdFSenxghGwSe1YcMIunFOdZDP9ae/xER4BSWpZnunJRzfyJhX0eeLDFp5s14A5Em92h\n+Ky+yRmKWZslEYgo3mC01C7F7UXjIIDRaDIaAyO0+IzixWcUD9P5p5ba9jR7U+WfaWVsjYrB\nGH2wL63xTujjd+LqRpzI1k+Db71sueS6rn02fqp/7IZ1Q7k0l5emgRzCaTcNFqGyqvBrzAWD\nwRjrYAcSlwFxAgkAEgEXQP/MRHsBIc4iFBGar9GoQn2K5knagUOigDNpjcwAAIC0ghQhyf9T\nfXUjBu2eKyJUMeRRjSkQQF2Bua7ADACUwtv7W90p1Sh1BaYkt472gOwNKwKHCs2iRWSPkWmZ\nUuX4yQ2zVI0SSg18f0vCdd00YwQj3Zt4DqdLRBM4vHBC4b5mn9pTrhI4vKB+/Dbu7Z1iq7hs\nYtHaI53xPxpCMLPcMa1Mp/KRwWDkOMyDY7wTXv++fnxdt+pBwvreSySkH+8n0oKluk1VpTPn\nYwv7RmEwGIMDAS4Evha4sqGrG3Ew4gRsNHElIpefuGzKY5OJrxhiM1qGHjYA3RR0G6BkOYn0\n8o9GseccBFANwJrdpAUhWDqhsMjSIxepNt80tzIv/tIvq+8eaFtzsH3LKfenx13/3tuyrdGT\nUtHC6AHPof6rGwBQkN6ltdMTbnGFKKUAMDmNl0dFgdliFIqt4ucXVucnlMMUWsUbekYYSUwv\nt984v2ZxfcHUUtusCsfcMnu52ZCuQ+0owul03nnnncuWLbPZbAihp59+OnUfr9d72223lZaW\nSpI0Z86cl156KeM7MBgjCZPexzvE1akb1xLihppa3X0MtXVDuTSXX+T42p2ev/yGat2LElxh\nif3Wu4ZyWgaDwRg+JK7IgB0ajVDQOCRxaJCVL4y+QAhNpXQ3QKJJhAmhKXaD4I26VZKQa4Dz\ngJqBprTMQhLiJwIYABy9pIQwYpgN/HmTitv8sjsU5TlUYBbzEgwICKUfHe7wJ+QXUAr72/08\nh2awNP7M0VBqy7MYUv01wmHlvqe3AsCEctu3rpg2pdoxozZv1/EevhEYo+uXdj2Y1RSYv7Zs\nQrsv4gkpDpNQYpf62QpnPGOV+Ckltmc+OPzetmZCKQCYJf66pfUXzKscvX+7lpaWJ554Ys6c\nOZ/73Od0dQdCyCWXXLJz5877779/woQJf/vb36699tqXXnrpyiuvzNQODMYIwwSO8Q5OUwyS\nWCRiv+Ia19NPpBqCOhJ8OonfH9m3W/O4DXUTxIln9PPqpvMuM5wx3f/aM+rxw8hsEaecabni\nC0garTWiDAZjPICRgFEmneeCUe2YK+iXNR6jApNQm29iUxEAADAjNB+ghdIAAELIClAKgDgE\n+WKhrEUUoiAAHgsiJwGZSyMbgCZMC5GAxAUAGWskOU4osYolekkETd6wX6964kB7YFqpjb1j\nMwXPoVuWN/xtzSFfQrlQVNHa2ruaBx1p9t39101//Obib1069bWNJ9/d1hTz7KgoMH1h+YSp\nVY74URxGZQ5j2aj13RgErmC0yRNWVFJoFasH5UL64Cu7tx3pdogLRtQn3j2oqOSys/StW3Of\nqVOnOp1OAHjvvfd0BY6XXnpp3bp1TzzxxJe+9CUAuOCCC+bMmfO9730vLk8MfQcGY4RhAsd4\nx7hoRWSLjpWGccnK+M+Gmtryn/+29b6faN7TXcowl/+Fm+KNSDz/eqbzkT8Sf5exn2nugpIf\n/UyorOrPAPiqurxv/nAovwKDwWDkCpqXyrtBcwGlwOUhaRpwfUywG73hXS1+cjrRv90vn/SE\nz6rOkwRmhwkAGKBCd+4scpLIJSRl4HxkPI8qh4G4AQBwHhIa+l+jRIHIqlulMgXKI4PIOTKr\nYY0BPGmaxSoaCUY1KzPjyBy1xZYfXTvz0wMdpzqDx1t9+467ff4oTagFkhXt+Q+PfO/6M69d\nUnv1opp2b8Qs8dbx3e+DUPr+/vbtp9zxv1O5w3jxjLI80wCqcg43+xLVjTgvrz9+0fxKnhuV\ndf0Y9zHsV155RZKkVatWxV5yHHfTTTfdddddO3funDlzZkZ2YDBGGPaFNN4xLbswsmVd+NMP\nEoPirAXmC69OjFjPu8A0b4H/3bfk48f4omLLknPEiZNim3yvv9z+m/sSdw59tqnxO1+rfeYV\nJLKU4FyCkND7r0e2riceF19aYVp5uWHanGyPicEYQyjNNLSuuxGp2kIDrcg4Hwy16Y6IKFqi\nugEAgCAY1Xa3+edVOtIdxdAHGZBh6iCO06gcUFoo7Sq21yAia36zUBL3jqUUIqpmHN+SUy85\nGhxL38g0ksCtmF4KAHf/daPXl9wPBQD2nexyO8YYleaNoxyNdHx8sGPbyR4FO82e8EtbG29Z\nXMfh/r4/Dzf7dOMhWW1yhmqKLUMdZU6yZ8+eiRMnimJ33taMGTMAYPfu3TF5Yug7MBgjDBM4\nxj0Y5991f3jdmtDHb2utTVxRqXHxuaYVF502ZuuGc+Ql1qTEcT7xaGpQaTzlf+dN22VXDcuY\nGQOHhkOuX9wZPbg79lI5uj+8fo35ks/bbvpWdgfGYGQXClTRIhpVEcICFjk06K9FQsNbutWN\n06enkW1IqIA06QBtAZnomTS2B2SVUL7fz+WMoRBSOuLqxmloSGm3iTXekLpmb/uRDr+qUaPA\nzarJW9JQOCDPyDFDkVnf/NJs4E2GcS39DCskpXFvV5yZuyagErr9VHJ/KwBwBaNHOwMTi639\nPE8vf1U6dv/gTqezvr4+MZKfnx+LZ2qHdPh8vs5OfStAiP3NBTZRZQwG9r5hAAAYl6xMrEnp\nPyQUVE6d1N0U2b+HCRy5Q+DlJ+PqRpzg6uekOYtZHgdj3KKQaEBxaTTuLIBMvNXED6qLk+YG\nGkkNU6qGo+0ycBpVMeIMWDLzdnRaQY4oJPUQAKAUZJXwbN44/BCqaFRnhZwC6fD7n17fGjnd\nRiGsaJ8e7jzpDN60uHYcWk4UW8Uym9TiS36Tl9lZquYwUl9m293TSTTGhPLcajZHKazf377h\nQHuHN5JnMcyZUHjuzLL+p04MEW9YUTT9e2mnX+6/wFFbor+nKHDlBeOuFzjq6y439B1uuumm\n1157bWDDYjD6ARM4xjVUUZSmk1xeAWcfbC50SqJHvzYxhhnqc9JT+2jQhworcO10wFx4Q5p+\nwOvXJAsclABQQGxmxRjjEEp80U4KiY/FNKT6MMISN/BUZJrc9QAAAJDfME2hFEAFAEK1iBaM\nkojDUIwRBwDpcgEQgGF01nuPOgjVMc6MsfagK5LSJLLJHd7d6J1ZNR4LiM6uL9ze5DnU0eV2\nSSlEVbKz0RuMqMsnFmV3bGOVyxfXvL2lMalZKYfRNUvr0x0y8hBK//eN/TuPu2IvXQH5SKt/\n86GOO6+cLo5IYVcvSsqARJYp1Y7JlY79jcnJIBfPrxrDeVsFBQUulysxEnsZy8LIyA7p+Oc/\n/9nW1pZu69Spgyk5ZDCACRzjFhLwOx/7k+/Vf1FVBQBx4uSi794jzZw90PNgo9FQ3xA9ejh1\nk3HGmRkYaB9QbfdasvND6mlDtkI8aQE37wLAWZiZU00FShGfCxZfVFv7orruJVC7DOFQUbVw\n+TeJWz9RUHMnJAd6TpAjb4PvFFAKlhJUtxIVsW8XxphF1oI91Y0uwmpgMAIH0nHsj3DFCkpe\naCVUC6l+i+AAgBKruK/dn5r7XGA2CNy4yxHICjh9UdJJp05mBwAc6wyOT4GDx4gQCEc1BIgm\n5Owf6QzWFZhrBtW0gtE75QXmn90y748v7W52dnVBLrCJ37hs2pTqHHoHbjzQEVc34hxvD7yz\nvfmy+f3ymx8iDqNglXh/REesrMwbwNsSAdxxzYy/vX1g4/72WETg8CULq687py4zA81Jpk2b\n9sILL0QiEUnqysbauXMnAEyfPj1TO6TDbDYn1bYk0mcCCIORjjGrRzJ6gWpa8x23el98JqZu\nAIB8aH/Td74a3v7ZIM5W8LVvpgbFhkmWc88f0ij7hFLlxd+prz5Iju2k7jZyYo/67v8pT/4E\nFP1H0mFC3vVZx91fa7lhZfMN57bfcXNk0yeZPT8J+IMfvuP55/8F1ryleZIfIFJ/WW3L2+pH\nz8XVDQCgHSeVZ3+O7Xm654/3A6ZtO8lnj4DnGBAVqAb+ZrrzKXr8w0z9IgyGDsQPajNoTki/\nij58aGkuqlGVJltp9APODlzyp0zF+pMQhXTl+ZsEbkpK+rTI4+mluZV/PobBSOD0mq0gwFFV\n/20Q68o5PjnhClIKhCY7Ehx3hbI0orHPjLr8R/7f2b/62sLvXjPj/q8seOyOZYunlWR7UD3Y\ndizl4SQWP9qHBUMGOWdScWpwUom1fIBdcm0m4btXTf/dfy78zhXT7rp25kO3LV61rH5sl6Rd\nddVVsiw/88wzsZeapj311FMTJkyI+4MOfQcGY4RhGRzjkeCH70b2JtsxUFV1/uXByof/PtCz\nWVeeD/f9puMPv1Y7O+KRojvuQcLwpjNoe9aS/RuTgqTpoLbhdW7ptYM+bXjrpsiuHSQcEhvO\nMC8/D/G9fUZC7692/+n++Evl+GHnL39gv+k2y1VfHPQAEgl+9F7nH36uebrqb7HFWnDbHdaL\nroSQh2x5gZ7aCdEwGO1o0tl45sXAGwBA2/Tv1PPQoFesrQt16uQBSmetAACghB58XefAY2tQ\n+Vww9Ld+dXgJuKinDZls4CjNSp4OI5OQEJW3gnr6PYlEJM4AoWZkB5H2mRWl39Tb6Uxn0eDH\nQILxCAH92yBJyBypyzflGYWjrpAvogocyjcZGgpMAqtPGUFMQnEg2twznQeZhKJ8S3t7iuUE\nABRY+m486Q8rh5q8vpBSmm88o8I+ZpYiw2lcY8IptTyMDMJzeEZd/oy6PhL+s0UgTQthf0g/\nPhxMLbMJHPpgf7s3rAAAz6H5tQVn1RcM7mwVBeaKMWS68dprr0Wj0V27dgHA5s2bY3kWV199\ndayD7FVXXbV48eLvfOc7Xq+3vr7+8ccf371790svvRQ/fOg7MBgjDBM4xiPpMjUie3ZQVe19\nSq+L9fyLLctXykcPax6PWDeBLykd8hj7hhzYpBvX9m8YnMBBgoG2n94TXP9RPGKoqSv92W8N\nEybq7k+VqPeJB1Pjvmf/alp5KbYNNX1UPrSv/X/uptrpp0YKJODv+PVPebvFcOINiPi74mEv\n3bGaNO/FF/8ANJW69asZTRNrFJdbOXqgR/D8q8UZ8wEA/M0QDegcRlTqPopKRqDaqFd8neSD\nJ+ixbV0vHaV4xS2oZkZWx8QYChoNfwIk4S1HZRrZghAHfOWIDULAhojepEzA+t0i+gZbkeVC\niB6lmhOAIi6Pw0WKFk7dMalXi8MozKmwD/KijDQQqkVJhFCNQ7wBSyi9LRSHRJuhOqJ5NBqh\nlHJYlDgHRsKcGuWtXS3JO2N0ZpV+Qlycdz5rfPajoyG5K0WortT69UumjI0ek2YD5wnraBxm\nZog7jslLI/nlWwd7Lx0UE4utE4utvoiiaDTPJIzttIsBcfPNN3u93tjPDz744IMPPggA4XA4\npnRgjFevXn3PPff84he/8Hq9U6ZMeeGFF6688sr44UPfgcEYYZjAMR6hShpNnRCqKoMQOAAA\nGURp8rQhDWugBL1p4jqtwvpDxwO/SFQ3ACB64ljLPbdX/+MVJOh8eSuH95GAPzVOlai8Z7tx\n0fLBDSOO7+Xnu9UN6F5s9jz1v8ULkhclaMcxeng9mrAIEAK9ZmZIMhb+zyPBd1+RP1unuTv5\nsmrTeZeLZy7sOlxLX9ej6ixgjijRiPbCfeDr6I54Wsmrv8HX/BBVTM7esAaCpqltTchk5hyD\nXE0aayineqgbp6HyPjSCAofImcJaQCVJ5qDIzA9Ba0AciBMRdKmiEolG9AQOiRs7a4MjAiW0\nldAOChEEBoQKOFQO0NuMOqIFgoo3XmqEETbzeSKXNlkdIc7IJ38859TkuYPRTcec8XuqUeAu\nmlnWewbH2j2tj79zMDFyrNX/82e3P/CfZ5mlUf/QVV9o3qrTj5NOKBwL8g1jcMyfWLT5kE6z\nz4WTMmY9G1VJh1+OqlqRVbL0+jmySbnghpZbeDx9PBg7HI6HH3744YcfHr4dGIyRZNR/1zIG\ngaG+QTculFdiaWDFitnEmiZX01Y4iJORQCCw5s3UuNLcGNq43nz2cp1DwmlLjmk4mG5T/4ke\nP6IbV5taAXTmybRpL5q0FJdPJE0HU7fiqinA8eYLrzVfqJPegowFaS0HTIP5e2YQuufDHupG\nDKLRDS+ha36YjRENABqV/S/+PfDas1SOAABfXm3/yu3SnEXZHleWoZpO10MAAOID0HqfuGYW\nu6EwpPjCWhCAAgCPDRbeweO+CxD6CY8NFsGRONMGAImzMIFjIFCV7KXQpSZTiFDaRKmLx9PS\nPcNESSSg9HigJ5T4FSeHS3g0sMnPyqklM6scR9oDgYiSbxEnl9lMfaUqvLZBp3W6Nxj9aGfL\nxQtGwnBxWJlZbm/zy02eHrLdzApHOWsWO46ZVZd/7syy93f2SHeaM6Fg+fTM5PNuPub65GBH\nrKURQjCj0nHe1BJpRPqzMBiM0QgTOMYj1gsudf/9Uc2bLOg6Pn9TVsYzOLhpS8jedXrxswdx\nNqW5MW65mkT0xDFdgYMvS/uoypdn4CkWGfRnWShdbwUlDADc8hvIM/cB6ZF5j8ob8OSFvV1M\ncqD8idR1KDluKkKOLJuH01Z9oYe2pIw293D97r8im9fGX6rNJ5333Zn//Z8bz1qevUHlNrQX\nZ4zMgwCbBYdZsGtUw4B7qWIYNBJnEbAkayGNqhhxIjZmUEAZDxDaHlc34lAIa7SZQ9UAoNIA\noWFKgcMSj6wAEFH1Cu4AImrAIvRRXZJKkVUs6nemPaH0VIe+wH2sTSfjb9TBYXThlJIjncGT\nrlAwqtmN/KRia8nIViIwcpBVVa/pqwAAIABJREFUS+vnTCjYcKCjzRMusIpz6gtmDdb/IomN\nR51r9nbX3lIKO095vCHlxkUj7NnEYDBGDUzgGI9wdkfZb/63/b4fRk8ej0WQIOR98av2q1dl\ndVwDA0+az827UNvyVo/gxLnc/IsGczZj2tSVdJv4skpxxlx5V7KhiVDTYDijj85Y/cF45pzI\nDh23FLEmzZKIvRQAcO104YYfq2//jXY2AgAgxM1aya+4sU9XTjT1OrrzSfA1JoygAM+4EYZh\nyjcwaLqGBQPvczGyRPfuSFQ34vie/PM4FzgQl0d16+SwDVBWFuUQl75X6NDhEG/iWVeUQUJB\nP7maUjeF8rDWqNHTyXQEOGQycpWq/tsLtDTxDIIAMAJN7+bE4bHjCDCh0DyhkGUhMXowqdw+\nqTzDXkKE0vV6xS8nnMETzmDNGPIBZTAYGYQJHOMUaer0qr+/GNq0PnriGGd3GOfMF8oqhvF6\nmkr8XmzPh4x6PvEXfBWfsZDs/IC628BWwE1eiKcsHtyphMpqvqRMbUv2kwOMjXPT5j7k3X6v\n61d3Rw/t7T5PVV3+9+/PiChgu+ZG/5uvqR09TEM5u8Px+Zthf0qrFIzRxK7UFVw73XDr76nP\nCWE/yi8DoX9ra6IVz7uNtu8C3ykgGtgqUMmZgLN/i0BFNfTgBp14WT1Ej1HNj7AJhHLAA+h1\nPzLIe7fpxtXWJs3ZzhXo9LQbLwhVED2QasOBxClZGQ4jl6FpuvlSUHuoGwAAoNFQWGtEkK5c\nYtglBoRQQ7ntQKOORdSkSuYjy2AMDHcwqt+gh0KLJ8IEjrEPK0RiDIrsz14Y2QIJgnnJMvOS\nZcN6Fa29Ofj8I9FdG0FVkSiJSy40X3ULMmWs7SiunY5rM5AuAQgV3f79lh9+Nylsv3qVoSZt\njQaXX1j0y0fDGz6OHt4LmibUTzItOQ+4zNyOOZu9/E9POP/3d8GP3wdKACHTgiUF3/qeUFlN\nOJnuXdOd2iBIeNEXUH4Pd0ZkKwDbABNEEUIlM6Ekt/qWo+krYOsbEO6R3Y2qS9CySTSwDmKJ\nHIhHxjNByq3psa6bL2eRpBnl0PYBjRQj+0SwjJynZi7BIeNSnTaxI+gwyhgtICRSqlfcQaUk\ndSOGRkMCNmuajiwy+P44A+G6pfW/eG67RnpkcVQUmJdOG4n+YgzGWCJtoiYCquenzmAwGMAE\nDsaworU3e+67jQZPm8PJkcj7r6gHd9h/9GdkyLmSXfM5Kyv+9Hjnnx+QD+wFQviSsvwvfc12\n6dV9HIawcdHyofdM0YUvLi35799QOaK0NvPFpdjYlaSAF1xPGxZB4y4a8iBrMaqbD6aUtUEt\nSEO7QHECYDAUI9MMGKWV/0Yrvvoe+u6jtP14V6S4GC2bCyhhAkNVGvoMYTMYqrMxRH2E6mRp\nzDijwn7BNGTgIHSQhg7SlrWocBaquTSzmU2jA2xCxrOBBID4AImA7TCcRSKM0QuGIgI6OeoA\nNkjx5ohhwLxMFNqzuo1DvJEfQKcPQmmHX/aEFJuRL7ZK/S8wmV6b9/3rzvy/dw60urucOBdN\nKb75vIkCn+1yPwZjtJFnMog8llWdStVSx+gxxWcwGCMLe6BkDCOh156Mqxtx1MZjkU/eMK68\nKitD6h3jrHlVj/2TyjKRI5wtV9KJkSgZauqTg/lVkF+V9olbbqSeNRAvOI820dB+lH8R8Gla\nz2QFSqj7EATbgJOQvQbMJel2REU16Ib7aOsRcLeAxQF5AVB0nEdp5ADKJYFDmr+UKynX2ppj\nL/lCi+OSGdBzmkQ7t4NUiEqz1FdFiYCmgJSxjKoBgy2AWXfJsQSB6HGquQA4xBeAUDX0qhCE\n7BxUabQxcTUXo2KK8oDoCxwYCw6DNah4oqSry7XImcy8HUF/JYZmT/idva0d/q7+2Q6j8Llp\npbX9zoc/sz7/gVvPanOHPcFoRYHJZhqd4jKDkW04jBY1FH64vz0pXplnin0ejzuDO5q87lBU\n5LlyuzSvJt/cV58jBoMx5mECB2MYUfZv14/v25abAkcMJIqcmHMJJgOAqtT7ESTZ6ZEw9XyI\nCvtKSBkQhGidLchkwZaBi0HBNrL/XxBsjb2iCKPSOajhsrQekwihsgYoawAA6l+jv4+mU/ee\nRZBBLPzRb11/+Kly9AAAmGZUgN4iMO3cPvICB23cQzc+T52nAABMdjznMjRlefYNZcc9KvFH\nSSehMgDisEnExRjl7L1IprQZIAjAIeQAKAXNS4Nr46IDlQG4fGQ+B/BQF1oxqkAoj9BOgAiA\niFE+AisFAoBSc9gRYA6ZEGCboZACJVTjEDcgncUbVv615VRU61409oSVl7Y23riwxiTy+9p8\n7pCCESq0GKaW2MQ0eRkYobJ8U1l+znkDMRiji8UNhZTC+sOdyumP5OQy2wXTSxGCT485dzZ1\nfe/LKvFFlKPO4BUzy/OZpMhgjG+YwMEYRmg0oh+X9eN9QIjW2YpMFmxh/Qh6JdoCJKwTV12g\neoB3DP0KVImG33gm/PbzsX8lV1ZtXnWbYfr8/h5PFLLnKYgkNEeghLZsAd6I6i7ox/Fp5uHZ\nacDRG3xlbfFv/hbZsVk9eVQs6gDQk2Bk1wiPih7eQN5/tPt1yEvWPo3czXjJF0d4JIxEoqRD\n1jriL1XiV0nAxFdzKAeN9Dop3Q/QZf5HaTtAIwRbk41jNRcNbUCWFUO/HgJTrClsQgSLXLGs\ntSXtaeCK4pkaaFD9cT474UpUN2JohH5wsB1xmJyu/O8MykedwRUNRb3Pptxh5bgrGJA1o4DL\nbFKFneXVM9JyoMn75tbGRmdI4HB9qfXy+VUlrBADYMnEwtk1ea3ecFQlxTYp32wAAGcwGlc3\n4kRVsu6I87IZZdkYJoPByBWYwMEYRviyauXwHp14+cC6l9OoHHz9H6G3uqbTfFW99cZvGabO\nycwoxx5aMO0mEgTIgMDh/8t90W3rui/YctL3+7tt3/qZYfaS/hxOnft6qBvxePMmVHtenzoF\nEkqp0qSzQUhb5JJNEJZmLYRZC+nx12mnXk4TN7JL9JSQDc/phPd8ANPOAwfzQcwOhCqyluo0\nQSNai5lvyMKAekNJVDe6UN2pbXEAANRWIEHAw6LRGHABBkEm7YRGAQAjg4iLeTxUBbzNpy/B\nt3rCJT0zMqIq2XjCddGUtJ+ava2+fe3+uBnicVeozCYtqs3H49B2h9EX7+5ofnnDifjLbUed\nu064v3nR5DMqcqVgdoSJKFqjOxyQVYdJqMwz1Rf1qGc86dLxGAaAFm84qhEDxxISGYzxC/v8\nM4YRafnlOlGOl5ZdMqDzeB+6N/jqk/G8D/XUUfdv7pK367QOZQAAcOkXfIacKw4AyqHdiepG\nFxSCz/+lv6cIdujHNVlX+EhGnAhcyhwGGZBxRn8HkBVsyUYqMZB9RKev1NMCId1aHkpb9o/k\nSBiJaDSg2zGA0Ghs9p5LdCarGwBA9Ju5AoC+8JEheGwz8w0WYbJFmGzmG4aubgBAunoW3Z4N\nnrDii+j/7p1BeW+bP6nVQ4svcrBjGP8gjFGKJxh9fdPJpKCqkWc+OTo+m4Xsafb+3/rjr+9q\n/uBg+8vbm57ccPyUu4eiEVH12scCUABdU1IGgzF+YBkcjGFEXHSe1tkS+vfToHY9/yGz1XLz\nHVx5bf9PEt27Vd6RomVoWuC5R8RZZ2VopN2QYEBzu/jScsSP2k+HoRywCEROjvP2jJiMKgd3\n6kQRaO1N2sbXUNQHBiOqmIwqp3VvJZqy8S11x8fU3YbshcKMhrTaKhb6HgHike18GtoO8lGI\n1eELZcg0D3D2zDL7AcqfCp3bqO9YjyhvQuXLR3QcavrZspLynhl+KFBF82hUBgAOSwZsH7ot\n5WiEQtoncpqqJmQVSnXfJ+nXS4a/P07/3UP7Q6ldanTrLA4bBP3ksoii2SSd3/GkW69UEOCk\nOzS5OKdvVoyRZ3+jV9Vo6s2v3Rtp84THW8eQY87gO/t6VJ95w8orO5puXlhrN3Y9JFhF/RsL\nj5EpzUeVwWCME0btFI4xSjBddpO44Fxlz2bN1cEVV4hzzkYDdNCIHthJ9WY8atNx4nNjW16m\nhho9vN/18G8ju7cDAOJ5ywWX5335W9g6Cv0+kIBsZ1PvB5DYJREJyL4sM+dX0s6QtXXPciIP\nAHTLq6huDj7/NuANoKmR/7tXO7o7tg/1u9Vgk2HhBJ3jjQUg9u8PjiRkPgvMC4AEAZmSq1qo\nCsQLVAXOATnk0YjQxBugbSPt2AqyG3gjsk9ClStAGNGpDrIVA8ZA9KbTeeUjORIA0KgcUptI\n3BCXeGXkNvMVGI07izgMaaW9XPtrICTQ1DVlQQTd6TyWgMul5k39YG5N3q5Gj6yQxC8eDiGr\nUf9/ZEzTtSEU1Vem0sUZ45mQrKaTdkNy+vSoIUAB9jd6GjtDAo/rSiw1RTnU0GrrSXdqUNXo\njkbPOROLYi/rCi0bj7tUknwzmlBk6X9TZwaDMSZhAgdj2OFKKriSisEfr0TTfVPR9DPtgRI9\nvL/ljq9SuWtlkqqqf/VL8r7dZQ/9HfH9yCnINaQ6xOfT4DZQXIAwCEXIMhtwZvz80yXgIA5x\nhu5bCj22lax/Fp9zs7Lp7bi6EUNzh7Q2L1eSXFeM6i8a4FiwTtaGfJBGdkFXVj8CwwRknAUo\nN/6JiEOli1HpYqBa1ixRRTOacBY9tD457ihDFVNHeCwhtZn0bPdDaDSktliEgdn0jAE4bEEa\nTyF5JsNjK4JcW43M12lfgjCIxSAnd3NExnmjLiXHJgmfn1/9zp7W1tNmHAVmw4rJxVubvdGU\n1Pc8kyHdSnK6Bivp4ozxTL5VX4tHAAVpNg0Fp19+fM2hY23djZbnTii4fH51myvksIhlBabs\nagSdAf10wsS42cCtmFT84aEOJcESuMQqLqorGPbxMRiM3IYJHIxch6+o1Y1js41zFGbqKu6/\nPxJXN+JEjx4MvLvaetGV6qmj8o5NmtfFl1VLC5djc7YXOqJh2tkImoKKqkFKMxjejuzLh+Pi\nhlmLuMJSrbM1KS4WWZImMnT/J7DkC9q+jaknie5oFGZZ+TIjaDIAgLkE1V+E8obsRiHvp+Ft\niUOA6GFKgsiyfKhnzixZbfiCz/4iUcL0ePcfCuVX4vO+AXhER6XRsK67hEYjGpW5HEq9GTwU\nqKz5NRoFQDwSRc6cbraPABv5yrDamKhxcMgocTnYDsCIUC2lPYutgEfGucB7aWQXaF4ABHwB\nks4EPmN36ZGkxCZ98axaV1D2hBWrJBRZRIRA4Ln1x51awoqxyOOzatLmp5TbpRN6pS7lrJEK\nI4WpVQ6bSfCFlKT4lCqHPdNNTwmlj7y9v8kZgtP5sZpKV68/+dw7h2I7lBeYbrti2oLJxZm9\nbv9J58KbFK8vNJfYxL0tflcoKvG43G5sKLaMMj2V0SuIVRsxBgUTOBh9QzVNc3Vy+YWIy8KN\nRpx3DvevxzRXsi2l8XNXQebGI+/amib+Ge04FXzrhXhKv//ZR+23/kCa2692IZmHUrLxVW3D\ny112CZjDcy/izr4e+JFLYkcG0Xb7/f5Hf66eOnI6hMQCk7EsJZlCkSHopkGfzlkI0U64DNc+\nDBE3cBIIGckuITSi07UH1BZQO0fpRGtYECR8/rdp60FoPUzVKCqoQjWzAY/0qnJS7kbSpjEg\ncKg0GlDaCVVjswgZ/BHNZxWKcRpPCg6ZzEKDQtyEygCIQyYB52z3hGqEbJSeBAgCcAAOhGoB\nDCBYkFDR5Ywz2hI3kkAICixigaX7fVjpMF48tXR/m98VinIIFZrFKSVWQ/p0jAq7sdJubPT2\nKN2xSfyUEmbAwUjGwOP/OG/SY+8e9Ie7b4zl+aYvLtMr5xwah5p9Tc4QQNdnlFJoavZFle7K\nqWZn6N4nttz/1QVzJmbne7PCYTyQkF2SGE+KmA38/JqMlSozGIyxARM4GL2hdra7H/tDaO0H\nVFUQL5iXfS7vP77D5Y/oFx4SJcedv/Q+cr966mhXCGPTuVdYrrg5Y9egNF21i3LyqLL1w8QI\n8bk9f7y36HdPc0VZaKiprX2ObHglYTQa2fxvCLi5S789ksPgymsdP3kkunuT1ngMmSx8tAMd\n+lB/V4OE7IXQcix1C3IUAyCQMlefr/khXb8JjQkcyaDSSVA6KYtz0F4qL1BWM1wyBO1SN6B7\npq/RaFDttAppbx0IsAGPlvxqB0Lpek6P2RIMi4GfVzWA2dRZtfkn3KFjzmBAViWBK7cbz2AG\nAYw0NJTZfnDNjA0HOlpcIVHg6kusZ00qwsPwbmnpaX/rD8iJ6kYMjdC/v30gWwLHgtr8o53B\nxNoTALBJwszKDPS5ZzAYYx4mcDDSorldLbffonV21VRTVQmseSOya2v5n/+BbSO6rshXTSj4\n2WPyrk1q4zFstgiTZqarWxkkCAnV9dGjB1O3EK8z9VGdRuXQh6ut1301k2PoD3KIbF6dGib7\n1uHF16D8kXWIxNgw8yyYeRYA0PajRE/gQEW1IFn5Wcu1/ZtTt/KzM2R62s347KY3WuGwEQFO\n7R6CgOORlJUhZRCFRLrUDb14uiQOxkBQKW2jEALACGwIFWV7PPrU5Jlq8jLjf8QYq1AKhzr8\nu1t9sf6mJWXWWZWOymErZRK4HqKJnKbP8YFGr0ZoVvS4Qot43dzK9/e3x31wGoosyycVM/8a\nBoPRH9gzFiMt3n89GVc34qjtrd6X/pF3y20jPRqOE2ctEmctGqbT265c1fnAz5KC2GhCik4F\nNQCozcn96kcA2nECNP2sftp8aKQFjgRQcT2acg7d93GPKMejpV8EAH7GEnJst7LxzcSN/NyV\n/OxzMzwObAXEg96sEjiWwppzIMASXxxWk81cjHzJaK9uAABddQMAAKjWL4GDjOE8iKFDqZfQ\nAwBd90MKrYg2YzwV9JvRRABi6e5WgFGvnTHGHp81ug91BOIv/bL6yZHOhTX59QXm4bhcQ1mP\nbmUpfUi6oJRSqtvFbiQosUo3zK8OyqpfVh0mQeLHQFofg8EYIZjAwUhLZMcW/fh2/Xg/UZ0d\nnqcfj+zfCwDS5KmOG7/CF2Z/5c1yweWaq9Pzz7/SaFeNA19cVvi9ez2//yGN6DQ/xGI2npJ1\nejP2Y9OIgFd8hRbXk21vgK8DOB6Vn4GW3IAKqgAAEDJc8XVuxtnqjo+oqw3lF/PTl3ATZ2d+\nEIgDcTJEdifH+SLgSzJ/OcaQMWA7JxgiWqdGIgCIw5LEFXKjP30DABBKJ08gvU00ooVUEsXE\nJyqHMfECJcDZkDgVhMrhHeioREtUN2JQCBB6BKPJSXtSegig5XR6FwKoQKiBiUeM3MEvq4nq\nRpztTZ7afFM6u82hUOIwnj2lZO2+tthLycD7QadrSU2Jleey/Ekxi7w5TZciBoPBSAe7a4xX\nKAmtXSPv20kjEaGuwfK5y5GUnAxJo/ptunQn/P0kvHVTy923k1Aw9jKya5tv9Stlv3rQOHv+\noM+ZKew3fMW88uLItk2a2ylU1hjnL0GiKE6fG9myNnVnw4x5Iz9CVFgFmAOSXCsLAKi0fuTH\n03MEGE0/l5t+LqhR4HhImcJx9dO5+unDPgppOgWAyD6A038loQqZsvDPyhYUfIR2UoggEBHk\nY5TrqSscMpr5qmyPIvPwSEKAaErZFEYcj3pYAhOqeaOdGlUMxG1W9ndXWmkeGloP4lQkDfsH\nZ3RBqStJ3QAAoEDBBUhNfLChdB9Ae4+doJFSDaEpIzFQBqMftPv1n7VklXjDSl6mW6jEWHV2\nXYnD+NbWxqCsWqwGjy+sKMnVgqtWZN7flMFgMEYAJnCMRzSvu/Nnd0YPdveb8L/wZMHdvxCn\nzEzcTaiqVU7qeEMKNXWDuy5V1bb7fhxXN2KQULDtvh/XPLca8dl/N/LFpZYLLk+MWFf9p7xn\nGw33GLNh8kzjopUjOzQAADBa8ZkrybZ3ksJowhxUVJ3ha1Ea3fqxsn8bDfq40mrx7Itxfv86\nxo1gPxc9EJJmgGEiaE4ADbADOFvfB40VNHqc0K6KDwoA0EGhgEMNuVjxQRUgfgAK2AZIp6yA\nAiFUJlTjkAGj7L6pBgNGnJHPC6nOpD++mU/2EPUrLo0qAGBSj+r4yMj7wFAHeFgy1UctehNC\nBACUgoy6H2xCPdWNOK0A9QCjvk0PY2ygpU/A7GXTEMEYrZxZdu7MMpdfNvDY6Y389vkdBxu9\nsa0mif/yhWecO7timK7OYDAYw0r2p5SMkcf90M8T1Q0A0Fydzl/eU/boiyih8sJ62XXBdR+k\nToysl10/uOvKe3ep7cn19gCgtrXIe3dJM4ehZmHI8JV1hfc/6nvqT9FdW6iqYLPV9LkrLVfd\nPPI9NWNwK24Gjidb347nceBpS7mVX87sVWgkHHj4J8rBHfFIZM2L5hv/n2FBNmSdQYAlwOPu\nyYxQV1zdSAg6EVgxykLHn/QQGj0AyuHTWTYcCA3IcEZi1UCU+CJqOz2dhsNjs5ErwXo6SC4j\ncTYOCWHNo5EoAOKxaOTzUtM3FCIDAEfDHNVdyKWgtoEh2ylauUX6/js9nmp00v4BAIACBJjA\nwcgR7JL+ozhCYBWH96aHAAqsIgBYjcJD316y94SnsSNgNxum1OTZzaNPVmYwGIwYTOAYdxCf\nJ7xJp+ZCc3VGPvvUuHhFPGKcvaDwO/e4H/sjCXcZbWKTOf/WO6TpswZzYaqqzo50G9UUN9OM\nE962KfD+22prC19aZllxgXHOgn4eyJdX5//g16CpJODD9uSepprX437iL6Gtm4nPY6id4Fj1\nJdPCxZkeewIcz624Gc+7lLYdBU1FJXXIkXl3ifCrjyeqGwBAo3LgqQcc9dNwYU5NlRndUHDq\nxgl15pTAQaO7QElMDdNAOUBBQYauDDKF+MNqS+IhKgkG6SmLUItGm3WCgI0C7q0Vgka7RBwE\nOqVnXVB9a+FxC0J5QFFqtgsCE5MtGKOOYovkMAqecPLHvDbfPJJNQxBC02rzptXmelUjg8Fg\n9AkTOMYdalsL0ORKy65NrU1JEesl15gWLg1tWqu2t/IlZaazzuHykvOr+0ALUN8GiJwCGuWQ\nJ91eXEHfPqPKqeORvbuoLBsmTJKmzexz/24o7Xzgft+/X4wH/P9+yXrJVUV3/hf0376L41PV\nDeXUicbbvqS5XbGXakd7aPOneTf9R8Gt3xnA8AYOsuYja/JgMgal8sb3dOKqEv3sQ+mCVcN1\nXcbQoDSaZot+gXd2oBFQjuvElWMgTAIkAYCs6Sg1hCoK8RvwiDaoHgHiDoIEiQA6k3YAAGwZ\n0TGNAiSEKik91TOIEU6yDEhXnobSb2IwRhqEYGl94bpjTleo+x5e5TDOq2JaA4PBYAwGJnCM\nO7A57bOy7iausNh68dWDvJjqpZ0vA+maX4n1Nr7AojqT04b5krLeBQuqRJ0P/tr3xstAuqQZ\n45wFRXf/jC/qV/JCYM2biepGDP/ql40z51rOv6Rfv0gaOh78dVzdiOP+x+OWlReIDWcM5cxZ\nhIYCSZ4jcbROnQojRo6AEJ+mpV8uVXYQj/4cHigQN3BlAFTTr9QAjURgzAkcHBI4xGtUJSBE\ncZ6BJN9PAJtAyKEEnBwBo2oKFkobKQQBOIRsGNUAJCXLSADlAM0pR1fm1oeCMe6xiPz5k0ta\nfRF3KIoxKjKLBaxChMEAAMzaAzMGwyhL92UMHb6ski/T6zvIceLshZm9FvVvjqsbAIB4XPyt\nlVjq8WSJjaaSH9/Xu8Oo88+/8/37xbi6AQDhrZva/uuOxEgvBN57QzfuTxPvJ1SWw5vW62wg\nJLj2wzTHUGXnhvCrj4defFTe+B6o6lAGMEwgyQic/r8DW9iyZ+6CIF/XTBSjYUv2GQxZ7mec\ng1iEvNg/LihMUJG1xzZsQqbFvVhOjGcQysd4JocXcXgBRpNT1I3YPpMAahIedTBAHUINIznO\nOCqhe1p9Hx3pfO9g++aTbm+EVR4xukEAZTZpaqltcrGVqRsMBoMxFFgGx/gDobzbftD50+/S\nnrNr++e/wheXZfhacmNSwDitsuoPN3pWH5OPuABAnDoj78av8EW9tecgoaB/9cs65z6wN7xj\ni3F231YaanubfrwtdWVvAGgBH9X0y+Y1jzs1SANe/19+qh7eHY9E3vyn5dZ7ubKaoQwj83C8\nMHWusmtj6hZhxlkjP5zhgoQAALAp2+PIGBgVEnBS2qMQDIEFo0x/rodCLykYXZsQh0TdJA4O\nS6nBMYCAxTyxJKh6VcL5DGcaqUeEMAaKuDwQagExdWMoYIQmANScNhy1ZOuxJyCrHx3pDCtd\nXxmesHLSE5pd4agvYP1xGAwGg8HIJEzgGI9IsxaU/P5Jz9//FN23i0ZlobbB9vkvGxeeE9tK\nVdXzwnOhLRs1n1ec0JC36iZDTe0gr0R0Vqj4fHPh1y5D+ef38xzKyeNU1V/pih451B+Bg7M7\n9OOOIa1sczYHkiQaiaRuEkp1ppSBp36XqG4AgNbWGHjkp/afPJouYyJbmK651XdsPw14E4Pi\nOZfy9VOzNaRMIh+i4R1AIgAA2ISMs0AcGy0qEI8mE2gntBMgAiBilI9RaW71iEUm4CtBTZY+\nga8C1CU2iVxBSE0WHzEyCHjMJhBxiLcJcXuj8mwOZWzCA+h/C4wY25o8cXUjBqWwvclbapNM\nAtOwGAwGg8HIGLk1rWKMGELthKJ7fw8AQEhix1PV6Tz19Vvkw4diL0ObN3pefL7kh/c6rrx2\nMJfh7aDq5DIAP4BCeiSkLZbuZVMipiXLw9u3pMbNZ69IDfYfJAiWFef733wtOW4QLcs/lxQk\nXqduToTW3qgc3CFMmTuUkWQcrqTS/l+Phv/9pLJvKw14ubIa6dyrDfOWZXtcGYCGt0M4QWYi\nIRpcj2gYpGnZG1QmwagYo95SorIOEmdT4EE9cbpcBQFfg8QZ8R0EbDXxZWGtg9KuLLNYm1iU\nU0oNg9Fvohpp9+skJRFfbhKIAAAgAElEQVRKW7yRCYUsiYPBYDAYjIzBBI5xD+7hw9L+25/H\n1Y0YVFHafvEz84JFQnnFQM+NTJOp79OUKEamyf0/iVBTj6124vembpJmzO7PGWxXXBf85P3I\nzq1Jx9quvL7/w9Cl8Fvfix49LB/YG48gg1h813/xpclrsKS9Gai++4DW1phrAgcAYFue+Qu3\nZ3sUmYZEILwXINmJk4Z3InESIOY7ODJwSJwFhkmgeQAAOEc8dyOOgG08thAqE6pxyIARq0hn\n5AiUUAUA4YHcLmSVpPOeiajp2wMzGAwGg8EYOEzgYHRDo7L//Xf14lH/mnfyb/rygM9omQGa\nF4Ld83/ABmQ/Z2AZHDyf/9XbOv/wi+Rzf+4SQ/3Efp1BMJQ98Kjv1ecDa95UW5v50nLLuRfa\nrri+d2fT/sDZHVWP/dO3+pXQZxuI32eonWC/epVQUaUzBknHAO/0prFjA5HrqB0ARKdkg2qg\ndoKQS14VYx5kAr63dz4CzCEjx5I2GLkClTW3rDkpEADAiJe4on6WTUk8TtMBGCSe1acMiUBE\n/fBA+/HOYFBWCy3ivLr8mZWO/vd/ZzAYDMbYgwkcjG40t5tGo7qblNaWQZ0SIftSME2GyClK\ngoh3gLEBcNqpfjpsV1yPjWbno3/QnJ0AgETRseoWx41fGcA4OM5+9Q32q28Y6KX7BmPbZVfb\nLuujky5XUYdt+cSX0gOS44UzZmV+VAx9elksZeuouQqVIXqEal5AAuKLQKjOLVcRxvggrLZF\nSXciIaFqSG0xcsTA9e3uIXC42Cq1+ZMNmziEyu1j0zp3ZHAGok+uPxaOdt2923yR1TuaT3QG\nL5894IRTRjoIpe3eiDsgF1qlQrvEbr4MBiP3YQIHoxtsdyCO0+0MwhcUDv68QhEIRUP8UrSc\nf4nlvIuUliYqR4TqWsSPtmoCzJmu+0bgb/cnhY0XrsJ5RVkZ0XiES5861I9ZCiMLqC00tAFo\nNCZq0OhR4A4h81JAYrZHxhhHEBpNVDfiRLQOA2fvj+I2p9L+0WEllOAzihDMqrQbmcPoEHh/\nX1tc3Yizu8k7s8pRy5xNMsGRVv8znxxtdoViL+tLrF84p748n6WdMhiMnIYJHIxusCSZF50d\nWPtRygbOsvzcXg5UWpqVUye4gkJDbT3ihu1xDWPd6o9hh9LA+2+G1r6vtrfwpRWW8y4xLRqM\n3aZh3jKbPT/08l/Vk4eAaFxJlfGSLxrmLc/0cBnp4fKALwE1pW2woRKwJRsDYvQKjdLQBqBK\njwmk5qLhrci0KHvDYow7VBrWjVMgGpU51HcWhtnAnz+55GBHoDMgRzXiMAqTiiw2abQp9bkE\npXC0I6C76Uh7gAkcQ6fZFXpw9V5FJfHI0Tb/71/b8+PrzrSbdXyRDjX7dp90+8NKid24aHKR\nzZQx76SoRgwc7ns/BoPBAAAmcDCSKP7+jyL796qdHYnBwq9/U6xv0N1faWps/9XPghvWxV4K\nVdXFd/3YfNaSYR/oSEGVaPt/3xne0mWVGj18ILT2fcvKiwvv+ikMvMyXn/j/2Tvv+LiqM+8/\n59w2faSRZtSrbclyt8ENTDEETAuhOCQhEMhS0gibnrDh3bTdZDe7aZCFbEIJCUsIOIYQIKEb\nMMXggiuyLFmyetf0du895/1D8mg0c0dlNNIUne8f/miee++5Z+TR3Ht+93l+z0rLt34FqkKJ\nioRcfgRNFTmw6x/hlkZASKxbbjjnYpg75WtG9HmpzoeM4/e+1OvHttxoE5tzyF1AtVpEy11A\nFUBzdv2iCg03gtIJxA/YAHwlEusBZcYHmJEeEpmETr5pAjxGy4rMUGROyYQYskrUBOatMR15\nGcnxwoGuaHVjFF9IefVIz9Ubq6KDikp//+qJ95rGbx2f29dx4/mL1y+ZRfIvgDek7GkbPjXs\nl1WiE7iGIvO6inyeOTMtKDC78jKSgQkcuQl1D6vdrYAQV1KNLLbpHyhWVNb85bmhB3/jf3+P\n6nZJixbbPnOL4Yz1mjvL3Z0dt9+oDPSPRzrau7/2xfL7HtKvybi2IMnheeaJiLoRwfvK8/r1\nZxu3bktyUI5HXK796dFgcPiR33l3vSL39/KOIk7xIdU/ehvi+8dO71OPFvzLf3FFsc1l5htP\nJ+3ZBwDUaEB6HVBKA0HwB8jIC3j1bWmeGyMOSnwJthCgfkDT8nec+Vll6t8FxH36VF4If0iV\nHmQ4n2kcCxacsCQKZUiLn5BCBtxBQqnDotMtjLIXkccGkfPHlagAQH7qcgcWMm0JEmRa+2Lj\n/9jfGa1uAEAwrD78SlOVw+RI1mXGE1R2HuwKntaqgrJ6oNPZ5Qp8bFUpZi6yDAZjUnJtlcUA\nORx89qHwO38HogIAcLx49hW6y26CaZtWcBaL46vfmnwf76svDfz6Z3JnR/wmqihDD/6m/N7f\nzXDeGYrvjZcTxF9KXuDIOVSPu+PWG8KtLaMvwx4PAHASL9mNo7chckfr8C++Z/+PNH8q6PDp\nFsg+P/X5xze42kENAZfLOTXZCEIijevpO4bcCUI14NSXgtPwiXF1A2Ds7MQJcguIdSk/HSMF\nqP1UaQLiBoQB5yO+AabX3GT68EjPIZ1KY11CRWxBkGY1gVL6ZtPA7qYBWSUAwGG0eXHh+UuL\nOJz7i8BVFXnvtgxFRygAj9HysrlRPzMeRaXvnxpu6ve4A7JVLywttpxRmZ/0J4EkSJChcT3v\nd38YV/gJoKj03eP9V26oTO7se9tHgnGZOP2e0PE+b0MxS4NiMBiTwUraco3Ak/eE33p2TN0A\nAFUJv/F08KnfTH8EEggEPzwaPHaE+P2aOzj/8nj3d74yqm5oXv2Chw/OcNaZizo8qB0fGtCM\nL0xGHnkgom5EUEOK4htvyhNuOip3tM7vvOJQY9cnp6GgJNrESB9CifZFilLq209dz0CoKfUn\nVXs1w1TRjjPSjNJMw3uAjACoQGVQ+2noDVA1lluzxMCX8RMFNQFbdHxRyk80U1462vvah32j\n6gYAqITubhp47mBXemc1P5xb51jsmGCfJPH4itWleQsygyOskj/tbX+3dWjYF1YIHfKF32oZ\nfGJfh5JAp5iSSru2NVVMXCV0xBPS3LPflfyFtdOpfQuaKM5gMBgRWAZHTkGGeuUDcRahAOH3\nXpS2fXrqWhWiDj/y4PDvf0sCAQBAOp3tM7fabr4N8eOfExoODd73y8hLzecClGR3+WvgvbeC\nh/cRr1eoquUseUq/xsKGs82qsjTH8L65SzNOAgpEWZGpvZ1CRc08zUkTXYI/AU4EkZmMZh7Y\nDNJSCH0YG6fK6L/U9x7CVhBSusjUdP2YJM5IIzRI5ePxUSofRpwjte2EMeKNfIVCA4QEASEO\n6bkMaOUTCKt7JqYwjHLg1Mi59Y6cX+fzHLpuQ2Vzn6d10OcPqwVGcXVlvlm3QO9s97ePDHpj\nhYZed/BQp3NdZX4SA160uvRQ23CM0YkkcBesLImOcBiJAhfS8j0xiMn/Xyiqti4T0fIYDAYj\nEQv0MpCrqF0tEJc6CABAqdrZzC/bMPnhA/f8bOSxR8YPCgaHfvtrdWTI8c27I8FQUyPxeCYf\nR7d0+QwmnUmQgH/g374T2Pt2JIL0es09jeddNPvTKZ1t4eNHqBwWKheJy1bPfsB0QdwaDRQB\ngNIJNyLIkGYRAdlX0NYXQY29BURFa5m9QmaCdCuBL6DBRlCHAQhQAkSNTh2joeMohQIHTSzO\nYtaUYU5RgLoBFEBGgGn/qskggNZqhwaAeFJeqAIAPNIDp31RSAvdzgDRvOgDdI0Ecl7gGGVx\nkXkxs24FODmobVrUOuRLTuCodphuv7j+z7tbh0/rJsX5+k+fu6jAPEHaI5Q2lFs/aB2OH2FF\nVfL91616YSBOrwEAq35BfKoZDMZsyD6B480333z00UffeOON9vb2/Pz89evXf+9731uzZk30\nPi6X66677tq5c6fT6Vy2bNndd999zTXXpGvCGYLa14nyHJyjHHjt/3R1ZNj550fj486//Nl2\n8228fWwJQULaiYjR2G7OVr/Gkd/+MlrdAAAaCADPg6JEB43nX2w8/+LZnIjKsut3P/O9+iyQ\nsbtzacW6/Dv/H1eY/oTnJOBLSpUhjVoexI+rBthsFevSrXwJRtTwCXp8B8jjOa4ofwmqYXYq\nGQxfikyl1PUsqE6NrapbI5gcxEO9u4D6AWt8SSKhOmUnYsRA2ylpATj9NYvsCDcATCM/YrK0\nmgWRcTPqhqBpVBNjlKAQ2ukMeEKyyHFFZilPzzrU5hqaORQAEO9kMX1WVuUvLbO2DXiHPSG7\nVVdlN0U7evS4g3tahwe8IUXieB4rE1uurK6xrayegcl9DA3FloHm2FpgjFADE7MYDMZUZJ/A\n8Z//+Z/t7e3XXXddXV1dV1fXPffcs3HjxldeeWXLli2jOxBCLr/88kOHDv37v//7okWLHnzw\nwe3bt+/cufOqq65K78znAa58MSCkmcThf/J/KaXYnKe/6hbpLI3lXPDDo1TVugoSEjxy2LR1\nbOEtVtUkOgUAcPk2+1e/bTzrnOTfQ/qgctj7yvOxQQCkKKaPXKG6hpX+XqG03Hjh5cZzLpzl\nuVwP/8r38jPRkdCR/UP/8R3HTx8EnH3OOJZLrwweORQf56Puoa23fAUJ6X/wgmx1aP1Xae8+\n8PWDYIC8GmSrT/ekGNMgUVPY1KXeUP+7QLwAAFSdOCwCqQH44lSdiDEB2kHJxDITOkBJAOFN\nU9eYoMQus2hBZNwUW/UIabthleSNZ5r0e0Lvd4xEmqce6YVqm3FdWR5rRpFLWPSCM6Ch61ni\nxCx/SGkd8HmDcoFZqpmoWcQj8HhJiQVKYuOtQ76XGvtGbwYliW+os3f1ePzecEhR7Fb91hXF\nW1eWzObz1VBsdgXCh7pdkftNkcNbFhfajOm/kWAwGBlO9gkcv/zlLxcvXhx5uX379qVLl/70\npz+NCBw7d+586623fv/73990000AsG3btnXr1n3jG99YCAIHthUJZ1wg730lJk4VdfRhDvE4\nfX/8Gchh6byPxh6sqW6MHq6O5y/whXbzBRd7XnkhZh+htKzoX36gW7EKG+bjtpKGAuH9b6l9\nndiaLzSs44orZj+mOjJEQ7GGWKOXZy7fVvjN78/+FKNQv8//8t/i4/LJ46Ej+6VVZ6bqRPNG\n3rXXBY8edj//1/EQwvqacg6CgLG4aKnl058Tl65K2fk8A+SDp2n/CZBDKK8ULb8EVcykwIfX\no/ItKZsMY37gi0DRcvwV4u67k4N4xscnKgABhEfFXGQ4A4RFqTkLIw5KtLyHqRdoP6CpMto4\nOyA90EBc3AEoyeaU2YVJx6+pzD9waiQmvqzUWmAaS4EJKurbbUPRTpOUQuuQzyBw7GF4LrGs\nxNI+rGHAubzEGv3yvZahFw/3RNI9Cs3S1WdWVBYaAWDQHTx0amTIG7aZxBWVeUXWhNVYFODt\nk0PRj7pEAddUWhHA9rXlqdIgNtUU1DnM7SN+X1i16vhFdpN+YbRAZjAYsyT7BI5odQMAamtr\nq6uru7u7I5Gnn35ap9N98pOfHH3JcdyNN974zW9+89ChQ6tWpW6Jlanot38JGczht56FUVWC\nAlVUdWKCov/ZP0jnXAZ4wnVCWlKfKDVDqm+Ifln03R+QcMgX5SupW7mm9N//my9O0UpDC/XU\n8fDe18hQL84rRHkO/ws7iPO0sxrH67ddZ7zmn2B2T6OwwZjoN4CNqTSPkHs6qKKdPi2fas5G\ngQMwV/z9H1suv9L76ktyf69YUWm59EqpvgEIAQSAUpmTQgdayMu/BHXsF0gHW+nr96MVl+I1\nH0vhWRiZBtIvo+E2IBOLzLER6RoSHDFDYkYGClQdezCeIGGNkQpCANplj5S60ZQCB2AknknD\n70N0A1dsRUIWWxrNlMtWlfEc3ts6PPoYAyFYXZF/6arSyA7tIwHNPhonh3xM4MglGootfe7g\ngY7xUj6EYH2VrbZw/LHT0U7X3/Z3Rh816An9YXfrndvq958c+vuBroil6AsfdH9kVcnFq0tB\nC6df9oaU+DgF6PUEU5hkYTOKLGWDwWDMlOwTOGLo6upqa2u78cYbI5GjR48uWbJEksbLd1eu\nXAkAR44cWQgCB/Ci7spbpQu2q92t4QNvBV//W/zdOfW61L5OrqRqwnHFJeaLL/O88FzMzqYL\nLhIrJuyJTeayn/1P4OCB4OEPqKJI9Q3GTWfPUlyYnMAzD4Ve/UvErgIAILrUU1UCzz/G2Qp1\nW2e1xMUmi1S/PNR4JH6Tfv1Zsxk5BpTABgUAMqGII2kM6zcZ1m+aEJqDchv63uMRdWM8ePQF\nqN0Elqx0MGFMCyQhyyU0cABCpwBUAA6kKqRfC6nqZDHJOBnQLCN3mfWFA+ch3QWgtFPiBoQR\ntgFXmtr+KSmHApwa8Xe7ggFZ1QtcuVVXkW9IesY8hy5bVXr2Env3SIACLbHq8ycuCD1aC1EA\nCMiqrFKBy+jfFWNGnF/nqC+ynOj3uIJynl6oL7I4JhqCvtWk0eE+JKsvHOzeM3ETofTFg90l\n+fqVWgalCknYykRWWJcTBoORZrJb4CCE3HbbbaIo3nXXXZHg0NBQbW1t9G42m200Pvlo9957\nb1eXduv4cDgMAKFp+GtmCMiUx9etVdpOJHz2qNXJtfi7P8SSzvXsUxEpwXLZlY5v3R2/JwDo\nV6/Vr16bovlOhnxkT+jlJ2OCmMegckQZfxeBV56epcABALbPf73325+nE/+jzZdfKy5eOsuR\no+HLq7HZSjwanUfEhgX04DEZAk460qERp4R2H0VM4MhmKFUJqBjxCBLoYliPjGeBcTOQAGB9\nihexXB5gA5C4BG/EA88+V3OHCKADiK0NBACErPHBBHDA12TLMn3QF3qv3RlpdekNKQPeUJcr\nuLHKNqkTwhRY9YI1gW8on2BchIDLPsenjKCx232k0+nyh/ON0urKvEWZlAhTYtWVWBPWZ/W5\n4uq5AADgZL9XM77nxKCmwGHRCRghzQ4+C6R3D2OewKwoiZEMWSxwUEq/+MUvvvjii3/+859j\n6lY0QVOlGHzwwQft7e2amxRFgawSOEbhKrTrxpGkw44yjbhOV3T3D/NvuiXUeAwIlZY2iFU1\nczzHqQm/H2spMgriMUQJHGpvBxAyy5QBqWFl6X2PjTxwT/DQPhIMCOVV1u03mi66YjZjxoM4\n3nL97c7//a+YuOG8S4QqVuo/GTSkUWA8tinsz5YVDiMGhQQDyqBKx75gRc6s5wpQQvdQBDix\ntWTyIGTYQL1vxLQdRfp1gNj9+hyCcC0lx+KiFkD2dExnbulyBfd2xPplAEC/N3RqxF9jm4sP\nNjhM0okBjeWr3SjhTHUZVVQ64AmqhNrNkpRJtguE0iffbT/SOVYG0jbgO9A2fGZtwZVnlGfo\nr3Iiif7HwwnSLgbcGuIjAEg8ri00Nsd9riw6oTwvg/ooMxiMhUm2ChyU0i984QsPPPDAH/7w\nh2uvvTZ6U0FBwfDwhHbcoy9H8zgm4cEHH0y0yev1ms1mi8UyiymnAaF+LV+5WGlvjonrtl6F\nhIRJ12JFVUxNSnohw/2a8RhjBySIKSmIEMqrHN//GQBQRUb8XDXSM267GhmM7kd+rQ4PAgAS\nJfPVN5iu+cwcnS4BFCjMaW1RykFGG2BOM/8ImXJwObQQUEjQK3dH94EIqx6VhMxi+XwXGvAl\nyHwpDR4CdQgoAT4fSSuAL5zXOSxAUBnCQMmJ8cauqAjhpRleZpIEhMLhHo3EvVF63ME5EjhK\nLLpis67XM2GlymG0snT6OTLzBwV498Tgq0d7R5ubchidVWe/YFkxnxmlNHtPDkXUjejgoiLT\nivK8tExpRlQVGo/3aLTW1gmc5kdTl1hdOmdRYUhRO0bGU0KseuHipUWT92RhMBiMeSArBQ5K\n6e233/7QQw898sgj119/fczW5cuX79ixIxgM6nRjSXqHDh0CgBUrVsz3RNMOxqYv/ND3x5/L\nx/aORThed8HV+o/enM5ZzRBstGj2d6ETXdOEpVPUy5D+DtLRCKEALqrEtaunXNjPnboxiuGc\niw1bLlL6e2g4xJdWIG4e/xidbeTki+DuAkrBXIpqL0K2OcscISo98jI9+R71DiNzIVqyGTWc\nn7ztqKBDFWvoqX2xccmIylfOcqaMtBBUh+O7XKo0HFY9Ipe8pqxSOaB4VaogQAKWdLwJTWfB\nzFmQkXXYmXdQGeKKgXoBZEAmgNxsgOIOyqHE9gRBOWEjsylp7ve2Dvm8ISVPLywvtcbYLgDA\n5hpbU7+3edAbUghGyG4SV5Varbq5vcYlx+sf9r1ypDfyUiX0zcZ+l1/++MbKNM4qwqH2WHUj\nEs8KgWPr8qKWfo+iTvjKLTBJy0stL4xoJEguLUuogok8vmx5Sacz0OsOKoQWGMVFhcaMzQli\nMBgLiuwTOCilt9566+9///uHH374hhtuiN/h6quv/r//+78//elPn/3sZwFAVdU//vGPixYt\nWhAOo3HgvELzl3+stDernS1I0vE1S7EtG+rJKQXfAPUPI4ONX75BPva+xi5RV2gk6QzX3pJw\nNFWRn39A2f9yxFsEly4Sr/0KKtSo05lXEOKLtC3K5w7ae4AefWL8tesUPfAA1H8MlW9KfFCy\nKGHy7E9p/8mxU/tdtK8FndyHL/ta0nWVeMOniG+YDka1lpSMeMutIM7Jw0/GXKMQ7RRohQZF\nSFLgCKo+r+yM6CZhEgyqPqtoxwnLXhhph4Mo0w1CPSrtJtSPACNk4nEZyn7VI+K7oUly/S9V\nQp893H1ycLwH0MEu5+aawo01E1JWOYQaiswNReaQQgQOZewqVFbJm40aOZuH2kfOa3A4LOn/\nDLgD2h3QhryhlkGfQeAKTaKQwdYmZfmGz5636Nn9XT3OscyLFRV5l64ulQTu4KmRXucEh45C\ns3T+8uLJByzP07OaFAaDkWlkn8Dxta997aGHHrrmmmsMBsOOHTtGgwaD4bLLLhv9+eqrrz7r\nrLPuvPNOl8tVW1v70EMPHTlyZOfOnembcvrhKxfzlVPblGQI1NlBDz1JXWOdzARzqVxTp7Q2\nRe+DjFaquAFUwFioX2385Jf48oR2IfLLjyp7X4yOkO6W0GM/1n3xlzDHaRqTQIa65V1/IT2t\nwAtcZb1w3rXIOPcJw0ShTc/Gh2nz31HxGuBTfPtIj7wUUTfGg90f0sY30LKtSQ4qmfC2b9FT\n79O+E6CEUF4ZWrIFROPUBzKyCppsc1aVKtHqRiToU5xmoSAVU2PMLSrtU8iYlzAFldKRsOoU\n8BKMsqxKNAajONkdV3KrxL3tI9HqBgBQCm+fHCzP15dpDSjxmbv2BoBeZzCRGUTHkD8TBA6j\nxI/4wvFxT0jddWIAAPQCt7Hatqgwcy9JlQXGL15U5/LLnqBcYJL04piydselS18+1LPv5JAn\nIJt0/Jpq27Y1ZZGtowz5wnvbhwc8IQ6jIrNufbXNLGXfOoLBYOQ82ffF9M477wDAzp07ozWL\nsrKyzs6x9TDG+Lnnnrvrrrt+8pOfuFyuhoaGHTt2XHXVVemZLmOm+IbIO/eBMu7nSj3d+nq9\nsvyG8ME9ZLAb24qE1WdLF25HvEBGBpDJisRJmzgqcoy6MTbsUI96Yh/XMAdpC9NAObw79MQv\nQB3r3kfaG5V9r+j+6Ye4bI5NRt0dIGv5dKphOtQIfh/19CHJDI56lJ+CfGDadiBBfH/yAgcA\nIISqN6DqDcmPwMgYOCSoVGPBwOEkrT3DaiC+5gUAQmrQJNBpFaowUkYYaD+lAYQkQAUAU6/6\nKMgKiW9nRhVySuSyuwzNIHJ2kzTg1XArT/oxeGOvhp/CaFxT4MhwNLtyjG0iSSqeqaWhzNo5\nrHENNejHbqcDsrrrRL/EF2d4XoPVIFgNEx7w6ATuijPKrzijXFaJZhJKU7/n5cb+yP/RkC/c\n1O/56MrSbPykMRiM3Cb7BI533313yn3y8vLuv//++++/fx7mk4MQNfzuC8qH71PPCC4sETZd\nyi+ev+oecvK1aHVjDDUslIvSxffEhLHNMeWA1NkPYe0ceNrXDukQOGjQF37qvoi6MRYMeEN/\n+ZX+ztj3mOJTK9q/CgAgB54EvxdGl4bHnkdVG/Ha65I3yxg9XVC78xwEPbMZlpFLSFyeX+kH\nOsFTEgGWcJKP6wkk8jKglJLEzVkYqYb2UtI4ah1KKQAgQDUIT6HhEuqOaWQzNhiEKA0glN1L\nqXXl1j2nRpxRZQ48Rg1F5tqCJB/4e4KKZtydIJ7hOCw6DiNVS8soyc+I//rNSwqP97jbB73R\nX1h6HW82RosF6GCXK8MFjknQVDfCCtl1YiBGgVIIfbWp/4YNVUw2ZjAYGUX2CRyMOYUG/f7f\n3q22j9WDqJ3N8gdviuddpfvorZMcpbqc7qefDLU0YYNRt3K1+ZIrEZfsKmJEu1NvwviUTFKE\nkqb6FLX5IA364uOk9xQZ7MJz6QyC9AWJnoKhkH98EyW07R1qLED1F83qdCYbdWs1wTGxzhSM\nMUTOTEENKsP0dNoFRoKBdyStRGBIeCCanWDHmAHUS8mRiak0FOhJoAZAJZMemdBrk4KS7eso\nHc+du6iw2xUY8cuEUqteqMjTz8YRQy9wmtYeyTl6pB29yJ1RU/Bey2BMvMZhKp+bFjMzReDw\nLecveq9l6Ginc8QnYwycwJkMselmg1p5OllNpzOgWT3kCshD3lChadJEWgaDwZhfmMDBmEDo\n5ccj6kaE8OtP8w0bEuVx+N9/p+9731LdYy3G3M/udD35WOnP7+dsSS1iE2aoTmbPNgnIakd5\nduociN+Eq5cnN+YsoZ6RhJvcwzCn1qdGB1grwRWrFlFCIO4umbS+zc1S4Fi8iXY3asSXbJ7N\nsIwcQ+LyBGxSaJBSBSNRwPrZtAgVOb1P0eh4KGIdq0+ZNyjt0iwUorQDTSpwIEhYmoRQLiyi\nEECZVV9mTc3j/b0LahQAACAASURBVFq78UCHM/5jvdhuSsn4889la0oRgvdbhiLJAsvKrOcu\ndfhCijEz7B4wQpsWF25aXAgArzYNtA5pPK5AmWrjmjSBxF1+/LNoAMRgMBhzAXucxZiAcnB3\ngvibmnHi9fZ9/9sRdWOUUPPx/v/6UZIzyCvXjlsrkhwQIeGiz8SHuWWbcXldkmPODmSxJd40\n5yaIeMUnwTSxkw4ngZZrGviHQdV2jJ8mqP4cVHdWbHD5hahm3WyGZeQeGPEiNklcnoANs1E3\nAIBDvJGP9evFiDMKWdDEMYfQWPUBANAE8dNgZNHUOBLFFzibagpscekDSxzmxY5sFTg4jK5Y\nW/aVS5d+YlPVNesrNiwqbOx23/vC8R/tPPyz5441dmt7jqQLe4LMBatOONHv7XEHNcttshFT\nYnWJ+YwyGIxMg30rMSZA3MOacXWwO/CHnyitxyAcxCXV0oXX8Q3rAcD3zhuqS6MtvO+t11WX\nk7POeEWBas+jnfuATKwfxjxadP5Mh4rArThbFCX57w/T4R4AAEHiz/6YcM61SQ84S7jFq5He\nRAOx/hS4tBYXzn3XWF0+3nAn7f0A3B1AKVjKaV8rDGsJW5gHPLuvCITw+bfSxZvoyb3gGQSL\nHS3ehErqZzUmgzEVet7MYymoehQiI4RFLOl5M2KC/jxAByhxAdBR6w0tpvxKwTyuVUgLjRoB\ngZ7H1SmaYk6hF7hPb6h8/9RI66DXG1Ly9OLKMuuykuxuNwMA+UYx3yj+6e22g6fGEx4H3KHf\nv95y/dnVqyrz0zi3aOqLTEd73L7whDsWBNDnDvS6AwBg1vHnLLJXZIaByGwoy9MbRT7mnQKA\n3STlx0lsDEbKwFlZbcdIO0zgYEwAm/OJVjWH2nJEJWNZiGrbh/4HfyBt+7R00aeU3m7tgQhR\n+nqTETjMxXjjbeTQk+A7XYVrLMSrPo7MUzRjnxyu7kyu7kzqGYZQANmK0/uNiSSDdO2Xg3/+\nGcjjeRPIaJW2//N8zQCjknVQEkmjkGirhsCBHHWQijxbVL4Cla+Y/TgMxvQRsChg1hRWG5Uq\nMgkRSnjEC1yKKndokKofAI3WuxEgHDs2Spi/FgEjk8itUOkAoX4EGCEjhwpnmdeTwwgcPqu2\n4KzaXPu0dw77o9WNCM8d6F5ZmZ8hnwaRw5ctL3775FCXKxAJKlFWnJ6g8o9jvVevLs12lwoe\no4uWFj1/rCfaicMo8hctLZrkqDTiDymvN/a3D/pklRRZ9ecudRRZ099mmMFgzA9M4GBMgF99\ndvj1pzU2kNgay9BLjwvrtmJzwidFnDU2S3yaoMIl3Pnfps528A+DwYbyKmabRxAZ2WwDc0pG\nmi3csk2Gr/xafuMptbsFCSKuqBfOvQbp05NUjEpXoKKltG+iWQYvoRUfTfm5aMcHtPso+J1g\nKcaLzwLr5HaDDAYjxfgUV0DxRgwyOIU3CTYh2aa8Eah6cKK6AQAUqAoTzGIFhGunNx7HoWIu\nQ1axjHTQ2q/dhMvlDw97QwUZoxdYdPwly4o8QcUVlD/s9ZwcjJ02ofRQl2tDtc0ZkPUCl6cX\nOJyVn+zyfP2NG6o+6HQOeEIYo2KLblWZVdRquZJ2epyBh19vCYTHblz7XMEjHc6PnVm+rnpq\ngZXBYOQATODIdYJe0n4MvMMovxhVLAN+irtY6aJPqS1H1M7mqUcmqnJ8v2HjFsRxVI2VP6TF\n9XzRLNaumEO2GrDVJD9CxoPyi8SPfT7dsxgF4c230ROvkZY3IOgGTkCOerzyY2CaugvvDFBl\n8sZvadfhsZddh9Xjr+K1V6OGj6TyLAwGIzEBxRNQJjRpVqnilgfzxWI8mxYz1As0kXcyB6AC\nYECFCNcBZH2uPmN+UNSE7hWTbEoXZh1v1vHvtWkX+XY4A637O0d/NojcpmpbTbKNgdOLXuA2\n12RBrtBf93ZG1I1RCKXPHeiqL7FkiFUtg8GYU9jfeS5DDu9Sd/0RTnckRVY7t+1zqGqyYgGk\nMxq//N/hd55Xjr1P3EPYXs4vWRPceZ/mztTvEUtKbbd8aei390wYRJLs37g7Ve+CMR9gHtVf\nxNVfBHIAeAnmoJsmOfrCuLoxFlLJ/r9gxxJUUJXy0zEYjHgCqsZTcUpJSPXr+aQzyEJAWhNt\nQ6gBcAGAwGpMGDPCkaCgQOCwzZShpg/xNZ0IAGOsRFmN+sPqq039Fy0tqsyfWeNblVBPSDFJ\nPJ/qBBBKYdATHPaG84yi3SLNpm9xJuDyh7tG/PHxsEJO9HrWVGWKgQuDwZg7mMCRs9DWD9R/\n/GZCxDWg7Pwp/9n/QnmT1kxyvLjlSnHLlWNH+T3Bp38DRKNLKy4oBoD8z9wqNSwf+cMDoebj\nWG/Qr1pru/3LQmmCZiiMDEeYq+er9OS7WlFKW/cwgYPBmAcoUEK1GzqqNNl+SbSHksbElqIA\niAfW/YQxc+pLLYVmadATiolvWFwgZGRZBAA4zLr+mAljTScrtL/DOX2BwxtS3mkdah3yUQoI\noMpm2FxbYNEJs58wAHQN+596v6NzeEwRKMnTX7W+oqowKxNMRvEGY51QI3iCs2oMx2AwsgUm\ncOQs6vvPaUSVMDnwIrf1xumPgwxmftkG5Ujs6hSZ8kYbqQCAYf1mw/rNyc6UsTDwaefugndo\nfufBYCxQJn0sm9wzWx8lRwFo4sM5QJnbnZeCElIHVOqjlGAkiVwBj7K1u2ruwWN007mLHn+7\nLfpp/Bm1tkvXlKVxVpOzqsza1O+JtuGMc9kdY8gXJpROJ1ciKKtPH+yOtC+hAG3D/j5P6Nq1\n5UZxtnbpw97wb19tDsnjumePM/DAq81fvqTeYclWS06zPqH0Y0m8icFg5BJM4MhZ6GC7dnxA\nOz4J+mvv8DsHo405kMmqv+FbSDezBMuFBpXDnr8/E2w8BgC6pcvNl16JhAV8cZVMEHRrxHVs\nRcFgzA9IwJJMYh+JA4CAk7FspKQnYlYKCAONTfRD3BKADP3SIzTkV9oonO4ORpWA4hNxocSl\n1HuIMQvsFumObfVNve6ekYAk4Gq7qSQvoz1czBJ/5crSN5oHInkcksBF6x3R0EmEwSgOdbni\nm7MGZPVgp3P2rXN2H++PVjdGkVXy+od9H9+YrZmVFr1QVWg8NeiLiesErq446zsoMxiM6cAE\njpwFYU7bhoub8X86MucZ7/y5vH+X2naMhoJcaY2waVu6Wn5kC+FTrT3f/rLcOSYnuZ/ZMfLY\nwyU//bVYWZ3WeaUNVL6KNms1oy1fPf+TYTAWJgbe4goPjqsSAAAgYEnikls3RhW6IwQQpXEg\nE8JLAGdoC0kACKq9EXUjQpgMCtiKUaZ06GAgBPUllvqSrFmXFhjFq1aXuYOyOyCbdUKPO/j2\nSY0sRZtBnGYvlW53UDse1Zg2aTqGNLwqAKBjUDueafjD6og/bBQ5q16Mzoa56syKh19vcQfG\nC1J4Dl11ZoV+1jkvDAYjK2ACR86CypbSxrfj47isPpnhMBbOvEA484LZTiszoIrsffG50PGj\nACAtXW666ArEp/RvgdK+730rom6MIne2933vWxUP/VmzJDcNeAepsx1UGfLKkXXOk37x6ivV\n3kbwDkYHUdWZqHzVXJ+awUgLFCjKMGdNAUt5ot2rOBUSBgAESMebDHzSq8eJX5sInW4KyyFu\ny6wmOsdQICqNfcA7ikI9IhM4soRBX+h4v9cVlHU8LjLrljrMmdCBFQFYdYJVJwCAUeQOdbm8\nodgUjDXl1mmOphLtZ1WJ4jNEexAK4PLLr3/Y1zXixwiV2wznNRSZdBm0ZPCGlF1N/SdO9xLO\nM4gX1DuqbGNpxYVm6c5Llr7dNNA+6JNVUpynP7vOnm9kZkAMxkIhg76tGKkFb76GtOwDeUI2\nMrIU4rUXx+9MvJ7A4YNKf69QVqFfvS63Kynkjrbef/mK3Hlq7PUzO5yPP1L843uEsopUnSJ0\n/Fio+bhG/ERjqOlDqX5Zqk6UJEQhB3fStncij1tRyQq87pMgmefwpHoLd/nd5PDz0H2E+l3I\n4kB156LaTXN4RgYjHVBKfIo7RPyUEoSwhA1G3oLmoDNRcvBYzBMdlBIChEOzugdAqJDSLq0N\n9tkMOw/QBGark29iZBRHe92Hul2jP7sA+jyhk0O+j9Q59EIGPaUXOHzpsqI3W4Z6TydiSDze\nUDWDNrE2gzDo1Sgrs6ViuV6ab9BM4rDq+Z89dyxSXNPa7917cuiz5y2qzAzzUUWlT+7vdPrD\nkYjTH376g65r15aVn7ZulXi8dVnmZpAxGIw5hQkcOQsqLOc/9X315Ydo94nRCF6yHl9wE0ix\nxhmeF57r//l/qCNjHpBiZXXR3T/UrzljXqebUqhniLz5Z9r5IQ36kb0Cr/8oXnz67VDa94Nv\nj6sbAAAgt7f1/+DbZf/7f6nKrZB7tG76Rzd1daRd4CAHd9LWt6IjtOcI2fMwPvfLc9vKUdDh\nddfAumvm8BQMRlqhlDjD/SpVIi+DqlcmwTzRkTkaBwAghDmY9XyQHVAB0JgMfAHhxbMdeY7B\niAdAmo+vMcplfT9ncAbkw6fVjQjekHKg03lWzWydKVKLRSdcvrx42B92BmQdz9lN4oy6wCwv\nsTYP+AiN/ayuKJluDsgkbKl37G8dltUJLiE8Rj2uYIx1SCCsPvHuqa9fviwTMlCP9bqi1Y1R\nCKVvnxy67gzmDZdb4AzSKxlZBBM4chlUVMN/+kfgc1LPEMorBp2G9O5/752e//et6Eiova3r\nK5+v+tPTQknmepVPAu1rVR7/IYTHylNpZ6Pa2Ug3fow791MAEGr6MNzcFL+KD534MNR8XFqy\nNHaDqiThWoJNCVMhsDndtcRhP217Jz5MB1vo8Clkq573CTEYuUNA9UbUjQgqVQKqdxaVIElC\ngahEQQjPMlMjMQjhtUDbKekACAAIgAoRXgKQ+SUeSMAWmcSukBFgHqf7K5oxDTqdAc3iik5X\nYJrdSeYZm0G0GZLJuXCYpa119t0tgyGFAAAFkHi8uaYgEJDf7nEbRK622GIxJKnK2S3SP21d\ntPO9joHTCSYFJmn9osJ/HNR4SDPoCXWP+Mts6VcQelzaviS9CfxKGAzGQoMJHAsAYx4yJmzU\nN/LowzERBED8fueOx+1f/vocz2xOUF/5fUTdiEDeewYvOwcVlis9nYlyFJSujojAofacCj7z\nkNJyhMphzlEuXXSdeMb508/v0K1cg00m4vXGxLHJrF+5dgZvZg6gnt74ZgdjuLqACRwMTahM\ng0dA7gLqB2xGYi1IdXOb75OdhIn2HXaYBA0wfytnSolPcQXVMZsJjDgTnydyekqpX/UpRAYA\nAYt63jBrlxAEqApxVQAEZp8SMo9IXDGhYZWOXywQYB1Xith9UUbS7Qr2e4NhlZolvsZmCMT1\n/hhFJTSsEh2fU099F9tN5fn6jpGAJyibJEGH0Z9eb2np9YxuFXl8xfqKS9aVJzd4jd30lUuX\ndo/4Bz1hm0kssxkOnRpJtLM7IGfCg69E9iOEUkozxeWMwWCkEXYhX+iETmhYRQBAqKlxnmeS\nGoJe2tWkEaeUnjyACsuxMWHzl0huhXLikPe+74I69hhW7Wnz/+GnanuT/prPTXMWWKcvvPPb\n/T/+fzFx+1e+g3Tp7i0/SZ58JqXQMzIIGqSel4CcFuxUJw3sB7kbmc5nGkcMNC6TfPL4HOGW\nB2UynsJNqOqWhww0z6/4yGl9M6QGA4rPKtn41OR3ZNm3BwLOwNfIxKlSPwUVgyTifMTqUzIP\nhdB32ob6POM+FE0DHrtRO0uIw0jickrdGEXHc0vsJgAIK+R7j+0fivpthBWy851TksBtXVmS\n3OAcRhUFxorTtiBmvfZfgSyrB5oHD54YLLYZNtbb9VLaVhB2k6R5h2o3SUzdYDAYwAQOBiS4\nFUA4y+5WR6FBX0JX8KAHAKQVa7DBSPyx/vnYZNatGOtXGvjL/RF1I0Lo9b+Km7dxJdXTnInl\nso+J5ZVDv/t16PgxAJDqlxXcfocu3ekbAICspcCLoMTWrwIAKqid//kwMh8aPDqubkRQeiHc\nBmJNOmaUuXCYV1VZMz5vcwiTYLS6EcGvuAid8MWuUtUddtqkwvmaWsYh4DwBEmY4MjKBIz2u\naHUDAGSV9niCCEG8bFiRp8/tJe7e5sEhj4bn6Av7u85fWaL51lVCj/d7hnxhALCbpCV20+S9\nZqrtRpOO9waj74Lo8HDQ4wl2dblHXz++q+XzlzesWZQeu5MVpZa97cOBcGwWz/pqW1rmw2Aw\nMo2sXMQyUoh+5Rrt+Or0L8WTABnzgNN++ICsDgDAekPBl74Rv7Xgjm8iSQcAxDWkdrdpHE+p\n0rh/RpPRrVpbdu+DtS+8XfvC22X3PpgJ6gYAACfi+m3xYVS5HszMcjx9BD3g7gWSkR0c5G7N\nME0QX8joOO0uA/oE8blAJhrrHwDQFH8VIiskVs9lMDIESuHUSGzN6Wi8zBKrZVh0wtqyHJer\nOge12xsPe0O+oIa0OuIP7/ig653W4aZ+b1O/962TQzsPdru19owgcPjjm6qinVA97rDbHYyW\nk5y+8C+eOjKYwAtjrtEJ3LVryh3m8SweiccXLi2qc8xlJzgGg5E9sAyOhY7tnz7n3f06DU24\nSvH2Iuv2T6VrSrNCkHD9RnJsd2xc1OMlG0Z/NF92lVBWMfzQfaHjxwAhqa7BdsuXdKvWjW6l\nQY2WaWObAto3FlOQeY+TUP2FWNCRY89B2A8AwAloyVa8VEP1YMwDtLeR7n2COrsBADge1V+A\nV10OfCbZNFKNdAAAAJpoIb1wEbHOyFv9ipueVhMQIANvEXC6a9PGoPFVRYSq7GaAkZmEVRLT\n4yOCScdvKyk6PuB1B2WRw8UWXZ3dlIH2oqkFJX6D8e+dUni1acAbmqBguoPya00DH1tVOslZ\n6kssX7u8Ydexvq5hP0JwcEDj5ickq7sO92zfkp4kPrtZun59Vbcr4AzIBpErseh0mdQemMFg\npBd2T7PQkZbUV9z3YN9//ihiumHcvMXxzbs5Swo6kKUF7oKb6Egv7WkeD4l67vI7wDDu8Kdb\nfUbprx4EQgAAJhbj4Hw78AIoGs83sD0T3LVSAkK1W7jqzdTTB0RB5mLgk3F3Z8we2nWE7Lpv\n3PZVVeixF8lwO/7IP2eQvQU2gqqlceCEjjYLGT1vFjl9WA2oVOUQJ3L6OWtiog2f0EgCaX6o\nMqp/7VxCAZxj3V4gD4DZbWQBPIc0S1EAQORwvkHcVLWwqhIWFZtf0oqX2AyGOFOMfm/IGdC4\nmRn0hYf94cm7uuQbxavXVwAApfT6/drJel2DCR8IzQMIQVmevixPn8Y5MBiMzIQJHAzQrVxT\n9ccdck+X0t8nlFfyhfZ0z0iD4Ptv+l54WunpwFab7ozNpis/hYQE12a9mf/0j8ix3bTzQwj6\nkL0Cr/4IaPaR0fIZQaJOXHde+L2XY+PmPGHlptm+jYwCc8g62TMcxjxADzwV39SG9jbS7qOo\ndEVaphQPEmtoQMNXH4nMtEUbDvF6Pm3J0iKn5xQ+vlst1SpKxYgT8EJY6nsobQSIrMc4hGoA\nckazzlk4hBwmqU/LdaLYrAMAZ0A+Oej1BBWzjq8tNOUlMMjMGdbU2qodprb+WFOkqzZWxu8c\nk7sRjSeoTLNtLUJI5HFIq22NTlgg2iiDwcgymMDBAAAAhITScqE0yTZjc43z/v/wvfTM2Iue\nznDjIf8bL9r/7T5sTpBmghBefg4sPye50+mv/TxxDipNH0Qi2GIzfPYupEt/+3dGThH2U2eX\n5hba15w5AgdIdaCOQLg1KoSRfjXwC9ecMpNBgCxioVcejliNIkB63oyR4A47Y3a2CNmarDcT\nZEoPAUQv9lRKmxESABxpmxRjeqwutb7WPBhTqFJTYCwwivs7Rva0DqunEzz2tA1vrLatq8hP\nxzTnCYzQnR9dtuOttnca+0ffts0sXbelZm2tht+nyCUSIKjEz0CbWF6Vv795MD6+IhtMPYNh\ntb3f6w8pVQ5TvjmTaj8ZDMacwQQOxjxBQwE61INMVmSZme126OB74+rGaZSOVveffpd3u4Zd\nqPbZ5TBCCPhpPdtBeqPpjp/IR/YoLUdowMuV1YobPoIklgbJSDWJzB0pgFYnjvSBkGETiDVU\n7gLiB2xGYg1wlqmPY6QJDvFW0SGTsEplBFjAIkYcAPAS71O8MpERAI8Fk2Ce5/KZ1KIQWaEK\nhzgeC2iykq6eierGGJR2IMQEjkzHohMurncc6XH3e0MhhVh0fJ3dVJlvaBvyvX1yKHpPldC3\nTw4VGqVKWy4/jTDphJsvXPLxs2t6RvxGnVBk1eEEXVGKLZLA4XgTEx3P200zWOp/8rzaY+0j\nwYldS+rLrZuXZfqfz7Pvtj/26glfUAEAhNCFa0s/u63elOtpPgwGI4vvbBjZAvU6w88/rBx8\nfbSOFpfUiFd+jqtqmObhgXdf14wH330dpiFwyIffDT73iNrbDgBcSbXuozcLy9ZP57zCio3C\nio3TnCSDkQySGXRmCHoiAaQXqVGHeAwGAooP+PlrvTE1fBHiWZ+dbELAogATstB5LFjFXHi+\nrRDZLbsUMqYDYoTNglXitJ1cKU3kD52UbzRj3tEL3PrK2M/t0V635s5Hely5LXCMYtTxi0um\nUJkFDm+qtr3ZMiH5AiE4q9Y2eafYGCodpn+76cw/vtJ8pG1YJdQg8RetK7v6rOoMt3R95p1T\nD/69MfKSUvry/q6+kcCPbl6f2RNnRIGZdywjGZjAwZguytBQ4MBeZWhQrKoxnLkB8WMfnnDH\nKe9rLys93XxJqen8C8XK6gmHyeHgA98l/Z2RAOlpDT5wt/72n+CKuumcl7g1iv8BQHWNAKWT\n9ygJvbYz8NTvxg/pOun7zb/qr7tD2nL5dE7NiCa09w3f839WOluxySzUrzZ9/FbOlulPbzId\nhHD9VnJwLEEJleSD1TD2gfY3kcP3oerLUf6y9M2PwchECCXO8DCJMq8hlLjCI3mSTcSaz6UT\nXSbYKieLcWnZZ04SX5jUOUxWvbC/Y2TQG0YICk3SGRV5M0rfGKW80HjXJ1YrKvEE5PyZHz7/\nqIQ+8XpLfPxw6/DRtuEVNVlQXMNgMJKGCRyMaTHy2B8G/ueXxD/m0CbWLCr94U90K1YN//HB\nod/cS+Wx+4mh/7234PY7bDfdGjlQOfBatLoxhqqEX35M99nvT+fUnE3b9JQrsE+ublC/N/js\nI/Hx4F8fFNdfMIclJ1PJLtmI57H/8f/jydGf1aBfGXwxtP8t23d/xVcuTu/Esh204hIU9tPG\nV8FmBOvEp45qkJ58Gq0oBondijEY4wRUP4mz5gUAv+wVJY2lF0JWSvu0RmJlVllMIoMJYWLc\nG1Q6Bn3+sFJk1ZcX5H5mRzxFZunSZcUpGYrncFaoGwDQO+z3+LWlruOdLiZwMBi5DRM4GFPj\nfv5vff/14+hIuLWl40u3Fv/L9wbv/Xl0nMry4P/8Qlq8xHj2eaMRte2Y5piJ4vHot1zkfe7J\n+Ljh3IsnP1BpPUZljd6WNBRQ2xr5+rXTnMA0kU8e9zz+23DzUVAUvmqxeftnpdWprnCRA9TT\nh3gdmOzzmbantLdE1I1REAAN+Dx/vDf/u7+at2nkJgjjM7ZD3bnkxB+ABGK3UpUOHkJl56dh\nYgxGphKpTIlBjmscc5oigM6oFiqjIISqUzktxvxSkW/odQfj49H1KbuO9r54sDusjMlhi4rM\n151VXcCcJhcAWp2FT2/SbDvMYDByCCZwMKZm+I8PxwdVt3vwgf/V3N/11JMRgSOhjSJRppnp\nINavsHziFveTDwMZf2QnLltt3n7zFEeGNG59RqGhuJXk7Aju3T3yi7tBHbPgkk8cHf7JNyw3\nfNF4xadScwI5SI49S0++NdZSVGfBK65EldMyE0kGVabHX6GdH1C/ExkLVC8GjIDE3hOEmw5R\nvw8ZMsknIl0E2qj7fQgPAuJALEZ5m0GcSbtlkx1Igo9raDglE2QwFjAYodWUtgD0n44YEVoM\nsBA6yOQsa8rzTvR7nBMLUvL0wpqysa7wbzX2P7tvQgJpS5/ndy83fePK5XzC9iILAmdA7veG\nwgqx6oUyqy7DrTSSoyTfYNTxo/aiMSwuY3/4DEaOwwQOxlRQGmpu0tyi9Gsk/VKAcHtb5CUu\nqgbYHb8bLq6efh2H+RO3SOs2+176q9J5issv0J1xlmHrpYCmuEHB9tKEmxwpbYhLiPuhn0fU\njQieP/9Of842bJ19JiQlex6i/cfHA0E32fsoJiqq3jTrweOQg+TVX0Tal9KQlwewrq92vd8W\nq3FQSvwejgkc7n3U+fbYz1SB4Cna24Hsl4O+erojIARYAKKRcAQce9jIYExAwEJI1RAEBTxJ\ncwQRoQaAJQB+ABFA246UkUVIPN6+rvy9tuGmfm9QVnUCV+cwbawuEHkMABTg1SO98UcNekIH\nT42codVUdSFAKN3X4WwZ8kaSGCw64axqW75BnPS47IPj0NVbah59+QQAjXbbqa/IW13L6lMY\njByHCRyMqUAIOB4UDRUc8Xx8nh8CwEZT5CV/5kfkN5+iAV+Mmxtf6KDH30FLNkyz1EJcskxc\nMjO3Ra58EVdVr546HhPnF63giitnNNTkyO0t6vBAfJzKcujIfv3ZH5nl+HSgeYK6cRpy7Hmu\namPK/T7I8Vci6kYEodCoK88Ltk8wfEWilAr5JstRvdS1Jy5K6MgupL9p+i6GyLqIjnyoscGy\naFbTYzByDh1n8Ct+QmM1ZSNv0tw/Cp75buQSOp47d7H93MV2WSUx1hu+oOLya0nGAF3D/gUr\ncBzucTcPeqMj7qD8esvg5ctKBC7X8ji2n1NLKd3xRmtIHvuu2Lys6PNXLEO5mLHCYDCiYQIH\nY2oMq9f63nsnPq5fudr3tkYPV+OmsyM/I3O+7uZ/De24hwyOrZkRAs4o4p7DpOcwer8CX/FV\nyJtJ70lK5fhm2gAAIABJREFUfK8+Hzq0j3jcfGWN6bJreUeJ9p4IGT/7L74Hf6R2NEdiXGWd\n4ebvzOB005mR35vEphkw3KYdD7rAPwzGVN+odR3SDItFlhiBQ7fpQiTk2mOfaUEJHToA3jaq\n+BDPA5UhPuFZ8YA8DMJ0/3dQ2VbqboWJz6WRZRHKX5qSKTMYOQNGOF+yecKu8OmkJw7xZtEi\n4AX5dcSIMxYFgEm6oOZkRUbHiP9Ql8sZkA0iV5FvWFueF98IllJoHtC4JwnISofTX1uQa8mY\nCMF15y3admbFyR63L6jUFJvLCnPtPTIYDE2YwMGYmsIv3uk/sDfSKmUUqXZx8b/+sONzN4dP\ntUbHxYqq/Otvjo7gyqX6f75HPXlYfflhcPdhgUOnr7t0sIM89yt8/b9H0hDkzvbQiUbEcdLS\n5bwj1vebeNwD3/9KuOno2Ov3d3v/9oTtjrsMWy/VnDm2Ocxf/5V86G2lvQkhxFXWCavOSnnK\nA+dIWAvD2R20aRd1dgHHo8JaVHnGlJU1GpDYB5XjxD3DnD007NOMY92E9G9hyQrz9V9M+dmz\nACVATj4GgbH6LAoACEAvISlucZXIVkMTXQFefjvtfIW6mkENgWhFjjNRUap9ahmMnIBDfJ5U\noFJVJQpGmJ+sOIWR3SiEHu5xdTmDAVk16/g6u2k6S3GDxNstugEtF9J8o/jkO6d6nX6J52qK\nTOcuK5b47Lbk2N0y+EGnc/TnET90OQPH+zzXrCnTCxMyZAOyGlY12g8BIFcwZ3vrWo3i2sWF\n6Z4Fg8GYV5jAwZga/eq1Fb95qO8/fhQ60QQAgJDl0iscX/s2ZyuseOixod/+j+eF51TnCJeX\nb774soLP3YHN5tghOJ6zl0JoCKTYjxwdOEV7m1HJEuL1DvziJ54X/jYaRxxnueq6wju+Hp0j\n4HzoV+Gmo9H1lDQcGv71j6UVazl7gi5oGAtrtghrtsz2t5AYrrBIWnFG6Mi+mLi0uIpvf44E\nXGNTbX4THX8Nn/t50M0wRzovgWOIoAdD6vNskcFG/c74OF+/3uQ4V+lsRQaT2LBWt3Fr7nXD\nnQ6097WIunE6BBAIAc/H5nHwM3QyE62o9hoEAEQGtmBjMKaCQxzHRS3hqIfSPqBBhAyAigEt\nxJ6gOUZQVv/R2OcetYqk4JfVfk+oyxk4Z9HUS9Zta0offeNkTNBmlp7Z26Ge9pNq6nHvOTH4\nhYvrs7e1So8rGFE3Ioz4w++2Dm2tc0QH43M6IvCTZLxkJ4pKOgd9wZBabjea9Ox6ymAsLJjA\nwZgWhnXra554Ru7tUQcHxOoabBqTMDizxfH1uxxfv4sEAlivn2QEOtKTcNtID5Qs6fvhXdEF\nL1RRXX/5E5Vlx7f+9XRE8b/5MsS5GtBw2L/7FfPVn07urQEAUKocP6B2toAo8dVLucq6mQ5g\n/eJ3R376bbntRCTCO4otZ5TDaXVj7Dwjp8j7j+FzPj+jwVFRA7KUUHfsLxAvPn8umsWiqvV0\nMPamEADwki3GkuUpP13WQZ1aThkUIKyAPiqJQ1cF3JSOAAlg6gaDMUOoehxI22hOFQUAaEZc\nHeDqtE6KMVsOdrvckUYYaOzq3zrsrykIlOdNdssBAGuqbQDwzN4Ot18GAASwsir/wy6XOtEt\n2+kL73j31OcumvF1P0OI8dSI0DLg2xr1nrwhxRWUTRLvDWlYqhWZcsp295UDXY+82OTyhQEA\nY7TtzPLPfKTOoGNLHgZjocD+2hkzQCguEYq1DS8mVzcAAMTEO4i68MnmWDsPBADgfnZnwW13\ncPk2ACBuJ03Q+VUZ0DBLnyZkuM//yH8qreOrVmHtuYbrv4Kkqd5RFJzNXvjjBwJvvRxuOgyq\nIlQv0dVV0T3x7XUR7T4KQffMkjgwh8/6HNn/GO1vikTQkgvQ0m0zGGTaoEVno6FW2vZeVAij\npRcipm4AgBoCNaS9iUal/op2VHDh/MyIwWAA6QbSGhOiaiNCFkAL3gg5m+kY0e7p3u6cWuAA\ngDXVtpWV+f2uoD+sFFn1RztGDp0aid+tucftCynGuAzTTGPYFx4JyAKH7CYpUn4SCGtXqgYV\nVaWUQ8gbUt5pHW4f8QMAh5EkxNbjlFv1jqxNYInnhb2d9z1zNPKSEPr39zq6B/0/uOnMBZl1\nmuXg7C4fY6SLTP82Z8wRqnMEG43z6RCJimpBMkDIH7uBF1BZQ+iNXdqHERJqPm5YvxkAsMkM\nHBffjRUAOEtektMixPe7H6pdExIW5ANvBATRcMPXZzYUxvpzLtafc/HoK9r0WnyLmdEt1DuI\nZlqlYsjHW75ER9rB1Q2CDtmqQZ/sW54ShPHGz9Cq9bTzIPiHwVSIqjciW9VcnS674ETAAhCt\ncmV9JZgKATgklYCxbvr9UxgMxiyhpDNRHHFM4MhigoqmZwRE+mJMCYdRSf6YFOLyaztNUAC3\nX85kgcMXVna3DHU6x+QeDqPVZda15XkAYJS0Ezn1AschpBD692O9kSwYldCgTEQej5akCBxe\n6jA3FMWVFWctlNI/vdYcHz94cuhI2/DKGvZtwGAsCDL325wxF1BZdj7xqPPxR9SRYcRxuhWr\nC7/8TWnpvDyZ5wV87g3kpd/GhPGm7aA3T2K9iU5vQqKkW7MhuC+unwvC+g3nJDcppeVwjLox\nSnjvq/qrb0PGWTQUFBI+XEJikpXhKL8S8lPZ4HaycxU3oOKG+TlXNkEpMtdQV1P8FmQ/FwwJ\nGvowGIy5JU46H4UmiDOyBKPIebRKKpITI8wJvBhQ4k2ZAKH0hQ/7R6K63qqE7u9wcgitKrMu\nsZsPdGjYZtU5zADQPOAdr/EZHY3QYFh1mMUL6hxGMddWAX3O4IhHO8uyscPJBA4GY4HAMn8W\nFn0/+PbQ/b9QR4YBgKpq4OD+zs/fGPhg7/ycHS0/D1/zHeSoBswBQqigHF/xVXTmFQCgW7ZS\n07ESCYK0dFnkZf5tX8VxyRrmaz4t1CZZPav2tmtvIETt034kOE1Q0VJtgwxTIVhm0haXkSGE\nXfTEDvLej2nvUYhLzkGOzUzdYDDSR6J1Wq6t3xYaiRqmJNfTdFm5lY9v6Q1QW2w2zYtBQ6cz\n8GrTwNOHul9s7Gvs89AEeZ4xdIwEotWNCIe6XZSCwyxtrI5dt0eCA17t1f6gN2wQcvCvg5CE\nv9NJNjEYjBwjB7/dchXae4IefQVGukFvQWXL0MqLgJvZA4fAB3u9u16OHVZRBu/974oHH0/d\nTCcDVa5E168EVQZKgR8vkBHKK83brvD8428x++d94jMRQ1MA4MuqSu573PXYA6FDe4nXzVfW\nmK+6Xr8++Q4piE9YpDPb+h1DHl5+GTk88R1hDp/xCVa8kH2EPeTwAxD2jL0MyCBg4EXQWZBU\nCPb1yFSdzukxGAscVAhUw2oRYfvoDzIJKCREgXBIFDkjYl/CWcKKEsugL9zlGnfiQAjWluUV\nGpO5QFsN4lXrK3buaSdR0oJZL2zfNB81mG+dHGrsG7uIDPmgYyTQPOC7ZFnRlB1Mhnwa6gYA\nhBTiDSlmHb++ylZpMxzucjkDsl7gqmyGZSUWjBAAJNJQKAUKNIv+EPafGHz9YHffSKDYZjhv\ndUmizq9FeXqjjvcFNbJ+aktmkZPLYDCyCiZwZAdk31/pvr9GXtKuY9C0m/vod0A/g+/rwL73\nNOOh48eIx43N8/jVryXNOL71r3xBofOJR6ksAwDW6fNvui3/0/8Usxu25ud/4Zupmgi/aAUg\nFH8LgIwWrrR6loOjZdtwXhk99g/q7AJOQIW1aOVHUV7ZLIdlzD+0a/e4ugEAQEFWQQ6gwk2o\n/Ly0TYvBYAAAAOJqKe0DOtGQElkAV1AgXnlAJuObAqrTJNh5lDuuijkMh9GFdfZTw/5OVyAg\nq2ZJqLMb8w3JP37YVGevspt2He3tGQlIAq4pMm9dXqwXU9+PLIaOkUBE3YjQ5wke6nKtqxjL\nS3UF5GF/WC9wBUZRiMo0mcQaM7KpyKwrWqrRCaXAKJ4Y0DjQZhRxllhuUkp/vuPwC+93RCLP\n72m/bGPlP1+zMv4dcBy66uzq/3sl1oajpti8bsnUrYUZDEZuwASOLIAOnqL7n4mNOnvJnifw\n+bdOfxwS0HYjH900rwKHFkiUCr7w1fwbbgmdbEY8L9YswoZkclBnBHaUSWdfFtr93NgcEMIG\ngRM44exLgCjAzfYPBJWuQKUrACjL2shqqLtVO+5qZQIHg5EBiIg/i6ongPQBhADpAJUhrhYA\n+5XBaHUDAAhVvHK/VSzPosfXC5wqm6HKlqR3VTwl+fpPbalJ1WjTpHXIpxk/OeRbV5HnDSm7\nTgy0nd5HL3BbFhXWn/b+dJi0xTiDyJmm8iJZ4jAd6nb54zqtrCmbM5/yVPPy/q5odWOU5/e0\nr6otuGBtafz+Hz+3VlHpU7tbw6cdatcuLvzyVcu5qTJlGAxGzsAEjiyAtu7TzDKkJ/fBebdM\npu1PRKys1oxjs4UryBRhG5st+tXr5vOM+u1fwPay4At/4nBIcpjwaOu15tfUng/weTehmpRM\nhl1WsxxV23tfu50Kg8GYCkKVkOpUaQgAOCRJXB5Gs7whERC3DLhlACTiL0aBhFSNhSWhqkz8\nIp5zDZ3BGMWfoO2LP6yqhD59sMsZGL+aBGT1pcY+DqPFdhMAlOXpiy26Xncw5tgzKvKnPK/I\n4UuXFe9uGew7bb0p8XhDla26IGWC0Vzz6oFuzfgrBzo1BQ6E0PUXLL5kfUVTpzMQVqscJlac\nwmAsNJjAkQ34XdpxJQRyAKbdksN0wcVDv71HdcY2gbdeuR1xc56fmblgTtp6tbhiHdnxfSBR\ntyC+EfKPe/8/e+cd2GZ17v/nOe/QlmXLeyfOcOJsCGQwCmFvKG3hdlB6S3cp9NLb0va293by\n67xtaXvpYJRVWkihaWnZBZIQSEJIyHC24z1lSdZ8xzm/P6zYsvTKU5Jl+3z+yvu860SypPd8\nz/N8H/Leb2Bxtpd6OLkGWgpZNPGDMxjP/mA4nJmORsNBtZNBbH1Vg4iiD9ikUpGkbD41EYZz\n+ynTINkTGAAAdMbVSU72sErGT1lWWTjaE4hXN4Z4q8kzKHAAwEWLi3ee6j/SPTD412yRhDOr\n8xcV28dza5dFumpZWV9QGXToKLKPqH/JfXq8xtnHR5p9jKVc4ytwmNYt4YbuHM4chQscMwFb\nikxCyTRKL9JkiN1Rds/Pu/7ny2rHsBzuuPTqgo9/dooDnFmwSEh541naehwQhOrF0vrLUTKx\nfc+PUDcGoTrb+0+8+NPTMUxOLlG6FrxHE4NIsGTtdIyGw5nRsJDWPaRuxEJAQ1q3U65Ob74b\npm4VN8ouDift1LqtR3sMfHDnuW1dSakZg3hCiqrTQTHCJJJz6txnVru8YVUSiMsiTbTgwm2T\n3ZNyZp12nCmG7Qspf9tx6ur12TCI5XA4MwsucMwAcN4ZbM/fDIww568df33KIOZlK6sfeSbw\nr+eVE8eJ02lZfZZ56bJUB+ue3vA7u7XeHqmyynrmOpRngyWb3nI09MC3md8zuKm+87ry2jPW\n2/4H+hIrPAdhvSn6yHLmEpi/CGovY80vDdekCCacdzk4Kqd1XBzOzEOjEcoMehxQpmk0KhID\no8RJQ1AUUDJM1pDSky3C4YyL6nxrfYkjwWe02GFaWZG37XjvOC9iloTSFJkgs5gNDSX7T3oM\nd/11exMXODgcTjJc4JgBoLuanHk93bl5RDC/gpz9vslczWRyXHr1mIf5nnys7ze/oKFY9bJU\nXll89/9YVs/w9Wqqhx/5wZC6EYt5e8KP/siSopYVyZx7mOAYgmXrsKCeeY9B1AumfCxYDNK4\n0oM5HE48DIzNCACAgYHwMUWsYsGA2pUQNAtOASfWZ53DmSIb57ur861Hugd8EdUmizUF1sXF\nDkQodhivHuVbs1RLEohqFlkQcrWpyjUbap945bjPqFduS0+QMYa5OnJOGuAP4ZxJwQWOmQGu\nvopULGX7XwRvB5jtWLkMGzZNvcdHKgKvPN/zv/fER9T21o7//Hz1I38RS8oydNMsoDcdon0d\nBvH2E3r95QSMOmWULsz4sDgzBZMLS86c7kFwcoTBlDr+YD1hEFI+sGIGnkkkYnHKZWGtX6VR\nACagZBbyTAJXJznTQFW+pSo/MXVoYbFjV3O/L8mGY23N2B6iU0HR6JtNnoOdflWnBLEq33JO\nXeFUWvBmCFkkZ9eXPL/bIM1WFglXNzgcTjJc4JgxYPF8vPATkz9fjeqnDjFPF+YXk+p6NI2W\nnet94uHkIA2HfH990n3b5yc/humG9nen2oUl9dCxHyIj/fZNVrLmyowPizM5aJj5doPSCYyC\nXITONSDmTfeYOHMA1sP0o8AGABDQgcIiQPeIA9RTTOsAFgFiR6kWBO5EOwKRmAmKyVUqBEWR\nZKQQUkSTQyoFYAwguTWszqIK9VGmEBBFYpcI1z44WUUkeO2K8n8d7Wn2hAYjZknYMN+9qNiR\nuZvqlP1lb1tPINZahTJ2yhPq8Ldes6JM05jOoNAu2+RcmSOsWug2FDhWLeDfrhwOx4Bc+fLi\nZBT96J7oX37FvLHpPToLTFd/UmhYl+p45cQx4/ixIxkZX7ZAa8pWYeguF67/On31IdbeGIuU\nLcLzbwEH//nMSZQu1v0M0OjwZvAQFl4GlvnTOizObIeeYvqh0xsMmI9pO1FYBqQSAIDpLLwN\n9J7Yfr2PqadAXoSm5dMy2FwFrWJxfBcVAEAgVrE4wxkxBku9Ub0vog87ICjUJxG7VSznuTmc\nbOI0S9csL+8PKZ6QYhaFIodJznBxSmPXwJC6MYSi0T/vbtN0CgCIsKLCde6CwlxouXLBqvIt\n25sONXvjgxZZvPWyxdM1JA6Hk8twgWNWwaIR7dRh5u8nxRVi1YLBxznadSryh++APrxcxvye\nyOM/sHzyHlK1yPA6KBkXJ6Occ7mLE0KYvwwtNhYOJsTRWSBULQRBJNd/FYL9zN+DjkKwF0zL\nIDnjgfW9OKxuxEI663sJK6qAl9ZzMoXKdAORl+mNSMoABKYcHlY3hlCOgFgCQnE2BjhDEInF\nIVdFda/OogAgoMkk5BPMdq21xsLx6sYgKg1E9X6TMBu+/ynTdaYLKGT/teVMgnyrnLUKkVZv\nyHjHaWWPMdjb6g0p2lXLy7MzpFEQCN5z29mPvHh0yxunIopOCK6qc3/6mqU1JRlMcuFwODMX\nLnDMHpSdL4eevI/6+wc3xdrFtg99UaiYp27fEq9uxKC6uvUZ081fMryUecWa4OsvJ8ctK89I\n65CzDZrM5us/Hf7jT4DGdSgkguXGzw0bmtjy0ZbZwlfOVFH7QDXyVKcRiLSCZV7WB8SZG9B+\nMDbI1ID5AAtAS9GMSW1FLnCMhKBoEac5P07VfYZxhfpmusChMzWgetXTKrBETDbJJaIEAP6Q\nKkvEPPeacXDi0WhiY75BEjKXjnYHegPRQvv0N9GzmMTbrlzy8SuW9PrCLrtJEqc/r4TD4eQs\nXOCYJaj73wzcP8IWVGs6PPCzL+d983e0o8nwFNpxItXVCj72qdDO7Swyoje7XF3rvOqGNIx1\nWpHWvIeUVEWfe1RvPYZISPUi86UfIqXV0z0uzkTQQ8wfBs8AAIDLhi5b3K7E9BwOJ32kbP8B\nTAMEYBHDnRrt1vUOk1A8ir8mZwjK1Ijeq7MQYzpBk0kokEhG1mlpiqYtzKiL7QyCMt2r9DA2\nrOOrNOoJ97xzlL74TkcgogFAVaHthnU1iyu4b9EcJd8inwSDn0sGicJHlz+SCwLHIIhQ5OIN\nnjkczhhwgWOWEH7uieQgHfBGtz6bssdS6t5LpoX1lfc+0POT70YO7gcAQHRcfIX7s19Eszk9\nw51WhIo668e+Md2j4EwWNUJffood2TscKS/ANfNh0A5NsKU6j8MxgEUB5fEaLqB1jF1oAqYm\nX42BqNJ+nYVs4nxu7jA6OosG1eYhew6dRUJauyy4LEJJ2u+VSm/CGV7QEdYH4tWNQZ7e3vvO\n8eEJbUtv8BfPHrrt4kUra2d2rsp4iKr6u83eHl/EaZUWlTuLnLPhSWaKLC1z7m3z6kl5HFQ3\nzuzgcDicGQQXOGYJevNRw7jWfFSurqfNjcm7SE39KBc01TdU/uYx3duv9XZLFVXEkvrJnjPj\nCHYx70nQgmAtRnc9kJlkWkFf+C07sjM+wto9oOu4cQkQC5grp2tgnJkEU5nWCForgAZAQChF\naSngWAuD6AR0AvMnxfMB7QAAYgUoI006GACCTpwAQFlUof0ymf3zyakQ0brjzUcHUXSvTPIE\nTPO8VCJ2lSa9mwAzvZGKSpWESIdHiVc3BqGUPbm9aUVtweyW3BrbfI+9ftIXir0mAsGLVpRd\nvqYyB//XjBkZ4WYGl0W6bGnpy4e7w+pwYppOWbziwQAQoITrQRwOZ6bBBY7ZQqpfRSTSOddo\ne15mwRGPcWiySuffOOZVBVe+4JqSIQUN+lE2ozRe3ywWCfufeSJ6aB+LROS6xc7rbxYK51bt\nOvO2Q08TMB0Ka7GgKt1Xp+z431nHTmB08NmFmfNJ/Y3grEnzjTJEwMOOvJkcZl0+8Cuk7mru\nMMoZBxqLbgUWOL1JQW9ntA9N58EYU2hEYRXT3447FwCdKKyM/VOuZ1o30CGrfwaIqpCnCbEJ\ns85CAFzgSAkDqjFj70ONBgVhUhMt5mGsCVgAQADMRzIfIHYdiTgkYldpIP5wgrJJcBtdaAaR\nuAh/ojOxX8YgfQNRz0DU7ciVAoS04wlEf//SUUUblsx0yp57p91lkzcszpVHC52yt1v697f5\nfBHVJovzC20b6gotEzdJUXW643jfiZ5AMKoX2OQza/MXjOrBOc9t+9BZNSd6A76wajOJnf7I\n/rYRrjQIUFdkz536FA6HwxknXOCYJYi19erhdwzi85ag022+7fvKX3+tn9g/GCTV9aZrP0UK\nSjM4IEpDLz8T3PIo7e8FQqR5ix03f0ZaNEavRLX1VNdXPqP1dA1uhve8NfD3p4q++j3LWedk\ncKi5gxqlbzzCju8YCmDNarLxI2BK23Iia3mVtccEgpgkFumnBx4lZ94B0gxI0mG9LcnP7oMg\nrgRLbXaHw5mZaKdGKBSDsCjTjqI0Vj9XtKK4EWgHY34AQMwDUjpcdYIS2i4A5bimHkemUiJr\ngkuL949IKhzgxJNcWDG8axQDlNGueJLRuK7nLMT0LiRnAMZahlvFCoX6FN1HmUJQFIndJBQg\nzGz/QhFlDdT4iKqlrDtQtEm9sDOEN4/0xqsbQ7x+sDtHBA7K2OZ3Wlv7w4Obgai2r813ojd4\n89pqu2kCj+jBqPaH7U39wViiijeknOgJnFlbcMmy0Z70TCJZUhr7LDSUOa2SsLu5f7BuBREa\nyvLOX1Q0mf8Vh8PhTCtc4JglmK/4oHr0XaAjnlRIQbFp42UAQIorzR//LvP3UU8XcRWhK+O/\nWP4Hfhx+9e+xDUrV44c837/Ddfu3Tas3jHJW70++NaRuxE4Nh3p++M3Kh/5KrLPfW4Fue4id\nHFl8cWoP1RRyyR1pugMbUjdGoIZYzz4sX5emu2QSIfVXljSzs8o5WYPRxM6gMfQeGFcCEAIp\nR0jVOpGAvFAhos7CBvuQr4WOBkEBgSSXqAAAGd97M5Iwo8eTghpjjYhnDW3LJE8ms8pr0yza\nI3ooXgsuzjP+5pRE4nbM5gKELp/BxxAAOn1hlht2OI2dA0PqxhCBqLbjZN9F9RPwnXmlsXtI\n3RhiV5OnvsxZ7R7X6gVB3FhXuLoqv2cgojFWbDc7zHyOwJluZrgjEme6mNnLFJwhpEUrHZ/+\nb1I4LNVLS9Y47/gBmod/2NDpFmqXZkHd0JqPD6sbQ+j6wKP3Aku5jqR1d0YP7E2OU78vvHtH\ncny2EexnJ3clh1nbAeZpTc8ttAgoSQvXg4S603OLDIPF80AwmucgwfJFWR8OZ2aSskdG2pay\nJWOjDZQIb0E9OmjYMAWBTKaRCus1TvhiPoDEqeBsQkTJKbtJ3MSgvtKe7zAoFN2wuFie1e02\nUzUTlQSSC+oGADT1GXf+ShVPxZHOAcP44U4Dl5lRsMpCjdtWV2jn6gaHw5m58O+v2YO07GzX\nkjP0jlPU5xFKKklh2XSNRDn4tmFc727XezuFIuOB6b0p59j6yLSOWQnztqUqvoD+VihIh3cm\nEQdtN4x2jdckZZoxWXHt1WzH5oQwrroE7NzaIM0w0BiLAAgEzbmx2JkmiB2oxyCOaUsCkkge\nAzWq9wx93BBEs1BGcKofNAYqZQqCRFCaVW/KacxiMdVUjQ47cSAQi1iGOOHHFTayTGPkPgWm\n/F7kMjIx55tKVRqlTCMoSsT02ctcv3/xSEdcssCZCwpvWDdD3Jcmy4JS51tHDTK2FpY5sz8Y\nQ6JGFTQAEFUnUM5GGYuoxvpsMDqzex5zOBzOJOACx+xCEIXKOqGybnpHwaKRlLsiI1Ix6YA/\nevwIMCrPX0RSu5kKrnymaVpPt1hYhNIsdZFM3bV3tF0Tu4UEzirwNxvscs1Pzy0yD1l3A7M4\n6PYnIRoEAJDM5OzrcM0V0z2uWQUDXaVtOu0b3ESQJKFcwFkiIaFQwzQDMxcUa9N4F5kUipin\nswBlGkFZJI4pOjswpkRpu85i67QETTKpEHD21O4xpqisn7GoREQR8yljDKiAskzyJqFuAACC\nOYVmjGO5yc4GEFAmw//N8gLrV29cua/J094fNolkQZmztnj2l/WdWefeeqiruXdENoRJEq48\no2K6hpRAnsX4kcZlncCjDkG0mURDLcOZ4vocDoczi+ECB2dKsIBPPbKXenqIu0RacgaaLQAg\nlBn3/kBRGkrfYJrm/cNvvE88xBQFAFAUnTfcLNfWKU2JJdMom/0vvdj+X3czVUVBsJ17QdGd\n/ymV5crTSbpAdy0IIuhJDyiIWJw2xYrMu4TuewDYiKUezF+IBQvTdYvMg7jyEmH5JvB2Mkox\nv2zSGuxcAAAgAElEQVQ0Y46ZCfP10f4ukleIrqLstQ2MQ9GPUzY8JWCgKvopWYBZonEQF8qr\nmLof2NAKP0FpMQhpznojKBFMT00KAz2sn4hPSaAsGtFPWoT5BGeAPfCYaNQTpe0x1YkBAAjo\ntAjVU8pSwSIAESD5S7Vobj78CARXz3evnu5hZBOB4Gcuq//nnrZtjd2qThFhYanzhnU1Zflj\n9YTOFg1lzn1t3uTi3WXlE/OFWVru3HkyMTENEZZO8DocDoczC5iLv/GcdBHd8ULoz79moZit\nA8krsP7bF+QV602r1gkFxbonseTEfM6lgwoIAPT96sf+v/xxaBfTNN+fHrauO5d0d9JQ3MwK\nUY9C9IV/xjZ1PfCvF8P79tQ8/GexaAL+WzMA2YLLL2fvbEkIY/0FYEvfrDKvlqz6BD3+d/C3\nADAQTFixAavPm3m57kSAgoqZNmgAAOY5Dt5m0KLgLMeShgQDLdrdOqLhUeVC03WfJuVZzcnS\nmT9e3RhC1TsEcVYIHAAgVCIpAr2TsSCiBYRiyO1UCI16jAoumEK7zUJt9seTXiiLDKsbp9GZ\nX6HdMpnK97yEZBmj+0doHGhHsmQK1+TMMCyycP3Z1deeVeUNKnazlGueIyVO8wWLil872qPR\n4b//5RV5KypcE7rOeYuL273htrgSJER4T31xad7sT1bicDicBLjAwZkkauOe4EM/jI9Qnyfw\nm2/n3f1LoWKe687ven/5Lb2zZWiv6YxzHR/83OC/da/H/8yfkq8ZenNrxb0PBV78W/TQuywS\nlhfUM9HqfeqPCYfpnj7PQ78rvutr6f4/TTNk9dXMbKdvPwNKCABAMpEVV+KyS9N8G0cFWfUJ\n0BXQImByzDxpY+aiRdjex1nPoaEAs5eQVR8Ce2wKx/x9kd98hYWGveJo69HIb79m/txPiDtV\nz470Y6huAAADhYGKk2lmkZOgCcSamfLXr7OQYZymiM8sNOY19AbSaP/UBA4ALEJhA6OtAAMA\nImI+YDn/0puDEMQCe472MFpZ6ap12xq7BvqDisMszi+0l01clTCJ5CMb5u1r8R7vCQSjmttu\nOqMmv4SrGxwOZ07CBQ7OJIm88heDqK5F/vWM7YN3iDUL3d+7P7rzVa3lBJqt0uLl8uKVQ0dF\njzQCpZDcpY0xrbvDffvdQ4HWT3/U8O6hXW+l4f+QGtZyELqOA9WhqAbnrcrWAzHikguFxeeD\nv4tRiq5SIBn7hAoyCLPZYy8HYQefjlc3AAACXfTtB8k5/zH4RqvbtsSrG7GzomH1X0+Z3vv5\nrI1zVFJ2QeJME7PhHWHMuKcJAxUMficmignJNPtScTijk2eRzq6dan4cIqysdq2sNk79YAw6\nvKG+gWieVS7Pt4oCl/k4HM6shQscnEmitzeNHkdRMq+/CNYbHTRYb2r088roiOd1Go0a3mUU\nH9OpEgnQf9zLTr07FMCyBeSK28HhztQdEyACuMr5o8dsQw2zjncM4qE+1ncUi5YAAG1uNDyV\nthjHJw8LAvUC6IB5QBIrtAlaDGeVCOLsSd+YaRA068yg3SPBXLESmBIoGAo1CALPtuBwpk6b\nJ/THrSeHzFbdDtP7N9TWV3B7Dg6HMzvJrVpEzgwCUzk7jsPx0bSwHtD4b8+0eGn8plxr3N1D\nnpepFTn6/H3x6gYAsI5j9O8/mx0rpZxpI9wHLEXbv0DMrYbRFAfoE+gXOBaUqftY5BWmvM2U\nvSz6GlPeAjZCRhQwDw2mzUwkJXy2OV1IpADBoJuSRIqyP5i0I6Jxz04BHVkeydxEo0xN55dM\nrqDq9HC7f/uRngOt3rBi3EV1LuALKff+ozG+lUzfQPQ3Lxw51ROYxlFxOBxO5uAZHJxJIi5Y\nrne1JselhcvHPFcocDuuvG7gb5sT4vZNl0kVIzqwuN57k//ZLUATH01c7//gBMc7Pgb62Im3\nk8Os8zjrPI6lCzJyU85cgKQuCDpdK0TKamnLYYNTy+elaxRM3Q/aqREhvYuxXWjaGBdCk1Cn\n6M2UDdXLEImUiKQ4XcPgTBQEySzURmkbZZHTEVEWymaHBCCgQ8Q8jfnigwiiLJRO+dpMpX7K\nooiCgFZhdiS8pI9Of+TdDr8vogKATRaXljpq8mdDUx4AONLh3/xWizcUq36ymsSr11SsnnIZ\nyEzk9YPdoaQOsjplL+zr+PimGdRAjcPhcMYLFzg4k8Ry2U3K26+z8IgVAJJfaL7w+vGcXnj7\nl4nZ4n/6CaZpAACEOK+8wf3Z/0g4zNywvPS/v9fzw+/qA7H0bGKxFH7+P2zrNkIGYJ72lPv6\n2oALHJxJYysCcx5ERkzhGAAiojv2dyVtuFp7+xXQRvoRCIJ07rg+U2PDVNCaDeLUA7QfyHBD\nUwTZJCygLMQgAiAQtPLilGmHoNUiLNBZiDEFUSRoNczpmKGYhGqBelTmoSyCKInokEgxTu0R\nRWfhiN5G4ww+JJJnFrjJaIyTntDulv6hzaCi7WzuD0S1hlLjhJoZRI8/8ofXTsT3JQlFtT/v\naHZapLqS2aAJTojmPmPf6OYe4ziHk0OQ2fMzx8kmXODgTBJSWOb80k9DT/xSPRxzFpBXbrC+\n/9NoG9ezEUqy+7N3uW66JXq0ESiVFywWi40X65yXXWVbtzH4+r/U9laxtMy24TyxKGMryVLq\nZXYpRw3YOTMDRKy/hr3z8IgYAFZvAFusyoAUV5k/8rXoX37F+rtiBzgLTNd8klSmaZGNDaSs\ntKL+eIEjNh60AmRnOZcCCwNaeNXkWKCAthzvaDtBht1eRFIgQtoW2BnQsNbCYMTCtUp9CKJJ\nmF0txicFZezddl9yvLF7YL7bZpFm9qRi+5FejSZ+11HGXm/snoMCB0A6vHo5HA5n5sAFDs7k\nEcpqHHf8gIUC1NNNCkvRPOG5kOAusrrHriEXXPnOq9O0iD0qWDwPZAso4cQdRMCK+iwMYE7A\ndPDvYeEm0IMgutCxHKzGTiuzDCxdDmd9kjX+HQbagDEw52HdJqw8O/4YYcEq6x336qcOMU8n\n5heTmiUoT6HPH9MheozpHgBAoQCkwtSDm6aHXxZh6kHQ22MP4EIJSg2AsyRJnpMKBqpO2yjz\nMtAQTAIpEjDNDi8a9SeoG4OotN8kFPPZnjesKka+G4xBTyBaPcMLVTp9ST/iAADQ4c2YPXkO\nU+22HW4zELOqi2aTVMrhcDjDcIFj7sI87aynBU0WLJkHlsmvaaDVLljtSVen4e2vKEcPMlWR\n5y+2nHcpSrmU4h7ysu4m0KLorob8suG4KJONH6CvPJhwOJ55NdiMW69NEdraSA9uZ74ezCsm\nDRtJxaJM3CWHoFHW+WdQemPrSWo/C58Ex3J0b5rukWUDLKjDDbeDrgJVQUoxhZBkYcFKgJXG\ne8eP7mMDrwCNFZExOA6CHWQZwMhsj0xHaTpTWHQrsKGpCAO9k1EPms4HnIKsw8ltGCiqfoiB\nenozqtFWij6JLEqj7kBZig5cQCnTCObS79F0kJzgMISeetc4iag6IprEaUvIElIotiLJXWEr\noumNnQP9IUUUSJnTXFdkT9dYz11avO1wog2HQPDiFeUJRzb1BI60+4NRrchpXjOvwGricwQO\nhzMj4V9ec5KgV3/h9/ToztimZBLWXU/WXZuuh0vd0+u558vKsUOxuwEMbP5DwX9+X6rJVOuT\nCcAofXMze+dZ0GM/9li3lpz/0SGJB1deTGwuuvWP4O0EALAXkPU3YsN5mRiK9tz9+q5/Dm3r\nO58VzrpSvPiWmbe6yCgLHIdIDxAJrZVgKUt5oHcHKL2Q8D8ceBesC8BSk/Fx5giCBELG51cs\nsHVI3YihB0DPM/BtEKoAkzTKzMO0k3HqxlBUYdpRlMb2KubMUHTaNqRuDEHZgM48AqatGzem\naNQ1+q65gyP13NVhnvy304EO/xsnegciGgDkW+VzFhQuKJrwd4ui08auge5AVNWpwywtKrIX\n2ydWIlpTZDvWNZAcr83VnIWW/vBLh7uiWiyn5mCHf3+7/7KGErOYhlqhPKv8ucvrE9rEvm99\nbU3cq6FT9uc3mnYd7xuKPL+3/QMbahuqMrK0w+FwOBmFCxxzD0a1p37Auk4MR9So/vofAQk5\n+5q03KH/F98ZUjcG0TpaPT/6WvH/PpKyuWy2oG8+xXZviY+w4ztpyEdu+NrQpBsXrBUWrIVI\nABgFS6bs1vT9W+PVjVjwrb+TysVkyfoM3TQjRHtp6xaI9gxuMQB0LsGKywGN3uvQUcNrsNAx\nnDsCRyoYoyffpZ1NIJlIdT0pmcILonlA7zeIKz60r2G0BWLOiwTEBShNk5E+7ZtYnDMroMyf\nKp5GgUNAG0BPcpygeTaZs04aiySUO83t/sSSDZdFcltTe1GNyvbjvW82eYY2+0PKln3tFy4u\nXlk5gUlyQNH+daw3osYSzYKK3umPLClxTMj6dMOiop3H+/zhETqaSRIuaJh6X570E9Xoi4e7\nFG1ExVDXQGTb8b5Ni9PjOFZRYP3i1Q3t/aFefzTfLpfnW0VhxCrDS+92xKsbABCKao+8duLL\n1y1z2Sb598DhcDjTBRc45hysad8IdeM0+lt/JWuvnLpfsd7TGd37VnJca2tWDu41LT9jitcf\nHfXAW5F/PkbbT4Iki7VLzNfcKsS32NQUtve55LNYxxHWfhjLR7psmDO7pk3ffc0wrr/76kwS\nOJhGWzaD4h0R8x8CwYRllxgcrxuXRoM+1+3cmadTeep/aeuRoYiw+kL5qk+COKnV1ITcjRHY\n0Hwx0CAABWKH6ZzsGRXLAABLEefMCliq993IMmPSCGgViVOjCWIKmrnD6GnOrMrfccrTHRiu\n5XFZpHW1BZMz5AlEtZ2nDETVrcd7G8ryEqbTo7C3zTekbgzR2DVQ6bLkjTu1xGYSP3XRoi1v\ntx467T1RU2i79syqQkcumoWf7AsmqBuDnOgNnreASkJ6Eo4QoaLAWlFgXBr55tHe5KCq090n\n+jYtT5mSyeFwOLkJFzjmHKzTQN0AAIgEmK8b86f6S6Z1tqXc1dGSUYEj8vwfI397KLahKurB\nnerhPbZPfFNacuZgjPV3JPbgHKKnCcqzaiPKfAariwDAvMbx3IQFTiaoG7G4dz+WXAAk6XlU\nsIFmtH4rzEln+yF0PfrY91lPy4jYnpdVSZau/MRkLoip19xQBiBAcuAFRweAwR9PxsbGFOrT\naJAyjaAsCy4RLcBU0L0AAIIL5rwvQ3ZANLHk0iQAhDQbr1iECgUtit47KKkIaDUJJQJa0nuX\nmYsskvPqCrsGop6QQhlzWaRyp2XSdsPt3jBlBuYdika7BiIVrnG97DpjXQMG5ikMoN0XGb/A\nAQAFdvmW8+aHFL1vIOqySVOpu8k0gxU9yVDGAlE935rxiipVo76Q8aNRT1KOD4fD4eQ+XOCY\ne4xWfpwG6we0pqxxJclepOmD9vdE/vFoYlTXwn/6pfSN+2NNIkZ7dsu27QVa7IZObmiZBjeE\nyRNNUU3ANFC9YEpqkWNbDL6dyYejbRrcVZmqRA/t17o7xOIyU/0ylKctEVc/sTdB3RhEe/sl\n6aIPg2niUzKxCFA+XYcSB8ogjt23KDugWMv01uTOtSjOMzx+UiiMeQEUBuawNqCdfkF0FlGp\nz6r1i9GmWMIICmBuQMsy3qo20whYqLHkv3YUMHWXn0mCMnHLxM1AQxBmnrdRVihxmErSkdeg\nprYmVY3ataQ60lAlAYCoNpnELqssWN253hFmFDdWs5SNryNRIAJBQ3NZ8wxvGMzhcOYmXOCY\nc2BZCqdPqxNdo1R7jreLuly7kLgKqNeTEEdJMi1bM85BTgLtyDtDvqHx0L5OvbtVKKkCAMwv\nB9kKSsjg/NIFmRubIWThmbT1sEF80dosj2RKkNTfIUbr4Zh3Fou2QaR9RDTvLDBXpHtkYxB+\n+82+n39P64glHIml5e7bv2o5Y11aLs6CfhaNkILxlk+zbgN1AwBAU6mng5RNvI0uCmg7iwW2\njZQPEG1rjb1RpgXiQnk1U/fHCTEiSkuBpEeCYayNsmaA2ORKJoRRh85if5bmaLOodscdrUN4\nH2NRtM6oD+AMRMBihhGdxaeqEZFUY8ZyK5A/6mSegtTOHQXjNnGQU8+0LdK43kTGwB9RB6Ka\n0yw6czhrI56qfOuOk33J/+ciu8mSFX0BERaWORuNWskuLs/LwgA4HA4nvfBf/TkH1izDynrW\n2pgQFzbcaJDcEfDQHX9mLfshEgBXKVl+MTa8Z9QcEABBcH38i54ffT0h7LjpNuKacCtKFgmH\nXt6injyCoiQtWGp9z+WQwqaUhVOaOLDQaTd1QSRnXE3feCLhAKxdhSXZ7vAirL1cb9zBOo7H\nB0nFIuGMS7M8kqmA1mrj5TYpD2QjYzkiYen7IHCQhU6CHgTJhY7lYEpsVpdplJPHur9xJ1OH\nExy0zvbub36x7BcPyfOm5Lip7N0e3vxbvbsNANDmtFz+b+YLrh3b2ia10QZOzoMDAORadDpY\neC9ofQAMxEK0rAQxbSaO6UGoQFIMtBtYENACpBgwPUXyjPVS1hQfIUjNgj+suygTCFNk1agW\nLHIEzMuA8CqGjIIiqSGskDIvgIpoJliAwI0MZzalTnOJ09yVVNEwv9A2fqGBIFbmWU71Jy5C\nEMTKcRS5dA9Et53s8wRjX+zFDtPGee7xyytphwEcbfe3ekIEobrQNr/EuPgu3yqtrHS90zqi\nXk8keE5d2nOaUnLlmsqT3YHoSPeTpZV59ZVc4OBwODMPLnDMQVC8/i79lYfp/tdiq7tmm3DO\nB8jqJEvI/nb9qW9D9LRw4Gmjrz6IbQfJpZ8b/QaWDRcWfe8+/yO/Vo4eBErFmjrnBz5mXnvu\nRAeqHjvY/+Ov6f2nva9e+Vvw2ScKvvIjocjACJ24U7ijIxL3sLEIrrmCCCJ96y+xPA4k2HAB\n2fCBiY4tDUgm+ZZv6zu26IfeYP1dmF8iNGwUzroqlYKTo5iL0bWMefcnhLHkgtTnINgb0N6Q\n0XGNjn/zY/HqxiBMVfxPPVp4139P+rLRN54L/uHHwxcM+kNP/p/e1WL7ty+MfiKpWWoYR7sL\n3VPIbRHd6Lhw8qdnB5RASH/+DmPtyUEEJmJUYVZBDyaXxgyeB3ofkMq0j4eTAEEbwRzt2cmZ\nBIiwvMLlDffET5LzrfIlSybWuGRFeZ4vonrjGqAQxNWVeTZ5DJnYG1b/cbBTi8v+6B6I/v1g\n5w0rywXEZm84GNVMklDmMOVPtk3MhPCFlEdeO3Gye9jvub4i79/OmWc1atB7dm1BicO0p9Xr\nCSmyQMqclrNq87OZgVKWb7nzqqV/29VytMMf1ajLJp9TX3zukhJe1sWZZqbc+oAzN5lRUylO\nujDbhcs/Tc69CXqawWTFwiqQDdzd6PYnhtWN07Bjb7El72L18tHvINcvL/zOr4BSxujkWsMy\nVe3/328OqxsAAKC1nfL+6jvub96bfLxYv4a4Cqm3N6GWRmo4izjz4wKIKy8Vlr6HedpAjWBh\nFZinz21RkISNNwgbb5i2AaQDLLsMTEWsd0esQ4q5CEsuRFtaer4y1ryHdTVCNADOUlK3EWzp\nSUBQjidmMMXix4zj44Lqoc2/Sw5Htz5rvvAGobRqlFNJaa2w8nx976sJcenijwDhlhCTgYFR\nJRqAgGO16khhAcDhcEbBE1JOeIIVBdZQVFM0nQGaJcEiCx0DkfnuCShZJpFsWlh8whPsGYhG\ndZpnFusK7Q4jUSCBfW0+Lam2RdHoGyc8AVUb2nWoC+YX2FZVujI6dWcAf3j1+KmeEU9QjW2+\nJ7Y33XqBcT1srdtWO5EXKu0UOkwfvWABA1A1Kqe2BeFwOJzchwsccxe054M9P+VuxljLu8Z7\nmveNKXDEIAQn69inHNyj93YaxA/t1bs7hOLEbi8oybZ//3rwd9+mvmHbS6FqoeUmo8VzyYQl\nE/c1yChaGCIekGxgMqrsyGWQoHstuteCFgAiAUlTHz4tQl//Des+3Ta1ba9+5BVyxvtxXjp6\n6KZaE5iCmqB3nGIBgxpmYEw7snd0gQMA5Gs/qxVWqls3QzQMAJhfIl36UWHJ2ZMez5yHGLah\nHZzl6EJq30FxwpV0HA6nxRvrjGM1ifFJCs394QkJHACACHVuW90Ez+oJGLRfAYA2f9gWNx7G\n4Hhf0G4WFxZm0M+7tTeYoG4McqDF2x9U8qevamaQqKrvON7X7g1RBuUuy7q6QsvpBBkE4OoG\nh8OZ6XCBg5MCXTX07ASA5LSOjNy/pyPVLq27PVngAAChZrHj679Vdjyvt51AURLmN8hnvGfU\nzim5gTLATj3Pek/LSdZiMu8KcNZO55DGgikR7eAuvaeNOPLFxatJfhEAgJjO50X67t+G1Q0A\nAARdpbv+KBQtAPtUTShN9cuUo4eM4uNT7oxgSooOxACxchimA6ZOthRE8bz3iudcz/q7QDah\nY25Ms5nKtCOgdwKLAFpRrAJxflr6mCA6GTNo8aMzGQAomlTRLWlJB5jmA+F1ExzOhAkpxl1O\nQupYOVNpIlXqlWH8ZF8oowJHly9ld9Uub3h6BY4Ob/jRN5qC0dj7cqxrYOfJvpvPrq3K+XYz\nnMyxe/fub33rWzt37vR6vdXV1TfffPNdd91lsw3/Gvp8vrvvvnvz5s1er3fp0qVf//rXb7hh\nRPbxmAdwONmECxycFIgyWPMgZLAijXklWbg/saWsHCF2Z6pdaLKYzr82MyPKDFSlBx+CcFwl\nTqibHnyYNNwCjurpG9ZoaIf3hB77Ke2PeTSiJJsu+6D54ven9SaMNb1lEKY6O7ULGy6f4tXz\n3vuh4Mv/oMFAfJBYbXnv+8ikrymUVAIRgCY95SOKxSI9cB9EeoGIaK/Gyk1gSdFghRB0G4h3\nsxOmsOjrwE7XkrAAUw+B3oWm9VPXOBCrGfMmJHEwkBnkE9QFlIltA0QOQfT46awOBNMCtJ4x\nxftyOHMTSTD+zKaKpx23XfZF1OS4QAwWOQLRzMouUuokiFF2ZQHGYPPuluDI/35Y0Z/a3Xz7\nRYuJ0WvFmfXs27dv48aNtbW13/ve94qLi1999dVvfetbb7755rPPPjt4AKX0yiuv3Ldv33e/\n+926urrf//73N9544+bNm6+77rpxHsDhZBkucHAMCO95Szl8gHZLUnDAUmIfYWohiLhoQxbG\nIC87A2UTUxKTToXCEqkm2x1PMgfr2TtC3YhFddbyCi69ZUKX0ltPKPvfYt5eUlQun3EecWXE\ngJ16uoO//Z/494WpSnjLA8RVKK9Nn5+lGjXu5gvAgp6pP4KJZRWlP7iv7+ffix4+MBgxLVrq\n/sJXxbLJu12i1W46e1P0jecT4rYLV5Ho3tiGpjDfMeY/SRZ9EBxpsSmZwTDt6LC6MQT1gNYC\n4lRfHAQrIcsZPclgUKVFxCIBa0US59tnWweWZaB5AADEAiAZXNHlcGY3JQ5Tmy+cHC91GDh8\nZYLl5XlNfSE6MmGDIJgkA0FBEjI7k59XbDfsd2uShOppNdro9IV7BqLJ/3lfSG32hGoLef7a\nXOSxxx6LRqN/+tOfVqxYAQBXXHFFR0fHww8/3NPTU1RUBACbN2/etm3bgw8+eMsttwDApZde\numbNmrvuumtIvxjzAA4ny3CBgzMC3eft/f5Xw3uGF8/lfIv7zErRIgEAyBZywcfAOdUCgfFA\nHHmOD37a/8D/jogKQt7H7xqjT+3MYqDFMMwGWhAS/FJTw1joL7+LvPQUUDoYCG95yPqBz5jW\np7/jrPLGP5NVJwSIvvp0TOBglPUehoFOEM2YXwuOSeUjCCIQEajRIpspPU9g8oLFZT97QO1o\n0zrbxdJyqaxi6n9X1g98jilRZfewV6jcsFSqdQw37Bh8P5lOm/9JGj45xdvNePQuwzCj3Qhp\nUH8QbEiWAegAUQCzcVYIsYPMdQ0OZ6pU5llavOEEIwyrLCwqytLnq9AmX1xfvO1EXyCqDn7V\n5lmkhUW2o70GRbUlGZZdnBbpPQ2lL72bWGl7+aqK6c3g8EfUVE8V/rBB/gtnLiBJEgAUFAwX\nxhYUFCCi2Rz7mDz99NNms/mmm24a3BQE4cMf/vCXvvSlffv2DWoiYx7A4WQZLnBwRtD7o/+O\nVzcAQOkPew4MlPzb1eiuwPpzwZq9pui2S98rVdUNPPWA1nQEBFFetMzx/tvE6hwzBx1C1wPP\nPxPdu1P3e6Wq+farbpSq5o19FjOuWwZGx3/n6PbnIi/8ecTZ0XDwkZ8KFXVitbFh+6TRO06N\nFg900b2PwUDsqY4hwYozcen1E2j0FR1gR/7BOt4BAcDoNcCK9P1YIpHKq6TyMew/J3A9k9n+\n8a9pF92onTjAFEWsqhPdGmt90eDQcDeoAyBNXwefnMBAwGIAyNL7nC0A8NpyDiezIML6moIT\nnmBzfygQ1S0SKXWaFxfZs1aiAgCVLsuNqyp6AtFAVHOYpSK7jIDeiJYgu5hE0lCastA1XVy2\nuqLAbnpub5s/pAJAgd10xZqKVbXTbK5kk1M+9ttMvB/nHOWWW275+c9//tnPfvaHP/xhUVHR\na6+9dv/993/uc59zOGKPKAcOHFi4cKHJNGwhv3z5cgDYv3//oH4x5gEcTpbhAgdnGK27I/zW\n1uR4tL1Trdpoql+W/SHJS1e5l/4s+/edKNTv7fmv25UThwc3o+++HXz+addtd9qvuHGMM62l\nAPuN4iXjTd8AiG571mhMNLrtH2L158d5kXGCkrE7GooSUI3ufgDCnuEoo6z1LRDNWH/VuK6u\nhuiOeyHcDwBoFpmuw8gUX6w7BwtzVeE6jVi7WKxdPPhv1v5qyjwcLTLXBQ60AjPIBgLkegSH\nM/OYXPeTeKIa3d/h7w1GKYMCq7SszDnKhNwQgWCpc0R2xjnz3Ue6Ayf6gmFVlwRS5jQvL3Na\npIxP5hHg7IWFZy8s9IdVgmg358Tzdnm+xWmRkpM1rLI4vbUznGlkwYIF27dvv+GGGxYvjnyO\nLl0AACAASURBVD263HnnnT/+8Y+HDujr65s/f8Sj12C6R19f3zgPSMWtt976zDPPpNobjRq3\nRuJwxiQnvnA5OYLa3JR618lpEThmCt4H7x1SNwZhmub97U/NK88SK0bzCsXi1ax9G2iJpctY\nsXH8d9e72wzjtLt1/BcZJ0LdMtj9r+S4uHAF6z44Qt04DWvZgYsuAzL2tw07tXVQ3QAAIATt\nZhbVQNMARMyvwkXvwao1Uxp99jEVGKsbKExHP2AGuh9oAIgNhLzxK2gZAoUqRvsN4mKO2uty\nOJzM0RtUXjzSrWixzL2eQPR4b/C8usJKl2UqlxUQl5Q4lpQ4dMoMDUczjdMijX1QtiCI16yu\n/OObTZo+vHhACF69uiKbuTacnOLEiRPXXHONy+V6/PHHi4qKtm7des899wSDwfvuu2/0E3Gs\nNoVjHnDXXXddccUVqfZ+8IMfHP10DicVXODgDIPmlFWpo+ziAKWh1w3KEJimhba95Hz/raOd\nK9nIkg/R489AqDsWEUxYvQndE5CT0GRlwQGDuDn9K+Hy2Rcrr/9N72gacSPZbL7iIxA4anyO\nrkC4H2xjW7ewvmMjthHRLAFI4CwnG+6Y7JDHQo3ELD8yALoWMdEKWqKVJrqXAcnuU6/ex0I7\nQT8tKAgutKwFMSNOtONFrAbmAy2+6ImgVA9kbrTI5XA4cWw72TekbgyiUbb9ZN8NKyvEdAgT\n06Ju5A6BsOoZiJYVWOuK7Z+5cNGrh7vb+kOMQbnLct7i4kKHaexLcGYpd999d09Pz65du/Lz\n8wFg06ZNJpPp7rvv/uhHP7p+/XoAcLvdHs+I5avBzSHbjjEPSEVDQ0NDQ0OqvR/5yEcmUODM\n4cTBBQ7OMKaFS4nVRkOJjlwoiublk1w5Z5oWfOW56LHDKIimJQ22cy6EsQTdGQcNDrCIgXs8\nAOg9xjaKI7BXkBWfYv5TEO4F2Y6OapAmlikqLVkT3fYPg/jSMyd0nfGAkmy//f+Ftzyg7Hhh\nsCWqWLfMcuOnhfJadqIp5WnC+CbzeorWfZoy8ZGOBWPs+Da6/58Q6AUkWFiLa96LRelu0COY\nyIL30eObQR0AABpWaPcAI3mCq0ZkLHufBTrAAi8Di3t5dS8Lvoz2y0DIeC16ahClFSBUMr0L\nWBjQimIlILf8HB1GmYJAEHNoWZjDmSL9YdVnZHIZ0WinPzLFJI45zrE236+3HDzU7AUAQvDi\nMypvvWTRdWsqp3tcnFxh7969dXV1g+rGIGvXrgWAAwcODAocDQ0NTz75ZCQSGbId3bdvHwAs\nWxZbihvzAA4ny3CBgzMMmkz5H/tc373/LyGe94FbhXz3JC6oNjd1fuOL6qmTQxHT0uWl3/6p\nUDCZq+UsaLGhJDPVYBJOXONbi0aCefMgbxympEZYrvyQ+u6b1D9CPhdrFmeiiwoAoM1pvekL\n1vd9Vu/tIM4CtMTkGCyYn9gTbxBrAZjHVY6B9iLmNyirQXvJpEebCrrrCXb4X7ENRlnPCfb8\nj8n5n8LKdBti2avJss+w3r2RV59VdrzDNA0A4JW3xNp664f+gxRn4ymTRQ+PUDdiUZ1FD6H1\n7CwMYDRIAfKUjXHAgCp6j0I9g015CMomoVTkehBnVhBWUvhtA4TVlLs4Y3Ks3X/Xb95UTr+G\nlLLndrYcbvH+7DMbprefCyd3KC8v37lzZ29vb2FhLKlz27ZtAFBVFbNgv/766x999NHHH3/8\n1ltvBQBd1x9++OG6urohA9ExD+BwsgwXODgjcFz9PqGgsP/+e9XWUwAgFpe6PvxJ+8UGDpFM\nU7XODrGoGE0pqlco7frmXfHqBgBED77b/f3/KvvhrzIw9mkDRdG8dmN4+ytJO4hl/flZGADJ\nL3J+9Zfhv/xe2fcGCweJM9+04TLzZTeDmMkPuCAKJSP7j7iqsXQF69yXcCAuHp/DKABWrWft\newx2VK+fzAhHwdfJjryaGGSU7vqTULk8/eYUgqwcaotu3REf05oaA7/6uuOr96Gc+dxgLYXR\nlz6GAVia0EH3AyAITuNerWPBQNWon4GCIIvoQDR2up3dhLUWnQ2n11GmhLUWi1ApkrltVcuZ\nFYzi+pkFQ9BZzCMvHFGSFKKmzoEX3267/Ky0dRDjzGhuv/3266+//oILLrjjjjsKCwu3bt36\ns5/9bNWqVZs2bRo84Prrr9+wYcPtt9/u8/nmz59///3379+/f/PmzUNXGPMADifLcIGDk4h1\n4wXWjRfQwADTdSHPYOFd7+vx/PZnwddeAF0HJJa1Gwo+9UUpyUozsv8dpel48unhXW9one1i\naXlGRp9GBnroO39jvU3AKBbWkJVXQV5pqmNd/36HcuSg3juiIMX5vo/I8xdnfqAAACTPbfvo\nf9oAWCSM5mnL5sUVN4GjlJ18FbQoAIC9BOuvxsJF4z0/vxaX3cgat8ROBwBBxsVXTOAK44N1\nNgIzSjcJ9MJALzjGtguZ4P1Y9OWnksPU06XueU0++2KDU6gOqgKmmZ6YzSB8gEX2x/JHUEbL\nCjAvnpCEpNI+hXYONQ1WgMikRCLT6h6SdTQWiFc3TsOitJsLHJxZQL5VyjNLvkhilYpZJAld\nUTgTYn+TgZEzAOxv8nCBgzPIdddd9+KLL95zzz1f+9rX/H5/dXX1nXfe+ZWvfEU8vUhGCPn7\n3/9+9913f//73/f5fEuWLHnyySevu+66oSuMeQCHk2W4wMExhtiNH5ppwN9xx8e07o7YNqPh\nt7Z2NL5b/stHxOKy+CPV1uZUF1dbT+W4wMHa9tOXfgV67GGL+Tr1k7vJBZ/A6tWGx4vFpaW/\nfHzgzw9F3nmL+r1i9TzHtTebV52VxSHHmEZ1AwCAiFh3Ec7fBGEPiGaQJ9x2DivPwqJ61n0I\nwh6w5GNR/TjLWyaGlrL3GFMjaTfGYKEB6u013KW3nUiI0I6T6nMP0uZDoGuYVyhuuFY867Kp\n+mwJBaAbNLgBIbO1ISy4E6JH4rYVFtoFLIqWleO8gs6CCm0fGaMK7SBoEnAOTex1muhTOwhl\nUQY6Al/i5sx4Ns53v3i4W9GHfUYFghvmudPiMDrTYQwOdvoPdvi8IdVuEhcW21dX5Y/HNlXV\nqWE8wc+VM8fZtGnTUL6GIS6X69e//vWvf/3rSR/A4WQTLnBwJob/mSeG1Y3TUL/P9/gD7i98\nNT5IrFYAAGawWEusuV03TnW69aEhdeN0UKPbHhYqlqXyyyRWW94tn8m75TPZGGGOgwjWKdis\nmJxYlWFjCGcKUw8ioCMDqQGjyBMjd9GmA9E//M+Q2Srz9ar/+D1tOyK/986p3B/Ni5l6ElhC\norKApiVTuewY0BBEjRrrhA+CeQmMr8xEo0a6DIBKPYIwhwSOQd+NFHvYdDf85XDSQJ5ZWl2Z\nd6IvGFKpgFDqNC8rddpN/DEVGIO/7ms70RvL4QpEtU5/5HDXwPvOqJLH6u1aU2w/1u5PjteW\nzKnvTw6HM7fgDkOciRHZ93aK+O6EiHnlGSjJyU/eQp5LXlSfibENQf39LBSY9Oms9ySEvAY7\nIgOs84hBnDOj0JsOhp9/JnywK9zYpbR6WVx9MtaeBVL6U2DQYhPKagx3ifNHNEhT//lAcisZ\nfd/rtKVxSiMgTrRdMKJhCnGg/XwQ8qZ02dHRelJMy/WUniBJUDDOtWEp4pOCAesDegpoGzDj\nRIlph6CZAVGpRdFtKrVSFpv1IYiIfAbImfH0BpWXjnY3dgcUnYkEEVGnzCTy1CQAgMYu/5C6\nMUT3QHTXKWP9N55rN9YmBy0m8dIzeRcVDocza+EPRpyJwZQU842kuJDvzr/lk57f/SIh7v7c\nl1DMTINDSiOvbgn99WHq7wcAoaza9v5PyivWTfg6kYHJ7OLMBJRX/xLZ8vuhTRpRtf6QeUEh\nschYvoys/UCG7mu+8pbg776VEBRr66Vlw7kqLBygHYkVK7FxHt9LqqYmC4pF6Lgc9H7QAyDY\nQcjPvMCdOulgtF0jwJSDHOfgKYMwgI5gATD62mEBpu8DNrTCiUBqUJiYS0gWUKkY1fJirxoD\nDUwCRmUhJGe4yCh30KiqM11AQSAi5ti7w5kiqk53NvcrI4speoPKux2+NZUZKFGcaRzrMV6w\nOdYd2DB/jJTDTasrPP7ooy8fG7IaLcm33PneFYV5ozmb6DoTBP4p43A4MxUucHAmhlwzP3ow\nsU0GAEi1dclB1wc/JlXX9D94n9J0HAmRFy0p+PjnLavXZmhsgcd/GXn56aFNvaPZ/7OvOf79\ny6YNl0zsQtb8lLtsc2U6MSuh/d2RZx9KCDKdKn1o/egdWJrBxCJpxXrbbd8IP/V/1NMNAIAo\nr7vEcu2/A4mbqCuRVKezaDgdoyAguEHIVpNmIfXMZJRdIyFoMzLXBGEc7VEZ66asCSBWa4ZY\nQrB25K+ezvTdwOJfWwa0iaGIZME4R5gFNKoG1MQkc52ZGDPLc8BsVaOqX/VpNPY+Cig6ZKdM\nMt97iJMtOvxRxcgqos0XWVnOxuM0MbtJ1UM3NL4Guu87f/57VpXvPd7XPxCtKLSduahQTtGY\nhjH24tttT29v6ugLmWVh+Tz3LZcsKndbJz90DofDmQ64wMGZGI6r3jfw/BbQ9XhvDQbgvPYm\nw+Nt526ynbuJqQogwUx2LdW72yKvPJMcD/7pPtO6TRPyaER3DeSVgq8zcYfdjcUGOg5nGDXC\nPK2ghLGgEmypdaJpQmvcnVwAAgB6bxdYizN9d2n5eqnhbNrbQYM+obQGLYkOrGjPB5MVogZV\nElg4A9OJBRdI5aC2J8bleUDG+8QskUKdeSlT4oMI0phdVBjrpizOAYQBgy7KwoQsHw7SrpHq\nxmn0U0DqcieJI6Iby1sam/0J/DrT+5U+FtfzSGeaL9qfb3KLJDOZgJysE1QMvpYBgDIWVnVu\nw+EwG78CTtN4PwJFeeaL1lSMedgvnj7w0p62wX8HI9qOQ117jvV+72NnLahwjn5i1jjVFWhs\n9UYUvbbEvmJeAWKufEtzOJycYq7/bHAmirxgcdFXvuu59x7dF3OpIBZr/m1fsJy5fpSzUEph\nKBjoZCdfZgPtQATMq8Z5mybdMkM9vM+w8Scd8GqtJ8XqiazHIpLzP06f/9mIghSTjZx/21Sb\nWcw+9DBEvSA5QLKz/S/SXU+DMjg/R1y4jqy/Gcw5ZCjLAr5Uu2jQLzgzn55DCCmuIJDiQVMQ\nxNUXajv+lhBGq1NoGO3zlbOg/RwW3AFKXEMl03y0TqC7EIJgFuoU2qVTLwMKQESSJ5NSHOvH\ni7KRXZwQAICBn7F+xJj0xlgqpx4VWBQwV5pT6onusKfjdFzrtzOasBZkSV/sDFhQC+TJOSeh\nciaHlNopc5Rdc4f6Emdjp0F5bH1ZOo1CG1u8Q+rGEFFV//0/G7//79PQEi4BVaP3Pdv4yt5h\nxXxBufPO65eVFvAEk1kNf+rmTAoucMxSGNMb36KtR4DqpKRWWH4OCGl7r23nXWRZc1Z453a1\nvUUsKrWs3SDkTybpnXW8zQ49BSyWmMqC3axrL1l1K7jmTeZqqdP79befFXxLsHol5I13lR7d\nNcJ7v8MOvsR6TgBjUFhLGi4CUw7N1acfxcfaX2C+066rHsaOHo/bzdjR7XSgl1z9FciZNRbi\nSrHsj0jyslW4MSrSxR9mAx79wPahCOYVyjfeieYJNtwNdjLPQYj6wJSPhQ1gKUrzQMcJymg/\nD/R+0DwACKJ7EramCKKJVACpYKCNqWucRoGU7qQDCEMT49RzJ8yhaVUqy4m5sHqpUtUwrqWI\nc2YixXbTAaN4nlkyiZn6JA5E1MYOvyeguKzSolJnvm1cfZ2mhXmFtjOq83c398cHFxTbV1W6\nPEGlZyBilcSSPLM8tddqzzHjXuaHmvtDUc063Xk0D75wJF7dAIBj7f7vPbH3J584W+QqGIfD\nGQkXOGYhLOBV/vj/aMvhoQi+/pTppv/Eoqp03YLYnbYLLpvSJbQIO/zXIXUjhq7Sg0+RDf8x\nieRwMUWXCkAQTr1F23aB8DhZex2ecc14ryhbcdXVuT6B0MKgeEF2gZj+3h+joUfosT+AOrSm\nxNjJpqSDkHUeZW0HsbIhadf0IDacjWYriyTWgIhL1qI1N3rmiZL8/rvoqYO06QALB0hxtbBs\nI8gTSyVgzS+y9u3D0mHb61j1Hqw4NwPDHR9CPghpWGwft7oBqU1MWfwuJPnMoPAfAG0AOTTb\nkQWTYZWKieRKjskwzMfYMWB+AAboRKwD5HkWnDFwmsX5btuJvhFuOwRxRXmmKiPePuV57t0O\nRYt9/l840HnBkpKNC6dJCB4H5y0sWlBkP9jp7w+pTrO4oMheZDc98Wbzkc6YO49FFi5uKFtd\nM/mPWyhiXCjEGISnW+CIKPpL7yRVOwK09QbfOd535qLcfeM4HM60wAWOWYj6l1/EqxsAwHrb\nok/80PyZn+ZOrhfzHAPdaIk13AeBLrCXTvSCUv1KobxWb29KiMv5Vhxc1tA1uuNJkleKC6Y/\n2TINRHpZ8z+Z/+TgFjrnYfXlYM5SGgLr3R2nbgBENNBSZMt3n4CcETjQ6rDc9MXwYz+Kz/ch\nRZXm935mGkeVDKlZSmqWTu5c1ruftW0dGdJZ80tgK0NXDhlnZhgZQBqyF40DEeLysNANWAgs\ncd0SyaLMjm6CmAVLhIQVOuILk6Bgk3Isp4x1MLo/brOfsV1IlgKOXfyfColIKlWS49yAY5ax\nvMyZb5GO9gYCUU0g6LaalpY6HJmZVLd4Qlv2tAHAkJWYTtmLBzrddlN9WUxS8YWUzv6wRRbL\nCiw5UiZT7rKUu2IrGTpl//fK0d6B4e+EsKL/dU+rQHBF1STrfFPVelhNoss2zZ6+Xf1hVTNU\no6GlJ8gFDg6HkwAXOGYbzNerH9tjEO9ppacOkXnLsj8kY1SDzgiDMCUwmbwJIjg//62B//uO\ndurIUExyWaxVI1Li2f6XMipwKIf2hre/qPd2CYUllo0XyfUrM3MbP218ELThRV3mP8kaHyRL\nbwM5K2ZgwdYRm7me6DKMuGyd7cv/p2zdQttPoskizG+Q11+RxgKuDMIUoCEgNsBRp3Y9Bh9/\nAIDuPTCHBA4kWEFZU2IUrIgjnFZQXM30Y0BPAVAAALSisAQw5x6XXaaCkBYMayGdaQQFk2Cy\niQ6SwToaylgXgwAAQ7AjlgCMKY5TRg8nRxk9gkLJpB82LKItrIcSbDgQ0CbmmLjDmTKVLkul\ny0IZZLpryu4mz+A/Eu6z66SnvswZiKhPbj+182jP4N+cwyJdf3bNusW59Z1wsM0Xr24M8erh\n7kkLHOcsK33kxaOhaGIex4Wry6e9ZayUuvpmlF0cDmfOMhMe6zkTgXk6Uu2ife05JHCYUpbi\no3nCVfqDCMUVrv/6lbJvh9ZyHA5vFTSvaE/MM2f9BlmO6YEx3/0/Cb0w3Kc29MLT1ktuyLv1\njrSbULDO7fHqRgwtxLp2YNUEe+JOkpFrKbIEomCcxFFUm5XxTACSV2i+8tbpHsVE0P0svAu0\nrtimVImWNUCMXTlYpD9F3DNzZKg0gFhBgFHWMvS3iugiuCBpUiOgsBiEhcBCAGLuGIsmYxVt\nVnGCViyTJKzTgwCxLCcGPcDaCFkyIvklGeYzSpkBAA2YF3CS7WwFFPJld3KbWJ7BMWl6g0qz\nNxxUNJFgnlma77aZc2mKmIWesH0BY4Oe3kCEMvbLZxtP9QzbDw+E1T/86xggrMulNIF2r3Fn\nJU8gGlF1c4ousKOTZ5O/9P6VP33qXX9oOGfqzEVFH7l4+jPaygosBQ6Tx0jTaZhCVQ6Hw5mt\ncIFj1iGn9GJAU3ZtGkYF8+uY7AAlyRjcWQnWKTxGIMor18sr11PoZqfeMThAzFSmZXjbC/Hq\nxiCh5zfL9SssGzal914s0Joi3pylSay5GAZODm8iYHk+a05K9S+ej5X/n703jZOjus7/z7m3\nqqv32feRNItmtEsgxCYQArMZsBEYbLAxYBPiOLZjG8dOPiROnPgfx/4nXpMQxxuxsWO8YMCY\nxSzGWGxCCIQWtKORNJtmn967a7nn92K2nu6q1izdPT0z9X1Fn+qquqhrquo+95zzFIymNk8R\nYQo/C8k+qVoHGQPouwbQ7GLm5lN0lHI0dSfQT5DRB5QA9KHcBMwPpIHeThQGkJGXA5+biQFi\nPccqohCADuhByKAOMEAZIAQQAPBCpm8ufIQ4Mq5ujKEKcZizjRmTtcwL+AHAQviYKhKTS5Vy\nXWgGGRy5xGRNxENaj0EaApOZ0yUVY4Z+sTZJvDMQOTU2N9YFxcOJ/qh6dm1RjopBChOHRcmJ\ng/P9J4eT1Y1xHt/Vfn5rxYLXiDe2lH/3Mxc//1bXqZ6wzy2vayzd2DJDaTK7IOLtly//zqOp\nvWgvWVfTWF0Y/bNsbGwKiUX0SFsksOpG9BRRJM0Ok0usoZCmmlzGNbfQvp+BnvQmrfjZ6vdn\n5fBYv9pE4CDAJbnqBxF78RmL+NNZFzjAwjYShHmRatbB8o008AaIpClNTRECUFcA9NHJDDZt\nYhd9uHAsVOYpFD8wSd0YQcQocQSd69K/j8XLKWKWplTckoPBqRR/BcTw2OcB0k+i1EBGF9Do\nUhtph4HXoHPTFGoccoGcUpNiBhG1AZxKykuqQFxRUH1G8wZBlMDUPTdOFETMkF6nkKX+kQUf\nR4nJEsgAENOHY8bQ+KkMQ1VFxCdX88ylWzYAEdVoT1v5NwQd7Q9vrJthXcN8pLHCe9xMxWiq\n9J7oNXFjBYDBUCIYVYvchXJPGG/GkUKpV5lZ+sY4Xpd8/YUWLdtniiFof9tgR3+k2OtYvbSk\nxDeTdaZL1tW4FOnHzx49PRgFAJci3bh52bZsD9XGxmZhYAscCw7O5WvuUh/6VkpYvvQD6BtN\n5NM7T2jHDwEJuXGFtKQ570McBUub8cK/pvZXINQJyKFoKS7ZDDw7LxC45jI4+CcYnGzq7vaz\nTduycvx0jIEe83ifZdHQjEFXJcV6TeLuqqyfyxxHCTZ+gNqfBHVscsvdeN57maeZ+k+CFsfS\nevBP1ZTXJhNGn3lcN7kAAABrN9PgAYhNzqbx1GD1udkeGZB6IEndGI2R3pb6PaOb1APoMJFj\nCgGiNoATk2N9RCpi5oSFBQqZZ+8DgJXz7sgmEnstzGy9gFlrDGSQGjOGU34XQUZUH/TJ+br7\njZ6U2HxTbweiqqm9UCCuawbJc91nIW+c11S2p30opYeFzylvaa14zsyqYwSysmaaC1bXFW0/\n0pvehmPrioJ77B7rDH7nkX2nekcVJaeD33rZ8hsvapjBoc5trTi3tWI4rMZVvarEtRh8sm1s\nbGaGLXAsQPi6LYq/THv2p6LrHSDByuuly27hqy8EAIpHg//7zfirz41/2XnuVt9dn2cekxy/\n6Ouvxve9JaJRZXmr9/KrUc7B2oXDi8256RkhK/zGL4rXHqLDL4MWBy5h40a2+VbwnnE5d4Yw\nr/naJvNnf2UMq86noQOpJrvIsCp/BjHobcAVH6PISUgMguxH71LgLgDAuhnaf2QHI0oDuyB+\nGoCBqwZLNwGf4/bvs8XcyBRS26CMIznZuj+n9hdo8AAkAqAUY/l6rL8YctGzwLAolUoXBvRT\n4FhbSHqBIOomGAbSEQfNvhAAGAawru6mOIggAAErKuTOHdMGJUuPXesXBhJHAUybAijI1mfx\nd1dFqsfzCJqIERDm/gLTBXUE4wNRVRckMyz3OOp8Tp6HvhHZQBeWP60uhMwLxWQt1zgkdteW\n5j8e7NnbPpTQhczZqlr/FWuqPYq0pMK8PK3I7Sic9A0A4Axv39z4+J7Oo6dHU05cDn7FmuoZ\ndxjNEcGo+qUHdoVjE0VqcdX48dOH/W758rNnaK5U7HUszvQ6GxubqWMLHAsTtmy1cvdXwTCA\nBEgTE5vgD/8tvvOF5G/GX/8TaWrx576aHBSR8Ol//EL0tZfHI4M//l71l7+utK7M8cCnBhGQ\nOLPlrdPDtt4JW++AyDC4/Lm2yHVuukg9tMckfs7F2T+ZpxabbqJTT4E2lmcre3HpNeCuyf65\nMsAk9DWDb86SgFKJtov2R8AYK3oKH6eh3Wzp+8GZ16XdLMOLQJiVDDDrF1muYMPV2HA1kIDc\nGW2QDmTedsFktkcaUKJghADNEPtGJuQIBJYT+pCFwCFIOwj6iTGNCUFahvLqOarByTIIXgAH\nQLozK89Un0Lm+USAVdltaEKWeh8QGYi5faXRBe3vDSXG7Co1Qd2hxHBcX1Pp5fNhMdmqmShD\ndBRSn9E84HLwazfUXruhNpLQ3Q5p/Ndb31BaU+LqHkpV664+u67QfmG/S/7QBQ0D4UR/KOFy\n8OoiVwH+iH/Y3ZWsbozz6MsnZixw2NjY2JwRW+BY0HCe/M5t9PekqBsjJN56Ve86KdVOlDL2\nffNfk9UNANA6TnXf+5llP38MlblcDzfajyae/LFx6jDpOq9tVK78oLT6jDkLCJ58NNl2X/W+\n2Gt/0o5OaoLlaF3rvurGXJwOS1aiv5FCJyExDEox+hqyVd0zXyFDdD4+oW6MoEdF5+Os+c/m\naExZAJUVpHWmhRkqU+ipkUMbUQAcub1YtIMxoVAeN4JOWKQbpGA+oSFtH+inkgOgnyDS0LEx\nG6Obc5Bhs6BDKboPwyZrBcewvgxm1V40HYbmY0BAq01ZpCsUH1c3xolpRk84UesrEPEuExUe\nxzsDmJ7HUeF1zAuBJhd4JndXlRh+8tpVP99+/ED7aPGdIvPrzqm/dG31XIzuzJR5lTJv4WYp\nnuwx72nS3hcxDJpz91mbeQArONnOZl5QKG+cNnlA7zhuuan9nXGBQ4TD4T/83uQ7Pd2RHS96\nt16Rq/GdCX3/q9EHvgZi9E3aaD8avf/LyjV3KJd/YK6GlAw6lLIv/Wf0qYdirzxn9J3mlTWu\nzZe7330zSjlrfccVLJ57/7YCgaIdoJm9SyUGIN4zj5M4pCp0X0CxN4HU0eIP5kLXuoVJ5wAA\nIABJREFUucDnPBUZQaoB3bxKJRVeBjleXZ86RAMT/52pC65ZwgIlQG83iRudQCsAF4L9CmIp\nww1Ep4hCAIToQ1yCkMGqgAPIFlpGlqf9DuaJwnB60o2De/JQABWMm6csBeJ67XxwcpA5W13l\nO9ATStY4/E6ppTyjAXDBE4jrHcFYVDM4ot8pLS1yWfmkTIVSr/Kpa1d1DkS7hqIeRVpa4fU6\nC+XeNe+wqhpjzJ632tjY5BD7rr2IQG75cydv0ro7SDd/jdNOncj6qKaKELFH/mdc3Rgn8czP\nHedegf5cddaYFijJnvd+0PPeD871QBYluvlKEQCQFsL5K3AAgKMR5VrQe0BEgXlAqobCMIxA\nx1oyBoEmt0VgPhApvwVHR+FYOOkp6QYEHE0SEMoAzFpjioBlSYsIAF8IAgcAIHgQV01DMcAq\noHSpCxGzvO7NUPJIZRF9IPlX4Ohw83yk6RkWfSYN694WhUaZ23H+0pKuYDyiGhLDYpdc6VXm\n9TJ6RzB+YmjiFhTVjP6Iuq7K73HMKqOnrsxdV5YF959FzmA4bhpvqS2yW4Ta2NjkDlvgWERI\nTStQkklPW2djTG6eaAzJ3Jbv6Bk25Rrj9AkKDJht0PWje+RzLsv7iGwKDOu5JUrz/z0VFZCX\nzvUg0kAnut5F2lEweoFUYD6Um4BXgHaC9GMgIgAceAU61gArnAVuCYAlt2glkgARYVzVRYA6\nxILpLDMfQNZCIggUTI4hWw6Y/dQAhXslpiSMkEEaApOZU+He/PSvdUo8nlaiAgBOeT4tRjs4\nayiZ/7dEAACI6+LkcGrfWV3QscHIhuqseffYzAwh6EhHwHTT8lr717GxsckhtsCxiGAev/va\nWyKP/Swl7r7qJlZSPv5Rrq2Xa+u1rrTlOMZcmy7I9SAtiZs3zwcAikdmckA1QsEekBT0V+e6\n/6jNzNECNPgqJXoAEJUqLL0AZPNOh+iuJ+4CI623guwHV357ry4qUELHKoBVk4JyI8qNAEZh\n9t1ELKfJTTGJOAFjrBVBBvACWCfIsCIANE/iyND2deEjITsPqJNoAEAF8CAuAcyVqsVRdktz\nkLVX6XEMx00qcSo9hdsEYWEzFFNJmKhboYSuGUKeRaGKzewJRrWoRVXXvEl5srGxmZ/YAsfi\nwnvTXcxXHH7kfykaAQB0uj3bbvdcc8ukLyGWf+ZvT//dZ8mYlLZdfNMHHcsa8znaZFhZDSCa\nOtGz8trpHUuLiT2P0vFXRo/m9LENN2JD/gxWbaYIRdqo+7Fxqw5K9FHoENZuQ3eDybeZjLXv\npo7HgEavWwJAlLD2mvy5k4oQ6H1AKnA/SDWF5Io6JxSiugEADJcZFErpM8qwAaHyzDujAlID\n6G2pcb4EcIGsis8UBKxHrJ/rYeSQEpdc73d2huLjDyKGsKTI5VfsV6m5QTPI6i6rCZIL9A5k\nSSCqng7EZc5qil2u2ZXYzIxwQt95crA7EFd1UeJxnFVX1FA287xdp4MjIpm9trnttiY2Nja5\nxL7FLDKQua++2X3FDXp3OwjBa5eatsD0XLS17r4f99/3zcTBfaTrct2Sktv/zH/tDfkf7zhY\nVCat2KgfeiMlzspqpOXrp3MkEi9+j/qOTQTiIfHaA4wENs5dfopNOmRQz9OpRqSkU8/T2HA3\nmDkmoK8Fmz9Kfa9S/DQAMlcNVmy2yvjI+nApvgcSRyeWprgfXRcUQCtQm3QcnJ1F1EUwDKQB\nuhFrLZtoGn2knwQRAVRQqh5xhCWQQH9nrM6FgdSA8irz3W0WFnV+Z6lbHoppCZ2cEit1yUrh\neXMuHqz+8RFgNn1G809MNR7f3fHm8cGR54dDYpevqb5kdVU+NfK+cOJ3+7o1Y7QIqycYfzoY\nP6u++PyGGWZLOR18zbKS/ScG0zed01Ix84Ha2NjYnAlb4FiUcEmqH83FIF2Pv/GKdqqNeX3K\n2rPlJaNx59oN9d/9CWkaaeoctt5IxnXLZ6P3/zP1nGASCl0IzWAlVa477wXr5qnp0OlDk9SN\nMcT+J3jj+Yt+yb2AoHgXGGbFR3oY4t3gslgodpRi3XVz8CsmDkLiyKSIEaToi+i9pnDcQ2yS\nYIj1CPWZ/+JJ3QfaOxMfjdOgt6NzM8orQWoCCgIQYBHg4nZoXmS4JO7ycQDQSRiCDBI8p37M\nOSChC0HklOa9OWypy8FZNL3Ja6nbIbF58/9GAD97qe2dJEdVVRdP7ekyiN61Jn/2tC8fHxhX\nN8bZ0zHcWuktcc/wFnfXNSv+7kc74+qkdODLzqpdtdSW/m1sbHKI/ea9qFGPHx78+j9q7SdG\nPzPmvfamkj//3Lh/F8oyygXh1wAA0Nfm9AqgMbXFXcKu+yusbZreQQZOmMejQxAdBnc+WvHb\nTAnDuuuKES2wV1eixFGTsIiB1g6OOSvsspkVRm+yujEWHCD1CDpWAToAy812m+8YAHEAxX49\nyIAqjOFEXB1z9VK4VOJwSvPB97I/qp4KxEfmsQyxxqfU+pzzRwpIRebYWuY50h9JNrhxy7y5\ndD7Vi53sCyerG+P86WDPJauqrJSajoHI7uODg6F4RZFrU3NZZbFrNmNI6KInaOJ4QgCnBqMz\nFjiaa/zf/sTmB545suf4QEw1akvd2y5quHJj3WyGamNjY3NG7DeYxQvFov3//DljsH8iJET4\n8V9zf7H/g3dn3lc7djC+60VjqF+qrndddAWvnF4XDIoGxf4XabAb3H7WtIHVrzjzLl2HxePf\nnmQTGx0Sj32d3/5v4J2GKkFk0gN/bJv1pjlHDdLpVyjWi8jAU4/VFwB35vaMsQHqeg1i/SC5\noaQJq87Oc3oLcq9VHzKUsm/NMCsoAZQw3yIC83busNghvdN8g9GR2lR1gRAnOgYwblZVirgc\nYFazpgWJJkRfLEpJfRITht4bj1S5vAWeEdETTpwYnmg9I4g6g/G4LpbPKzkghTK3Y2Od1B2M\nRzSDIxY5pWqvs7B/h1EEUWcgHohphzvNrUYSmugLxmvMlIuHXz35zO5OMSbrPP76qRvOX3bV\n2TMXDhJ6ulX2KKbOQVOnptT9t7eeBQC6IaR5VTdkY2Mzf8mmwLF9+/avf/3rXV1dGzZsuPfe\ne5cvXz6+6cknn/zYxz7W0ZFmzGEzd0Rf+sMkdWOM0OO/9t9yF1gtRpEI3P+t6LOPjgfCD//E\nf/sn3Ve9b4rnFYd2aE/8D4xZnxgv/pqvv1S67uOZrUzo9d9NUjdGSETFnmfYRbeY7WEOFteb\nT5sVb8Gmb9DwETr+MAgNRho8BNuo/03W+mFwTaEn4szO2L2L3nkcxFj/i963qPt1tvZOkHKs\nqiTjrAG5CLS0Nz+5GJyF5oqS4dKdb23uFjIEIgKkAfOb9nBJ+7q5aAXCZJ1z/qMS7QZQkyKD\nRLsRzwGwLUImEdISlOYCIYjCmlrkKMR/q7guVENwxA6zJfqBqFrrU9zzriFnEso8NL4NxrUd\nJweDcR0AekLTu6XsPNr3+zcnvU7rBj30yomlFd6V9TPsOeV2SJxherEPAPiz1BDUVjdsbGzy\nRtYEjjfeeOOKK64AgIaGhgceeOCXv/zlT37yk5tuumlkazQa7ey0WA2zmSO09jQjAAAAEMFh\nY3iQl5pnX0efeyxZ3QAA0tTA/35HblolL09d1aThXmPHb+n0CZBkrG2RLtxGiYj26H+AMclp\nz9j7ApbW8IsySSTUe8J8Q4/5/4UVWLsWi2oo0J0SZyuvgMKsozZUOvHYiLoxgRYRbY+x1WdI\ntJkhsQE69rtxL5JRgu3U9jS2bMvJGU1BxqqvFV2PgJH08sedrPragmuVgjLwYjCGTbZIdiu1\nwkA7SbE9QCPXEgOlBZU1Z2iPghaTVcyjzJcviDomqxsjaETtiMtNdljEqOlS+2hcLzQxKGGI\nzlA8rBoAIIh0s+krAIQS+rwWOOYdguiVE4PhxOgSgsdCQVBkVuE3udu8dKDH9PsvHeyZscAh\nMWwq9xztDafEHZw1zsJIxcZmtmRc+7SxsSJrM7ovf/nL1dXVhw8fPnLkSFtb25YtW2655Zaf\n//zn2Tq+TdZB2bKoEq2XoaJ/fMIkSiL6pydTYuLwTvV79xhvPCM6j4iTbxuvPqr+z6fFq4+l\nqBsjGLufO9Nwpxm3gnF2ySewKqkohsts7bW48vJpHmgKkA6hvTTwHA08B6G9qZLBFI8RPA56\nzGRDtBviJs3JZw/17TcdKvXuzXcVj7OWLbsLS89H9zJ0LUFnNXCfOP0Udf8W4l15HcmZQOf6\nsWsxaQohVYOUvxZxNpaoxyn62pi6AQACEocptiPzTihZVN6Nxgu4om0mmCfJA5jIdosdi9o5\nq5K6ucIgOj4UDY/3dyTLh6WF7mGTK3pDiXF1AwA8boffY9LsbKtFA46+gHnGR++w2avClLmo\nqbzKN0lPcUjsXSsqnbb4ZWNjM9/IWgbHrl277rnnnsbGRgCor69/4oknPvGJT9xxxx1EdNtt\nt2XrLDZZRFm3EX71v+lxubGFeS0cEwGMXvOJpXF6cv1RIqY98V3QJy0JUjSkH3jFdHcK9IOh\nZ/BDwepmOrbLZEP19FcX3SXs0r+ioXYIdIHswtJl4MqBk6g2SH2/Az048okiByG0GyveC/I0\nHde01BWVcUgLoXOG/m2ZSFhMdYwE6HGQ85sJzF1YdjGog6LjF2CMvr2RNkThY1i+FUs25XUw\nGZCq0bOV4rvBGPnX46C0oLJ6jkdlAwBAFN9vEta6wBgAXma5H68CqQH0E5OCrAgkJHoZQAdw\nItYA1GdxqWDusJrj2nPfVGTG9DSzCQBwFNhK41Bc0wwaXwNABmjxc3ochTXyPNATip8YjEZU\nwyXz2iLnsvxWuIQSk+zPEaC5vvjU6dDAmHLhkNhla6ovXV1lurvLIQGYFNC5lFm90isS27ah\n9lhfuDsQS+iizONYVe13zYW6EU3oHX0Rh8zqyz12YYuNjc0MyJrAMTg4WF4+UdTAGPvud78L\nAHfccYcQwuWyG5UVHM4N5zrPuTD+xquTopwXf/SvMuyFbg+Eg2bxSX0fRdteiJnNzOMW03XZ\nATzTcxTP3UZtb4Ex6bUAXH624aoMe2U6YMkSKFkys32nAFH/78fVjVH0IPU/jTUfnN6RZMvs\nULTeNCusJAzkIM1NAjb1/WFc3ZgIDryI3haQcyBOzQypEr1XAyWAVGDegqujWbSIcFLuxmT0\n/kwCBwAqZ4FUTdpJoDCgE3kZSQMAfWPb40RtAEOI6+f/z+0GMPFxALCz01Pxykos5UkEgABe\nqbDcgmOaIJy4LhGAc9SNVInD4+C+2U2M5x1vdAy3DUx4kHcGYqeGohc1lrFc9ibtCsZPh+Ix\nTThlJqWdiHNsrPPXVnpWlnmK3I7aErfbWnVas7S4Y8DEQ33t0tn2EUOAlgpvS8WctfGOq8b/\nPX/0iddOGQYBgN/tuOPKlivPsXCFt7GxsbEga8rokiVLjh6dZJSIiN/97nfvuuuuj3zkIw8+\n+GC2TmSTNRDL7/2a/wMfRWU0KVFubKn8//7Lefb5GXZSNphvVc66IPkjhYemNRbWtCHzDAGr\nmtgNfwMlE90lsbaV3fz34PZP60R5Qu0FbcAkrvWD2jutI6G/EbiZrOCqBGdOXCqxzNwhAktb\np9SaMeuIOEXbTeIkKJLm4jnnoALMN/+nuwuITHVVUygz4dXoPB9dl6PzIpIIIHVmCzCcJHnM\nVxDrzC5aRLQNHVNROC9TXMmGKRJj5U53AdrEpvyiEmcTJQ8EAOBXpNYyz6K6W3UGYm1p6kBP\nKHG0z0QyyApEsLtzeP/pYH9Ejaj6QETtCSc8Cse0v7hit2NjY9nyKl8GdQMA3r2xvqIotTfH\n0grPpWvnfUXk13+957FXThpjMlwwqv7Xb99+fMepuR2VjY3NvCNrsv2WLVueeOKJr3zlK8lB\nRPz+978vhLj//vuzdSKbLIKKs+j2jxfd9jG9p4t5/cx3ZrHAd9NHEm++YgxMmqU7Vm5wX/Lu\nSUf2FlsdgS1dJU4dnBRyeqTLzlzHhEvW8Nv/fxrogPAgFldDSXXhTiN107XQkU1BcEzH/YQ7\ncdl11PbopHkaV1jDe2c+PFOMBET7ABDc5Vi3mTpfAUr6B3b4sPnaLJ9xqgOLWebJG9H8DmV6\nkKaqB940Tnew0grHyg3MZ/lHYZNDuBdQAkoXJgD4dH8Rc92WaBAxV5ZG+cKHuIroKMB4jyQJ\nsQWgYDKkCgmXJDu5lBCGQUJC5uBSAT6KXDIbmpy6hACyxLwSK1VkQeR2cJ9jceVuAECHRaOK\n9uHoisqcZC50BGJ9kdQOvpwxWSJVn/RoO6uuaCoXkscp/f37Nzy289SuYwPBqFrqVS5YUXHN\nOfWyVHAS27Q42hF4/bCJWPzgH49dc+4Szgvwj8zGxqZAydqz7c477+zp6Tl27FiyOywAIOIP\nf/hDv9//6quvWu1rM8cwJtVMNQOQFZeVf+3+0K9+GH/9RREY5BU17ne9x3PdrSkFJqxxPTg9\n416w42DlUvlD/2i8/qSx6/cU6APFxZrOki6/HYumZjbBOFYsg4plUxztVBE69R6EcB8oXixv\nAdes/WKZdSlHhk0WYOkadFdR98sU7QHG0FOPNReDnL1XMTLo1AvU/tKoVwt34JJLcPVt0LWD\noj0ge7C4GZddBtIcFZpxDyAjEiZvN5Jls5g5Rz24O/ijfx/vWYNuj+8Df+F61/VzO6oRCERC\nH9YpTiQ4cyi8mGNhZddnFQ6O5ZA4lBYunn4LWDOVBABgJv2DC48KxBKAIYA4gBOgGMCk8WHh\no4qAZgQEaAiyzLwKL8mFFI6ITuumUYVAqVPui2paWruQWq+yCHWNcWKaed5WTMvVX3Fv2Nxw\nutTtCMX1iKojQrHLsb7GX+Gd6uuBW5Fu3dJ065Ym3RC56FKR0EXbYCQY1xTOq3xKtZmZS9Y5\n1G7e0jgc0zoGIstyIz/Z2NgsSLL2kLvkkksuueQS002I+K1vfStbJ7KZGRQcNPa8IPo70VvM\nl5/NGtfO+FDMV1T0Z39d9Gd/DYZh2ThDccvXflz77WRHWJdXfu8ngUv8guv5BdeDlgB57k31\naLBNvPVziIxVlDCJtVyBrTNs7TGKUgNMAZH2WsOcoNSY7XAmnOXYuM3kJV0kIHqMtAByD7iW\ngjwTaYaOPU7dr098NlQ68RzWbcb1Hy2IFRPmQHcTRI6lxlFGT4EaWBr9p4e/cS+pE+unFI0E\nf/xNVlymbLxoDgcGAIK0sNYlxjIaDCOhGmGXVK7wgqz2ygboXEcgIHF0IhVIqkTXedOf97oA\nTJOGFkyTKQlgXhsbU0Tv1MW4sK4bRkwTIY+8BBdCI9jpwRCbi10dYzaxACAzrPU5F4m6kdBF\nZzAeUXWJYbFLrvY5ESCqGpLFO0vuvEISZi1pAQARrllVpRmCMeRTbv8hiA50B7sC8bhmlLgd\na2v9Je4sy9OdgdiOk4OqPjrsAz1QX+Ta3FjGzSxdsohhbecjbKcfGxub6bAonnM2xp4/qY9/\nD8amW/pLj/B1Wxw3fjpzX88zY7a7OHVIff6Xous4SDKvXsG8bgr0I5ewfgXffAMmt8yYrG5o\n3V0D3/t27M1dIhxyNDSVfOij3suvhlw2/QIASITEzh+AlpTIK3Rx+PdM8eGyC2d+WJSwZCsN\nPJMSxdKtgNn7o4sep4HnRso0CACQQdEmLN48vYMkAtRtYk9DXTtw6dYMLU7zCVZeTp2DoCbZ\n4iLHqitByvLwjOGBxJ7Xjf4eqbpOOesC5pnhklHs+ceS1Y1xok/9as4FjqjeJ1LrNSim98vM\nzbJ4cRYWiM6zwNECxgCQBrw4c29R66PUEKW3fUHEeV/6vjBQRTBJ3RjFoLhqDCljv7hqiIQh\nZMYUUwfOKSOIdEGAILMcT/tmgYOzpmJ3whAJXUgMnVKup6iFQm84cagvPD5hPh1K7O0MDoQT\nI2kajIFTkuTJdUX1RbmSKRWJmeZwKBIHAHk6+RcxzXh8X/dgdLTgpTsYP9QT2txUtqYma/J0\nTDNeaRvQJwsKHYHY3u7A2XW5rbJsrDFPyVRkXlvmBoC+QFzVjOpS92K5jm1sbGbKQn2dtZmA\n+jvV3/4XGJPSL419L+oV9dLW92f3XPrOpxOP/vfEx+E+4JLzji/ylrMz75g4crDzkx8Vsej4\nx9P/9DdF+3ZX3HNvdkeYArXvnKRujCGOb+ezETgAwLMC5WIa3jHaVdRRicUXgMPc9W0maMPU\n9wRQ0s9KAoZ3glQE3jVTPwyFOsw7XJCgUAeWrpj1QK0RcTAiIPkBz5QJL3nZ0jso8BZEO0jE\nUanA4o0gZ/llK/L0I4EH7qOxi5AVlRT/xRdcF1w6g0PpHcdN41p7fruiUgJEAIADG/1HJhC6\nMC1BJ01EFL6gGy4wD7BZKmJ1AFGA7qQIR2wByK93so0FmjB36dJEWOFlCUOcCsSCYw6dTokt\nLXLNIJ2BAAKqGlK1kfsmQyxyyD65cMt5FM6UxeS1qRoiWd0AgP5wIhidSCYVAqKq7gLuGGta\nUeZxtObMOqTa6xxI68EBANW+aWewvto2MK5ujCCIXjk+UF/iKnJm5wo8ORTVzdIljg9Ezqor\nzqmusL6xtKWu6GhnqlH9ey5YuvNQ3w+fOtQ3HAMAl4O/f2vzzZc02g6yNjY2VtgCx8JH37s9\nRd0Yjb/5h+wKHBQNJZ5M6yZr6IlH7nN/4QeZczH6vv21cXVjnMBvHvRfd4PSam7qkRUodNp8\nQ6QXSADO7vHpqMLKbbM6gjUU3j9J3RiPB/fgdAQOIOvMzwybZonaS8Mvgtoz+tHdikWbgWec\nJSLH4nOg+JwcvWDF33x1+Hv/nhwRgaGhb/yj9PX75WXTL4SxKM5HKV+zINIpsQ+0tlH1CiV0\nrAJHqzC7Zkb3sN6UP4whShwGIwDoQLkKHCvmxrjHEkRsBagmGgRQEV0AVQALuH3JPMPqGibQ\nDUGHByLJDSniujg6GFlR5vVMszZhIJ6I6hM5UIJoKKESgd9RuBrHoqIvrCarG7pBwZiW/rWE\nLkrdDrfC6/yupjJP7rJFa4ucA1H1dGjSUkqVT6mbZs6IIGrrN7F6GYmfVZ8dxT+cMO80pOpC\n1YWSyz6miPj3Hzr7P3/79htHRluNco7vvWBZRZHzqw/uHv9aTDUeePZIR3/k8+9fn7vB2NjY\nzGtsgWPhQ4Pmc3gK9GVqojF9jOP7wCwtn4b7RHcbq22y2lGEw/F9b5luir76Uk4FDmAWfwLI\ncl4dM0s0CyNebdA8bgF6ayxkDETvjNqFnBGtn/oeneRqET1Cai9WfSCb9TvTJPLEr9ODZOiR\npx4u/vjfTPdojtZ1iTdeSo/LK9bNZHDTh+KvgZ70h086JfYhGMxhmZKDcy4lJA5R7K3xfCLS\nT0PiOHovB1ZoHS78iAu2X8m8hqFkmN3OEOS+qJrebpMIukPx5aXTyOvRhEhWN8YJaKrPIRf2\nM2OxENcn6VxxTTdNUhSCNtYXV04/jWK6IMD6Gn+1T+kOxWOa4ZJ5tddZNf3zJnRhmlsB1qrE\nDLAqmUGE2RV1TYkSn/KPH954vDvYdjrkdPCWuqIyv/PDX30+/ZvP7+68aUtjY3XhNhq3yQ5s\nrt9MbOYndn7XwgedFgvjspJZ3SA1brTt1/e9LLrbprSYH7X0RiXrTQAgomGr4xuh1GTF7IJl\nzdbxwn5ZtdICpqsRuMqw3CTjA6s2gJKTggUKvm7i2akPQ+RALk43RbT2tmnFM+O67L28shYm\nF/+gw+m94SMzONq0MYYmqRtjkHoEgSTmBpOqJJRnW74xO4xgsroxighR7M05GpANAGhAYQDz\nFokFiMzMhScH90csPDKs4lZYNYwkgnQBxWZOSJmiZ3h5ydDVMutUepUNNUUXLC3dUFM0A3UD\nABycMYt1F7cja5PAGgvDlCqvM2+dL5pq/JefXXfRmurKYldHXzhgVuADAPvbprecY2Njs3iw\nMzgWPqxlI7z+dHqct56TYS/9rRfUJ++n8Ki+wJetctz4KVaZyU0WSyotN5Vm6j3BS0rR6aS4\nSfaHXLckw45TRY9Q9wsUPgFaGJzlWL4JS9eP6BdYezYe306BjknfZxKuvDYL580l6FxCkcMm\nG1xLp32oFe8DrlDP+EwSsWYTNufsXyDRbRqmRDd65yzjFGXzQgOr+BmO5nSX/v1/hH5+X/y1\nP45E5KaVvjs+K1VU0L4naKgDJAdWLMfmzTlZnRAW2T2kgxF0S+XJLioj43VL5XPcYVRrN+8F\no3UACFuLzzcUIDoEFAQAAASsRdZSIK6xggydVCLiKEls0p+nzLwOXqIaQ5ODPgcrJjLJ7Qfz\na26GUDYPZjNzytyO4wOR8R9DtqiqQIBid0Fc0lOEM1xW6m4bSL2SEaGhLGvydKVXaSh1nxic\nVDIsczw7SyUw00XTLXVDW1K0sbGxIidvtC+88MKDDz548uTJ+OQp6wsvvJCL09lkhrdu4ivO\nNQ6/nhxEt1++4sNWuxgHXkv8apKzr3HyYPxHX3Tdcx86LZ+jvHEtFlfQcF9qfNkqVprJZQBl\nh+/Ka4O/ezglzjxe76VXZthxSiSGxLGfgD72tI71UPsTEGrDZTcAADDOLvxLcegJOvUaCAMA\nsHgprr0Ri6ctE+Qb7yoI7R3tYDoKAXNi8fR7o3IFV7wPl26lcBcAoq8WnKXZG2gaVr0e5rQH\nhLLmbL273SS+9gz9ca1gJeVFn/yS/6Of13s7eUk5KyqlrgPGY18CdfRSpOOv4eE/snd9GtxZ\nf3HMMNEihrLPsSRhBHQRIxKcORRezHGOG0kQmeibAAAgQCQKr0plQUMBEruSEjcIqJNEENl5\nc640RfVgTA8R0UiCncwUr1zCk7Q5F6+UmU8zAgI0BrLMvBLzAoBb5gGzNH73NBtwyMxytmy1\nySbPeBx8aYn75NDondYpc0ViibR5cmO5x5Uza9gccWFTWV84kVKQcs6SktIAz4HGAAAgAElE\nQVSsOsVe0FBW4VUO94ZDCU1mrMrnPKuuyKvMjQJeW+6RONPNtIyGKrs+xcbGxpzs37Duu+++\nT33qU6Wlpa2trYqS8+JGmzOD6Lj1b/TXn9Z3PkWD3ejysZaN8hW3oc9yEqu+YNaPIDSk73pO\nvti6a6YkO2/9fPxnX6Xw8HiMldUo7//sGcdY/qnPa50dsTd3Tuzo81f9w1d4yWxn2tT9/IS6\nMR4cPgClG9DXCAAgu9i6m2HNjRDpB6cP5HliiIAcq2+i4R0Q2g+kATBwLcXSrSCXzPCArjJ0\nzcQ7c9rIZaCa9YVx5OXsFvhuuiO24wURDiYHeVWt59qbZ3NYdHvkhlYAAC0mXv7RuLoxAg13\nidf+j132ydmcwgRmdQ1wYH4AQGBOXgJ8ppdKDkB0WqgyDJj9HMkrRMdMylIoJERHWHdrpAIR\nZ7KL+xSeV+EppoeiehBgonxQE4mg2l+iVCVXFErokqTUgVW4Hb1RNb0kodozvatL4czJeTyt\nb7dXlq3KB2zyT1Opu8gptQ/HwqohMVxfV3RiINqXZNa6tMR9cdNcPnFmhk+R3r+x/q2O4c7h\nWEIXJW7H+tqimiLzopIZgwDLy73Ly72CaM6varciXb2p/onXTqXEm2p8G5rn3y9oY2OTH7Iv\ncHzjG9+4+eabf/azn9nqRgHBuHT+tdL514IQcMZVJiLRbW5yKTqPneE8S1e6Pvdd/bWnRNc7\nwCW2dKW86UqYgnME83jrvvOD8PY/xN7YKSJhR2Oz/z3v48VZmIBRyMKYM3gMRgSO0RFw8GXP\nwzU/MAVLt0LpVtDDwF0F5jdhCfo20ECawIEyeqZj/pJteFVtxde+H7j/O/HdO4AIGHNfdIX/\nzk8xd3a8A6lzPyRMkuSpaz/EQ+DM6koULwVeCUZvatzRModtXM+AvATi+0xyT+T6Oc8aWHTQ\nsGk4YfSoYrTxsC7UkBgwyOeW8mctHDNMejkZpCeM+BmlFpmzllLPyUAsNtZ0Q2ZY73f5p78u\nXe5UBhNqcqtRnywXK7aZTmFR5naUJeU1rK8t6hiODURUhljpU6bYBUMQHekNtw1Gwgnd45CW\nlLhWV/mTW1H0hhNDUZUASlxylS/LQoMpDs7OW1YKy/JwKphzdWOEP79ulaqLZ9+YKCVe01Dy\n+fdvyFtPEBsbm3lH9l92e3p6brvtNlvdKFCmmkNr9tggmIqxCDrd8tabpjeq0T3Ru/UK79Yr\nZrKvFUIHYdFd3LBKiZ+HSNmZhOcJVzMWb6HADqAx6z7JjyWXAc/G/wUJIGFpjpMRqXZp2Re/\nQfGY0d/Dq+pQzmp5dsSiHRoRRYcwuwIHALrOp8Qe0MZXvZgmFQtJcVCM4dRW3UkDEQZUgOUl\np4n70bWBYqNuSjRyD2JedG3Mx9lzB2kggoAcmA9gXkiQZFXihGlpHVE9pHAPz4tqJsgQZF5y\nb5AGcOar2iPzVeXeiGokDENmzOPgfEbzN4ZY7lQ0IatCIICD8Ty4S9jMnvpiV33xNHKOBNHz\nR/vG8z4CcS3QrbUPx65srZQ5S+jitZODvUlZIeUex/nLSudL2YsggoKRMEw53D785tH+wWCi\nrsJz17tX3HhRw8FTwwnNaKrxrWssK+CB29jYzD3Zfy/ZtGnTO+9YrJnbzAsQeX2LcfJgWhzY\nktbkgBjoiT/3kN7ZhrJDWr7W+a4bUSmwUnkmgewFLWyySZmbjlk2AADedehqhkQnGBGQisFZ\nn4XMgnC76PgDRLqABDhLsWYLls3EkxWdLqm+YbaDSUexbF6DjhzYl6ADnZvi3IMiQICCOQk5\nUCxmHHfxpjNoHJSgxN4JcYT50XkW8IrsDzIFZRVKlZQ4DEYAUUGpCpSV8yUvyQyD1AOgtY2W\ne6CE8iqQzW2bCgkE9ACZ3DMNMFmg1kScZ0WazAsI4HVwbzaUJpkxu+nGwuad/khyVcsIgZh2\nsCe0vrZo56mh3slb+yPqaycHL12e+1vl7Dg5EHn5+EB/OAEAFV7loubypaWFVZkrBP3no/uf\n2tkuxmrKHnjmyD03r7/mvGx0nbexsVkEZF/g+OY3v3nzzTefddZZl19+edYPbpMf5HfdYvzv\nP6UEsaRSOmcivUJ988XIj/+NtNEHvHZgV+LFx32f+TdelclpJf9gyXrqfSUtyrF47VwMx2YM\n7gZ3S7YORkOH6PhDE36A8QFqexRivVhfKHchrFkNTErPJ8KSOvCeqZA4NkQDR0ENg7sCK1YC\nn1JqiS6GDTTS8mKEKnqcvMF6P4Oi20Ek9SIRAYq+hO4twMunct5ZwcvQvTnnZ8kLlHgD9K6k\nzzqp+xB0kFfM3aCmBOIyordTgkQsYZiIwmIqDuJTRhdRVQQEqQxliXkcbKL+hSFnyIVZK+IU\nLxUbm6zQGYhZxZvKPD0hkyTQ/og6HNOKXYVrzvJWx/AfD09UL54Oxn+zu+PKVVVra/NXa3ZG\nfvNi2xM7JjXdCMe0rz24u7l2a21ZYWkxNjY2hUn2BY6NGzfefPPNV1xxhdfrLSub9OJ+4sSJ\nrJ/OJhfwlrOdH7438bsfUKB/PKJs+zg6RlfwKBKK/PQb4+rGCGKoP/J/3/J/7htnOHo8DIob\nME9rX1h9McT7KHh0IsRkrL8GFOsGH9F+OvUShU8jd0BxAy69GLj9Al3IELU/DWkTLep5FSs2\nZvqh84mnlJ11vXhzslWQ5MDzbsu8Hx17jo4/P66MkKsE130AS8+cCGCAuS+mYeGXOYp2apK6\nAQCAAIISB9B9yRlPajOKCE5SN8Yg9QjKywu9VgVrkakk3klqNeoOGRXCbNg8eyk2MaN33OHV\noLgmQhoLuqV6HCuZ9EhFIS2l1Itk5nSwfPQ+sFlsqBYupKohQmaOPCME44UrcCR08dKx/vT4\n9qN9K6v8Ei+Uqo8n01qKAoCmi2ff6Ljzqtb0TTY2NjYpZF/g+OIXv/j1r3997dq1a9eutTtx\nzF/46gvcreeInlMUHsKK+hSfV+3t1ymeak0CAPrRfSIwwIrMVqS1hHj9t7T3DxAPA5exYQPb\n8iEozn1fT5Sw8f0QfAdCx0ELkxYFPUYdL0DfHqjYiCWrUr5Op3fTwd+MWMYSAAwepa7X2ca7\nIT8OIzYzINYPatAkTkTBNqwoDIEDAFdfxcqW0f7f02A7yApWNLMN14M3U1oEtb9Gx56ZFIoN\n0Zs/xos/D84zLbhZLq1nWnInY8B8g1XcxhRh0XIFDBBBa5ubjIekIYJhAA3AxbACzQpGsgY2\nIK8GGgJQAdyAZUwMgZG6oM2QO7JkpKKLyLi6kRSMqsagwkfvvQp3A0BED4zncTi515PHLqc2\niwqPQxqIqOlxr0PK0N5SKuDCpa5ATDNTbRK66ArECqRQhYi6BkxeLwGgo8+s3NjGxsYmjewL\nHN///vc/8YlP3HfffVk/sk2+kWRWZ75QLIZNFgHGN5kIHMIQD3+VusfSKAyN3tlldBzgt34Z\nSmqyM9qMoL8Z3LXi8E8g1jcSocQQBI9D+QZsuH7ie2qIDj0yom5MEB+mQ7/Fs+/KwzgXF/Eg\nRQfQVQyu2WkQwuQd9Myb5gKsWoFVSRUKZED0IGn9AIRyObhWpLScoFMvmxxFT1Dn69h8hna8\nDBVTKYNhZt05gyxC5u2HbUyY9M9IyJDGmnfOpKZD6HSEksxNBHVzbGBYOctRZsQJOHFz9kol\nggwt6Q+KIffJpZilS0ITJg4pAKCK4LjAAQAKdyvcpZNORBJKmK9MQJu5IqGLjkAsnNA5Q79T\nri9y5q0vZmOp59SQyUy7scxT6nZwhumuwwyxzFO4+Z6qbp6TAtbpKvkHEZ0OHjPLkXFP3/PI\nZv5T2AmPNoVK9m8WsVjsqquuyvphbQoK9FvOSJnPZBMdfnVC3RgnERWv/Ipd95nsjs0K6to+\nrm5MBPv3QMkqLGoZ+3gIDC1tV6DBY6hFQM5BM8jFSbhX7PsN9Y9eEliyDNfdhEUz7d6ilACi\n+bzRWcB5N/owDT0DxmjuCcWOQGQfllwN0ninA4JwmtXrCOGeMx5eYiWa6KM05wsZM+WMICsy\nn38zv61uTAM28iOiIfkM7gVkAMSExrUAMv90D2ZQJ6VatwqD2hB9OAX3kKyAyIoclQkjposE\nAXEmO7knW+oGAAgwz/knSo+jhLJ9MS4G+iPqgd7QuI7QF1G7gvGzavzOvDiV1BY511T7D/QE\nk58ty8u9jWUeBFhT7d/bFUjZZVWVT5EKV3TLUDtTUGU157SUv7Q/zUse4JzW0Qau/YH4Iy+3\ntZ0OSYy11hfdcFGDt5DGb2NjM+dkX+DYsmXLW2+9tW3btqwf2WZ6CMN481ljzx9p6DT6StmK\n86TNN4IjO1nN8upN6FBITW0wLjWsYKUmi4p0ar/pcaziuYCGD5lvGDoMYwIHJMwqHQAACBIh\nW+DIDvGg8fJ/gTqRa0pDJ+mV+/glnwPPjPrPS24sXkVDB1LjSgn6m6Z9NDIgcojUPkCOSjW4\nl+doYk/Dz4+rG6MYIRp+HstvHDsjAuNgurA2BR9cBFnhDQmjnWBcs0MHq5Qy10fIy0A9DJSa\n+YIOu/J5OrAS4OU6BzHRHgIFcwilQoY4g+nZjhClKrMjYUH9HPNqK6Bwl5KlmpQUmMUyHebF\ng9amANEFHUxSN0aIacahvvBZ+eqIub62aEmx68RQNJzQPQ5pSbGrwjuaAdda4XXJfH93IKIa\nAOCW+Zpq/7LCqPKwosrvrPQpvaHUN7faIle5t4Aqyj/67hVvvTMQjk1abTp7efmWddUAsOtI\n37//ak9cHc203XN84Jk3Ov75zk2N1Vl2W7exsZm/ZP/V4b777tu2bVt9ff0NN9yQ0mTUJn+Q\n0H7xr6Jt7+inRNTo7xCHdjju/Aq4smDpx/wl7ls+Ffm/bydXc6DH5/nwPeY7aKkP1KR4vvLe\ndfOqzkkmsg6rBySCY95YIRY4om17sroxip4QR//Azrp1ZsfEhuvAiFGwbSLkLGXNN0/bZFTt\no/4nQR/LqggBOKqw4j3As/3aqvWDbtamQR8ErR/kUaEHS5upz0yYm0KTUQDg6HFLrTqFiBII\nEkcv4pnSp1FB18UU3zXRahQldKwGeelUzmgzDjlWCWpLjxvUzjC19U/mIxFYVVpZ3FfnIRLz\nqandbQEAZJaFSYtBhAB5K22wyQoDUVVPqwEBgKGYphrCwfOUKFHidpS4zW+bS4pdS4pdI6Ms\n5MSNZK5bV/vYns7k3iLlXuWatdUZdsk/Syq9//2Zi3/wxMFdh/tjql7qV66/sOHmrU2IGFeN\n/3hk/7i6MUIgon77N/u+88kF4sBlY2Mze7IvcDQ3NwPA3Xfffffdd6dsoqz6ydlkwNi3fVzd\nGIcGuvSXH5auuCMrp1A2Xy0ta4k9/Uuj4zg6FKl5jeuaD6HXYl2l1KLRhqtIvPIbUNxYvwqr\nGrMyMEtkHyRSm9gBATgmMsaxfCWZeXlCSYMtcGSNoZOmYRo6MfNjcie2fhgCxyDcDkIHdxWW\nrpm2ukEG9T81rm6MovbQ4HNYcb3FPjPFMO84MLppXOBouYoG30ktm/LXYe3ZUz4Tk7BoehIi\nL0HP5aD3gwgBU4CXA9ouFdNGgLnHpKAIgACY+nQIATiAiT3qQipOlpnXwYpUMSnnn6NT4aUz\nPiYBBFUtpGkjXrYOxkoUh8IXzj/awiZh3TAioedP4DgjjnkibYxQ7JI/fP6ywz2hnmACAar8\nzhVVvgKU/qpL3f9w+zlEEFd1V1LrjX1tgwGzzq8nekIdfZH6CjvN1sbGBiAXAseXvvSlrB9z\nugQCgXvvvffhhx8eHh5evXr1F7/4xfe9731zPai8Io69YR4/uguyJHAAAK9r8t5171S+yVZv\nNd54ctI8jYh0QcN98PJDAADI2Nqt/Mo/A56rhGQsXUvdL6ZFAcvWTnxU/Nj6Hjr8GFDSq5XD\ny1bcmKNRLUaE6VTNOj5lsGg5FC2f+f6JTtBTa6oBAGInwQgDz6rCxazzgZM3+evZ+Z8QB38L\nI+oP41h/Hra8e9razfTHB1IlQE57WC54LKdnBAKnIXAAwxJBJn2dGRSKQ1BWcEnVkvCqYliQ\nxlCS0Kvw4tnk9/XH4zF94q6iCtETi1e6nE5b45gPyNaupXLBqBvzEYa4qtq/qrCSNsxBBNfk\nxqKDwbjVlweCcVvgsLGxGSH7k8l/+qd/yvoxp4UQ4rrrrtu7d+9XvvKV5ubmH/3oRzfffPPD\nDz98ww03zO3A8krMwkzLKp5riqvYtZ8Sz/4A4iMDIJGyOENC7PsjuHx864dGI1oM4kHwlE2l\n3cBUwJqLINxOoROTg1vAOyn3HuvOR/9SOrWdwqeBO7C4ARsuBSlPnfwWBUW1YJasgUV1eR/K\nZEzVjfFN2RU45EpgCoi0EgOmgDzZO9lfx87/BOhxSITAXTo7aYNUMaCLCIHO0OFgpRzt18Fc\ngeg0daRBkHGaT16GS4iCKYUqDMsRi612mafIzCuz7PyhJQwjWd0YZyih1rjt+/k8oMztYIgi\nLfPXp0jOeZU0YZNFin2WawMZNtnY2Cw2FmD7rocffvjll1/+8Y9/fOeddwLA1VdfvXHjxs9/\n/vOLSuDAIotmjcUzauIIYHQe13a/KIZ6WWmVvPESXtMw7SE1b+J1K+noazTUTcEBOPhq+nfE\nnuf4llso2En7H6GRQgZkuPR8tvJacMx6JsZkXHE7DOyDwFFSQ+gsw4qzwWM2qfbV4JpbCi9n\nc4HAGrcYp3am1gEhw+ZL52ZAE2Ow7k9xxtYV0z6XhL4LKfCnFD9R9F0Ipl0VJSdIs6oTITCi\n+glBo5KKoIQuQg5WpvCqzDvazAyGpQBdkGYOwtm082IQFImtN6iDaJhAQ3AyrGY4w5v5IiFu\nYXupCWEQ8QJMyreZjIOz5eWeo33h5FukxHBFhV0uaoIu6O3u4KnhaEQ1fIrUVOZZWVmItSez\ngYgqil1uRYqmmcjWV3iW2heGjY3NGFkTOMLhMOfc5XKFw5Y5Al5vPu4+jz76qNPpvPXW0W6F\nnPPbb7/9C1/4wt69e9evX5+HARQCbN1WY88f0+N83aWZdhMCiCAtfTf22P2J538DYvR9Mf7s\nr5xXf9B5zW3THpbTi+suRwBj+4PmX0hERdfbtOdnE7NfEnTyVTF0km25B1gW8oqxbB2UrVtY\nD/35hreKnXe32PsriI512VR8bN1NWNIw+pFI7P2D2PM8DZ9Gbwk2n8MvvBEcuV90ddYDcqC0\nVV/uBUf56MB622mgG4vKWNUykGbnS+dajlIRhXeB1g8AIJWj7xyQJ89+KQGkAvPOvhFvwugd\nVzfGUcWAxHwcC7rz/zwFQZLZcl200UQrUORYwXFmqeESx4bMVwGBQEDbzXcEMs2fGd1kMz+o\n8zt9inRqKBpSDY5Y7JQaSt2F032jcFAN8fuDPcNjtiMDujoQUduHY1e0ViyY3rp/2tP9vccP\n9AfinKNjsk+wW5E+c+O6hfI/amNjkwWyJnD4fL41a9bs37/f57PseZ6fJqNvv/12S0uLokzk\nqq1btw4A9u/fv4gEjmVrpK236i/+OrmvAV97Cd90ten343t3BX76P9o7hwFAbmop+vDHnWed\nN7JJ2709/tyvJz04hBF/6md8aYu85ryZDQ8l6/Xwtu3pPT4p2EWndgAoEOoBVxFWrwKvbdAz\nj8GKVn7p39JgG0T7wVWCpU0gjf3BEumP/Du98+bop0SUBjrpyA7ptn8Bt9/yiFmBe7DoPBpO\nyS1CLN0KgKLnZOKR/xanRj1NsKRSuf4v+IpNszqjXIEl15hv0k9T7E0wAgAAyEFZhcrq2dSn\n6GYWFQCgixDPukeMDQAAMPQ6+BpBw4JiiBIDP2JOdDpNDKuiX5AKABxdCq+0i49kZj4NZoh2\n+sY8wq9Ia6tnfucPJfT2oVhE1Z0yr/E7yz3ZzsUrDN7uDg5PNlUFgNPB+LH+SOuCyGt49o2O\nf//lnpH/NgyKC0OWmNcl1VV4Vi4pvvmSppJCsrm1sbGZc7ImcHzrW98qLy8f+Y9sHXNmDAwM\nNDU1JUdKS0tH4pl33L9//+nTp003xeNxmG8uMPzim1jrJmPfdhrsQn85az2XNZrrO5GnHx38\nr6+Of1QPv933D39V8om/9V7zPgBQdzxr+jKo7nhm5gLH0jXw8q9N4hVLKdRhuovY8wiExpKD\nuMzWXgXFXgh3AgB4arH2ApDsSdq8gstY0QrQmhIWh3eMqxvj0HCv8cpD/Iq7cj4q/yaUyyiw\nE7QBAASlGos3g6OKwoH4D79IkQmNgIZ64z/9V+fd/8IbVmd/GFonRV6cWGkmA+L7yRhCzyUz\nPSKRuQ0HUFoNRZ4gFfTTIMLAPCBVLVyjFsawlOVyQp0welQx8XQzKBbVT7p4vcRyLAhmC0qA\nMQCUAOYFXjYdf5lMuDjniEbaU9srS7a8sUg42hc+2BsavwSOD0SWFLvOriteeAJXx7C5Z1PH\ncGxeCBzhuP76sf6eQMyryCvri5ZXT1ooJaL7nzqcEtENEYionDGnLB0+NXzBarvW0sbGZoKs\nCRyf/exnU/6j0MAzPdOuv/76tra2DF84o0RSaGDlMuny2zN/h+Kx4fv/Iz0+fP9/uLdezdwe\nY6DbdEfRbx6f0sDqV7LW88SRnZOijPNLbxP7f2q+jzHRYA8loMDrEB57Dx4+Rj2vs5UfAt+S\nGQ/JpkBIVzcm4nkQOADA1YiuRiABOJHtr7/2VLK6MYowtBd+zT+Sfd8oir9lkkevdYLeC9LM\nnE0QgZtqHNNteJkdtHaKvQnjJTMoo3MDOJoy7mNjgiA1Wd0YJ26c9s4LgUM7TtoBoDGVjflQ\n2QgsC+4wgihd4PDKUpFjYa7h26QwEFEP9KS6cbcPx4pdclPZQstvSlh0nMlgtVs47Dkx9OBL\nx2Pq6OPp2b1dG5vKPnxJEx8Thk8PxQaSzVMQOButvBkKJ4aOJXYf67/2/KWf2rYm30O3yT2C\nFmCzSJs8sABLGcvKygYHB5MjIx9H8jgycPz4cbIgFAoBwEiKygIjcWifiEbS4xSPqQf3AAC6\nzOV/dM3qFYG/56/4lltBGT0IVjdJt/wDNmxAn0WBupH0nlpVDClVuHpMHH14krerzfyE4uZN\nfCiW+qqaW5Al9zIwOo+Zfkt0HM3+qSkOhnk5Cem9Mz6qxIos4nmfBhtDFN0ByQ1BSKPYLtDN\nE+hsMmBQ1DROoAuy9FMsFPQOUvdOqBsAIEIUfwXSmsVMF0HUE4urYtITAQG8srzgFu9tzGm3\nSGqwis9rPA7zSaDXkR1HZF1QdzB+rD/cHYzrIpu5zIPhxE//9M64ujHCm8cHnn6rc+Lsk2Ua\nxkzWK5987dTe44OpURsbm8VKloWxcDj8wAMPbN++vaurCxFra2u3bt16++23ezz508vXrFnz\n0EMPxeNxp3M053nv3r0AsHbt2ryNYR5BZurGCCISAQB59Sbj5OH0rfLqc2d1Yi6zC25gF9xA\n4UF0uMZbSGLjFtrzy7RRAqijL8GoyCCbPbMTQxDuAN9Sk0028wf0l5u+PVkaA80tuShby6TT\nmZeZTAWFVwiKGpMnvQqv4LnpCpEBUo+Z9nmkxFGUZtaAc/FCYHm1ZNhUIJBmphuSBvoJkFfM\n5sghTU8vTiGAoKqVOxVViIgmdCKO4Ja4025auRCJauZ3y6g687towdJc5ukLm8iCTeVZqE/p\nDMR2dwTiY6bLTplvrCuuLcpOUeEb7wxoZuknrx7pu3Zj/ch/V5e6XYoUG3NOsWqb+uqB0+ub\nzrCQaWNjs0jI5nN9586dy5cv/+QnP/nLX/7yzTff3LVr1y9+8Yu//Mu/bGlp2bVrVxZPlJkb\nb7wxkUg8+OCoT4dhGD/96U+bm5sXT4fRaSHVWpZ1jGxSLnsfr05VDXhdk+OS67MyAPSWJhtk\njJrCsiTpjckQiY/PJMlU3RjZlAhkZUg2cwhbs8UivjXPI0mG1y03jbMlqT1EsgBzWbnSokUW\nxlRA4G6p0cmrJebj6JJZkVtqdLC5kI0s8lPAog2qTQaYtYExw4Lvumfxi9OsrwTVMJ/EJgxj\nWNW7o2pQ06O6EdKMnpg6kEjtzmhTUGgGDUTV/oiqWhRimCJz82mwvBD1rJYKb3P5pHVERFhX\n66+btQwxGFV3nBwcVzcAIK4ZO04Opvc0nRl9QfN0rWBUS4xJVLLEtm1uOOOhhsPqGb9jY2Oz\nSMhaBsfg4OD1118vhPjhD3+4bdu2kWqOvr6+Rx555O/+7u+2bdt24MCBoqKZv51PnRtvvHHz\n5s2f/vSnA4FAU1PT/fffv3///ocffjgPp56PyA3LHa1r1CNvp8Qdy1c6mlcAADrd3r/+dvzp\nB7XdL4qhXlZS6dh0mXLlB9CRq7dnbLmC159D/ccgEQJvBQQHxc4JW1nM8Iojz4NmWnOAGqHj\nL1GgCyQHljbisvOzYribI7C2lW+9zXjxF8kGQGzFBWzTtXM4Kun8a7QdT6S24WBcvvT9OTgb\ngtIC8dQ/SWBukGfZZQZlVirDXK9xWXrBLMCJR66R0MPQMeKfMinO/AiF+2c+hlW9yGzrSKwS\nqwggoKZ21Q1rhsKY11o6t8kbBNAViHeH4jHNcEqs0uuMqcaxgbAhCAAYYlOZe1WVbyrWp9U+\nZ1fApEqr2lfwwt/0QYSLGsuayjynhqIR1fApUlOZp2xGljEJXTCckIGO9UfS8xQF0dG+8LlL\ns9Arx2VRRMMZytLEE+HOq1tV3fjtyycMQQSAZHKTqCjOdzaijY1NwZI1geNHP/pRIBB44403\nVq+e8BSoqKj42Mc+tnnz5k2bNt1///333HNPtk6XAcbYE088ce+99371q18NBAKrVq166KGH\nbrjhhjycep5S9jf/0v8vn9dOvDMekZc0lv3NV2DsHQIVl+v6u1zX3wVEkJ/+464SXDJWAlOh\n4dHtNDRakElxFQ0DeNpDUfZihiajoT5x9GUI9oDTj/XrsH6x1CtRzxjlpNMAACAASURBVCGx\n8yegjRbq08md+M52dtFfgisfauO0MaKAjJ33Xmw6W+x/AQa7wVfKlv8/9t47TK7sPO98v3ND\n5aquzg00QiMPgBkMBpMDJ4qZI5EiKVFLiZS5lmg9z66stf2sH1uWrXVarb2PrcdaU5YsaVeU\nqEBJFCma5DAOh5w8AwwwGACDjA5Ao1PlcNP59o9O1dW3Gh2qqqsb5/cPUN+te+7p7grnvPf7\n3u9eGjiyvvOiaCL4uX9jfeX/kUMz5VrU1h149pca0kIFoOCdzDasilIOrY3CD62lTWwLoXfB\n10xklf6ptzkU0raVvGFZ4VuhUzSobVnHOS0XrQOezyuBxFq7gJuaKPsncfh/f+VdTwkc6w4z\nTlxPTxRm1LqS443lrErHB8l8caJgufKe/rZbjtafCA2lS1WFGxFT39e9aW+E9MWDffFVpmww\ncP5m7sRwpmC7AJJh8/4dyf62UK1MjXS5PhkcB7Ymnn/Hx33pwNZEpYylCfr8Rw4++/DOs9dS\n3z0xcvJStbMyET1+V19dpqRQKDYBdRM4vvnNb37yk5+sVDfmOHz48Cc+8YlvfOMbzRE4ALS1\ntX3hC1/4whe+0JzLbXT0ni29//mLxRe+bV84y2Bz78HwYz9But9rY126q2mG+In/Tb71Vb74\nEqQLJjhx6GVwxfqVNNr97ILClgr43R/KV/8C3sz3MZ/9Pu04Kh7/JWib3ZzZKcnX/3hO3ZiG\nszfkiT8XD//yek3Kn/w7nHkVXgEA9Di1Paw98en1ntMCRO+O0Od/S968xpOjlOgQvTugN64d\nA1HoXph74Y1D2tASMLas/bZ2a8AEnUHVd9kpQIE71mlKGxtBgYi+y5U5j8tEpFFYo43RJIKM\nA+xNoMorRERhrNVKKWYYeceVi249CxKLvTkA1Nc3UbE6RrKlOXUDANf4uwylS/u7Y5Fb2WcS\n4aEd7VemCoOpUt52Q4bWFw/u64rqDW3avGF57erU6RvzKYqpov3c2dHH93TV+m1Rnb6P7uhP\n3LUjeepaqjIYMrWfvM/nftWWjvCWjvDDh3p+4/97452r86cIQZ993/7dWzZC3yiFQtEU6rbB\nO3PmzKc+9alaRx999NHf/M3frNe1FPVH08JPfiD85AfWex41MCPi/p/DvT+DwhRCcegBlFM8\n/EPOjwCg6Bbqfw+C/jf9ODUiX/5SlXEjXzshT31DHK2PjUjLwqNnYPu1yLl5FlYe8PjqDzk7\nDIDi/bTzCQRutT5gD9njXLoINws9QeF9iB1Ze1oBp19Gdt6mh90sJr5FySJi65y4UQ2R6N2J\n3p1NupyWgLbyRBs5we4lyBzIgOggfV8tR491gQs/gjPCAEHMC6Z6D4WOQYTXdWobGtJFXMdG\nW99r7RR8hO2T82Ycej+Zh7Hm4hqNqCsYnLIsZ7aRikaUDJh5Rzp+9Ss17BoUTWVsYbYF11ad\nUiU7Yt66HoEIuzoi001hPebLk8WXrk4VHS9saFviwV0d4eWUuqwjticHU8Vs2Q0aoi8WTIYb\n9UmeK7vv3FhsfEOvXkvt7AjXSOKomyb4i0/u+dG5sRfO3JzMlYOGdmBr4tn7trVHa1YShQL6\n//X3H/jeietvnB9P563+rugH79++qy9Wr/koFIpNQN0EjlQq1dVV06+up6enqnWrQrFihIbY\n7GssmKQ9P7WctQlfesW3LQVffAmbXeBAKeUfZ5ajJ3Hl23Dt6dswnB3iG2/Skc9QclfN0djh\nm38Ne3zmoT3O9jhKl6n7o/4aB7ugZXzCeAVkj1cGpv+snH6ZogdBxq1HUMzhnmdntucRlyCz\n7I1Q4BFQa2RlO4NwRjDzJ5aYrqNmQIQhWmOGiiajdVDoKXARXAbF6vh+D2iiNxyyPc+VrAky\nhRBEHnslPxenkK7qU9afKg9Rrv0FzxJFxyMgWMtKdCGu5JevTWXLM/YrOct9dzx/M289uCOp\ntarGMZIpvXptynIlAAZOUWZPZ/TYtmQjpnsjW/aVK8qO53n+QkbZqVuTJiHo8YM9jx/scT3W\nl6c1EtEz92x95p6t9ZqDQqHYZNRN4LBtW/ctapi+jK5b1lo72ysUqyE/USM+BZagDehrWLjJ\nuesAUXwrwkt2wTBrZ6pf/QE8e0GSqWfzmS/Tw/+k5u8k+9a8ujGHdQP50wtSLdjj8Vd58jjs\nLLQgxfdQ35Mwat9dsa5X56jPjOPAHkNArWCWDefZOb8oaLN9mgIPrseEqmHn+qIQAMAZQWht\nbacVGxoKg+qfv0NAQNMCFdpF1NAKrmct3EgbguLGZi9X3AgEdA2Yt4AVtYsgLk0VLk4VABia\n2N0evqXxxNWp4py6MUe65FxLlXa1t2LiWMF2X7wy6c3msBDAjAvj+aipH+ipf6pCjRItBsjh\nmg2J6j6NZaobCoVCcUvq+aV+8uTJYND/a+bkyZN1vJCiZeFCVo5eBVj07qRIa9hY1trkm6GN\np264ZT73t3xz5t3EIOq7h/Y/C61GV9HuAxBaZTuSmXikE65P6QrKaWSG0LbDdzQuXakVpzmB\ng1le/nPkr8089MqcOs25y2LvZ2HWsIXj6nXnPLL2IcVivFH/tGE5Abj1/bRfJYuafcwgVXs/\nRTMgoCdk5hy34HgOs04U1rWEqat9VSvQFwtUeoISQRPkLdp7G4LmQo4nz43nHcnbl+ygcTPv\nf4NtLGe1lMBRsNyxdCkRNq/nrcU/OICLE/lGCBxtId/MKSIgHjBSRZ8v4pAy5VUoFC1MPZe8\nv/7rv17H0RQbDM+1v/sl58WvwXUAQNOMhz5svvfTjTRiXBa07U5+94d+8buaP5k1wu/8BU+c\nqwzwjTfBHh36Gf8Twklx8EPy9NcWBDUTux7E4Pf9L2HnZtb60kVxAmYU5mztgPRpuVcV5/Q7\n8+rGHG6RR1+g7TUKgozaXUuXONQ02OObbyI3COki3EO998NoUQdHriUfgMH2ssqFGk0tlw2t\nRX+lis0HAXFDVykbLUhvLDhZdEYypbmIqQtDUM5yp51hCTA0EVhkL3o1VdwaD2q13UOdGnUW\n9hJd55tLKm/92Q8vv35hJuG0IxHYPZBsW5SZMv2rqHtVTW88kAwbqWK118ZAZ2Rne+RaqrT4\nlG1tLSQMKTYxcgP0O1e0InX7jv+jP/qjeg2l2IhYf/d77mvPzT/2POfHX+ViLvDxX12/SQEA\nbbuLdt7DVxe4PCDcJo59dJ1mtFoKYwvVDQAAg0dP0u73I+ifL0P7nhbJbfzudzg9Aj1AHQPi\n4IfgZGUNgYMCMbhlvvAcD74y06cm2ksHf5Lad0OPwc34nKNXWBvmamR55C7XXJKZPTB7YS9q\nFBcagL7etmFWWp79E5Rmq5ymzvHoq7T349S2Z12n5Q9RqIbtmwDVNGxrJmQOsHXBJ24MNH8y\nGx2GtLyMJy2ANQoEtARtsC7CeearwLS1YZRoJzacSaqi3hzqifXGAjey5aLjhXStNxboigbK\njpcuO8xwJF+a9Mk99CTnbLctWNPAJWyIgp/8G75VK5bmULa9f//lU+OZ+VsFkxkrderm/Xf3\nxaMLbhFpghrhGSKIntnf/f3z45MVv6ZtydAjuzpMTezvjr07lqt8fkfEvKMBiSQKhUJRL+om\ncHz2s5+t11CKDQfnUu7r314cd0/8wHz6U5Tsbv6UKiDxxOf5wo/53POcvoFwgvrvFHc/i2BD\nTA25kPVGB8kMiN4dZNQze4XzN3yiBIA5f4NqCBwAqGsfde1bOFYbggmUF6kVwSRiW+Vr/w3p\niiyM/Ci//vu493+myAEuD/uMHzkw/0DWsNrxlrLgoa4P8MS3YFX8gMEd1PHMEqc0B770d1ya\nWLCedMt84W/onl+F1hKSwQK0PjhnfAxNtL61t6WoD1oHhY5y6eSCSRrbEDxQ+xyFDx5bBeeG\nnC2Pd1C0vEzE6NPFLcwIWoZx5rMVFVUp5jTRfqBnPSelaAE6wmbHwnYhQUPrNTQAN3I1sggB\nv+a/8/S3hcYLNmZsjSviiVu3YmkCL5werVQ3ppGSL11LHT204B3RG2vUGzweNH7yzi2DqeJk\nwdYEdccCc84md/bF++LBy5OFvDXTz2Vne6RVvVkVCoUCaImqbMUGgfMpeeUUspOU7BG77kZw\nPqtcXr/kv75g9oYv6EsLHE6Z08NwypToQ8S/1etaIaJ9j9G+xxoy+Cxslcpf+0PrxW9ASgAU\njgU//JnAox9a06DS4cEXePwkymlotZdiKzUTIY0OfpJP/vEC3UEPiEOf5PGzC9SNaVjy+W/S\nQ/8LrFHkT1cOhPhRhCoarwRqFJXUik+jRann4ygPwh4DBAI9LeEt6uQ5c8lnFecWOX2BOg43\nf0a3gIJkHmF7oXwg4mQcWr85LSJwgPQ+ti9D5iBCpPfD6FvvOW08is6YXGj+x5BF92bc3I7a\n1owtg2S+sMgvhpkvEnW2ihinaD0ipv+SlYDIkrkYW+LBVMm5OlWsPGWgI9Ibawmd+oJPi1YA\nSGcW3BgwNHFkawOtzYiwoz28w8+UpDNidkZaqN24QqFQLI0SOBTLwnvzOff7fwJ75iYDRRL6\n+/++OPDAGofl88/L0/8DzkyFJ207Ku75JIIbMvWx8If/1j375txDLuZKf/k7cJ3AEz+1yhGl\nI9/6PcwlbjiFmZ6aVZBG8f6Vjk3JXfTQP+JrP+TsMIgo3k873gMzhrNf8z8hMwzpUvuTiOzj\n4kW4OehxiuyH2bNw2Lt4/LXFfXmp4+5bzym4HcHtK/1BGojlV49zy0Pri9ZPwXZ2r0DmQAaJ\nDujbgRYz09USFDq63pPYwHhse35+K5JdV5Z1scqb0sxSwhOkU8MlkhxQXe0PAHCBDNACzjuK\nliQe0NuCRrpc/eLpiQVM7Rafcod6YlviwRvZcsnxwobWFw/WcNZcB3z9RKcJ6qLsSk1Qbyx4\ntL8tFlCLdoVCobg16rNScWvkxePut/57ZYQLGecr/8n83G9R9w5vatxNZTzb03SBKpcvIq2/\nplsBX/ihPPFXCyJDJ2RhUjzzj9fa30R6PHIK2RvQg+jaQ+1LbZs5NYTJawDQsZOSK1YKpnGv\nnKlUN+Yof+tLgcc+DG01bzQeeQWVZSkEEC3OlKEdj8FYld1XIEb7Ply9lanZuIQhXQgdga20\nRHpFsJO2fZiHv1XZF4M6jlLnvauZ4fri/1tlgKC3sL8ahVsrZUNRb2TtxkNLHFoCV9oFN+3M\nvmeDWiSsJ0QDm0wtMUlf4UNx++JKtlwvoGu6IACHemNnx/JTxfnvl55oYF/nsgpOkyEj2TKi\nRiXbOiMnLk0uju/sjn70rq22Jw0hVEmIQqFQLB8lcChujff6N32i0nNe/rvyWL704+dmdt2C\nzFjArMj51O9+gpI1aqqZ5ZnnfMJTg3zjHdpy56pny+lh+fIfIXtzLkI77xf3/RzEole7XZSv\nfJEHT8w/c8cx8eCnYaz4Fqh3dZH95/RkijlvbETr82+8ujScOl8d0gQgwbMFx1qAdj1N2x5d\nxeA1ifX6x0NJ6Muq/qXkYYru5PRpWCnoUYrvRnhLPWfYNAJJhLtRHFsYJQi9NU1GFbcJoraZ\n6Cp8Rl3pZOxxrsgNK3sFR9ptge6GpXIs8UnSEp4Iilag6HgXJvJTs609OsLm3s5IyNCO9MUz\nZTdvuwTEg3q0Rt3KBuKJO/u+c2KkZFc3dP/AsX4At0xOUSgUCkUVG/6LQbF2rHfPFF9/2Zua\nNHYMxJ56n4hVW9nzxJDPaYz897/jpCpy9SXbmTIAMxaA0IwH3m++/7M1r1pKo+xfd8pTg6sX\nOFxbvvC7KKUXDHj1NRmIirs/VvVc+eIf8cjbC5557U3JUrznl1d83SUszhbVaywX189QTRMI\ntos7fgYgRLp9VJu1QX1H+eJ3Yeer4ztX4mBiRKnrwTrOqiF4JZ58kQtX4OVhtFHiCCXurkod\nErs+Is98sTIbBQBt/wmYLVpFxfCYHSKDlmNkwGV4GUBCawOpjeWGQaOAIH1xsgaRtgqT0aKb\n5UWVbx47llcIag1xYgYiQBSo/pABwkCLvrMUi5HMjcsrKLvy+Ei6sr3rZNHOjbj3bWszNZEI\n6ong5lm+JqPmr/3U4T/8zvnR2Yas4YD+iUcHju5ujCuZQqFQbHY2zzeEYjVIOf5//9vM3/31\ntC8mgKnf/53uf/Z/RB5+fMHTNJ+sTrfkLFA3ZnHKiP2Df6b176H4Onw388ipKnVjJn7pRdz1\n7AI5IHuzSt2YeebgCeQnEO1c0XW1/t2+cQqERNcq/TIpmPTtnELhDsQa5sFphMS9n5On/hz5\n2RQYodHAE7TjkUZdcV1wc3LoS3Bnt1j2FI//AIUrtPVjCzwaY9vE3b/CQz/g3BCkQ+Ee6n8P\nYq1kFDKL5LItr3s800NRo1hAbCGqZQsn2XoH9sVZO1KCMUCBO0HqG2FjENa7886NhZY8FNY7\naeV+Kw77dzhypB1smN0n0R3Mp4FSRSxAdHAjOKTe7pRdOZgpZS3XkxzQRW800BMN1P3PNpQu\nVqob09ieHEqXdndEfE/Z0OzdEv/Xnz727kh6NFVqiwT298ejtbveKhQKhWJp1HL2tib1pT/K\nfPXLlREvnRr9F/94x5e+pvfMtzYQOw56qdGqcz3Lv1iarTJinbdWN0JtCMZ9kziofTUFHTNX\nz1bPcwbXQjGFaNf8M9MjNQdJDdMKBQ597xFtx37v2rtV8cB7Prj6ZrG9xzBxxifec2xZp0sL\n1gRIg9kBsZKlUnyreOQf8sQFFMZgRKh9AKHNZvvHky/OqxtzweJV5M5TbP+CaKCN9ny0xXdd\nzHbZu8yYz3D2OFfyLoX0veT3Ic/lk3AuVwbgXGa2KNTyeTcKAIAuQnFzW9lLebLMgE6BgJ7U\naupZS1Ij+4yXbry5VsJE9wKjzFkARDGgr+XccBWLKDrembG8N/vasFx5LV3KWu6+eosO6ZK/\nUctih9Emw4ys5RRtL2Ro8aBexxwWXaND25OHtifrNWAVnuShdCljOcyIBfTtbWFDa/FvNoVC\noVglSuC4rckuVDemYcvKfuvv2j/zS3MR7ZGf9t59DaUFG0IKxYDC6q9NJA6+Tx6vngC176C+\ng6sfVTdrrsq1hav/JYrVV1H3QRT9pX9Z/LPfdk6/OjsIBe7oM7tu8sRp6lxNS1Fq348dT/Hg\n89NFLgwQCep/lLpuNRq7PPkSp49jupGkMKn9QUreu4K7o6RR1wF0HVjFtOexhtkeBbtkJBHc\nvfLsAAa7oIbcxeLCFf8DxcuoEjg2AjaPV6ob0zBcR06aYpEJDltw/H58dwQyB6FqBDYGgoyw\nvmQH7uWhCcOVPj1Z9BWpoqtBAFuINqZBz+3KYKbkVSlfjFTJyZTd+taMSJ+GYQBQu99IM5gq\nOm+PZvPWjPgSMfU7+2Id4Q3QPzVvu8eH02V3Jld3LG8NpUt3b020qTwRhUKxGVECx+0LO45z\n47rvIefagv0PtXWbn/137rf/UF46CTBIiEOPmNvvKf6nX198LoUiWt+ycvhp7+MCkG9/vbpN\n7Frc+7v3+sfjPQgtaCBPXQMg4WOQIXTqHFjFlSmWDD9zzN1elKkiGZroiIqgDnb40lcpvg3m\natrX046nqPMQj59COU2BNuo6jGjfLc/im9/m3Nn5x9LmiRcgbepoVpkJO5z+HqzhmUcA8ieo\n7SkYy9uPuSlOvwRrGOxBC1P0CKJH1tpYpwrpn5YPr0a8tZHsLzX6x72MX7fh6UMpJXDcboS0\naE5OVQUJFNQ2YSGAYi0wI1telFhBAJAuO/UVOKKmXlxkugkgajasbupWFGz3taFUZUvXgu2+\nNph+dKC99bu3vn0jO6duTGN78u0b2Ud2tqsGLYpWRnKrv7kUrYl63dy+kK6TYbLjc+9OhKuX\nttTeZ/zsP4dd5uwkJbuhGWA2Dxyxz52sembkQz9L+nLvCdDex7WdD3B6CE6ZElsQWattB3UM\n0PZjPLiwYysJcfdPVz81GKc7nuYz36ke4eBPILCqlT1LnnhbS4S1RGUPUYZ0eOId2vLwasYE\nEOmhyE+s4Pn21AJ1Y24eqTcoeR9EM+41cfalOXVjBi/Pqe9S1ydunZHhjPPY34Jn85C9Imde\nhnWdOj9UzykaCdjVmzoAMNrqeZXmUaPKwDeuVrOKCgJaWLJXaTUqSIsZ7Uv0alHcnki/siWP\n2XK8K1PujWypLWgMtEcCek0x2vZkquh4ktvCRthY6gXWnwiOF6yq6xGhP7FudsiXJ4veogQS\nyfzOaO7+7W0tJRN4zGBoYmZKecvNWT4lPyXHS5ec9o2QgaJQKBQrQgkctzFEoXsfKL78o8VH\nQvfVKMU3g9Q5a29JlPjVf5374n8pvzSjEVAgGHn25yMf/rmVTcMIUleNtItVIR74BU5uk2e/\nDbsIgJLb6ejHqMunr6c4+lEOJeSpr8MpA4AZFnd9mPY/ucoLu0V4i9UiAgDLx/e0QXDZx5cU\nANiFNYZQ/4JgaYJzQ5A2Qt2U2FmfvS87KF/yicsirEEE/d1Y589OvzyvbsxRvobyNQRXb85S\nBcUP88QLi8MUP1SvSzQTQhDwUyrJr6eGaAPIXxPRNpvZimI5hPRYQAs70pLsaaQbWrBhDWJv\njWS37E25ssTsCTIDWsLQYiXXsqXLzIbQw3qgpTaTtw+aIEOjSu9Px5VFe2bnbLsyW3avZ8vH\n+tsSiwofGHjnRvbU9YwrGQABe7qi925rM2r0QE0EjTu6Yxcm8nOXMzWxryuyjrkSmcXZKwCA\ndNl583rmYFcssn7ZJXOM5a13x/LTckYkoO/vivbGAlW5G5WUah9SKBSKjYsSOG5rOj//D4dP\nHpfFBXns4fseij721HJOF5F44vP/PPrxz7lDl8kM6jv3iEh1i1mw5MwIipMIJSmxte5tTf2m\npdGBZ7QDz6CUgRGAXrtvIgm64xlt/5PIjQFAvGdNdRBaACSYpc/SW2/mTaeaNcoMnp8be3z1\nOb75xlyRDse2iT0fRXAlW1xpwxmHtGAkoc9ao3n5Wp1x2c3cal8iYfmXTXF5mOoocLQdgz3B\n2QoPV9Kp60kEumqf1LoYotPzFvv1kkF+KVFkwtwN++KiUbZDNKgtqKLVEaQFtPCtn9dgPLbz\n9gjPNPeBx1bBnbBsWzJh2ofIs/NuqT0QM5vwVaJYRHckMJKd6V/OzCWnes/vSn7nZu7hHdXf\nI6dGMievz7ddY+DCeL5oe0/vq/mR2xMNtIfNVNG2XBnURTJs6qJFhS3LlWfH88e2JJqsvDme\nPDmUvpEpM7g3HuyIBc+Nz3ul5S33zeH0Hd3RjkjNHA1T+YwqFIrNiFoi3NaYu/du+4M/n/id\n/1h8/RW2La29I/HTn0p+6jMQK9jna529Wmev7yFOXeWTf8m52c4m4Q5x10/TGq0rb4ljwQgA\nqDLdqInQkLi1scUyxjEoMYC0T/ICJZvnW0mBbn+FgwSZ861hePB7PPragifkhuS5PxNHPr+U\n/WolhXc49xrm7AkD/dT2HmixJYpQanctnZuWN9u4dPGhuprnk6CeDyB+GIUrcPMwkxQ7BGOR\nPLdB0CgS0LbZ3vU5q1GCEdC2+GdwABS4i2HAPo+Z5xPM3RRYjRWuQlFHyu4kL/wEsGVYzgqz\n0/9I5pSV7w4l1jHN5LZlayxYduVk0QbgSunbaSdvuQXbq0xn8CS/M+rTMW0kU5oo2J21t9+G\noO5ooA7zrgdtIT1TthfnOU5XgpRdL2M5zfTsnMhbf/3mcHa2rcy7ozldE9u7osGFiSTvjhee\nToSCulicx6ELSoZUfYpCodiEKIHjdsfYtqPvt/4LpJTFgojW1V+wOClf+W9wrQWR1/5APPqr\nlOivfdpimG+8waNvoDQJM0btB2jHE9AWLXqskvvjv5Jv/5ALGQrFxIEHtCc+ReGmbllp5/v5\n9B/CLS0I9j2AaBM7BQS6KbyTi1erwpS4C9psIol0efQNn3NL45y+uCw5pniOMz8GVyz2rGGe\n/AZ1fRxaFHob3MVVOYTA1kXBqqcY0MLwij5H9Pq7Y1BoG0Lb6j7suqBTm6bHPM4zO4JMQVFa\nqukmUeAgzD2QGTBDS4CasouQebjDLAskItC3QGxURUnRGNiRCz48GcJjnx2jx9LynKCm9mbN\nhgh72sM9ETNjueN5u2j5+IACsD0Zwfw2O11y3BrtTyYK1hICR0sx0B4ZyZSrfhAimMbMJ+0S\nlSCN4OunrmcXNs11PTk8Udi9JVap/UnmVMk52BN/63qmUpEi4I6eWMsmxSgUCsVaUAKHAgAg\nRJ3VDUBefmGBujET9fji9+nYLyx7GObTf8JT7848cstcHOeJd8TRX4ZRYQXqWPYf/zqPDc6c\nU8p5J74rL580/t7/2VSNI9QljvwKD/2AM1fgFhHupr6HqGP1XW9XB/V9GGPfm7caJUGJu6nz\nPfPPsFLwawwJAIWbWIbAwbkTwKJbWW4a5csI7aX4Qzz1XHUuRvgg9CTcHIrn2c1Ai1JoAOai\n/OTIIWRfX/QjGQjvu+WsbnMImk6JFdzVJhNaE0ty7HfZOjP9qmAA1hmYByjQ7HeHomVhcFWF\nHdcW6dyFdXAMONL1pNSEMISmkjsaSiygxwK6IWg8X/Z9whI+oxuXiKk9sD158kYmPyvraIJC\npqbN1qU0UywYy5XHcz49v2zXK1leeKFTiSu5JxZ4eEf7xclCpuwAiAX03R3raWiiUCgUDUV9\nuikaRmbYN8zpoeWPweOn59WNOcpTfO17tOfZuYD35nNz6sb8uZlx76Wv6M98ZvmXqwNmjHY/\nu86LaxGg3g9S+4NsjYMEBXugL1R5lihCEcuoT5EWfBwfAIDtcQrthbmVOp7l3GtwboIl9DhF\njiC0D/m3OfVj8EzZNmdeQ+wIJR9bMLX4MfZyKJybD2lhSj6FFjAIUKwe9yZbpxeGGPZZaG3Q\nm5jfpGhhCIIgFpSo1HDzASAqJAxbumm74MrZbSeJhBkJas0rYsq5dQAAIABJREFUFrg96Qib\nhiYcr/pvlAhWd0hJhAxdkOvxYt2pY0O18GgLGY/t6nxlMGU5Uoj5NiUABNFia9XGkS35O54C\ncFyJhQl50YAGIGxqd/WppDmFQnFboAQORePw2+azf7gmi9WN6WEm36WKvijycnW32tn4qZVc\nbHNhtpNZwzE0mIQZh+0jUlBsOUaetf+Ecx5rRie1fxBgsAfSAcAe5annFz6bkXsLRgeilbfx\nBSWfQuQwrCH2SmS0I7SnOd1tFY2DnSu14qQEDsUsphazvHkrSkFSQMpFeRwEBGb1C5flZDlX\n2RHZY5mych3BuDIibSi6oMO9sVPXs15F4UNAF4d6q7NBdUEHe+OnKkxGp9mSCHa1jMXGMhHA\n/s7IufF8VcnNzmTIaGIGR7B2k11toW9oW8hopvKiUCgUrYD6+lc0jLZtmLpcHSRQ20p6YTil\nGvGFNg22f6IsrIJ//HaHaPvTfPEr1dH2OxBbhj2KMKEnfFuikNFdFZhRNwDOn130dADgwjsU\nXVSnYHbD7G65LHNpARKimT1xNguyxjtR5v3jivVDspd38o60JEtN6GE9EtSa9JoP6u0eO66c\n/3g3RdGS0bnN5LRCHjPD2mzHq4JT5kWtoxgoOGUzoLoCNZauSOCRgfarU8Wc5QqitpCxIxn2\nrdQ4sjUhCG/fyHqzHha7OyP3bU8ufmbr0xE2j/TFr6ZKOdtlRtTUtreFmmkvartyvGB3xIMS\ncF2vUHbl7G9V10RlfUoiaNyzdXlu6wqFQrGJUAKHolGIXY97Q69VKxRCpz1Pr2CUYA1ryeCC\nhRElezFyfvGzqF3dHPaHuo5AM/nqc7DSACB06nuY+h+71Xmzp8fuRep71VGjA8GBmue41bfv\nZnAW25G2HqXLnHl55kfQIhS/D5HGm0d4k+xegcyDAqT1wNiJpXxDW5tavXVq99xRrAsuu2lr\nUs7WhrjSydppW7PiZv1dfhdDEFGjz5EFV5aYPSFMU8QkKGeXbOky2BB6VA8GKspPHOmfq2/X\niCvqS1DXDnTf2sCLgLu2JPZ3x6aKtiu5PWxEzA28/oya+uGeOtuWLZOpov3dd8fKrtR1AcDU\nRShgpHOW7XpEeP+h3vZoIF12mNEWMnpigZa7T6BQKBSNZwN/wShWjTd4wTn5Y5kaF8lu49jj\n2pbam9K1EGoTD/0Kn/oyp2fdMaI94q6PU3wFPVmp+26+/qpPvPdo5UNx9Gnv9AuLn6bd88wK\nJnybQe13UPsdsNKQDoLty+0OO01oD7Hk7CuY63oQ3EWJR0C1d+C1trKi5VOU86c5PfvqYsAr\ncOp5uBlKPNS4a7J9Bs68ZsfeTbiDFHykiYoAAxJYyauiNqT3sDfuc0Drqcv4inqRd7JykfNF\n2SuFZNhoVqWYISKGmPeQFkBy5bkYal/XggR00Rf3712tWCYvXp6satdChLZYIKaLR/Z09sSD\nAHpi6/mtyozrufJYwSo7MqCLjrDZnwjOWbEqFCtCcn0WIYrbDSVw3GYwl776B9bzX4GUAMAo\nf+/Lwff+bPCDP7/8Mbzrl61v/LE3+C48R/QNBN77c/q+o77PpMRWevRXkR/j4gRCSYr1LrUB\n9iW+jXa9n698Bzzfjo667qStD1c+S2w/qL/377nf/yLc2a5pQtMe+ai4Y8HTFD4EVntjNryP\nQrvgpuCVYCSh3eJ2FoV2cGlRyRKA4PZVTqA5sMvZV+Yfzi3ScicRvRNaY3LgvalKdWMGmWbn\nLJl3NeSKlXCG3bOQaUCCwqTthta/1g2jsRvOIORC2xcRJVM1x2ktbM+nNQMAy7OaJnCsCFPT\nfZM1DE0tbxSbjXTJSZWcxXEiPLC7o6cFxCNmnB7LZsozb8mi4xUzpcmifVdvXLWkVSgUTUOt\nAG4v7OM/tL7/1/OPCZCy/K0vadv3GYcfWM4I7pnXiv/vv4X0pmuhvatni7/3L4If+Zz5+Ef9\nTyBCrIdiq79PS/2PUvs+Hj2B0gQCMbTfQe17Fz9Nu+8DYu8xefZlTt+kRJfYey91bVv1RW9P\n+MZpHj6OYgrhJG07Rr2HbnEC6TC6sMyUgsgdyJ+FPbogqEUpcf/qZluBhJuBLEJL1F9xcCZq\ntNSVsG4g7PNSXDvsjfgfcEfQaIFDjrPzxny3Ti6y+zY4S/qtXgxLQzqFn2D7HJwhcAkUhNFP\n5h2NTUhxR9i9Ol3jA62XzL31SkjZrHDtriWMmofWl4geLLqW5AU2HASK6corR7HZKNreKg41\nk5t5a07dmKPoeCPZ8o429ZZUKBRNQgkctxf2q9+pFV+WwCFl+W/+K6SHhfdzy9/8Y+OeJyjW\nMMOwcDftet8tn0Vt3dpDP9moOWxuWMrX/5iHT8w8nAQPvUnb7xXHPo16ZZaSRj0f5eybyJ+B\nl4cIIbyLEg9hjf6F1jBnX5pvWxvop/gjt0wnWQFcu5Kffe6k1emiNUxz2Vp5I6IVXtk9g0WW\njfAGoe0ArU08IoPMvaxp4BxAoPCK87lWAlvH4c4Wx3EJMs3eMAUfA7V8SVRTcKXlyJKEp5Fh\nioggDQCRECQWl6gA0FZUwtZENBIdgXjGLszlcRhCS5gRfTkdrxWKDUXIqPmZGardV6WZTJV8\n7wdgqmgrgUOhUDQNJXDcXsjJUf/4xI3lnO6NXpPpCZ8DruNeOGnc88QapqZYT/jaq/Pqxlxw\n8A3uPkDb76vbZUinxANIPAD2Vmb5UQt7lFPfRuW9ZWuYp75OnR+vW2qAXruEx6jRhXftUI1M\nYwo01luAS2DfdicMOb7W7Bg5xs4JwJtVaCZYDpJxP6gBXn3ezXl1Y34CebbPUuBuAAAzjzJS\nYAcUJOol3D69BrjgTlrefP+aEtIRo8MUEQBBLVR0q18DBAo0q5HKKjCE1hmMu+x5UmoklLSh\n2Ky0hc14UM8uSpEI6KK3BepTALhykT4OAHBqxBUKhaIRbFhPfsWqoFDEPx5e3h6jVLPrKpdU\nu8cNDA8f948PvdmQ69XpbjDnj2Nx5rxXQPFcXcYHAC2KkJ8Lr9kNs1EGmaTV6P6jb23QFWdZ\novHEGvOfXXZPTQ8yr9Cwxc6ptQ3rD7s1FFtvOu5KeUryZeYUI888IeVpyVcbMZMqJLsu2+tb\n7lH2spXqBgCGzDsTHjsAIkbM1BYkuRAoZiZaNoNjDp20gGYodUOxiSHg4YEOQ1uwdBdED+1s\nbxGHi4Duv60I1ogrFApFI1AZHLcXxsH7vMELPvFDy7pLTx01d3Sio3f101KsN1yq0cO11No9\nXB2/xhwAO2N1XOtR8inm76BckRFg9lDH+xqYTKF1wNgD5+KCoIiTcUejrjgNhQDhoxkBIH9t\ndLnISbBf6jJnwIW1Du6Df5r09BwkDzGqBVnmEaZ2QrzeM5mdkCwW3Sk5W/EU1OIhvY3W4x5D\n2cv5hdn28iE9SaA2s93yyra0maUm9JAWYsi8M+ZwmZk1MkJ6whR1/5MpFIpb0x0N/OThvlM3\nMhN5m5nbI+adfYl4sD6LeduVb16dupkpaxr1J8NHtreJihrV0XTpm8dHBicKDO5vj3zgnq1b\n28PV04sExgs+H7/dUVUbqFAomocSOG4vAk9+zHnrx97oguRtbdte87GPLOd00dal7z3iXjhZ\nHW/v0fccqdssFU2HAlHO3fQ5EGzUfq9O1Mp6rWs2rAhQ54dhjcAeA0uYnQjuqOf4fpB5GFoP\nu1cgc6AgaT0wdjUq586bgjfBbJOIQfRBLrI4pSBE95ouUctVZPpQ3QUOWlhPMbNGZyAIgNmv\nzg5gniBqyAveloW8M1apiJW9rMd2zFgHXVjWsJXxKuIBLRjQZjLeXWnlnFGefU95bOed8aBm\nhfWGlWgpFIrahE3twR31f/cNTxX/8rXBXHnGW+r41amXL058+uGd8ZAB4PRg6g+/f9GbrTRJ\n5e13htKffnzXsV0dlYMkQ8b2RGgoU6r8Du6NBnqVwKFQKJqIEjhuLygUif6j/2x9+8/t4y/I\n1Ljo6DHvfSrwzCfIWG7/v+DP/lrpv/8r78bVuYhIdIZ+/p9Cb2Q3BEWDoS1HeOKST3xra+tW\nRmd1WxYAABmd9b9WYCsCja4QWYjWRVpXg68huXwczrXpBwxAhKHHgIqb/BQm4+ha+48sYe3Z\nANdP0vvZuQQAJACaFxbIAJcBu4ZVa428jzVTdFOLr+fIsiNLhmi2twVB+NbIUA3P16I3xYsU\nw7KXDWhRjVqxcayi1WBgJFMazVklx4uYen8iqO7ntxqux3/1+ry6Mc14rvy3x4d/4ZEBT/Jf\nvHjVW+ijIZn/6uVrh7e1BRb6m25vC3WEzbGCVXZlQBMdYTNRpwQThUKhWCbqQ+e2gwKh4Ed+\nMfiRX1zd6SLRGfm133aOP+9dO8euo23dZdz/XjJbwt1KsWpo16M0eprHzs+HGNR7gHY+tH6T\nqsAa5cxrsCdAGgJbqO1B6DEAFDnCiwUOEUTowDpMcgPC1pk5dWMGWYTjUuhe5hzgEcWg9dWh\nu6roAHQfjw+KrrU5i//lkmQeZOdcdQ8gLrD9KowwyFfLaMh2XbJXK2nClVbzBQ5DhGzp46bk\nOxNm6UrLdxxHljRNCRyKW+Axvz6UnpwtW8iW3RvZcn8idGRLi6cHtgSeZK0p5hpXJvKZkk9f\nsCvj+UzJmcyWs35Hi5Z76Wb+YH+1Q3PE1AbM6uoVhUKhaBpK4FCsHKEZ9z5t3Pv0es9DsSqk\nA2sS0kOwA7NZ6BCaeORX+OrLPHyCi5MU6aD+Y7Tjgbr1iF0LuZM8+fx81YmT4uJ56vkYAn0I\nbKPEE5x7BXK2AsLopPhjEEpxWw4M54pf2IYskbGvrtcyyDjMzsmF1UM6GXfV9SqVF9wL74qP\n8YfMEjq4KlmDAQJRAxJ/ZkavdWAdOguE9DbHLlUlcRgiZAqfDckSfqi+3WQViiquTBUnF5ky\nDGdK3VGzrzUaf7Qgjidfujjx9nAmW3LCpra/L/74/u6w2UAD3XSxZv5aqmAXrJr+04Vyw9ql\nKxQKxWpRAodCcRvBE6/z6I/glQGABHXeR73vgTAAgIgGHqaBh+t/VWnBnYAsQ49D71yZN6dX\n4KkfVW8RpcOT36MtnwaA0B4K7oAzDq8AvQ3GCse/nWHb3/gTYJmr/y9RbCEzzt4lyCwgINpI\n29OI+pQZuFzrpyNPgxZhVGQxEIi2NshhVJBOpDH7tKHRxTpkQGhkJMwtRTflyBJDCtKDWiyo\n+f/sRBqBFgkxDJBGav2guDU3sv7+OzdylhI4fHEl/8lL125kStMPi7Z34lrq0lj+Fx8diAQa\n9aYLGTXVk5CpGaLmB3W7qjZSNBKpNqqKVaFeNwrFcuGpQYxdhFNCYgv134WN1o+Qx17iG89X\nPJY8/irsDO38WAOvWj7HhRPg2Zs8eifFHobWttzTS9fgm95vT8DNQE8AABkwa/RV3XAUh7k4\nAmkj2E2xvahhi1AnlngBN+argaKkN83VZQmJRghxhPkGIwW2QSFCH1F1lnUdCWrxkpuqCmpk\nGH5JE01AkB41ugAwmJYUBAlkapGFbWUZIIJQjVQUy8Fy/TN9ys4aO09vWt4aTM2pG3NkS86L\nFybee7hRtsQDXVFdI9erzilLhI3uWBCELcnw9VSx6mhnPDjQ3YAaQ4VCoVgbSuBQKJaB58rX\n/pQvvTwfifeKxz5HyW3rN6cVwi7ffMknnDlHpZsI1WwAvCasS5x/bUHEneDMdyn5LJZpTyir\n13nzeMUZgWNzIB2+/j84O2+DwoEO0f8sAg3zGSUdWju8Kb8ja+uZ0gpQEBT07d5CIgkQ0RbC\nluak+4S0BFiWvOxcLpIuAhG9c2lxoQksZwJhvV2y68xVgYGItKjeWcuUVKGoJKALX40jWDtl\n4Dbn8riPRQ6Ay+PVza3rSCSgP32w97m3b1QGhaAPH9k6Xaj6C0/s/t1vv5uuqDaKh4zPPLFb\nNMUiRKFQKFaEEjgUilsj3/rbBeoGgOyo/MF/1Z79V9A3SH5maRyyRj1CcYQaI3Bw8R2fqCyi\nfHm5PqA1MucBQN9UHnU8+r1KdQMArEk59BWx+++hYbUAFLiTiz9Clc+CvhUN797SDMjYz3Z1\nT2uI5Lr8dCE9GdBiLlvMUiND3zg2MQQRM3ptWXRlmSE1MgNalBrUsVix6eiLBbNln515X2yD\nfHU2Hdv1z22plQtTLx7c3dkTD/7w3NhopqQJsa09/NTBnu7ZMqK+ZOif//RdPz5789pEAYxt\nnZFH7+hWKpVCoWhNlMChUNwK6fLFH/vEiykeeosGHmj6hFZHbTtDbpDToYSX9r+gN7Xcmz6h\nHdBC8BblcQS3QdtEGfLS5oyfGGSnOX+FYnsbdV2tk8JPsnUK3gTAoACZe2E27HJNRttOJtg5\nO2/GofWTcWi9XFoE6eaG9a0wRdjXhXTtuDLryIyELWDoImaIZCOuolgvBjrCEwV7cqGH5dZE\nUBlw1CIZNgcnq4tBACQjDbfsGeiKDnTVLDkxdfHUnX2NnoNCoVCsnY262FJsYFzb+fHXvAsn\nOJ+mzi3Ggx/U9h5d7zktSTENx98mjTM3Nkx2ZrATpMHP6bBB6RsAAVRDWFn2r02Y1Pk+Hv/G\nTPqJZC5YkKZou7des2wJ7IzvnwYArEk0TuAAoLVR+D2ABDsNtPxcL7TtpG2FzAEOKAZSe6rW\nouyNODIz/X8Jy/XyjsyE9O0qQ2TToBE9sCM5nC7dyFllx4uY2tZEqFelb9TmyPa2k0M+9waO\nbl+2d5VCoVDc3iiBQ9FUuJAp//4/k2PDM4/Hh72zrxmPPGt+6HPrOq8l0YxaR0hbhyYIq0SY\n1HmMx1+rjke3I9LfmEsS9A64Ez4HVmTxENpJWz/D2ZM8eJLPnEG5DMA7/m9o2xHx0M8h0l6v\n6a4novZHsaj58qvvDDahujGDBqE2Bg3E8ixHWsysCT2khZbvzTGdu1EV9LjoyElTrKaMiFUL\npZaEgG1toW1tofWeyMagPxl+7+He75+9OWf5SYT7d3Xc2b9JPscsV9qejJiaaIU+9AqFYjOi\nBA5FU7G/86fz6sYszotf0w49pO08uC5TugUsIW8ikkCheiEOAL37mz6h1UN9TwHEE6+DZ0p5\nKXGA+t/fwCuG7+bs96qTOLQ2BHaubCAtglyEjx+vjPHQSS8zqv3Uv1xCgdowmEkYcTjZxUco\nsmNZI7AN9ya4AApD79m8asUGhuG5MiPZJtI1imobP52EwRk7ZXvWzGMPRTcfN9vM2k0lK3HY\n5wUPwJG5FQkctvQytmV7LgBdaHHDDOkb/zNBcRtz7872Pd2xs9czUwU7aGoeo+B6Xz99oysa\nONKfCOob1fliomC/czOXt1wARNiRDO/viurKplShUNQbJXAomor3zsv+8dMvtaLA4aR4/Ouw\nJ2lHjM9UCxy04xh17V6Xea0SErTlaeq6n4vXwS4FexDsbOwVzS0Uf5ILr8PLzUQCOylyH2jF\n6zN+62s+0exNvvQq7Xt0bbNsCaj3aR76SnUweTcCy/gbOUNsvTXvNEEGBY7AWJ4yomgKrsyW\nvRuMuUKkMVO0B7RGNX1sDnknO69uAAAky4yd6gh0i2XkcbBvB2hA1oj7UvLcyfK8YYEjvUmr\nFGcZN5TGp9jAtIWNh/Z0DqdL3z570/Fm7kmMpEvnRnMfPNzbFd14L+/xgv36UGrO8osZV6eK\n2bLz4I52pXAoFIr6ogQORRORkgv+t+w4n2ryXJYB89jX4UwCoPYo7tyOK2OcLwOAGRYH30sH\nn1nnCa4OI0aJJiaemP1kboWXhSxDS2B1zSOkx5NDvkd4/PImEThie7HjZ/nm8yiPAoAepc4H\nqX0Z9jTeFJdfX5Amww6X3yQRgdZgAUuxPCTbJW+kKpXJllNEpik2ao0Vg8uuTxdnyWx55ZB+\na0dSIr0qu8uRYcuLSdZzTs4UWsIMGuIWQknaWjwHztlWRDc01chWsZFxJf/g/NicujFN2fW+\n/+7YzxzbOC3qZ3l3LL/Y0Hyq6NzMWcqTRaFQ1BclcCiaiBAUbeO8j3sWxSt2Yl4JTg5mAsvL\nc24U5ZFpdWMaSoRx907yJFxJuz8PvabTuGIRBC0BLbGGEZbo89KgFjDrAEW2065fgHTADrTl\ndqxg55LfL4HZvkghJXC0BI5M+75QHZnauAKHZMl+PxQB3vJSMAxKuJiXvEtem+3NfK4yc9lz\nrVK+MxgJaDUTvhzpeT5NoIgBy/PCuhI4FBuYG5ly0a42n2YgXXImC3ZH45uq1BFPcqbs+B6a\nKtpK4FDUQvJGLchSrC9K4FA0Ff2uR52Xvu4bB4DymBz9Dooj00GK76Oep2DEmznDeRy/Fqea\ngBBwU7eDwOGlJtyhyyIc07cNkJOVl55H7gb0ALUP0K4noDdxRSJ0Sm7h1MjiI9Sxs3nTaA7C\nAFbiIOD5Z0VB1ogrmo6E7R9n//iGQNQ29Fymz6guYgYnHZkC4LExp27MwUDaLveEajaElrXl\nTdmo7tcKRZMo2j5C4fS7rmC5G0vgWOL9uMS7WKFQKFaHEjgUTcV45n/yBt+Vwxcqg+YzPyf6\n98KalFe/NNMNFADA2fNcGhW7fhHaeqj7tbpXEECb3MFO5jLZL/5O6UfPgRlA+OC26D19mF2g\n8Ph5XHtVe/hXEGleggAd+RA//3vV0WgH7X6waXNoUWoamqj7Hq1CraanG7oZKpEwhOlIH40m\nsOzku6DWp4uYI9O2678amc7R0Gp0W9BrF7AscUih2BCEzZqf4aHah1oTQxNBXSu7Pt3Q4wG1\nE1EoFHVGfawomgoFw6HP/5b7+nfcC8c5lxadW4wHPyi27QPAE69g8VrZyXLqOHU+tJqLSY8H\nX+bJS3BKiPWKgccQ7ljB6cF+kJhrODKPFoK5ki6nS8MS+TG2MhTuRLi9Jbocskz9h39qnz89\n/UgEjchd3ai6/VJOy1NfFg/9gwZcXsKzoFU3FKSB+4RjyTf+ClZhJtK7TzzyGSgrQa0T3qRP\nXFf1Ka2CRhEHPhlhmqiZm7AhiBrxtDVZVagS0sP6Snob6xTVtajjWSVYvk+QtQUOjSio6WWv\n+ka3LkRAU8ubTct43jp9PZsp2QFd60+GDvXFN2XD0b54KGhoZadaFIgHjc7WMBnNlJ1M2QUQ\nD+htoVu86wc6wmdv5qqCAV1sSWz4flIKhaLVUCsARdMRmv7A+/UHqruTcrG6fewMteJLY+Xk\ny1/g3OjMw/Hz3tWXxJGfof5jyx1BiyBxP9KvcKXqwKDk46iTdx1PXuQzX0FhHNMF+u27xMGP\nIVo/9WRVWKfemFM3AAT620jz+Xl54iLsAsz67dDcDKd+jPI1sAcRQPQQxe+DmM/CpX2PagPH\neOIayjkkeqm9vyX0oDriXWd5DVwAghBdpO9ezkc0mXvZuQYuL4wGyGyIlSwjCy4BOlF8ZaU0\ntzGGSDgy5XGxMkjQAmKd3+xrxBBGe7Az7+RsaTNLXehhPRpcpE4uh1oJFwQs3UWyPRCaKBdt\nOb8J1El0BEKb66NhY2N7ciRTztuuLqg9bPasbXN+Yjj9+rWpOcl9MFU8dzP3kcN9QWODJTXc\nEl2jx/d2fffcTa+iisPQxJP7utb95e14fHo0O16YFyU7IuadvXHTb7Uwza72sOPJy5PFuXKV\nWEC/e0tCtYlVKBR1RwkcipaBfXIXl4oviTz9N/PqxkzIlaf+Uuvcg+By3S6p7UEYSaRfgZMC\nCGYXJR9FaPsq5uNDepDf/ANUrMsxdVm+9rvi0X9UT9Vg5TiXzlQ+FDVvyzCXM1SvqTopvvll\nyNnVkrSQPc7lEer5+AI5yQhR34H6XLHFYOctyOuzjyx4WZY3yHgIdKvNAAUo/CRbp+BeBxgg\n6H0UuAtU59tijCLLC4z87GNN0HaiLfW9ymYlrO+wvAlHphguQWgiGhA9YuMXu2mkJ8zk2scJ\narogqirUZyCiG7SkjimIukORkuvY0mPAEFpYN9SGqXUYz1tvj2bd2S36ULrUHjZXva2dLNiv\nX52qMm2YKtivXp16fG/XmifbcuxsD3/ynv4TQ+nxvCWIumOBo9uSkRaoT6lSNwBMFuxTN7L3\n9rctcdb+rui2ttBU0XY8jpp6Z8TcjJk3CoVi/VECh6JVoGAX5/M+BwIrX7V4No+e9os7fOMU\nDTy2gqEi+ymyH9IBEaie7xe+/P0F6sY0dp4HX6I9P1HHC62YhZZfsuTvfA4QBevm/8qZV+fV\njTnsmyi+i8gd9bpK6yLHKtQNzGSmcJG986TfeevTRZhCDwIeZBEUru3KsRY8Kd/BArNMT/IV\nAZ1oY6chNAsKaF0BrYshN7T1RoMQRB2B8JRVrOyKEtT0hLksnS6kGyGVT9R62J48NZr1Fnyn\n8FTRfnc8f6gntooBL08UfC0pL00UNqXAASAeNFrtRys6XpW6Mc1U0c5bbrS2p4blStfjvlhQ\nU1kbCoWikSiBQ9EytN+L/JXqIOnUfnTFQ1l5H+0AAMCl9Gq+V1dSUr5MODPkf8A3buXh2og0\no6OksWtBaYM9kmFPLq5Soc7dMOvXSqbs/9vg8iDdBgIHyzH/A/ImsAyBYwYNYjV7huXAPAa/\nViCShzUlcKwEpW7UIqBpPaFo0XUc6QkiU9NDykdjg3MzZ3nVTTIIwGi2fEd3dBXGGb6NRQA4\nnrRdaarGwE2hUOOvACBv+wscU0X71PXsdKdYArYnwwd7Y0vUsygUCsVaUKsHRatA0QFs+QDf\n/AG8WTcBI0Z974e58l29GQZRtS/m9FUCLdPetVbXtIW2pjxySp78CnLjAGCGxcH30b4n62UC\n4kvg7geNnXudqzOdbrySnX/jWuy+nai85RKMi7s+Uc+rco08EVlzIbXh8QpcPAs3BRFAoIad\nSK1fS9NhFGocKQESatOuqAeCKGpspOaXiqUpLTLInMZjtlwZWrlrRq3uIYYmDKVuVDA4UXjj\nytRkzoqF9H198aM72utYDLKEMuV7aKpov3hlaq4AjYHUO6ACAAAgAElEQVRrqWK65Lxnd8em\ndIdVKBTrjhI4FKuEXdc+97Y7OqJ1dJn7DolIHYQDaruTYnu4MAgnB7ONIjtunTrhWPBsBBfe\nuNaD1LmXx88vuoCg3sPAtIjAjcnkXy6U6Ofxcz4HEtvm/stXXpGv/cn8Ibso3/oKZcfEfZ+6\nxej5KU6NwAxRez+MFXoxCNH+T/9D5vf/Y/nNH08HykN54/5Dof4wsjegB9CxS+x+asXDLo2R\nZHvCZ6Vj1KG8vxUpX+LMj8Bz8k0cQb93EK3Gr7ExLLEMVStURaMoulbBtTz2BImgZsT0EKkd\n0cZB12r+sYxV3b0f6IicGPJpSDTQEWnBl4UneV1qMb799o0fnZvPCnxnOHPiaurnHx2oV4ZL\nImgsdswBQATfXipnb+YWPzlTdobT5e3J1vmOUygUmwclcChWg/3u6anf/jfO0ExFiUgkk3//\n18KPv68OQ2shii+r+wOPnOFX/pInhwBGOCGOfpgOzqc2iMMf8178HdgLTD3E/vczlXjoT2GN\ngRlmB3U8TNG9dZj2yqGBJ3jifHUbWiNE22d74rKUp766+ES+/CL2P4l4r/+45bx86Ut88dXp\nxiwww+K+j9Khp1c0N9HWkfwn/969fs29dokiUWNgv4gt15l1dVD0Tkz9YFFUo+jBhl53ffAK\nnHlhgXuuU/IXOMTWpk1qaQgxxqhvXAkcigYxZeXK3kwSk2QvL72ya3cGE+qu70ahMxK4OOGT\n/JUMGaszGe2KBu7Zljw+lKoMJkLGgwPNqN9cJo7kK1OFsbzleGxooicaGGgPN61XyJXxfKW6\nMc3V8fwL58aeOVxj2bBCdEG7OsKL/7IDyYhv1clU0T8VcaJoFxz3Zs4quzJiatsSoR3JsHpz\nKxSKtaMEjtsCdmzr1e95w1dgBIy9h8y7HlzLaN7E2Phv/K+yOP/dJjOpyf/4GyKRDN59/5on\nuyz48uvyu1+Yf1zMyBf/lKaGxWOfmYlEu7Un/3d5/tuYvMROieJ9tPtJGGUe+TJmWk0A9gTf\n+Bo6H6Nkk6a9gPZddPen+exXUc7MRGJ94vAnEJhx7uTsKMrVTeNnDo1doHgvFwZhTUAYFN46\nU8jDLJ/7bb55af6pdlG++KcCWKnGAUDfskPfsmNZT7VznLkCO4tgO7XtgbbyJPPoYbgZZN8C\nZhUfEaT2p6AvZcm+USlfru4N5DooZRCKLxALZjrFtgREXcTXFxWqEFGdmgopFAspefacujGH\nyzLnlBJmeF2mpFgp8YC+vS00mC5VBnVBB7pX7xZ0345kf1vo9I1MquiEDK2/LXTn1hZqNepK\nfnM4PVeb43hyOFOaKtrH+tuaM8m3B30yXACcGkzVS+AAsKs9EtC0i5N5y5UATE3s7oj0t/mk\nYzCDfeuFgbGcNWcqnC2775RzY3nrvu3JVvlbKloAyevfM0ixEVECx+bHvXY+919/05uYufta\nAow7jsZ/5V9SeJUrjPw3/7pS3Zgj99dfbJLAwSxf/nOf8Nkf8uFnKDl709uMiMMfrTjsycu/\nO/3fyq9PnnyJ4oehrcOKmXoOU+d+ZIa4nKFIJ+JbF5hrLFrcz+Pm5dU/RXFk+hGDKHk39T7N\nI2cWqBuzyON/px1slHMHj77Kg9+DN+NAyWaMdj1LyRXnxVDbI4jcgdI1lgXS2xDeA1HnRqct\nAnt+fhblAhwLbUcBGxQg0QXR1/SpLQEJcUjyFebx2UhQ0C6izahAKVqAsudjajsdT0AJHBuG\nA92xtpBxNVXMW56hUXvI3NMZWYX7RiV9iWBfokW/HQbTpcXOI0XHG86Udiab8brNlf1XDtma\nDdFWydZEcGsiOP3DLvEHJUI0oOesajstQeQtEj7GC/b1THlrq/5xFQrFRkEJHJsctq3s7/yG\nnBqvDDpnT+S/+NuxX/711Y1pX17kbTEdv/Tu6gZcMZmbKKT8D42cRbJGVn95FLLsE2ePS8MU\n3Ve36a0IzUD7Lt/7FRTrhtB82sEQMV1BsbL6hjl1AsLAmH/GB0pZ5CYQr3+3C546y1e+Of3f\nGeHIzvH5v6Aj/wDBjhUPZ7TDaN/0d29IBPwNZj2P9EP17UZcVwxB+0C7GEWCAQRVcYqicfje\n9QWwuJhf0eL0xoK9sdtly5oq+gtzU0VnZ1McpSI1urRGgw3po7wcrWp3Z+StkUxVkGrks4zl\nLSVwKBSKNaJMpzc5zunXq9SNaaw3XuBijc3wrSBR4/tMNOnlxK7/AgIAah/iJRpSyNoDriNG\niHb6ZMRQzxZ4+cVxTv3/7L1nlCTXded57wuX3lVl+fa+G0AD3Q3TaBKOBAgBNCAAaSRxuBRH\nlLSSDiVyZWY0S+7yjMCBpFmOpJ3d5ezRocQVSXEpkViKIiiCFgTQhG2Y7oZrtDfV5TKz0kdG\nxHt3P1RVVpqIqsyszCwXv9MfOm+YfJUmMt7/3fu/r9Q6enQeuvr83H8rblaERWMvdnkkqwnN\noaxDG17B6kYZGSEE4HXVDZeOIjlknEnd+qFxqcTgIlk0EwXDqTFKN7ma0Y+Ppo9dnj6XyDcu\neHVHGrMcnsWqaJc7XTTPJgvvTOXHsqW2D2rvsL1h1r6RzhppLcCmqG9vf7DSbzWgyZKD2YbB\nu30b4+LisvZY+TfTLkuCj1+x3yA4n7wqb2qlSkXddU3xhafr49qe61o4WwtgKA5Mtm8gGh1y\nPEqJON1I4Ept1cEO/KKwDLp4DABmUiQwtgl3HKTpl2z2FiZEHJImPAEI9HZkiEUb+QwAoFBr\ncuYyj9ID/mshf6IqyDQMLskcx8VlLeGVtbxVqo/7JK37g1nPEMGlTHEsNz8Vj3mVLdHuuWZW\nYnLx1JnElfS8qcdro5nbt/VGfQulJ1xMFd6ayGVLpoTY49euGwqFO5POAAA+RSoYNhqQT5EA\ngAs6fjUzmplJJiUADGrygeFwwCHtogV2DoYObokdO5esDA5EvHfu7W/qPILobKIwntVnHEA3\nx3xLScPZEQ+MRLxTecOwhF+T+wLq0fPJjG5zF+dbWvmSi4uLC7gCx5oHvY41n+jxt3bOwH0P\n5b73TZ6omtmiooZ++ddbO2HTqF7ccZjerhNZwv244RrHo5QI+jZS4WJtXIuD11EWmaWYpfEz\noOcgNoR9W5a4dk3JUXH0mzR2Bkhg32Z268PY5+DlKans8Mdp13tg6ixwAyIjOLCHknbqBgAA\n4MgejG+myfM1cXb9/Z3Kr3Fqtcvca8tCYPBmUAcofwKsFKAG2hAGDgJzG+a5uMyiMjmk+DJm\noTLolVR/extUuyzGxXRxLFelNCWLpiXye+Jt6A3fLM9fSFWqGwCQ0c0nT09+8NpBp4yA10bT\n70zO5jxaRONZ/cenSu/a2tMX6IhSNhTyTOVtckKHwh4AeGM8O6duwMyNRLZkvXh5+vatPW3s\nDfTAoQ27hkIvnkkkckbIK+8cDB3eEW9KkDK5OHoumZ6z88iVrPFsaVPUd71DekgjeBVpQ4UL\n6UjY+4adjbqtU6mLi4tLU7iTkDWOsucAIEJdEqQUH5T6FpvVz0F6ofi9r5lvHKNsivUNe27/\nUN+j/z31f/9v+rFnZ59l07bob/+xuq2h9q5tgR35VWEU6NyxcgRjI+y9v73wvBr776Or/wz6\n1fmQ2sMGP7CwYEGvfE+8+G0wZ29KcHAnu+sTEGnRjZzOvmJ9+wvAZxcuKD0pzrws3fc7bM8R\nx2HHNkJsvqgBfSP2qShyALQYu/dT4pmvzL8yisYOfAivu6e10S4KhjbR1Il5A44yoc0desa1\ng7YJtcaa1Li4rEsCiscjKQVesgRnyLySqkmdWnhfzwiijFnSLYuTkBjzy2pAUWcu6FzQRN4m\njyZTsnKGFVC7eg9pWOJc0s7gvGSNpvUNdhPjjG6dnqyt6BREr16ZvmdXcxkNDdLjU7fG/OdT\n+XJJCkPcEvPFvIol6HLaxgisYPCJnDEQbKfgsmcovGeodTHi7Ylcus6s9EKqMBT2tEsY2hz1\npXXzyvwLQoi4Ox6Met3vuIuLy1JxBY41jtQ/7L3n4eIT/1QVZZL/Vz8JjS0XiOmpzH/5tEiO\nzz7MpHKnT2q33B3/3F/xqQnr6mWpt0/uH+qaAccsssbu/l0aewfGT5NlYGwEN92w+BhkP9vw\nK5Q7DfoYgACtDwO7Fu4tQid/LH5e1bGFrp7i//JfpF95FOTmm6EKzp/4m7K6MR/80d+ybQdB\nbWxl0juIoZ2UqbV6xf7bARC8IXb370J6nJKXQfVi7ybQWkzVaQQcuZ1Sp4BX3wF7erD/UOee\n1MXFZZ0gMynE1nvPFEGE2L71/bqTTxTzJomZ81tCpA1d51bc4wOAosWFQ21n3uBdFjhyhpPB\nBaSLpq3AMZ7TbY/I6FbB4D61I9UQm6LeeECdyhu6yT2KFPerM06cBZM7+YDkSha0VeBYIhVp\nJtXxtN4ugQMRrh8Kj4S947mSbnK/Ko+EPW0s1XFxcVnPuJeStY//F39L3rC98PjX+NgllGR5\n217/L/6mvGV3g4cX//nvyupGmdJzP1Rveo+y54DU2/7GHI2DAztgYEeTt32IgR0QaLSJqXj5\ncZtoZpJOP4+7393cMwPQ2FnKJW026Hm6/CZuvaHB8+Dw+0F9lhIvAlkAAEoI++/CUEUjmHA/\nhjuyPFWLt5dd8+t0/l8pfQ4AABn2Xoub7gapefXHxcXFZQWgW+JKRi+YHAH8qjQc8qjS8jib\njmb0c8l8weCIGPEqO3r9bTePyJgla07dKFPiVt4y/bKCzumNHZNcHFmgyEJxeIMsbicoEACC\nJQRAp+wefIq0sU5wcSqiAYAr6aIisY1R7wqxbi5Z9k6fJavNFrO9frXX794tuLi4tBlX4FgH\nIGqH36sdfi+ZBkoSOPVAccB47VnbuHn8WWXPgXaMbwVTykN2ynYLTV5oQeCAomPnGipkmriz\nQRn73o3xI2CkgCmghJoeSRvx9eHejyEvgZEBLeq6b7i4uKxekkXzdDJfXmjPmzxRMHf1+oNd\nX1t+ezJ3ITVrQUJEyYLxwiXjhqFIeyeEOrez6wbQueWXFa/CJERul3cQ6Ez6wwKEPIpflfNG\n7YARYDBkn/9onxGAwBB93U0/AQC/KnkVybYNTc7gJ8cyo5niTRui0nK4t9bgVVjezirV4zqA\nuri4rAbcdmvrCFTUZtUNEJx0m5JXABC56TaMaYWzQPXKgoUtjoQcW5lgON702ZCB1rPM6kYZ\nSQNv3FU3XFy6AIHFKWOJFKc8QDeaX64TONG5VKFmOs+JzqYKDkd0irzBL9Y9KRG8NdFif3cn\nnIomZuIMcShkU5IQ8yrL0u3ixo2RyoczQ9/dHwx57H96BkMej2zzY70h4l2WLjB7+hZyZk3m\nzbPJbn/S6nn9SjpXtJe9hsOtO4AaXIxlSxdTxUTB6ErHXhcXl/WLK3C4LAiTWNi+8yiLteiy\nuZpQvRAbtt2CA9tbOB/GN2B8o0083IfD3bNodXFxWb2YIlG03tb5hZK4rPOzRX5GkH3BvEtT\nWEJMFkqWneeEbom83cJ750gWDNs5YMHkhbaORHYQ6+W5eoqhoGdTxFtOK0CEwaC2LeabyJde\nuZo+ejH5/OXUW1M53aGoob1sjPru3d3f61dnRhfU5MObY4c2OjZ6lxke3tzjrdZi4gFtKd1A\nlsJgyHPzxqhjNhDCmIP5RXewBH39+Yv//MqVty6n86VajWNHPNBy9tD5ZOGnp6eOXZ4+MZZ5\n7kLq6XOJehNTl+Xl6NGj9913XzQa9fl8e/fu/cIXvlC5NZ1O/87v/M7AwIDH4zlw4MBjjz1W\nc/iiO7QGkez+W9X/2vIxaAF3udVlEdSb3qP/4Bu1Uca0m+5cjuF0G3bzQ+Jf//eaIMY349aD\nLZ0Ppfd/kn/zUcpWOHH4QtIHfq/p5BoXF5f1h0XThhitjAgq6vycV96JHTMUWGOYQhdkIjIZ\nNYYyABBAqmTkTLNoOM7STS6gizkLtjrLDI62ny3hkxXDrhjBX+GiPRDQ+vxq0RQCyCtLMsPT\niXy5d6zJaSpvpIrmdf0hf/N1KyUudFOoEnoUqZGcir6gdt/eAU7EBTXijdLjV9+3u/9CspDW\nTUXCHr825FDP0h16/eptW3t+fiGZKtjM8DukE01kSxenC3mD+1VpU9QXdzAKff5M4txkDgAE\n0fmxbNSvBnyKLLHhsHffUKjH16K6cSWtvz5elXmULVkvXJy+fVvPcrnbuNTwjW984yMf+cjh\nw4c///nPB4PBs2fPTkxMlLcKIe6///7jx49//vOf37Zt25e+9KWHH374sccee+CBBxrcwcWl\ny7gCh8sieO//t/zSO+abL8+HJNn30G9Kw1uXb1DdA7ceYvd+UjzzNZg1B0XcdZgd+UjTeoQ+\nSkYSmIbRIfnf/Vfxyg9o7AyQwP4t7MD7OtroxMXFZc1gisn6IIFliZTCHCvgXGbgZObNKYtm\nZ+YI6JHDXikyo24ALNRbTOvuTGyBBh9vTuZ6ferGNhVZBBTVFDxvzU22CRAhpHpUqWoADLEs\nXmRLVlndKMMFnU0Vru0PNv7UuiXOJPOp4uxT+xRpW8wfdig2qUFClBrSQwAAZIbbeh1/ZLkg\n1rkuNQ54ZSkFNgKHR2nzx4wAXrqYOj013yv3ncnczr7gwZFI/c5vjKbnW74TpHJGKmcAQFCW\nWlY3AOCsXXNfg4vL08WtPe7Nz/IzNjb2iU984oEHHvjHf/xHZteO8LHHHjt69OiXv/zlj33s\nYwDwvve978CBA3/4h39Y1i8W3cHFpcu4AofLIqCqBT/5qPHyU+brL4lMUurfoL37PmnAps6i\nUQSn5DtQmAI1gJEt4FmeTNHGwW03SlsO0PRV0PMYHQRvk54XRlKMPwH67KIroYKxm9nNH2z/\nQF1cXNY45FSN4lapLAqByJrjgqyKCBWtaSCWN2en7pqMjKGoS5HwKZK3u5YTvX7VI7P69XxZ\nYgYXo1k9WTT2D4QbS3pYhKjm9cmqzk1OJCHzy4q8YM/1siRRQ1qfOUNDQ7IEHR/LGHz+DyyY\n/OREZv9AqDvdZwng1Hj2+Gg6o1sSw/6g56ZN0Z5udfQYDHlsW7G2PbvkfCJfqW7McGoi2xdQ\nN0RqGzBndcv2zcs6vOONQABZ3d7RI1NXBeOyLHz5y1/O5XKPPvooY0wIUa9xfPvb3/Z4PL/8\ny78881CSpI9+9KN/9Ed/dPz48euuu66RHVxcuowrcLg0AKJ68Hb14O1tOFX6onjjn6Aw25qE\nmIJb7sLNd7ThzB2FSRgbaeVAMsWVb4GVqYxQ4hlgKkYabQrbQQrjlL8CwgRfPwY3L/domoYm\nz8LEabJ0jAzhhuuByZC9SrlRYDIGh8G3Phe0CYpnyJgEQFT7wLsFnBs9uqw2EADtXUXdN3kx\nDF6oVDfKFKw8waxsjYgRn5TKW/MmiASKhNtitVPBTiMhXj8Uee1qurLpBmOozllm6pa4nClu\niS5pYDq3LCEYoiZJYanRqfXC5TMNJlaMZUuV6sYMRHBxuri3r4k0kJY5eibx9pxjKxc0mi5+\n54R+757+wXA3ClgGgtqGiPfSdLEy2OtXt7T7k3bewbX0XKJQL3D4VEm3c3jRZGk0rUsMI15F\nszNtXQAEQESysxVdoAmxSzd56qmnRkZGXn311Q984AOnTp2KRqMPPfTQX/zFX0Qis2k+r7/+\n+o4dOzRtvrLp2muvBYCTJ0/O6BeL7uDi0mVcgcMFAACKGUpdAm5hbAP4Y516FiMnXv07sCpW\nLYRJZ54ANYBDhxyPIg5GEgBAjQEutoZm6nTlbUhPwIxtp7KcpbYAQNm3q9SNcjz14jILHMKi\nC49T4ng5QIENbOuHQV3pCTWzcFP8/P+hCy/NPCIACMaxfxDyl2YjiDh0E+58//pq7GJlKPF9\nMOcERABQ+7DnF0By04DXCBJ6OdnMWCRw3+JF4GTYxgmqptmajH1BOWcI0yJVYmFNGQxoy9K5\nM+SRj2yOXc3oF6YLJYsYw5phJIvmFkd7zUUwBE+ViqaY/dsRIKhqIcXemqEGpzIKiaHScCFP\ntq7b62y8K6v6ibzxdl0/GkH07Pnkg/uHujAAALhuMDQY9FxKFwsG9yqsP+gZ6YC2Ut9Vd4H4\nroHQs2embHYW4sVLKQBgiDvi/l19waa+D1GvkijYfPtiPqWZ07h0itHR0enp6Y9//OOf/exn\nDx48+Nxzzz3yyCMnTpw4evToTDZHIpHYurWqLD0Wi83EZx4uuoMTf/Znf/bjH//Yaath2F+0\nXVwWZT3d+q85KJMUU6PoC7L4CEitZs8KLl77Z3rzJyAsAABE3HYrO/iLnZAG6OqxKnWjHL/0\njIPAQZR4gSafBWEAADAV40ew55DTYiWdfVn85G8hl5p97I+yuz6O21pzA20TJZuCeQAAKwu8\nCFLrHdeWCF38fqW6AQCQuyROf4Pt+Y2FytBXDOLlb5XVjVmyk1RIYDw0O34iuvI8AODudVQC\nSoknyurGLMYEJX+I8XX0IqxtFNbP+XkBjIgBMQIuMS6hKjObinqXauyvbAytmhVmxjDkkQBg\n0OdVFizW6DQMcTjsTRRNATbT0ZbdRgXRlF6obBBLABmjJCFWeos60etTL6SKvG5Nvs+vNf7j\nYbukD91qenylOnWiTKpgFAy+gAFKe4kH1Higs0UxmsyytX4pM3Gbv/HWHb3npnJj6ar7tIhf\nDc0pEYLo7YkcAOxuJstmZ9z/3EWj+uOGQU0e7kqyjMuiCCFyudxf/uVffupTnwKA9773vYj4\nmc985gc/+MG99967wIGLWtcsusPOnTunp6edtj755JMLH+7i4oQrcKxKKJsyvvs31omjMw8x\n3Ku+/xPyvsMtnEq8/C166ycVpyY6fVToOXbHb7dlqFXkxu3j+QkgAXXN6mj8p5SomMQKg8Z/\nCryAfTbFMjRxXnz3r0BUZFfmU+Lxv2b/5nPYv3x+qA4d+BbZ1Gm4TonXbOKFccqex9CWrg+o\nSbhJZ35uFxegm+Cdv1+k0Rdx2z2gdDu9fHkwJsC0E9RKo2ClQG51qddlJSGhn6DX5PMzECFA\nk+NujcqiyEwDu/6qEiphRZmuWyr0y/LyqhtlPIpk61bgabJYoEzBMoWdvpA1jUYEDlViu+KB\nU1O5ylqViFfZHG1CsverctLO2WGJBhzZkjVVMHRLeGTW61OdGrKaztqQKQSsoYZEw2HvVL5+\nGZxss0U0mX3syJYXzyXfGc9mdTPgUVDCULUvCQGcmcrvjAdYw2shMZ9684boyfFsbvZjjEMh\nz97+YONncOkoPT09AFCpZdx3332f+cxnXn755ZlgT09PMpmsPGTm4UyaRiM7OPHggw8++OCD\nTlv/+q//uqk/xMWlzIr48XZpDsvUv/TZsroBAJSeKn3tz/hbLzZ9KiNPp35WH6bLr9H0laWM\n0R6nziMo2WQNWHlKHKvflxIvArfJ0KZXvl+lbswgOL3y/eYH2j48Dsmuai+whpKBO4KeBHLo\nRVecsI+vKPJJsByyzWs8+UhAfjX8RW3BclwGAdN5k8uqomilDV61vkoEOTNBTt9olzlU5pPt\nrro+ORpSlZimlqdbCBBSlZhn+S7R1fR47UWHfodmn4ti1v9WAgCAVWevanBxJaOfTuYvpouV\nxSMxr3JwKLI56uvza0Mhz96+4DV9wQbtRWcYDGq2XWBGluCyeTZZeG0scyWjJwrGlYx+fCxz\nLmXvQBFy6NUiMeyOxWnX2NUXjNU1QIkHPNvjAdv9JYa3bOv56K2bf+euHUd2xkN1rqsIYAlq\ntpKox6/evrXnzu29t26O3bMzfsNwuFkvD5fOMWOTIcT8j8jM/8tuo/v27Tt16pSuz//0HD9+\nHACuueaaBndwceky7vVl9WGdeEZMXKqPGz/8WrOnotQVG1FghsSFZs+2OJHNtmGMbKpffqTi\nqH2yKgkqXrWJTzkMeLIDf0jDYGA7eAZs4r1Huj+Yiqd3Xp5aFY4VzmuMNjfYy5gp02XQeemV\ndak1gEun0UWtcQAAEYiSqG2U4FJPUOnXpPnUeoayX+43hZwxSwxh0Ocd8HkHfN4Rvy+iqsu+\nuEwEo1n92Gj6dDJf476BAEMhT8sCB6KDVW01E3nj2Gj6XKowli1dnC4eH8ucruj3qUg4EvLs\n7PVvjfpi3qbNFFSJ7esL+ip60ygS7uoNRJo/1QyTeWM0W639AcyIHfU7b4r5PHZtcXbEA8vi\nt9I5JIZ37+rbPxyO+lRVZjGfesNI5K4d8YayJ5x3ac0f1KdIUa/SuFGLS3d46KGHAOC73/1u\nOfKd73wHAG655ZaZhx/+8IdLpdLXv/71mYec86985Svbtm0rG4guuoOLS5dZDZMZl2rEpVP1\nQQIQV8+BZSww92uS9v/GY//1dPEo5KrlCSbh1nts9l5gQdJ2k9PM3CltpL0IHUQBpBBgzTCQ\nDT1IU09R5vXZG0o5hPE70b+9G6NywhsH2QuWTREyBhpoAMwzlD0GxgQAgRLH4IFulz/4ohDq\nh4xNxRNpStUHV1IhONitYS032iCgDPV9IpgKav9yDMilzRAJonpJGgFAUOt9HNcPCMwv9/jk\nKCeTgaRzSpT08kvHEMOqxy+vFOPDd5L52YasBAwRGRBASFUCmtTjU5aSaKAyyfYnXq3ogFIw\n+TtTuUoVhADGsiWfIg0F22OdENTkG4bC2ZJVNLkmsYAm2+Z0NMhE3s5qAmA8Z/TUpTCoEnvv\nrr6fnpqs9NrcGPXdvLljJuvLB0Pc2x/a299kh3uAqIPYpEgsoK2dKh6X22677eGHH/7sZz+b\nzWYPHTr07LPPfuELX7j33nvvuOOOmR0+/OEP33rrrb/3e7+XTqe3bt36t3/7tydPnnzsscfK\nZ1h0BxeXLuMKHKsPssu5mL0pEM1lKWNsAzB51l60ZlNvB4wYmMQOfIJOf5+uvgQzBcDBIbbr\ngxC2mVSjJ+60wISePpvo4A4YP2sTH9rV8ngbwmHmm4cAACAASURBVJqi7HPz3geebRi4EVhF\nKbLkxf73Yfx2KCVA8oISWf6cAmQ4fBddeLw23Hs9eOMARNOvQ+4sWXnUYhi9HjwV0+PSZUp+\nH8qzLCtN+lmMvAe8XTU6YQcfFk9+sVbq8qpYvSiHW94DbKVMVzoO0zB0E6Vr3UkwfGud7uay\nOllo0XW5ryqrBwQmo2ZwnixV1S8IolSpKCPTWjbtbh+ZkpUqmkBzrYEBcDbvgjZFlupO7ZOV\nnGkY1fcSCBCuMBcfz5Vsf4LHsqV2CRwzTxrS5JCDU0ZT6Jb9/U+J26ep9ge1h28YPjOZSxVN\nheFg2DMUXjbb75VJzKf2BbSJXK1y1JQBh8uq4Ktf/eqf/umf/v3f//2f//mfDw4O/sEf/MHn\nPve58lbG2OOPP/4nf/Injz76aDqd3rNnzze/+c0HHnig8R1aRqwhQxyXbuLe9a4+pKFttrWP\nLD4CapO3HYoX99xFr/+gJoybDkHYprCidQpjlDgBpSSoIRi+ge38ABQToAZAta8CBQBQYxjc\nTtnTtWML7QLFppUpO3g/f+so6NV52p4AO/T+NozfCStBqe9B5ZqqfobMKYx9sHZKyTzgHe7g\nSJoE4wdA9tClH4GRBgCQVBx8F/YfBmGIi9+EwuWZ3ahwiVLHse/d2DuTqSho+mdQs4ZMgtJP\noWcDYPekBBy+lt39P9Gxb1LyAhCBJ8T23g0eogtPzwp2ig+33o0jN3dtSCuC4PUohynzPJgp\nAAClB8O3gKeBrByX1QACykyzhM1KtcLcfgTNkXPw8clZhtbh/lYEkDGsvMUtIhnRL0shVa6Z\nL866itbNIvMmtwQtJdNhhl6PL22U8nMvgsJYRPVUKjtOekHRcihrXW6cXhPZwSlWEMgMd/U3\n0Q1kHXLjxujrY5kLycKM2qVIuLsvuLXHbUq91tA07ZFHHnnkkUecdohEIl/84he/+MUvtryD\ni0s3cQWO1Yd0/e3403+kdG2vcuWOh1s4G7v+AZI1cfL7wE0AAGS483Z2w4eXPs4yNPoUXX0K\nyp7tEy9Bz7W4+UOLtiPF4fth9AnKvDUfCe/FQbt6FgAI9ki/9L+In/wdXX5z7vDdeNfHIdiz\n9D/BCcq/WjvbBwCeBv0d8O7p3PO2BYzuxeheMDIgLNCiM28HTRwtqxtzEE08jf7N4B0AYwq4\nXam/KEHpapcn0ti3HX/hP4BlgFUCz+xNKm66EwoTgBL44l2qTlppeLegdwuQBYALma24rE58\nUiwjxmoMFBTmU5i7+NwcpkPCo5MBZ7sggPGiUeKzz24STRtWwRIDXrXyJ9G2y0nFpqUKHAwx\nqnkimmYJwZDV+4M6OYbKK3XpPuZVbJ0va/xBcoZ1PlXMlCwi8irSSNjbV+ej6VJGZrh/KLy7\nL5jRTZmxoGdJZUQuLi4u3cEVOFYfqHo8v/6fjG/9N35hbibv8Sn3fFS+4c6WTsfw2vulXXdS\n6jIIjtGR8lyxLVDmHI3WNmqhxAnwD2PfjYsczDQc+SCWbqXiVQBE7wBovQvtHxtmD38G8tOU\nmcBQHPydN4Yw7RvfkjGGK17gmEWtqsul9Jt2OxFl3kTvAAgb245ZFtjUUWS1yndGUiC4gjJl\nlg2UQRRJfwN4EoBAiqK2B5hzwpTLKkFmWkgdLFpJU+gAgCh5pJBXarq63sVpltaae2LjZE2r\nrG6UMYTImFa4wlbDI9urkzLDNno0IqDiIARHvYqtq0XLJqCdZijkSRSMnFGlTwU1eTA4b8Wa\nKppvTGTL0lHB5KemcnnDsyW6PlqJt4oms3irjrYuLi4u3ccVOFYlrHfY85uPitEzYuIS+sNs\nw070Lm3qovqwf2ebRldN4oRtmBLHFxc4ZtB6cWFdowZ/BP2RJvZfCo5OqKu0ZSOB5dCLwcwC\nAEjOH7MFNrl0H2uSCk/PG47yaTIuoO9WUBz6FrusHmRUg8oAAAkSzE3SaRVNkg27ZA1Phw04\nik6lH1xU1l72eJXLaTRr27ZCn1/rzgJ6r18dyylpvcq8VpFwU2SFagES4nUDoSsZfTJv6Jbw\nyKzPrw6FvJUJB2fnSi0quZLRBwKa166piouLi4vLasQVOFYtiGx4OxteqBmHcfrtwvNP86kJ\neXDYf8c9ct8ytJMgY9p+Q8khvrqQI/P2olXx7nYVqcRI0fRLZCQQFfCOYPRAM9YYCJIXuF0u\nhuwDAFB6QI6ClardKgVAXTfNSpYBAlEA9DRcckJUfKGunYqg4osov9+tW1kr4KpTNwiACyGx\nFZHjHlTUgmXyapFaQgwonV2pdqo94dVxieHOXv/pZKFUIYjE/epIqEtmKwiwry94JVMcy5VK\nlpAZxrzKpqhPW8E9PhnihrB3g4NXqG4JJwORlG6uZ4GDaNGKYRcXF5fVhCtwrFGIkn/zV5n/\n7+vlvirTX/2b2G99Onj/Q10eCMpe+7s5eWWsAnETJLnlemb07aP0k3VRBT0dbt3iAGVep4kf\nztiCEADkz1PmOBv+JVtbVlswuIOmj9tsCGyn8WOQeptKKcQi+VX0zk0DmIbR9yx/a5g1CRmk\nnwDjHIAAQJD70HsDsMXqEXgKRN7ubCXgkyC31T/YxaUBOIlpo1S0ZtMBvLISUTVpWS8aDLHP\n66selRxRPU7eE+1CZmjU5WUAgFL3vAFVvq4/mCqaBVPIDEOa7Fe7OglnCDN6gSBaA10zuN3L\nvuimNYwl6J2p3Gi6pFvcI7OhkGdHPOBabLi4uKwBXIFjbZJ74juZb32tMkIlPfHf/lzdvlvb\nta+rQwnvhNRb9WGMdKYipkFI0KXn6PxTUEiBpGBsK+66HwL9ix9Yg7YFg0XKvQw0l8crBTH4\nLpC66zHODUqcgvwYZV4CTYLyChsCmBma+BEONypsYd9tVLgERlWOBkb20/mfQPbizEMCgDxA\npB/6NoEaR/+1sMQODoJDMQWeMEgVySZWGqxpkPwgx9aresIp/1Pg03MCHIE1TrkfYeC9i2gc\nZFM5P4vQ2ztEF5dF4UQTxXxlhkLRMg1u9XuXudmkhKxH85Lm7WZeiV+WCnZVKn67DAKG2ONT\nO2iUbYcgKnLDEpwhapKiMnkNqBsAoMkMEWwTaNZh+oYl6Oj5ZG7OllW3xNlkYSJnHNkSczUO\nFxeX1Y4rcKxNst//tk2URO77/9xlgQN7roXkScqcrYp6enHwSDeHUQMd/3/p6quzD7hBk29R\n8gy78bcg0nwfEO9e1DaDMQ4iD1IY1KEulwBQ4hS9+RiU0rOPESHih+C84kCFC8h1kBrTIGQf\n2/prlHiRcmfAKqAWg+gNMH2prG7Mn3Z6HAfeg8GlCVWlrDj5L3TpJRAcELFvD9v/YfColPoZ\nlK7MDSmEkdvWY69T4zzwdG16EVmkn0TfrQsdiM7vtdtro0NYV8g6DyIP6EF5AJRtAKt4ymQJ\nnZOBgDLzsCW3f86ZJV43reREWbMUbra1eQdA506incAnSyGFMmZVBVlQkfwOrqJdRufGtJEv\n19FkzaJP1iLqWmgLKjPs9amT+dr2wKrEoivVObUTEIDJxYVUMVfXdCZnWGcT+Z1x11HLxcVl\ndeMKHGsTa/Sybdy8UjtNXYRSgfJpjPS13nETGe74FZg8RlOvQSkJaggju3HwCLDlu59InZtX\nN8pwU7z1L+yW323lhMwHni1LH1crFKbo+FdAVNymEEEqBxIDX7m3CAHPNypwAABTMH4rxuen\n0OLM9+33TLwO0SUIHFaJ/+yvIDfX8JiIxt/gPz3PrtkCSsVfZGUo8T2Mf2iV2XxQCXBJxfxk\nTdhvsOxsXyqRosCCILK1ceYFOb6UIbVEgugiQB5AAggjbgHwAgBQmvhpoDQAAgsj2wHYzv5N\n3YRKL4E1d8mlAhlJsC6h57Zm7G9WCoKsgjVhVTRF0qSwV+5ZSl/SErc3PqiMl7jIGZwL4ZGl\noCav7RXkqCb7ZFawhEVCRuaT2QoxtuAkUqU8VTchLlglGVlAWYnaqG5xhqg2/Opti/lNTtMV\nzqmazHb3Bjpdl7RCKJj8zfHsRM4QROjwJ0/mjZ3d/5VwcXFxaSuuwLE2QZ8f0nVOkADM36gw\nL0bPmP/6JXHxLQAAWZEP3i3f9SvoaWkZBxn23VjfM4UmTkPyAgBCzyaMb2vlzDXkRmnyJOjT\n4Ilg/BoI2DeMoKlT9odPXwRLB3n5VxQbh668UKVulMkWKgQOAGlpjieGQ2sVo24K3Qx09pl5\ndaOMWaBLl3FrtU8ECcocw973L+XpugSVyHgT+GUgC0ACeRjVvQulVCyE/bTQOT4P+m6m/FNA\nBgDQzNwUZfTeDNDVeRTReYALc484wCRRAvE6EDniJ6E8jxITJCZRuh5Y82Viyw6/Oq9ulBFZ\nMt9C9drlGNBSoLx5lVPVEneJpwHQK7deJ1EzYa6Pj2b1sXypnOThVaSRoOZT5DWcKq9JK0XU\nqKRglWzfrLxVWlECBwFcShXOJAsmFwDgVaSd8UB/A31MZYbX9AeTRTOjm5zAr0p9fnVtFOAs\nSt7gR88nzbkWxeRgdmvx9WhH4uLissZwBY61iffQ4ey//JNd3DGz3XjlaOnZH/HJUdbTr27d\nSS/9C/C5VQ7LtJ7/nrh8Svv1R6EtLfRKOXH072j09XIAh65hR34NtNYTI+ncD+jyM+W+rXT5\nGRw5glveZ7Mrr81QLZ8DuLG6BA7Ij9vHzfk5MHo3gLS0e1M1AEU77wZ1SUvuNHnaPp4t1N1v\nEhgOf+mKgkzSn6ow+ORgXSQ+id47AdWFDrSFhQBGHeKLIcUweB+V3gKeQiCQIqjtblVnaZki\nQH3KmCB6B/g0VM+jCAj468jiXZZglg5ZV+03WFdhtQkcpihaZNTP9gye9sgxbDWJQ2aSKWxc\nJxQmAcB4vnQ1V+UaUzT5O8kCAGgSGwl5Ip7VlwjTGkXOC5bFBckMfbLc6Ya19XCHxuecBAG1\n/AFoO29NZC9NzycZFU3+2mh6b39wxKF/Sg0xrxJbTzUpM5yazJXVjQXospGti8vCCHInqi6t\n4H5u1iaRX/314rM/s6aqUty13dcE3vcBm72Jsn/zn0vP/2T24cXTxitHJY/s6fFX3s+IK6f5\nyWek/bcvfXjimS/R1TerhjB6Uhz9O3bXJ1s7IU29QZeeqg4JuvQ0BEewt85zxOewFKl4QV1t\npafM4StcXpKSfNj33iU+CfbspctP2WzoWZqfizAd4vUrSCvlxnphyDxj076EisJ8GZSdDKNN\n/SGobqXSO/X5GqjtaPB49FzX+NN1gCTYLt1TBqA27QgBAAygNODytVhuDXIQTBewel2pCDt1\nAwAISJAptSDSAQBAQFbLnUpq4gAwXueJUKbExZlUYXPY2+Nr8alXEQm9lLfmvhcccqYVUJSY\n1tU/3OnyhAArR90omPzytE0v83em8kMhT006xlTBmMobOheqxGJepd+vrY90DRumCo5ftEo2\nRFZQqo6Li4tLa6yytTKXBpFivYP/51eD9z0ohSMAIMf7Ix/5jYG/+O8o26xalJ778by6MQfX\nLTNfe4POz59sw+DSYzXqxgw0+jpk7FfpaeyUeOK/8n/4NP/GH4mffhHSY7V7jL9s/1xjNnEc\n2G+bpoHDN66+bh0RB+8PbwA8gxi9kW3+OKixJT4JDr0Lghtqg/0HcSkGHAAQtO9Xil67e3pl\nwbJgwSE3CeZytwgRDu4YPMHplCWOExSaOBsLoP/W6rQLCT3XgrJpKWPsInbFU2AvesxtctC8\nVjJOeTFrzc+19XmhJklRzVs582SIMc2rSpIlaNFV5ctZfc0nzedMa17dmA+ahbpgR9Ek+7wG\np/iykCqatnU0Jhe5UpUc/E4ifyZZSJeskiWyJevCdPGNyaxwKM1Y8yzaChcRdsYD/cElWUe5\nuLi4rATcDI41ixSJ9fz+f+z5/f9IhoHqQqtApReftI1bBVOpKWq1GloBWBhKO2R0A9D0KIZq\ni/DpzZ+I574+//D8y/zicXb3J3Fo73xQtzEcAQDSkzZ35aofb/gf6PjXoTRvIYED1+LOexv8\nE1YOOHwTXXkeCtVOFpLGrvkE+NtnFCapbN/HaeJlSL5FRgY9PdB3AKON5RE4g1uP0Pmfg6hz\nlBisN2JgGDpofxYjL15/nC48N3Me7N2G+x/C8PASx9Yi5DQbIQAgKHLxtsz2N6Esy4MYvA+s\nUeJZZF6Q+4GtonYGDjP/BWREXH2iAMojZJ232SCPdHsoS0Z2EGUQJWlphql+WfFKcpFbXAiZ\nMY8023m0EZMNS5Bu8rXdyLNe3SjHfXL37tM8kqpJSolX6YyIGFKW5uLUVoQgJ7mtsllPomAk\ni7WCac7go9nSSGhVFaK2iYAqp3UbBTnqU/yK7FOlwaAW0NxJgYuLy1rAvZatfZARTZ0Do4iR\nQfDZpH+LTNL2QKrzmmLxdrTqlJw/dfXLRHpWvPjN2qCwxM+/Ij30n+cLMWSHeZFDHHu247v/\niK6+CtkxUHzQsw1j7XA57T6Syg7+Jr3zrzT+2qz/SHQr2/mBdqobMyDD/kPQf6iN6b0YGmQ3\nfUy88o9QmjMxlVS27/24aR+lngRjrsBK8mPkXaDZaRbcFE/9H5SZN6qgqTP05F+xOz61PBoH\nC4JI14dprmMoQUlQimEzfo0og7JxJbbjIB0oB6gABh0kmx4ABaDulhr7ABNAdbU8GFqVjVSk\nXlB2gfl2dbAPlaUqgN1HQk1hfrOuzMorLTULDAAYor8uhZAh+hSpYC7im7u4c8Aqx9H8YrFV\n97YT04J5s5i3SpwEAmqSElK9cnd7ny+M0yQcAQIV/hH16sYMqaK5PgWOjVHviau1rwlD3D8Y\n8qvuXMDFxWVN4V7U1jj0ztPilW+DMXPDirjlRnbol2q8PFnI/uYVperJrOaTbrhr6UPC3q3A\nZJveH5KM8dqCCxp9E7jdbUp2iqZHMTo7icXYTkqft3munl2O45A9uOGWxoe9clGDuO+XcM+D\nUEyAFl7EJJXnKfcyGBNAApQeDN4A8nJaHuDQfim+k8Zep9wkeqPYvwe8YQDAvofAnAIzDZIf\n1Dig/ZWKLr5YqW7Mwg1643t4+Dc6Pfh6UN5M9Q01AKhi/ZPApnp8lUE6WW+AmKsUQw/Ke4DV\nN/GVEfcQvVmtcQQRd4A0QvxloIqSIvSitL+jo+4cqO4BeYDM80A5QC9KA6sxfWMGn9JfspI6\nT8+kHTGUPXKPypozJyIggxuCLIaSKmkL2zcMBz2nk/kF5vEI4Fl5DUfaC0O0rd3qfoMPBAgo\n3oDiJedOostLxKsENTlbqr2FGAx5lIrPiemgDTVitLkm2RjxFgx+Npkvp7koErt2IOiqGy4u\nLmsP97q2lqFTT4kX/qEyQOdeENlJ9r4/hoobF+3QbcZrz9YfLvvn61Mw0qd++JMYbMdkWPPj\nNffS8e/WhNk1vwBqXfq94exZUJrvXYpDt9DkCchVF7/4B3Do8NLGunpgMvgXa7FpjFPi8Xmb\nAytJ+hmM3Ane7Z0e3UIoXtxQnxiCoMQX8d0AoCmHPiwO/Vk6jtSL6n4yTlY4g6JQgsQqvkqr\n3vlIkPkCUEXnYNLJfAUVBFZvqhJFvAngKlEOQEIMA/QDAKCC8rtBXCJKAwBiBNgIwApaJW4a\nFkVttdmj2oGAHrlHk2OCDATGms8dKnE9a2YEzX4FGLKgEtYkR+E1pMnbY/5LmaJu2c88e3yq\ntHb7xc7gk2TDrr1XN+tTaliZ6gYAIMD1Q+ETVzPTFQUX/QFtT19V/pfqIIqp8mq/ArfO7r7A\nSNgzkTdKJver8kBIc3qVXFxcXFY1rsCxdiESJx63CU+do9GTODzfv1A7fLdx/PkaJw5l36Hg\nR3+fX3wdctMYH5F2HASlbXbu7Lr7yRcRr34H9AwAgDfE9n8Qtx+x2dXvmBqNgd75B5LKrv9N\nuvQ0TbwG+jR4Ith3HW64DdgKzOxfNmj6Z7UmjiQo/TRqG4F1u0mB8epR49kf8MlRFulVb3iX\nduQXgDV/p8UdPC+c+rN0AWULygNgjXFxHpBI0qg6uxuxgSavKxl+tUrdmIOsd1C1dY2VATbY\nzZUkYJtX6BRqZUMggAg7WTWAgBK24jVoCTNtVDkiCRJpIxXVehXnq3FIk/fFgyYXBc5HM6XK\nipWYV9mwDgoKgqpS5FapOrnAI0kBxb1Js8GrSDdtjCYKRka3GELEq4Tregn3+JSEXd+QHlsT\n63VDQJNdow0XF5c1j3uZW7sUUlC0sQMAAJo6XylwAGLwf/yseui20rM/4hOjUm+/duMd2uG7\nAVGOD3VmcIjb3yVtfxfkkwALqhhDe8AfhXythygO7IJAtZEBU3DTXbipDUU0axMrBZadFasw\noHQZvFu7NxKi3Jf/3HhhtnEPv3rBfPNY6YUfBz/5KKpNzqlCAzD6Wn0YQ/XlEh2ATLCmgWkg\nBatM79ALyhakCKdTNWnnDHsRVlsr4mqIph02ZAH46s7C6DpE3BQFARZDRWG+RbN7TJErWklB\nJswUj0hRVVpZelnBqrNWAQCAopVX1MjCxyoSC0ss3KtkSlbB5AwxoEq+Ne0tWgYB+rzembYp\nliCZMb8su+oGABBBweQGFx6Z1RjN9vjUBfoHRz3KQEAby1U1g4t4lIGA2yXExcXFZY3j/nyu\nT2xqU7VDt2uHbu/2QJyljVkkhd3xW+In/xcUM/PB8AB798c7Oq6VBFH6TShcATJBi2N0f4vZ\nFtzZ+kF01RXCeOXpsrpRxjp9Uv/RN733faSpU7HNt/B3fgp1qd24/Y6ljHBxRIlyL0Lh7dmv\nkhTE0C2gVXVvZRhF2MPpAsHMlE+WcIhhV5QXl9WAwTNFK0FzBpqIkk/uVZzdLko8XbTm+yUJ\nsgrWpCDLI7fBAbRdWA6NhKxmGgCHNDm0/haZESCoKEHFzTqcZ1o3z08XS3O1S0FV3hL1Nt5P\nZ1PEG/Uqk3mjZAlVxphXjXnX5stbsgRDVCQ3Jc7FxcUFwBU41jK+CHiCoGfrt2BsU31wxYJ9\n26QHH6G3f0aJCyApGN+GO48AWx8fXSsvLn4L9LFygBIvsJEPga95C0PJub3oAps6gPHy007x\nZgUO8MXY4U+IY/8Axbm0Aklhu9+HG29c2hgXhij1BJgT8wGepdQPMXp3jcaBGJLxWgBOwBHW\nSF40YqheHyUARP/S0jccWz+uKgj4eeJXgAqAHmADKG+zfVksUSxYk1VHEs+bE0FVsa0NISDd\nsml3pfNpVQoxBxfe7uP8Fq6BN7fNWEIQgMzWur9Iq2QN61QiX9H4FbKG9eZk7tqBkNLwa7a2\nxTICGM+VrmR0SxAAaDLbGPauVRHHxcXFpXHW7HXfBZCxa+4VL/1TbTi2AUeutT1i5aJ68dp7\n1+FdIF19olLdAACw8uLyd9j232jaXkQOgxIHc7I2zrz2HVg7hlNbYpFOtHA27Nsl3f0/0/gb\nkB0HTxj7dtn2Qm4n+oUqdWMOyr6Emq10KOFaKtyQhoCfAapy/0UAlFqzqhWWuCooQWAgKAyj\nMhtatT9MRObzIOY+3pQDfprEGKqHAWq/rSVuWz9IJZ7xyTbeulzoZN8slTjpDDtS90RAlhAM\nUcJG/XEUppp2DjhK111+Vg4EUOLCEiQhahIyxJxpTRuGIAIARAgralBVlv0HjgC44IAoN/x2\nd5TRTInqxFRT0ESuNLwObFka4XyqMJGfT2AsWeKdRH5L1NfnX79fN5c1hqA1dPvk0kVW6X2k\nS0Pg7ruYEOL4d8GaLUPFkf3s5l+BlXH74rIIvEDZMzZxK0e5cxja2ez5MHoHJR4HXjE1RQWj\nd0LzjRKWAgv32McjvbbxxZFVHL6+9QE1CZnj9husFAij+3atXUdC5SayToAoC1IKyjtBakEm\nE4Z4i+a0EgKT04TgaUXag6vxt4lfmlc3ylCOrLMo1/ar5mRjf7hAHICcUlyofha4ZIgoYxbz\n1mwfXxmlkOrzSItfKHyyX+dFQVVaDEPmk7uaJrZyKHGRKJnllqUMwctYoaL3ORFMG4ZFIqYt\nmzcEAeTMYs7UabY3MAspXp+8zF4VecO+3ClncNv4yiGtm7mSJTEW8coeuVPTM90SlepGmUvp\nYtynrtQeOC4uLi7dYBXeRLo0AeLeu6Xtt1LiIphFjAxDaLFOog1DuXOQvwi8CFoPRq4F5y6A\nLi1iZGzdUgAATDu70EWRY9j3byh3AoxxAA5KL/qv63J9CgCoB283XnqyPr4MFjCtsdB80r7P\n5VoDfajcDJQBygGowML1GQqNwGmKqLYPNEGJi6sy29COgXYVEjZ5PQAAYgKgVuBAh5INpzhD\n1WnCInVAU0uUsoaYn15axJOlbEwLeKRFnouhFNV6skbGELOqusrUgBqWOtnzZcXCiSZ0Q1Rc\nMARBnov6tzJnWiFFkVvoJNUO0ka+YM2bcQoS00aegPyy+7PeHEWTvz6eTc51b2GIm6Le7b2B\nTqgN2ZKD342ggsn96nr8xrm4uLjM4Aoc6wDVj4N72nlCYdKV71QmF9DUczh8Pwa62IljPSA5\nL6BJ3hbPiSoGD7Z4bJtQ99+qHfmF0tF/rQwquw943vPQcg2pKVCJ2Ssckh/YepoPYAiW1vJW\nkH2bJ0FpgNUncAA4+WjarLLKzMOrzXFnEjRktP8IMZQV5jdFbY8SmXla6+e6ADo3K9WNMhmz\nuKjAAQASyhEtJkhw4hJKbB0nDOZMLuwuFgQSQm0aQkmIZRE4LMEr1Y0yGbPokzUnxa0L+FV5\nWrf5Tq3YqTsRvHIlnatIPBFE55IFhritp/0LCcJZal9gk4uLi8t6wBU4XJqGJp6qLZ3gRbr8\nHdz+G7Be85CXBJH58pPmiZ9TaoL1DCgH75L33QwAoEZBjYFRl/SODP2buz7K9oHo/7efVm94\nV+nZH4jJKyzap1x/RLv5vbCicmqNKcqdBjMNSgQDO0Gt8PXwbIXcy8BzNUeg/7qujrBRBCzW\nfHT5cEo1X+kp6A54AexSq9BXH9OkqCHyDOkilQAAIABJREFURPN/KQIwlDXZsZeqT+krmOOm\nmM95kZnHJ7ctKa+MYWeiAQCW4IKINfY9ZcjWs7Qxg2ErbziwXHNSWzELAIjIFFxdPj/voaCW\n1s2aV0Vm2O9foX1eJ/OlnF1ZzZlE/tRk3quwgaBne69fbpOrrFM3GXTe5OLi4rJOcAUOl2Yh\nmj5pExYGZd7C2DJnB6w+LLPw5Uest47NPOJXzpjHjyoH7/L+8qcBEQfvpovfBKqa72H8CChL\nWjlfCSj7blT2dbTXSetQ4iilXoA5HwFKPos9RzA6N1qUMXovpX8279iKMvr3g29fRwcF1jTw\nHEgBkCMN9KQgsM6TdQ6oACgDi6OyF3A28UdQhtMEkY6oMAxJ2L8sIgiCBlCrEwEAOmQxrHBQ\nGiYxahNnNu4kDOWgMly0psqChcICXrkHnd8IBOZXBi1R5FQiIBk1mdlIJx1mbTS7WXZsxAx1\nmepTVuxaf1CTd/T6z6eKBp+9FPtVaWvUt2KboTrVjAAAEeUNfiaRH8vqRzbHFKkN73VQk32K\nVDBr5eAen+qkoeiWyBsWQwyo8op9GV1cXFyWjitwuDSJVQBhk84KAGC0ZAyxvjGOPl5WN8qY\nx34i7zmkXH8b+jfh1l+jiZ9R4QqQBVoc44cxsK255yAOVgqoBFKk+44bqw7KnaLkc9UhTlNP\ngdaHvrkmKXIEez4IxjhY08A0UPuho1NNc5KyP4dyl1A5isFbQelb4AgyjgG/OvfABD5KYhK1\nI4BBS1zmNNuah0gXlOWQUKTd3ff1ZNjLKVE/Y2bYqt3s8sLiIO0AfrpqziiNgGRfbsNQ8SuD\nBEKQxVBpsBZAZl4ZWq1QawzZwS/DTcpoFo/ECpZNOlL9O61JUlsmvS2gMMfVfqdPQteIepTw\ngFwwucHJIzNfY4kJJhfL8mI2kpmRN/iZRGF3XxvaHiHAzh7/mWQha5jlz1SPT9kStbk+WIJO\nT+XHsvrMtUlC3Bj1bor6XJHDxcVlTeIKHC5NwlQAtF/1YSs0cXQlY772tH381aeV628DANB6\ncMODrd+FFE9T9nkQxdmHnq0YOgyssxOkriKKYEyAKIESBcWmv2bTpE/YxzMnwFfZBRZBHQB1\nYOGTEZQscYUoRyAYeiUcZM36VvAMTT8BVFE1YKVo+gmMfRCksMMhk/PqxvxQTDLfJGVvWd2o\nGKTOxWWZbW5uYEuGYVBmGyxxueJ6ghL2S2jfZ2flg/IOkPqJjwLlAb3IBoDFFjkEmIQrq/OO\nV1azZpFTrWNuwLWcbJKALGVMy6ouVEEAj8x0a/7lZSiVOFzM6QrDsCr7O9Z3wxaVySqT6wtV\nfLLWYDlSR5lJN2hkT4OLM4n8ZN7ggmSG/UFtS8yvtKkepBHCHgev5epGR+O5UlsEDgDQZLa3\nL5AuWQWDM4SAKjsZlLw+nkkW5n9EONG5ZIEItsS6nwXm4uLi0nFcgcOlSZgCvhEoXKrf4pqM\ntgBl6iw2ZuMJ27gNZh6sPGgxqC+WLp6m9JNVEf0s8TT2fGgF+zI0Q+5Vyh4Dmrs1VwcxcjvI\nDtP+xiBz2j5uTDd7pywoZ4pT5dYqgrKCsjIblnCwifEUXq9SN2ajFhVOYvCI/SFO7Tz4pJDt\n06w4pWTY3Pio2oWE/UyKCEoSlQBUCSNo51ixItGJLgBkARAghLgJQAUAwBDKq7uCDAF7tGDK\nyJtzk14E8CvegOIKHM2BCANeNVmyynkcCsOYpngkZgrF4MLglLE4n5v+moKmdNNQKdrYlL5d\nRLXAtJEvVTSv9claWF0t30QAAIOLly5Pl+ZkI0vQlbSeKpiHRiJStzSOmE/t8amJQq2pMK9e\nDzJ5m5tthTU5rC30gcnoVqW6UebSdHFj1CutABnLxcXFpb24Ase6hjgvHH2ydPY08/q8+w9o\nuxsyEWADd4nz/wDVRnQYuQZ8NqXmLguDoShMT9rEg4ss/AIAZC+Ki9+H4jgAACL2HsCRuyr7\n9VLuFZuKeTMB+gXwbFnKsFcE+ZOUeb4qYlyl5Pcw/ouAS7iyOSQiIRmU+t5MpQ/69oKyuLmj\nRRfqG8daYpRJPQgNL9qbNh+PheIA84pPLcK50wdfLjtSBE3CwdVm7DBFdLLizU0TXUW8HmB1\nSxtlZCbFPaESNy3iDJgqyZJbnNISEmLcowiSTUESYtkcQWFMYWzatGtfYlhBWWqXFWVjg2Q9\nWtAQlik4AqhMlp3rVlYmF1PFklV7sS2Y/HJa32RXstEh9g+FT0/lLqWLMzkbBMAF1djHNlho\nU0+iYFzJ6EWTy4yFvfKmsLfBShwncxBOlDd4aEFxxMXFxWU14l7X1i/GmXfGPvfHxrn5fijB\n972/79//r6guVmni6WczxhD5C8AN0GIYO4TRldlCYqWjXHeEXzxlE9//rkWOzF4Qp75aNsIE\nIpo8RoVRtvvfwcxURJSAp23vkcmcwLYLHDwHIg/MB1KgOzaElHvNJmploHgWfDtbPi36N1Np\n3ObpWAnNcQAAa5pK59F/A/ivX2h4YBAVbbcISkvYjmoaB5D5yLYPCWrgIKwgyGskqacbcKI3\n66Qri+gNxFuWZ0SdQZMUDWqz7ol4iac5lQBQYh5NCi1gjLqiIKKMWSpYFichIfPJSkhRsSvL\n1wxRq/N0NAVZDm1WilwEuy4xzNSqdPlJW6amoU+yaC/dpopGNwUOmeHuvuD23kDesLIl/tqo\nTSfsDZFWxnM2WRjN6jP/NwUvZvlU3tg/EHIbpri4uLjUs2p+zFzaC5VKo//+k9ZYled/9onv\nsmAo/qn/sPjxahRHHkAAIA7L7UO2qlHf9QHrrZet01VzdeX6dyu7ttPo98hIoRyAwDaM7KtR\nDcSVn0JdkTzkr1LqDYxds9jTttU430pR/nkw58oi5F4M3AJyAxkoS0Ho9Y1aZyArsZQpC0YO\nUfZtqClUkSTUqoQ/yr+K2iaQo+CEYxoFACywqQ6lByy7eiXZ2YlTGgHzVH3yCEgbJYzVe3AA\nAMMOv19rimmHRJgCQA6gPdX1KxNLFPPmGM19tEyRN3jarwyuNBuRegTRhJ63xOzIOYmsWdK5\n2efxd0fjqIecr8M1i/4ziQDdzOlYsRRNfiZRSBUNLsijSBvC3qGwBwG4Q5dd3kyn3nYhMwx7\nlLBH0U1+aipXObRNUe9I8wJHzrDK6kYZS9DZVGFfX3DRw4MOORoSopNnh4vLCkGQO1F1aQX3\nc7NOyf/8ZzXqxgyZ7z7W+9ufrpnLLYSrbiwRWfX91iPmiz80X3tGTE+xngHl4F3yoBAX/mFm\nOwFA5m2aPsE2PgxsbjWVBOQv258wexFmBA6mgRSwVQGwLWacM4g8pZ8Aqqg6tqYo/QRG7gep\no+n6C9zrL20aIHnYxo9Q4lnKvg08D3IAJQ6ap+60BKULCwkczlM+hCbseNF3Delna+USlNDn\nLGOhF9UDZL5WZd4hDaGyE4DJbNgSo5UiF6JPtmtl6uJAbY19BaU1LHAQUMEapwrhjAAEWQVz\nIqiOLOPAGiFnGmV1o4wpRM4ygsry2GPLzsJKWcswOE0VzaLFCYAhhjUpqnXTN3NlkStZL4+m\ny5pF0eSnpnLTurmvP+hTJL2uYSoA+JZ1Ar+9198f1K6k9YLJvTLrD3piPgcj0gWxddAAgOmi\nWZPJYkvII8d8arLOHGRDxDXgcHFxWZu4Asc6xTh/1jZOum5evaJudu1CuwiictM9yk33zD4s\nXBLnv167T+ESTT2Hfe+ee0zgsGAFFcUJ6L+eMs/U7iBHQNu8xCHPP1vxjSp1YzZqUvF1DBxu\n17PYwDSQI2DZGIKiurg7xmIn92D8TozfCcSBTJqqezsAAIBEcUGVRWYYEVQ7QgSZ4awNKqc0\nUREAGfoYOizESWGM3EOZo8Dnsp2lEAaPgBxZ6E+QBpHFgF8mkQNUUYoDm834kHCQsRCnCQId\nQGEYkjDenaqitcIC2QpruZOUJYqiuvZp5kPDqSTIZNjKzK1r6Nw+bUrn1nIJHAzRJ0v1fWRl\nRK8sAYDBxeVcqZyCIIhSulW0xEhgLX/MbBFE4znjXDJfn5ExkSsNhTzDIU/9BB4AhkLL7Iwb\n1OSl90wxHfJQCMASpNZVP9Wzrz9o2yZ2iQNzcXFxWZm4Asc6hXkcf/UX2NQpMuOUvASSij2b\nwLekFhhrAEq/aR/PvDkvcKAEnhjodh1YvH3z//ftRuCUfWl+JV8dxvC725l304IFZpvA4I2U\n+mFtVO0Dz+b2PYcEiIAS2HlaIFvk7lBmm0xRqnbikGW2FUAiMgxxTlChvIFhUJU2o+01WenH\nngfATALPghQAuQcacXxEDeRttne+iH4ZV7/L7LIRAVDt8jh8azh9AwCEc9WVIGuFCxzCoR5E\nOCnFXaFHkwWRXtFWQ2YY9ygzX9uEbuPRoVsia/Dgeqos0C3x1mRO56LeRnSGZMHY1uPf1uM/\nlyyU31CJ4fYev2Pr1lWFR7a/4EuIDfqMygx39wW2xHw5w2KIAU1ev4lALi4u6wBX4FineG+4\n0TauDG+QB4a6Nw49J577Gp17afahJOO+e9iBDzU0f1urWPbWEmBmKx9h/y104Xu1+8g+7Kl2\ne/XtQ892sKZA6CBHO2CN4TQ96Py0wbsV4W7K/Bx4fjbi24mhw+1ORmCgbQb9jE18MSUFQVHZ\nXk4JohwBZ+hjGJ+RMAxxVlRbkArKmvyiKjklTzFQekFx9t1w6R4S4h6ikwCVspeMuHfZRtQV\nmLMwiiu+VlFGZtW70gDIy/pbwxD7vWrREroQRKQy5lfml+PrkztmKFrrS+A4myqUuFjgsj6j\nA22MeON+dSpv6JbwKizu1zQHXWDV0etTL0wX68W4uP//Z+/NoyS77jrP7+/et8QeuWfWvqtK\nKqlK1mIt3m2wjbeRLdswBprmtOEMDG04xsy023S7Dz20B7rpMXQfPANYCIzxMC10gINtbONN\nsuVNsvYqlWpV7bln7PGWe3/zR26xvJeVGRmRGZn5Pv9I8Xsv3rsV8TLevd/3+31/1orudrYh\nbKPb7XIiIiIiVk8kcGxR7CNH0299Z+GrX6wNkpQDv/axNRwF62/8MY+eXgwon5/7kgbEne9d\nw2G0hFbu09/xL50l0zL232LefEfbjixD8gKMZO0rGrwTfpmvfQd6/rFqbEDsewBGk4GZsGF1\nzGHB6AuxwFwT08r4forthZqBdmD0QnQk+YhSd7M/WV8OQ5S6G3I52UYkaQBUJ0xoLuqgBiuK\ncwyPmvpWtITPXAQ8UJxumFbARegcwKAsxI0t69aJ2cVel6zr+onuYb4I5AEBZIj2oD1fXPdi\niDhBcJNMIMjsfpPRpGEGVqkkzfUfedwQ8aBONGHJJevhm7luuEovdDklosaeqwBqGq/GTdla\nj5IuxzbEof7k6clSrcaRsY29UY1JRERERBCRwLF1Gfq3/9E6cNP0X31WF/IA7IOHB37tY/G7\n7pndysVplPPUO4KO1Sfz6Jk6dWMhfuKfcfyd6OLnDOr6xeKf/Ed17ZWFiHnznakPf4IS7UhQ\nz9yEmeeaw5RpbH1K215H/ce4cBF+GbF+yuxf+8wXit/MzrmmCg5B8aNrNQLRcTFFxKn3f0Ll\nJLtXwQ5kDyVuWc1JmZ3wTVW6caq/hhoHl0AWRD+ocULPfE3zK3OKAIOQEeIgEDjv99l9HqrG\nsFbuIPM2dFO5geYpzZcYFQCEmKBdgvrXe1AAYkStdyPeiBBE3Bgo+2P1QUoYHex53C7ihpnR\nOu/N/+kxQMhYdkx27yzIEuQGiRnW8qoSNgduff2Opxo/ECloaAuYkgwmrYxtXC86ZU8Zgnpi\n5mBynedIntLLLJCJiIiIWGO699Ye0WnIMHp/9hd7f/YX/bFRkUiK1NziXF884X/lszx2EQCI\n5O1vMd70s4h3oLZ88mJw3Hc5d43697T/jG1B+cXP/Ac1VtfExDv5VOmv/zD14U+s/vCU2o+e\nY9ygccQGaeA1AXtbWeq/bfUnbR3ZQ5k3c/H7UPMVNCJJqXvWKINjzSCBxFFKtEm1WUqHutF8\nUU+z+zR4vioHgsybYBxa2M48qrnOQpiRV/pFKe5oPji7z0LVd1NSVxiKrOAStrVH83XFFxZe\nMqqKTwNVQVHbl3XAkmkprKo/rdgFYIhYTPYJ2hgTiYxlxw2z4ns+a4NE3DBN0dXLs4xtTFQa\n22cQIbOV6lOMmu/INEhDqBrJw5R081B605SiLI1tiD1dkJ9ScvxvnBg9eS1XcVXSNo7v7n3D\n4SFra3wFERERG4WNMS+J6CjG0GLXCX3xhPf534GefyDPrJ7+Z339nPUv/xNEuydVS/Un6177\nK+/UMw3qxizujx/j4q9Rqg0mqbT97UgfwPSz7E7DSFF6P/Xd1b0dec1t1PMe+ONQRcgUjMHu\nHWp3ICgFULNNCUEKWjLlmB12f1DX/BWavZcIFow9868D+wc7zONE9f1luNKobsyiroNLoGTA\nprVGKQ6QQRVfETQc3b/WBUl20hxZ71G0iCmEaW2Yp/09tuFrnnEWK2ukoOG4aWwle8iYIRKm\nLM+1gCXbICVIswbTrp7Y9kx8fc0ylebJilfxlCEoEzPS1ib/USo5/p9860x+XncrOf4Tp8fP\njhb+1RsORNkcERER3cMm/y2OAIByni+fQHEK2SHafdvSJSfqW19YVDfm4Wvn1Ikn5K2vC3xL\n6wyG+CmacepZQ6PTFaJGAxeQALMavWy0Q+AAQOlDSB/qommsdgBAhFw8JGGOtNOCQDtwJgCG\nPRDqrKFcvvQYj78AJ4dYLw3fTjtfsyG0FYJpiGFfX2+IG2LHDaQ9dale3ZiD/XM0J3AooBr4\nVkap8dA6H3oinYNcf4GDuYggY0hAay4I6l3rAUVEtIOyr1zNBFhSxJuWhYq54CpXsRRImjJj\nybKvlWZTipQpt5K4Mce+3sRL40VV0x7FgNzXm1j3Go1c1T87XfYWMkryGEhY+3sTSz272eA8\n/vJ4vimraDRfffL81H0HIw/siIiIbiESODY5/Pw39He+AGe+G2WqT/zEh2nv8ZC9tb78cvCW\nSyfRboGDBvbSrmN8qclvYv9rvZefF9leObKr/Wkjq4bs0BzRJTZtYMpnePq78KYBwMhQz/1I\nHVnxQVijfIKdq9BVmL2UvBVGyOqUNU99j6efxGxPSpLUexf13deoXPhV/cyfoDxvB1Ae4/Nf\n5cmXxPF/tSE0DlNsE7A9fY3hAhAUM8R2STdQx1gXQjbMCgFihalP3Z5C1exnWUNwg4mIiG7G\n1zxe9Vy9eGHbUgzGTDm/Ji55arTsLXhJTsNPmXJ4hc0yNhkpSx4bSV/JVwuu0sxJU25Px5Lr\nXafjKX16sqTqTU8nyq4lxa5sR+yu58/LVaUsKez1yJg4NxZ8Dzo7VogEjoiIiO4hEjg2M3z+\naf31z9aFilP6H/8v+bOfQu+2gDdoFerb7gc8N1494g2/pJ96lF/6NlgDUEpWryn3O//P7FY5\nvCP1sx8xj97ViVO3jHnTcQgB3dRKINsnt+9t66kYpZNcOQdVgMxS8jDiB9p6/GWQf4anvrX4\n0s/zxD+Rn0PPPSs4iK7w5D/Cm8KstZ97jcsvUeY1SAa01eTxb3LumZrXiqd+AFWhoZ+s2+3y\n44vqxuJoL/K1J2n7Ssa2fkjRJ0UfQxGw3OYgoeYdC9KGICQY5eY9CE3tUUQPIIJSJAiiK5Ij\niGJh7YapyVo1IqL7aVA3ADhKT1a9obgFwNd8vew23ISLnjKrXn+sMUFOMRNoiyR0WFLs67KO\nIZMVTwXNl8ZKTocEjqqvX5mpzFTnJmNJS+7tSaTWVuhx/GDROSweEbFKdLe0TovYYEQlc5sZ\nfvqfAqK+p5/75+A3SJP6g4QPgIY6Y/lpxsS9H5I/8wfi7b9JP/HR4qlp98KiP6IavZL7w094\np1/oyKlbRQyMxN7yYHM88f5fQRst61jx+D/w9LdQvQhvGtULPPkVnvxqs3FDB2GPZ74bEM79\nECpgCR16mNz3ZtUNLCQGsOb8d+HnGndVJc49G3SE5+HXPTjiyVPBJ5sKiXcrBLn81qckQrqH\niL6Fj5Zod9BZkkRNj9fIghFUJmbsA3XwCeTyIcQpKKuFkCasfwVNRMSKcJR2m5RxABWlfc0A\nCm7gkhl5ZzFfiYG8518qOZdLzqVS9VrZrapobbkOVEOW9L5mvwONfH3NJ8aLC+oGgJKrTo4X\nK96a5rL1hzSs6U9uGGubiIiIrUAkcGxmeDLELWLiUthb5Kvf1RykRJoO3uF+9XPVhz5ZfeiT\n7lf+kkvh1fstEEvRtiPOyRf11ETjJuWX/+Ev23kuAJ4DN9inYJkk3vvh5L/4mBjYBgBCGLsP\npX/996y73tCe4c1SfA7OlcZg5QzKAY11O4VzHSooc4cVnCBzykBYo3p+mXGujoYoOMzVescK\nP8RpwluB8rLxkNshmhf8gszDCy+I+gUdBqyayIAQtwRWnZB5hMxbalL55HykWzDoYEPuCSEl\nxaGw/SMiuhY/LEES8DQD8ILkDwCKeWHJPFn1ph1/oYbF1Xq04paj5+drzhK5M51IqxktOl6T\nkqWZrxRCm453gjv2BCf33bl3c/VNi4iI2OBEJSqbmjADCxn6vcs7fhKVgv+dv4XvzkZoYId4\n9burf/xbXCnORtSZZ/wffsX+hX8nd6/ciyEc/9zJFcVbgM/+WD3+//LEJTCob5t47QfE4fta\nORCRfd9b7fveypUiDJOW9G1tDa6cDYtT4qa2ny5kEH6oFYNedskSV+fcNJq3qGLT4Zf94CvW\nA2emOUyxzT3NEmTdx/5J+BfnPiuRJfNWiLp/NdGApH5GGewRJWrFjiYIxgEy9mHW3UOklp9O\nslaYhjjKPMMoMkBIRt6iERsUAmar9AI2EQCIEHdKmt/B1brkBzyxn3a9hBE9Ql9TsjHzWsFp\n/j4zthH2Pa6Gght8Gy04wfEOcevOnvGC852XxxckNkOKtx4d2dXfXQVEERERW5xI4NjM0I4j\n/FJAiQHtWEqYkK95n7jtDXzpJBdnaGCn2HNL+dP/ekHdmIUrRedv/mviNz/TrKGw76nrl0ga\ncmj7EkpKACEPr5q7urSGfuZr6muLjiQ8dVX9wx/idaPi3gdaPibFU+0YWhBhNSCq1KkzNmOG\niwVLbGqAbJCY9Vhp3CIap0RkD4UpHGTXtTil4Vdx7kLAfiOvWu7Aug+Go/Q1jRIAgaQU2whN\nixYyyTwG8yh0CWSDwlY1REgu2ypUBCWGdBFEPYSeVRxggnkK8IAE0TagxQIchq+4DFaC7Bs0\n9N20BK/PI5aDLQWBmn/iBGHWMDJhypmg9WrclLMfemA1Cs+WRTAbm7h7R/eRtY3euDld31JE\nEO3OdsQeKCz7Zw1rVud4083Dt+7MnrpWyFXc3oR1dEdPNtHGDmoRERERbSASODYz4tUPqHNP\nNZZjZAbp+E+GvGMOyvTT0dfO/r+6+BJPNfazBMDTo+rSy3LPzYsh5Ze//DeVL/01O1UAlEwn\nH/jF2Jveg+XNuuTug3jqsea4sbcdCQueo779181h9cTfimNvRiLThlO0F5kISnAAZMcklWaM\nLOJ7UHmlMW5vQ73csBQkYe9G9ULzBsT2Np0xTZlbOH+icdf0zTDrviMauQPFK3z1h8D8mosE\n7X4j9W7U4gXNOU+fXbD8VKgoNWWKAyK4tYqE6L6LthvRzM8DkwuvmS8S3QQEmw0tgafHXD2+\n8AVJSlpip6B1blS5ZjDPKL7MKANElJS0m7CGv0WbAkmUtYyZpkfxPZY5+1OfMETKlMV6VwVB\nGIjNTdUCV7m0sCnSN9aWQ33J0ZJzreC4SguijG3szsbiZkeS4BKmzAeJX8nOnG5pBtOxwXRX\n+DRFREREBBIJHJuavu3ig5/kbz7MV2adF4kOvVq84edgreAJA+cml7mp+Pn/Vv32Py5uLRWK\nn/8jXcon3v3zyzlR7A3vrHztES42unvEf+p/Xv5oQ4d6/SzcSsAG5ekrp8Shu1d/ijm0i8pJ\n9qZAkqxhxA+15nRD8YNwm7qEzMbXEBp4O49/CdUa0xZ7Gw2+Y2UHyd7P3nhj7kn6zsA0EBr6\nSZDJuefmH00RZW+jwTc270gH30ODx3niRTgziPfT0HEkR1Y0sG6CfX2hqaGJ9vUFSx4LWLiw\nC1WETIZncEQAAPP5WnUDAKCYXyLKAitIwfD0hKtH649ScvSFuDy0FZaVmq8pXhQ6mfM+vyjp\noKAQ19uIELKWYQiacf1ZH0pTUK9lxo3Fe8Rw0oo7/ozje5oFUcIQ/XHTnDd1MEPcHQgwNmY/\nFc3wtTYEdaKsI+h0PFF2y54CUdKUA3FrNaclwkjKHknZmrnT4x9O2WMlV9dLXARsS0e3gIiI\niIhGIoFjk0MDu+kD/x7VIhemKDsEa8WiO6VCM8NrN6mJ69XHvti8T+VLX4j/5IMUu/FaQqR7\nsr/5n4sP/xf/ldMLkeRP/4p1rB1dP71wI65A4aM13Gs883XouZQZrryM0gvU+3bIlXd8SB2D\ncxnVi3XBxGEk1lTggIzTyIOovAJnlKHJGkZi38oPkqbBD3LxaThXwA6MXkoeg709eGcyaOgn\nqO/VXB0FQLFhGOGpCtk9lO1Mf5+1RXOREWBrwvA0FwXVuGzqApeehDdvQGsMU/JuyNWUb7QT\nhsJca5gu4VpQkJlHiVZwJXt6vDmo2fE5Z1C3fPgdw1fc7EvNii8I6tsK+k57SRoyachZ09Bm\nUYKArG1kbSMwISNuSIP8ZrPS1HwNywbC1zxR8RbSVeKGGIxbliRX6auFasFVvuaYIYaTdl+8\nPRUQZU+dnS6782U+k8BoyTnUm7SN1drtC6KSp67kqiVPEZC05M5Mm1M5YoY4PJA8P12u+no2\nadEUtKcnnrGjaXxEREREI9Ev49YglqJYi+nEcvdhSvdyYbohTuleuWfRy8M/+2Jg+iy7jv/K\nafPw8eWcy9h9sOfffcY//5K6fkkLsW4CAAAgAElEQVT0DBj7jlC8TYXuvaEZ6dQXstJeKezz\nzDcW1I05/GnOP069b1/YCeVTXL0AVYKRocQR2LtChiVo4F0ov8yVc/DzMLKUPBxQ07FM/Cku\nPgt/GiRhDlPqOMRK6oTjexDfs6oJtLAosxKhyshQakuVYCxh2lqzSVc5/5W6a8wf5fxXKfsO\niHWuF/B0ztVjmj0AgixbDBnrX0SjATdk0wpkTYbPCHby01zd9At8zfmm3KJZPEYpKlRpjRvm\nWwRuJ2Aobo1XXa+mEWnSkL12t5sg5By/5CnFbEnRaxtS0OWiU9tOteLry0VnMGGcnSwvNMst\ne+r8TLngWntWbWzha65VN2ZxfH1+pnxkYLXX8ETZPTdVXvjHOBU9XfVu6k/2xNr5vWRs47bh\nTMHxq76yDZG2DLkx03YiIiIiOk0kcETcCGnYD36k+lefWuirAgCGZT/4kToPURVuBeovu+MG\nACJj/83G/ptvvOdKoN4R2nMbv/J8Y3zkAG070J5zOJehg1ZNzmXoMkQC7PPkl+DOP1L2Jrhy\nDslbKPu60GMmbmpDz5TKy5z7zuISxRvn6mnqfQfMKL28e1jCymFxE1dPNipoANjlyguUvLcj\n41oerh531GKOg2a3oi7bGLbE+l5jAhAhi/NuXxC2CQesQatcHIa3aVii0VJEZzAFbUvYFV95\nmokQk8ISq01A6AQFVxU95Ws2BMqectXC8l9NV72MbdSqG7No5utFTzU9KZkouwMJq2WziZKn\nLuUqJS94ilLyVNXXsVUkcSjNF2YqDYNmxvnpyu0jZnsrVwQhGzOy0dQ9IiIiYkmiX8mIJgqj\nPHEGTgmZERo5CiHlTXckfuO/ud/8//SVMwDEjoPWmz5IfXV+B3Ln/uCjEYVuWluMd/6a//f/\ndd6OBABoeJ98z2+0LcVaFZfaJBJcfAbutcY2BKUTsPcgtrs9Y2hGVzj/ROMaTzucf5z6W28f\nE9FeBCUJNqOxkIpgC6qpb/LHg50E/YACCgCABkqADyTQ3JClTTCUoyaa464aN0UvteRB0z76\ngbkPh0EACAyAVmIeQTAEWZoDkkEkrbz6bG3Q11mfAs9KribJ/RB7W/utI4qFtmqgyGhwHSAg\nYXRPFVgjDFwrueV5QaHscYNzBDNKrqKgpT+HNAvJVb3WBI6Sq05NFcNakMziqFUJHAXXV01i\nDQBX6ZLnp6wVTLMZKDl+xde2IVKWXBtfkoiIiIjNRyRwRNTAWj//d3z28bmmngykh8TdP0+9\nu6lvxH7wI0u81dh90Lz5Du/kjxvi9n0/KbLLbinaUZJZ40P/gc89o6+eBmsaOSAO3rXMDi/L\nQoQ/hJ91gqycm33RsJGrZ6lzAodzBRz0ANabgCpApgM2RawDZIh9nj5T/7TcMMT++gtGB1+v\nQV14gevMZ2pqNIaJDi2ZKtIiSpcDmxUytOKKsa4SANFB5pzPwueEZgmASJlkmrSyHyVTjDjq\nYkNQUlJSV/4F6UusXqx57bE6BS6TPNrCwQhpQpybinqIMtRqw92IG1L1tatZEmKGkBtqoZtz\n/HJNuoReovXLsuPNaR3L5EqhesO3rrK9bnMqynI2NVNw/JfGi8X5VilxU940kOqLOrBGbG00\nRwvViFaIrpuIRfjU1/jMt+tCxTH9xJ/It34C5o2TnNP/y28XH/4D9+nvLkTs+9+a+vnfaPs4\nVwHR/lfJ/a/qyLGtHSARsNQ0sjCyAKBLzW8C0NhepL0EVs0sbIoEjq5BUMqStyo9yigBICSl\nGKaGn2jZAz8gVwJGs8/lKPOJpkiJ6O62u0JycA3I7Da93iUMcZ8PujUWoczSZQ2Mm2Jw+Ucx\nKAu521XXFrxgDdFnie7s2sOsXg4I60sQe9GK3kRSHFL6ZcZieRQhKWltDY83LyVP5VzlKS0F\nJQ2ZMOR4xa3Ou0UIQl/M7Nk4XpK1bW554T/1vwOsiYJyJsLECEu2mGFRbGrK24ApKGGtKh1m\nibHZTZuqvhotulVfWVL0xs0Fk46qr5+5mqszJfHU89dzd+zoSXfBV8/czodBEREREZ1m/X83\nI7oG1mcfrwvM3s+cAl/+Me17zQ3fL1LZzK/9jn/xjH/hZUhp7r9ZbutYYkIXIpOUupsLP6gL\nkqTMa+f+XySggywPRScfcctwl9aWz8sK1L0J0hsXgmGIHUvtEDvMzrkmUwmiWKNnDfP5oAMU\ngXFgaHXDbESEt6pdYtPawFCuDmh07epRQ/StqNuLQVnDyGh2GUqQ3U2dYurhQqhnLU+tVOBg\nzmvMAL6gQUAwqgARUoL6o/4pbWG87OXmF+Ge5qqvp8irXedrxkTFE4TMSood1pHaeg1C8GWi\nWUuIZjUjJqm51FMQ9bXk1smBqWU1ELA7G1/ldZy2DNsQjt+o8yZN2dBI5Vqhen66vPDxXMlX\nB5PWTQMpAq7kKkGmJHhlpnLr8Lo9h8hVvdMTpVzVY0bKlgf6k4PJqCttRETEBmBj3C8j1gK3\nDKcQuIXz15c/AzB2HzR2b9Une8nbyOzn4tPwJwED1jCl7ppL3wAQ349CYwkPAIp30qPE2gmy\n0GwfYI2suHktK1Re5Opp6BLIhrWTkq+CaFObm6DzQZUgbFCUozuP7KX0G7j0A+jyXIRsSt4N\no0Gz8IFy05sBgDlP1GaBQ1JMUkJx4xkNSgpqf0XMitBcDjEZZc3llReY0LpLNssgPKFmqU0B\nOys+q7lWHpKS9ouV2JdELE3Z17mmFAPmgJSH6aq/UQQOKai2yUugzS8DfTEz5/oLq3pBNBA3\nU6Zwlc47i5+JJNrbEzdbyuAgIGaIapP0AEAQ0paxPR1LrLqZKxEO9iVfnix5NS1abCkO9Nfd\nHIuuf3aq8UdyvOQmrerOTKz2n1xLoboSj/a2MlpwnruWW/gi81X/6Su5mwZSe/s6d9OPiIiI\naA8b434Z0RFY88wY5yepZ4iygxAGQMEPPGS0wlw21vaQvrNMZDOZ4Pr5SuIIYns6OB5hU/b1\nnPtWnROHTFImvHVLMMz5r8Ebm3/lwDnL3hXqeWf7M1DY5+IzKL8wN2ZziDL3wmzzsnyjYu6g\n7HvgXYcuQiRhDiNARFjisWWLpexLE5c7K+pyrcYhKRkzlspGWStC/71LVdZsaCgZ+ktOK2iH\nqfhKvboBQCk+S5SijhnWbjXCWns0Zz14mjXfuLlsN5AyZa2mIEkwNyYn9MXM3piRtY2yP9tp\nheLzViOH+pIzVS/v+Io5ZsiBhGWu4p89lLQv5hrrNKWgWwfTRvs+zZQljw+nR0tOyVVESFnG\nUNJqsAgdKwa3rB4rOjszS3jZrM9XzoyXxgvNPyJnJkvbs7GWK4YiIiIi1oZI4Nii6Esv+f/0\npzw2Z5sn9t5m/NQvUe8unm400gMAGPqr/51z1ynRQ7uP09E3Q3RrenZ3worH/h6ViwAgDAgB\nIog49bwR8b0BO0/+iHMn4c3AzFDmCA3cA1rFn2psL5kPcul5+FMgSeYIkreuOC3CObeobiyg\nq1x6htI3Ll9aETz9VbhXF197Yzz1j9T7DljdaXmw5pABa+eSe5hADGhqKAtQZ0wxiYyEsdfn\nouYKQIJixkrW0p1jiaW42PCrdM1cBQmCXb8KMiG2Q19p3J0yWIm1quamv/e5k04QdYN01QqO\nUmXfU6wlUdwwY3Kdp0CB3TfC2AjiBgBkbaPsqfKCxkEwpJAAA4rZkqIvZqYtCUAQUkEJFD2x\nRXOKVTKYsDylr5echaofW4q9PYk2qhuzSEHb00t57lb9YDFrVgzK2MZMJSBZIxNbn0u04PjN\nRTcANPNU2R1Z8l8aERERse5EAsdWhK+f9/76d+Av3k31hee9z/1748H/FU/+JXT9bdjK8BOP\nzL1x+ipfOUGnnxDv+t9g3dh2NGKOwvNz6gYA7c8/OXbgN9UEsa8vfAGVa3MvnUke/y4XTou9\nH1qqS8sNkWnK3N/62wGuVRxq8ZrWUavEuYjmc7Hmwg+p/z1tPtfmhWgv80tN4UTbDThqMSi1\nohyBNUCQLSmpuM7Hl+fKZ9Znjs7Qmj2AJJmtLlq1r68oHptN0yCYhtglasQLkrcwNPS1xXdQ\nD8njKzmdDjPy4CDhbEMw41aL3uJT9JLvJQyzz17PG9nyl9n2xukZSsD2lJ13VdFTvtamEGlL\nBgoZa8P2dKw/YRUc39ccM0TGXk1GSOsYIjjrYfYa2JmNX81XG2w4BNHunvW5Pn0dmuC2otYw\nEREREetCJHBsRfwnHq1VN2bh4gyfPy1e92v87KM8cwkApEXDt/Bz32ncc+IV/eN/EPf+9NqM\ndhPA5dNhcUrfVheZemZR3VigOsaTT9LgqhSK1cJhtoVtrhBmt+mfP4s3BvZXlcmy7jiTPPUU\nO5MkY0jupt7bO+nVup1IMZ+v6TvbS3QE2HKpxbbc7aiLtRqHQUlbrov/MVf96aqamRcmRMzo\nMwSUntRwCKakjCEGbyhDePqs5tziQeF5+pwhtKSB+ZgkeRxiH3gGUKAMVmycIULME4CudVdd\nkorvFT23wdui7Hu2NJLGutVgpkw5E2K+0EDfOj3Jb5mMJTOr607SRmwp7MQ6+wH1xc2xkhMY\nB2Ab4vbt2VPjxUJNm9jDg6mGFiqK+eJ0Zbri+ppTlrGnN5HszIccDxekVu9aEhEREdFpNtgt\nM6It8JXg9ba+/LJ83QfozR+DV4FbQqJPf+dzgbXcfO5JRALH8lHBjo8B8eLZwB25eHadBQ6Z\nWVm8ZTikKH12UxcKHN44nKvMLhm9iO0L0yx4+hm+/s+zXYQZQOE0Tz8r9vw0jM710NlFNALM\n9tRIAt2VW7FmEIyY3K+4oLkCQFB85d6i7aHsj7tqMWmLoSv+hBSOQQ4ARlVzQfG0LQ8uISJo\nLtaqGwsofUXKgboQZUCt/3kSZZmnm+MCzT2JV0zJU2VPMSNmiMyadMEsz2r6TdpR2fc6JHDk\nXVXxlWZYkrKWEZisETNEX8ycqjeStA1hCiq6c7+EpqCBuJmMVpUbnIGk1VsypyuN3/VCjkba\nNu7c2VNy/aqnbUMkrcZLpuKpJy/PVOZ9W2Yq3tV89Zbh9PalLDxaJG7K/oQ1WW70DUlYsje+\nzlJRRERExA3pvtVCxBoQlnzI83EzDjMOAOWAmTQAlGc6MKzNi0zBC1gqQDauOVmFpH/Px7ly\nCc4YyKDYCOzhdg5yScg+wJWTzWoXxQ61+URGT3D+q0hAdJlpAivOPYbyy3OvABhZ6n0LzMHG\nPd2ZBXVjEWeCR79JO97VySGawApsFzYxktLrpWvMotmrVTcW49pi6dDibmVPj5oi0KgYABjN\nbTRn4x7DaaP9p6TdPhdqMoAAQFAf0coFDi6DJwEXSCoauJT3antGxA25pzO2hYo55/qOYgAK\nFOi9qsJT8Vdz3qtF15nvqVHykHP8oYQVWKbRFzOSpsg5vqvZIEqYc4kPKs6uYklkyo1SmxJx\nA24ZSl8rVEcLTtlXthR9cWt3T7xWxiAgZRmpEAHh5FihUu9Kq5lPjBb6E5ZttP/P59aRzNNX\nZ/LVmj9VU96+LRtdjhEREd1PJHBsRWhkH58JWG/TyL7GUDJkLpvsbfegNjOUvImrl4Lihxsj\nZoarowF7mln4BX39S6hcno0wQOkjNPRWiBs9fqye5eq52aYbFNuH1iQJo5fS93PxBzXdWAix\nmxBr/CesltgBFJ+CbkzlpeTRNp9o1XDhhwvqxhx+jif/iYZ/uqGzCRdONaobs/H8y7RddbJQ\nZb1gzUXNVSIpkKQN0Fq14/g6WLtkELMkWly3KJ4xESpwaJ4KP0k7a+MJcUPcpvmS5hlAEWxB\nI4JWLKqyehn6/MLYNMeUOgQsppZUfHUhVznUl2zvuslReqzq1tgFEGAKeESNNgdtPS0ATFQ8\nR9X9vWvGWNmNp2MyKI/DlmKoqYBCEsWNaCm5qSBgezq2tBdpGL7myVJAHxbNPFZ0dnXAqsM2\nxD27+0YL1Zmqz5ozMWNbJrZhnGAiIiK2NpHAsRWR975Hn3u2yUw0Lu96e8OetP/VfOJbzUeg\nA6/u2Og2I6mjqLyC8pm6YPIIUkca9+y5FYWgAqKeo/rq38Op0z648BJI0PBPhZ9Yc+4bcBa0\nlRl2r6B6nnp+ohUvBvsAmdvgnGdVgIiTtRPGSgv7l4GwqfdtPPMtqPx8iJC4Bclj7T/XamCN\ncrOLJ6DLqJxHol738YKfuoN9qAqMTVU8ornq6Quz9SAAADLEoCk2at+N9rFc9YHDfW00T2sO\nqXeDbHv3VoIt6aAkoMG7YvnoV6DP1QYMqu5JnXg5f6evF5XZqq9Lrkq11U1gwvGazRA1TMke\najSOuNHmWRAzFqpL6k7NKHoquyb1OBGbDyfc2zOw3UlbIGAkHRtZz9S3iK2O5s33BChiLYju\ntVsRseeo+cCv+199iItzlSbUv8N4169QZqBhT9p+hI69jZ/7Sl1w+IB41bvXaKybAxI09C6U\nTnP5ZfgFGBlKHkbiQMCO6UMYuJcnflC7HKK+O8nKshOQ2cH5kzTwRsiQpzeVMzXqxjzuFVRO\nIX5zK/8QkUD8aMef4JhDNPAgnIvsT5OIwdoGo/syhnQZOuB5GgD2Zxo/ojCjDRKQm6zfnnbV\nWUbtJ8O+HiNIQ2zpLr8yvMBK1Ht5UngLZxVkijF3fBrqZCPRFo/M+pXmoCTVa42NV+s0r6pS\nqfbZl7pah60HGUTzv66WkCmzzaqQzxy2EI3aT0S0jCVD/waXX5/CwGTJLbo+AWnb7Eusm71u\nREREREeJBI4tirj5PuvA7frKac5PUu+w2HkYInhyKe79ad59nE9+G7lRJLK05zgdeT1oy/Vi\naAPJQ5SsLw9hDXcM/gxkGvbwrIMmDb2eMjdx7iW407CylDmC+HbOPRtyUIY7ifjO4G3OheB4\n9QK1JnCsGSQR20doqpnqHsLtTqlpE6UP8tjjAQ4mqQMrsU1lX48rnmF2iWxJvYZoVCTXHcUz\n9erGHL4e3+oCB9mGiPu60hR3UV80IWkJOS8suYOMcNuO9UMjJN/Elo1x0VZ1RoUrCQYJEEsS\nccNMmVbbNaHwdSjEurQnjdgUmFL0JaypJtdPQTQYZNrBwNV8dbRQrXo6bsptGbsnbr14PV9r\nf9MbN48Op80O2N9ERERErC+RwLGFseJi37LS/mn7EdreVEzREux78DyKJ9pytI2Nc40n/hne\nJAAwYKap701I7AeA2AjF6leDS9g0LLFCblpNzcfDstxvhK6geob9HESMrB2wtrV4nE2AiMHs\nn/v6GrCbyjHsARp67bzGMb/IMdM08uZln0876vRCeQKzN9tNw5L72/rcnhlMq2glW1OZUn9c\n+AyPsKUfGCaN4bI/7umahrVCSqrz5hCUMsUSPhfBHyBRrJPpGy0T7OsJgLnxGmtvfcoSvpw9\nsVi8kys6QRQ3RCWoaiBpRLnWEa1z81D6ycvTtQUpBBweTMWarivN/PSV3EIPlKLrj5ccS4qG\nP8XpindyrHhsW7tboUVERESsN5HAEbFGuC89l/uL/+6eOQml5PD29IP/IvmWdwVngiifp6/B\nq1Lfdtida6K5rvh5Hn0Uev55LAF+gcf/kUY+ADtANaD4jpDeIjbspp4di1tDSldkSwJT9TwX\nvot5gwAuP4/YPkpv3XQeytzLU19edA+d1S7iByAkF38AXYRIkLUb1g4ANHAfJXbx5I/YmYCM\nUXIPDdwLsdx+e74eazRfYCjkfT1liDY4oShdrahJXzsACzJjssdqsQHwEhfDWqzANfu+Lih4\nAoYpUoK6qKMhkUyaI4pdpR0QGWQLMjUP+HqC4RBMQdmlv01JfYEmo5K6s1cOgfrAASJgyc/U\nin0DCau9XVQsQaagZhMOSRTr/PPqwbh5uejq+lKVvpixRJVBRLtQmsfLbsVXgihpyv5E+5N0\n2gUDZVdVfRUzZGIZF0fSkq/Z239+qjRd9nzmlGXs7UsEdlm+OFNp7vDqKi0FNbiETpbdqqdi\nSzYh9hQbgmrfx4zLucpMxWMgGzN29cQj89GIiIiuYuMJHI8//vhf/dVfPfbYYxcvXuzt7b37\n7rs/+clP3n777bX75HK5j3/8448++ujMzMwtt9zy27/92+973/vWa8ARAKo/fHzyP/9bqDnr\nNTV6deaP/0/vwpmeD3+0YU8+8bj+zhfm2tOSoGNvEa/5aVjtdwhfXzj/zKK6sRjVnHuShoL8\nTcxeyh5vLlShgdctkdxBsX3sXgmI2/tXOmCoAhcea2wFUj3PMkvJV634aJsDeyf1v4fz34M7\nBjBkjFLHGT7PfHlhF66ehr2P0q8FCImdlNjZ2jRQcVPDZgIAzTlgtQKHp8sl73pNnwuv7I8r\nduPGiktgBAUrkkQx6vztxtW5qj/G85YWVTVhy76Y7K5CHkmWlIuyi6CU1dQuOgxBPZIGFY/X\nB9Ny5c1N1gaSh9ifRr3JiEK2oofmGisLGk7a/fH2p/YMxMzRilerMhBhIGZ62neUp1kbQsal\n3YmFmSXF7rQ9VfUrvtIMS1KvbSSWXEMCKPnKUVozTEEpU0bNYVug4PrnpssLXicTwFjZPdSb\n6MIqjHzVPzVRLM4XjKQs4/BgKhO7wY+kIejQwI1/LsYKjW3IZmFmNF1XpSaBw1V6puK5Puer\n3qVcpeopQdSftI6OpLMxs+yqH12aKbpzI78EnJss37WrJx0Z6EZERHQNG+/36Pd+7/cuXrz4\nwQ9+8Kabbrpy5cof/dEf3XPPPV//+tdf+9rXzu6gtX7nO9/53HPP/e7v/u6BAwc++9nPvv/9\n73/00UcfeOCB9R351oV55qFPL6gbC5S+/Gjq7e8zdu5d3PH5b+ivf7bmjZqf/Zqevi7e92/W\nZKBriDseEh8LewcNvhlmD099f66FqpGmgddTesnSodhBuFdQPV8XtPcgvuJOsVw9G9joFJXT\n2LICBwBrmAYeAPtgFyIB9zLy32jcxzkPc2iV/XR5sTtvfTzUlGEFVPyJ5joCR+UsmZErzICQ\nlBGUBk9JlAV7AGkYPtKm7LhDhOJqxb9eH2NHTUqyTbF52gAYYo/gHsWTjCrBEtQjqbsUnDqo\nh4x7WJ0EzxpaC4jdhjx4uM9QzMwwOmZLYQmxI2HlPTXbsdUSIm3Jgleq+ItPtgtU6bGSMdn+\nNB9D0FDCDCspakAzj1XrOsvmPX/AtuLLNo+MAKCYa9WNWSqeeiVXOdjXXamgFU89cy2naoZa\ndP1nruXu2tlzQyFsOTR0KV4g0P+2QUo7PVE8NVZU9R+jZh4vOo+ddV+zr+/kWHFB3Zil7Kkf\nX8m9fn9/pMlFRER0CRtP4Pj0pz998ODBhZfvf//7jxw58vu///sLAsejjz763e9+9+GHH/6F\nX/gFAG9729vuuOOOj33sY5HAsV7416+osWsBG1g7zz25KHAw6+//bcBeF5/nyydpZ3ebYraN\n8BkCCeq9i3rvhJcDSRjLWbMRZd4Iex9Xz0EVINMU2wu7JefOxaat9egSWC1lEbIVIGPWCYWd\nc4HbuXqOVidwEJnMAeadhNWuzTT7OqQvqa8rcuVrP5tirKcXFBMJX8Jtl18ss+PzJLNDZAjK\nSMoubHLVTOBbXD2zmQQOAIKyouYf3u1Qlox7AR/sguILP3GSqNNFS4Kox1qc5BS8Sq26AUAz\nTzulobgh17XObsrxG1akmjHhuNulHeVxLJ+84wf2qck5vq+5c1JaC1ycqaimoSrNZydLg0nb\nlJSNmasZsG2IihfQq7h5gmEIqk0bOTtROnG9ENYUWjM/ezVX9gLUk6Ljz1S83g6kYkVERES0\nwMZ7PlCrbgDYv3//3r17r169uhD5u7/7u1gs9jM/8zOzL6WUP//zP3/27NnnnntuTQcaMQ9X\nQ6wuAV2psRXIj6MUvETBtdPtHtR6Y4d0lAiLL0Iwe5anbiwccw9l30R976Hsm1pUNwCEPckn\nudXVjVpq/CPr48VVHjjMZEGK1ZovMIKf9S29KRyP1UtN+SCa1QsrP1Qjvp6oqpd8Pap4xtcT\nrjrnqnML5Q86SAACoELimxoFzoOLaOUb7BAGKLG+TqhlPyBvn8GB8TWDgbIfsBbVjHKQU2lE\nGG5I2gLCMxrWi4LTmJHHgK/5Wr763LXcU5dnHjs3cWkmdOJ0Q0ZSwc2PmzWT/f3JhSotZpye\nKGLJv9Ji08gXKLmBkkpERETEOrDxBI4Grly5cuHChePHjy9EXnzxxUOHDtn24u/7bbfdBuCF\nF9oww45oAWN4O8ngXCFj557FFzr87hi6iVGa5KlX4LXaFmSdoMztELGmqEHZu9djODeGmjuD\nzGIFd6jdolDwtDLgu14hhuhv7h5qiEG56if5goywCa2klT+O05NA0AyYy+BVqTyaq56+3Ojm\nwDlPj869CHkIv5qmMBsQzeoMe99g/wn2v8PeN6EvrveQugIGq8AiOyAsvjYoHVg3AAB+6JaI\nAJbIdjG6LBGm+XtVSnPN1+1rPjlWeHmidGKs8NTV3POjhcv5alB6SjC7ehIDycZnEtsysaMj\nmfh8CUzSkreNZHZkFu9NFV8ttmgJORdz6CdpRh66ERERXcPGK1GpRWv9S7/0S5ZlffzjH18I\nTk5O7t9f56HY19c3G1/6aMeOHbt8+fISO9zwCJsMXS7nv/wP7pnTIpGM335H8nVvbO04lEjG\nX//W8je/1BCXQ9tid9y3+DozCDsBJ0iqGNrbHOOxU/rZR1CYNa0g2n2nuO0B2BskF10maeQD\nPPl1OPPJR2Yf9b8FVnhLlPXF2gV7N5z6xZKwKXXXOg2oGyFrN7tBvyHWrjYcW+5V3Kd4htkV\nZEnqE7Rcc8qljgthyZSrCg1xQYYhWihcXyJdwgFaH7DiqcBJt9JTptgGwKCkj4AMGkNsoabU\nrF6ErvUV9lidIPgQK/cV3lwQKLhjLdDxUpklWaIQIUqNWxEZ2wj8imOGsLvMzSRtG7WpEHXa\nRg0Xp8sxSwKoeCpX9caKzpK/MEcAACAASURBVLGRzHJKV4hwx46ea/nqaNGpeCphym2Z2FDK\nBjCStmcLeZqPQ6EvFjEkmVI0J8sYgvoTXdSyKmLTwNEPYURLdLXAoZQqFBan3ZlMRojFuxQz\n/+qv/upXv/rVv/mbv2moWwmEbiThP/zww1NTAR34AFSr1Xe/+92zQskWofLMj6994mP++PzT\n0c99NnH3vdt/79Mi1YqC0PPhj+r8TPWpJxYixrZdfb/1u2TW3BGlIV71U802HDS4h/Ycawjy\n+Mv6u/93je0l88Un9cwV8ebfgtggv4ZWP237ILwZ+DMw0jD71jd/+4ZQ9k2onOLyCagChAVr\nJyXvhOwu87Z1JnYAzgV4V+tqmI0+ih9ty+ElZSS11r11KeLGALPy9KK2KMhMmsOtLPxoiYZH\nq+qFFGhBAoDhzjYctWTW0znFdeUGgoxYO9rodj8Vv+z4E1kZ0DWJ1VkSe6L1si3Nqgqwm7Hl\nehoHCCJbisAairix1b+yFWFJsT0du1Ko1gYFYU+267qw7c7GRwvOQu5OiL6BhuSesqdemakc\n6FuuYrstE9uWCUgeDJNIYqaMGbIaVDC1wN7eRE/CfOZKrmHENw+nu8rlJCIiYovT1QLH888/\n/6pXvar25a233jr7/8z8K7/yK3/2Z3/2l3/5lw8++GDtu/r7+xt0itmXN5Qn7rjjjrBNxWIR\ny5BI1hG+8iJfewnVIvWM0MH7EVtVIoMuFa/+77+upus+xvKPvj/2B58a+eR/auGAFE/0f+K/\nOM8/5Z58Vlcr5t6D8fvfTEbjtJLueS9pxU99EfPTUNp9m3jrLzdrFnziy81NPTh/jS89SXvu\naWGE64bZA7NnvQexTATiN1P8ZkBvguq2FeAXeOZ7qF6GdmENUPZOxMOsTIiyb0H1NKpnoIsQ\nSbJ3I3ZLl9uUEETS3Obriq8rDJZkmTLV4mNt6gNsoMnUgHpAy5mX+4qvMucBRRQXtJ3mkz6I\nZODzd4KcVZMIImnudtSEq/IMRRCGSMXkIFFX3+baQt6dqapKjBrTcOZR4DyaSpy2Ghkz4ep8\nw6IxJq1YBwQOzTzj+lWlFcMUlDZlMlyt6LON0YrbUICQsQwzWjGukJGUnTDltaJT8ZQgpCxj\nezoW67L0DQAJSx7fljk1XizPWoESBTc4aWKi7C5f4FgpBBweSj17takl+Tw7s/EjwylBlDDl\nqfHiTMUDkImZNw0mo/SNiIiIrqKrZ36HDh16/PHHF14uFJ4w8y//8i8/9NBDf/EXf/GhD32o\n4V1Hjx595JFHqtVqLDYnXc/aiy6II5sN5elv/SlffHr2FQN49kvidf+SdrfevLP4+Lca1I1Z\nCl/90tBv/bZItHh/tW+7077tzqX2IBL3fwDH3sLXz8B1MLCLgopTwMxTFwIPwJPnN5jAsSHp\nuvliB3HH+Poj0PPpA9XLXL2M7Kup9/6QNxBiN1HsprUaX9swRNwQq3/UKck4xv4zqO1fSzGS\nt93wnYyKr08svJG5onla0m5B2wAIygITze+q7SdCEDE5FJNDDEVbJmHB015VVXCDBLAN5+bA\nzHlGFTAFZdoyVzGEHIxl8265qj1mliSSZixpzM0TFLOjfMVsEMUMYzV1Kz7z9bKr5pesjmJH\n6YqhB2LBSoolxLaEPeP4VaU12BIiY8pElL7REhnbyNhdPbOdpSdu3r2rt+j6FU+VXf/0REB5\nXfNDNa/Dbql7+xJEOHG9sFCE0pcwMzEzbsqBpNU3r2L0xM17dm91wTQiIqKb6erbQDKZXGj+\nugAzf/jDH3744Yf//M///Od+7uea3/Xe977385///Be+8IVf/MVfBKCU+tznPnfgwIFjxxrL\nHDYH+sd/t6BuzOGW9bf/TL7v/0CyxTuQdynYl449z79+1dp/44KgVZHqo4OvXmoH1qGPO3hj\n+nh7Jb78GBcuQjmUGKYdr0Gqw+adfpVHX0R5Anaa+g8h2a3eHzdEV+GNQ1dh9MIcaPvhefKb\ni+rGfAy5HyF1BOYWqllbAdRP5utYXQDnQYKoB8urj1B8oU4WAQBWfJGoj2BLykjqVTxVu5An\nmKbcFjCELaNuAHD1XL6MhzAvW0I77FrWDEZB6XOMuS4SiqWk3YKGV39kSaLXTgFgcK2EUfDc\nvOvwQm9jV/TZMTvEG/uGzDi+arpDlXyVUjImg9VhgyhM/ojYrAiak2M0W9cKTnODEqvpammO\ntJ09vYmd2Xje8T2lMzEjFgltERERG5CuFjgC+ehHP/rQQw+9733vSyQSjzzyyGwwkUi84x3v\nmP3/9773vffff/9HPvKRXC63f//+hx566IUXXnj00UfXb8gdhfn0EwFh3+VzP6Tb3tbaQUUy\ndDbcmgdHmxGSMsOcv968hbIhzT66mfKofvFh+HOzea5O8fQp2vNW2nbvDd7ILrxx6DJkBubg\n8hMrePwkP/8/4M51tWCStP+NdOitXW4CEkDxOS48CfZmXRhg76CeN0C27xJVFTjXmqIEMCrn\nI4EjHIvkSnNYfObA1GhmniYaAWDJvUqnfZ7QXCUyJWUMMUIb8C7WXhYK+H1OuJyyqKlbjdgJ\ntD+BnJkryvVZCyJbmGabzI8Yrq9fAmqlaqX4PGCKkGbJLVCrbpR9L+fWGTco1hPVynAiaYS0\n5lmaigrW2cu+ChM4IrYygujOHT0vjRdGC3NipSUFCLKpL8lgU2+UTiAF9cZNALmqN1pwNXM2\nZvRFRSgREREbh403Nfze974H4NFHH63VLHbs2LHQAEUI8cUvfvHjH//4pz71qVwud/PNNz/y\nyCMPPPDA+gy307gVOEVwwMqUC2Mtr1aT994/LgR0YzKkfeCQMdSGx2irhw69mZ/668aonaY9\nS6Z+dCX63D8uqBtzsOZXvkZ9N8MObwJaPcPFH2L+4S2MXkq/dlkpDOVJfvpz0DUPi1jx2a8j\n3ks7N9SnV3qR89+bf0EA4FzhyS/S4Afa5nmhq2FbWFU3mhrU1XBgc9m5TYtpHVL0S2wJ09Dl\nY4jF+3hB70mJSzbl5wMEsZPkzW0/qaO8GbdU22A1acSyVhvcATSP1asbC/FrbRQ4ail6Af61\nDC55XtYKafwcDgNh7TyX3+YzYqthG+L4tmx1UBUdZUmRsuWlXPVSrm5ikLaN3WtlmOprfu5a\n/lp+8Q7Yn7RetT3bbf1oIiIiIgLZeALH97///Rvu09PT85nPfOYzn/nMGoxnnTEsBCkRALCK\nuaa1/2DPBz408zd/VRsk0xz82CdaPmZ7oT33CK+iT3wJ/twKn7Lb6c6fhbnRWkJ6RRQuBcRZ\n8czLNHx38Luci5x/vC7iT/PMV6j/fbiRjQJf/lGdurEQv/jExhI4uPhMQNTPoXoO8UPtOYdM\ngWRg3RMZ4drTMuFR5kvgEsgC+kns34g/yO2CYM6lxjTDM4wEUYd0DZf1JaAICKIe0I4NZzFj\ni5ggoVkD0Gzk1T6DqibKpozFjBFQ+/scKdZTTpHrv6ySX5UkUmZYmczyCeoUDnBQD+C24DX5\nVc/FdSsFjwRIouYSFYR3r4iImCVmyIWSkD098b64OWuYaknRGzeHU/aaXUDP16sbACZL7lOX\nZ+7fG+UtRkREbAC27nx6kyAM2nYzX3mxeQvtXJWp6tBH/03sllunHv5T95ULImbHj98x8K9/\n0z7YRdaJdPCNctedPHEOTgHpYRo4gJbSidcZL3g2DwBe6ISey88HRV2unKLk7Tc4Y2ksOF4M\niXcnugrVlIoPAGBvgtolcAgTiYMonWqK20gcWM2BWZ8Az3f0ZBcosholeTdCbRTWB80ziq9p\nrhAEUdoQO6hTI5SCejRPN29g5DXnCX2CjrS5ioonWT9fY2t6HbhE8o4lvgXmnOLLjDJARGlJ\nuwjrLKoSUdbqzbszal6J8zkmZY9t9nSo6KziuxwkRZX8ajsEjrAxd2pxF6Krtd43LWnKvBsg\nIi/RSCUiohYGZqpexVMZ29iRiSXNNb1yHF9fzQdkL05XvJmK1xOPzGIiIiK6nUjg2PDQ3R/g\nsXPw6lIZ6cA9NHJ4dcelzE+9O/NT72bXJdNEd7bItdO04/h6DoA1T/yI82fgFcjqQf/tlD2y\nsiNY6dAJthWeI+AHrAMBwJ+88RnDeiJ2oFdiJ1nigmzntUp9b2R/Bs7oYkjYNPA2yFWkCvP0\norqxSJX1aRI37jayZvj6iuI5CxKGZp5y1YwpbhLUESMeQfuYK4y6iTVBz36dzFOMq0RtNNnx\na9WNeUqsXyQR3OxJ8zXFr2C+JpB52ucZgw4TrXOnZ1NYfbFBR1V87RMJS1im6GDBvB/i5axY\nN5h3tgAhBQT8jlFnrjoAtpAVFaBH2K26ivRYhqd0pabhBQG9thm1fY1YDhVPnZkqz3WQBQAM\nJKx9vXGxVtOwQpPdae2mSOCIiIjofiKBY8NDvTvkA5/UT/4tXz0Jr4zMsLjlJ+im17Xt+Fbk\nLBWCcvTZz6My53XKzhQK59B7K+1+zwoOYsSp5yDPnG6MS5t6l8iXCZvoLCOHpe8grj7dHKa+\nDjfHaS/ChpGFH2BLSVZbbWJknLb9DIonuXoF2oE1QOljkKt6aM88HrIhJL4eMBzFDT6+BLDP\nFy062okzEixDHNN8XeEa2CUAYKrR/pjH2ylw8GSTujEbnwIcoNl8wVN8aW6kNXsrPm9Q6z25\n2wWBYjKxNt1jlkixWKW6AUDQkObRBp0LEJJ2rfLIYWSsWLVSashJMYRImi3e+wgYiltlX1WU\nVsymEClDRupGxHLQzKcmS45fVzY1UXYNQXt61siAYwklZc1EloiIiIjVEAkcm4JUv3jjLwOA\n1hAbsEyj8+jpcfdH31TjV0W237ztHmNPG2pteOwJVK43OLzy9AvIHqHsCo5P+9/FJ/4C1anF\nkDDpwHtghhfPm4Nwm1MAQObQjU+3/Q6++D3kL9dFjRgdarHnznpB6bt4+uuNUXMQsb1tPxVS\nt1DqlvYdMMDUEACgAL0GHhCMEvPE7DKeaIAQcKVpzgUmFjFXGC51oCsHAEAI2g491bS+BQAO\n/dxaI9RBFlwFNQocmnNAgFkDw2GU171QZS2xpVmaNz+qxWpPFpiU4hbNFzTP/SQSklLsDbxK\n24IpxFA8MeNWnfnuJ0nDzFqxVa7kEoZMRDUpEStkpuo3qBuzjJXcXdnY2ugL2ZhhCPKbTHEJ\n6EtE6RsRa4rnR+bMEa0QCRybi0jdCML57pfL/+Mz7MytZypf/nzs9e9OfPBXV1l3w7lTQNDT\nzNxLWInAATsrjv8qj/4I+VegHCSGadu9sJdKeqfk7exdA+s6eUWmEF/GeYUUr/5lPvM1vvQD\nKBckqP8gHXkPUjcWR7qL+EECOP89qHkfk/ghyt63EZrdhvkUWGugbmh+hfnKgnjBfIVoh6A9\nTTuGOyyyD+pkYhcZgTVbhPbOrcOPRoGbQtO2wf4GuOjaR0xatjQdVZf/QqBMmzyeCZakmyT5\njCpgUkA2TYsw4GstiRoWiqaQg7GkZlbMhohyLSLWjYoX/MOrmR1fx9fEjEMKOjSYOjlaaIjv\n7UuszQAiIiIiVkkkcERscvyLZ0qf/zRqPe21rn7r7+W23fbr3726Q4eYgIbFl0AYtO0+bLtv\nufubQ5R9Gxe/B39mLmLvotS9IQuzJowYHXk3HX4XnBysFMSG/R2IH6TYPvjT0A6M3lVWjqwZ\nRCPMFwLyI2hbp0/NPMl8uTHGlxmphjYl4atKoqbshvZC6OcgFwa0t5EK9QMiICmDUghKx1jK\nXbXDH0gX0menil615Fc1MwBbmhkzYbZqWhGCQUi161iaecZ1i96cSmUJ0WtbtqwbsGgSPiIi\n1pguKQ/Z35ewpDg1Vqj6GoApxaGB5N6+jXGHjYiIiNiwC5uIiOXhfu8rCOrY53z3y6sVOMw0\nVECeNsxOmeHVYY1Q3wNQBegyZAZi5TMPIsTW2RyxDZCEObDeg1ghlCJxmPXLdatr6iWxqs4s\ny4ER3CuHMUaokw8EZQkGN6UtCOpBh50eiAYJk8x1GgchJdrpMArAJnGIdUOLHEl0c8ioMsQ2\no/FPnijbxhSDjQKB0mY8bcYVa0EdTHlQrB3lKK2kkLa0JLVy7TEwVqm6Nf3UXa3HKtWheNyW\nUdpjRBeRsYOn5bYhbGNNr9Wd2djObKziKWbELRkpfxERERuISOCI2OSo8avB8bEAD4sVQT23\n8PXHAjb0dMSCMXAIkBnIzFqdLqJ90C6SfawvA2XAIuoHjazFeTnYeIK52lRkIQ1xwNdnazUO\noqQhmotZ2o+gI4wxxhhzhWAT9RNtX37xEUMDmm54g6PdJDLM58BFQIB6SBwAwpz8hBSHlH65\n1gqEkJDUcVmqm5GdbM5d9qsFt7hg/0mglJlIrrwQpuz5terGLAzkXHco3l2NmSO2OElLDiSs\niXKj39Du7Bo5jDYQ1aRERERsRCKBI2KTQ7Hg2TDFV2tZR4P3oniRixfqg/dQet8qjxzRVjRY\nLbd4Z01JklhdL+dWCP7ND9QCBKUteZviceYKIASlBfV1eHg1Q6IhwtBKvS0Ulx01qrgCgGBY\nst8SfUspI9RDdMdyh4SUIY5rHmOUASKkBQ1sBM+XDYmrvbxb5wLA4IJXMoRhS2shspy+LY4O\n9jVYcBWdOxpz3nMrylNaG0IkDCtlWtG3G7HG7O9NxE15tVBVmgHETbknG8/Goul6RERExHKJ\nfjEj2gdrPvkYX3iKS9OUHqTDr6V9d673mGAevct96tsB8VvuXu2hhUEHPoTpF5B7mf0CWb3U\ndztSu1d72Ih24U9z8UfwroMVRIISRxG/BZ184Nz9EPUy5wPjIe+QkkY2yhLe14WKurzonwrf\nUaOaqzHZxtoWKTpvlRIBoOIHZxuV/YokkfdKnvIYLEkkzUTCWOr5dlCRIlDvgqOZx6olfz7R\nw9M651aryhv4/9l77zhJjuvO8/ci0pRr78b0YCxm4AZmAJAASIAgQQK0Eu2JokSRlLndpbTU\nSUfuUsRn9063pNydbk9aaSlzx5WWlGgFAlrRSAQJOhCE9278DMa3rS6bJuLdH+3KZFZXd1e1\nm/j+1fUyMzKqukzGL9/7vUQ68u2vmBVrSUIa2w5DSyHClg53S4frKS2JLGN6azAYDIvECByG\nFhH6+pt/zOcOTT/iiTN88mnac5N4w6+18g4nMx95iM8fgldEz1Zx2euQ7Gp8hPuqO7wHvx0e\neb4yKLp6k2//YEtmRD1XoeeqhZ8hhygd53CCRALuVtgrdyf8IiUY4clvgWfv0Ooi5x9FcIG6\n3rCq01pliDYTjzCKAOZa8BDStCEW7Z4+X2/dGuisLfokxVUihEAOCIF0pLeoYbUIdXTbmkCH\nY+XJuboVxXrKz/sq6HZji/XsmP5ilfFc4IV1ZSyeUsXQT1tVPYMCrSf90lz2hyutHidhLb6F\nWag50CwJtjRLWEMExiDGYDAYloYROAytgZ/59py6MR88/FPecR3tWnauxDTlvP7On/Do8ZmH\nJ55Qz98vbvtluuTaRkdJq+M3/7D8na96D35Lj1+gdKdzzc3Jn/2I6FxBicE7yxPfRTiFmeUX\nIbOful9jktvbB+cfnVc35vBOwD8DZ0vkEQgnoQvztiZhAGsNFrYsEynE1ZpPMY+CPMAl6hc0\n3G7f0BVAc6C5tnB9GsWFGIHjNPORiv6vfUT74pv4GlYUismMYNb1CRll5fk6cET0BzZtW1NB\noOsSOTrs+f1LKlpPKYdhpcARsh4pFzTruW9vT4UXyoWhZKb5VI5Q82gpyM82BLUF9SfttDE7\nMBgMBoOhFRiBYyPAo8f5pe8jew6JDtpyBe17HRZ/N2m5czj6WFy8VQKHfviL8+rGNEFJ//Bz\n8r2fQSKicQmHYe6+Lxd/+gM1Nmpt3pq568Opm28j26nfcymERaginB4s6OqvSzz2TejK5guM\n/DOQKXQ0W/xvWBysEMS0C/HPUL3AEZzj/MNQ2Zl98qy+/4Q+fYY6esRVt1mvfQ+c1TF4aw9S\n0HbQSniFrix1DV8X2HSGuaaFyhjz00Q3AubG6erjCMdXQX08ptwEvvLjBA5JNJBwxzx/LkeD\ngC7Hydjzl0AcU8eiq0+Y833NXKNNa+Z84HU5TUljzDid94IKlSbQfK7gb864qZVtk2EwGAwG\nw4bECBxrGxVAyMauAfzMN/UT94Fnrtv45FN06Mfizf8rnBVNt+ZSRGE/ABSzrTlB6PHxJyLi\nQYlPPEn7bqsJ62Lh3Cf+lX/4pZm9Tp8sPfZQ+g1vGfh3/weWWTJdPKXP3Y/yBQAgQd3X0OCt\nkPGXtsVD1erGDJx/lozA0SY4iF0Hcd2SKRznqe9WpntQhuQb9/NXxzk3oR66Tx950vnw78G+\n6FqBri8EOQBF/t9FVBtX5uNRwxSAEWCoxZMzLJ60lSyH5bA6D0uQ0BytZMUpFNO4Um5OJcuh\nClhLooSUNQkXkoSqz/mqaxPjxxTO1PiVNiAXhEFdDgoD4+UglTFfMgaDwWAwLBcjcKxNmF96\nSD9yLybOQkratIde+/O0KaIZIY+d1E/cW2OhxmMn9eNfFzf/wkrNFgAo3R2tcaTjzAsXSTGL\nmCtLzo/WKxbZL/23OXVjjsL3vpV+7RtSr3n9Mqbxij7x5Tk5Cax54kkunxM7fiFOh+JgInoo\nVYD2IVqUUWKoRLgQCegIk0Kyak1buPRsfTELJRxx1U71yIsA+MJJ9fg/y5t+pk2TNbQIskV3\noGs/boJsS9RneAVAXMfcHJEROFYfIupNdOeDQkl5zEyghOV22OmR8nikliHFAtczBCQtmYyp\nxkrbtu9FiBTp6jq1RiJKc5TDaIEmLm4wGAwXLYEyX4yGpWDyIdci+sdf0t/+c4yfBmuEAZ96\nUX/ld/nYk/V78vHHIw3i+egj7Z9mFbTrVdHx3dHxReOm4xwryI1o+Fp88HuROxd+HB1vEn3h\nh6i/f1g6y7mXo3YHAFDMZbdmnjrK2ZfgxygghqVDSOyNCjtwd9UGg9HoIQa75/7WR55q2dQ2\nAj5QaMVar8Uk5CZLVDlNCnKTcpsxu1mnCBKdTsdQsn8w2TeU6u9yOgSJVFS6nCCRkMtKf0hb\nTo2ZKIAux3Vl1Re4I6L1kTgf00Wx5j5RBoPBYDCsQ0wGx9oje54f/2ZtUCv9wN/KndfWXqkX\nY9bGfhGhB2vl8l1p/5vozIt8qqpZCV1+O+24rjUncNM0uJsvHK47saDhq+t3V5PRr4yeGFv6\nHFijeCZ6U+EUOi+P3EKJrZx/um7/Ens+Ju4BgwnUs5+23onlXaAbKqH0dawLKB+ZD4kUdd4G\n0bSFZMVHjb1iKye3fuEx1i8DBQCAAG0nsWstCeWUlMNKlBQXmZWkhCU6YtQNG0gCpYghKLYZ\nh6EexSrQITPbwrIWyqFYMqIiPy7jpBXrspqv+5MkutxOsex2rT1uImXZhdCfKIVBAEcKnwjV\nth4Z2ymGtWVuBGSaLmFzpAAiUkVc00vFYDAYDIZWYASONQeffD4iRwDA1AgmzqGnuptjMuZa\n3E5i7maUChF6iEpzaCXSFm/9bT70EB9/EvkxdA7SvtfS8FVV+zDz6Uf55EMoXIDTQf176dK7\n4DQ7Mbrp5/lb/xeCqjUJXf0WdEd0uJR9Azqfi4gPLCf5XMc7O0SXzwBAYgfcYXin5iOFIpdn\n2z0QAPDEs1Bl2vm+ZczNUA0J6rwNycvgn2Uuk+xBYicoyoPQ6oVfqA/zyLx9DPW2p4uqmuTy\nUegcRIqcbbA3teUsrYIvsK6U6jT4GOs8iYZtjFYcSUlJC5vCEu1gfrEunAEGmjtPnvkCUJru\nRAN0L3zExoLBOb9QDOe/kBPS7XQyoqFp1PIhULfb6evAVwGztoSVsFxqUZJOoHB01JurFjmT\n8zpd6/LBDntWfHCE7HOTk35ZzeZOShI9bqL5DI5OR056YVhnw9GTMNdjBoPBYDC0APODuvbw\nI24qTsN+qeYijrYf4Gf/uX5P2nEAIB49xo9/jUePgTWSXeLKu2jf7Y0tS5cH0aW30KW3xG3m\np/+Ozz0z86A0zq/8lM8/J27+t0g25dNBfZfId/2ufuJePn8QXpF6h+mqu2hbRPoGgPTtd03+\n7Wcj482cK2YGFpye6IqSRKNFEfW/lXOPI/cMOIBm9iKaWfLUISpfQGJw6dMz1GMPwh5svPSh\n1FXsn65ttOGH+rmjc4/kdXc0e0YO4b+CMAuRhLN5puNsJMVnufjUnGTGpReR2EOZmxvWUzCg\nV6ulK+vaPtAAwCPgCVCLrHZWlM1EzHwEmLshP0C0t5l6FubjwMn5/x2fBjYTRVVFbVxyfr4Y\nVvmYlJWnfd3rroTW4wg7rmfKktGMl0ZyNV4YU154eKxw+UBmLpK07IS0PK1CrS0hXCkXJa8I\noi1p50IxKM/WlguivqSVMW1iDQaDwWBoBUbgWHt0x9zFJUFdtatfGthF+9/Mz367Kto5JK5/\nN595QT/wZ/PJIKWsfuwrNH5C3PKRlk+5GXjkpXl1Yw4/zwe/Rdd8oNlR0j3i1qbm3/XeD3rP\nPlF64uHKYOd7fiF5/U21u5Ym9ZEHMHUGQqJ7u9h9O+zY27/Uez2fu786xpAp6roq+oCZwyzq\nfDU6Xw2V5+I5THwlci8unicjcKw81gB13s75h6Fn8jh4PBf+4CnOlwFA2tbrPyC2N/z/zuGf\n5dyPoObyQQTSV1H6+og9g/NcrHPVKR+G1YfEvqih88xHgUmAAZdoG7BlgaW4fw7hBIQDewgy\n02jPpvCAuDqdCWApAofmMNB5xb6AZYmU1Xz1UMvYQjQE5IEASANNNgOeAE7UBc8CHUB7Mn3W\nHpp1jboxja+CQId222pV2spkOYh0+hwr+oHStpy/N0BECWktWWl0pBjucMtK+4otgmsJuez6\nGoPBYDAYDNOsy6uQjQ1t34+OPuRqrSLo0lchUbdKCTxxw3t46xX8wvc4e44SHbT1SrrqTkhb\n3//V+lIXPvow73s99tpHogAAIABJREFU9e1Y5iTVyLncP/z34MhLEMK59MrMuz8oe/sXOGak\ntqfJzJRGXmzHlR05ztDv/Vn+e98q/fSH4cgFe+u2zJt/NrG/ti0rn39BP/7foWZTKkYOqpMP\niZv+NXVuiR629wBUiUd/Ot93w+0XW97aqE1sJTITXSUxM7q5xl0lnGHq2YxwDLoA2QEuyMs3\n8ZYL1LNJXH4z9TRXOaKLnP1udRtajcIzEGkkL6vZl70jiILLRyhC4MgyP11RHuUxHwZyRLXD\nzhBmOfsDBBdmHpJA+mrKXL88r81YJ3NmvYR3rq9ypXCU54ZVE47sSFkDs5PUK2XtIYHaxjqN\nYT4XEz9PdLEIHGFUR9Vp1q/AUQ5jn1QprBI4WkJCioRJ2jAYDAaDodWsy6uQDY7liHf8tv6n\n/wdTI3MxGr5c3PHL8/sEnnrkH/XT30VhEm5a7D4gXvfzItM7v0Mpi+zZ6PHPvoTlCRzlJx4a\n/6NPsT/j8eYfeqHwwDf67/5j54prGh0WxJTehB60Qow1/bIgytzx1swdb43dIfT0U1+cVzem\n8fL81Jfott+OHXXgNdS9n4snEZbg9FJm56Kqfig5xCTr+5ICoFS0qmJYCUjCnkmfoYF+ObB9\n0SOUDlerGzNw8QWqEzig8tGD6AjjGOYjUeYv54EtQF0JDIc88W2oinFYI/8UyEa64Sd0ARKA\nBUR4zRA1Tg8ZB6afbMdcoodivxheqNnPVzlJtiXKSo8wfEAK6rTEMGGtme96MfHY6sKNRwNF\na/3KtA3SKCxjAGowGAwGwzrBCBxrERrcIX/pj/jln/DISVgObdlLu66bv27UKvzKp/nMbD28\nV9Av/Egff9r64O9R52waRRjh8jANh95yrtQ48Cf+7DNz6sZMsFSc+C//aejPv4IGRmup3uh4\norst6kYT8OjBaHfJ7CkURpCOt9WwOxeoSWmATNDAq/jCQzVh6tkPN+YlMqwHWE1Gb1DZiHwE\nqm1IORuvX8yHQITqAQCYANscngDnAIdkP+RWlI9XqRtz0ys8R+mrl7H8JNA28LG6eBIUV1fl\nMb8IZCsi3URXALYfNUMAml8J9dx3l9I84aspR15OaCpDKuSs5iLAAklLdLdtrR33u9liS4i1\njCUsAnGU6bLdamuMFaM7YVOUlOhaImUMMgwGg8FgWCcYgWOtYjl05e2Rl+f6+R/OqxtzFKf0\ng1+Vb/k3Mw9TPbBchBF3GqlrWZ0a/Bee0pPj9fHw/Bn/6MvOnuhWqQBo83V89IH6qhkavmE5\n81kW5am4LVzKUgOBY3nQptshXT7/IHQAACRp4EbadFubTmdYIShmCUQSzPBe4nAcAFl9cPeQ\nM8z+yYidneG6UGzaPIfjCJ6f24HVSYjjCGOmoUtQRcild1MisZt1CD41vwakDhL742pJ6tQN\nAJPMLxJdraNSXSQFguqVWRXq07bY3XhujLCsTmiedwkJ+IIrtgtqva8HUR9zZLfpvpafa81C\noLSdyge1AnHSSlirJFgvH9cSw13JV7LVjbqA3b2p1ZqSwWAwGAyGxWIEjvUAszr8JJ87BmGJ\nbfv4eJ1V5/RelXFp0e6b+eXv1+6U7KJty+rpqKLUjWn0xGijIzODdOV7+MV7oebXNjR0Je16\nw3LmsyzcjrgtlIjd1AKIaPAW6rse5VGwQnKwWQsPwxqG7E1cOhixwe7nyfvmalIYh1F6gTpf\nD3sTgnMAzycayE5K7a873gFkhMzBGsHp2vvNehwcv7yMk2CahUhcBmwDTwIB0AHqjc+SyNep\nG9NMACWKquqSFKF6ANAcOU4VnjpVqW4A0Ox7+kRSNtUVZZFsAi4ANQk7KaJtTY/AniqFHBDI\nFq4t1loNTlNk7JQgygcFzYwZySOZtte3FrC9O5l25CuTpWKgiKjDtXZ0Jztcc6VkMBgMBsO6\nwfxsr3V48oL/1f9bn5pfOMn+/sgLdvarPO3Fgffoco5PPD4fyvSLW38V1rIupmVP7F1K0btA\nygMN30h9e/jUoyiOwMmgfx8NxLgkRsIaxTEuT1KyF6kGK6tmof69sJP15iDUsQmZoWUOvjDS\nRXpr289iWDESO1F6EcEIUKFakIT2oKsdN1SOcz+m7rei9DJ7h6GmINJwLqHU/igPWgI2Aadr\nw0pHZdMDFOMQYXWhNW1K0qBm0kDiWq4AKNgiVV+lwhT1dABAV8lAdTACxRE1L5p9xQW5gEVI\nM4TTugyQAHoAm+hq4DTzeaAIJIB+okuabN8bcpDzxxTPu5k4Itnh9C6q1egaIWUlJcmy8pjZ\nlnbKSqzHZ1FDf8rpTzmaQRvgyRgMBsN6JlCxBucGQwOMwLG2Yfa/9If6bFXdO+cmKKoemHqq\nDfylLW79Nb7sDTh/iIMidW+lSw5ALrc62rn8Wtnbr8ZrkzWszducXXsjDlBB1UmTPXTpnUs5\n8eQJ/fw9yJ3F9Kque7u48t3oWF7PAjshrn6ffvLvoFVlkK59f+wh/gjnnkUwAZmk5HZkrljP\nnnqG1iKo+y4uPInSSzMJF/YQpfZx7sGIfcNxhJNIXk7J2KquOYh2MfvASEXMBTLRIoJlwRmC\nf752kMyNTT+RltBgtS9tkbFELtTV82cLESUqILiNP2WaYy2HNHvLFjhGmA8Bc9klFtEeYAgY\nJqqvJ1oABk/5o7raY9jXpUIwmbGX0mp3FWHwpDflzZo0l5VXDIpdbqezbj04KrloTUW9UB8d\nK2TLgSDqSdq7+tLyon0tDAaDwbA+MQLHmkaffLFG3QDAgYYl6y/4xTV31I9AA7sxsLuFlydk\n2z0f+w9jf/BJLs8nPoh0puc3/2NVMxEV8vPf0S//APkxOCka3i9ueA/SS72Cz5/Xj/5VZW0L\nJk/oR/5SvOa3kFhci8caaMu1snOLPnQ/ps5ASPTsEJe+MbZ0Jfs4T/wY4Om7yVw4iNxzNPQu\niBjDSMPFBtmUeRUyN0LlIRIgG97x2J1VDlaTtrJC8TbFFlGBGCQ6LNrBOBy3M/XcwbknUHxp\nprer7KDOm+Auvi/MsugERFRzWTnd+SVjb/JU1lNTmgMiaYuUKzeH+sX6tBQpFkoNi28oS8ut\nyskx10wpZH6ZKFHfXJZ5UmMKCAlJQQORP6+BKuuoDkplVUzbXQ2eyBok5+e96hZUivWkN9Wf\n6BWm4/X6ZLTgP3JyYu6W6dmp8rHx4s07ek2RjsFgMBjWEeZHa02jR07VB1mz8kOZTs97iJIQ\n179ZXLMSZhZq9Fzpwe9YA/16YpyFlP1D7lU3dLzrF0VXhXjBrL/zJ3z2pZmHfpGPPqxOPy/f\n8Sl0LMW5k48+UKVuTBMU+fiP6LK3L+2JzJMZFNd9YOHd/JFpdQOVd5O9szz5EPW+brlzMGwo\nCHJWI4vrlgKgaeeFsjoT6GnHBxcAtC/oREoOIqwzGwYgB0AJ6rwFHTcizEI4kB2rkWdkE13C\nfHzuMU/n/NOO2eQOcmW3K7sry09I7Ar0icpmtJIGJC1QLyYoQbA4ooUtyaaqaWJhrnM5mQ7z\naaJKgUOHfJB53pVD8WmL9hB11xxZWZlSMR5ArLSyGnShWmMwuBRlYq1Ze8pLWsZUaP2hND/+\nykRNQngpUE+cmnzd7v64owwGg8FgWGsYgWNNQ3b0EogDLd/xm5g8x5PnKNNLu66lwR0rMB//\npafHf//j7M2bfagzx+23vq9K3QD4+GPz6sYcXl4//nVx+/+8hPNy9pWY+MkVW7dx4eVoy4PC\nSzAChyEOexDkoL6GglxYTYl9Iedn1Y15NHs++461E2F1hhc5ZF85+7cNe3WXJduJEszHgTIA\noiTRTqD+Wc9/iAX1OLJD8zhzmcgW6KSmFApy5BZP1XalccQgLbd1a0Qb6fq44hOV6gYAIAz5\nkE3X1vaOJYrwE6HpLesp60GzjuwRCyCMSlExrH1GC345jCh3nywFk8WgO7URKo8MBoPBcDFg\nBI41jdh+BYSArmutmsyIHfthHVjR2TBn/+qPKtUNANB66m//JHHjraJj/n4mn34+eoCYeBOn\njjEZ4jhXwjag8jHxEjgEmY+SIQqyKHNjnQ0HUebVTfY0CXWEfSaAgHOufRVEH4dHwXnAhhwg\nay9oTbXkGCIaAgKAmvy5IViSBhebcWJRl5C7fX1OcRGAINcRQ5I6lzDjauJSKirjrDmygZTS\nPC6q008c4RainpskS66r75AG/pvC2BKtT0pBrDJ1eDR/wyXrzCPGYDAYDBct6+mK6iKEuges\nm94e/uQfa+LWG38R1krfTgnPnAzP1N4jBcBe2Xvm0eRr3jgfqutLMhsvN+6GEAd1DnNxLCq+\ngl1IREz7Q+HMqBsqx+VDUFOgJLnDcBrMjRFOAQRr+Qsww5rH3U2ykwtPIRwDAKuf0tfCaja3\nIqryAgB4uthBbia5PKvdlWAlvqwEpRJy12yaVavW2N3AVFS8Qs9FENHEd2ZTuSYiyU7ITDnM\n10wwbS/LS2jlESRsYQU64s3pSONJtC5xrdgKqbFirI+vwWAwGAxrDSNwrHXsOz9EvZvD73+Z\n85MAqHezfecH5eU3NTiEA0+fPqKz46J/s9yyCy3KfNa52jz52E2dMTXznYNLW3jQztv4/HOo\nyXyWDu24dQmjLQ1K7eapJyI2pPYAQOklzj8yN0MuvQB3B3W+rsp4FQAYuac5+wi0BwAySV23\nIHNFW2duWH2sAep609IOpZhvadHUDf8p5lNAEbCJuoFt8SkJG4ZWpg8QDTOfA2pWdzbRtorz\nxWbiRG7K2N2WsIvh1LTbqCWctNVlN23IsnbocDIT5WxNoUrKStjCXFesS/rTDs15aFfjqxVM\nljQYDAaDYXmYC5E1D5F1413WjXdxbhzSplRMg49Zgmd/Ur73Lzk7k+8gt12aeN/H5Jady5+I\n7Iu1+qvZJPbcop77l6req9Pxfbct8dxd2+jAh/j5e1CeVVLSA+LK9yDVt8QBl0BiKzquQe7p\nqqDVRT2vQTjOuZ/WOnR4x1HsQ/rqyhhP/Bi5p+YfqxKPf5d0CZ3Xt2/ihnWNLboCPVEft2jB\ne/4nmWccOpgBTALniK6dcSptPa1NnVgj2ETXMR8B5opQ+oh2V7+GktDBiKgkqjcZnSYh0wmZ\nZtYgalDrscZxhN2X6M4FBV8FDLZIpu2UsRddv9hS9KWd0UJEskbC3vDCqMFgMBg2DkbgWDdQ\nx8IdJcPDT5c+/4eVyoJ65VDxLz6V+cRnqSP6Urt55MAmZ99+/+Vna+Kiq9e9+saqUNcmcetH\n9E/+rrJWhfbeSldENLJtEhq4jG79BGdPojRJqT50bYNYZgPIxc+h7/VIbufc0wjGIVNI7qDO\nGyBszj8f6T/K5UNUKXCofK0+Mr1b9hHK7De9Zg2RSEo5ot/Xo7VB2bjIpVDZwWQ2i6vMfISo\nxRlDiidCfU6zB0BQ2hZbxPJ6l7QWhgr1OcVT4IDIldRviUUJowmiKwEFlIEEopIypNge6hdq\neuIKGiRkGoxLteldi4OhwZpW1bnDElaP2wWAwetXqTHMsW+wY/RYRDXo1q7kyk/GYDAYDIal\nYQSODYX/va/V501wMec/9E33ziY6oS5E17/51PhnfkuNnJuLiHSm+9/+R3Jr79rRrlfLzZfx\nkYd56jySXTS8nwaWnUUiberdvdxBlklqF6V21QZj/Uer497Z6D4sHMK/gMRwK+Zn2IC4ctAS\nHYGe0OwR2RZlbLGgXjka/WbD2NJ8cOII9NlQz38haM576pAjd8iY5IUVhhF44UGerTFhLmo+\nqTnryLpP8QJIIFa1IWQscbXmk5qnAEVICNosaLB2MqzKaiLUZYaW5Liy2xJLyXcIdaGkLmj2\nARBkQvY5cpUNII26sTHoTzu7+9NHRquaBPWmnL0DjaQ6g8FgMBjWFEbg2FCoU4cXFV8s1qbh\ngT/+QvFfvu4feo593965N/3m94jOmGvrZBdddedFcdkbVz9fk5TRqHuiaaxoaISkpJSLuInK\n9Y1pZ9BAALQmXYjhh/p8fThQp6TVtRbKVUJ9jmsdNKA4q3iytRIMISFpryTE6UeKg3xwmme/\nBDQHgS4krL6EXNw0Aj1VDM/OPWSokrqg2E9asSWEa59SqHJBEGgtiRKW7LRtsa6a5m4krtrU\nuakjcXy8WPDDhCWGOhLbe1Lmv2EwGFaFQMV0UTQYGmIEDsPiIMdNv/39cbcyeeK8PvECl/LU\nv1XuugZypatIZijnefQkAOrfhsQCriXLh5xhLh2M2OBsq3poxyfGN9hkMCweIjemh7KI62mi\n2NPsASQpIaipviea89HFWQg0lwWtflq70tm4uFykstA00WvBUjjCdRJnORx3RLrJV3tmHDVS\nH/T1pMs9gtZlmduE5+eCYPpvxez7uhCEm1JJaVbVq0R/2ulPr8v3ksFgMBgMMALHBkNesjd8\n6fGI+JYdfO4gJTLoHGqXdQVz8L2/Dx+8D2qmcSANbHPe/TGxZTeKEzx2FF4OHZtocG9dY5GW\nokL9yD389LdnpiEtuvpO8er3Qi7prR76/NS3+NiTXJigriG64nW075aIBYy7He4l8Cp76DJE\nmjIHqnZzBpDYivLp2sNTl0KaBGBDa+kHjkepDwP1b2BmVVLnAj1XUUWO7E7KiD3rD4zfuCaS\nkjh2GtHNd9s2DR3qyObZHOiC27TUotibaQ9cR8hFZx0KHJ7Sc+rGHIr1hOf3J9ZfZxmDwWAw\nGAyrjhE4NhTuHT8XHnp6TmKYhtyEPPVDffZHAJDuFTe9ny65ruWnDh/6H+EP/6EywiOv+H/3\nGefN7+SjP4SauYSlzk10wwepu11+E/pHn+fnH5h/rEJ+8pvaK4rX//KixyoX9Nc/zeMzegQX\nJvjMS3TsCfHm36hf+FHXG1B6mUsvQ2UhUnCGKX1dfekK9b2Fx76D8on5UGoP9b5h0XMzrGM0\nvJOsJohsWIOwa50aWkSKaBfz0WqNI0UU4T1RDE+HXLn8Zl9NEJCQC8yNKHYJ2mDTSkJkM3tR\n8RWdXgMlSDcSiSJGij/FumzkWVKReg2VwrBt7X4MBoPBYDBsZIzAsaGQO69IfeQ/lL/+WT02\nY/snuzqcgQTJ2QV5YVx/97+KO36DLrmmtacOf/pP9UGRCPnQdysjPHWOH/wLeeenYKdaOwEA\nKEzwC9+vD/MLP8CN70Rm4TY0lejH7ptTN+aHOvIoH36U9ryqbndC8jJKXrbAoDJJgz8D7yz8\nEZCAMwhnMevb0OeJ0yjlqGcLOho30TCsScJxzv0QagpzS1V3O2Veg7b0whgm6mY+DRQAm6gH\n2ALU5k8pLlWrGzN4atKV/VS3fyWSOojcegVBUhfFFMKsMBb1BXymPi5pcd8Gy4RIAhQpT4jF\n/OsF2XHjyHWYvgFAx+gyDGhm48RhMBgMBoNhsRiBY6NhXXZ95t/9hTp3grNjVB7BU/fWp5nz\nk/e1WODwipwdrQ+L/ij/Cy/HJx+j3be1cgIAAB45jujLZeYLR2mRAgcfeyIuHiVwLAZ3M9zN\niz2ID/9U//TLKM54CtD2a8VrP4j0KndPMCwCDnnqAeiqDgXwTjAlKPPq9pwyQ7Sv8R5KRyQ4\nAABYsycX8NEgR+z09bFKjUNQxpaXLHKe7cISg5qLiicrYmSLLYLaILDGQxC2SAU1/3qAQLZY\nREtdgnREh6+nauKCHEss/Rkx4KkgZCVArrRlW6sIq4kz2hBERt0wGAwGg8GwBIzAsR4oZnni\nDJwU9W5tyktCWnLrbmzdrX/6RY66ROTxVxD6sFp3x0/aIKoVFwSRGz1bzp5py6VrgyTtJeRv\nl3PR8VLt6mIF4KOP6e/9VVXkxFMqe16+53eXaC9iWHn8U7XqxjTeYaSvb08SRxM0+igu/DEV\nlEzIy5We0CgBJCgtqat1k1s+5Midiqc0Z5kDIteiPqKlNGddJklrQAeBqupuQ0lrYFEZHAAS\n1hCHHOj5bydJbsrasuSeNYEOJ/xCONtfnIC0ney0V8ggNmVZWb/WgwNA2jJfa63BV7oYajAn\nLJmwVk66MhgMBoNhtTDXEGubck7/+O/55Z/M5CSnusQt76d9r2n2cI7vrtRg0xKwbLFljz59\nqPoU8fuLtrzxaGBHTP420eDORY+W6a0vUQGAjlXoeMJP3BcRnTzLRx6mvU2/Hwyri4qRxlhB\nFyBXRxeIy9EgCNmsUQVJ0btKDZOaQlKnpM7VnYMg2eEMe2oq1CWGluS4smtR/VOmIYiUtUVx\nKdQlgAW5tli6RbFmHvPyuuLngIF8UBJEGWslZCBbiB7XmfT8ym9tV4ouZ02UOK1rmHEmXx4v\nBXOvbZdrDXckpDCpMQaDwWDYyBg5fw3DWv+PP+aXH5xfsRez+v6/5JcfbHIA6otJFO8chN3i\ni1f7jb9Q25+FmcOoVQ+D+ne39uwzZHrp8lvrw7TvNUtwrKC9tywq3kZCP1pqAfjCsRWei2Hp\nNLhXv1rpG4Ak1xYRpWSu7FtyUoAhBnJlV9relLG3JK3+JagbsyhB2pGuK3uWo24AKClfR4nd\nhaC8nGEXRYdtb0olO2w7IWXKsvpcdyiZNPUpy+d0vjxWDCqVo6wXnphauf+swWAwGAyrghE4\n1i58/Cm+cLQ+rh/+h/pgJLTzVUhHGE+Iq9+yrJlFIXZd7X7ofxND22ceS2nd9Dbxmo/UN4Wl\n3u20tcUWp/PTeN2H6Oo756UWIWn/m8TtH17CUHTtW2jHtdUhQTe+k7ZevtxZtpJ12TrhIsXe\nFB2XnViMEUPLSVqbXdk7J2cQZNIacuWK2nAamoMVnwr046F+NtTPBvpxzWeXM1yoo5vOKtZx\n9p/tYDqPYzCZ6E+4adsklraAUPNEKaiXKPN+WArXRAtng8FgMBjahLmSWLvw+SPRG3KjKGaR\naiKh3XblXb+lf/w3fGF2KDshrvtZuvS1LZtlBWLHVe5H/zPnJ7k4Jfq2THtD8Gs/qp/+GqbO\nAQAJ2nGzuOrt9apHy5C2uPUXceBtPHoCDBrYvnQbTmmJt/02H3mUjz+J/AS6h+jy22gwostm\n27EcdG/GZMRKhgZ2rPhsDEvF6kViD8qHq6NE6RtXZz5zMwAl5IAr+zT7AElyTO7G2kTxMc0X\nKgOKTzBCSduWOmTsP9qkUKxfAqWPZ8txAlUp1ElrLZeUGQwGg8GwLIzAsYZpZJnZtING1ybx\ntn/PI8cxeRaJDA3sRCKqs0nroEw3ZbrnHw5cKt/4Oyhl2c9TZgByRXoZpnuoRe1FaPeNtHuV\n158AxIF31JiMAkDnIO1uU/cNQ1ugzM2QPVx6DroEEKw+St8AezGtgtsGQcjVcN9sI5wDsgAD\nGVCb+g3pFUuEZHjV6sbsDPispC3AUpasrrQKUTkcjrDIiFzrE6X5pbFCoNmK9Npg8381GAzr\nhlCZPGXDUjACx9qF+rdHf6xT3Uh1R26JG4kGdmJg0S6brSTZRck11VthnUF7bhKhrx/+KryZ\nNhy09Qpx64da2QrHsBIQkpdT8nLoMshaReuNjY5i/QL43HyAeklcBTRpm7oADD9QZzRPMRTB\nsUS/JQbbnfbCHNPXCZq5QEtyUU1IxxGWX1eo0rFSXVQMLedC0feVJoAjs3AIKdukbxgMBoNh\nI2Mur9cutOt69GzGRG1hgrj+HSZ7+CKELrtN7rqRR4+jnEf3ZuodXu0ZGZaBWOlcCYYO9YRG\nmSAkpVe9q0hbYf1ilboBgMdZP0OiBdlYzJ6nDjJmRAGGH+gzinOu3LP8wZc8qSUf2et25IJS\nMZypaLCF7LRTrjRNTNYreT8EwAAzU92lQk/CdqUxXzMYDAbDRsYIHGsYacl3fEI/8P/xK8/P\nRGxX3PBOuvpNqzotw+rhJGnLmrI4XXcwVBa6CNmBqNYhGxXFBU+dnFuTBxiVlHbldlpSXcMa\nRjOYoGrVjWl4EpwFLTeVLNBn517JihPnFE/IdhXCAABRKk7HIEoteVhB1OWkOp2U0koQifYZ\nJFUQ6DDQIQG2sC2xwd6Eq4yefZMozSCIikKVwbQzlGpNEpPBYDAYDGsWI3CsbTr6xc/8ex49\nifFTcNM0uAvJi2hVZjC0kuA8Fx+Gmpp5aA1R+lWQG79yiqEr1Y1pFBd8dcaVSzanXFsozvv6\nnOYSAIK0kbSoFJXXkAeW+x/XPBUZV5xrr8CBFFE382RNXNAgsNyECwJWRmjQrLN+zlP+XCRp\nJTqdjLH8aBVJS+RnX13FrBSIGIwO19qUNuqGwWAwGDY+JlNxHUD9l9DeW2j7NUbdMBiWSDjG\nue/OqxsAwvOc+w64vHpzWiEUT9VnHAAIOcto2q54DRPyZFkdm1Y3ADCUj84Akd+WC//kKfYC\nnVfsxdV9xL5o3PbumxbtEdRXGRE0KGlHC08RauWpQDXvY71IJr2pSnUDQCksT/n5Np3uImQg\n5VRrRcwMBoaMumEwGAyGiwOTwWEwGDY+XH4O9etSXebyQUpevRozWjmY/dgt7NO675zCvoro\noBxwUqIoqFLZIVAje2bF5VJ4TrE3/VCQk7Q2WVRrt0nk8Ow+1fEVWEBaki4VNMxcAIgoTWjZ\nv89TQTYohnpGpnGl3eWkLGplWkegQ18H9fFSWO6w0ytTHbPhSdpyV0/qRLYUzharSKKtnYlO\n11zvGQwGg+GiwFxPGAyGi4BwdHHxDUXs9zy1dPm6Kmj2IvNTwKRrCjdoGIhtDqI5KASvqArl\nQrNfrI5MY1F/1ABkVedWtA9CUlC/oL4Wqhu+Dse93Jy6AcBTwVg5lw/Kk34h6xdKYZxMtgjC\nunYtFZvanv9y8dCdsK8a6NjZndrSkdjRnbxysGMgZfptGQyGhfn0pz9NRJs2baqJZ7PZj370\no5s2bUokEgcOHLjnnnsWu4PBsJIYgcNgMFzMbPwW65KiS9sEJWjZ3g1rgJhiCgLPPzuLxF4S\n+xqM4umJ+toTBntqvCZoiQFL1Ggc0pHbVySDo13kg1L9J0GxngqKxdArhN6Enx8pZ9tXumIs\nOFqLFNSbtDceV7YKAAAgAElEQVRn3L6kYwvz4hoMhoV58cUXP/OZzwwNDdXEtdZve9vbvvCF\nL9x999333HPPzp073/ve9957773N72AwrDAmZdFgMFwEyB7oUkTcWqG77quIINcW/YGuyVUh\nR2xZ6NA88wjgESWBwQbpD6sIkQtQpFAl5RVEFsBAcsEFtNLRbiwqwqWFbLFNUq/iKSAkuFL0\nrHepyI/PrZgj0GrSL/S5S7GCYiDQSsfcUyGQLczViMFgMKwaWutf+ZVf+fCHP3zo0KHnnnuu\nctM999zz4IMP/s3f/M2HPvQhAHfdddeBAwc+/vGPv/Od72xyB4NhhTEZHAaDYeNDySsBql0E\nk0Pu3lWZTyvQwHHmR5l/zPw4cKZBNoojNrtym6AEQAQhqSMp90hKNxid+TDzo8Ax4AzzEeaH\ngVNteBbLhSCtqM6vglxJGSAJpKLUjUDxsVA/E+onQ36ZkVtsLo+gtC0222KbJQaXrG5oDstq\nohheKIVjgS4ubZCVZGn+o6UwOFfMXygVJryyjupMnLFTpouKwWAwrCJ/+qd/euLEiT/4gz+o\n33TvvfcmEon3v//90w+llB/84AePHDnyzDPPNLmDwbDCmHsmBoPhIsAaosxtKD6KuWWk7KH0\nTRCpVZ3WkgmZHwcKsw995iwwSrQ/LlXBom5LdgNcuQMjIMgopfsccLI6opkPEmWARj6dq4Ij\nt7JSinNzEUEJV1wS91IwCqF+EQhnXg32Qp4QYkhFuUDItpmw+jpfCkbm6mI8NWmLdMoeWpWl\nvi0sT0XYf9YTaiXlIm6NlFU45s0nTzFbGjRn/ipIZOx0ylrvTrcGg8Gwjjl69Ojdd9/9+c9/\nvqsr4obB888/f+mll7rufBnm/v37ATz33HNXX311MzssmUBthF5vhpXHCBwGg+HiwNlG9maE\nY+AiRCes3vVb9898okLdmGMUOA/UeoNVM/2UdaDPh3oEUJhJRhgWNK/1MJ+JOe9ZatiIZFEw\n/ECd0ZxjKCLXonpvi6YgiITcobigucBgQQmLOhv8c5U+jhlf0vl9LIyG6K6z4SBX9i5hSgui\nOSgGF2rSRgJd8MKJhNWWMzYmYyWaFDjKKvB1aAmZkE4zn59cUOXSygBDapYdtp22HEuse5tb\ng8FgWO/82q/92p133vnud787cuvY2NiuXbsqI729vdPxJneI47777nvooYfitobhwrWTBkMk\nRuAwtBNm0HpdQxo2IGTBrnXPWp+MREaZR4kaCxwA4KmjuiLfQXPBUwdduUdQZjYW5VcCAC0r\no2Aue+ogQ809DPgVzXlH7ljagJLSjYtuZgkYufookUpaSU+FVW1i5ZBsj3Wor/ORRTGenkpg\nFQQOV9o9bibrF/VCFSiFcMaUxCLZ46YX9M7wY+6/hQyjbhgMBsOq89d//dePPfbYCy+8sNgD\naaEr/AV3eO655x5//PG4rVqb9A3DEjECh6ENaBX89Fvhw9/SY2cp3Sn3Xu/c+YvU0TOzMZ+b\n+vLnyk8/qrMT9radmZ/5ueSrbl3d+RoM642Im+3MIFr4JrziqUp1Y+7oQJ9x5ZwjiQXUtked\njbeGQJ+eUzcq5jahuU/EtH1pCdE9ZQEwhEDG3q7Y0xwIsuWMfenCQyoONStJlqBmXx/N0f8p\nZsXQtBr2WEnpJJK2r0LF2hLSV+FU0EjPClmNefmhRNeCl7AGg8FgWJuMjo5+4hOf+OQnP5lO\npycnJwGEYcjMk5OTjuOkUikAfX194+NVDcWmH06naTSzQxx333333XffHbc1mVyL1uaGdYEx\nGTW0Guby53/P/6e/1iOnoBXnJsLH7y/9l9/kifMAwvNnzv36+3P3/n1w7JAaHy0//ejof/r4\n5P/7n1d70oaNCAcIRhBOoH29LVeNCM8Couh4DVHqxnS8UNFyNfqihKhVTWdYxUxD8VSLThEN\nwY6WLQgEByBJCVt0SEo0o24EupT1z2T907ng7KR/Khec1zwvoDCU5pziCc21GTHxEgatirox\nc26QK+2U5TrCytiJPrfDERaBiKJFDM26pPzGYzoxhh2OSd8wGAyG1ebUqVPZbPZTn/pUzyw/\n+MEPLly40NPT89GPfnR6nyuvvPLgwYPl8nxPsWn30KuuuqrJHQyGFcYIHIYWo154WL38WE2Q\n81n/X74AYPJzf6rGa9pVInffl/yXn1+h+RkuBjjk/KM8+vc88U88fi+PfRmlg6s9p1ZCFF1o\nExevJlbu4dmiCaLtUVpJB7BgZ9km0XFdS+rTOlqNJaJtRKSgxdWGhNrLBRfUTC4GAQh0aSo4\nN23kEeoRL3zeU4d9ddxTL3nqEPN8Uowd424bF18VXGn3Jzo3p3oGE10c02Qm5AX+Xx12RI2P\nJEpb67uxrsFgMGwA9uzZ80A1Bw4c6OnpeeCBBz75yU9O7/Oud73L87wvfvGL0w+VUp///Od3\n7949ZyC64A4GwwpjSlQMLSY8GF1Np15+DFqXH30wcmvp4R84+65s57wMFxE89UN4J+Yf6zLn\nHiQOkbpi9SbVWrYB2WonDgK2x2VeoGq/6CwPgk3zLTwdohuZjwIjgA8kgE1E25ejifvK83RZ\ns5ZkJa0kwYqsFhFomecFIwz0hGKPICSlbTFjDi9oJ3OZq3xGhKTddT+IDB4HCoAD6q5XfEpq\nsl6m0Rx6Km9TEOhT1fG8p48k5GXTr6ElUrbIBDpfuQ9BJK1W5ci0kgaNXRbs+ZKQVl8iNemV\n5/rLulL2OElhClsMzeEr7StOWMIS5j1jMLSYTCZz++23V0Z6enpOnz5dGXzXu951yy23fOxj\nH8tms7t27frc5z733HPP3XPPPc3vYDCsMEbgMCye0jhPHIGfR2qA+vdBVN+IK0eXbbNX0oUc\nB9H5zCo72fJpGhaLnhwLTx0lN2kN76LkGrqTvDjCsSp1YxYuPEnJy0AbI22NiPYDo8yjQBlI\nEm0GOps5UoqeUJ+tT5So6GDCwAjzFCCI9gADwLKqCRg85U96aj55tRQW0nYnYbxuXyFFz3LO\nNUfIuXJ4Zu5pBpgM9HjSuoQgCY4lrtZ8npFjKEJK0CaCUz3pLOvnK1rVCBK7QDurTqEjbUoQ\nag8iwgWW2VN6QooZCSNtD/kq6ams4kCQsEQqIXubd/FYSQSRLWSgI5I1XLlwIkZSWolUJtBK\nMVskbLExPoOGtpP3w+MTpUIw88brTdrbu5NxRU8Gg6FNCCG+8Y1v/M7v/M7v//7vZ7PZyy+/\n/Gtf+9o73/nO5ncwGFaYtXgtZVjDMB+9n0/8ALNXupzoEVe8Dz3z1/3UG50kL3qGRKZDpDO6\nkK/fag0u3PrB0D64mC98+S/KD34bzAAokUr9zC8l73zvumyCE1yIjrMPNYnV6MHZNvqJFt1X\nlWA5cpevTjDm1EayRJ8lpj+5HvMzmO0zwgwgRXQ1sHTBqxgWKtUNAAwuBHan3akx77hBkLa8\npFZoWBIMVQ5P1/R8DblUVmeTcnj6bII2xbfUDVg/We3kqlkfJuGAtjZxfl1ZjVK1AaVKrciR\nnY5sSpZadTrt1LiXq8lXSUrHWaiLyjRkTDcMi6QQqBdH8rriPTdeCgqB2j/UIdfjD5PBsE64\n//7764Pd3d2f/exnP/vZz8YdteAOBsNKYoRwwyLgUw/zse+h8j5eeUI/87fw5v0CrevegKgL\nWeuGN4JE6rY76zeRZaVufVMb5rs+8ad45Bk+8xMefwl64aYYLYB56r/+7+UffwuzdfZcLha+\n8helb395Jc7echpZisYYCVxkCMokrMsducMSm2yx1ZX7bLFt2kWC+QXUdlEtMj+7nJeuHEY0\nnWVQwAOu3GOJQUm9ttjiWlfIaHeMRRPqHNdZjVBMPAI+E92nRldlBkkRrcXExdc1rrT7Ep1z\ncoYg6rRT3W6m8VEGw5I5PVXWdd86Xqgv5BfwtTUYDAbDRY7J4DAsAn7lJxHR0OOzj9OO26cf\nicFh992/4f3jX8Kfv2drXXObfeu7AXR96Nf9owf9l5+b20SW3f2vPm5t2dbWma8X+MyD/Mr3\n53QNdrtp989S187GRy2T4PDzwYtP1seL3/j75JveC2u9fUvEuRiQhOxa2amsDoxAcQlgiSRR\n3GJbSOqRtfdBi8BE1M4FIAssUX1QHN2ZVbES1NOOprBxTVgBZg6IFrD5YI7IMgMATDeambkx\nkJRdOV2qabZCJFzRGeiE5nL98YLSjU+9lnGE1Z/oZLBmlhuk1Muw+jAjHyhfa0GUtERitgIl\n50V/b+T8cHPrnHoMBoPBsPFYb0sXwyrCGsXaBigzFM5XPrIOvEHuviZ84nt65BR19Mh918td\n+6c3iXRm6P/868L3vll+8hGdy9rD2zNvfa+19ZJ2z31dwCNP84nvVIW8SX75i3Ttr8Np48o8\nPPZS9HxKhfDcK9Zwe+WV1uMMwR5AUGeCkLwca9LgoKVoT58N9cRcwoUlehyxpenOoxGpFhWb\nlihwCBI6Kq2mff1QiSKSyBigmE11xPZwrZQzbJFMWwNFNcGznUQkOWm7X5C0xGZfHasdlJKt\nSlEBAATgPMCgDFpR19MkBDIFAoZWUQ71+aIfVKRqdDhyMOVQfM5YXEMfg8Gw8QjCJpIuDYY6\nNvzlvmHZsOaxxzF1mIMcOpMo+/Dr7qvUlWFTV5/9+vdFD0gifcfb03e8vQ1zXd/w2YfrY1A+\nn3+Ctr1+FSY0PYH1B1HXHTz1IPxXZiMCqcspc/1qTmpF8NTpUE/OrcEZCPUEWLuySQ2xwS/C\n0n8sXJEoqQjvYVe26zasRZl6DwwCJCWpiSdC1M18KmpDd02+hiszjkyF2tOsJNmWmHlGkrod\nuSNQp3m21EVSty2HsVDPkeZg1kfBx2c7/hJomMSly/SCNRhWGM18tuCrasUi5yuLgr6knbJl\nZBJH2jHvc4PBYDA0wggchoZoXx/5OxTPzjx0JJwkvAC56uzrnl0rP7UNSKneHZMAoBjjmtki\nrO2XRsYpmZKb1mfpkEhS9xsRjiEcB1mwBiA3vlkAIwh5snIFPf1nyFmbfRFbq1JJB2BH2U+I\nJadvAEjbHb72FFf14HBlwpXRDWuXjyDHEf2+rso4IwhXbq7bl5Ue1ygCEEhJ0QsQaBPoBLjG\ni4SIdtefiyBskayPS+qRVrfmEqCIEoSFu400CetD4Eo3EAa/wjogsb9VpzAYVoB8oFRUPkbW\nV71Je3PGrRc4JNFgegN63BgMBoOhhRiBw9AIPv+TeXVjDteGF87ncXRuo6FrVnhiGxOygKiq\n4+b6FCwZe+/V9t79wcFna+LJN/8cWS1bla0CVl+sH8dGJNL0YWYTSqKpKgZBdCnzCzVRol1Y\nxvpckOh1+wth3lOeZiWFlZSppNXePsSuHJQi5atRzR6RkJRyxKCgqmfB7Hn6KFe8bsTnXbGb\nyCVxPetD4DMzSUyUIboMtNgWtiSo5U8zBL8SEeZzwC5gHXt8GC42fBWdIaiZleaepL2zJ3Uy\nW1KzBSwJS+zqSZk2sQaDwWBojBE4DI3gbLQ7AxwLfghh0dZX0a43wRjOtQLqvIQnDkZs6Nze\n5hNT54f/l9yf3+2fPjcTEJS45prUm3+uvec1rEU2EbnMR4EcwECGaCew6Ga0NRCJjN2ZWVm5\nzKKMZTXK3PH1ca5WhZg9Xx9z5WWATeIKYB+4CHKwdkwNOYe4RjCcxbJNTBlMramjMRgWQMS/\n0aY3Daad3qQ95YW+0klLdrhWg0MMBoPBYJjGCByGhoSFyDD176H9dyHZg6Yc+5YBK84+heJJ\nVmVyeqj7ANzB9p5x9aBtr+fssdrWsKlBGri2vSdmzT/6XHrYTQxs1UWfBIm0I6wJ/chXxS0f\naO+pDa1DUBKI9uaTi8sj6CG6HmCAN3Arcc0lzRHOINPx2cwLiTY0eVke7bLFKYXlfFBUrAiw\nhd3hZOw2544ZLnKSloxMWnSlELNGtpag3uR6TiQ0GAwGw4qzYS9eDa3Bjrm4d7qQ6m+7uqGK\n+uTneeT7XDiK8hmeel6f/AJPPNbek64i6c3iqo+gY3jmIQkavE5c8aF2l6jw6Rd5/BQA6Vp2\nT8rqSgpLAuAXv48gturBsNYgWLaIKMmxRO+SDCBoY/9AMOp9SBfetPpQR7xTaeeSR53y81k/\nN+2TwoCvg7HyhKf8JQ9oMCxI0hIZu/YqgoB+o2gYDAaDYRls5OtXw/KhrsujN3RfsQJn59Ef\nwB+rifHoD+uCG4j0FnHVr9KlP4vuTUg5XDrCp76Jckx33lYxHtUwAoAKOXuuvac2tBRHbHLE\nUMUXO9li0BVbVnNOaxVq1HNkLbdpsEFR1r80CFqik26oVTGM6BA85eeXNqDB0CRDaac/aU83\nHiYgaYnhDjdpzXyD+UqPFP3TufKFgl823SINBoPB0BwmAdXQCBq8iQsnkZ927OfpO4fUfyN1\nrEDbFOb8oZj4Qeq9uf0TWB349Hd49NGZB6rMU4c4d1Ts/gDSbWtoIuKXcyZHfZ1Bthi0RL/m\nMsCCEg2X8Rc1gtIEyVB1W6RcqlKwMpDYy1qAT1a0id1CYu+SB/R1dKaGYqVYyXZn6hkuYgjo\ndq1u11LMgqgyN2m05J/Le7MGozhf8AZSzqbMmnHDMRgMBsNaxaxeDA0Rttj9AR5/BlOHOMiR\n04O+aymzYyVOrbxaN4o5wo17X7E8wqN1NTis9Ol/EXt/pV0n3RyzNEp0ULe5+b/+IIhFmm5c\nHHAZnAU0qAOUAYQthn19omYvR2xd87mNROJSYPuM4Sh1AMtquMtRrTrnNxlbR0P7kVXiBgqB\nOpOrqhRj4ELRT1iiO2EKWAwGg8HQCCNwGBaEqPca9F6z0le5wgFZ4Ki2qXLDtkLk3NFoE8HS\neYQFWG154tR3Ce1+NR95uCYuXvUeiDW+0jMYmoFZHYQ+Md9/RAyRvFKKXpecQJ+ddhsVlLLF\nZtHq9A0Gl8JioH0AFllJKy1a03bKAbWmC7KMyeEiwKRvGFaF8VL07Y3xcmAEDoPBYDA0xggc\nhrUKCcrs4Vx9n1qizKWrMJ+VQcW7G6ryvMDBIUjGew0uGnH7r+iuIX7m2wh9AEj3iFf/T7Tn\n1a0a33BxwIqLDJ/gSEot8/3JCAmteZOzOgh9rCqkzzP7ZL1aUMaVbfo+YYbSTJPeuJoVaj2g\nqApddo8j5zLtGTjFfAYoAS7QR7RrhX+aXelIEoprPQ4SVqK6aMBgWCF8Fe244RknDoPhYiKI\n+SowGBpjBA7D2oX6X8flcwgmq4J9N8MdWK0ptR2nJzpOEnYnwDz+DF94CN4EhEXpYdpyBxKt\n6JsrLXHDO3Hg7Zw9T9JBZ38L1RPDxYDivKdPM8+4ORAcV26RS+mxqgJ9XulRhgJIUpctthAt\np/BeQZ+MCPMEeBzUu4yRo2EEvjob8hTA5bBLcdUNZ2aeCib75CCBAGZ+Ghif3VgCTjGPEt0A\nOK2YCSsdEonGiRgE6na7Jr2p6S4q07jS6bTXtBeJYQMjYpQ1IcwPk8FgMBgWwAgcG5fSBL/y\nKOdHkOikgX00sHQLulXDyojtv8QTj3HxJFSRnD7qPoDkcKNDymNcOAfpUnoz7PVXyUJdl/KZ\nJFRtRwPquRLC5lPf4rEnZ0I64NwxPvjfxK6fR+aS+V2Z+fxRnjhHmR7atBv2YlaGwqKerct8\nCoaLEM3lsjpeWV3F8MvqRFLuEbQoewj21OHpgpHph4ontcq5ct/SNQ4uIMJJdHrTVMsFDkZQ\nCg8zQgDMokbdmEaz9pXnygRwoULdmKPMfIxo37KmwTof5krhzCspyeqwOyvSRmqxhdWf7CmH\nXqhDIrKF7coWKCw1hFqFHAKwyLIaeBsbLno6HJnzIwpUOxxz1WowGAyGBTA/FRsTPvmwfu7r\nUDN3U/nIA7TlWnHdLzTql7E2IZt6b26qZ0pQ4GPf4LEXph+xsGnra2j4dessE0EmaMe7+cS9\nCAtzMcpspy1vQun8vLoxByt95l/E3l+deXT+mPrnv+Lzs9n46W55x4fEvg3bccawRgh4NMo7\nhgM94spFdP8J9XiFujE7ClSgzzpyx1Jn1+AboPVfDr6+MK1uANDxLWymcyWY4zpejwI1Asck\n8wWgDCSJhoDOxtOY9MeDCpNmxeGkP97t9DbQOAiUtJZlVtoAZp4K8qWwPBdJWolOJ0Pr6/t5\nxVHMhUAFmi2ilC3tiyZ/oTfpTJSDUqi54lPqSDGYar3uZjAYDIYNhhE4NiK58/qZr6KqoJr5\nzFPcuZkufdOqzaq9sH75S8i9Mh/QAb/yfYBo+HWrNqklQZntdNm/5olnULoA6SJ9CXXtBcC5\nY9EHlC7M+I8WJsOvfBrleWUEhUn1T39GboZ27F+RuRsuUjSXo+OIjsePk1tUvCkoDVhAlF0x\ndS992Bg0z38ABcVkjgCzPqMxjaKqZ8v8MnC64uEpYJgo1jrEU+UgqgVVPsz1xgscbSXr58rV\nBkOlsMzM3e4CSs3FTM5XI6VAz/a4oVLQm7R73Ivisk0QdvekLhT8iXIQaJaCul1rKO1aF43E\nYzAYDIYlc1H8Ul5s6FOPotYujgDok4/I1RM42Pf02eM6n5WbLhF9m1o8ePZYlboxFz/zE9ry\nGoj19j6XLvXfWBvUDfxHPVhp9dT9VerGzFFKPXyvZQQOw/og2k6MY+LNIUjuYVVnVywGQV3/\nP3vvHSfZVd37rrX3iRW7Ovf0JE3SjGaURhGhAEhIIIJBwiBMEnB9wTg/g43B+D3b4toX43c/\nxs/vOiDAj2uLa/CYJMsiiSAhEIOEpAmSJufOoeJJe6/3R3dXV1ed6u7qrq7q6tnff6SzTtpd\nVXPO2b+z1m8t47DhUMm1F0EyDCSVX38Q0GBTQoNd5TCl8YFSdWPqJABnANoAwt2Ipjq2VBJI\nn4AanzQhSDhh9smOcAUJ1aglFFfIwfyc75EARgu+zjCmXxSfGEPsjZm9MVMSVbPkUCgUCoWi\nklab+CkWQ76yqBsAAApjQBLq06GwNryffbfw1QcpMz61qF9+o/3Lv87aOut2gtyF8LhwwRmD\nSD1sOJuOUcUsgGmgJwAABsNTPGjw5EoNSaEAAACOEUnlxjEAwDBS03EQzdAuyTUaeVTuvxmB\nkTgykzGBwDYiD7UlcoEuEOUADMSOJTh0MDQFzWZPGDzrBkmaqynE9MRUBgdiL9HZyoMg9hX/\nn2gg9EREFxDDBY6wj7C4FzW+MYovw9JnZlZxflFM12sl7Yan/0y6wUUicBRR6oZCoVAoakIJ\nHGsRvcpbQc1qjrrx9A/yX/x0acR//idy6Fz8D/4WtHo1tF/7D0CY3EEXIhCUOxRg6vLpFJVq\nT4Hq6VCxwujYGcAEzfXyRGB6lRl4NTTsCGC4coau4bLFULYRWf+04SjGAMKuPHSB5AvF8hCi\nk4C9yHYD1HDZ1FiHENniIsfA0sZ9ESWIAIDG9IgW01nx7AnEHURH56au9AKUWilXK/OpWv6j\nYfidnSFnzbgFzGuCoq5O4XgyPGvJE/PoVwqFQqFQKJTAsRbB7l106smw+GWNHwwAuI/8c2VQ\nDJ7xnvmhcd3tyzw4jZyTg6cQK0ozptBssDuWeYrVAjfZ5nvlqa+CP+tHgImtuG76M8S+bXB0\nf+V+uK5qrX45RHT6KTr/HBQmINqJm27A3ub8ZhStBaJh8S2uPFe0CGVom6yf4eIdAQsAWURu\n8PW+OF+ilaDGujmry79iDjiP40Oe5MFybYUGSEaQbV38OTRMEOv15GDxUBzRNtu0qn4f6xHb\niQYACgAGYidAWa9ojShUpayqDluanQuykspTAKJacxpLlQg6lavUQ0g41RJtlAeFQqFQKBTz\no54t1iDYuxt7d9PAwTlRM8523d34wZBbEIMh7hgAIE69BMsQOCiX9r/59+LQtJRjXL8V26M0\n920hrr8V1lKBd3QD2/kBGj8IzhAwE2KbMH5JcSW76g75zKOUHZ+zC+P8ZW9e1MGFL3/89zR0\nZPoTnDhL536Bm29ke99evz9AsWZhaNl8qySPwEMwKqSNIaJJAIEYBeibe+vxiV4CGJw+DjCD\nb5CUIPAQdI4JXGZ9yuIgeSG8toPOA9QgcACAzro0TAaUIfAZGJwlcIFbbQRxS7V1iJ0A6Srx\nKrsAthntaX8imLEaRcCIHrObJHAwZBHNzgflRUxR3W5KRsk8EEAghSTQGWtuZUREY3k/pErF\n1tbQHU2hUCgUihVACRxrEmTXvpdO/ZhO/phyw2DEsXsn2/laMOPNHthcaBmptkTeQ38hz8wa\nB/rPnOCXruP97dPzc83C9a/EvhuWO8jVBjOw4+rwVVaM3/fH4tsP0qkDUwFM9fE73ofrQr0G\nyqEjj9HwkbKEcTr5E+rdjeuuWNaYlwaJNSVOXRwwNADKpA2f6HmA6bQjIgA4g3gZQNtM5ABA\nqSonEU5x3DzPnH9lCPEQAQAAB0DWVKUCAIiGjvXKHdsAMFz8AGdIAvTPs4/GtHaz05OekD4i\n05nRXC/PhBFjyHJ+noAAAAFjeiSq12bRstLkA3/Sc8TMjSmq6UnDapbMkTC0tBeUFaRwxHZL\nPbYpFAqFQjEf6k65RkGGm2/GzTc3exyAps26++XQucpVfOOiSycqkCcPlqobAEC+DA6cFacm\nzPd9HPUoRLqBh6XHS58Gn4bcBUCE2Drsurr1eqxUB1N92lv/iCaGYPwCxFLY0Q9ssbMaOveL\n8PjZZxorcBANP0/nH4fCKDANE5tw06vBrs3KQbF6IHqpYnLuEx1CvAGAA0zOVTeKnAHYBNDI\nOXm16wCvVd2oNxzxGoDTREMABQAbsQdgw2JGZTAD2HxVQoJEIAVDpjG+0nYYMT0S1e1ACgBo\nwOlqJRf4426hLOJL2W03K+0F+mPmWCFI+wERIEBU5x22rvqkKhSKiwdfuQ4plsTamdpddEgJ\nrIbHbhp7kU5/H3KDwDRMbMRL7mxYbxHrrreXmYwCAOvq1/fetuRjygvHQ+OUyZJvYfv60LWQ\nG5AvPHY/LYgAACAASURBVATe5PTi0DN0/km26x1grRWfDgAAwLZuaKv5yyUnJA0eAMCZDI+v\nDHTiERp4anpBCBp/iSaPs8veDfGNjRyGok4EAKNhcR9gFKC7QvsoIgDyAI1LOkPsIAorpqtb\nIsZyYACbETfX8YiCxKSX9cR0I1KOLG7ELG7W8RSVIOCqNd1IeyGNbD0pHBFYvDlj5ohdEb0L\n9EASV61EFAqFQqFYHKv0UUNRDfK9zFcfyn3768HAeZ5qt2+4NfnOD7BENfu6mb1OPUanvju9\nIDwafYHGjuCed2OqttrypWFcdzv4fuHrn6P89GRG27k3ct9vob54A8IK5nnYq7aKpHzpy7Pq\nxhTOmDzyb+zy/7r0kSwZGdDgC5AdBiOKXdsgUnNDyvqCVpzcsNmmNY8vY73JD86qG0VkIE88\nwq74QOOGoagbTvWmpdVKQpoEdgF2AQ3PjerIlp5otmohojFnQtBsnw5BcsJNp8ykGZr4ttYR\nRKWfRimeEM0SOIpcDFkbWU9MuL4npM5Y0tQSpno6VSgUCsUSUbeQVoKCYPiPfsM99OzUohgb\nyT6yr/Czx3v+6vO8vXobRWeCTj9WcSxBR7+O1/1OYxqsGje9Rt97qzhzhHIZ1rOB921a5gFZ\nf/isA+0Y66hSmp45A07Yy+TsecgPNSyfZQoaPSF//s+QnZlNcZ3tuAN3vaaRYygD+6+iyfMh\n8fVVLD8WT2aITv6MMkMQSeGGq7Bjc7UNaeJY+IrcBfCzoMeWOxJFo5nnFjPVWaOafMYBGl0a\ngOxKoLMkzwDkAXTADmTbAVY2qaEp5IUTOp/P+fmLU+CoLsNVX7FGkUSFQPpCWhq3tAYVZ53L\nOKMFf2ZJjDt+0tQ2Ju21r+soFAqFYgVQAkcrkX/skaK6UUSMDKUf+mzq1z9abS+aOAqh76YK\no1AYa1gXVbQi2vYr63U0tnEn23qVPFZuG6Hd+hbg4XX75E5UOxq549hIgcPNyB//Pfglb7CF\nLw8/wqw4XvLyxg1jLrj9lTj0Io3MkRhw0/XLNOCgw9+Vv/h3EMH04sFv4Y5b2XVvm6OsFY5T\n9iD4ExD4GLMpG/ZuP3CUwNGCWABRgMouzggwlbKUAOioLGNB3NQM5wsE3IB8AwA1RvltFsXu\nKmX4MmjwSFYJHBlHFGG+10YtpaCtzrjjn0k7vph+YIgb2qakba6wzDHpBiXqxmxwrOB32FUb\nDCsUCoVCUY2L6M69BnCefrKm+DSBU33VKssSrwXzbR/Wrr2zWJCCVlR/7fu1m95YdQfNrrYG\nq69aCejUU3PUjRnk0R80chjlcIPd8hvs6rdhz05M9GLf5exl/4Vd847lzPRo6Kj8+ZeL6gYA\nAEl68ft09InZwNh3afhhKJyEYIIgB8kIdiehLCWbaWAmlzwMRRNB3B52o9kIYM1ssBtgXcnP\njCNuA9jcqAGGspbVDZgvK+Fiy1eYJaaHpOrojFnaWptjjxX8w8PZp89PHhjKXMg4xa980g2O\nj+eL6gYAZLzgpbFcqO5TRyaccLltvEpcoVAoFIr5URkcrYTMZWuKT2NVM3dAsFLLHVMTMSP6\nGz6overtNHgKDIt1bwTDmmdzTGwkboKocJLToxCbr+Fi3aH0hfAVmSGQYvF9T+oPMrzkJrzk\npnodj479OPRdOB19ArfdDABQOAnZQ7Pnn/qPrkE8ApOzr/2x83Jga22aEY4cIDkBECAmgPc3\nto3ICpFEvJboBMAEgASIIm4EKE0c0xB3AmwByAIwgJi6Ma00OtMdCPHU1C6Sf2VhxHUDgNKe\nRzMqj8W1lGkhAAFJkgzZauv8sgSOjOYGs9Nffd4XEwV/KOft6YlzxAvZkHchnpCjea87uoKF\nWr4MF1BKpRaFQqFQKBaPeo5sJbS+9fDMT8Pj1cHUNjLi4JX7R2LHTtCjIPLgnAaRBS0J1kZg\ny3+OITr3HI0cg6CAiT7cfCPokWUfsyoYTeKWxdVQcAs330XHvj53f4aXvA6wsdPIan0EGANc\nW0lV2ZHQGQFlR6b/pxBuuoG2TjNusJjcgpub6U7SIMij4Ocgp3umEgCIo6jtBbaAhXArYCNe\nttA2xkzRimLFsbmVw7ysKF2MreS1evUT182oZnhSSCKdMZ1xQWLCTTszsrjFzbgR4y17lR4r\n+EV1o0jGDc5OOpva7LwvQvfKVYnXi2oWqtrFVBykUCgUijqiBI5WInrH67OP/HuloUb0zl+a\nbzdusMvuk4e+NEfjiPfjjjdB9gBNPgk0kwjKbEzdCvYyWqv4efnkZ4s+DgQAL36HXf8e7L60\n9mMR5I6Qex6kj0YnxHYDW677HXbvRbuTznyfcucBGMb7ccOrINq3zMPWPIzObXQypKoIO7bO\n1x2mYcg8yHEgATwJbHmFIUaV+ZI+UxMk8uEbcA2794JmQWITppbw42k9KDhQVDdmQg4FT6Nx\n22rO4yCQQjoEkqPJ8OJ9/98sCMgTgSDJkRlMw8VdQBhiu9mW9jLejBkHQ5YwYrU6jAoitshT\ntggMsdgzRZIcdSZKZSBHuK7jt5sJvTVTXUZyXrX4praG1mmWkjC0tFth/kIgiQ6PZDnDqM57\noubF0EpGoVAoFHVBCRythLH9stQHPzzx4F+TN/sSJnb3vbHXvGmBPROb2HW/QwM/h9wAMA0S\nm7D7CnBO08QP52wmCzT6bexJgl69J8u8yGf3lblUgpeTT32B3/mJ8rmuCIKBszI9znvW8/au\n8gOJAg19HdzpUg4CgMn92PU6sNYtbWCzxDfiZe9u7oMSrr8aj/2Axk/PiTINd7+uSSMqIsl9\nHrxjs6X42jq09gIuMa8H1+2mM+VGsACA/Xum/49X8Q3VErixup3K2oM8kINhcQfkMLDehg9o\nUbhi3BUjBNMzQJ3Fbd6NqG4rDcIV/oSXK/ZD4ciSRsRanEihMd5utQUyCEgwZDrTFl9/IYkm\nPT/r+1P1Z5bGU4ax9t635/yQJBciOeZMxvRItAWzXTxZrRWuBICIznNeSLJGVF9ZgTVl65Nu\nkPHmahwIrpAA4EtyAjnhBlvbIg3r6qJQKBSKlkY9ibYYsbvvta69Kf/9R4Nzp3hnt339rcal\nuxe1Jzexf463AmWfD9tOUvYgpm5byuACj84+ExL38nThedx0QzHg/uLJzBc/I4an9Qtz78vj\n7/5t3j7bx4RGv1tUN6YRORp+GPvvXwteDIyzm39NHnyYTj4JUgAApjbilfdi++bmjovc58E7\nOicUnKeCh5El/R4AcOvL8PhPaPjoHB+OWAfb89rpDSI7KHsgZM/ojqWdsWUpVPV3pMoWJKsC\nV4w7Yqg04suMJD+mL7cJtGIxBFKMuVkq+dkIkmNuttNKGNWK4CrQmKbV+BhAAIMFx5+ZKhNA\nIRCucHpte429Y/eq9JohoIyf44xbvMVaCJs8XCAwNAYAfTHr6Fj51cbgrCOysp2DEeCSNnus\n4I87vickZ8wNRNnVUEg6n3W2tLWeqKRQKBSKxqMEjtZD6+5LvPX+OhzIHwuPB1XiC+JMQpUu\ng5QbKT75egd+NvHXfwRi9k2R+/QTwflTHQ88iIYJACAdyIdZM4gcFE6skamvHmFX/TJccQ9l\nh9GKgxFt9oAAyAfveEhcjIAYBb6kdsJMY3f8Dh3+jjz2Y8iOgN2GG65iV75+9u+1+iFxDaR/\nPmcvsx8T1y7ldC3MPJfiVarouaK8tysACHICmdPYKvg9r3VygUNholjWd9rNFWyonPV9vyIR\nQBJNel6H1WIT/vkJ/XiL5P1CywkcnRGj0oMDALoiBgAkTW1LKnJmslB0/Ywb2sakzRtShNRu\n6+22DgDDee9CNiSRJOsJQdSYwSgUilVCoMyGFUtCCRwXMdWcNZecYa5XbWKC+mx9b/bf/6lU\n3ZhCDJx1nnjUfuUbAQCCdNW32cFkeLxFYRwTq6b6QGYAqtxI5MQSBQ4A4DrueS3f81ogCnUY\nwbabwN5M2YMQTACPon0JRHeu+T6d5WAUMAJU6UiCwJZYL7aiSPIJwq0HBTkaKIFjxfFl+Ocf\nVFGZ64VTcfWeP966aKgFVX7kABBQ6/29KVvvi1sXMnO6pSRMbX1y+t6dsvSkqRV86UtpaszW\nmuD+E4Q2VSEABCGJ84vs1qBQKBSK2lECx0WM2R+qF6C5VJ8LM46pjeXWEgCACD0zPRRI+sdf\nCN3bP3poWuCYp5NLHZq8KKoxz4NjPZ4p53nzZq5b+q9urYDabvL3l0t7fAugyspW1MIKv+Km\naqVUK3rWZhDRbaeyrfgMLTrP3toe6YjoAxnXCYTBWXvE6I3NuasyxKjBm2hsbIRKGAiIVfut\nKBQKhUJRinJsunjB+DXAKnIutAREL1/6Ma+8F3h5Rj1ue8VsngJBZReY6TXFtGctCXoq9PBg\nq/L+FYMnqibvLDl9Q7F4WBcaLwfWCcABEDCO+tWordIOMgx1rJICxrFqJpeijhg8/F/r4g04\nloZexUy0Wrx1MZieNOLVusQYNXacWT20WfrOrthVfcnLuuNl6sZqIGHqoTpG0tSYqk9RKBQK\nxSJYa08kihrQ4th9L1ibZt5FMYjswK57ltONFds389v/APuvBCMKTMO29ez6+9nlJU1eGNM2\nhLeh1TfPmmtgx6tCKmjargNteV1LVyEkQBaaPYgpOBo7Q8La+uU2i1UsEkygfj2ad6F5Fxq3\nAGt0A+PFQUB5kGMWJirXcbSVAUdjiGpW5XwPEWPVSwUXCQGJKjI0AMR0LXSWGdfXYEKorVmd\nVopj+ZMSQ4y1YBeVlkBnuD5ulWkclsbWxZRyqlAoFIpFsQafSBQ1oCWx83VAAYgs8ARUPMYt\nhVgXu+F9ADBdNVtB9PW/Mvm3f1IWZG0d9i2vmV22NmDfr9D44+CeBwrA6MTkdRDZVofhrR7c\nAZr4EbiDAAQ8gom9ELuyPl/BkjEuRdTIPQTkAQAAA2MbmpctsNeCkKTh5yA7AIgQX4+dl7Vs\nfnfDWK3Ssxyn4ABQBgB0AMJ+F+yiF6POErbWDUAADoALEAFo1bfcqx+OrNNMTPp5V0w3+zCY\nljQiWjVzpUXgySDt5T0ZwNQcXrOiul32b1VnrMMyx1xPzhSrIEDCMCLa2nyc4Mg7rFTWzxUC\nd+qnbnIjYcT4Mj5nxfy0WXpE56MF3wkkZxDVtXZbV/cMhUKhUCyStflEoqgN1EBrW4njhkat\nG15JhXz2f/+dzGWmIvqWXYlf/QOMzHX+Nzqw55cAoJpQ0to4p2jom7OmniJP44+DO4SddzV1\nWAD6VtQ3g8gASGBxwGW38CiMyIP/AvnZfqIU72e73wlGfLlHVjQYmiT/p6VOtAad0zEi9WuA\nGEODoQ4wTvQSQLHZZBfidgD16nVF0BjvMOOCpCDJkVUmGtSEK/wxN1O00pBEab/gSVHZkyWi\naRbn+UAEJDVEi3ONMU/6WT8fyAABdKbH9KjGOAD4MvCEJ4E05JZmYgtezBmyhBFPGHFBgiFr\nyp/gS3IDSQCWxsJLOJbBuBNMur4nSWcY03mHbTTR7MIXJIgMzvpWX/mMQqFQKFoCJXAomoD9\nitdZ19/mHz8sJka1dZv0Sy6dN3Oh9R6IF4TGHw9pWZJ/CdwrwWx6XxUOvF6CF8lDD5WqGwAA\nmXP0wpfxivfV6RRrCxojOQDgAESQ9QGuouIgCo5V/miR8pocBD6VXTVB9Iu5dpPDRFnE65vo\nWbjmWb60MUXaz1cahTrCc4VvVjgrMcRYSU1KPiikvWxxUQjXFW7KTBaEWwhme3Zk/FybEW9d\n94qmZG0QwVDeG3f84rfTZmk9EbNeGsSZtJP1pzvCCElOIDOe2JS0Gt+QNeMFgzlvqkktAqQs\nvSuiK98NhUKhUNSKEjgUzQEjMWPPdaURMXgu+28P+kcOklPQNm2LvvGdxmV7mzW8lUXkwR8L\nX+WeWQUCR/1In4HcYGWYJo5jYRRs5V1aCpE4BPLM7LI8BewS5Dvm2aeh0Hh4WI5PzfuIToY1\n0ygAXABYv6JDUywTSVSt76wng0qBY+6+MuPlyoIEMO5maK4iJkmOe+kuq53NVWSmql3UVDaU\ngZw74c5p/TvhBFJCf7wOCQ6TblBUN4q4Qo4W/O5IQ3WotBucy872rCGAMcd3hdyYUPlfCoVC\noagNJXAoVgXei89OfOoj5HvTi4ee9g49Hbvvg9G776u2i3/yJf/F58gtaOu3mFfeCLx1XhGT\nX3WNDNbSMz4VRudZhUrgKEWeK1U3AACAQB4HlgLsas6QylmwE2hI22kAIJpEVALHqoaqf7lU\nrTHsDK7wQ3enyiQ1ACIqCDeq2VOLuSCY9LxAEgDojLUZhq21zpV85QkklakbU6S9oEvoBl9u\n5k7WC1e1Mp7obqyJ6lDeqwzmfJHzRVRXPwmFQqFQ1IASOBSrgszn/++iulEk95UHrRtfxdu7\ny+LkuenPfdr58beLEW3D1uSHPqH1b17pcdYHHgPUQ2UODO+P27Lwqu8YsfqqixOS56vEzyFf\nHQIHJoBGwuPTVJsJV23JoVglcGQMUYZpGVNWGvMQKmTMg5DTM/ZJz5v0Zi+DvpTDjtNumrG1\n2JBlaThB1c/WEXL5AkdQRb0SckE1s54EkvwqZywEUgkcCsVFiy/U84NiKaxWr37FxYQYOh+c\nP1UZp8D3nv9ZZTz70P8sVTcAIDhzbOJ/fKxSIlmlIIdYWGsSHoVIeA/dFgWTm4GFzVU0G+L9\nDR/OKqdKt2BaJV2EAfmWsLCGfNPM/4cbxyIqQ9kWIKKFaI4MmbWQZUatzhRTJp2CKO2FiLwT\nntfQufUqZ4Uz+qr5leq8oamE83zjCyYQKRQKhUJRhhI4FM1HZtOLX0WeU/jRI5VbiqHz3rM/\nqfPIVgxsezlE53oraEnsen0dupasKvQIbr69uFR8UMWtd4cLHxc11b76VfOTYJ2oXwVYMt3F\nGOrXAU4XySNuDNtNB1jXiOEplkdcj5RpHBxZuxlb0BrD4HqoyylW2XHKZNQVInTmKok8EV43\ncRFiVc/RyPvybMYdyHm5ChONxZM0w6/D1eIrhMawmqepqanHVIVCoVDUhppjKJoP7+wBRAh7\nUcO7+soiYniAPLdySwAIzp0yr63/8FYE5NhxF8SvBOc8SReNDrC3Qk0vQv0CZc6DDDDWC9Yq\n6rVRBq6/BexOOvldyA8hIMT68JI7sS00F+DiBruBQpQ+ZOUlWs2ErUOjG+Q4kAMYAdY+9xVz\nF+IuoqMAxTfzMcRdAK3aNeOiAgHajGhUM10REJDGuMX1xbRERcCkmZhw05Jmc4k1xhlyT5Rn\n1ZncMLkBEHq9n2ZVvbIXRFnf9yUxBIvziNbomX+7pY855akuCDA+Exx3/IShrYubC35VkgAB\nSpWEqM47bX2kMOf4CVNLWQ3VVRGg3daHK2w4DM7iql5JoVAoFDWi7hyK5sMSKfPy693nfloe\nT6bMK24oC6JedbKEZqvZOhi9YPTWnApMRCceo2PfA+EBACFi/3W48/WghbnNu3n586/D2YOU\nT2P7OrziTtx89fIHXhPYsQs7doEMALE2EediAvlmokGgzNxoO7DVZs+pAZvHE6QPsRMgDeAC\nRACSa7LN8xpGZ5pee3aVwfROq70QFHwZIKLOdASY9DIhW840ZNFY1Tfz86xqMLkgGHPcouCS\n9QOLB12W1ch+L91RgyGMOv6UJIQAVKEBpb3AKrAOu6oqkfXFmON7ggDA4KzD0orGFl0RI25o\nk27gCalxjBtarBmeF522LonGCrPdcG2NrYuZqrWOQqFQKGpFCRyKVUHi/R8Z//TvB2eOFyMs\nlkh+6I/Rssu25F19vLNHjIQ0HzV2NXrq3hTo2HfoaIkFCRGdfQqcSbz2/eWbZkbEvgcgO92S\nlnLjdOYgXnkXu/kdjRpsCSKg3ChGUmA01p2/ZdBQuxHkCZIDQHkAHVkHsEtbsJBQB1D9cS46\nGGJUn/2nPe6Gt9RxAy+qRQDA5MxgzJPlBnK2xrXVMakNJJWqG1M4Qkx4XspsXFISAnRFjHZb\nL/gi64ucL70w171JN6gmcEy4QWmOhifkhZzXZevFOhRLY5ZW/78okDSYc7OeEEQmZ10RIzFv\n5Ut3xEhZesGf3j6ivEUVCoVCsSSUwKFYFbBUZ8ef/mPhR//pH3meXEfbuM2+/Y0smgjZFDH+\n9l+f+Js/LgtbN9+lbdreiLE2F+HTiR9UhmnkRZw4DW1zTBDkj79UVDdmt3z2Udp2A/ZuW8FB\nlpEdkfv/lc4+N7WEfbvYdfdBoqdxA2gZOEAc5CmgACAgeRZgELVdwFdbEodCsQCl5SqliJJ4\np2WNOE6pxmFx3rFqEvHyQRBaLJMLgkYKHFMUfHk+64nqhT1+hVQ0hSQarahwAYBRx48bWhWP\n0TrgBvLoRL7YjSWQIjdZ6IoYfbH5vl+dod5Y+w+FQqFQrD3UjUSxauDcfsXr7Fe8bsENzetu\nTf3+pzP/8rfB2RMAwKLxyBveEb3rLSs/xDpAIIlcRA2XZh6ZG4SKsvbpI6fPYqnAQUQnngnf\n8sTTjRM4nLR49FNQmHWXoAuHxaOf4nd/HKLtDRpDq0AZ8p+em37uU/AcogWss2mjUihqh4XZ\njpbFNYa9EbsQCE8KADQ5s/iKvLTP+MIVUhLpjMWNxWaIBFU0GkkkiRb0Xq0jgaRzWWf+zq3V\nTDoLQWjzX5AEjpCRFbPwPJ91K3vNDue9Nku3lW+oQqFQKFYSJXAoWhJjz7Ud/+3zlM9KJ8/b\nV5MLY3UIAk8OBHJ8apGhabB1HGMrdT7fARHy4g4ASuWGlUYe/l7I6dycPPgou/7tDRtGS0Di\nVKjBIomTqAQORUthcdMNk2Ktima0tsZtWJau4Uua9AJPSgQwOEsaWlHCEERDBc+bmWkXhMz4\nQYelR7WFz1hNwsDqq1aItBvMr24AQcwIf5ybZ79w5aMeSKKsF4SuSruBvQLlMAqFQqFQFFEC\nh6KFwUiMR1ZMIKgz5IgTkpzisiTXESctvrk2jSPaA1wPVS4w1kfp4+COg57A+EYwLDAj4OZD\ntoyvsEUCUXDmRDB4jrd3sgsvhm8zdGRlx9CKUIgp43xxhWK1YmuWKzxHzOl4ZXAjopXbKi2T\nfCBGnFlnSk+KnC+6bWOqweq4G3hztQECGHV8K8qqpTwUsTlPQ8iV1lqEOFJfKp1KytA17Kpi\nwKFXr0IxVszMVVBVYaUyrUOhUCgUivqiBA6FohEENFmqbsxAnhy0eS0CB9dx82107DtlYWzb\nIM99E9yJ6eNqEdx4F25/GR34bvkRGMftN9ZwxhrxTx+b/IdP+UcPTY83Zscv6za6y/9GEuHv\n9y5uqs03Wi+j2xUZR2QE+QyZzmybp5hqoHOR0WYmHOEWAkeQ4MgtbtqhnZ6WAQGMueVOGVMS\nRn/UJIB8IEL3ygcyvpCHpcl5TNey/pwrFUNMGY1OQJgnYcTgLG7wTluvto3JmaUxJ6g0c2UG\nXzgPJZAUEGmINdl1aIgMMTRDZDEnVSgUCoViOSiBQ6FoBIJyoXFJeQCqqZUmbns1MEbHHyvm\ncWDvHgrOglvyKB/k6cTX2OVvkSOnaODobJxxdss7oa1vCX/CYpATY2N/9tsyM9tAQWQLE/tP\np27crLfPaZ6Cqf4VGkMLg+0AoyFx1mJmJVl/2JPTP3gphUtZTxYSei/HJfnOKFoWi5sWX0HT\nUEfIUN/NgMgVkiNWyxYIFled0W6aJucZ3/clcQSL86RhLJj6UXdiOh8thOSSRHW+MbGwZtQb\nMS7kPFeUmrmynsgCMo0n5ZgbFPcyOWs3tUUmfSBC0tTGK8xNGULSUhcBhUKxWPywplEKxYIo\ngUOxkkgJK5YE22rULy8XEbfegRteRpnzIHyM99HwT2HoVMUJJY39gt3zCXrxcTp9AJwMpNax\nPbdDaqXUDQDIffvfS9WNaSTljo60XV9igIoMd96+csNoUZBvInkGyjN9dORbmzOgJeHLQlHd\nAJjW7ohEIRiP6a1hl6NoFeZxkZA0X3XG4rOJopoW1Zr8pBTRecLQ0l55LklPdFG5JBrDDXEz\n5wtHSACwOIsulL0SSBoszCnucYUcLHh9trnIVI51cdMVMu+Lon7PENfHzXm+FIVCoVAo6oIS\nOBT1R+Zz6S89mH/8u2JkUOvqjdx2Z+Kt70WrzqXXrQUDG2A8JI5mTekbsxhR7Jhui0v5wdBN\nKD+AiLjzFtx5y1JOUTv+8XDHjWCyZNJuRtm1b8PuBvapbRXQQP1lFBwGOTAdYR2o7QaMzLvb\n6sKXhZriilZEkMj5hUAGiKgzParbuLTr2PKYJ5mCM2AIFmdO2AtAu+E+GsukP25GHDbmBJ6Q\nHDGi8+6IbvAa3h9Edb6grlEk7YfYmkqCtB+0m4tKweCI21KRccfPekIQWZx12Lpey4AVCoVC\noVgaSuBQ1BmZywz+3vuDc9MJBcHQhfSX/6nwsyd6/vKzF7PGobE2Xw4RlBtP6Ni1/IMjsvD3\nmKvH9cCMsls/ALkRiHZg76VgRJs9oNUK2qjvBQiACoAWLK2XcFMhCE8oJSACaso0WFFfCoGb\n9jJFH0lXeIWg0G618YZfcEzONMTKehOd4VQxRbupDxS8skSPNkNrxTyClKWnLJ0IGlAi44rw\nW0q1eDWmxlyPESkUCoVCsViUwKGoM5mvPlRUN4r4J49mvvnlxFve3ZQhLQ154jnx02/S8Bmw\nY2zzHu3l94K99I4tCNzim115tsRqlBmsW2OpOow1tgEyJ0NOGttQh4PXgr51p/vMk2HxXbjx\n6gYPppXRAOPNHsMSYVVuKwy5UjfWAJJkqboxhSA56WXajKQrSUjSGJq8ERICAnRY+rAzp5iC\nIXbOTKp1husixqQXOEJKAoOhrXFXBOfzPgMwOU8aVe05VwOekBNu4ApiCLbG2xZbILJSVO+O\nolAoFArFakEJHIo64+z/cXj850+2kMAR/OBL4vF/m15Ij4jBk/LAj/T3PICp3iUfk6Ft822C\nReyxfAAAIABJREFUcpJcRI1jBCvfz5MHIABrS3XBnutp5Bnws3Oi3MC+m5c82qURffWb8o/u\nK7fh4FrszS3z1SuWicGjBTFZaTpjslaVbBSluMILneU6As7lPUk05bmgM+wwdXPlSxIszros\nfcT1i/1HEaQrWNEOkyMWqyrSvj/uzpbLeVLmg6DbtvRV6RWV9oLh/GwH3EIg017QHzN1hllf\njDu+J4gjRHXeUb2LytIwOHphmVhmvT8oIhjKu+MF3xPS4KzdNrqixuoVnBQKhULRCiiBQ1Fn\nZD4bHs9lGjySJUNDp8UT+8qDucngW5/X3/aHyzs2coxxDMsECQbIfR5kGgAADTR2grFtsfYc\nWoTtfA+d/k+aPDYdifazTa8Fq2N5o60Zlmzv+MRnJj/7l95LB6YivLsv+b7/w9ixp2Qrogv7\n6cJTkB8BI4btO3DT7aC3ks2EYh446jGtIxeMlk6DDRaxtWQTR6WoF4JCJr4SNF9aU6rW1DXL\nlzTkeOsi5kr3HJFEo44riXA2AuOuxwCj+pwnnEDSpOuV7S6Ixl2v265z/9rlE0gqVTcAAGg6\nyBHGShqUFAI56Qabk3YdszsSupbzy8tRECBh1POhURK9NJrL+dP9v3wpcn5hwvG3t0dXcVaN\nQqFQKFY7SuBQ1Bmtb31w/kxovPGDWRryyH4IM+eXx58F3wV9Bboe+mfIeWp2kTxynwOZRuua\nxR7BbMftv4JBDpxxMBJgJOo/yMWhbdzS8Sf/b3D2RDBwjnd0aRu2ol6aqEJ06CEaOTS95IzT\n+Z/SyCF29QfAbFv0SSQEaUAduPLyWI0YPKYxy5VZIX2GXGe2zi5e/501BseQd/hChrTzkAQZ\nX7TNTIldISe8wJUSAAzG2gzNqkd+Rz4IQjvFpn2/TOAoiCC0vsIRQhKttkKVSn1hSjrKByKo\nsE31JQ3lvXWxut2bdIbdtjHqznqNagzbTX0e7xIhyZdkcFz8JzmU84rqRpGMF4wUvK6Futgq\nFAqFQlENJXAo6kz0jjc4Pw9xYYjd+cbGD2ZpUL6i0ekUUpCTw/oLHETu8yFh/yQY24DV8t5b\ni0JsFcz5EbUNW7QNWyrX0OiLs+pGES9DJ76DO9+y8JEpoOwzkD8AJAAAeAITN4K5caHdFI2G\noWbzthq6cSpaBJMbCFhWpSIhXKrwZqbiuUCMlCQdTPUc7TD12KL7elTDk+Gmtr6UNDcFLlQH\nKa5abQJHpXNqCVhZApbxypWCZWJxti5iukIGRBrOZ6pSCMS5jJufkSraLH1dbFFmIRMlP4my\nuBI4FAqFQrFkVmPdqaKlidx8e+Kt90NJpS5qWvJdH7SuuamJo6oJTHSGr9AMjKxAZoTMAlXp\noBmM1P90zWUsvI8sVYmXbzbxXcg9O61uAIBI0/i3wDlRr9EpFIr5YcjiRnmRXdW5LAIAEMCY\nW95ACgDGPb+yF2ntLFaY0ObpKbvK1A2Yd7SV6gYASCI5nyayFBDA4iymcWs+dUMeGy/kSxIx\nJhz/2Hh+MYMJaUU7b1yhUCgUisWgMjgU9Sf5rl+L3PLq/BPfE4PneW9/5JY79A2XNHtQNcB2\n3giPPQSi/OUS3/Uy4CvxT2aeN28hs4LWJnCqxklCWPb7LN4FcEOqnyjzM7Ra6QemULQ0Ec0y\nmJYN8oEUCGhwnQmtEIRcxyzGAMAVMnS6KwlcKe3lFaqYnGXD8gAMVj4ntzUNXa9yHLbGV1v6\nBgBEdT5S8CtHqyH6QJWyDscaakPqyGDOrfxyXSHHCn7nQlkYBmduRbnNVLxu41MoFArFxYcS\nOBQrgr55W3LztmaPYolgsku7+78G//EPpRoH9l7C73jPypwvBsAAwhKtWdOsNFYKK8xogwDs\ntgXUDQDwBsLjIg0ip/w4FIqGoTGtrcTox+TSCco9IzTEmK4BgCSAsDk5ACw/6SCiaWnm+xWF\nKkmzfHbNEdstc8xxS0+pMdZuroCt0rLRGHbaepnGoTHssvXT6RAtKWE253EuX2GiMUXOF1Uy\nIWdpt/WMFyLid9gV/cUUCsVFSaUZkUKxGJTAoVCEwK94BduwU+z/Txo+A1aMXXI5v/KVwFbG\nUQA10DeBX1FnweKg9azIGZsHdl9FZ5+AskYMCNizd8F9KVQDmmaeVQqFYmUxGOuxjTHX92aK\nC2yNtRvTlpQ6w2qFJPMWYiwKBOi2rQnXywXTU2WNYcowbR5yuY5qmhlhGT/wpGQIFucxXV91\nyRszJE3N1ti4G3iCGIKtsTZTZwg9EWMwP6cdjK2x7khzRIFqClUxTgCTTpAPhCQyOUtZetGe\nozNiZH0xOvdv6Y4abZYSOBQKhUKxdJTAoVCEg6le7dX3N+hc1pVEAQRnZl9zsja0r1+DLjnR\nHtz2Bjr2MMjZF3fYeRluvHXBXVFLhT9LM0Olb9QbSXSeaJTAQ7ARuxG7mz0kxarG5KwvYgaS\nBJHGsNTVQmdoMOZJWZbHYTA061GMwBE7LDNFhi8lQ9TZfMfUGEtVJHesWgzOeioKPdptPaLz\ncdf3BGmIUZ0nTG3c8bO+CCTpHOOG1mZqjRFuTI2FJnFYGgOAQNKZtOPM1KFkQIw5wbqYGTem\n5afNSbvd0scd3xPS5Kzd1mN17USrUCgUiosQdSNRtA4kgCSwNfluh6N9PYjtIEYABLAkaL2L\nN89rLbDvOkxtpYGnoTACegzad2D7jkXtaW4CHgeRKY9Hdq9BJaiZBFI+T5CfWiDwiCYRxhju\nbO6wFKsfjaEWduHqsvQhx/PlnC07rXoKDQzRDMvaWJNYGuvTpitrBNGZjFP0a/UEjRb8vC/W\nxcwG3EK6IsapyXKTbIbYbusAMJjznLkuG5LofNbd1mYXfUsTptas+hqFQqFQrEnUTUXRAlD2\nJF14DAqDQBLMduy5BVO7mz2oFYCngKeaPYiGYLXj5jtq3gs5pu6iye+DX2wugxC5DGNX13Vw\nFzuSzhbVjSJEowQjiAuW1SsUIWgM+yJmzheelARgMhbV+dpUcBvOuBNUdqMpBDLjBYmwbAgC\nCGQgSGrItWXXXSZNbV3cHMh6RTsVg7P1ccvgTBKFWmwIoown2iz1/KlQKBSKFUHdYBRVye9/\nyjl0ABDtPVfYV1/TrGHQ+AE6/fXZZXeMTn8N3BHsva1ZQ1I0Da0NO34JvPPgjwPTQe8FLdns\nMa01iMbC4zCKoAQOxRJBgJjOAS6WJIuGkQ/rXwMAOV8mKlJkXOFPeDkx44JkMC1pRPXlyRyd\nttFm6llP+FKanMWMaZONoIqFLAJUmsIqFAqFQlEvlMChCCEYHTn/sQ/nn/pJMRK7+ba+Bz7F\nkw2fTJKk898NCQ89iR1Xg77mmowsFfe5/c7+x8XYiNa3PvKK12r9m5o9opUDwegHo7/Zw1jD\nhHXdnC+uUCiaRlijVYCwDjWeDMbcDM2NjLrpbivJFmxiNS8aw8qMDF7eqHdRq+bBCeTZyULO\nDTjDpKX3J61V2N9XoVAoFE1HCRyKEM7//u/mn/5ZaST7+A8u/PEfrP/rv2v0UJwhCHIhcZKU\nPY2pPY0ezypEiPG/eSD/w0eLgezXHkq869dib7iv/udyMzRyFJxJiHZg16XAW8arT7F4EEyC\nkMRygNXYTVOhuMjROYogJFVCrxARsn6hcjtJlAvcuG7XfWAc0dZYISgXYGZyeWpjOOseHsqK\nGdVmOOedSztXrUtOuZkqFAqFQlFECRyKcpwXD5epG1Nkf/h9/+wZff2Gmo8YePKpr8sXfkLp\nYWzrYbtezq69G/jivEKlV3WVcMsjJMEdpmAStQSYXYAXRS509uF/LVU3AIACf/IL/49x6eXG\njno6ldDJJ+Thb0Iw87HbbezKt2HXpXU8hWI1gNhFFKIqInQ1fjAKhWJ+4jp3KkQEAIhXGHB4\nMryYxZOhgmYd6I2ap9JOWS5JZ8Qwauye4wt5eHhW3Zii4IuXhrNX9KlEToVCoVDMQQkcinK8\nE8errXKPHa1Z4PAKwb/8nzR8emqJhk+L4dPy6H7tbX8M2iI0DqM9JDjVatDqmBN0zsvBb4E3\nOrUe9DbsfjVGNtY22hLE2AhJqXU2uzumN0aDP6D8WZAemJ3YeT0mdpWuzz/2HyF7kcx//5E6\nChw08Lx8/t/mhAoT8mef47f9PkQ7quykaEkQ1yGky5w4ENcjtjVrSIq1hyckQ9SWVKqgKCVp\naq6gdImdJwK0W7q9ClIbLI1tbbOH814+kJLI5KzD1qO1p2+M5j1RYaQKAGN5zxek/GoVCoVC\nUYoSOBTloFG17gCtmnPU5f7/KKobRej8Efnst9k1dwMACI+GXoDcMJgJ7NwO9txJlB7DxHZK\nH5k7DgCrE6Ml4oU/Ic99BaRfGqHz+3DDO8Cs+bVz9jsPT3z+b4ORIQDgqfa2d30wfveboSm1\nvvlz8tT/Bpp5cnUG6ew3oOMC9ryquEkwdCF0VzFwro4DoeM/CjuHL089wS57Y8gqWQAKgMfr\nOAZFo0CGuwhGCcYAXAAbsRtBfZWKOkAAwzn3QtYNJAGApbH1cStprcnm342jO6LHDZ7zRSBJ\n5xjXeWiKhMG4E+bYYbDFPgrmfTFVcmJpbH6dgmb6nGsM+2JLqW7L+2Ik77lCagy9sBSVqbO4\nQuhcPcoqFAqFYhZ1V1CUY1+5FzWNgvKcVWZZ9p4raz2aPPZ0tTi75m4aOSJ/8SUojE9Huc52\n3Inbbi/dEje8nk7tg+yp2ZDVyTbfCyWmaDTx8znqxnRU0PjPsPfumgY8+a//NP7g3xQXxfjY\n6Gf+WzA8kLr/QzUdpy7Ige/Mqhsz0OjPse0KMKf7WbBoXBTKm3oCAIvX0xGWMuEyCqQHyiPO\nCZr8CYg0AAAzMbYXYnsAmv8uUVEFAvAAjJn5yDSIHQgqN0dRN3wZ5INCzve9gAyNCY8RgBPI\no+P5zW12h60MfZaFrbEFUzZiuu0KvywRgiFGNRMAAkmFQEgCk7NKYwsh6VzWzfmzRS4RnffH\nzLIcHAIYK/gjec8TkjOMGVpf1NBrLEgBgAtZ90LGKR1q1NJyTkgpTa3VLgqFooXwq4ibCsX8\nKIFDUY7W0dF+/38Z/Wy5n2jnB3+TRaM1H84NswgFgEIWCuPyqQdBlLhsCF8efphZSVx/bcmA\nbLb1HZQ+BvmzQALsHkzugrmW7+QMhp6E3MGa8i5kPjfxv/6hMp7+8v+X+KW38VRj53tBDsL/\nLqLscZwROKxrX577z32VG1nXvryeg6nWR7Asnn+BJn4wuyhdSj8JwQS23VrPwSjqgyPpBNE4\nAAEwxC6GmwDUu3RF/cn6+ayfAwCGYOlg6TJqsNGsNvXoejbttNuGKjNYaQympcz4ZEWbWIZs\nOO+NOn7R4yKq876YWWpTWqZuAEDeF+ey7qaEVRo8PVmYdKdliEDShONn3GBrKlKTFWjOE+cz\nTlmQMzR15vpzZjtJS1MCh0KhUCjKUDcGRQhdH/rt3k/8qdYxPYXWenr7/uwv2t/z/qUcKxFe\nIYJt3fL0T+eoGzPQ8R+GbJ/YCtBJZ0/RLx6WT32WTj0JVPqgU59nY+/Fg+RWeJcCUBC4B5+d\nExI+VDFsqxsyZCQzZ59dlXjr+3h3X9l66+ob7Ztvh/qB7VvC4x0lcZKU/mnIRvnDEEzUcTCK\neuAI+SzRGEzPaSTRoJDPA6zwr1px8eFLf0rdKMXgMm7NzoQdX/3wGoHF9W472Wkl2s1Yl5Xs\ntBI648N5b6Tglzp45nxxtiSBwhMyF/YF5X3hltS8ZL2gqG4UEUQD2er3sjDGnHBz8bJMEI3h\njq5YTUdWKBQKxcWAyuBQhIHYds9b2+55qz9wATnXupZutMl23ypOPR9yht23wURIHAAoU1H1\nACSfeYjO7p9eygzQ0At4dj+78QNTnUrR6iXnfMhZrN6aRiu9qs9h0p16oUR0ar88+AhkhoEx\nTG3AK9+MXVtrOsti0eKAHCjsud+YdSphbe3df/VPmS9/3nnqh2J0WFu3MfLqN8TuuqcsyWWZ\nsB13iaHDsy1Upoh24qabZheDcZDlr92mcc+DpiwqVxGSzsJUL9g52mCBaACxvzljUqxRnCD8\numobNDlzwVBZyA0DAUtNNwhgLKz0wwlkzhMxgwOAK0IMPqdwA2nO6A4ZL1ylyngB1fIWwqty\nOs6wM2pkXaExTNra5lREpW8oFAqFohIlcCjmQ+8tTw2oFbb7Zho4Kp8u6WOKjF3/BrbtGvn0\n4Sr7lP8s6cLzs+pGMTh2go49hjvuAgBsu4bSB8p7yqKGqetrGq2xqapUYWzeBgDy2a/RC9+Z\nDklBoyfpe/+D3fR+3HB1TSdaFEzHxE6aPFge5zbGt8/ZMBpL3v+byft/s/5jKBLvYS//TXp+\nH40dBwBAxHVXs8veCFqJe1yoFjONej27uiCaDI/DJIISOBT1RFC4fMGRsNgUS81Um4QvZFkb\n1yKOkDHgADBPrxsssd8WVY5DAJKIL9qou1pvHY2zy7uV27FCoVAoFqC1HykeeOABROztLX9L\nPzk5+aEPfai3t9eyrL179+7bF+JQ0HqQdA8+m/3WNwpPPSHTkzRwhJ75D9r/NTrxNFR5qlgd\nIL/9vdqv/Cm75m62/Tp23eu1dz7Ab307AGBHuJqAneVxuvBs6JZ0fiauJ1j/W8HsmV1ndLD+\ne8GozTVD610XueGWyrh1xV5j6w7IjdKL36tcK5/5ClR5gl8m2HsHRIp9eQkAgEdw/RuBW9V3\nWikwsY69/Df4ax5gt32Ev/bP2d53gpWYs4WWrHpJ0cLa/SqaSZVf7HwqlUKxFFiVbDJJOHXr\n6ogYXPWLXX0UvxJbY6HqBCJE9Nkvt1o+hcZw8eoGALRV6arTZqp3cgqFQqFYmBa+Wxw+fPiT\nn/xkT09PWVxK+brXve6555775Cc/uXXr1gcffPAtb3nLvn373vSmNzVlnHXBO3Fk5K/+1Dsy\nnfLATCO5syO2KTW1iF2b2Z0fgrbayjEaCfbv4P07yoMbrsMTPyovSOEGXvra8v2dTOhhyUnP\nLlg9bOM7wBslfxK1BJidSzPm6PzInwx/6hOFp56YPfBV13V99AEAoKGXwoWMwiRNXsC2FXjv\nzU22+e2UfgnyZ0C6YHZh2xXAl9Jyr27oEdQj4auYCZHtkH+xJEQACHoHmOsaMjjFYkG0icIK\n3bHKl6tQLBWLm/mgUBkv+AgAKUvfkGiCYquYwuBMYzjVtbeMYlsWhthlG0P58itGp22UKhdt\npjaYcytfuLTbtVkXJ02tw9ZHC3M6o1kaWxdXvxOFQqFQLEyrChxSyve///3333//kSNHDhw4\nULpq3759TzzxxBe+8IX3vOc9AHDXXXft3bv3wx/+cOsKHDKTHvzor4uJsdmI640/e4FpPNKf\nAAAaPike+Wv+tgeqtrpYnTCN3fTr8vDDdOapKeEA2y/BPfdgomIyXJYpMAPaZZ1QEYxONDqX\nNah4oufP/to9+Kz7wgGSwrx0t3XFNdPr/OpOaVWKzOsCJnZAolweaj7+GKWfAm8IQILehYnr\nwOjG5M1EAgpHZzZCMHowdXu9XGAV9QKhj6CySgURV69OqmhRDK5HNLtC42AxPdob1aN6S922\n1iJdtnEhV34Li+o8UvLVdNi6xnA47/mSAEBj2B0xknNTKgzONibss2mntFYlaWo9kZpF+c1t\nkaTlD+c8R0iDYdLSe6JGeBqJQqFQKBRzaVWB4zOf+cypU6ceeeSRe++9t2zVV7/6Vcuy7rvv\nvqlFzvm73vWuj3zkI88999wVV1zR8JHWgey3vlGqbhRJHx2ZEjgAAMbO0bnDuGFPQ0cWCkk6\n9jhdOAROBuJduO2WypITkILOHYaJC2An2fY74fJ7ITcCVgJ0O/SQuO4qOvd0WHwFnC8AAMDc\nfaW5+8ryaLyK2SoixsKbxaxZCido9NHZMgdxipzT2P4qiFyKqdshdgV4g0AB6J1grm/qQBXh\nIHYw2CzpNICcsUHQGG5FqL0VtEKxEAkjZnIjHxQCKTgygxtR3cYK3bMmK0pFTRBAIQh8SQzB\n5Nxgs+UkbZaGCEN5byqPAwHaLL07Up52kTS1pKkFMwJH6FmSphbtiI47vhNIjWHc0KZsSqfI\n+2K44DuBBABLY10RPaJVlbdSlp6qUquiUCgUCsU8tKTAcfz48Y9//ONf/OIXk8lk5dqDBw9u\n377dNGffGFx++eUAcODAgRYVOLxjL4bG/bRDBLOvNMbOQdMFDr8gv/83NHZ6enHsFJ3aj5fd\nxS5/Q3ETGjouv/uPMHZuelk32fX34FUVZSklYO8e3Hgjnf7JnGDnNtz6irqOfgGw51KIdUF2\nuDy+/iqwLibnM5I08YMKEweiiR+hvQVQB70L9ItM8WlBEPs5dhKNE3gIFmJ7i94RFC2ByQ2T\nG6Grcr4YyLqFQBCAxXlPzEgY6qdYTzwpRx3Xl7MX7ZiupUyz+PiQNLWEqXlCSgKDz2eZUU3a\nKN2gKxLyRU+4wYWSfrF5X5yaFOtiZlI5aygUCoWirrTkfeVXf/VX77zzznvuuSd07ejo6JYt\nW0oj7e3tU/H5D/u7v/u7586dC10VBAEA5HK5pQx3+fDwVxyIiFjyxksLf3ZsJPLAw7Pqxgx0\n6FHq242dWwAAChn5jb8Ep+ST9F35xEPMiuPOm+c5MrvyrdR3OZ15CnIjYLdh7+W4/jpocMIq\n4+zmX5VPPAiZwWIMe3awa9/e0GE0HX8IRD4kLj1wz4G1udHjUSwdE7FXvTNXNJEJxz+dnm0v\nXQjEyYlCX8wMnSQrloAkGi44ZS1Osn7AENuM2Q8ZAcwV62UjiQZzIaY/gzkvbiyomSgUCoVC\nUQOrWuAQQmQys+6SiUSCMfaP//iP+/fvP3ToUK1Hw4Umw52dnfl82LRtRuBgrDlNZ8ydl2e/\n9Y3KuJGyZ9UNROzf1dBhhUFnnqkSf3pK4KAXfjRH3Shu8ItH5hc4AAC7d2F3k/9GTK7jr/kY\nnXmaJs4C07BzK/Zd1twhNQHpVF+1gl4kCoVijUEE57MhF42BnJuydDXxrQsFIUIbuGb9oFTg\nWFHyQXgzWkFUCITyYVEoFKEEYkV6FCrWPKta4Hj++eevvvrq0sXe3t6PfOQjH/3oR6PR6MTE\nBAAEQUBEExMThmFEIhEA6OjoGBub41gxtTiVxzEPH//4x6utymazn/vc52w73CFipYnd8br0\nvv/lnz0NpSXKCMlLZ6sA8PI7VkEXFYJCOnxNYdrOkEbPhO85dhakaA2TVMZx03W46bpmj6N5\n8Or1OPOsUigUirkUAhHav4MIcr5QxQt1obQypRRJFBBpDUmEDFU3pphwA4ZgVzfjUCgUCoWi\nJlb108P27dt/9KMfFRe3bNny0ksvTU5OfuxjH/vYxz5WumUqlXrPe97zhS98AQB27979la98\nxXEcy5ruKPbcc88BwJ49zfanWCpomr3//e9G/+en849/b+pJRO/uTl3Zb9ouAEAkya57E+5+\nZVPHOAWCFQcnTOMotkGpJmEgA2xOgkxViGjsLKSHIdaBHetbQ3xpDHoH6J3gj5THtSSYTVfZ\nFApFyxCaWTC9Kkz4UCyBSjPXIg276erVE2DTbpB2g6Sp9cVMlbGjUCgUiuWzqgWOaDR6881z\nyha2bdv22GOPlUZ+7/d+78SJE/v27evtnZ5ZvfnNb/7nf/7nhx566L3vfS8ACCG++MUvbt26\ntUUdRqfgnd3dn/iUGB/1z5ziqXZt3QbkHNw8CB8iIU6robj7f+B8/xti4AxLtBtX3GC/9j40\n69xVHjdcRUd+GBbfO/0/fTvocNgGfTvmGGoEOZASjKalA9DQCfr+52n45PRyqo+94r24bmez\nxrNERIEmngZ3BJCB1YdtVwHW5588dryaRr4JwWwFGfAItt/ZwAdmBQB5AC6gvcqv5ApFNeYx\nfTA1dTGpDxZnlR2hAUBnbDmNV51AOoHQGbP1hY9ja8zkzK2ebT7pBnoVd9J58AUBgq5KmRQK\nhUJRQos9FsdisVe84hWlkVQqde7cudLgm9/85ptuuum3fuu3Jicnt2zZ8rnPfe7AgQP79u1r\n8FBXAp7q4KmO2WUzsvh9Mw/+d/fH35r6fzk+Epx6yf3p95J/+BmWaKvjCNme18vhYzQxx6sV\nL30Vdk13isUdN+Gzj5YXqjCON0y3+6WxA3T2e+BNAgDoMVx3G3btreMIF0VmRH7tL8ArzEbG\nL8iv/yX75f8LOzY0ejBLhfKn6cI3Zv0ysi/R5DOs/y2gp+pwdC2FPW+H3CHyBoEIjS6I7QFU\nLf0aBaUpOAhyfHqR9aG2C7DOeqVCsdIYnMUMnvVEWdzSWET5MtQJk3Nb44Wg/ENOmUs04HCF\nPDVZyLjB1KLOcH3CbrfnXP89IRliqYvK+rh5JuN61TWOCTdYvMAxmHGPj+WdQACApfMt7ZGe\nmLngXgqFQqG4GGgxgWMxMMYefvjhP/zDP/zzP//zycnJXbt2feUrX3nTm97U7HE1E+/Az4rq\nRhExdC7/1c/H3v279TyTEWGv/n068n26cIgKk5joxW23YM+lsxtwjf3SH8gnvkQvPgFAAIDt\n/Xjru7FvBwDQ4E/ozLdnN/azdOph8Caw/1ULn5oE5A+TNwhAqHdC9DLAJT69yWcfnaNuTCF8\nevphfPUHl3bMRkM+DfxHuRuon5YD/8k21KnnC2oQu0K9OGsClCXvJwDBbEReIH8CjVtquaRP\nEJ0GyAFoACnEzWvydqBY/WxI2CcnCqXTb5OzjQlbXVvqSKdlpT0/4/tTXhgGYynTMKs0aJsf\nQfTSaK5Up/AlnZjIs/+fvfsOjOu+7kT/PbdMH3SABQAb2ItYJJESpchqVLPVLFmWixzbkZ1E\nSZxdb5w8J3nZ3ezajv3y0jb79JzYsmzHdhTTcpEs2bK6RFGFkkiKnRQrSJBEn8HUe+/v7B/o\nM3eAwWAADIDz+Uv33PYbEBrMPfP7nUOBCp/JQFs8fTGW7l185NG1ef2NYD26tqTC350Qs5s+\nAAAgAElEQVSyO5LplO2y/shWrJjzmVRyqjN+vGOwJHzScg5ciKZstaDCvVBaylaK2Wfq8ksl\nhBCzwbT/RPvss89mBysqKh5++OGHH3548sdTmtLv7sgRfxXFTXAA0HRacQOtuCHnAf4y7cbP\n45oHuOs8+csQ7p+TotJ89sXsw/n8TqrbDDMEMGKHOX4YTgR6mPxNCK3pWxNhd3H7U3D6Vkxw\n4n3E3qPKm+GpK+QlXDzuGuYc8RLE8dNw3LoaJ8/B6oJZzGk7YpKxfWxYdqOvikECzinoTfld\n4yTz0F/mKPN5ok3AGCaFCVEUpkbLqgJdSTtuO8wImFqF15zkDuAzHgHlHrPcY9rMGjCelSkd\nCct1FkZLT6rCZ56NJDuS1kAw7ahT3YmGsK93fgcBFV4jZauUbWVfgfLodgfAcvhkZ9Y3EMCx\ntlhHPG05KugxGiv8Ya8BoDWWPtYeS1oOAF2jhRX+xoqArGgRQoiZbdonOEQ+uMd1BS5UtBvM\nmJLPkh4/1S0eGuBYC5TLhx6w4p7TVLmS255G8lRf0I5y6hziR6n2DpDBnc8PZDf6OHHufI7q\nPlpI7dIx1bZjBeVAL7HVGVY01x62o1RwgsNOcrydfOXwhAq8ghg/7l+ZwgzHgur76pvtIxSY\nDxq12VOM+URWMM18lGh9MccpRN4qfEaFfCCZeOPvmRLLWk/UK2E5CVsNzW4MOB9LVfrNgRsH\nPbrrYYH8ZlhEUlZGTxZmOMzMfLEnBaAzYZ2NJJbXhLyGdvBiz8BhjuLjHfGY5ayuk25fQggx\nk8nniVlBq6zNGS+db8pcsxt9u2zEDg9mNwakz6NnL/yLYbW6nOVEkG6Bt37MI6lbjPNHs8OZ\nGZnWE/zGj/nCMSgHZXO0jR+k5VuRu179pNJzPuVS7l0jSXSp/T/nlj19F6laRGvvofKx/2xF\nESgAYIY1fAmSinPsBQreONrirLYcObwOwAGk8IEQoo/D3BpPxy1HMfyGNnKNjJjlnvuwFads\n5euvGhsy9bBHjw5PlGhEdfkV4Mhur6PAnJXyONLa43Mr43IhmlpQ4Q955NOvEELMWFKlfFbw\nbnFfMOK78sZJHskIyO+ehendxYkcy0YSx+H0uO4CkDmtIz/a+ptgZtVr1A3a+MHB+555T/38\nK3zuEBwbzOg+r178tnrtRwXcLhduP6Oe/aZ67C/VT7+q3viJS1mQ3CiwAJrbpBKzEp6aMQ/F\nSjo7/tdAdgMAd5xUr/0zei6M+VJi/CgMAI7tsosTnD428tnMuTKJDOROMgohZpmkrQ63xy7G\n0j1pJ2457QnrcEdMy7HAI2DqyN30N2PORX3YNyfg6e1+ohFCHn1xuc+XX9+coCczbcFu92Ug\nZbsXNO1KyBudEELMZJLgmBWMxSuC9zyI4Y3ozVWb/B/65FQNyYWnnCqWu8RDjQjMg8rxeO/E\nQblrp2sF9ZUoq9Pu+NNhDVMq5mof+i9Us2AgoHb8ACrzwxPvfw5dLYXcMQvve079x1/x4R3c\ndprPHeJdP3d++H+hO++Egu6n2usyp5OQoc25qZDBnNyBRGdm1E6pI79xO3yqsQMVH/2waYv0\nxQDA7l+Wwmkb5fSc8zs0oMC6vEKImac5mrRV5syIaMr2uDX3nRfy5spQUFbTXwKq/ObSysCK\nquCKqmBj2Od6TVdBj1ExvGNL7ryKu6wpIEIIIWYUmaQ3W/hv+5i5bnPypV86F85o5VWedVu8\nm68rofUpAABafCeO/5S7h3wFHV6gLfkwAOgh4KLLOXoInhroAThZz7TkgWd+gSOZu5Tu+2tu\nP4NIK8I1VN0Ifcj/LNE2RNwGw8zN+6liXmE3HXL9dvXqDwZqK/SJdaqXvqvd8ad5XoPK1pGn\njjt2cqoVpJF/PlVthVlewHC4M7tkAwBwe4lVXbXaOfo60ucBhuZBYC0FLwHNuHc5rZaMdWzt\nyrHSJEfiAwznFDsnwQmAoenQjeHvALWS8hZC9Eo7Ku625MRhnhfydSXTkeFtYst9JgM+Q0tm\nzZuo9Js6EQBbcVs8nbSVriHkMSp9ZmH1PtfMCe8/H+1yK+QxVK4PONlzQIQQpcnK3VhaiBHM\nuI/+IjejsSn0yS9M9ShGpPto2ceo5wz3NAOMwDwq6yt7QYFlmatUGCBQYDmgUfk13PkMeOj7\nIFH5Vmjj+EZa06l2EWoXZe9hK/dqkYyyCAXhk++6LkDg5v1IxeAN5nsh3xyaf1cRkljKbTUE\nAM6Kp85x4iicCPQQ+ZbAt3D8N8+XdZE7fjk4r0Gl0fMOpy9Q1a2TN4ZJozdCPwXbbUaPVuZ6\nBlvvQp0f3FYOlGLDQ31PGEGiZRMwUCHEtGSPNM+Bl1UFk7ZK2o6pa35D623LQsCicv/pSHJo\nZqTCZ84PeQFEUvaJrvjAZS/G0mGP0VQV0Mf+RYtH1zbWl3fE09GUDaAtlm6Pp7MPqwt522KZ\n8aBHr/LLVDUhhJjJJMEhcrPT6Z1POycPspXW5y3yXH07hSsn476hRgo1Zgb9TQiuRuzAYIQA\nfxNCqwHAt5Bq7+XIW7AughU8tRS6tMAesXmgUA00PXOGRa/yuWO7Fis+8Rqf28uJLgpW04LN\n1LCR4+5db8CMePdAgoPbj/G5vUh2IlBN9ZdSRdYPrVjC89B6JDtMZcMmyHD3q4jtH9yMH4G/\niSpvmJzCqxx902XVRvosUqfhXeB2xvRG5nK2L/Tl+QZp5HHrFKvahmU3+jAphlZHVAnMl+kb\nQogBRu7JFb3zLnyGlr0mxaNrSysDPWk7YSuNEDB1v6EDsBUf74w7wxeTRNP2me7kooqC6l4D\nVQFPVcADYG7Y9+aZzsTw+SaLKgNLa4JHWmMt0cFvHcp8xuq6cIlNXRVCCFFkkuAQ7lTHhfi/\n/KVq66soYR94M73jSf8n/tRYddlUDYkqr0VgKccO900Q8C+Ff0hbE6OSqgopMFEIj5+atvDR\n1zLjwUpacMkYruOk1asPDyz04OgFPn+Azu2hQIP7d2dECFQAACu198d8+o2BPXziVVp6vbby\ntjHcPW/aoqucU6/ByZwPTEs+MLiROD40u9EffB+e+QiunohRDcMKafcCJZw+RzMxwQGjjnyb\nOPUeBoqGkpd8G6G59ABm5dZmCICyiNaWSusfIUTJ8Oia39ATdmbWWCcKj9aCJOQxQsMnSXQl\nLcetVEZnMr2Afdr4Ug5eQ9u6sOpEZ7w9lk47KujRF1QEaoIeACvrQg0VvkjSthWHvUalv8Qa\nugshhJgAkuCYWZwonC6QAaNqpNKbeUj++J8Gshu9OBlP/Oj/Df35t8iX9xKJovM2kLdhyu4+\nhHbVJ1Qiws37BkPhGu3Gh2CMYe4rH30hu4wFN79LaxdDN10SCgvWwRsAwKffGJrdAABWfPRZ\nrlpMdavG8jryE6zRLv8dteexwVKjpl9bffvQe3HCvXkHJ47SJCQ44OQoSOG2jmbGMBeTMR/2\nBXAcWgj6HFCuj++5fggM2IB86BdCZGoo8x3vig9ty0pAfdirj71yRirHQnrFsBz2GuPNseoa\nLa0OLq12+XAS8hjSFFYIIWYVedOfKVSSY28idbJvkwzyr0NgXWEX40iHfWyvSzwetQ+9bW64\nptBRziAev3bbF7l5H7ccgZ2iqgZq2jKm7AYAPrvHPd7xvnbNp9RLjw5bBROu1q757b4Dmne5\nn9i8a0ISHADVLtev+zNuO4pYG3wVVNMET2jYESrmfuYITXyLiEzoQTguYyDdZUbDzEFemKPP\nTyEK5pgT5JHshhDCld/QVlQFW+PpmOUws8/Q64Ieb97tToYaodBGAekSIYQQYgSS4JgZmCMv\nwB4yC51tjr9LQGE5DtXdnqvxGnfmmOs+K1HDWmpYW/DpnIq670hFaPUH9LlL1e6n0d4Mj5/q\nV9L6W2D2zcrh7KatveIdBQ9mdLqH5qzJuTdXR16toPXViWMcPwgnAs0HbyOFNiBnc9M+FFjF\n0ay8j+aBf2khA5hhtPnAUZcGK9pMXLwjhCgSQ6N5oXHNBu1V7jXOuv25C5r6CMU+hBBCiAJI\ngmNGsM4Py27048R+CqzJrB2YOs7pM3Bi0EPkbYKnPvtECro3YgBAoUL6jApX5CvnZMRlh68c\nAKrqtesfdD/RDHCiy2WHZ8pWD5FvMSdPu8SH1knJD3e9gOT7fRsqAbuTk8ep+g5ogZFOC66H\nE0f84OBaFT1I5dfmzLzMKuQjcyNbe4Ah6560eWRI9kcIUTRxy+lI2ilHEcGra9V+06drAPym\nXhf0Xoylhh6sETWWF1hhVAghhMhFEhzTjXL49AHuOIdgudawEsEKALDb3Q/mNJwIBqfoK468\ngPTZvi27jVMn4VtKoa0Z52lVc/R5i5yWk5kXNDzGik3FeBmzGltpMj0AqGEjd53JPoAaR/sh\nz1mDyDmXE0eYYTHRAsuROI5U38vp6+3hmYPgGOcQpc4MZjcGOD0c3UXlI6+NIirbisBKpM6x\nSpJRAd8ikLzF9dPqyPMBqLPMMcAkrRZa1VSPSQgxc3Sl7LZEXwqVGQlbNUdTc4OekKkDaCzz\nhUz9fCyVsJ3eSqX1Zb7CFrwIIYQQI5BP/9MJtxxznn6Y2/syFI7p1a++T7vsgyOeNGTyZ+LQ\nYHZjQPIYzHp4F2aEfR/5o/g3/4JTyWHB2z9LZfJQVCBOxpO/+qG16wUV6aBQuWfjNb5bP0at\nR/nCwaGH0eKtNH/9yJfSll6nLuzjyLAqsFS9lBo3F3/c+dKo+lbED3P8CJwI6SHyNyGwBjS2\nz6+ccpkGAgCpU3mdb1TBqJIZz+7IA32x/HCEEEXnMLcnMgtjA2iNW8Fyvfdtp9JvSh8TIYQQ\nE00SHNNHPGJv/xqSQ8ooWinnhe/DH9aWL3c/hTzQwwNbnDrJbg0hOX2SshIc+oIVwT/9/1O/\n/oFz4iCspDZ/ifeG+/SFK4vwQmYlTiV6/v6LTkvfUzr3dKdeecI68Fb4S/9IbUe4ZS/iXQhW\n08ItNCePH7Lh067+Y3XsBbTs4UQnBWuo4XJadBU0fWJfxigIgZUUGN8viUrmiKcG5oUIIYQo\nKQlbuRbucphTjvLJTA0hhBCTRRIc04Z678Vh2Y2B+FtPamu+AXMOrAsZuyhwybACHCru/nTo\nxDMu2XuWVl7jv++PxzVo0S/9ypMD2Y0Bqv186rmf+G7/DC24bMxX1D3aipux4ubijK906KHc\nccluCCFEKVI5CpMDcM98CCHEaCzbvcO0ECOTnPq0wW3uU/e5rRmsqOw6eJsGo2RS8DL4Vw87\nNFc/i94429zzNrf+O59/hC/+kCOvQaXcjxdjZx18e0zxWYt8S90TGf5lkz4WIYQQeTE1l8+T\ntuKU5TRHEqe7Ez3prC5OQgghxASQGRylTanka7+2Dr7L8R6vGXNPR2k6iAAPha9CcCPsTpAJ\noxKUudKVvAvYrRwpeReCHe54ElZb/33jiB/g1BmqvlOaUBQFp+Lu8YTLrJxZzaym8BaOvgkM\nSdt7Gyg4Sl0SIYQQ41fYUkCfoZkaWf2zNRicspVSDMCxOWmrrpRdE/DML0bTWSGEEGIEkuAo\nXRyLdv/9n9knDvdtV/j8c8PZh1H98sFPI1oAntytNH2rkT4H68KwDzDeRfAuQvzAYHZjgBPl\n2B4Kbxnf65jO7AS3vIXYRegmyhdR3SUFr5LQa+Y7p4+6xOsaxjfEmSi4lrwNnDgEuxtagLwN\n8I2516wQQoj8Ocxd6XTCdhSzThQyjTKPJ/8/eATMC3paYuneHIflsMpamtIWT5d5jJBnamtF\nCSGEmOEkwVG6Yv/xzcHsBmB1J72Vfs07/J9MN/SrP5rvFUmn8puQPMqpU1Bx6CHyNsG7CACn\nmt1PSZ3BbE1wcOcxPvQfsPpnXrS8xWd3ams/BTN3Cik3z9Zb0u+85BofzyBnLKOCwldM9SCE\nEGJWcJjPxxNOfx0Nh7k7bSUcZ47fn3+Ow6NrC8K+qGWnbL4Yd1/i2pWyJMEhhBBiQkmCo1Qp\nJ/XWC0MDzIid6fbVBs1yP8AAqHaBfuNnqD5HCxV3BN9y8mWdotI5hpEjPuPZCT74GOzEsGC0\nmY89QauyMkp2mvf+ik+8w7EOCtfS8q206joMX5BsLN/gv+vBxJPfhd3fSE/TfTffb66/auJe\nxGRgG+kWOBHoQZjzoJXY9GMVQ/I4O5G+mSBG7VQPaCJYcFqYe4i80GpA5VM9HiHENNOdTjtZ\nVULTjopZdsgcwwdFIpR5DHhwPubeDytHrxUhhBCiaCTBUaJUT4RTmZ8PlK3iLVH/yg8Ett2J\nQAWFq4p2PyOc3YQFAIyyot2iRNjdiB1muxt6kPxL4J3rehS3H8rMbvTG2/aTnYQxpC5JOqF+\n8TXu6JsCw8kebj1BJ9/Vbv1iRo7De/095ror07tfVW0tWvUcc92V+rzM7rzTTPosR3ZA9ZcR\nIQ+FN8M3WdVAlcVdh5FshxGkskXw1WQekDzCsbfAfZXtOPEefMsodMWM6saiLrD9HjiN3qwn\njkBvJGPNjHqNQogJlrTdK4AmHWdMCY4BpqalHZf2B4Ymb01CCCEmliQ4ShT5g9ANOLbLrrJq\nmrOk2LdbwYljbvGVxb3RFIvu4a4dg0+8kbcRWktV17o8DSY73QutsUKqe2iCQ+15eiC7MXjU\n2QN85FVaeU1GXKud79t23/heQ8lwurn7uYEfJgBwmiOvkhaAp36ib87Rk3zi50hH+jZJozlb\nqOHGwSPsDu55o++pf0DyKPRKzJjfak6w9e6waqxgOKdBAehFfosQQsxguToxKmbLUed6Uj1p\nWzF8hjY35A17Rv/oWO41WuMuM0ArvJnlz4UQQojikgRHiSLT41l7WXrP61k7yLNha/Hv55lH\n4Su4560hz6uE4Fr4lxb3Pur0QfuNp7jtLAJl+tINxpYPwpisjzup89z5cmawZx88NQity4wb\nuRceZ9TgOLXb9Sg+tTs7wTGTcPzgsOzGYHw/TUSCo6eVI+dheKmiAaT42GNwhnx6ZsXnd8IM\n05y+kjGcOpaZ3eiLH50xaTtWza4PJuycJklwCCHyZmiUdlwXj9D+1p6B1StpR0VSdn3YN3e0\nZih1QU/McuLWsL8RNX6PFOAQQggx0STBUbqC9z9knzisIp1Dg/5b7jMWFDnp0H+/teRrROI4\nOxHoIfIuglntfqSd5Pd3cGczNJ2qF9HiK6Hl9ZHFev5H9ks/HthUx/fa777g/ez/oMBkLITh\n2EH3eM8BykpwUNUyPq6Bs54eQ/PhGdbLhpM97vdLRgsb57ThdLnH7U73eMGSEbV7O5/b07dp\neGnJ2mHZjX588a2BBAecHD//XPHpSOXoMcxxQAHufaWFECJDyDA7HJeyoF0JK7s2x7lostJv\nevWR3mF0oqbKQEfCiqRsSymvrlX7zVAeUz+EEEKIcZI/NqVLr6uv/B+PxJ/4fvrAOxyL6vUL\n/dvu9VwykT1N9HKENo68QJbbT6od/4pEd9/middx+AX9Aw8hmCMb0k+dPTY0u9F3eusZ+9l/\nM+94qPAx58/uHkPcX0ON1/DpF4cFNVNbenvGgRSq5rjbo344qyTETJPj0y0V9bmalXrtX7jr\nzGDETqHtCFy/P0x1QtnQDAAgT47h5YhPR5TrDVyX7IYQIn8h07CUilrWQIQIZaanNRrPPpiB\nSMquDYzyXkpAtd+s9suaFCGEEJNKEhwljUJlwY/9QXCqhzFI2WrnIwPZjT7RC+qN72nX/+eR\nT3UO7HSP739tkhIcuRp85IjTohsRbuAzLyN2HrqXyhfRom3wZxZ2peVX8sX3XU5fNgEriUoJ\nmXM4fdZlhzmniHfhCweHZTf6wznGpA1MJiLPfE6dcDlm4uuDTBrSatg57bJDGyXbKIQQGSq9\nnqBpJGzHYWVoWtAwXKuE9rJy75pkjuK2RDpuKQABU6vxe3SpYyrETGHbpfJWI6YXSXCIMeCL\nRxHrcIm3vo+eNoRGmrPAPe4rFzgZRzoJj891bzZ7/xvWgTe5u12rnmtu3qbXN+V5IvkWctyl\nkCp8OVuZUPVKqh6lXgOtupYuHOejrw0JabThNmrMqusxwwRWIXkYzvBVEmRScEMx79KZnd0A\nJ9MUcvmFofDCwcqw3iVIvg/r/LAjtAAF1hdzeFNLmwOtGqp9eNQgY4YUGRFCTCaPpnk82tBN\nypFOHnl9ylilHdWRtFOO0gkBU6/0mpRfjiJuOSe6E05/69loGu0Ja1G5P2BKpQ8hhJi9JMEx\naygLdhRmWe5p7Xlwy2704lg7jZjgoFCle9wXzDe74djx73/d3jc4EyS98ynvjfd7b/p4XqcH\nVyJ2EKlzw4JGmMo353V6LqRp1z3Iy67kE28j1oGyOlp2JdUuzhg573mGT+1FIorKebT+Jpq/\nfFw3LQXkoYoPcs8bSJ3qi5hzKHwF9ImvqBK3OGVRRjV+zaD6G4aOj8pvROIAJ4/C6YHmg6eR\nAhug5ZtKmw6IzMvhHGfnFDgF6NBqyFgJKqFZX0KIaUrXqNxndiWtzDhRua9oC0+6Uvb5ntRA\nGiWadjqT9sIy36gNZRk4HUkOZDd62YpPR5IrqoMyi0MIIWYtSXBMV9b7B+0zx8kfMJeu1qtH\nXBSQ7uTW5zl+EgBAFF5ONdfCCBVyV0+gkF0AAH3NVvvVn7rE116V583TL/98aHYDAJRKPfND\no+kSvWnt6OeTRnV3Ifou9xyAHYEehH8xVVxRlCdealhDDWvc9yWiavv/5Pb+VrIXT/LhnbTl\nw9qV94z/vlNMD1L59eA0nCi04ITkDqpc59cwIkzrruSLu6AsgBBu1BpvRmDu8MM0+NeSfy3g\n3vN3RtCgLyV9KWABxsx9mUKIKbCgzJe0neSQWeIa0aIK/6jZhzxZis/HUhmTRNKOuhBL14dH\nadQStxzXRTRpRyUsRyZxCCHErCUJjunHaTsf+de/SR/s605Khhm45SOhex+E5jZl1OpWzT+E\nk+zfZo4e5kSLtvCBAh5HqW4ZdI9LA4tAJVWMUtpAm99kXPtR+8XHhgXrFhg3fCLPu1vvvOAa\nT7/9vD+fBAcA0lF2GZVdNplPvGrHY4PZjX785k95yUaaMz17eaokNM9gGUvywJioog9Ut5Kq\nFnHHyYy4tuJmariaGm5AOgIjAG3krxNnw2O/VPITQhSZqWura8Nt8XQ0bSvFflOvC3jM4q1P\niabtrCYtfXHF3pGzKJZ7X1sAsFTOXUIIIWY8SXBMN47d9bd/Zp87NRBg24o9+UMyPcG7P519\nOHe+OSS70c+OcNe7VHXlmO/uDWkb7lJv/8ewoKZrl30sn94Z5nUf1Zeut994mtuaESjXl200\nNt8KPd9fQtXV6hrnzot5XmGY5Em2WgEis3aEMhzjx0ffdIsyH31jmiU42OH4XiQOQqUADZ55\nFLochvvKo1xXQPII261gRUYV/CtAo3xHByJt6+fVnsf5zK6+iOnX1nyQllzduxue8kJey4zD\nSCnuYk4TeTWqIMygZjFCiKlDQG3AM2rPlMLYOTIRDDiKNX2kDMcIs0iKNcFECCHEdCQJjmkm\ntfv1odmNAbFfbw/e8cnsZAEnMucO9MkVHw0tvUYrr+f9T3HnGWg6VS+mdR+i8vnDBnlwb+xX\nP7XOntaravyXXhncdsfA7BKtcaWnscAKiBQo40TMJR4a4yOuE+WO38DqS5cwAM9cqtoGbZRV\nNoWwLaRcxgwAPW7NZUsXc/dvkGrpnwyhkD7LnReo8rZ8p284UY48Cyfad7n0aSQOUtm1o3dd\n8QS1yx/A2ts5cg6Gj8rrYYyWFpkxOAVEwAwqA+WccuVwi63O9VUDZADNhtaoU+2kDVMIIQqg\n564mOuo0kaCpGxplp0hMjQKGrE8RQojZSxIc04x92q0PCMDxHqf1vD63IXOHst2PV3bBX3BQ\nbRNd+0e59nZ//+HIfzw6sJnY+WLsuV/W/vd/JH/+6QPmc/u44zRAVLWA5q/pXWJgrN2Sfuln\n2Ucba64Yy/AxNLvRJ32eO5+n6g+N6Tp5MUx4A0jFXXaFKop/u4mTOo10S+ZSD7a5522quCmf\nC3DPjoHsRn8oxdFXqPKuvArf+ivIP61+YuOl2DkMdbq/iQFBayB9JZD5wV1xp60y+vUqW50i\nza9RQaV2hBBiUoQ9+kXXP4+mro3WSYUIDWHfqUhi2CIXQl3Qm2cTFiGEEDOSJDimGz339xJu\nu8hbzXbELT5Sx5M8OR2tPT/5jnX0AFtpc/Hy0F2fcnqiQ7MbvVIH93Y/9kjFp/8wr4vGu9Rr\n3+L2k71bDFDNEm3r78Bf7r3ho/aht9WFYX1DjTVXmOuvHsOgrdbM7EbfKM/C7oZR/PUOtGwL\n73OpHkLLthT9XhOH0y3uO9IteRU0cXpgua0kUnFY5+HJSszNeuwcgBo6zYqhzjBs0jPb3Drs\nvnRLcaskOIQQpcyja3UBz8X4sMJehkZzgnmtiDF1MjTNUczMTNBAukbtCavca8gqFSGEmLUk\nwTHNmMvcq2nqVbV6zVyXHRWbEDuRGSSNyjeMcyTpQ3s7/uZPOJno3bRbziTeeNFckfn01Sv+\n8m/yTHConY8MZDd6cdtxtfNR7fo/pkA4+J/+If38dvvAm6q7Taue59lyk3n5Nozpyxo798IQ\nu3MiEhzaVR9VLUcz6ozSlrsHCnBwOpl+/sfW3h3c2Uo18zyXXuf5rTvzL00ySdh9KhCgwAo0\n2nxglWOdDgDVU/CgZq4UMidlAABUC7RloGGToZhTrpdQyCq+I4QQJababwZMrT1hpRzWCEFT\nr/abIyxdGaotYQHQNRqaZHeYO5PWBBUNEUIIUfpK7CFKjMazcoNn7WXpfbsy4qF7H3R9zqfA\nItRt47aXoPq/IdEDVHcjvONbn8/c/S9fH8hu9HEc6/AeUP+c+qF72i+CedRMBHed5bbjLvHW\no4icR9lcMr3emz/hvTnfxisuKHezCZqYz0P+sPbxr/DuZ/jUXsS7UTWf1t9E9QxmmLEAACAA\nSURBVCt6d3IiFvvnL6kLp/s2W04mn/yOtf+N4O99taRyHGSUudeC00OjZzcAaFlVM5QDx4JS\nbL+LQDeFN430TzPbcMTl/6K+Xd0ZCQ6Q5nosoWidDoQQYuL4Db0hXEjVjKH9a4dK5IgLIYSY\nDUroCUrkhajiC3/ds/3b8ed+BscBoFXWhO//fd+VN+Q8o/wSCi3l+ClYEZgVFFzk8rQ5RvbZ\nk/a50y47HEfTNZX12UKvqMprnkU0Zz8UjpynMrcpKmPlmQvSwU5mnDzw1BXh+q50gy69jS69\nLXtP+sWfDGQ3BjgnDqTfeMaz1eX4KeNbithesJURJv8KPv02N+9GvBOBKlp0Oc1f53K6XgE9\nPFiDw07D7s+42Z2IvM2xw1R3N/TwxL2CaWUMk5I0lDlIuMRJ+ssIIYQQYrqyLElWikJIgmP6\nIV8g/Mk/Ct37O/bZUxQIGnX1IxXm6KUHKLyqiGNQkdwLPdwezQJX5cy/DGPknkNRrMYZmo/C\nl3HkjYwwlW3Jq9RlsdkH3JrIAvbBN0srwaEFqPw6jrwKNVARjuBdrt7eyefe6wu0n+Qz79Ci\nzdqWB7J/Dyh0JUeeAztQajC7McDp4a4dVH3LRL6G6YPK4DoVCgBlVlrVtbnK6WQM+5ESfDpN\nWMJOCCFKgN/QoumsrysAvyHz14QQYvaSvwHTFfkCZtMqY96C0bMbE0CryrnCxbvpyoyIuaip\n7OOfy+eyVLMEuts6BcND1YvHMsARhTZQ5TYYvQ+KBLOKqm5BcHXRrj8WnHCvQMHx0qtM4amn\n6g9T2dUIrKPQZVR1By6kB7Mb/fjkm3zqbZfTzblUcQe8i8E5fmOTJ11m1sxSHmgLXMJaPcif\nESOYpr5Kp+r+93NdpzqPvkre3oUQM1u135P9lYpOVOmTBY9CCDF7ySfgGSfSzPsfU2/+L373\nET71Uq42seNkzG0wm1ymhGjlVdVf+mrNX/2d//KrjfqF3rWbKj79h3P+7lEtlN/SA9OvrXXp\n1aqtuwOmb5xjHsa/hOo+SvM+S/M+S7UfgW9hMS8+FlQ5xzWuVc0Bpzi5m2PPcuxpTuyA494s\nY1KRCd8yCl2GwDoYVXzmHdej+IxbggOAHqbwb8G7yH0vK6jZXReTE1DNUMehzpO+DFrTkLdo\ngraY9DWu5xFMQ1vs1Td59A1efaOhLcjuJiuEEKWGAZWj3FA+/IbWWObz6NrQyMJyn7RQESJ/\nr7zyyu/+7u+uWrUqGAw2NDTcfffdu3fvzjimu7v7oYcemjt3rs/n27Rp0+OPPz7WA4SYTLJE\nZUbhky/y8d+AFXpnt3cc5XO7tEs/D0/xSxtUPPQXHV/5z07H4FM3+YMVf/iX5PX5L7/Kf/lV\nhV2WVt6gBavUvicRuQAAZXO1dR+ihmE9X1TzUefIOxztpOp5xvprKFxZ4GsogaqWnstvSJzY\n7xLfeg3HngH3rztQcbbPk3ctPCsmdXwj4kS3+454jjgAgPSQ+wda0qFlTk+YWRjqAnOEyABV\ngKqG7VPH4BwH+pebKj/p66AvAkcBBoWB0YvgkrylCyGmg4St2pNWylYMmBpV+UxTo7RSpqb5\ndC3/3mhBU19S4U/ZylLs0cmry/d2QozN17/+9dOnT993333Lly8/e/bsP/3TP23ZsuW55567\n+uqrew9QSn3wgx/cu3fvV77ylaampm9/+9v33nvv448/ftddd+V5gBCTTD4NzyCxCwPZjUHx\nNj76FK35aNHvZtQvrP27H8R+tT19ZB8sy1y8PHjbfVpF1ehnjoYaN+qNG2ElAWRO3GBO/+Kb\n1lu/hup7mdZzP/Lc+XvGhmvHf99RKAuRZk52kb8aZfV5tQ7Jg3n5NufM0fTOpwdDmua98X5t\nTgpOZqEKTu0nox5aqCi3Hj/yhTnW7rLDXzbSaf4liLyR+YsKwN8EmrmfTbmHnd3gHgyU1tDq\nSL+k701YnYFzbPjxCbbfIePqjDyIEEJMdz2Wcz42+Acu7fDZaHIg8W3qNC/oDZr5/pElwGdo\nRZ3kKcQs8g//8A9Lly4d2Lz33ntXrlz5jW98YyDB8fjjj+/YsePRRx/97d/+bQA333zzpk2b\n/uRP/mQgfzHqAUJMMklwzBx8cZ/LQyPAF/fTajURj47k84fueqDol+3jtibF2vmk9cbTQyOc\nSqS2/5M2v0mra5yokQDcdogP/xzJLvQ+nQbrtJUfRkUxFrYQ+e75A3PTtdbe11TnRa1mvrnp\nA/q8Bu55wnUgsFvgWVaE+xYDNWzk9pMu8caNI51mlFP51dz16uBsBQBmJVUUOOtnOlDsvAOO\nD49dZBwkfR0AVqfczrLBZ0FNkzFAIYSYLG2JYQ257OHLVCyHz0SSi8r9PqkVKsTEG5rdALBk\nyZJFixadO3duIPKzn/3M5/Pdf//9vZu6rj/wwANf+tKX9u7de8kll+RzgBCTTP54zCCpqHtc\nWbBcukhOR/Zbv3GJKsd++7kJvGvkDO/9t97sRp/YRbX7ESQ6inUHffEa352fC3z6L3wf+ow+\nf8ngypQszKli3XT8aPkHaM7yzGDDelq4eZQzQ2tpzn0IroZnHnyLqOIqqvvoTF6fotoysxt9\n8XOABTA45noec+nVmhVCiHFIO2poRsNRnN0xioH2RGZXciHEJDh79uzJkyfXr18/ENm/f/+y\nZcu83sFuhuvWrQOwb9++PA8QYpLJDI4ZxJuj0IZmwizRR0eOR8gfRt7LbVV7i3u87WzxBpWJ\nT73s0t3DSfOZHbT89gm5JfkAbdgEh4E9WmBC7lgYzdA+8Ed84jU+8y7HOylYTQs308JL3XsF\nZzCrqPLaCR9hqXDPXwAMjoPKAQ1w7SAjOWghxIyS8YctV43RhC1NtYSYbEqpz33ucx6P58tf\n/vJAsL29fcmSJUMPq6qq6o3neUAuhw4deu+9zGZ8AxxH3gREgSTBMXNQ7Vo+8Xz2KhWqW1Nq\npQ040WO/8O/Onhc5GYfHp6/aYm57gMKj1xogr59tl9kN5AtOwDD7cPScezxydqIKtZMBYx7s\nrKwNGTDmT9A9C0RES66iJTN4dUlRjLCY3AAAqgS3Ze8jKcAhhJhZzDxbnEgjFCEmFzM/9NBD\nzzzzzGOPPZaxbsUVjfb15KgHfO1rX3viCdcV2QBg2xPSCFLMBpLgmEFCc2jJNn7/18OCgRpa\ndtsUDSiHdDL17T/n1uaBTWfPS+r4e97f+1sKVYx8qr5so737Rdd4kQepUkh3AARPdc7PWdlv\n3Ikod7YgWEnlNeP8dEa+DRyPQkWGhHTyXQqSSmrTEPX+FmV9VUk+UAAA6cvZ7sycxEHl0Eos\nnyWEEOOjE4VMvcfqe7vTCI7bLA6/Ib2uhZg8zPz7v//73/rWt773ve/dc889Q3dVV1d3dAxb\nlN272TtNI58Dcvnud787wl6/v0Snn4vSJwmOGYUWXUtVTXx6B8cukieAyqW04CpoU98MdSj7\nzacHsxv9ONphv7zdvO3Bkc/1bPu4c/RdFesemjzQF681LvktALDTfPRF7jgN5aCyQVt+LTxj\nn9nBDnfs5M63wTYAaCaqKnCu3eXhtHzB4Ek9Her576nDr/ftqmnQb/wsNa4e890Hr+6j4A2w\nTrDdBljQyslsQkmtTxH5oyC0RqjTmWFtVV8ijMrI2MLOQXAnAECD1kD6MvkSUwgx89QFTBXj\nuKVAICIC99bhGHi/I0K1r7Q+uggxgzHz5z//+UceeeS73/3uxz/+8Yy9a9as2b59ezKZ9Pn6\nvmPbu3cvgLVr1+Z5QMEsWaomCiIJjhmnrJHW3l/KT0XOcfflds7xvcM+zihlv/ucOr6X41Gt\ntsHYfCvV1FPlHP8f/n361991Du3iZIzKq83LbzavuRuaxt0t6uX/jXh/KdBz7zlHX9Ku/jzV\njj7Lbii++CxHhlRFUhb0bpSH0TW8hqsZoMb+dRlWyvn3v+bO84MXaWu2f/xV4/6/ovmZNTjH\nQoPZROa0aqJhtcLpAnlg1s3koqFjR/oqUJjV++AkQKAy0lcMawFLZWRsAWxwChSQ1IYQYqbS\niOaHvHHbSdpKMQgUSVtJu2+BrUfX5gU90kJFiMnBzA8++OCjjz76ne9855Of/GT2AXffffcP\nfvCDH/3oR5/5zGcAOI7z/e9/v6mpaaBDyqgHCDHJJMEhJl06OWqcEz3p7/13de793k11fK+9\n69fmrQ8al99M5dXe+74IAFYK5mDFZn7ju4PZjb4LxtXrj+q3/VfoJgBEmvnUyxy7AN1LFYto\n8XUwsp7Are5h2Y0B5X6oACIX+jcXaivvgresb3jvvTg0u9HHsZ0d242P/HmOn8KM40Q4ugN2\na98m6eRfh4D8bRtA0BpJawQsQMtdlcMAyduyEGLmCxh6oH8dSrXfSDkq7bCpkdfIs0qHEKII\nvvjFLz7yyCMf/vCHA4HA9u3be4OBQOC22/pWuN99991bt279whe+0N3dvWTJkkceeWTfvn2P\nP/74wBVGPUCISSafpMVko5r5OHPILV4/8N/WM98byG70cRzr6W/pS9ZRdX9VgiHZDUTOc2fm\nshcAiHdx6zGau4rP7OCjTw1UYOXIGT7/rrbp8wjWDj2ck+5dWsCKVt9DCHCyiwLV8FcN/YKd\nzx1xP+nsYferzTxsc+RZOEN6mrLD8d1EJvyrpm5YpUnmXQshRCavrnml7IYQk27nzp0AHn/8\n8aEpifr6+ubmvs/Vmqb98pe//PKXv/y1r32tu7t71apV27dvv+uuuwYOHvUAISaZJDjEZDMu\nu8l593mX+OU39/2XUs6+V13OdBxn/2vGNfdm7+F4Z877xTuQ7ORjT2f2l0n38OGf06aMqh+5\nOtYBBATqKFjnskvlWCKo1PBlxTNX+tSw7EY/TuwnSXAIIYQQQpSk119/fdRjKioqHn744Ycf\nfrjgA4SYTLLEUUw2rWG5ecdD8AzpBqIb5g2f0Fdd0bvFyViuZSzc7dJKEwC8oZz384a57ZBr\nDoI7j8OKD42Q1y1/AQCUexeobmHu+CzIbgBs50gwqThUjhVJQgghhBBCCFFUMoNDTAHj0hv1\nZZucg69zx3kqr9FXXk5V8wb2kjcAw4RtuZwZLHe9IFXUI1iFWEfmDtNPdcv4jNt8EABgpHtg\nDmlN4qmmYBPH3s84jsrXQs/ZwURbd73z1pNIxjLjm2/PdcqMkzuPM1oXdCGEEEIIIYQoCpnB\nIaYGlVUZW24zb/2ssfWOodkNANB1fdmlrmfpKzfnuJymXf6JvmKiQ4OX3gfTP1ANNPus7F00\n91YKD11VQVS+nmqvH+G1IFhu3Ptlqm4YjHgD+rbf0ZZvGemsGYTMHNNb9HKQ132XEEIIIYQQ\nQhSVzOAQpci85dPq7BGODJuRYVx9tzY/Z89UmrNCv+Uv1P6nuO0EWFFlI625lSrqAVDNKtZ+\nCZU5JYSql8PwZV5I89Lc26jqCk5dBGnknQPTfdrIsEvNW2p8+uvcfIg7ziFYoTWshD+c54ud\nCTwNMGoHW6j0o+DGKRmOEEIIIYQQYhaSBIcoRVRR5/2Df7RfeVy9v4fjEa22Ub/ydn3phlFO\nC9VoWz7lEveW0aq7+eDjUPZg0F9NK+7MeSlPFXmqxjZoTacFa2jBmrGdNUMQlV/PsXeQPNZX\nqFULUmgzPI1TPTAhhBBCCCHEbCEJDlGiyBc0tz2AbQ8U52pzN1JZI5/ZwT3nSfeicgk1XglN\nGnYWD3kpdCWCl8LpBnmhh2dJgVUhhBBCCFF0tqVGP0iILJLgELNGoIZW3CnP3BOLPDBqp3oQ\nQgghhBBCiNlIiowKIYQQQgghhBBi2pMZHAL2qSP2ySNgNhYuNxavmOrhCCGEEEIIIYQQYyYJ\njlmN4z3R7/xt+p1XBiKejVeFP/MlCs6mDiBCCCGEEEIIIaY/SXDMatFv/U16z86hkfS7O6K2\nVfafvjZVQxKZoi3cehDJbgSqae56+EbvWTvDsQWVhh6c6nEIIYQQQgghSoskOGYv5+LZjOxG\nr/R7bzotp/V5CyZ/SGI45sNP8clXwH1FpPnoM7TqTmq4fGqHNWWsC9zzJqw2ACATgTUUuASk\nT/WwhBBCCCGEECVBiozOXs7Zk7l22c3HJ3Egwh037+ITLw1kNwDASfP+nyByduoGNXXS57jz\nV33ZDQBsIbabu5+f0jEJIYQQQgghSojM4JjF9Jz/+mSYkzmQsYm08vG3ONJKoSos2khVDVM9\noInCzW+4RRU3v0mr75704Uwx7tkFZLVDTzcjfRae+qkYkRBCCCGEEKK0SIJj9jKXrIJhwLYz\nd+iG0bRmKkY0On7vN+rN7XAsAAzgnZ/TJbdqm++Z6nFNjFjb2OLjwTaohN8NOA27Pcee8yQJ\nDiGEEEIIIYQkOGYzCpUFbrk//uS/ZcT9N39EK6uYkiFlcDpa7VPHoOnmwqVaRRW3HFY7fzTs\nCKV49y+5qoGWbpmiMU4kwwsr7h4vFquTu15Fshlswyij8AaE15XiyjV2cu6KHuJYlCrWwVs9\niQMSQgghhBBClBxJcMxqgbs+rZVXxX72HY5FAVAgHLjzU/4bpn75A6eSke//c/z5J6AUADLM\n4G0fCc5xf/DmQy+NNcHhtJxKvfykc+EMBcvM1Zd5r9gGoiKMu6ioZjmfcVulUrO8ODdIX+AL\nj4P7p/DYEe58GalzVHNrca5fLGzDjoA84LTL3kQHJ89yxy6qu5aqLxvLdW3gPHMM0InKgdpx\nDtPhtMO2RrpBHqDkfp2EEEIIIaYRy85amyxEHiTBMbsR+a6/03ft7U5rC8B67XxoJfHtfdf/\n9z+Tb7w0sMm21fOLH6rlC0MNnuyDufvCmC6efP6n8Z/8C1TfpID0rhdTrz4V/qOvki8wnjEX\nHTXdyBcPIhUZFi1vpPridFHhzlcHsxsD4seQbIavZCqb9Ozjrp1QSRgeGFn/+kpxKgkArPjC\nCxRogH9uftftZD4IWL0bzGeBcqK1hb0l2pyKWe1Of/5FJzNgVJmav4BLCSGEEEIIIQpWEk+z\nYoppmj6nXp/TUCLZDfvcqaHZjQHxY6eV5bJUgTxjSEw4507Gf/LNgexG3x3fP5D4xaNjHObE\n85VrW79A9ZfC9AOAN0xLrtc2/y60YjRGZRuplt5KJpl7kqeLcP2iiO7hjhegkgBgp3trrwxy\nbI52gwdeAnP3/vyuazMfGMhu9OtmPlbAGBXb0fQFRw3OLnHYiloXbdf5JkIIIYQQQogJIzM4\nRMmxjh9236HYjqY8VVnpjMa1+V88vevF3mUvwxBSbz4XuO+h/K9TBNF2tevnuHgcSqF2oXbZ\nnajImn3gLaN1HyUAThq6y+yVwrEFsPtKClUiT+bM3W8NC1gp2BZ0nS0mO8UpKzNBY3Xnd+U2\nIGvqCgC0AsuAseWPkk6EobJ+kJy0u0PmeJe9CCGEEEIIIfInCQ4xnVBZHdAzJMAI12obPpj/\nFVRnq2ucY1FOJcg7ScsKuPmA+uXfwe5PJXQ0O8fe0G76A1pyqfsJxc1uANB80Hx9kyOGI7Oy\nyPcqjN0NlcgMsoKtyCFOuWVh9Lz++ZhdXjUAQAEpYGwrlXLN1LA5NabrCCGEEEIIIcapJJYk\nCDGU2bTKfYdueD72f9OG2xCoAABfmFZfr9/9VzAJ8YMc3YX4Ydcn9qEo7N4ghrw+8vjGNe78\nsVLPf2swu9HLsdULj2QGJxAhtNolrHkQKFIR03Fil+UzfXI1TAkvzefCNFJDXMn5CiGEEEII\nMV3Jp3lRcox5jf6rbkzseDYjHrzlHq2yFpvvxeZ7Yaf7Sk7G9nHnG73FMhmA5qXyq+Bfluvi\nnvVbk7/5cXbcXL910hqp8MWTiLa57EhG+dwhWnDJ5AyDyq9guwfxI4MhPUDV2/KcBzHhjDJo\nXiiXeRBUcSnHIxkLUqhsOYVz/rsPl2uKShgY80wZg7w2XNJqBhWvm68QQgghhBAiD5LgEKWo\n/PN/poUrYr/5KRwHAJme4O0fC9/zmcEjerMbydPcvWPYmSrFnS+QUYEc5Q+MpjW+6+5KvvCz\noUGtqi5w94NFfg0jSERy7orn3pWNFdiGVujqFdKp5mak1nHyDFSKzCoElhd+taIjncIbufv1\nzLhnDoLLtKaF3LqTe96H1QNvFVWup4p1eV86CDQAzcODGlFeE0Ay+PRwyokyMgq7kN8oL+Bq\nQgghhBBCiIJJgkOUIvL6yj79x6G7H7BOv0+abixcqoXKsg/j2D63s5lj+6ni2lwXD9z3kLly\nU/LFnzktp7WyCmPVZf5b7p/UHrHB3EUuQvnVv0i3cucrSJ4FOzBCVHYpwutBBa04884n7/xC\nTpwE5ZcBjMjbg+1s/Yup6jqAoHlpzrU059rCLkzUBJQxnwZigA5UEC0BCpm6opER9syNW232\nsDax1TqVTKpICCGEEEKI2UESHKJ0aeVV3nVVIx1hd+WId458ZfOSK8xLrih0XONFtQtQOR+d\n5zJ3hKpo3goASHRy12k4FsrmUVl95mHJc3zhJ+D+Trd2D3e8hFQL1d42zoHxhePoOAtPgOY2\nIeherGRyEZVvRmgt0hfBaZg1MEf8fRibWqLidDkxyFPmme9w2mFbI8Mg0709jRBCCCGEEGIi\nSYJDTDjVft45dYTTSb1+sd6YZ5WE/OSqFjlSFclSQNq231NP/D9IRAdjnoC27fegaerAE3zi\nZai+/AXVrdLWfxS+wQks3PnyYHZjQM8RhNfDl5UNyVOkVf3mX/jMgb5Nw6NtvpM231kSD+p6\nAP5FUz2I0enkkVkbQgghhBBCTKESfw4U05xjJ37+7dTLTww8rvuuu9qzdT1RCkaY/MvgXzyu\n63vnu07WIE+hz/mThWoX6Z/4htr9K1x4H8yoW6StvwWBcnXwSX7/haFH8sWD6q1HtKv/uK8G\nqkojdd7tiuDkaSosweFYzk+/js6WwYidVq/9WDO8tOnWQi44jdgXOX0SqgdagMxGmKX+myOE\nEEIIMRvYVtb3eULkQRIcYgIlnng09WJ/OU+NAh/aZK6shHUaANIXOH4MgWVUdWPB0wQotJET\nx6ESw6JGGYJrxzPsSeINalvuGRZx0nzi1ewDues0tx+jmmUAwFbOC6oCW8zysV3DshsD13v7\nSX3jLZPWXGbyceJtpAabyHD6BMxGCl5VEvNWhBBCCCGEEGNUUFVCIfLA6WT6lScGNj3rGs2V\nWcUs40cRO1z4PfQg1d4N36K+32TSEFhONXdNXisQZfPZXXzoCT7yFF/Y39uptmAca4OTI0nR\n3d/yQ/NDc+8/SoXWp+C20+47Yl2I5ahyMgNYzUOzG/3BM0gdnYrRCCGEEEIIIcZLZnCIiaJa\nz7E1+LhurnSf/M+JYxRcWfht9DBV3QxWUDFowQI7iRQm2qLe/R7i7QMBrlykbfxteIIFXnCE\nwZM+eExoLSJvZx6g+RAstL7JSPedsXMZOH3KPW6dIu/ySR6MEEIIIYQQYvxkBoeYMNqw3y4K\n+dwPc3pGuoiTQGQ3t7+ArjeRupDzMNKghyc1u6GcjOwGAHSe5H0/LviSFKyFmaNbbdWiwcMq\ntyK4YtheI0R1t0PL8RMe9b7zlrrvKK8rjV4qE4Pj7nEVm9xxCCGEEEIIIYpDZnDMMsx8/l10\nnYCVQGgO1W+Bt2z0swqi1zVQIMzxvkYhHEuhOuRynJbjkR5A7Ci3PwuVQu/aj66dCK+j6utK\noUQCdxzLzG70xi8epFSkwJ+qpmsrblb7fpoRprlrqWLBkG2dam9FeD2SzawS5KlBcDnILOSO\nvddbtIHmLeeWzPUa2taPFHzNaYBy5INyxYUQQgghhBClTRIcs4kVV7sfReRM32brfj79Kq3+\nCNVNTElO3fDd9snE9od7t+wjLcaC6uyjKNCUY7Rd3Par4f1QGdG9MCtQtqnogx2zWFuOHYx4\ne8FpI1r8W5pmqkO/RDoGAJpOi67WVrq1MvHNh29+cTI9RNqd/0W98iM+8BKYASBYqV3zcVqx\ntSiXL01k1rPV7BZvmPzBCCGEEEIIIcZPEhyzCB95cjC70ctJ84EfU/lCeMMTcUfvNXeQx5f4\nxSPc053afcpYNtdYWDPsCN8CBFe5j7bnwPDsRn88+h6VQoLDyP09/wi78kALr9AbL+eeC3As\nCs2BOSkTCnwhbdvn8Fsf4/az8Aaoch70mf7m4FkM6wysc8OCehW846gII4QQQgghhJg6M/0Z\nRgxQFl98zyXupLl1HzVcOUG39Vxxk+fy652LZ5FOanMbyT7FiWOwozDC5F+GEcqL2jn6d1hd\nAE/5KhWqbmLSwCpzh68coTnjvbqmU1lWx5lJ4AtR/YrRD5shiILXIH2c0yehotCCZDbCu1wq\nEwkhhBBCCDFNSYJj1kj3QNnuuxKdE3tr3dDnLez7b+8qyjFlI1OuohKaOeXZDQDwVdCS6/n9\nZ4cFSaNVd05qrdNiUWk4EejhXD1oZyiCp4k8ORZJCSGEEEIIIaYVSXDMGoYfRH0VFjLk6twx\npci/kHv2u+zwL3QJTgVadhPCc/jYs4i1ggjlC7QVt6GiVIaXLzvCna8gcbxv09dAlR+AWZXv\n6Y6tzh7hrotUVqPVL4M5q/IjQgghhBBCiBIiCY7ZgRWSR7BwAakEp2109SCZ7ttFRDX5TamY\nZMFliDYiObxoiOaliqumaEAuaO56mrseygZp03PiRoIvbIczpDFqspkvbKe598MYvU6qOrXf\nfuqb3NHSu0ll1cYtD2rLLpugwQohhBBCiFnCsrNWgguRh2n4SCbGSiX54o+561XyKvi9VB6k\nBXVU3ff4SguuKULNiAlBNOdOqtwKPQQAmgfB5TT/EzArpnpgWTRjvNmNWAufeoYPP8Ynf4XI\nqSINa3Qc2TMsu9FLpTiya/RzO1qsf//qQHYDAEfare1/y+eOFXeQQgghhBBCCJEPmcEx83H3\n67Dah4WIUFMOqqL6a0p0+kYvMlC+mco3Q1nQTABgBbsT0GCUlUQljmLgehiMWwAAIABJREFU\nM8/z2VcH6pVyy+s051Ja8qHJeIGpc2OLD+G89RTsdGZUOfbrvzA//MVxj0wIIYQQQgghxkYS\nHLNAwv0bdVq8gcpKOLsxlGYCjJ49HH0HbAGA5qPw5QiunuqRjRd3HeXmlzODF95GqJ7qJqEb\nrksjXgAu3WGyDzl/Ikf8uGtcCCGEEEIIISaUJDhmOragsr5m75W9NqGEcfdriO0b2IJKcvcr\npJIIT0IWYCK17nENc+ueyUhwmNVInXePj8ptVQ4ZmmYqtfMHFKqmhZtQVjfuIeaJ4Zxkpxkc\nA/mgzSFjmby/CSGEEEIIMatIDY6ZjkyQCbg1T9FKsXmKO6cHsaEdVfrWbnDPO+Ac6ZtpglNd\n7juSOeJFReFL3PMUZRtGP7d+WUZEC3n12jIyLD70otr1E+en/5Xf+1VxBjoKZutNtg+Co4AC\nx+Gc4PSr0/13QwghhBBCCDEmkuCYBfxLXKs5kL9p8sdSoPQF9xwNO0i3TvpoiokMv/sOM0e8\nuMwaqrkNenAwovmoehu89aOeqm/+IPnDA5vkM7Uy/7BfNGWrXT/h5n3Z5xaZcxaqPTPIcXak\n3KkQQgghhBCziEzhnvmo/EpOX4A9fEZA2WXw1E7RiAowQkmIad5BqnI5Oo9mh6ly+SQNwL+Y\n5tUjeQZOBHoYvgZovnzOo3CV+cB/s5/+V3XmEAAt4HE9jI+8TA1rizng7FuoHEkudRHordLi\nMHcDKcBHVC6JXSGEEEIIIWYkSXDMAnqA5tyH6B5OnYGTgFlJoXX5fEVfQoyqQnZNB1S3idv2\nZbaGDdTR/KsmbxCaB4FCpvNQ7QLzU3/NXa3ceZ7ffgzRiy4HdV8Y7/BGZ7mH2QLA3Kr4xOAx\n7NVoKVHpNRsWQgghhBBCjI8kOGYHMlB2KeFScBp2J8DgNMj9K/dSZFbDW4/U2cy4f+mw5RXT\nEena6k9xyxvcugfJDngrqHo11V8Fbbr86xBV1FFFndr/JEfd9nuy1trYSSQ74S2DWaR/O3JZ\nzsMAUYDRrfjo8PVNKcUHddoATMoiICGEEEIIIcRkkQTH7KE4tgfx98AOAJAG/xoKbgTpUz2w\nvFDljdz1IpJDZjr4l1LFNVMzGqWYmfQi/ehIp/lbaf7W4lxtqtSvQatL41iqXzO4kYrw8ae4\ntb8qR7hBW3YHQvPHeWfSGtg5kxkESG9QfM6legsrxec0mj41aIQQQgghhBB5kATHbMHRN5E4\nOGRbIf4ec4rCk7gUYjw0H1XdAqsV6VaQBrMO5hQsTkns3tX57f+dOnIAij1LV1R99vf9l0/z\nxESRaGu2OcffRGT4KpXyubTmxr7/dlJqz78i2Tm4N9qsdv+rtvH3EJwzvntXkrGS7cPDchl6\nA/QFrN5xOZ4AjrkV3hVCCCGEECXBspypHoKYliTBMTuoJBKHXOKJIwhsmE6rPMxamFNWGzX6\n9M9bv/HfBjZTh/a1/Okf1Hzhz8ruvn+cV+buQ4iegB2Ht5qqN8JTPs4LTgFPQL/9L9S7v+Dj\nbyDZA9//ae/ug+wq6wSP/557+y2dpJN0IAmvA4kJkDclRMSMgjo4IOzMJIizsWYol5J1pqwp\nxiosV0aonaqBcsdaZ3a2pmRnrcEM4DAqdkVU1sLaWdTBIL4M5EUUBOVFEoFO0uStX27fs390\n6HSS20mn0923n/Tn8xf3OeeefmKOl8u3z3nOzLTw0tLFvz/4OJji5ccPqxsDqn3F8/+aln7w\nZH96eWEqzSv6fx3F/kgtqTQvSnMjoubzgyLCOqMAAHDqETimhsprtR+zGhGVV3MKHPVT9PR0\nfu6zR493/u//OeO915ZmzDx604hUe4tfPlDs/dWhH/Ta4+msq1P7ylEesI6aWktvWx9vWx+V\n3mg4ag2RIxZSfUPx+vNjcy1FmpEaLjhyLM0sigO1dh7t3xcAADBZ+TXmFDFM3TjOJg7pfmpL\ndW+NVTSL7gPdW/591Ictdnx3aN2IiKhWipf+T/QcdbFDRo6uGxEHF385WnUcrz8spbMjjl4q\npamUTnbhDwAAYLIROKaGhrnDXqvfcNrETiVXxf59w22q7ts7+sPu3FJrtL/YvW3Ux5ykWhfU\nHp8xzPgxVPdGz0+L/T8ourdEf+cxd51WKq1I0Tb4OqU55dKKiMYT/qEAAMDk5haVqaHUGi1v\niu5njhxvWRhl1+qPSMNZ59TeUETj2eeO8qDV3uivdQNFRPTuHuUxJ6t05luLlx+Lat+R42e/\n48QO1PN00b05ojrwquj5WTQtStNWDftzY3oqrYjoLaInxTQfegAAcKpyBcdUkWa+PaZdNORv\nPMW0C7J5hMok0PRbC1uW1VgXo2nxBc1Llo7yoKXGSMP893Z52iiPOWm1tKflN0TLnEMjDS1p\n8R+k9iMXzjiWymtF9xODdSMiIoro/UX0Pne8dzalmKluAADAKczX/SkjldPMy6J1RVQ6I4po\nmBvlGfWeU2bm3f7fdvzFn/c+d+hCmMZzz5v/Xz8TpVGHwpTaFhVdP6+xYdaS0R5z8kqzF6bV\nNxe7n4sDndHUlmafH40ntsBt0ffL2uO9v0xNC8dijgAAQK4EjimmPN0zU0atYf4ZZ3/+X/b+\n67d6ntpaFEXzBUtn/M77UsNJ/Z8onfmeYt9LUTlsgY/U/paYPswdMZNHUY3u54q+nSk1RvOZ\n0TR/RO8qNZ7YJRtHqO4rai4nUx39MigAAMCpQeCAE1EqzbjymhlXXjNmB2yaU7rgPxe/+V6x\n57mo7I/m09Lpq9PsZWN2/HHS91rR+XBUdsfgY3haF6c57x72jpuxkhprL5abLBoKAABTncAB\n9dbQms66apiH3ExKRaV47aHoP/yiif3PFKXmNPvycf3JqWFB0ffrGhsazhjpIaqdRd8zUeyJ\nKEepPTVeGKllDGcIAADUi8ABo1Td23XgoX+p/PKpoq+34dzF0963vnz6iP8zO2sHfnVk3Riw\n72cxa834XsTRdH70Ph/9rw0ZKqI0I7WMbJ3Xyi+KvqcOvbF/X9G/PTW/PUqzx3qiAACMXqVS\nPf5OcBSBA0aj8vzTXZ/9RLFvz8GXv/x5z/cfnvmRv2ha9c76TmwCFJVdw22I/j3RMKf21rFR\nSjOuiJ6ni95fRnVflFqi4azUsjxS0/HfWuwv+oau5zpw0Uyl6NuSmk/9vzUAADjlCRwwGnvv\n/sxg3RhQ9PXu+cJn2y+6JE1rrdesJkZKDcWw2yZgLYxyNF+Umi+KqL3e6LD6Xzn8+bJvqO6O\notuNKgAAkLtRP94Spq7+7S9UXqrxvNJi/56+p3488fOZaM1n1x5vmDWxzx4+0XVLeofdUgy/\nCQAAyITAASes+vow92hEVHfvnMiZ1EfT6dF69KNeS2n2O8b0x+yN2BHxakT32BwvDXdlTYo0\nbWx+BAAAUD9uUYETVprVPuym2XMncib1ktrfHU2nFa//JKoHIiKa5qVZa6L5zDE6fG9RPBXR\nOfjTIs5JadGJX7JxuNL8SI1R9B05Xp7nKbMAAHAKEDjghJUXnNNw9sLKS88dMZ5mtDUuXVWX\nKU24Usx4c5rx5ujfH6XGMQ0ERVE8GTF0fZMi4oWiSCktOqkDp8bUeHHR+5OIyqHB0szUuPKk\nDgsAAEwOblGB0Zjx4f+SZrQNHUmNTTP/08dTS51WGK32xf7tceDVKCb2kVrl1lHUjWLPc8Ur\nm4pXH4/9vz5q467D68agl2ovEXpCyvNTy7ujYXGUF0T5rNS4IjVfbnlRAAA4NbiCA0aj4dw3\ntd/5T/u/9S+VZ58q+noafmvJtPetL5+2oA5TqVaK7d8tdjwWRX9ERENrOuvd6fR6X0iyf3ux\n86fR2xVNs9KcpTH9jIPjvburz2+M/S8PvCoi0qwL0rm/F6XB57zuHeaI/RH7I056EdPUkhov\nPNmDAAAAk4/AAaOUZrRNv/4j9Z5FFL96sNi57dDryv7i+W9GtS/Nf1vdpvTS/y1+symKgw+T\nLX6zKc2/LJ19ZRRF9VcPxIFXDtu56+fxUmM69/ffGDjGQhsntwYHAABwShM4IGcHfnNY3XhD\n8fJ30rzVkcqjP3K1L/r3RUNbpFJEFM/9qHjhydi/O9rmpQvfmU47b7j3FbueKnZ8//Chotix\nKaafFY2tR9SNg9t3b0tnXhkNA3f3zBrmwI0Rdbr9BwAAyIHAARkr9r5Ue0N/Txx4JVrPqL31\n2Po6i53fiQMvRESkUkxbUvx4W/HiYEbZVjz1ndKq30uX/EHtt3durj3+2pPRvqT2pqKIns43\nAkdbxGkRrx2xS0rnu4IDAGCK6Oub2HXlOFVYZBRydowlRUe32mjfzmL7lw7WjYgo+out3x1S\nNw4eufrjrxU7nq79Y3t2DzO+K9LwRXXIppSWRZwzJGc0pnRhxNkj/SMAAABTkis4IGOpdX5R\ne0M5Wk4bxQGL3Y9FtXfogYrtu2rv+cxjaUGtKzLKwzyUpKElzTiniBRx1JTL02La/EMvq70p\nzo7S+RH7I0oRrVIsAABwXAIH5GzGuTHj7DjqRpV0+iVRbh7NAbuPem5rd1/tPfd21hxOs99U\n7H2hxvisJdE0J512SfHaj47cdOZ7Blb6iO5fFPufiOr+iIhSa5p+cTQvOpHZAwAAU5ffi0Le\nSos+kGa96dDrlNK81emcK0d5uKJy5EjjMCuVTmurOZzmXRqtRz0ut3VBmn9pRKQz35vO+t03\nltuIaJ6Tznt/an9zRBT7nyj2fv9g3YiI6v5iz6NxYOso/hAAAMAU5AoOyFzjjLT4g2n/9mL/\njig1pulnRfOckzja3Oh5eehAmjereOHIJT8jIp13ce0jlBpLF95Y7Ph+sXNr9OyO5tmpfVla\nsCZKjRERKaXTVqfTVkff65EaDpWOanfsr9Eyiv1PppYlkZpG/ycCAACmBoEDTgmtZ6TRPTPl\ncKntLcWrhweOhfOKzr2xr/uwwUWXDhs4IqLUkM68PJ15+bF+UuPhF4BUXo2otSpq0R99r0XT\nmcedOQAAMMUJHMAQ05ek/j3Frk2H7lWZNqv8+x8rnn6qeP7JYv+uNGt+uvCKtPiyMX5oa9E/\n/LZjbAIAADhI4AAO13ZJar0gup+Pyt5onBPTzotSU7pkSbrkD8bxh5ZnjWYTAADAGwQOmKwq\nr0dqiHLr8fcccw0zYsayif2Jc6JxQfTtOHK86awo117NFAAAYCiBAyabIl5/otj9g6h2R0Q0\ntKU574zpi+s9q3GXZl5e7PnuYY2j8cw04x31mxEAAJATgQMml6Lzkdjz5KGXldfj1W+m6u/E\nzBV1nNVEKLWkWb8bfTui8lpEiobTonF+vecEAEAdVCpWYWM0BA4Ye9XOl/t/9qNi96tp7hkN\nSy9Lbe0jfWelK/ZsHjowsJJnsevf0oylkcpjPNFJqHFBNC6o9yQAAID8CBwwxvr+35d7//VL\n0X/wKSR93/qnpv9wU8Pq947ozd2/jihqjFd7ove1aHZFAwAAQG2lek8ATimVLY/2fvuLg3Uj\nIore7p6Nn6u++PSI3l9Uag0OvwkAAICIcAUHjK3K49+qMVqt9j3+reZzlhz//Y1zo3jjvpRB\nKSJSNI74PpdJo+j6eezcXPTuSo0zY9YFqf3iSOn4bwMAADhxAgeMpeprv649/mrt8SO1nBnN\n86L3lSPHZ1wY5WknN7WJVrzwYLFr68F/7n4t9vyy2LW1tPCDUWqs78QAAIBTkltUYCylpkMZ\noiiiWqlW+6pFtUjNI8wTKc37vWg+47Cx6YtT+3vGcpbjr+j62WDdOGTfS8Wrj432kJWodkZ1\nexSvn9zUAACAU5MrOGAslRetrL76UkRU+/or+/uK6sEVQ6svvNj02vbSaWcc890REdEwM53x\nH+PAC9H7SqRyNJ8RzRk+VWT3z2qNFsXun6X57zzho/W/UFR+HtF38GWpPTWsiDT9ZCYIAACc\nYlzBAWOp8V0fSG3t1b7+vr29g3UjIio7Xnz9f3yi6Dkw0gNNOzdmrY62i7OsGxFFZW+t4RR9\nNcePqf/XRWXroboREdWdRd/jEZ6ODgAAHCJwwFhKbe3TPvrf+0utR2+qdv6m57FvT/yU6iI1\nzKy9ofGI8SKqe6P/lajuG+5QRf+ztUYPRP/IljUBAACmBreowBhLM9ur+2pfp9D/q5/HFRM8\nnTqZfVHs3nb0cJqz9NCL/p1F7xNRfWNNjfLc1PTmKLUd/o5qFLX/xyyKLk9kAQAABgkcMA6K\n4sTGTzlp1pKY+5ai84nDBmecl06/9OCL6utF96OH3WbS31l0/1ua9p5ILSP8IWM0WQAAJpe+\nvmq9p0CWBA4Yayk1nPOmyvM/P3pL+beWTPx06iWdfU3MujB2Pln07EpNbdG2JLWvGKwSRd8z\nNRbRKHqLvmdT07IhQ6VIbTWfnJLS7HGaOQAAkCOBA8Zey/s+uPd//eURg6U5pzW//XfrMZ26\nSTMXxsyFtS+0qO46eqyISNWdRx6kYXHR9+OjDj0jymeOxRwBAIBThEVGYew1vXnNjBs/WZp5\n6BKDhjctn/nnn0ktNRYfnaJq3a1TO4WU5qfGiw+7b6U0PzVe6uMLAAAYKtcrOB599NE777xz\n06ZNPT0955133oc//OFbbrllcGtXV9ett97a0dGxe/fupUuX3nbbbdddd10dZ8sU1HTpexrf\n/Pb+F35R3bOrPP/s8pnnR7JmxBDlWVE56skpRUSp1o0npTNS0/wo9kTRG2lGpGkTMEEAACAv\nWQaOL33pS3/0R3/09re//c4775w5c+Zzzz33yiuvDG6tVqvXXnvt5s2b77zzzkWLFv3jP/7j\n9ddf39HRsXbt2jrOmSkoNU9rWLyi3rOYpFLj4qKyI+Lw5aNSOTUuHOYdpUizrCsKAAAMJ7/A\nsWPHjptuumnt2rVf/vKXS6Ua16h3dHQ8+uijGzZs+NCHPhQRV1111apVqz7+8Y8LHDCJlOak\nlkuLniejOPDGyMzU9JZI0+s6LQAAIFf53cS+YcOGvXv3fvrTny6VStVqjacHbdy4saWlZf36\n9QMvy+XyDTfc8Oyzz27evHliZwocU3lBar0ytbwjNV+SWt6Zpr07ynPrPScAACBX+QWO7373\nu2efffYTTzxx4YUXNjQ0zJ079yMf+cju3bsHd9i2bdvixYubm5sHR1asWBERW7durcN0gWMp\nR/m0aDgnynNz/DgCAAAmj/xuUXn55Zd3795944033n777Zdccsljjz12xx13bNmy5dFHHx24\nY6Wzs3PhwsNu429vbx8YP/aRH3rooZdeeqnmpu7u7ojo7+8fmz8DEBHV3dG/K6Iapdmu3QAA\nAE7SpA4c/f39e/bsGXzZ1tY2cFvK3r17//Zv//ZjH/tYRFx55ZUppdtuu+3hhx+++uqrj3G0\ndLxnWHzmM5958cUXa24qiiIiyuXyCf8ZgKMVlaL3J1F5+dBI+fTUfMlhz4IFAAA4EZM6cGzZ\nsuXiiy8e+nL58uVz586NiKEt45prrrntttt+8pOfDAzOnTt3586dQ48z8HLgOo5jeOSRR4bb\n1Nvb29zcPHCrC3CSit5/P6xuRET/q0XPD1PLO+s0IwAAJpG+So3FFuG4JnXgWLx48fe+973B\nlwM3nqxcufKRRx4ZurzowD8PPlFl2bJlDzzwQHd3d0vLwd8GDywvunz58gmbOTCsojsqv64x\n3t8Z1V1RmjPhEwIAAE4Fk3pVv+nTp79jiNbW1oh4//vfHxHf+MY3Bnd78MEHI+Kyyy4beLlu\n3bqenp77779/4GV/f/+99967aNGilStXTvQfADha9fXhN+0ZdhMAAMAxTeorOGq6/PLLr7/+\n+ttvv33Pnj2rV6/etGnTZz/72auvvvpd73rXwA7r1q1bs2bNzTff3NXVtXDhwrvvvnvr1q0d\nHR11nTUw6Bhr2Uzq5AoAAExm+QWOiLjvvvv+6q/+6p577vnrv/7rM84445ZbbvnLv/zLwa2l\nUumb3/zmrbfe+ulPf7qrq+uiiy564IEH1q5dW7/5AkOUZ0eUI45+JlHyLBUAAGDUsgwczc3N\nd9xxxx133DHcDrNnz77rrrvuuuuuiZwVMDLl1HRR0bv1yOHGRZGm1WM+AADAqSDLwAHkrfFN\nKTUXvdui6I6ISE2p8YJoXFjvaQEAABkTOIB6aDgnNZwTxf6IIlJrRKr3hAAAgLwJHED9pNZ6\nzwAAADhFeGYBAAAAkD2BAwAAAMieW1QAAACYRCp9/fWeAllyBQcAAACQPYEDAAAAyJ7AAQAA\nAGRP4AAAAACyJ3AAAAAA2RM4AAAAgOwJHAAAAED2BA4AAAAgewIHAAAAkL2Gek8AAAAADumr\nVOs9BbLkCg4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAAAIDs\nCRwAAABA9gQOAAAAIHsCBwAAAJC9hnpPAAAAAA6pVKr1ngJZcgUHAAAAo9HV1fXRj350wYIF\nLS0tq1at6ujoqPeMmNIEDgAAAE5YtVq99tpr77vvvk996lMdHR3nn3/+9ddfv3HjxnrPi6nL\nLSoAAACcsI6OjkcffXTDhg0f+tCHIuKqq65atWrVxz/+8bVr19Z7akxRruAAAADghG3cuLGl\npWX9+vUDL8vl8g033PDss89u3ry5vhNjynIFBwAAACds27Ztixcvbm5uHhxZsWJFRGzdunXl\nypXHeGNvb+++ffuG21oUxRhOkilF4AAAAOCEdXZ2Lly4cOhIe3v7wPix3/iBD3zgwQcfHMeZ\nMVUJHAAAAIyZlNKxd7jnnnuOEUGuuuqqsZ4RU4XAAQAAwAmbO3fuzp07h44MvBy4juMYZs2a\nNWvWrOG2Tps2bUymxxRkkVEAAABO2LJly55++unu7u7BkYHlRZcvX16/STGluYLjBHzta1/b\nunVrvWcxcb7+9a/PnDmzXC7XeyJMIa+//npRFMco+jDmqtXq9u3bzzrrrHpPhKnl1VdfbWtr\nG7oyH4y37u7u5ubmyy+/vN4ToYbHH3+83lMYjXXr1n3xi1+8//77b7zxxojo7++/9957Fy1a\ndOwVRmH8CBwj0tDQcNlllz3wwAP1nsiEeu6558rlcqnkMh8mTn9/f0TIakykoigqlUpDQ8Nx\nbxiGMVSpVEqlkn/JMpGq1WpEbNq0qd4Tobb3ve999Z7CCVu3bt2aNWtuvvnmrq6uhQsX3n33\n3Vu3bu3o6DjJw771rW995Ks3j8kMqZd6nc/JM3ioqVqtlsvl73znOzI/E+mmm27q7e295557\n6j0RppBNmzatWbNm4Beb9Z4LU8iKFSv+5E/+5M/+7M/qPRGmkL/7u7/7whe+8MQTT9R7IpxS\ndu/efeutt3Z0dHR1dV100UW33377ddddV+9JMXW5ggMAAIDRmD179l133XXXXXfVeyIQYZFR\nAAAA4BQgcAAAAADZEzgAAACA7AkcAAAAQPYEDgAAACB7AgcAAACQPYEDAAAAyF5DvSfAJFUq\nla655ppzzz233hNhalm9enVfX1+9Z8HUcs4551x99dWNjY31nghTyxVXXLFs2bJ6z4KpZfny\n5Zdffnm9ZwEwjlJRFPWeAwAAAMBJcYsKAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAA\nANkTOAAAAIDsCRwAAABA9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncExR\nnZ2dt9xyyxVXXNHW1pZSuu+++47ep6ur66Mf/eiCBQtaWlpWrVrV0dFxojvAsf3oRz9KR3ns\nsceG7uM0Y/w4uxhvPuWYAL7UAQwSOKao7du3b9iwoamp6b3vfW/NHarV6rXXXnvfffd96lOf\n6ujoOP/886+//vqNGzeOfAcYoU9+8pNfGWLJkiWDm5xmjB9nFxPGpxzjypc6gEMKpqT+/v6B\nf/j2t78dEffee+8RO3zlK1+JiA0bNgy8rFQqK1euXLRo0ch3gOP64Q9/GBFf//rXh9vBacb4\ncXYxAXzKMQF8qQMY5AqOKapUOs5f/caNG1taWtavXz/wslwu33DDDc8+++zmzZtHuAOM3IED\nB6rV6tHjTjPGj7OLieRTjvHjSx3AIIGD2rZt27Z48eLm5ubBkRUrVkTE1q1bR7gDjNAf//Ef\nt7a2Njc3//Zv//bDDz88dJPTjPHj7GLC+JSjvnypA6YOgYPaOjs729vbh44MvOzs7BzhDnBc\nra2tN9100+c+97mHHnrob/7mb15++eWrr7566E2/TjPGj7OLCeBTjsnAlzpg6mio9wQYd/39\n/Xv27Bl82dbWdtxLGY8hpXSSOzA11TwPly5d+vnPf35wcP369StWrPjEJz6xdu3aYx/Nacb4\ncXYxhnzKMZn5UgecelzBcerbsmXLnCF++tOfjuRdc+fO3blz59CRgZeDgf+4O8BQIzkPTz/9\n9GuuueaZZ57ZtWvXwIjTjPHj7GLi+ZSjLnypA6YOgePUt3jx4u8NsXDhwpG8a9myZU8//XR3\nd/fgyMBCU8uXLx/hDjDUCM/DSqUSQ9ZLc5oxfpxd1IVPOSaeL3XA1CFwnPqmT5/+jiFaW1tH\n8q5169b19PTcf//9Ay/7+/vvvffeRYsWrVy5coQ7wFA1z8O+vr6h+7z44ovf+MY3li5dOmvW\nrIERpxnjx9nFBPApx2TgSx0wdViDY+p68MEHe3t7t2zZEhE//OEPW1paIuK6664b+LXSunXr\n1qxZc/PNN3d1dS1cuPDuu+/eunVrR0fH4NuPuwMc1x/+4R9OmzZt9erV7e3tv/jFL/7hH/5h\nz549//zP/zy4g9OM8ePsYgL4lGNi+FIHcFDBVDX466OhDhw4MLjDrl27/vRP/3TevHnNzc1v\nectbvvrVrx5xhOPuAMf293//929729vmzp3b0NBw+umnr1279gc/+MER+zjNGD/OLsabTzkm\nhi91AANSURTjX1EAAAAAxpE1OAAAAIDsCRwAAABA9gQOAAAAIHuvvrhWAAAHAklEQVQCBwAA\nAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgAAACA7AkcAAAAQPYEDgAAACB7AgcA\nAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4AAAAgOwJHAAAAED2BA4AAAAgewIH\nAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAAAIDsCRwAAABA9gQOAAAAIHsC\nBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgAAACA7AkcAAAAQPYEDgAAACB7\nAgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4AAAAgOwJHAAAAED2BA4AAAAg\newIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAAAIDsCRwAAABA9gQOAAAA\nIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgAAACA7AkcAAAAQPYEDgAA\nACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4AAAAgOwJHAAAAED2BA4A\nAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAAAIDsCRwAAABA9gQO\nAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgAAACA7AkcAAAAQPYE\nDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4AAAAgOwJHAAAAED2\nBA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAAAIDsCRwAAABA\n9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgAAACA7AkcAAAA\nQPYEDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4AAAAgOwJHAAA\nAED2BA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAAAIDsCRwA\nAABA9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgAAACA7Akc\nAAAAQPYEDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4AAAAgOwJ\nHAAAAED2BA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAAAIDs\nCRwAAABA9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgAAACA\n7AkcAAAAQPYEDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4AAAA\ngOwJHAAAAED2BA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkTOAAA\nAIDsCRwAAABA9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZEzgA\nAACA7AkcAAAAQPYEDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA2RM4\nAAAAgOwJHAAAAED2BA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAAANkT\nOAAAAIDsCRwAAABA9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAAAADZ\nEzgAAACA7AkcAAAAQPYEDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AAAAAA\n2RM4AAAAgOwJHAAAAED2BA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidwAAAA\nANkTOAAAAIDsCRwAAABA9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIncAAA\nAADZEzgAAACA7AkcAAAAQPYEDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACyJ3AA\nAAAA2RM4AAAAgOwJHAAAAED2BA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAAsidw\nAAAAANkTOAAAAIDsCRwAAABA9gQOAAAAIHsCBwAAAJA9gQMAAADInsABAAAAZE/gAAAAALIn\ncAAAAADZEzgAAACA7AkcAAAAQPYEDgAAACB7AgcAAACQPYEDAAAAyJ7AAQAAAGRP4AAAAACy\nJ3AAAAAA2RM4AAAAgOwJHAAAAED2BA4AAAAgewIHAAAAkD2BAwAAAMiewAEAAABkT+AAAAAA\nsvf/Aev72QXh8k6fAAAAAElFTkSuQmCC"},"metadata":{"image/png":{"width":720,"height":720}}}]},{"metadata":{},"cell_type":"markdown","source":"#### Evaluating the stability and consistancy of the clusters"},{"metadata":{"trusted":false},"cell_type":"code","source":"# Silhouette plot\nwithr::with_options(repr.plot.width=12, repr.plot.height=25)\nplotSilhouette(FG,K=4) # K is the number of clusters","execution_count":7,"outputs":[{"output_type":"display_data","data":{"text/plain":"Plot with title “Silhouette plot of (x = kpart, dist = distances)”","image/png":"iVBORw0KGgoAAAANSUhEUgAABaAAAAu4CAIAAACYeIg0AAAACXBIWXMAABJ0AAASdAHeZh94\nAAAgAElEQVR4nOzdWXwT9f7/8UnSNBS6sITSvUipgiwWBAQpi7IIBwo9shcqRcXCoRwF0eNB\nkHoEjsoiB2QTFEGOssgiUFZpVYQWpFD2sghU1kLZSgvd87/I78x/TNs0aZtMp3k9r8LkO/N9\nZzLk0XzymRmVwWAQAAAAAAAAlEwtdwAAAAAAAICKosABAAAAAAAUjwIHAAAAAABQPAocAAAA\nAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAAAAAAAMWjwAEA\nAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIHAAAAAABQPAoc\nAAAAAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAAAAAAAMWj\nwAEAAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIHAAAAAABQ\nPAocAAAAAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAAAAAA\nAMWjwAEAAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwKHbTk5\nOan+JykpqdzLYTdKfAsKCgr+85//tG/fvnbt2mq12hh+6NChlqzbt29f4/iQkBBb56wUSnyD\n7CwrK0slcfLkSfEpB9l77AFbMxgMTZs2Ne7GQYMGyR0HAADg/zjJHUB54uPjV6xYcfDgwRs3\nbuTl5dWrV0+v1wcGBrZr165du3YdO3Z0dXWVO6PMtm3bdvjwYePjNm3a9O3b19oB1YA9X+PQ\noUM3bNhQjhV//fXXuLg44+N33nmnUkPhT6rNMV9tXki5sQcEQVCpVG+//fbo0aMFQdiwYcPh\nw4fbtGkjdygAAAAKHNYoKCgYNWrU6tWrpQtv3Lhx48aNEydObNu2TRCEFStWREVFyZOvyti2\nbdvSpUuNj6Ojo0sscJgfUA3Y7TUmJydLqxs1atTw9PRUqVT169cvc91//vOfxgcBAQFDhgyx\nUUII1eiYrzYvpNzYA0aRkZFTp069efOmwWCYPHny7t275U4EAABAgcMaU6ZMMalulOnNN98s\nLCw0Pvb29rZBKDg68cdkQRD8/f1Pnz5tYQ/RoUOHfv31V+PjyMhIJyc+Dao/PpHYA5VFp9NF\nRETMnTtXEIQ9e/acOHGiRYsWcocCAACOjq80lsrJyVmwYIH4T19f34EDBwYGBgqCkJ6efuzY\nsX379mVnZ5usNWfOHLumhOPJysoSHz/55JOWnyG1fPly8TFn0TsIPpHYA5Vo0KBBxgKHIAjL\nly//z3/+I28eAAAAwQDLHDp0SNxpNWvWvHXrlsmA3NzcDRs2JCYmShdqNBpxLelTFi5/8ODB\nP/7xj6CgIJ1O5+XlFRkZefny5RLjHTlyJDo6+umnn3Z3d9dqtQ0aNOjZs+fnn3/+6NEj6bBL\nly5J3/0bN26U9tTt27dNpjhx4kRMTEzz5s09PDycnZ29vb379eu3fv36oqIicUz//v3NHGz1\n6tUrc4C1M5pnsj9v3Ljxt7/9LTAwUKfT+fn5jRkzRroHzL81lu9nq15jaSyZ6O233zYz0fr1\n681s/9GjR25ubsaRjRs3lj6Vlpbm4eEhbmfVqlXStYKDg8Wn3nnnHUteS+Uq7Q2S1h8FQViw\nYEGJ4y05BrZs2TJp0qRu3boFBwfXq1fPycnJzc0tODh46NChW7ZsKTPS7du3x48f37BhQycn\np65du1bK8VCa7OzsqVOnPvnkkzqdztvbOzIy8ty5cw8fPpROceLEiTL3Xm5u7pIlS3r06OHj\n46PT6WrUqOHn59emTZvXX399yZIld+7cMVTSgV3pquAeqODxc+fOnUmTJjVq1KjMj/2LFy++\n9957zz33XL169bRabf369UNCQt58881Dhw6ZjLT8s9SS/SDl7+9vTF63bt3c3FyL3jMAAACb\nocBhqb1794p/g/r5+Vn4HbsiBY41a9Y88cQTJn9De3l5XblyRTpFfn5+TExMaX9zBwQEHD58\nWBxsUsUwnj5d4lPSAkdhYeG7776rUqlKnOLFF18U/+qtrAKH5TNavv+/+OKLevXqmWyqfv36\nJ0+etOQts3w/V/B7oOUTVaTA8eOPP4ojo6KiTJ5duXKlNLBYzps0aZK4vEWLFjk5OeZfy/Ll\ny4MsNn36dPNbMyrxDVq5cqX0aBGrG4ZyHQPPPPOMmR0bHh6el5dXWqR169aJX/kEQejSpYvt\n6gK3b98uflKAq6vr1q1bpUvK/Hr/+PHj9u3bmwm5Z88eQ4UPbFscDFVzD1Tk+Pnyyy+LnzhT\n/GO/qKjoo48+kq4oNXLkSHGkVZ+lFu4HKem1e/bv32/JuwYAAGA7FDgsdf78eenfeQMHDkxI\nSHj8+LH5tSpS4CjtXINXXnlFOsXYsWPN/D0qCELdunUvXLhgHGxSxUhPTxe3Y6bAYfJFWq1W\nm2Tr1KlTfn6+ofIKHJbPaPn+d3Z2LnHSxo0bS9siSntrLN/PFfweaPlEFSlwxMbGiiM///zz\n4gMGDBggDhg6dKjBYDh48KC4c5ydnY8dO1bm/p81a5b51yL15ptvlrlBQ0lv0MaNG6ULpdUN\nQ7mOAekXVHd391q1apmMj42NLW0Kk2u7du7c2XYFjj59+pS4TZP/LGV+vRfPMjCqUaOGse9A\nXFIpBQ5bHAxVcw9U5PjRarUlbtzkY3/y5MlmkkgLHFZ9llq4H0p7Wz/55BNL3jUAAADbocBh\nhWeffdbk70itVvvMM8+MHj36u+++e/jwYfFVKlLgEATh2WefXbFixaJFi6S/6dWqVaugoMA4\nXnrijCAIbdu23bFjx9GjRz/++GPpX6X9+/c3jjepYkhPtCmtwHH06FHx1z+VSjVr1ixjWefg\nwYMBAQHi+C+++MJgMKSnp1+6dCkiIkJcHhERcel//vjjjzIHWDujeSb7Mzw8fOfOnTt37uzX\nr590+fz5882/NVbtZ0teY2msmuju3buXLl2SftXp2LGjOFF2draZiXr37i2u9csvvxQfkJGR\n4eXlJY7ZsGFDs2bNxH9++umnZe58g10KHHv27NHpdOISk+qGoVzHQFRU1OzZs1NTU8V9mJ6e\nPnHiRHFwvXr1pD1cJlNoNJrw8PB33nknMjIyPDy8IseDGT///LN00meffXbTpk0///zz+PHj\nTXZsmV/vpbti48aNxpdWUFBw6tSphQsXdu3ade/evYaKHdgGGxwMVXYPVPD4KfNj/9ixY2q1\nWnxKr9fPmzfv2LFjZ86c2bx585AhQ1599VXjSGs/Sy3cD1LSm6e8/PLLZb5rAAAANkWBwwqn\nTp1q0KBBaX+Ru7q6xsbGmrQVVKTA4efnl5WVZVweHx8vnev8+fPG5aNHjxYX1q1bV1pkmTFj\nhviUSqUyNmuYadMo7ano6GhxYWRkpPTVSe8p065dO3G5dJXo6Ojie9L8gHLMWBrp/nzuuefE\nLxWFhYXSclX79u1LXEV8a6zdz5bshBKVYyLp98Zu3bpZOFFISIi41pkzZ0ocExcXJ46Rllc6\nd+5cWFho4USVTvoGzZ07V/rzePHqhqFcx0CJ8vPzXVxcxPGnT58ucQqNRlNiwah8x4MZ0g3W\nqVMnMzNTfGrUqFGCRJlf71966SXjErVaXdrlHmz3QspNWXvAwuPHko99aZOXRqM5cuSIyVz3\n798vHtWSz1Kr9oPRkSNHxO106NDBwr0BAABgI///VyCU6emnnz527Njf/vY38eqMUllZWbGx\nsSNHjqys6caOHSt+eTNpHrl//77xgXibT0EQBg8eLO09fu2118THBoNh//795Yuxb98+8XFK\nSkpficWLF4tPJScn5+fnl28K+8z46quvij9mqtVq6VegI0eOiHeOLJEd9rOdJ8rIyBAf161b\nt8Qxf/nLX8QvSAUFBcYH7u7uq1atkv6ALKNJkyaJty5asGCBmWuXGFl4DBQVFa1bt27w4MFP\nPfWUm5ubRqNRqVRarfbx48fi+OvXr5c4RURERKdOncr9iiz322+/iY8HDRok/VCSHiqWEHtz\nioqKnnrqqU6dOo0ePXru3LkJCQk5OTmVktYWquweqMjxY8nH/i+//CIuHDBgQKtWrUw2Il4k\n2NrP0nLsB+mnh/RTBQAAQBbcJtY6DRo0WLhw4dy5cw8cOLBv377ExMT9+/dLr9j/7bffTpw4\nsfjJLOUgvXieyVnc4rfNmzdvigtNrkjaoEGDmjVrPnr0yPjPGzdumJ/OYDCUuFy64okTJ06c\nOFHisMLCwoyMjOKXxysHG81osn8aNWokPs7Ly7t//37xy0+KKnE/m2e3iaS1IWl3hok5c+bs\n3LkzLS1NXDJ79mzj3ZGrgqKiIuODF198sczqhmDZMZCdnd2nTx+T0x+Kk96dV6pnz55lxqgU\nt27dEh83bNhQ+pT0dVli3LhxX3755YMHDwRByM3N/fXXX8VCm5ubW3R09PTp06XnAVURVXMP\nVPD4seRjX1ocadmypZlZrP0sLcd+kH565OXlmQkDAABgB1XiZ1jF0el0L7zwwgcffLBjx46M\njIw5c+ZIn/3pp58qZRbp1QpLu1q+tCpR/Dr55p8VJH8xC4Jw+/btMqcwLzc318KR5tloRpPN\nit+NrV23HPu5Ck6k1+vFx/fu3Stt2OXLl9PT06VLpD0mZVq6dKmfxaTXPbVWfHz89OnTyxxm\nyTEwc+ZM6bfTli1bRkZGRkdHR0dH16hRo7RNiaS3ULEpM/9NrDq2BUFo1KhRYmLiyy+/XPw7\n/MOHD2fPnv3mm2+WJ+KfVfrBUDX3QAWPH0s+9qXMfw5Y+1lajv1w9+5d8bHJFXYBAADsjw4O\nSxUWFqrV6uJ/TTo7O0+cOHHBggWXL182LjH+/GUf3t7eYt/yxYsXpU+lp6dLO6KNF4w0+a1e\n+itiaT/uSaf47LPP3nrrrcoIbo6NZjTZP9Jrjjg7O9euXdvCSJbs53Kz20TSryJ3794NCgoq\nPiY/Pz8yMtKkNX3VqlX9+/d/+eWXLZnl4cOH165dszCS+MIt179//x9++MH4eOrUqQ0aNJBe\nxKQ4S46B77//Xlw4fvz4+fPnGx8XFBSsWLGizEh263Ro0KCBuG9NLqBj8k9LNG3adMOGDY8e\nPUpJSTl79uz58+d37tx59OhR47Nff/31vHnzpN/Py6HSD4aquQcqePxYwsfHRyxKHj9+3MzI\ncnyWWrsfpOVRChwAAEB2dHBY6sqVK02bNl2yZIn0ByujtLQ0aSew9LdxWwsNDRUfr1u3Tlqw\n+PLLL8XHKpWqY8eOguTcbKPDhw8bH+Tn54t/iJuQXlDgu+++K/FaFTdv3jx48KD4T+mdDqXf\nyS0cUI4ZLfHVV1+Jv+sWFRVJv2y0bt3a/I+l1u5nwYKdUFkTlY/0TpZnz54tccy0adPELzbS\ney5ER0ebtHXI5b333nv99dfFf44dO3bjxo1mxltyDEj/L7/44ovi4927d1ekA7/M4+H8+fM/\n/k9p1Uaptm3bio+///576Yly0kPFEmInV82aNZ9//vlRo0bNnDnzt99+E4toubm54m4p34Ft\nC1VzD9jo+JHq3Lmz+Pj7778vXuMQi+zWfpZatR+Mzpw5Iz6WfqoAAADIw84XNVUu8SdBrVbb\nqVOnCRMmzJ49+7PPPouJiTGpaCQnJ4trVeQuKtLlhj93Gpd2+9J27drt3LkzJSWltNvEGgwG\n6QUUPDw8Zs2atXLlyuKXRRTvopKcnCxdPnjwYPFi/vfu3YuLi3vllVdcXFymTZsmTvHhhx+K\n4729vePj4y9evHjp0qU7d+5YMqAcM5bGpGzRt2/fuLi4uLi4sLAw6fL//Oc/5t+CcuznMndC\nicoxUfnuoiL9nfmtt94qPmD//v3irnBxcTl37tygQYOke9LCiSqdyRuUn5/fq1cvcYlOp0tI\nSChtvCXHgPQ6Hb17975//35RUdFPP/3k4+MjHb9p06bSIpUYu8zj4e233xYHDBgwoMz9YHIq\nXOvWrb///vv4+Phx48YJf1bmPUTefvvt8PDw1atXp6WlGe8yU1RUtG3bNukhl5GRYeELsZuq\nuQcq9/iRriI+lZKSIu0lrF+//vz5848fP56amrpt27ZXXnlFvE2stZ+lVu0HI2mFMS4urhLe\nVwAAgAqgwGEpC3ueTb5h2rrAYfjzLQNLVKdOnQsXLojj33nnnRKHOTs7S/8pvYPshAkTTAa7\nuLiY3EpGWm7YsWNHiVO8+eabFg6wdsbSSPdnife+EQQhKCgoOzu7zLfA2v1c5mssjbUTla/A\ncevWLfGVPv/88ybPPnz4UHrSyqxZs4yrSFvQv/jiCwvnqlzF36CHDx9K73rr7u4uvXGmtcfA\n3//+d+lTGo3GeK1HrVYr/ene2gJHmceDtQUOg8HQu3fvErdp8n+5zK/30gsrODs716tXz+RE\nG+ntP8t9YNtCFdwDlXv8SDclfeq9994rMYPRyJEjxZFWfZZatR+MxKucOjs7i7enBQAAkAun\nqFiqZs2avr6+5se0a9duzZo19skjmj9/fvFfLEUBAQG7d++WflmdPHly8Qsu1KlTZ+XKlaVt\nZPbs2e+99570N8PHjx9LG8KFP3917NGjR/E7F0qVOcDaGS2xZMmS4u9gvXr1Nm7cWLNmzTJX\nt3Y/l/kaK2ui8qlfv77Y+JCUlCS9e4sgCBMnTvz999+Nj9u1a2f8jlS/fv1FixZJx5hc0kIu\nrq6ucXFx4tU9MzMze/fufeHCheIjLTkGPvjgg+DgYPHZwsLC7OxsjUazbNkyd3f3cocs9/Fg\nxsqVK59++mmThTqdzsz/5TLl5eXduXNHevleLy+vZcuWif+0xQsptyq4B2x0/JiYOXPmtGnT\nLLkKabk/S8vcD4IgXLx4UTxBpl+/fianQAIAAMhA7gqLkhQVFe3fvz82NrZ3795BQUGurq7G\nX+eCgoIGDhy4du3awsJCk1Xs0MFhlJycHB0d3bRpU1dXV61W6+np2b179wULFkh7E0Q3b94c\nM2aMr6+vVqv18/OLjo6+du2aSYuKtIPDKDU1deLEia1bt65Tp47xhTdu3Lhfv36zZs06c+aM\nyeA7d+6MHz8+KChI+lOq9DfeMgdYO2OJTPbnrVu3xo8fHxgY6Ozs7Ovr+8Ybb1y/ft38KhXZ\nz5a8xtJYPlH5OjgMBsOmTZvEFRcsWCAu37Ztm7jc2dn51KlT0rUGDx4sPtuxY8fix7ytlfYG\nnTx5Uvr96oknnrhx40bx8ZYcA3fv3n3rrbcCAwO1Wm39+vX79+9/4MABg8EgvZGwtR0chrKO\nB2nnTkxMjIV7Iysr6/3332/cuLGzs7Onp+fgwYNPnDhh8vW1zP6FP/7446uvvho9enTbtm0D\nAgJcXFyMh1znzp3//e9/37t3z6oXYmdVcA9U4vEjfRXFD60LFy68++67bdu2rVu3rpOTk16v\nDwkJGT9+/MGDB01GWvhZau1++Pe//y3G27Fjh2XvGAAAgA2pDBbfRg5QHCcnJ/G6eomJie3b\nt5c3T5VSUFAQHBxsvPtP+/btExMT5U5kE0o5Blq2bGm8tmi9evVSU1Ptea1ioHzEgzY4ODg1\nNVWtpicUAADIjD9HAAfl5OQkXi4xKSlp//798uZxZBkZGSdPnjQ+njt3LtUNVH179uwRb/fz\n0UcfUd0AAABVAX+RAI5rxIgRzZo1Mz7+9NNP5Q3jyOLj443NdN27d3/llVfkjgOUTTw5rlWr\nVtIz1wAAAGTkVPYQANWUWq3esmWL8Vqh/AAro4SEBEEQXFxcli5dKncWoGwGg+Ef//jHu+++\nKwhC48aNpRcxBQAAkBHX4EB1ppTrL8B2OAYAAAAAB0GBAwAAAAAAKB5N6QAAAAAAQPEocAAA\nAAAAAMWjwAEAAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIH\nAAAAAABQPAocAAAAAAD8ydWrV1UqVXh4uNxBYAUKHAAAAAAA5dm6datKpVKpVFOmTJE7i6Uu\nXLigUqmGDh0qd5CSpaamjh8/vnnz5h4eHs7Ozr6+vv379//uu+8KCwvljmYRChwAAAAAAIW5\nffv26NGjXV1d5Q5SffzrX/9q1qzZ559/npub26tXrxEjRoSEhPzyyy8REREvvPCC3OksQoED\nAAAAAKAwb7zxhlqtnjBhgtxBqomZM2dOmzbN09Nz165d58+fX7t27VdffRUXF3f79u0VK1bk\n5ubKHdAiFDgAAAAAAEqyYsWKzZs3L1u2rG7duhXZTlJS0uDBg318fHQ6nbe3d8+ePdetW1fa\n4G3btqlUqtjYWJPltWvXbty4sXTJjh07evToIW42NDR01qxZgiB8/PHHwcHBgiCsXbtW9T+r\nV68WV0xMTBwwYICXl5ezs7OPj8+IESNSU1PFZ1NSUlQqVVRU1O+//z506FBPT0+1Wp2UlGRm\nRgtdvnw5NjbW2dl59+7dPXv2lD7l5OQUFRW1d+9ey7cmIye5AwAAAAAAYKnLly+/+eabo0aN\n6tOnz7x588q9nSVLlowbN06r1fbr169x48a3bt06fPjwokWLBg8eXJF4q1atGjlypJeXV//+\n/T09PW/fvn3q1Knly5e/8847YWFhWq120qRJ7du3HzdunHF8x44djQ+WLVs2ZsyYevXq9e3b\n19PT89KlS+vXr9+8efPevXufe+45cftXrlx57rnn9Hp9r169srOza9SoYWZGCzOvWLEiPz9/\n5MiRLVq0KHGAUk4FosABAAAAAFCGoqKikSNH1q5d+7PPPqvIdo4fPx4TE1O7du1ff/21adOm\n4vKrV69WMOHSpUs1Gk1ycrKPj4+48N69e4IgNGvWTKfTTZo0KTAwcMSIEdK1zpw5M27cuB49\nemzatMnFxUUM2bFjxzfeeOPYsWPiyPj4+JiYmHnz5mk0GuOScePGlTajhX799VdBEF566SWr\nX20VQ4EDAAAAAKAMc+bM+eWXX3bv3u3h4VGR7SxevLiwsDA2NlZa3RAEwc/Pr2IBBUEQNBqN\nk9OfvmvXqVPH/CqLFi3Kz8+fPHlydnZ2dna2caGPj0+3bt1++OGHtLS0wMBA40K9Xv/JJ5+I\n1Y1yzyh148YNQRD8/f0tX6Vq4hocirFx48bx48d37NjR1dXVwhsLWXjbJDPDDAbDpk2bunXr\n5ufn5+Li0qhRo0GDBiUmJlbolQAAAACA9U6cODF16tQxY8b06NGjgpsyXrqid+/elZHrT4YN\nG5aXl9esWbOYmJjvv//+5s2blqxl/JLVpUuX+n/2ww8/CP8rQBiFhITUrFmz4jNKGQwGQRBU\nKlWZI8PDw2NiYqzdvt3QwaEYM2fOTE5Odnd39/X1PXfuXJnjxdsmZWVllXtYTEzMokWLPDw8\nwsLC6tWrd+7cuY0bN27YsGHFihUjR44s/4sBAAAAAGsYDIbIyEgfHx+rLp9Zmvv37wuC4Ovr\nW/FNmYiJialTp87ChQsXL168cOFCQRA6dOgwa9Ys8VobJbpz544gCFu2bBHPT5GStplIz0Op\nyIxSPj4+qampf/zxR5mrvPjiixW8sKtN0cGhGLNnzz5//vz9+/fnzJljyXgLb5tkZtjFixcX\nLVqk1+tPnz79zTffzJs3b/v27Rs3bjQYDFOnTi3PawAAAACAciksLDx27NilS5fc3NzEW5AY\nv8jMmDFDpVK9/vrrlm+tdu3agiBcu3bN8lXUarUgCAUFBdKF+fn54hklouHDhx84cODevXs7\nd+4cM2bM4cOHe/fufeXKFTMbN55x4+Xl1b0k0vNxSuyzKMeMUqGhoYIg7Nq1q8yRf//7302u\nHlKlUOCwlHhLnitXrkREROj1ehcXl7Zt227fvt0+Abp27dq4cWNLuoYEi2+bZH7YpUuXBEFo\n166dtEYYFhbm5OSUkZFhZXwAAAAAKD+1Wv1aMe3btxcEISQk5LXXXuvUqZPlWzOuuGPHDstX\nMV7VwqRqcPToUZOSh8jd3f2ll15avHjx22+//fDhw/j4eEEQjNfOKCwsLDHPmjVrLM9j4YyW\nGDVqlFarXbNmzYkTJ0ocIPb7V/FTVChwWOfKlStt27Y9e/bs4MGD+/Tpc/To0bCwsH379smd\n60+kt02qyLAmTZpoNJrffvtNehLX9u3bCwoKqsH1dQEAAAAoiFqtXl7MkCFDBEHo06fP8uXL\nrTqJ/m9/+5tGo4mNjU1NTZUuN3MXlRYtWtSoUeOHH34Qvx89ePBg4sSJJsP27NljUvIw/jxs\nvHBGvXr1BEH4448/TNaKiYlxcnJasGCBSVUiKytr7dq15l+L+Rkt0bBhw9jY2Nzc3JdeemnP\nnj3SpwoLC1evXt29e3cLNyUvrsFhnfj4+ClTpvzrX/8ydlKsXr06MjJy1qxZ5ouF7733Xpkt\nDx06dHjttdcqntDC2yZZMszX1/fDDz+cMmVK06ZNjdfgOH/+/K5du/r06bNs2bKKRwUAAAAA\nWbRo0WLBggUxMTEhISH9+vULDg6+c+fO4cOH3dzcEhISSlzF1dV17Nixn332WUhISFhYWF5e\n3p49e5599ll3d3fpsGHDhjk5OXXp0iUwMFCj0Rw8eDAhIaFZs2Z9+/YVBMHd3f255547ePDg\nsGHDjL8oh4eHN2/evHnz5kuXLo2Oju7evXvPnj1btWpVWFiYmpoaHx/fsGFDYx2nNOZntNDk\nyZMLCgo+/PDDnj17BgcHt27dulatWrdu3UpMTLxz506XLl0s35SMKHBYJyAgYNq0aeJ5IsOH\nD4+JiTl06JD5tdasWZOWlmZ+TE5OTqUUOCy8bZKFw95///1GjRqNGTPmm2++MS556qmnhg8f\nrtfrKx4VAAAAAOQyduzYli1bzp49+6efftq8ebNer2/ZsqX5C3nMmjXL3d396+J/4twAACAA\nSURBVK+/XrlypY+Pz2uvvTZ16lRPT0/pmOnTp+/atevw4cPbtm3TarWBgYHTp08fN26cePXQ\n1atXT5gwYdeuXWvXrjUYDA0bNmzevLkgCK+++mrr1q3nzp37008/JSQk1KpVy8fHJzIy0nx1\nw5IZLfTBBx8MGjRo0aJFCQkJ27dvz8nJqV+/fmho6LBhwwYOHGjVpmRjgGWOHj0qCEL//v1N\nljdr1szZ2dmeSbZu3SoIwpAhQ4o/dfz4cZ1ON2bMGHGJsUHj/fffL8cwg8EQGxurUqnefffd\nS5cuZWdnJycn9+zZUxCEf/7zn5X6mgAAAAAAVV3//v3HjRsnd4pScQ0O6xivtSvl5ORU/Aox\nsjBYdtskC4cJgrB79+7Y2NihQ4d+8sknDRs2rFmzZuvWrTdv3uzv7//pp5+W2ZMCAAAAAIDd\ncIqKPUyaNKnMa3A8//zzb7zxRkVmMd42SRAENzc3k6dmzJgxY8aM1157bfny5RYOEwQhLi5O\nEIQXXnhBOsbFxaV9+/br169PSUkJDAysSGAAAAAAACoLBQ57+P7778vsdygoKKhggcN42yST\nhadOnUpKSgoJCXn22WeNV0K1cJggCHl5eYIg3Lp1y2Rwenq6IAg6na4iaQEAAAAAqEQUOOzh\n8uXLdpjFeNskk4Xz5s1LSkrq06fP9OnTrRomCEKnTp2WLFny+eefjxw50s/Pz7hw69at+/bt\nq1mzZocOHWz2UgAAAAAAVU5ubq6rq6vcKUpFgUMxNm7cuGXLFuF/d2Y+ePBgVFSUIAh6vX72\n7Nm2mHHIkCHLly9PSEho0qRJ3759GzRocObMGeNdkefMmWP+9isAAAAAgGojLy8vNTX1yJEj\nERERcmcpFQUOxThy5MjKlSvFf16+fNnYGBIYGGijAodGo9m5c+fChQvXrFkTFxf3+PHjunXr\nhoWFvfXWWy+++KItZgQAAAAAVEEHDhwICwvr06fP4MGD5c5SKpXBYJA7AwAAAAAAQIVwm1gA\nAAAAAKB4FDgAAAAAAIDiUeAAAAAAAACKR4EDAAAAAAAoHgUOAAAAAACgeBQ4AAAAAACA4lHg\nAAAAAABUZ1evXlWpVOHh4XIHMWf27Nkqlerw4cNyB1EwChwAAAAAAEDxnOQOAAAAAACADXl6\neu7bt69evXpyB4FtUeAAAAAAAFRnzs7OoaGhcqeAzXGKCgAAAACgOrPpNThSUlJUKlVUVNSV\nK1ciIiL0er2Li0vbtm23b99ui+mWLVsWHh7+xBNPuLi41K5du0uXLuvXrxefTUxMVKlUL7/8\ncvEVmzZtqtPp7t69Kx08YMAALy8vZ2dnHx+fESNGpKam2iJz+ezYsaNHjx4+Pj46nc7b2zs0\nNHTWrFnmV6HAAQAAAACAMHToUJVKtWTJknKse+XKlbZt2549e3bw4MF9+vQ5evRoWFjYvn37\nKj1kdHT0zZs3X3jhhbfeemvAgAGpqamDBw/+9NNPjc926NDhqaee2rZt2507d6RrHTp0KDU1\nNSwsrG7dusYly5YtCw0N3bdv31/+8peJEyd26tRp/fr1bdq0OXjwYKVnLodVq1b95S9/OXny\nZL9+/f7xj3+Eh4er1erly5ebX4tTVAAAAAAAqJD4+PgpU6b861//UqlUgiCsXr06MjJy1qxZ\nnTp1qtyJ0tLS/P39xX8+evSoS5cusbGxo0ePrlOnjiAII0eOnDx58nfffRcTEyMOW7lypfEp\n4z/PnDkzbty4Hj16bNq0ycXFxbjw+PHjHTt2fOONN44dO1a5mcth6dKlGo0mOTnZx8dHXHjv\n3j3za9HBAQAAAACAMHLkyM8++6xjx47lWDcgIGDatGnG6oYgCMOHD/fw8Dh06JA4IDw8XFpx\nKDdjdcNgMDx48CA9PT0zM/Ovf/3r48ePxW6RyMhItVptrGgY5eXlrVmzxtPTs3fv3sYlixYt\nys/Pnzx5cnZ2dsb/+Pj4dOvW7fjx42lpaZUY2KgcW9NoNE5Of+rJMFZwzGyNDg4AAAAAAITe\nvXuLJQBrtWrVSvptXKVS+fn5nT9/Xlzy4osviqeHVMTRo0djY2MTEhIePnwoXX7t2jXjAz8/\nv27duu3Zs+f06dNPP/20IAhbt269e/fuhAkTxISJiYmCIHTp0qXEKW7cuBEYGFg88IULF2bP\nnh0fH3/z5s2ioiJxeVZWlvFBRkbGzJkzt2zZcvXqVb1e37x587Fjx/bv378cL3/YsGEHDhxo\n1qzZkCFDunbtGhoa6uXlJT5b2tYocAAAAAAAUCG1a9c2WeLk5FRYWCj+8+9//3vFZzly5Eho\naGiNGjXGjh37zDPPeHh4aDSaH3/8cc6cObm5ueKwqKioPXv2rFy58pNPPhGKnZ8iCILxCh1b\ntmwRz0+Ratq0afHAcXFxQ4cOHThw4LJlywICAsReFdG1a9eef/55QRCmTJnSsmVLg8GQkJAQ\nHR3dp08fJycna19+TExMnTp1Fi5cuHjx4oULFwqC0KFDh1mzZhn7a0rbGgUOAAAAAABsKzw8\n3M/P7/PPP6/IRubOnfv48eMtW7Z0795dXJicnGwy7K9//au7u/vq1atnzpx59+7dHTt2PPPM\nM88884w4wMPDQxAELy+vtm3bWhL42rVrw4cPnzVr1pgxY0obP378+Nzc3NTUVLHW0759+9Gj\nR2s0GqFcL3/48OHDhw/PzMxMTEzcvHnzl19+2bt371OnTvn7+5e2Na7BAQAAAACAAly+fFkQ\nhPbt20sXxsfHmwxzcXEZPHjw9evXf/zxx//+978FBQXS9g1xC2vWrLFw3i+++KJVq1ZmqhtZ\nWVlbt24dP368SSeLXq8v3uthFXd395deemnx4sVvv/32w4cPi79YKQocAAAAAAAIO3bsmDdv\n3okTJ+QOUqpGjRoJgrBnzx5xybffflvid/6oqChBEFatWrVq1SonJ6fhw4dLn42JiXFyclqw\nYIHJullZWWvXri2+taSkpE6dOqWWQhCEixcvFhQUGC/5USn27NlTUFAgXZKRkSEIQs2aNc2s\nxSkqAAAAAAAIK1euXLt27eLFi1u0aCF3lpLFxMR8++23w4YNGzJkSGBgYEpKyvbt2wcNGrR+\n/XqTkR07dmzcuPH69evz8/PDwsI8PT2lzzZv3nzp0qXR0dHdu3fv2bNnq1atCgsLU1NT4+Pj\nGzZsOGTIEJOt5efnz5kzZ/78+SWmun//vsFgqMSXKQjCsGHDnJycunTpEhgYqNFoDh48mJCQ\n0KxZs759+5pZiwIHAAAAAAAK0K5dux9//PGDDz7YvHmzIAht2rTZvXv39evXixc4BEEYOXLk\n1KlThT9fXlT06quvtm7deu7cuT/99FNCQkKtWrV8fHwiIyOLVzcEQWjZsmWHDh1mzJhRWrBG\njRo5OTmdOnXqr3/9a/lfnsT06dN37dp1+PDhbdu2abXawMDA6dOnjxs3rsSroooocAAAAAAA\nqrOcnBxBEHQ6nflha9assfyyFKKQkJAS+xdSUlKs3ZQlunbt+ssvv5gsHDFiRPGRU6ZMmTJl\niplNhYSErFq1ypJJX3/99S5dukRFRQUHB5c4wM3NrW/fvgsWLIiJiZFehuPOnTt169Ytx2U4\nxowZY+aSH6XhGhwAAAAAgOrs3LlzgiD4+fnJHUSpmjdv/v7773fq1Onrr79++PBhiWPmz5/v\n7OwcEhKybNmyQ4cOJSUlffzxx82aNZPeK9fW6OAAAAAAAFRPJ0+e/Oabb7799ltBEAYMGCB3\nHAWbOHFicHDwRx99NGrUqFq1akmfysrKEgTB398/OTl55syZH3/88dWrV/V6fatWrb777jsn\nJ/uVHShwAAAAAACqp5SUlPnz5z/55JNz5sx5/vnnZUySm5vr6uoqYwBrGS/zIRUWFhYWFpaV\nlZWenl7iWTmenp7z5s2bN29e8acq9+UXz2ZEgQMAAAAAUD2NGDGixOtT2FNeXl5qauqRI0ci\nIiLMDHv++eenTZvm4+Njt2Dl4+rqalWpwsKXXylUlX43FwAAAAAAYPTTTz+FhYX16dNn5cqV\nZV7otPqx58unwAEAAAAAABSPu6gAAAAAAADFo8ABAAAAAAAUjwIHAAAAAABQPAocAAAAAABA\n8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAAAAAAAMWjwAEAAAAA\nABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIHAAAAAABQPAocAAAA\nAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAAAAAAAMWjwAEA\nAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIHAAAAAABQPAoc\nAAAAAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAAAAAAAMWj\nwAEAAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIHAAAAAABQ\nPAocAAAAAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAAAAAA\nAMWjwAEAAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIHAAAA\nAABQPAocAAAAAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEocAAA\nAAAAAMWjwAEAAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAUjwIH\nAAAAAABQPAocAAAAAABA8ShwAAAAAAAAxaPAAQAAAAAAFI8CBwAAAAAAUDwKHAAAAAAAQPEo\ncAAAAAAAAMWjwAEAAAAAABSPAgcAAAAAAFA8ChwAAAAAAEDxKHAAAAAAAADFo8ABAAAAAAAU\njwIHAAAAAABQPAocAAAAAABA8ShwAAAAAAAAxaPAAQAAAAAAFM9J7gAAUG0ZDIb79+/LncJq\nTk5Obm5ucqcAAAAArEOBAwBsJT8//4svvpA7hdX8/PyGDx8udwoAAADAOiqDwSB3BgCongwG\nw7179+ROIQiC4OLi4uLiIncKAAAAwIbo4AAAWykqKjpy5IjcKQRBEIKCgp544gm5UwAAAAA2\nRIEDAGzI1dVVlnldXFyeeeYZWaYGAAAAZMEpKgBgKzJeZFSj0bi7u8syNQAAACALOjgAwFYK\nCws3bNhgu+337dvXy8vLdtsHAAAAFIQODgAAAAAAoHh0cACAreTl5f373/+23fbfe+89nU5n\nu+0DAAAACkIHBwDYUH5+fvlWVKvVGo2mcsMAAAAA1RgdHABgKwUFBZs3by7fusHBwSEhIZWb\nBwAAAKjG6OAAAFspKio6e/asVat4e3vXrl3bRnkAAACAaowODgCwIWuvkaFWq22UBAAAAKje\nKHAAgK0UFRWlpKRYtUqHDh3c3d1tlAcAAACoxjhFBQAAAAAAKB4dHABgK/n5+Z999plVq7z8\n8suNGze2UR4AAACgGqODAwBsyNrPWJVKZaMkAAAAQPVGBwcA2EpeXp5VHRyenp6jRo2yXR4A\nAACgGqODAwBsxWAw3Lt3z8LBbm5uWq3WpnkAAACAaowODgCwlaKioiNHjlg4+Nlnn61Tp45N\n8wAAAADVGAUOALCh/Pz8Msf07NlTo9HYIQwAAABQjXGKCgAAAAAAUDw6OADAVgoKCr7++uvi\nyz09Pfv162f3OAAAAEB1RgcHAAAAAABQPDo4AMBW8vLyFi9eXHy5t7f34MGD7Z8HAAAAqMbo\n4AAAG3r06FHxhRqNRqfT2T8MAAAAUI3RwQEAtlJQUBAXF1d8eYMGDTp37mz/PAAAAEA1RgcH\nANhKUVHR2bNnBUFo1KgRLRsAAACATdHBAQA2ZKxrqFQquYMAAAAA1Zxa7gAAUG0VFRWdPHmy\nUaNGzs7OcmcBAAAAqjlOUQEAAAAAAIpHBwcAAAAAAFA8ChwAUPny8vKKiorkTgEAAAA4EAoc\nAFD5Ll26tHHjRrlTAAAAAA6Ea3AAQOXLzMzMy8vT6/VyBwEAAAAcBR0cAFD5srKy7t+/L3cK\nAAAAwIFQ4ACASpaTk3Ps2LEHDx7IHQQAAABwIJyiAgAAAAAAFM9J7gAAUE2sW7fu0qVLarXa\n19dXo9F4e3t37txZ7lAAAACAo6CDAwAAAAAAKB4dHABQTosXL05LSyvt2SZNmkRFRdkxDgAA\nAODQ6OAAgHJ69OiRmWc1Go1Op7NbGAAAAMDB0cEBAOUUFxdn5llvb+/Q0FC7hQEAAAAcHAUO\nALBaSkpKTk6Ov7+/mTF16tSxWx4AAAAAnKICAFbLzc01f36KIAhardbV1dU+eQAAAADQwQEA\n1rl69erPP/9c5jBfX9+uXbvaPg4AAAAAQaCDAwAAAAAAVAN0cACApR4/fvzRRx/Vrl3bksFB\nQUEDBgywdSQAAAAARnRwAIClCgoKLP/MVKvVGo3GpnkAAAAAiOjgAABLbdmyJSMjw8LBAQEB\nvXr1smkeAAAAACI6OADAIo8fP75z547l42vUqKHX622XBwAAAIAUHRwAYJH8/Pz09HTLx3t4\neFDgAAAAAOyGAgcAlOG///3v9evXmzdvLncQAAAAAKXiFBUAAAAAAKB4dHAAQBl27Nhx4cIF\na9dq2LBhWFiYLfIAAAAAKI4ODgAAAAAAoHh0cABAqR4+fDhjxozyrdukSZOoqKhKjQMAAACg\nVHRwAIAgCEJWVtbZs2elS1xcXAIDA1UqVfk2qNFodDpdZUQDAAAAUDY6OABAEAShoKDg3r17\n0iX37t27fv16uTeo1+tDQkIqnAsAAACARShwAHBcX375ZUZGhiAIer2+WbNmrq6ulbhxFxeX\nStwaAAAAAPM4RQWA48rMzCwsLLTRxrVabeVWTAAAAACYQQcHAAd18eLFxMRE223f19e3a9eu\ntts+AAAAACk6OAAAAAAAgOLRwQHAsSxatOiPP/5Qq9W2vsVJcHBwRESETacAAAAAIKKDA4AD\nKSwsLCoqss9carVao9HYZy4AAAAAdHAAcCDnzp3bt2+ffeYKCAjo1auXfeYCAAAAQAcHAIdw\n6tSpzMxMf39/u81Yo0YNvV5vt+kAAAAAB0cHBwCHUKdOnZycnPT0dLvN6OHhQYEDAAAAsBsK\nHACqv/T09KSkJDtP6uXl1bhxYztPCgAAADgsTlEBAAAAAACKRwcHgGpu+fLljx8/tv+8DRs2\nDAsLs/+8AAAAgGOigwMAAAAAACgeHRwAqqFbt26tWLFC3gxBQUEDBw6UNwMAAADgOOjgAFAN\nPXjwoLCwUN4MWq3Wzc1N3gwAAACA46CDA0A1dOrUqUePHsmbQa/Xh4SEyJsBAAAAcBwUOABU\nE9nZ2Z9//rlKpWrfvr2zs7Orq6u8eVxcXOQNAAAAADgUTlEBUE0UFRU9ePBA7hT/n1arlb3I\nAgAAADgOOjgAVAcGg2HdunUqlUruIP+ft7d3586d5U4BAAAAOAo6OAAAAAAAgOLRwQFA2YqK\niiZPnqzT6dRqtdxZ/iQ4ODgiIkLuFAAAAICjoIMDgIJlZWXpdDq5U5RMrVZrNBq5UwAAAACO\ngg4OAAp25cqVffv2yZ2iZAEBAb169ZI7BQAAAOAo6OAAoDz79u07c+ZMw4YN/f395c5SKldX\n16ocDwAAAKhmKHAAUJ6MjIzMzEy5U5ShZs2aXl5ecqcAAAAAHAWnqABQmP/+978uLi5ypyib\nl5cXBQ4AAADAbujgAKAkBoNBpVLJnQIAAABAlUMHBwAluXbt2t69e+VOYRE/P79u3brJnQIA\nAABwFHRwAFCM/Px8JycllWVpNgEAAADsRklfFQA4uJSUlPj4eLlTWCooKGjgwIFypwAAAAAc\nBR0cAKq0rKyspKQkQRD8/Pw8PT3ljmMFrVbr5uYmdwoAAADAUdDBAUAZrl69evXqVblTWEGv\n14eEhMidAgAAAHAUFDgAVDmLFy/OzMwUBMHd3T0kJMTV1VXuROWhiHvZAgAAANUGp6gAAAAA\nAADFo4MDQBVy9+7dNWvWODs7yx2kEgQEBPTs2VPuFAAAAICjoIMDAAAAAAAoHh0cAKqQa9eu\nLV++XO4UlSM4ODgiIkLuFAAAAICjoIMDQBWSn5+fn58vd4rKodFodDqd3CkAAAAAR0EHB4Aq\n5Ny5c6dPn5Y7ReXw9vYODQ2VOwUAAADgKChwAKhCatWq1bx5c7lTVA6F3t0WAAAAUCgKHACq\nEK1WW1RUJHeKyqHVauWOAAAAADgQChwAqpAzZ85kZmbKnaJyeHl5eXl5yZ0CAAAAcBRcZBQA\nAAAAACgeHRwAqooNGzb8/vvvcqeoNEFBQQMGDJA7BQAAAOAo6OAAUFVUv48jlUoldwQAAADA\nUdDBAaBKMBgMn376qdwpKlNQUNDAgQPlTgEAAAA4Cjo4AMgvJycnKytLrVbLHaQyabVaNzc3\nuVMAAAAAjoIODgDyU6lUaWlpcqeoZB4eHhQ4AAAAALuhwAFAfufPn79165bcKQAAAAAoGKeo\nAAAAAAAAxaODA4DMTp06lZiYKHeKyhcQENCzZ0+5UwAAAACOgg4OAAAAAACgeHRwAJDHzJkz\nMzMz5U5hQ02aNImKipI7BQAAAOAo6OAAII9Hjx7JHcG2NBqNTqeTOwUAAADgKOjgACCP7du3\nV+8Cq7e3d2hoqNwpAAAAAEdBBwcA+zl+/HhSUpKzs3ObNm00Go3ccWzL1dXV399f7hQAAACA\no6CDA4D9BAYGurq6yp3CTrRardwRAAAAAAdCgQOAnZw7d+63336TO4X9+Pr6enl5yZ0CAAAA\ncBScogIAAAAAABSPDg4AdjJv3ry8vDy5U9hPUFDQgAED5E4BAAAAOAo6OADYg2N+1KhUKrkj\nAAAAAI6CDg4A9vDzzz+fO3dO7hR2FRAQ0KtXL7lTAAAAAI6CDg4AtpWTk/P77797eHjIHcTe\natSoodfr5U4BAAAAOAo6OADYlkajycnJycnJkTuIvXl4eFDgAAAAAOyGAgcAG8rIyDh06BCX\nogAAAABga5yiAgAAAAAAFI8ODgCVLyUlZdeuXRqNpn79+nJnkY2fn1+3bt3kTgEAAAA4Cjo4\nAAAAAACA4tHBAaCS7dq1KyEhQe4U8mvSpElUVJTcKQAAAABHQQcHgEqWl5dXUFAgdwr5aTQa\nnU4ndwoAAADAUdDBAaCSHTt27PLly3KnkJ+3t3doaKjcKQAAAABHQYEDQKXZuHGji4tLnTp1\n/P395c4ivzp16sgdAQAAAHAgnKICoNJkZWXl5+fLnaKq0Gq1rq6ucqcAAAAAHAUdHAAqwd69\ne8+dO+fu7i53kCrE19e3a9eucqcAAAAAHAUdHAAAAAAAQPHo4ABQUUlJSceOHZM7RZUTEBDQ\nu3dvuVMAAAAAjoIODgDld/369Vq1atWsWVPuIFWRWq3WaDRypwAAAAAcBR0cAMrvt99+S09P\nlztFFRUQENCrVy+5UwAAAACOgg4OAOWUnZ197949uVNUXTVq1NDr9XKnAAAAABwFHRwAyqmg\noID2DTM8PDwocAAAAAB2Q4EDgHWuX7/+zTff1K1bNzg4WKVSyR2n6nJ2dpY7AgAAAOBAOEUF\nAAAAAAAoHh0cACx18uTJNWvWNGjQQO4gytCwYcOwsDC5UwAAAACOgg4OAAAAAACgeHRwALDI\nN998c+rUKblTKEmTJk2ioqLkTgEAAAA4Cjo4AFgkMzOzoKBA7hRKotVq3dzc5E4BAAAAOAo6\nOABYJDU1NTMzU+4USqLX60NCQuROAQAAADgKChwAynb48OGioiJXV1e5gyiJi4uL3BEAAAAA\nB8IpKgDKlpmZWVhYKHcKhdFqtZSEAAAAALuhgwNAGX799de0tDS5UyiPr69v165d5U4BAAAA\nOAo6OAAAAAAAgOLRwQHAnFu3bi1evFjuFIoUHBwcEREhdwoAAADAUdDBAcAcg8HA3WHLR61W\nazQauVMAAAAAjoIODgDmXL9+PS4uTu4UihQQENCrVy+5UwAAAACOgg4OAOZkZmZmZmbKnUKR\natSoodfr5U4BAAAAOAo6OACYU1hYmJeXJ3cKRXJy4gMWAAAAsB/+/gZQsgULFqjVam9vb7mD\nKJWXl5eXl5fcKQAAAABHwSkqAAAAAABA8ejgAFCCbdu2Xbp0Se4UytawYcOwsDC5UwAAAACO\ngg4OAH9ivCks9zetFCqVSu4IAAAAgKOggwPAn6Snp69evVruFNVBUFDQwIED5U4BAAAAOAo6\nOAD8SX5+/sOHD+VOUR1otVo3Nze5UwAAAACOgg4OAH+SmZl59OhRuVNUB3q9PiQkRO4UAAAA\ngKOgwAHg/+Tl5a1evfrpp592dXWVO0t14OLiIncEAAAAwIFwigqA/2MwGO7fvy93iupDq9VS\nKgIAAADshg4OAP/n6tWru3btkjtF9REQENCzZ0+5UwAAAACOgg4OAAAAAACgeHRwABAEQdix\nY8ehQ4fkTlGtBAcHR0REyJ0CAAAAcBR0cAAQBEEoLCwsKiqSO0W1olarNRqN3CkAAAAAR0EH\nBwBBEITk5OS0tDS5U1Qr3t7eoaGhcqcAAAAAHAUdHICjy87OPn78eO3ateUOUt24urr6+/vL\nnQIAAABwFHRwAI5Op9M1aNBA7hTVkFarlTsCAAAA4EAocACO7tSpU7///rvcKaohLy8vLy8v\nuVMAAAAAjoJTVAAAAAAAgOLRwQE4tEePHn3zzTdyp6ieAgICevfuLXcKAAAAwFHQwQE4ND4B\nbEqlUskdAQAAAHAUdHAADm3dunWXL1+WO0X1FBQUNHDgQLlTAAAAAI6CDg7AoT148KCwsFDu\nFNWTVqt1c3OTOwUAAADgKOjgABzaqVOnHj16JHeK6kmv14eEhMidAgAAoS4prAAAIABJREFU\nAHAUFDgAxxUXF6dWq+VOUW3RGgMAAADYE6eoAAAAAAAAxaODA3BQRUVFX331ldwpqrOAgICe\nPXvKnQIAAABwFHRwAAAAAAAAxaODA3BQmzdvTktLkztFdfbEE0/069dP7hQAAABwFLNnz37n\nnXd+++23Nm3ayJ1FHhQ4AEeUk5PD2RO2ptFo5I4AAABQ3WzcuDEhIeHIkSPHjh3Lzs4eMmTI\nmjVr7BkgKysrLi5u8+bNKSkpaWlpWq22RYsWUVFRr776qsn1+62Kunfv3gULFiQmJt6/f9/T\n07N169YTJkzo2rWrzV+PWb///vsHH3ywd+/e+/fv+/v7DxkyZPLkyTVr1jS/liUvvLCwcMaM\nGYmJiadPn759+3aNGjUCAwPDw8PHjx9ft27dcgemwAE4orS0tOPHj8udoprz9vYODQ2VOwUA\nAEC1MnPmzOTkZHd3d19f33Pnztk/wPLlyydMmODs7Ny6desWLVqkp6cfOHBg//79W7du3bRp\nk7TGYXnUf/7znx9//LFOp2vfvn2DBg1u3769f//+Fi1ayFvgOHnyZKdOnR48eNC3b99GjRrt\n27dvxowZe/fujY+Pd3FxMbOiJS88Pz9/2rRpXl5eTz75ZLt27bKyspKTk2NjY7/44osDBw4E\nBgaWLzMFDsCxPHz4MCkpyc/Pr3nz5nJnqeZcXV3ljgAAAFDdzJ4928/PLygoKC4uLiwszP4B\n/P39Fy1aFBER4eHhYVxy+vTpF154YcuWLWvXrh02bJi1UVesWPHxxx936NBh/fr1vr7/j737\nj7K6LvA//p65XGYGZxixUe44MALj2GyCzZKbVrN+2SwcO4vtBufo0q+xH+fU2fPtlGYdtR/a\nSTtrWrsny0oN7eSS7oamIX1LEcKUH4JiiSiSWGr8/jkIDDOX7x+okSHCcO9938/9PB5/4TDz\nuU9K5sib1+dzW/Z/MJ/Pb9mypai/kDf08Y9/fOvWrTNmzOjp6dmf9KEPfWjmzJnXXXfdl770\npUN84eH8wmtqatasWXPgQUZfX9/HPvax22677aqrrvrhD384uGYHHJAudXV1bW1tsStSIZvN\nxk4AAKg00e/amDp16ms+8pa3vOVzn/vcpZdeOn/+/AMPOA4nta+v77LLLjvmmGPuvPPOkSNH\nvvrx6urqN73pTQVKHoxly5YtXry4s7Nz/+nG/qRvfvObt99++w9+8IPLL7+8qqrq9b72cH7h\nVVVVr5lpDB069JOf/ORtt922atWqQWc74IAUWbt27aOPPvqamwMpkuOPPz6Xy8WuAAAghBAu\nuOCC22+//YYbbvjUpz5V8IvvX3PU1NQc6RfOnTt37dq1H/zgBxsbG2+//fbf//73dXV1Z5xx\nxrvf/e5DnCCUwNy5c0MI55577oEfbGlpOe200x577LGnn376zW9+c8Ff9Gc/+1kI4a1vfeug\nr+CAA1Ikl8u95psUAABwNPbt2/fjH/84hDCIW2aWLFkSQnjTm9502mmnHbhceMc73vGaTUeJ\nPfXUUyGEvz3FOOWUUwp7wPHZz3529+7d27Zte+SRR5555pnTTjvt8ssvH/TVHHBAWqxcufLu\nu++OXZEibW1tf7tgBAAgio9+9KNnnnnmu971roJf+corr1y4cOEHPvCB97znPUf6tevXrw8h\nfPe73z355JMfeOCB008//dlnn7344ot//etfX3DBBQ888EDBaw/Ttm3bwivLlAMde+yxIYSt\nW7cW6oVuuummnTt37v9xd3f3Lbfccvzxxw/6ag44IBX6+vre/OY3X3LJJbFDAAAggnPPPbcY\nW+brr7/+yiuvnDhx4owZMwbx5QMDAyGEqqqqu+66q6OjI4QwYcKEO++885RTTpk3b94jjzxy\n+umnF7j4r+Xz+c985jMHfuSiiy4aN27c633+vn379gcXKqC3t3ffvn3r1q2bP3/+F7/4xc7O\nztmzZ0+cOHFwV3PAAalw//33e1/YEmtra5s2bVrsCgAAiuW66677/Oc//7a3ve3Xv/718OHD\nB3GFESNGhBA6Ojr2n27sd8wxx7z3ve+99dZbS3PA8d3vfvfAj1xwwQXjxo3bv93Yv+M40Ost\nO45GVVVVLpc7//zzx48fP378+AsvvHD58uWDu5QDDqhwDz/88PDhwydMmDBhwoTYLelSW1sb\nOwEAgGK54oorrrzyyne84x1z5swZ9B/49z/JYv99Hwfa/5Hdu3cfZeQbGjJkyP5RxkHD9j+J\n40D7HxRyyimnFCPm1FNPbW5ufvzxx7ds2bL/6OdIOeCACjdy5MgtW7aU4Jsjr9HY2NjU1BS7\nAgCAwrvooou+/e1vT5o06Z577qmvrx/0dc4+++yqqqqVK1fu3bs3m82++vHf/e53IYSxY8cW\noHVQ3v3ud4cQfvnLX1599dWvfvDFF19cvnx5S0tLkQ44duzYsf+hJEOGDPKkwgEHVLjVq1f3\n9/fHrgAAgJjmzJnz1FNPnX322Ue5a87n85/61KduvPHGc845584776yrqzuaq7W0tPzrv/7r\nrFmzrrrqqiuuuGL/B3/xi1/MnTu3qalpEE8tLZSJEye+/e1vX7x48Y9//OOPfOQjIYR8Pv+F\nL3xh/y//wGdw3HLLLVu3bp0+ffoJJ5xw+NdfuHBhXV3dge8Iu2nTpk984hMDAwNnnXVWQ0PD\n4LIdcECFe+973xs7AQAACmPWrFn73xnw+eefDyEsWrSop6cnhNDU1HTttdce4gtvvfXW22+/\n/YYbbjjKA47rrrvuxhtvrK6uPu644z796U8f+FMTJky4+OKLjzT1O9/5zrJly6688spf/epX\nEydOfO655+69995sNnvTTTcdc8wxR5N6lG6++eaurq4LL7xw1qxZY8eOXbBgwdKlS88444wD\nf40hhK9//eurV6/u6up69YDjcH7h8+bNu/TSS8eNGzd27NgRI0asXbt26dKlu3btam5u/sEP\nfjDoZgccULH+93//d8OGDQdO3Sil1tbWyZMnx64AAKgoy5Ytu/XWW1/9xzVr1qxZsyaEcNJJ\nJx36gKNQNm3aFELI5/MzZ858zU+dc845B/7h/zBTTzzxxCVLlnzta1+7++67H3nkkeHDh7//\n/e+/7LLLiv140Tc0fvz4pUuXfvnLX77vvvvmzJkzatSoyy677LLLLnvD0crh/MLf//73b9y4\ncd68ecuXL9+yZUt9ff2ECRPe9773feYznxnc0zf2qzroA0UAAACABLn22msvueSSJUuWRD8c\nicWCAyrTli1b/uM//iN2Rap1dHTsH+MBAAAl4IADKtPw4cO/8pWvxK5ItUwmEzsBAABSxAEH\nVKYtW7Y88MADsStSrbm5uaurK3YFAACkhQMOqEw7d+4cP3587IpUO5p3RAcAgCP1zne+86tf\n/eqJJ54YOyQaDxmFyrRjx47+/v7YFamWzWadcQAAQMlYcEAF2rx585w5c2JXpF1LS8ukSZNi\nVwAAQFpYcAAAAACJZ8EBFWX58uU//elPjz322KqqqtgtadfW1jZ16tTYFQAAkBYWHFBR8vn8\nwMBA7ApCCKG6uto7xQIAQMlYcEBFeeKJJx5++OHYFYQQQmtra3d3d+wKAABICwsOqChr1671\n5illora2tqmpKXYFAACkhQUHVJSNGzfu2bMndgUhhNDY2OiAAwAASsYBByTePffc89vf/raq\nqurtb397bW1t7BwAAIAI3KICAAAAJJ4FByTeL3/5y1WrVsWu4LXGjBkzZcqU2BUAAJAWFhwA\nAABA4llwQLItWLBg9uzZsSs4iI6Ojp6entgVAACQFhYckGx9fX3eF7Y8ZTKZmpqa2BUAAJAW\nFhyQbGvWrPnjH/8Yu4KDaGpq6uzsjF0BAABp4YADki2fz9fX18eu4CDq6upiJwAAQIq4RQWS\nrbe3d+/evbErOIhsNuvsCQAASsaCA5JtwYIFmzdvjl3BQbS0tEyaNCl2BQAApIUFBwAAAJB4\nFhyQbNdff/3AwEDsCg5i7Nix5513XuwKAABICwsOSLB9+/Z5j9iyVV1dnclkYlcAAEBaWHBA\ngm3atGnWrFmxKzi41tbW7u7u2BUAAJAWFhyQYDt37tyyZUvsCg6utra2qakpdgUAAKSFBQck\n2O7du9etWxe7goNrbGx0wAEAACXjgAOS6plnnnn88cdjV/C6crncySefHLsCAADSwi0qAAAA\nQOJZcEBSLV68eNGiRbEreF1jxoyZMmVK7AoAAEgLCw4AAAAg8Sw4IJEWLlx41113xa7gUDo6\nOnp6emJXAABAWlhwQCLt2rVr165dsSs4lGw229DQELsCAADSwoIDEmndunXPPPNM7AoOpamp\nqbOzM3YFAACkhQMOSJ4//vGPa9eura+vjx3CodTV1cVOAACAFHGLCiRPX1/fzp07Y1fwBrLZ\nrEMoAAAoGQsOSJh8Pj9nzpzYFbyxkSNHnnnmmbErAAAgLSw4AAAAgMSz4IAkefTRR++6667q\n6urYIbyx9vb26dOnx64AAIC0sOCAJOnv7/d7Nimqq6szmUzsCgAASAsLDkiSu+++e+PGjbEr\nOCytra3d3d2xKwAAIC0sOCAxVq1a1d/fH7uCw1VfXz969OjYFQAAkBYWHJAYw4YN27NnT+wK\nDlc2m42dAAAAKeKAA5Jh3759ixYtil3BEcjlcrlcLnYFAACkhVtUAAAAgMSz4IAEuPnmm3fs\n2OEtOZJlzJgxU6ZMiV0BAABpYcEB5S6fz1dVVcWuYDD8HwcAACVjwQHl7p577lm5cmXsCo5Y\nW1vbtGnTYlcAAEBaWHBAWdu4cWN1dXXsCgYjm802NDTErgAAgLSw4ICy9uKLL65fvz52BYPR\n1NTU2dkZuwIAANLCAQeUr+3bt7/00kv19fWxQxiMurq62AkAAJAiblEBAAAAEs+CA8rXxo0b\n77rrrtgVDFJra+vkyZNjVwAAQFpYcAAAAACJZ8EB5etb3/rWjh07YlcwSO3t7dOnT49dAQAA\naWHBAWVqYGBgz549sSsYvEwmU1NTE7sCAADSwoIDytT69esffPDB2BUMXnNzc1dXV+wKAABI\nCwsOKEcPPPBALpeLXcFRqa+vHz16dOwKAABICwsOKEft7e19fX2xKzgq2Ww2dgIAAKSIAw4o\nO5s2bVq8eHHsCo5WLpczwwEAgJJxiwoAAACQeBYcUF7uuuuup59+OnYFBdDW1jZ16tTYFQAA\nkBYWHFBe/JasJFVVVbETAAAgLSw4oLx873vf6+3tjV1BAbS1tU2bNi12BQAApIUFB8TX29u7\ncOHCTCZz6qmnDhni2LFCZLPZhoaG2BUAAJAW/igF8WUymREjRoQQ/vSnP8VuoWAaGxsdcAAA\nQMk44ICY9uzZ861vfauzszN2CAAAQLK5RQUAAABIPAsOiGnFihUPPfRQ7AqKorW1dfLkybEr\nAAAgLSw4AAAAgMSz4IBoNm7cePPNN8euoFi8TSwAAJSSBQfE8fTTT48aNSp2BUWUyWRqampi\nVwAAQFo44IA4/ud//id2AsXV3Nzc1dUVuwIAANLCAQdE8Nxzz7300kuxKyiu+vr60aNHx64A\nAIC08AwOiGDo0KEDAwOxKyiubDYbOwEAAFLEAQeUVG9v73//938fc8wxsUMoupaWllwuF7sC\nAADSwi0qAAAAQOJZcEDpPPvss3fccUdVVVXsEEqhra1t6tSpsSsAACAtLDigFPbu3dvX1zds\n2LDYIZSUwywAACgZCw4oheeff/6OO+6IXUFJtbW1TZs2LXYFAACkhQUHFN1vfvObcePGxa6g\n1Gpra5uammJXAABAWlhwQNEdc8wx69ati11BqTU2NjrgAACAknHAAcUyMDBwxRVXnHHGGZlM\nJnYLAABAhXOLCgAAAJB4FhxQFNdcc01LS8uQIX6LpVdzc/NZZ50VuwIAANLCggMAAABIPH+9\nDIX3/PPPX3/99bEriKyjo6Onpyd2BQAApIUFBxRef39/X19f7Aoiy2QyNTU1sSsAACAtLDig\n8NasWfPoo4/GriCy5ubmrq6u2BUAAJAWDjig8BoaGkaPHh27gshGjBgROwEAAFLELSpQeHv2\n7HnppZdiVxBZNputr6+PXQEAAGlhwQEFtmjRomeeeSZ2BfG1tLRMmjQpdgUAAKSFBQcAAACQ\neBYcUBh33HHHsmXLstlsQ0ND7BbKQltb29SpU2NXAABAWlhwQGEMDAzk8/nYFZSR6urqTCYT\nuwIAANLCggMKY/bs2WvXro1dQRlpbW3t7u6OXQEAAGlhwQGF8eKLL1pwcKDa2tqmpqbYFQAA\nkBYWHFAYGzZs6O/vj11BGWlsbHTAAQAAJeOAAwpg3bp1W7dujV1BeRk6dGjsBAAASBG3qAAA\nAACJZ8EBR2vFihX3339/7ArKzpgxY6ZMmRK7AgAA0sKCAwAAAEg8Cw44Wj//+c8ffvjh2BWU\nnY6Ojp6entgVAACQFhYccFS2bt3q3WE5qGw229DQELsCAADSwoIDjsrmzZv/8Ic/xK6gHDU1\nNXV2dsauAACAtHDAAUfs5z//+cqVK0MI7e3tJ554Yn19fewiylFdXV3sBAAASBG3qMAR27lz\nZ19fX+wKyl02m3X4BQAAJWPBAUds4cKFa9eujV1BuWtpaZk0aVLsCgAASAsLDgAAACDxLDjg\nCDz88MP33nvvkCF+4/DG2tvbp0+fHrsCAADSwoIDjkA+nx8YGIhdQTJUV1dnMpnYFQAAkBb+\nIhqOwPLly5csWRK7gmRobW3t7u6OXQEAAGlhwQGH5Xe/+93u3bubm5tjh5AYtbW1TU1NsSsA\nACAtLDjgsLS0tGzdutW7w3L4PKsFAABKyX9/wxvbtm3bvHnzYleQMLlcLpfLxa4AAIC0cIsK\nAAAAkHgWHPC6Zs6cuXz58urq6lwuV1VVFTuHhBkzZsyUKVNiVwAAQFpYcAAAAACJZ8EBBzFr\n1qxVq1bFriDZ2trapk2bFrsCAADSwoIDDmL79u39/f2xK0i2bDbb0NAQuwIAANLCggMO4qmn\nntq2bVvsCpKtqamps7MzdgUAAKSFAw44iHw+X19fH7uCZKurq4udAAAAKeIWFTiILVu2xE4g\n8bLZrGMyAAAoGQsO+Ctf+9rXcrlcdXV17BASr7W1dfLkybErAAAgLSw4AAAAgMSz4CC9rrnm\nms2bNx/4kaFDh2YymVg9VJj29vbp06fHrgAAgLSw4CClBgYG8vl87AoqWXV1tfMyAAAoGQsO\nUmrFihUPP/xw7AoqWWtra3d3d+wKAABICwsOUmrt2rXeKoWiqq+vHz16dOwKAABICwsOUmrI\nkCE1NTWxK6hk2Ww2dgIAAKSIAw7SaMOGDQsWLIhdQYXL5XK5XC52BQAApIVbVAAAAIDEs+Ag\njTZs2HDvvffGrqDCjRo16uyzz45dAQAAaWHBQeXr7e3du3fvq/84dOjQYcOGRewhPaqqqmIn\nAABAWlhwUPl+9atfrVq1KnYFqdPW1jZt2rTYFQAAkBYWHFSyzZs3v/TSS/YaRJHNZhsaGmJX\nAABAWlhwUOFWrlwZO4GUampq6uzsjF0BAABp4YCDCrRnz56vfvWrJ5xwwt/93d/FbiG9BgYG\nYicAAECKuEUFAAAASDwLDirQhg0bfv7zn8euIO1aW1snT54cuwIAANLCggMAAABIPAsOKsTl\nl1/+6iMPamtrq6qq4vZAe3v79OnTY1cAAEBaWHCQeOvXrz/mmGOcaFBuMplMTU1N7AoAAEgL\nCw4Sb+PGjfPnz49dAa/V3Nzc1dUVuwIAANLCgoNke+yxx/wlOeWpvr5+9OjRsSsAACAtLDhI\ntpEjR+7atSt2BRxENpuNnQAAACnigIMEy+fzjz/+eOwKOLjjjz8+l8vFrgAAgLRwiwoAAACQ\neBYcJNUTTzzxi1/8wpunULba2tqmTp0auwIAANLCgoOk8q8u5c8BHAAAlIwFB0n1yCOPzJ07\nN3YFvK62trZp06bFrgAAgLSw4CCptm/fvn379tgV8Lpqa2ubmppiVwAAQFpYcJBUO3fuXLdu\nXewKeF2NjY0OOAAAoGQccJAk9957729+85sQwrhx40aPHh07BwAAgHLhFhUAAAAg8Sw4SIxr\nrrnmuOOOi10Bh6u1tXXy5MmxKwAAIC0sOAAAAIDEs+CgrL360A1InI6Ojp6entgVAACQFhYc\nlK99+/bt2rUrdgUMUiaTqampiV0BAABpYcFB+erv7589e3bsChik5ubmrq6u2BUAAJAWFhyU\nozVr1jz44IN///d/X11dHbsFBqm+vt6bGQMAQMk44KAc7d27t7e3N3YFHJVsNltfXx+7AgAA\n0sItKpSFO++8c9GiRft/3NjY2NraGrcHjl5LS8ukSZNiVwAAQFpYcAAAAACJZ8FBTJs3b77m\nmmtqamqOOeaY2C1QYG1tbVOnTo1dAQAAaWHBQUz79u3zbyAVzFNyAQCgZCw4iGnJkiWPPfZY\n7AooitbW1u7u7tgVAACQFhYcRLBixYonnnji5JNPPv7442O3QLHU1tY2NTXFrgAAgLRwwEEE\nf/7zn1988cXYFVBcjY2NJ598cuwKAABIC7eoUFJXX331m970Ju8CCwAAQGFZcAAAAACJZ8FB\nES1evHjWrFmv/uPIkSOHDPGvHGkxZsyYKVOmxK4AAIC0sOAAAAAAEs9fp1MUl112WT6fj10B\nMXV0dPT09MSuAACAtLDgoPD6+vr6+/tjV0BkmUympqYmdgUAAKSFBQeF9/TTTz/55JOxKyCy\n5ubmrq6u2BUAAJAWDjgojE2bNt10000hhDe/+c25XG706NGxiyCyESNGxE4AAIAUcYsKhTEw\nMLB9+/bYFVBGstlsfX197AoAAEgLCw4Gb/369d/61rdCCDU1Ne3t7VVVVbGLoIy0tLRMmjQp\ndgUAAKSFBQcAAACQeBYcDNJTTz01d+7c6urq2CFQplpbW88999zYFQAAkBYWHByxF154oaam\nprGxMXYIlLXq6upMJhO7AgAA0sKCgyO2ePHiDRs2xK6Actfa2trd3R27AgAA0sKCg0O58847\n+/v7D/zIxIkTa2pqYvVAgtTW1jY1NcWuAACAtLDg4FDGjBmTz+cP/MjWrVtjxUCyNDY2OuAA\nAICSseAAAAAAEs+CA6BY+vrCV74SO6KEpk0Lp58eOwIAgLSy4AAAAAASz4IDoFj6+sI3vhE7\n4siNGxc+/OHYEQAAcIQsOACKaO/e2AWHZ8iQUFUVOwIAAI6CBQdAsezdG2bMiB1xeN773jB2\nbOwIAAA4ChYcAMWyb1944YXYEa8YOTJks7EjAACgaCw4AIpl376wbl3siFe86U0OOAAAqGQO\nOACKJZ8Pzz0XO+IV7e2hri52BAAAFI1bVAAAAIDEs+AAKJa9e8P3vx874hX//u+hujp2BAAA\nFI0FBwAAAJB4FhwAxdLXF7797Wiv/oEPhPb2aK8OAAAlZsEBUCz79oUtW0r0WvX1YejQEr0W\nAACUIQsOgGLJ58OyZSV6rdNOCyecUKLXAgCAMuSAA6CI6uuL/hInnBDGjSv6qwAAQJlziwpA\nsezbF7ZuLfqr1NSEYcOK/ioAAFDmLDgAimVgIPzsZ0V/lXe+M7zlLUV/FQAAKHMWHAAAAEDi\nWXAAFEtfX/jGN4p4/fb2MH16Ea8PAAAJYsEBUER79xbryplMqK4u1sUBACBxLDgAimXv3jBj\nRrEufsYZ4a1vLdbFAQAgcSw4AIolnw9PPVWYSzU0hFGjCnMpAACoSBYcAEVUU1OY6wzx3RoA\nAA7JfzIDFEs+Hx577Kiu8J73hOHDC1QDAAAVzS0qAAAAQOJZcAAUy9694b//e/Bf/i//Ehob\nC1cDAAAVzYIDoIgG/S22qqqgHQAAUOksOACKpa8vfPvbg/zanp4wcmRBawAAoKJZcAAUy759\nYcuWw/rMIUM8TBQAAI6KBQdAseTzYdmyw/rMxsbwD/9Q5BoAAKhoDjgAiiibfePPaW8PJ55Y\n/BQAAKhoblEBAAAAEs+CA6BY+vvDLbcc5OPt7eH//J9SxwAAQGWz4AAAAAASz4IDoFj6+sI3\nvnGQj59zTjjzzJLXAABARbPgACiil176y49rakImEy8FAAAqmgUHQLH094fZs//yj11dobk5\nXg0AAFQ0Cw6AYsnnw1NPhRDCSSeFYcNi1wAAQEWz4AAoopqaEEKoro7dAQAAlc5/dAMUSz4f\nVq0K48aF2trYKQAAUOncogIAAAAkngUHAAAAkHgOOAAAAIDEc8ABAAAAJJ4DDoDCW7QoPPJI\n7AgAAEgTbxMLUHgnnBAGBmJHAABAmlhwABTYrl1h5cqwbVvsDgAASBNvEwsAAAAknltUAApm\n8+Zw3XXhpJNCdXVobQ2TJ8cOAgCA1LDgAAAAABLPggOgYJ56KsyY8fKPOzpCT0/MGAAASBUL\nDoDC2LUrHPgNNZMJNTXxagAAIGUsOAAKY/v28Jvf/OUfm5tDV1e8GgAASBkLDoBByufDTTe9\n/OPjjw8dHX/1s/X1YfTo0kcBAEBKOeAAGKR9+8Kzz77uzw4bFnK5EtYAAEC6uUUFYJDuuy+s\nX/+6P9vS4oADAABKx4IDAAAASDwLDoDB2LcvfPObh/qEtrYwdWqpagAAIPUsOACOzEsvhf7+\nUF8fqqre4DPf8BMAAIBCseAAODKPPBJWrnzjT2ttDd3dxa8BAABCCBYcAIfjiSfCihUhhPB3\nfxeOPfawvqS2NjQ1FTUKAAD4CwccAG/sxRfDn/98ZF/S2BhOPrk4NQAAwN9wiwrA61q8OMya\nFcaNC6NHx04BAAAOyYIDAAAASDwLDoDXWrcufPvbYdiwMGLE4C8yZkyYMqVwTQAAwCFZcAAA\nAACJZ8EB8Fpf/WrYs+doL9LREXp6ChADAAAcDgsOgL/YujUMGRKqqwtwqUwm1NQU4DoAAMDh\nsOAA+IsdO8LChYW5VHNz6OoqzKUAAIA35IAD4C82bizYO8IezQOE1kYcAAAgAElEQVRKAQCA\nI+UWFYC/2LYt5POFuVQ2G+rrC3MpAADgDVlwALxs5cqwdGnBrtbSEiZNKtjVAACAQ7PgAAAA\nABLPggMghBC2bw8zZxbygq2t4dxzC3lBAADgECw4AEIIIZ8PAwOFvGB1dchkCnlBAADgECw4\nAEIIYeXK8OCDhbxga2vo7i7kBQEAgEOw4AAIIYTt28P27YW8YG1taGoq5AUBAIBDsOAACCGE\nnTvDunWFvGBjowMOAAAoHQccQNrNnBkGBsLxxxf4slVVBb4gAABwCG5RAQAAABLPggNIu9tu\nC5s3F/6yY8aEKVMKf1kAAOCgLDgAAACAxLPgANLojjvCsmXFfYmOjtDTU9yXAAAAXmXBAaTO\ntm1hYKDor5LNhoaGor8KAACwnwUHkDobN4Znny36qzQ1hc7Oor8KAACwnwMOIEWeeSYsXx5a\nWkJ9fdFfq66u6C8BAAC8yi0qQIrs3Rt6e0v0WtlsKY5RAACA/Sw4gBSZPz/s3Fmi1xo5Mpx5\nZoleCwAAsOAAAAAAEs+CA0iL738/rFtXupdrbw/Tp5fu5QAAIOUsOIBU6O8PJf5uV10dMpmS\nviIAAKSZBQeQCgsWhFWrSvqKra2hu7ukrwgAAGlmwQFUvh07woYNpX7RYcNCLlfqFwUAgNSq\njh0AUHRuFQEAgIrnFhWg8s2dG/r6Sv2iuZwFBwAAlI5bVAAAAIDEs+AAKtz//E9YuzbC644Z\nE6ZMifC6AACQThYcQGXauTP09YXhw0N1vGcNVVVFe2kAAEgbCw6gMt13X1i5MmZAW1uYNi1m\nAAAApIoFB1CB+vpCb2/khmw2NDREbgAAgPSw4AAq0O7dYdmyyA1NTaGzM3IDAACkhwMOoAL1\n9YX6+sgNdXWRAwAAIFXcogJUoL6+sHNn5IZsNv4hCwAApIcFB1Bpbrst7NoVOyKE1tYweXLs\nCAAASA0LDgAAACDxLDiAyvHkk+G220I2G7sjhBBCe3uYPj12BAAApIYFB1A58vkwMBA74hXV\n1SGTiR0BAACpYcEBVI4nnwwrVsSOeEVzc+jqih0BAACp4YADqBy1tWH8+NgRr/AWKgAAUEoO\nOIDKMXRoqKqKHfGKMnkUCAAApIQDDqByPPlk6O2NHfGKXC7kcrEjAAAgNTxkFAAAAEg8Cw6g\nQuzYEW64IXbEAdrawtSpsSMAACA1LDiAClGG38zK54EgAABQ8Sw4gArx0EPhwQdjRxygrS1M\nmxY7AgAAUsOCA6gE69aV3buWZLOhoSF2BAAApIYFB1AJNm8OL7wQO+KvNTWFzs7YEQAAkBoO\nOIDEmzkzHHts7Ii/MTAQuwAAANLELSoAAABA4llwAMk2MBBmzIgdcTCtrWHy5NgRAACQGhYc\nAAAAQOJZcABJNW9eWLQoDB0aqqtjpxzM2LHhvPNiRwAAQGpYcABJ1dcX+vtjR7y+TCbU1MSO\nAACA1LDgAJLqiSfCM8/Ejnh9zc2hqyt2BAAApIYDDiCpcrlQWxs74vXV18cuAACANHHAASTV\nkCFlfQ9INhu7AAAA0sQBB5BUDz0Uu+CQRo4MuVzsCAAASA0PGQUAAAASz4IDSKQXXww/+Uns\niENqawtTp8aOAACA1LDgABIpEd+6qqpiFwAAQGpYcACJNH9+WLQodsQhtbWFadNiRwAAQGpY\ncACJtH596OuLHXFItbWhqSl2BAAApIYFB5BImzeHnTtjRxxSY6MDDgAAKB0HHEDy/OlP4dln\nY0cAAADlxC0qAAAAQOJZcADJ89vfhuefjx3xRpqbw1lnxY4AAIDUsOAAAAAAEs+CA0iYyy8P\nAwOxIw5DR0fo6YkdAQAAqWHBASTGhg1h2LBQVRW74/BkMqGmJnYEAACkhgUHkBibN4d582JH\nHLbm5tDVFTsCAABSw4IDSID77w9794ZcLnbHkRg+PIwbFzsCAABSwwEHkAA7d4a+vtgRRyib\nDfX1sSMAACA13KIClLsbbgjDh8eOOHItLWHSpNgRAACQGhYcAAAAQOJZcABlaubMsHx5GD48\nDEnmN6q2tjB1auwIAABIDQsOoEz194dEf3+qrg6ZTOwIAABIjWT+xSiQAv/7v2H79tgRR6G1\nNXR3x44AAIDUsOAAytHWraG3N3bE0amtDU1NsSMAACA1LDiAcvTSS2HdutgRR6ex0QEHAACU\njgMOoBw9+WTo64sdAQAAJIdbVAAAAIDEs+AAys7ChWHJktgRR23MmDBlSuwIAABIDQsOAAAA\nIPEsOIAy8vzz4frrY0cUSEdH6OmJHQEAAKlhwQGUi+3bQyYTqqpidxRIJhNqamJHAABAalhw\nAOWitzesWBE7onCamkJnZ+wIAABIDQccQFm4447Q2hrq62N3FE5dXewCAABIE7eoAGVh69ZQ\nYd+NstmKOq8BAIAyZ8EBxLdjR5g9O3ZEobW0hEmTYkcAAEBqWHAAAAAAiWfBAcT005+Gxx6r\nzMdVtLeH6dNjRwAAQGpYcAAx9fdX2qM3XlVdHTKZ2BEAAJAaFhxATHffHTZujB1RHK2tobs7\ndgQAAKSGBQcQx+rVIYRQUxO7o2hqa0NTU+wIAABIDQsOII7hw8Mf/xg7opgaGx1wAABA6Tjg\nAOJYtSqsXRs7ophyuXDyybEjAAAgNdyiAgAAACSeBQcQwY03ht27Y0cU2ZgxYcqU2BEAAJAa\nFhwAAABA4llwAKXW2xu++93YEcXX1hamTYsdAQAAqWHBAZRaX1/o7Y0dUXzZbGhoiB0BAACp\nYcEBlNrWreHxx2NHFF9TU+jsjB0BAACp4YADKKnly8OuXaG+PnZH8dXVxS4AAIA0cYsKUFK7\nd4ddu2JHlEQ2m4pzHAAAKBMWHEDp3HJLinYNzc3hrLNiRwAAQGpYcAAAAACJZ8EBlMju3eE/\n/iN2RAm1t4fp02NHAABAalhwAEW3eXN46aUwcmTsjtKqrg6ZTOwIAABIDQsOoOhWrw6PPho7\nouRaW0N3d+wIAABIDQccQNGNHp3G9xNJ4S8ZAAAicsABFN2QIaGmJnZEyWWzsQsAACBNHHAA\nxXXffWH79tgRMeRyIZeLHQEAAKnhIaMAAABA4llwAMU1c2bo64sdEcOoUeHss2NHAABAalhw\nAMWV5u8xVVWxCwAAIDUsOIAi+uMfw8yZsSMiaWsL06bFjgAAgNSw4ACKaOfOsGdP7IhIstnQ\n0BA7AgAAUsOCAyiiNWvCn/8cOyKSpqbQ2Rk7AgAAUsMBB1AsDz4YhgwJ9fWxOyKpq4tdAAAA\naeIWFQAAACDxLDiAYvnJT8Lu3bEj4mltDZMnx44AAIDUsOAAAAAAEs+CAyi8H/0oPPtsGJLu\nbzDt7WH69NgRAACQGhYcQOHt2hV8a8lkQk1N7AgAAEiNdP8FK1Ac8+aF3t7YEbE1N4eurtgR\nAACQGg44gMIbNy7k87EjYkvt++MCAEAUDjiAwqupccARstnYBQAAkCYOOIACe/LJ8OSTsSPK\nQC4XcrnYEQAAkBoeMgoAAAAkngUHUEgvvBBuuy12RHloawtTp8aOAACA1LDgAArMN5VXVVXF\nLgAAgNSw4AAK6emnw513xo4oD21tYdq02BEAAJAaFhxAAbz4YlixIhx/fBg9OnZK2chmQ0ND\n7AgAAEgNCw6gAIYODSNGhP7+8OyzsVPKRmOjAw4AACgdBxzA0brxxjBqVOwIAAAg3dyiAgAA\nACSeBQdwVLZvD3fcETuiLLW2hsmTY0cAAEBqWHAAAAAAiWfBARyVlSvDLbfEjihLHR2hpyd2\nBAAApIYFBzB4e/aEgYHYEeUqkwk1NbEjAAAgNSw4gMF76aVw332xI8pVc3Po6oodAQAAqWHB\nAQzGY4+FVavCqaeGqqrYKeWqvj6MHh07AgAAUsOCAxiMMWPC8OGxI8pbNhu7AAAA0sQBB3DE\ndu4Ms2fHjih7LS0hl4sdAQAAqeEWFQAAACDxLDiAI/aLX4QVK2JHlL22tjB1auwIAABIDQsO\n4Ij5tnGYPIEVAABKxoIDOGIzZoT+/tgRZa+1NXR3x44AAIDUsOAAjkxvb9i6NXZEEtTWhqam\n2BEAAJAaFhzAkdm9O6xbFzsiCRobHXAAAEDpOOAAjszmzWH9+tgRAAAAf80tKgAAAEDiWXAA\nh2vBgvD446G+PnZHQowaFc4+O3YEAACkhgUHAAAAkHgWHMBhufbasHFj7IhE6egIPT2xIwAA\nIDUsOIBD+f3vw9694eSTQyYTOyVpMplQUxM7AgAAUsOCAziUP/0p9PaGZ56J3ZFAzc2hqyt2\nBAAApIYDDuAvduwI3/veyz+urg7veEcYMSKMGBG1KbH87wYAAKXkFhXgL/L5sG1b7IhKkc16\nxxkAACgdCw7gL+bNC3/+c+yIStHSEiZNih0BAACpYcEBAAAAJJ4FB/Cyvr4wY0bsiArS2hrO\nPTd2BAAApIYFB/CygYGQz8eOqCDV1d5bFwAASseCA3jZokXh97+PHVFBWltDd3fsCAAASA0L\nDuBlW7eG3t7YERWktjY0NcWOAACA1LDgAF62fXvYsCF2RAVpbHTAAQAApeOAA3iZBUdhDR0a\nuwAAANLELSoAAABA4llwAC+bMcOCo5DGjAlTpsSOAACA1LDgAAAAABLPggN42dVXh+3bY0dU\nkI6O0NMTOwIAAFLDggMIIYQdO8LevbEjKks2GxoaYkcAAEBqWHAAIYTw3HNh7drYEZWlqSl0\ndsaOAACA1HDAAYQQQlVVqK+PHVFZ6upiFwAAQJq4RQUIIYQdO0J/f+yIypLNOjMCAIDSseCA\ntHvuufDLX/qjeOG1tIRJk2JHAABAalhwAAAAAIlnwQFp95vfhAceiB1Ridrbw/TpsSMAACA1\nLDgg7QYGQj4fO6ISVVeHTCZ2BAAApIYFB6Td3Lnh2WdjR1Si1tbQ3R07AgAAUsOCA9Jo7dqw\nYEEIIbzlLaGxMXZNhaqtDU1NsSMAACA1HHBAGu3aFf7859gRlW7YsJDLxY4AAIDUcIsKpM6e\nPWHOnNgRKZDLOeAAAIDSseAAAAAAEs+CA1LnJz8JW7bEjkiBMWPClCmxIwAAIDUsOCB1/KYv\nmaqq2AUAAJAaFhyQLs89F37609gR6dDWFqZNix0BAACpYcEBafHCC2H9+nDSSbE7UiObDQ0N\nsSMAACA1LDggRTZtCps2xY5Ijaam0NkZOwIAAFLDAQekxZAhob4+dkSa1NXFLgAAgDRxiwqk\nxZ494aWXYkekSTbrRAkAAErHggPS4qGHwurVsSPSpLU1TJ4cOwIAAFLDggMAAABIPAsOqHwb\nN4b//M+QzcbuSJn29jB9euwIAABIDQsOqHDr1oXjjosdkUrV1SGTiR0BAACpYcEBFW7RorBn\nT+yIVGpuDl1dsSMAACA1HHBAJfvTn0J7e+yItPIWKgAAUEoOOKCS1dSEvXtjR6SVh54AAEAp\nOeCASvbb3waP2Ykllwu5XOwIAABIDQ8ZBQAAABLPggMq1t694Uc/ih2RYq2t4dxzY0cAAEBq\nWHBAxfKbO7qqqtgFAACQGhYcULEefDA89FDsiBRrawvTpsWOAACA1LDggIq1bVsYGIgdkWLZ\nbGhoiB0BAACpYcEBFWvlyrBjR+yIFGtqCp2dsSMAACA1HHBAZVqzJmzeHDsi3cxnAACglNyi\nAgAAAIl37bXXXnLJJUuWLDn99NNjt8RhwQGV6f77w7PPxo5It9bWMHly7AgAgArS29s7e/bs\nu+6667HHHnvuueey2eyECRN6eno+9rGPVVdXl6ZhYGDgqquuevjhh1esWLFhw4ba2tqTTjrp\nX/7lX/7v//2/xx133JGmHubVYlm9evVXvvKV+++/f+vWraNHjz7//PMvu+yyYcOGHfqrZs2a\n9cADDyxbtmz58uU7d+48//zzf/rTn/7tp3V0dDz11FOv+eDIkSPXrl076GALDgAAABLgP//z\nPz/3uc8NHTp04sSJo0ePXrdu3UMPPdTf33/eeefdeeedpTnj2L17d11dXS6XO+WUU0444YTe\n3t6lS5du2LDhxBNPfOihh0466aQjSj3Mqx2mwi44fv/73//jP/7jtm3b/vmf/3ncuHELFixY\ntmzZmWeeOXfu3Lq6ukN84emnn7506dLhw4fncrmnn376EAccq1at+vCHP3zgBxsbG//rv/5r\n0M0WHFCBNm4Mt90WOyL1xo4N550XOwIAoIKMHj36e9/73vTp0xsbG/d/ZMWKFf/0T/909913\n33777f/2b/9Wgoaampo1a9YcePTQ19f3sY997Lbbbrvqqqt++MMfHlHqYV4tio9//ONbt26d\nMWNGT09PCCGfz3/oQx+aOXPmdddd96UvfekQX3jttdeOGjWqra1t9uzZU6ZMOcRnZrPZW265\npYDNDjigAo0YET75ydgRqZfJxC4AAKgsU6dOfc1H3vKWt3zuc5+79NJL58+fX5oDjqqqqtcM\nK4YOHfrJT37ytttuW7Vq1ZGmHubVSm/ZsmWLFy/u7Ozcf7oRQqiurv7mN795++23/+AHP7j8\n8surqqpe72snTZpUmsi/5YADKtCGDWHBgtgRqdfcHLq6YkcAAFS6/ROJmpqaQ3/aBRdccPvt\nt99www2f+tSnCt7ws5/9LITw1re+9dCfdpiph3m1opo7d24I4dxzzz3wgy0tLaeddtpjjz32\n9NNPv/nNbz76V8nn81dfffXq1avr6upOO+20adOmHeWTRxxwQAUaNiyMHx87IvXq62MXAABU\nun379v34xz8OIRz6Vohi+OxnP7t79+5t27Y98sgjzzzzzGmnnXb55Zcf4vMPnXqkVyu2/Y//\n/NtTjFNOOaWABxx79+498Jd58cUX//CHPzyaJY4DDqhA1dXhjc6FKbpsNnYBAEClu/LKKxcu\nXPiBD3zgPe95z6E/86Mf/eiZZ575rne9q1AvfdNNN+3cuXP/j7u7u2+55Zbjjz9+0KlHerVi\n27ZtW3hlcnKgY489NoSwdevWo3+Jj370o//wD/8wfvz4xsbGP/zhD9///ve/973vffjDHx41\natQ//uM/Du6aDjig0jzzTFi9OnYEIRx/fMjlYkcAAFSu66+//sorr5w4ceKMGTPe8JPPPffc\n19xwcZR6e3v37du3bt26+fPnf/GLX+zs7Jw9e/bEiRMHl3pEVyugfD7/mc985sCPXHTRRePG\njXu9z9//NqyHeADH4bv00ktf/fGpp576ne98p7Gx8aqrrvrGN77hgAN42cknh5NPjh0BAADF\ndN11133+859/29ve9utf/3r48OFRGqqqqnK53Pnnnz9+/Pjx48dfeOGFy5cv/9tPO8zUw7xa\nYeXz+e9+97sHfuSCCy4YN27c/u3G/h3HgV5v2VEQH//4x6+66qrFixcP+goOOKCi7NkTjuJ9\noymktrbwNw/PBgCgAK644oorr7zyHe94x5w5c4r0h+0jcuqppzY3Nz/++ONbtmwZMWLEgT81\niNRDXK3ghgwZsn+U8Rr7H7Gx/0kcB9r/3i6nnHJKMWL23/+yZ8+eQV/BAQdUjj17wtCh4ZJL\nYncAAEDRXHTRRd/+9rcnTZp0zz331JfHc9137Nixfv36EMKQIX/1R+zBpb7e1Urp3e9+dwjh\nl7/85dVXX/3qB1988cXly5e3tLQU6YBj/vz5IYS2trZBX8EBB1SOJUvCb38bO4JXtLWFadNi\nRwAAVJB8Pv+pT33qxhtvPOecc+688866urrD/9o5c+Y89dRTZ5999oQJE46mYeHChXV1dQe+\nh+umTZs+8YlPDAwMnHXWWQ0NDUeUephXK72JEye+/e1vX7x48Y9//OOPfOQjIYR8Pv+FL3xh\n/6/rwGdw3HLLLVu3bp0+ffoJJ5xw+NdfsmRJTU3Naaed9upHHnnkkX//938PIXz4wx8edLYD\nDki8P/0pLFwYTj45jBkTxoyJXcMramtjFwAAVJbrrrvuxhtvrK6uPu644z796U8f+FMTJky4\n+OKLD/G1t9566+23337DDTcc5QHHvHnzLr300nHjxo0dO3bEiBFr165dunTprl27mpubf/CD\nHxxp6mFeLYqbb765q6vrwgsvnDVr1tixYxcsWLB06dIzzjjjNf87f/3rX1+9enVXV9erBxyz\nZs26++67QwjPP/98CGHRokU9PT0hhKampmuvvXb/58yfP/+SSy5pa2sbO3bs8OHDn3322cce\ne2zfvn3nnXfeax56ekQccEDi1deHceNCPh/WrYudwgEaG0NTU+wIAIAKsmnTphBCPp+fOXPm\na37qnHPOOfQBR6G8//3v37hx47x585YvX75ly5b6+voJEya8733v+8xnPnPg8zIOM/UwrxbF\n+PHjly5d+uUvf/m+++6bM2fOqFGjLrvssssuu+wNhzPLli279dZbX/3HNWvWrFmzJoRw0kkn\nvXrAcfbZZ3/yk59cuHDhsmXLtm/ffuyxx77nPe/5yEc+8sEPfvBo3qKl6qAPFAGS4oUXwuOP\nx47gYE44IbztbbEjAABIjWuvvfaSSy5ZsmTJ6aefHrslDgsOSLaWltDSEjsCAAAgNgcckFTP\nPRduvjm0tsbu4HW0tobJk2NHAABAajjggKQ66aTwta/FjgAAACgPDjggqW69NTz5ZOwIXl9H\nR+jpiR0BAEBqvPOd7/zqV7964oknxg6JxkNGIal27Qp++5azTCbU1MSOAACA1LDggKS6//6w\na1fsCF5fc3Po6oodAQAAqeGAAxKpry+0tcWO4JDq62MXAABAmjjggEQaMiTkcrEjOKRsNnYB\nAACkiQMOSKTnnw8LFsSO4JBaWsKkSbEjAAAgNTxkFAAAAEg8Cw5IjN27wxVXhBBCJhMaGyPH\n8Iba2sLUqbEjAAAgNSw4IDHy+TAwEDuCw1ZdHTKZ2BEAAJAaFhyQGEuXhkcfjR3BYWttDd3d\nsSMAACA1LDggGdavD319sSM4ErW1oakpdgQAAKSGBQckQz4f1q2LHcGRaGx0wAEAAKXjgAPK\n3TXXhIaGMGZM7A4AAIAy5hYVAAAAIPEsOKCs7d4dbrwxdgSDMmZMmDIldgQAAKSGBQcAAACQ\neBYcUNa+//2wZk3sCAaloyP09MSOAACA1LDggPKVz4fdu2NHMFiZTKipiR0BAACpYcEB5auv\nLzz0UOwIBqupKXR2xo4AAIDUcMAB5WvVqlBfHzuCwaqri10AAABp4hYVKF/bt4eBgdgRDFY2\n63wKAABKx4IDytTGjeH//b/YERyFlpYwaVLsCAAASA0LDgAAACDxLDigHP3oR2HHjtgRHJ2x\nY8N558WOAACA1LDggLKTz3v0RiWorg6ZTOwIAABIDQsOKDurV4cHHogdwVFrbQ3d3bEjAOD/\ns3fvwXbX9b3/32vvrOy9w84NNmSHJJtcCEQbIAIqc4iWesGg4mWCBydWxVKVOtOL00Odas0/\nhTJT8cSZzpHoaBM9VcrpgWI1yDDVonYAscQglBqtipdwCLeEEAKstfdavz/wVwWBZCfru97r\nu76Px1+QrGSefyQryWe/Pt8FUBkWHNBbfv7zqNWyI+iE4eEYG8uOAACAyrDggN4yY0bcd192\nBJ0wd64DDgAA6B4HHNBb/t//i5/+NDuCThgfjxNPzI4AAIDKcEUFAAAAKD0LDugVV14ZjUbM\nnp3dQYcsXRrnn58dAQAAlWHBAQAAAJSeBQfku/nmuPHG7Ag6bdWquOii7AgAAKgMCw7It29f\nTE5mR9Bp9boLRwAA0D0WHJDv+9+PffuyI+i0sbFYsyY7AgAAKsMBB+QbGorR0ewIOm1kJLsA\nAACqxBUVyLdvX0xNZUfQafW6cysAAOgeCw5ItmdPfPOb2REUYMGCOOus7AgAAKgMCw4AAACg\n9Cw4INknPxkPPpgdQQFWrowNG7IjAACgMiw4INnkZPhd2JcGBmJwMDsCAAAqw4IDkv3DP8Rj\nj2VHUICJiVi3LjsCAAAqw4IDMt1zT9Rq2REUY3Q0lizJjgAAgMqw4IBMIyPup/Stej27AAAA\nqsQBB6T50pdiaio7gsKMj8f4eHYEAABUhisqAAAAQOlZcEC3PfJI/PVfx3HHucLQ55YujfPP\nz44AAIDKsOCABH7bVYQnyAIAQNdYcEC33X13bNuWHUHxVqyICy7IjgAAgMqw4ICu2rcvJiez\nI+iKej1mz86OAACAyrDggK566qm4887sCLpibCzWrMmOAACAynDAAV312GMxOpodQVeMjGQX\nAABAlbiiAgAAAJSeBQd01U03xc9+lh1BV0xMxLnnZkcAAEBlWHAAAAAApWfBAd3z4x/H//7f\n2RF0y8qVsWFDdgQAAFSGBQd0ye7dPjS0WgYHY2goOwIAACrDggO65Mc/jl/8IjuCLlq4MNau\nzY4AAIDKcMABxZqais9+Nk47LebNi3nzsmvoIp8HDAAA3eSKChSr3Y6f/CQ7ggyzZsX4eHYE\nAABUhgUHFOv++2PHjuwIMoyPO+AAAIDuseAAAAAASs+CAwr0z/8c27dnR5BkxYpYvz47AgAA\nKsOCAwrkt1fF1WrZBQAAUBkWHFCgL385/uM/siNIsmJFXHBBdgQAAFSGBQcUZf/+aDSyI8hT\nr8fs2dkRAABQGRYcUJR9++Kee7IjyDM2FmvWZEcAAEBlOOCAovzkJ9FsZkeQZ2oquwAAAKrE\nFRUAAACg9Cw4oCif/axPUam0iYk499zsCAAAqAwLDgAAAKD0LDigEJ/8ZDz2WHYEqXxMLAAA\ndJMFB3Te1FQ89VR2BNkGB2NoKDsCAAAqw4IDOm/v3vj617MjyLZwYaxdmx0BAACVYcEBHfbo\no7FrV9Rq2R1kGx2NJUuyIwAAoDIsOKDDRkZieDg7gh5Qr2cXAABAlTjggA575JG49dbsCHrA\nokUxPp4dAQAAleGKCgAAAFB6FhzQST/9aVxzTXYEvWHFili/PjsCAAAqw4IDOsnvJ36dZ80C\nAEDXWHBAJ117bfzoR9kR9IYVK+KCC7IjAACgMiw4oGPuv2AdnZwAACAASURBVD8mJ7Mj6BnD\nwzE2lh0BAACVYcEBHfPkk/Hww9kR9Iy5cx1wAABA9zjggI556KF48MHsCAAAgEpyRQUAAAAo\nPQsO6IBHHol//McYHc3uoJcsXBivfGV2BAAAVIYFBwAAAFB6FhzQAVdfHXfemR1Bj1m1Ki66\nKDsCAAAqw4IDjlS7HU88kR1B7xkcjKGh7AgAAKgMCw44Us1mbNuWHUHvWbgw1q7NjgAAgMpw\nwAGHr9GIT386zjwzlizJTqH3zJ+fXQAAAFXiigocvnY79u7NjqBX1es+WAcAALrHggMO3/79\nccMN2RH0qkWL4pxzsiMAAKAyLDgAAACA0rPggMP0P/9nNJtRq2V30KtWrIj167MjAACgMiw4\n4DBNTobfPbyAgYEYHMyOAACAyrDggMN0zTXx+OPZEfSwiYlYty47AgAAKsOCAw7H3r2xf392\nBL1teDjGxrIjAACgMiw44HA88UTs3p0dQW+bO9cBBwAAdI8DDpieL3859u2LxYuzO+h5M2dm\nFwAAQJW4ogIAAACUngUHTM/nPx+PPpodQRksXRrnn58dAQAAlWHBAQAAAJSeBQdMQ6MRGzdm\nR1ASq1bFRRdlRwAAQGVYcMChuu++GB7OjqA86vWYPTs7AgAAKsOCAw7V5GRs354dQXmMjcWa\nNdkRAABQGQ444OAefji+9rWYmIjR0ewUymNkJLsAAACqxBUVOLipqdi3LzuCsqnXnYgBAED3\nWHDA87r//vjEJ2JoKFaujFotu4ayWbQozjknOwIAACrDggMAAAAoPQsOeF5XXx0/+EF2BKW1\ncmVs2JAdAQAAlWHBAc9rcjL8/uCwDQzE4GB2BAAAVIYFBzyvr3wlHnggO4LSmpiIdeuyIwAA\noDIsOOC53XdftFrZEZTZ8HCMjWVHAABAZVhwwHN7/HEfDcsRmTvXAQcAAHSPAw54DvfdF3fd\nlR1ByY2Px4knZkcAAEBluKICAAAAlJ4FBzyHO+6IW27JjqDkli6N88/PjgAAgMqw4AAAAABK\nz4IDnsOXvhTf/352BCW3YkVccEF2BAAAVIYFBzzDrl3RasVRR2V3UH71esyenR0BAACVYcEB\nz7ZzZ3YBfWFsLNasyY4AAIDKcMABv3LTTTFnToyOZnfQF0ZGsgsAAKBKXFGBX3nssZiczI6g\nX9TrDssAAKB7LDggIuL66+MXv4jh4ewO+sjERJx7bnYEAABUhgUHAAAAUHoWHBAR8aUvxY4d\n2RH0l5UrY8OG7AgAAKgMCw6IdtujN+i8gYEYHMyOAACAyrDggGg2Y+vW7Aj6zsRErFuXHQEA\nAJVhwQFx//2xZ092BH1ndDSWLMmOAACAyrDggJgxI4aGsiPoO/V6dgEAAFSJAw6qa/fu2LQp\nli2LY4/NTqEfjY/H+Hh2BAAAVIYrKgAAAEDpWXBQXf/yL/Gzn2VH0L8WL45Xvzo7AgAAKsOC\ng4pqtaJWy46g3/k1BgAAXWPBQUXdfXd89avZEfS1FSvigguyIwAAoDIsOKii/fuj0ciOoN/V\n6zF7dnYEAABUhgUHVdRoxPbt2RH0u7GxWLMmOwIAACrDAQeVc+ONYbdEF0xNZRcAAECVuKIC\nAAAAlJ4FB5Vz/fXx0EPZEVTAxESce252BAAAVIYFBwAAAFB6FhxUy9RUXHZZdgTVsHJlbNiQ\nHQEAAJVhwUG1PPlktFrZEVTD4GAMDWVHAABAZVhwUC233RYPPpgdQTUsXBhr12ZHAABAZVhw\nUBXf/nbU6zEykt1BZYyOxpIl2REAAFAZFhxUxcqVsXdvdgRVUq9nFwAAQJU44KD//fjH8a//\nGgsWZHdQMcceG+Pj2REAAFAZrqgAAAAApWfBQZ/bvDn27cuOoJJWrIj167MjAACgMiw46HN+\ngZOoVssuAACAyrDgoM99/OMxNZUdQSWtWBEXXJAdAQAAlWHBQT976KF48snsCKpqeDjGxrIj\nAACgMiw46GePPuqjYUkzd64DDgAA6B4HHPShT3wi7r8/Tjst5s3LTgEAAKArXFEBAAAASs+C\ngz70D/8Qjz6aHUHlTUzEuedmRwAAQGVYcAAAAAClZ8FBv7nmmvjud7MjIGLVqrjoouwIAACo\nDAsO+s2TT0arlR0BEYODMTSUHQEAAJVhwUG/+da3fDQsPWHhwli7NjsCAAAqw4KDfnDjjfGz\nn0VEvPSlMTycXQMRETE6GkuWZEcAAEBlOOCgH9x/fxw4kB0BzzRrVoyPZ0cAAEBluKJCP/ju\nd+ORR7Ij4JkWLXLAAQAA3WPBAQAAAJSeBQeld9NNsWNHdgT8hhUrYv367AgAAKgMCw7KrdmM\nGY7p6FW1WnYBAABUhn8aUm7XXhv79mVHwHOZmIh167IjAACgMiw4KJ9HH42bboqBgTjjDPMN\netfwcIyNZUcAAEBl+Nch5TNzZixfHhHx8MPZKfD85s51wAEAAN3jgIPyefjheOCB7AgAAAB6\niSsqAAAAQOlZcFAyjUZ86lPZEXAIli6N88/PjgAAgMqw4AAAAABKz4KDMvnKV+Jf/zU7Ag7N\nqlVx0UXZEQAAUBkWHJTJU0/F1FR2BByawcEYGsqOAACAyrDgoEy+/e3YvTs7Ag7NwoWxdm12\nBAAAVIYDDkqj3Y6ZM2PJkuwOODTz52cXAABAlbiiQpns2ZNdAIesXo/R0ewIAACoDAsOyuFb\n34p7742BgewOOGSLFsU552RHAABAZVhwAAAAAKVnwUE5fOMb8f3vZ0fAdExMxHnnZUcAAEBl\nWHBQAu12TE5mR8A0DQzE4GB2BAAAVIYFByXQbMbWrdkRME0TE7FuXXYEAABUhgUHva7RiPvv\n93hRymd4OMbGsiMAAKAyLDjodQMD8eCD2REwfXPnOuAAAIDuccBBT/u7v4tZs7Ij4LCMj8eJ\nJ2ZHAABAZbiiAgAAAJSeBQc9at++2Lw5RkayO+BwLV0a55+fHQEAAJVhwQEAAACUngUHvejG\nG+Pmm7Mj4MisWhUXXZQdAQAAlWHBQS/asyf8wqTs6vWYPTs7AgAAKsOCg170ve9Fs5kdAUdm\nbCzWrMmOAACAynDAQc85cCCGhmJoKLsDjoxH5AIAQDe5okLPmZqKffuyI+CI1esxOpodAQAA\nlWHBQU+4+ur42c8iImbOjKVLY2AgOwiO2IIFcdZZ2REAAFAZFhwAAABA6VlwkO/BB+OTn8yO\ngE5buTI2bMiOAACAyrDgIN/UVLRa2RHQaQMDMTiYHQEAAJVhwUG+22+Pu+7KjoBOm5iIdeuy\nIwAAoDIccJBv8eKYNy87AjrNR6gAAEA3OeAgX70eQ0PZEdBp9Xp2AQAAVIkDDvLddVc89lh2\nBHTa+HiMj2dHAABAZXjIKAAAAFB6Fhwk27UrrrsuOwIKsHRpnH9+dgQAAFSGBQdpJifjqadi\n1qzsDihMrZZdAAAAlWHBQZoHH4zPfz47AgqzYkVccEF2BAAAVIYFBzmefDIOHMiOgCLV6zF7\ndnYEAABUhgUHOVqt2L49OwKKNDYWa9ZkRwAAQGU44CDHPffE6Gh2BBRpZCS7AAAAqsQVFXI8\n+mi0WtkRUKR63SkeAAB0jwUH3bNrV/zN30RELF0aM/zSo99NTMS552ZHAABAZVhwAAAAAKXn\ny+h0z+23x1e/mh0B3bJyZWzYkB0BAACVYcFBl7RaMTWVHQFdNDAQg4PZEQAAUBkWHHTJk0/G\ntm3ZEdBFCxfG2rXZEQAAUBkWHHTD9dfHSSdFrZbdAV00OhpLlmRHAABAZVhw0A0veYn7KVRO\nvZ5dAAAAVeKAg8Ldc098//vZEdB14+MxPp4dAQAAleGKCgAAAFB6FhwU6+qr4+c/z46ADCtW\nxPr12REAAFAZFhwUqNXyYFEqza9/AADoGgsOCvSVr8R//Ed2BCRZsSIuuCA7AgAAKsOCg6I8\n9lg0m9kRkKdej9mzsyMAAKAyLDgoyt69sXNndgTkGRuLNWuyIwAAoDIccFCU738/JiezIyDP\n1FR2AQAAVIkrKgAAAEDpWXDQeY88Ev/n/8QMv7iotomJOPfc7AgAAKgMCw4AAACg9HyRnU76\n+tfjO9+JmTNjYCA7BbItWxZvelN2BAAAVIYFBx0zORmNRnYE9IzBwRgayo4AAIDKsOCgYx5/\nPG66KTsCesbChbF2bXYEAABUhgUHnXHzzXHccVGrZXdAzxgdjSVLsiMAAKAyLDjojJNPjiee\nyI6AXlKvZxcAAECVOOCgA265JX7yk+wI6DGLFsX4eHYEAABUhisqAAAAQOlZcHD4rrsubr89\nhodj1qzsFOg9K1bE+vXZEQAAUBkWHEzbgQPx1FMxOhoznI/BC/LYXQAA6Br/QmXabr01/u3f\nsiOg561YERdckB0BAACVYcHB9PziF9kFUBLDwzE2lh0BAACVYcHB9ExNxUMPZUdAGcyd64AD\nAAC6xwEH0zA5Gffckx0BAAAAv8EVFQAAAKD0LDg4uD174tOfjlotFi+OwcHsGiiJhQvjla/M\njgAAgMqw4AAAAABKz4KD5/ZP/xS33JIdAWW2alVcdFF2BAAAVIYFB8+wb1/88IexeHHMnp2d\nAiU3OBhDQ9kRAABQGRYcPMMTT8SPfxw//nF2B5TfwoWxdm12BAAAVIYDDuL//t/40Y8iIhYs\niFWrYsmS7CDoC/PnZxcAAECVuKJC7N8fzWZ2BPSdej1GR7MjAACgMiw4Ku2v/zrmzPG4DSjE\nokVxzjnZEQAAUBkWHAAAAEDpWXBUyw03xDe/+cv/Hh2NmTNTa6CvrVgR69dnRwAAQGVYcFTI\nU0/FwEB2BFTGwEAMDmZHAABAZVhwVMjdd8cdd2RHQGVMTMS6ddkRAABQGRYc/emRR+JrX3vG\ntyxfHgsWJNVAJQ0Px9hYdgQAAFSGBUd/GhqK5cuf/Y27d2ekQFXNneuAAwAAuscBR5/4u7+L\nu+/+5X/PnRunnRa1WmoQAAAAdJErKgAAAEDpWXD0g0YjPvWp7AjgmZYujfPPz44AAIDKsOAA\nAAAASs+Co/R+/vP4X/8rOwL4DatWxUUXZUcAAEBlWHCU0uRk3HlnRMScOXH88Z4nCr1ocDCG\nhrIjAACgMiw4SqnVij17IiL27Imf/jS7BnguY2OxZk12BAAAVIYDjlL6+c9jdDQ7AnhBIyPZ\nBQAAUCWuqJTS449Ho5EdAbyget1BJAAAdI8FR2n8zd/Erl0REYsWxdFHZ9cAB7NoUZxzTnYE\nAABUhgUHAAAAUHoWHD3te9+LL34xImLmzBgczK4BpmPlytiwITsCAAAqw4Kjp7VaMTWVHQEc\nloEB55IAANA9Fhw97Yc/jG98IzsCOCwTE7FuXXYEAABUhgVHz7n33vjOdyIiFi2KiYnsGuBw\nDQ/H2Fh2BAAAVIYDjp7z8MNx773ZEcARmzs3TjwxOwIAACrDFZVuaLfjz//8kF65YEGccELB\nNUBXjI874AAAgO6x4AAAAABKz4LjSG3cGI3GQV6zcGEMDHSlBugZS5fG+ednRwAAQGVYcAAA\nAAClZ8FxqK68MqamsiOA8lixIi64IDsCAAAqw4IDAAAAKD0LDoCiTMbkP8Y/ZlcAQGe8Ld6W\nnQDwQhxwABRlIAbOiDOyKwAAoBIccAAUpRWtHbEjuwIAOuDUODU7AeAgPIMDAAAAKD0LDoCi\nNKO5OTZnVwDAkZoRM/4g/iC7AuAgLDgACtQO77EAlF4tatkJAAdnwQFQlEY0NsWm7AoAOCIL\nY+G74l3ZFQAHZ8EBUJR2tPfEnuwKADgcs2N2PerZFQDTYMEBUJRWtLbH9uwKADgcZ8QZ82N+\ndgXANDjgACjQaIxmJwDACzkzzpzhHwVAX3BFBaAo7Wjvjb3ZFQDwQubFPM8QBfqDw1qAokzF\n1LVxbXYFADyvt8XbnG4AfcOCAwAAACg9Cw6AojSicUVckV0BAM/rL+IvBmMwuwKgMyw4AArU\njGZ2AgA8Wy1qHiwK9B/vawBFmYzJ6+P67AoAeLaBGFgf67MrADrMggOgKK1o7Yyd2RUAEBFx\nTBxzXByXXQFQIAsOgAINxVB2AgBERLiTAvQ9b3MARWlFa0fsyK4AoNIWx+KXxcuyKwC6wRUV\nAAAAoPQsOACK0ozmptiUXQFAdf1h/OFIjGRXAHSJBQdAgdrhPRaANLWoZScAdI8FB0BRGtGw\n4AAg0YfiQ9kJAN1jwQFQlHa098Se7AoAKqoe9dkxO7sCoHssOACK0orW9tieXQFARR0dR58e\np2dXAHSPAw6AAjWjmZ0AQOUcHUe/PF6eXQHQba6oAAAAAKVnwQFQlMmY3BpbsysAqJahGHpn\nvDO7AiCBBQcAAABQehYcAEVpROOquCq7AoBqOSPOWBtrsysAElhwABToQBzITgCgzw3G4FAM\nZVcA5LPgACjKZExui23ZFQD0uYmY8JkpAOGAA6A4AzGwOlZnVwDQ5xbFouwEgJ7ggAOgQDbD\nABRtIAayEwB6ggMOgKK0onVX3JVdAUCfe1m8bDRGsysA8nnIKAAAAFB6FhwARWlGc1Nsyq4A\noJ/9dvy2J4wCPM2CA6BA7fAeC0An1aKWnQDQoyw4AIrSiIYFBwCd9d5479FxdHYFQC+y4AAo\nSjvau2JXdgUA/WMoho6NY7MrAHqUBQdAUdrR3h27sysA6B+zY7YDDoDn44ADoCjtaD8QD2RX\nANAn5sbcM+KM7AqA3uWKCgAAAFB6FhwARWlG87q4LrsCgD6xLtbNjbnZFQC9y4IDAAAAKD0L\nDoCiNKKxMTZmVwDQJz4UH5of87MrAHqXBQdAgQ7EgewEAPpBLWojMZJdAdDTLDgAijIZk9ti\nW3YFAP1gVsx6Q7whuwKgpzngACjKQAysjJXZFQD0g4WxMDsBoNe5ogJQlHa098be7AoA+sFI\njAzHcHYFQE+z4AAoylRM3RA3ZFcAUHpnxVkrYkV2BUCvs+AAAAAASs+CA6AozWhuik3ZFQCU\n2/viffNiXnYFQAlYcAAUqBnN7AQASqwWtRm+JAlwaLxdAhSlGc0tsSW7AoASG43RDbEhuwKg\nHCw4AIrSjvau2JVdAUBZjcWYT04BOHQWHABFaUd7d+zOrgCgrGbHbAccAIfOAQdAUdrRfiAe\nyK4AoKyWx/LsBIAycUUFAAAAKD0LDoCiNKO5OTZnVwBQVu+P98+MmdkVAKVhwQEAAACUngUH\nQFEa0dgYG7MrACifN8Yb18ba7AqAkrHgACjQgTiQnQBA+QzF0GAMZlcAlIwFB0BRpmLqlrgl\nuwKA8jkjzpgf87MrAErGAQdAgUZjNDsBgBI4PU73PFGAI+SKCkBR2tHeG3uzKwAogbkxdyAG\nsisAys2CA6AoUzF1Q9yQXQFAT3tZvGxlrMyuAOgHFhwAAABA6VlwABSlEY0r4orsCgB616lx\n6lvjrdkVAH3CggOgQM1oZicA0LsGYsDHwQJ0igUHQFGa0dwSW7IrAOhdJ8VJ58Q52RUAfcKC\nA6Ao7Wjvil3ZFQD0qFkx6+g4OrsCoH9YcAAUpR3t3bE7uwKAHnVMHOOAA6CDHHAAFKUVrZ/G\nT7MrAOg5r4/XD8dwdgVAv3FFBQAAACg9Cw6AojSjuTk2Z1cA0ENOipNeF6/LrgDoTxYcAAAA\nQOlZcAAUpRGNTbEpuwKAnjAQA5fGpdkVAP3MggOgKO1o74k92RUA5DsqjhqKoewKgD5nwQFQ\nlFa0tsf27AoA8r0oXrQoFmVXAPQ5BxwABRqN0ewEANKcFqeNxEh2BUBVuKICUJR2tPfG3uwK\nANLMiTmDMZhdAVAVFhwARZmKqZvipuwKAHKcHCeviTXZFQAVYsEBAAAAlJ4FB0BRGtG4Iq7I\nrgCg286Ks14Xr8uuAKgcCw6AAjWjmZ0AQFfVojbDFxEBMnjzBShKM5pbYkt2BQBdNRRD7453\nZ1cAVJEFB0BRWtHaGTuzKwDoqqWx1EfDAqSw4AAo0FAMZScA0FW1qGUnAFSUAw6AorSitSN2\nZFcA0FVLY2l2AkBFuaICAAAAlJ4FB0BRmtH8YnwxuwKAYq2LdQtiQXYFABYcAEVqh/dYgD7n\noRsAPcKCA6AojWhsik3ZFQAU6wPxgdkxO7sCAAsOgMK0o70n9mRXAFCggRiYF/OyKwCIsOAA\nKE4rWttje3YFAAUaiZGz4+zsCgAiHHAAFGo0RrMTACjK6XH6zJiZXQHAL7miAgAAAJSeBQdA\nUSZjcmtsza4AoMOOiWPeGm/NrgDg2Sw4AAAAgNKz4AAoSiMaV8QV2RUAdNjiWHxxXJxdAcCz\nWXAAFOhAHMhOAOBIzYgZHiYK0PssOACKMhmT22JbdgUAR2p5LD8jzsiuAOAgHHAAFGUgBlbH\n6uwKAI7UoliUnQDAwTngACjQUAxlJwBwpAZiIDsBgINzwAFQlFa0dsSO7AoAjsiyWLY8lmdX\nAHBwHjIKAAAAlJ4FB0BRmtHcFJuyKwA4Iv8j/ocrKgClYMEBUKB2eI8FKKta1LITAJgGCw6A\nojSiYcEBUFKzY/YH4gPZFQBMgwUHQFHa0d4Te7IrADgcM2LGnJiTXQHANFhwABSlHe2fxE+y\nKwA4HEfFUQ44AMrFAQdAUdrRfiAeyK4AYNpWxaplsSy7AoDpcUUFAAAAKD0LDoCiTMbk1tia\nXQHAtL0l3jIWY9kVAEyPBQcAAABQehYcAEVpRGNjbMyuAGB61sSat8fbsysAmDYLDoACHYgD\n2QkATE896vWoZ1cAMG0WHABFmYzJbbEtuwKA6VkVq06JU7IrAJg2Cw6AorSitTN2ZlcAMA1D\nMbQ8lmdXAHA4LDgACjQUQ9kJAEyDyykA5eWAA6AorWjdGrdmVwAwDfNj/pJYkl0BwOFwRQUA\nAAAoPQsOgKI0o7kpNmVXAHCoFsWid8Q7sisAOEwWHAAFaof3WIDSqEUtOwGAw2fBAVCURjQs\nOADKYmbM/GB8MLsCgMNnwQFQlHa0d8Wu7AoADslRcdT8mJ9dAcDhs+AAKEo72rtjd3YFAIfk\n2DjWAQdAqTngAChKO9oPxAPZFQAcxOyYvTbWZlcAcKRcUQEAAABKz4IDoCjNaF4X12VXAHAQ\n/z3+u89PAegDFhwAAABA6VlwABSlEY2NsTG7AoDnUI/6X8ZfZlcA0EkWHAAFOhAHshMAeLaZ\nMXOGr/MB9B3v7ABFmYzJbbEtuwKAZ3t5vHwiJrIrAOgwBxwARalFbUksya4A4BnOirOyEwAo\nhCsqAEVpR3tv7M2uAOAZ5sf87AQACmHBAVCUqZi6IW7IrgDgV46P438nfie7AoBCWHAAAAAA\npWfBAVCUZjT/Nv42uwKAX3lpvPT0OD27AoBCWHAAFKgZzewEgKqbETNqUcuuAKBwFhwARWlG\nc0tsya4AqLq3xFuOi+OyKwAonAUHQFHa0d4Vu7IrACptfsw/Ko7KrgCgGyw4AIrSjvbu2J1d\nAVBpwzHsgAOgIhxwABSlHe39sT+7AqC6XhwvHoux7AoAusQVFQAAAKD0LDgAitKM5ubYnF0B\nUFFvjjdPxER2BQDdY8EBAAAAlJ4FB0BRGtHYGBuzKwCq6IQ44Q/iD7IrAOgqCw6AorSjvSf2\nZFcAVNFQDPnwFICqseAAKEorWttje3YFQBUtikUvihdlVwDQVQ44AAo0GqPZCQAVsjJWHhPH\nZFcAkMMVFYCitKO9N/ZmVwBUyFFx1MyYmV0BQA4LDoCiTMXUDXFDdgVAVcyMmW+Lt2VXAJDG\nggMAAAAoPQsOgKI0onFFXJFdAVAJs2LWpXFpdgUAmSw4AArUjGZ2AkAl1KOenQBAMgsOgKI0\no7kltmRXAFTCu+JdwzGcXQFAJgsOgKK0o70rdmVXAPS/OTFnTszJrgAgmQUHQFHa0W5EI7sC\noP9NxVR2AgD5HHAAFKUVrR2xI7sCoM+dGqfOj/nZFQDkc0UFAAAAKD0LDoCiNKO5OTZnVwD0\ns/Wx/vg4PrsCgJ5gwQFQoHZ4jwUoSi1q2QkA9BALDoCiNKKxKTZlVwD0rT+NP53hb7MA/P8s\nOACK0o72ntiTXQHQt46Oo7MTAOghzrwBitKK1vbYnl0B0J8GYuBV8arsCgB6iAMOgAKNxmh2\nAkAfmhkzT4/TsysA6C2uqAAUpR3tvbE3uwKgDw3EwNyYm10BQG+x4AAoylRMXRvXZlcA9KFX\nx6sdcADwLBYcAAAAQOlZcAAUpRGNK+KK7AqAfjMQAx+Nj2ZXANBzLDgACtSMZnYCQL+pRz07\nAYBeZMEBUJRmNLfEluwKgH7z7nj3UAxlVwDQcyw4AIrSitbO2JldAdBXjo6jF8SC7AoAepEF\nB0CBfI0RoLNm+OsrAM/DnxAARWlFa0fsyK4A6BMvihe9KF6UXQFA73JFBQAAACg9Cw6AojSj\n+cX4YnYFQJ94Z7xzIAayKwDoXRYcAAVqh/dYgM6oRS07AYCeZsEBUJRGNDbFpuwKgH4wFEN/\nEn+SXQFAT7PgAChKO9p7Yk92BUA/mBNzfH4KAC/MnxMARWlFa3tsz64A6AdnxpnzYl52BQA9\nzQEHQIGa0cxOACi91bHa6QYAB+WKCgAAAFB6FhwARZmMya2xNbsCoPTOiXNOjBOzKwDodRYc\nAAAAQOlZcAAUpRGNq+Kq7AqAclsZK18fr8+uAKAELDgACnQgDmQnAJTVSIzUopZdAUBpWHAA\nFGUyJrfFtuwKgLI6L84bjdHsCgBKwwEHQFEGYmB1rM6uACiroRjKTgCgTBxwABTI384BDpv7\nKQBMiwMOgKK0ovXD+GF2BUApLYklM/xNFYDp8JBRAAAAoPSciwMUpRnNTbEpuwKgfE6L014X\nr8uuAKBkLDgACtQO77EAh8MDOACYLgsOgKI0omHBAXAYTovT1sW67AoASsaCA6Ao7Wjvil3Z\nFQAlU4/6gliQXQFA+VhwABSlHe3dsTu7AqBkZsUsBxwAHAYHHABFaUf7gXgguwKgTGbFrDPi\njOwKAErJFRUAAACg9Cw4AIrSjObn4nPZFQBl8qJ4hix8EgAAIABJREFU0dlxdnYFAKVkwQEA\nAACUngUHQFEa0dgYG7MrAMphWSx7f7w/uwKAErPgACjQgTiQnQBQDvWo16OeXQFAiVlwABRl\nMia3xbbsCoByODVOPTlOzq4AoMQccAAUZSAGVsfq7AqAclgYC7MTACg3BxwARalFbTzGsysA\nymE4hrMTACg3BxwARZmKqRvihuwKgHL4nfid4+P47AoASsxDRgEAAIDSs+AAKEozmptiU3YF\nQK87PU5/TbwmuwKA0rPgAChQM5rZCQC9qxa1Gb7eBkCH+BMFoCjNaG6JLdkVAL1rJEbeGe/M\nrgCgT1hwABSlHe1dsSu7AqBHzY25s2N2dgUA/cOCA6Ao7Wjvjt3ZFQA9qh51BxwAdJADDoCi\ntKP9QDyQXQHQi14Tr6lHPbsCgL7iigoAAABQehYcAEVpRnNzbM6uAOg574/3z4yZ2RUA9BsL\nDgAAAKD0LDgAitKIxsbYmF0B0Ft+P37/xDgxuwKAPmTBAVCgA3EgOwEg34yY4U4KAEWz4AAo\nymRMbott2RUA+U6ME18SL8muAKDPOeAAKEotaktiSXYFQKbBGHxpvDS7AoBKcEUFoCjtaO+N\nvdkVAJlqUZsX87IrAKgECw6AokzF1A1xQ3YFQJr/Fv9tWSzLrgCgKiw4AAAAgNKz4AAoSjOa\nfxt/m10BkOZV8aqVsTK7AoCqsOAAKFAzmtkJAAnqUc9OAKByLDgAitKM5pbYkl0BkOB98b7s\nBAAqx4IDoCjtaO+KXdkVAF1Vj/qCWJBdAUAVWXAAFKUd7d2xO7sCoKtGYsQBBwApHHAAFKUV\nrZ/GT7MrALrklDjFI0UBSOSKCgAAAFB6FhwARWlGc3Nszq4A6JJXxitPi9OyKwCoLgsOAAAA\noPQsOACK0ojGxtiYXQHQJZfGpcfEMdkVAFSXBQdAUdrR3hN7sisAumFmzByN0ewKACrNggOg\nKK1obY/t2RUA3TAe46tjdXYFAJXmgAOgQL6eCVRBLWpONwBI54oKQFHa0d4be7MrALphfszP\nTgCg6iw4AIoyFVPfjG9mVwAUbnEsPiPOyK4AoOosOAAAAIDSs+AAKEojGlfEFdkVAIV7Y7zR\nggOAdBYcAAVqRjM7AaBYM2JGLWrZFQBgwQFQmGY0t8SW7AqAYq2P9cfEMdkVAGDBAVCYVrR2\nxs7sCoACDcXQ8lieXQEAERYcAIUaiqHsBIACzYyZ2QkA8EsOOACK0orWjtiRXQFQoKPj6MWx\nOLsCACJcUQEAAAD6gAUHQFGa0dwcm7MrAIoyGIMfiA9kVwDAL1lwABSoHd5jgb7l02EB6CkW\nHABFaURjU2zKrgAoyh/FH43ESHYFAPySBQdAUdrR3hN7sisAOu+oOMqnRAHQayw4AIrSitb2\n2J5dAdB5q2KVD08BoNc44AAo0GiMZicAdN6xcWx2AgA8mysqAAAAQOlZcAAUZTImt8bW7AqA\nDlsdq8+Ks7IrAODZLDgAAACA0rPgAChKIxpXxBXZFQAdc2Kc+I54R3YFADw3Cw6AAh2IA9kJ\nAB1Tj3o96tkVAPDcLDgAijIZk9tiW3YFQMecGqeeHCdnVwDAc7PgAChKK1o7Y2d2BUBnDMfw\nsliWXQEAz8uCA6BAQzGUnQDQGS6nANDjHHAAFKUVrR2xI7sCoANGY/TcODe7AgBeiCsqAAAA\nQOlZcAAUpRnNTbEpuwKgAz4QHxiN0ewKAHghFhwABWqH91igH9Silp0AAAdhwQFQlEY0LDiA\nPrA0ll4YF2ZXAMBBWHAAFKUd7T2xJ7sC4EjNilnDMZxdAQAHYcEBUJRWtLbH9uwKgCO1Mlae\nECdkVwDAQTjgAChQM5rZCQBHakEsyE4AgINzRQUAAAAoPQsOgKJMxuTW2JpdAXBEjovj3hRv\nyq4AgIOz4AAAAABKz4IDoCiNaFwVV2VXAByRt8Xbjo/jsysA4OAsOAAKdCAOZCcAHL6BGPAB\nsQCUhQUHQFEmY3JbbMuuADh882Lea+O12RUAcEgsOACK0orWztiZXQFwmObH/PEYz64AgENl\nwQFQoKEYyk4AOEwz/EURgFLx5xZAUVrRujVuza4AmLaj4+jz4rzsCgCYHldUAAAAgNKz4AAo\nSjOam2JTdgXA9CyOxRtiQ3YFAEybBQdAgdrhPRYon1rUshMAYNosOACK0oiGBQdQOktiiQUH\nAGVkwQFQlHa0d8Wu7AqA6Tkqjpof87MrAGDaLDgAitKO9u7YnV0BMD3HxrEOOAAoIwccAEVp\nR/uBeCC7AuCQvDhefEKckF0BQMdceeWVl1566Xe+850zzzwzu6VLHHAAFGUwBs+L87IrAAD6\n09TU1OWXX37rrbfec889Dz744PDw8AknnPCWt7zlD//wD48++uieaujsy7rgRz/60caNG7/2\nta/t3bt3yZIlF1544Yc//OFZs2Yd+s/w5S9/+U1velNEfOQjH7nssst+/buuu+66f/mXf9m+\nffudd975+OOPX3jhhX//93/fkWzP4AAoSjOa18V12RUAB3dKnPLieHF2BcD0PPnkkyMjI+Pj\n4yeddNJxxx23f//+O+6448EHHzz++ONvueWWE07oxirtEBs6+7JDdNgLjrvvvvsVr3jFo48+\n+sY3vnH58uXf+ta3tm/fftZZZ339618fGRk5lJ/hwQcfPOWUUx5//PH9+/f/5gHHmWeeeccd\nd8yZM2d8fPwHP/hBBw84LDgAilKP+oVxYXYFAEB/Ghoauvfee3/9n/2NRuP3fu/3vvCFL1x+\n+eWf/vSne6ehsy8r2sUXX7x3794tW7ZcdNFFEdFqtX73d3/36quv/vjHP/4Xf/EXh/IzvO99\n7xsYGPjgBz/4l3/5l7/5vVdeeeXixYtXrFixbdu2888/v4PlDjgAitKIxsbYmF0BcBAfig95\nqihQRrVa7VmjhpkzZ773ve/9whe+8MMf/rCnGjr7skJt37799ttvX7NmzdOnGxExMDDwsY99\n7JprrvnUpz71kY98pFarvfDPsGXLluuvv/4rX/nK8zWfc845HU3+FQccAEWZGTMdcAC9bEbM\nmBkzsysAOunaa6+NiNNOO+2FX/b2t7/9mmuuueqqqy655JKshs6+rFO+/vWvR8R55z3jQXKL\nFi069dRTd+zY8YMf/ODkk09+gR9+7733/vEf//F73vOeN7zhDZ/4xCeKbf0NDjgAijIZk9ti\nW3YFwPM6KU46Lbr0N2aA4vzJn/zJk08++eijj/7bv/3bf/7nf5566qkf+chHerOhsy8rws6d\nOyPiN08xTjrppIMecLRarXe/+93z5s3btGlTsZXPwwEHQFFqUVsSS7IrAJ6X0w2gP3zmM595\n/PHHn/7vdevWbd269dhjj33hH/Lud7/7rLPOOvvss7vc0NmXFeHRRx+NiLlz5z7r2+fNmxcR\ne/fufYEf+/GPf/yb3/zmTTfd9Js/vDsccAAUZSAGTo4XmvABAHDk9u/f3263d+/e/Y1vfOND\nH/rQmjVrtm3bdvrpp7/ADznvvPOedQujOw2dfVk3Pf0BrC/wAI677rrrox/96CWXXPLa1762\ni13P4IADoChTMXVD3JBdAfDcZsQMn/QE9I1arTY+Pn7hhReuXr169erV73nPe+68887ebOjs\nyzru6fHF0zuOX/d8y46ntdvtd77znccff/zHPvaxogtfgAMOgKLMiBnviHdkVwAAVMhv/dZv\nLVy48Hvf+96ePXvmz8/5iKhDbOjsyzrl6UdsPP0kjl/39EeinHTSSc/5o6ampp4+f5k9e/az\nvuvyyy+//PLLL7744s985jOdz30mBxwARWlGc1PkPGAJ4AVMxMTb4+3ZFQCFeOyxxx544IGI\nmDEj7V+7h9jQ2Zd1yqte9aqIuPHGG//qr/7qv77xvvvuu/POOxctWvR8BxwDAwMXX3zxs77x\n3//932+77bY1a9acccYZr3jFK4pr/i8OOACKUo/6B+OD2RUAzzYQA9kJAB1w2223jYyM/Prn\npz788MO///u/PzU19cpXvvI3pwS/7qtf/erOnTtf/epXn3LKKV1o6OzLCnX66ae/7GUvu/32\n2z//+c+/613viohWq/Vnf/ZnrVbrkksu+fVncGzdunXv3r0bNmw47rjjBgYGfnOg8YlPfOK2\n2257wxvecNlll3WhPBxwABSnGc0tsSW7AuDZlsWy10baE+AAOuXmm2/+8z//8+XLly9btmz+\n/Pn333//HXfc8cQTTyxcuPBTn/rUC//Yz33uc9dcc81VV111hAcch9jQ2ZcV7bOf/ezatWvf\n8573XHfddcuWLfvWt751xx13vPzlL//TP/3TX3/ZZZdd9qMf/Wjt2rXHHXfctH7+66677p/+\n6Z8i4he/+EVEfPvb377ooosiYmxs7MorrzyScgccAEWZETNeH6/PrgB4tkWxKDsBoAPe/OY3\nP/TQQzfffPOdd965Z8+e0dHRU0455fWvf/0f/dEfde3pG4fY0NmXFW316tV33HHHRz/60X/+\n53/+6le/unjx4g9/+MMf/vCHR0ZGOvLzb9++/XOf+9x//e+999577733RsQJJ5xwhAcctac/\n6wWAjmtF67vx3ewKgGd7SbzELRWAvnfllVdeeuml3/nOd84888zsli6x4AAoSjva+2N/dgXA\nM5wdZzvdAKAvOeAAKMpgDP52/HZ2BQAAVIIDDoCiNKO5OTZnVwD8yiVxST3q2RUAUAjP4AAA\nAIB+c8stt9x0003ve9/7jj/++OyWLnHAAVCURjQ2xsbsCoBfWhEr3hvvza4AgKI44AAoSjva\ne2JPdgXAL82LeR4vCkAf8wwOgKK0orU9tmdXAPzS2XH2SIxkVwBAURxwABRoNEazEwAiIk6I\nE5xuANDfXFEBKEo72ntjb3YFQETESIwMx3B2BQAUyIIDoChTMXVD3JBdARAR8Zp4jQMOAPqb\nBQcAAABQehYcAEVpROOKuCK7AiAi4oPxwTkxJ7sCAApkwQFQoGY0sxMAoha1Gb6sBUC/80cd\nQFGa0dwSW7IrAOLYOPat8dbsCgAolgUHQFHa0d4Vu7IrAGJOzHE/BYC+Z8HB/8fevfvWed95\nHv+eQ5GHpKirJVmRQnl8j236fnecxHE2jpOJ7YydScbObCa7c4UHg9nrZICd5RZbsGQZVabS\nuHOQhiq2WSDFuiMETCWk2A0wLCgMQBYBi/OQ59liJ3DitWxR4o/f53nO6/UXvBsfC19+fucA\npdRRb8RGdgVATMSEAwcAnefAAVDKKEa/il9lVwDEiTiRnQAAxXmiAgAAALSeBQdAKVVUF+Ni\ndgUw7p6L556Op7MrAKA4Cw4AAACg9Sw4AEoZxnA5lrMrgLH2hfjCG/FGdgUAHAQLDoBS6qg3\nYzO7AhhT/egfj+PZFQBwcCw4AEoZxWgt1rIrgDE1HdMvxovZFQBwcBw4AAqai7nsBGBMPRvP\nZicAwIHyRAWglDrqrdjKrgDG1Ik4kZ0AAAfKggOglN3Y/SA+yK4AxtHb8XZ2AgAcNAsOAAAA\noPUsOABKGcZwKZayK4Bx8ffx94MYZFcAQBoLDoCCqqiyE4Au60d/IiayKwCgESw4AEqpolqJ\nlewKoMvuiDu+Ed/IrgCARrDgAChlFKOrcTW7Auig+Zj3K9QA8DEWHAAFeQ8PlNCPfnYCADSO\nAwdAKaMYXYkr2RVAB30rvpWdAACN44kKAAAA0HoWHAClVFG9H+9nVwBdczpOW3AAwP/PggOg\noDp8xgL7rxe97AQAaBwLDoBShjFcjuXsCqBrXo6Xn46nsysAoHEsOABKqaPejM3sCqBrjsSR\nyZjMrgCAxrHgAChlFKO1WMuuALrmsXjsVJzKrgCAxnHgACioiio7AeiOY3HshXghuwIAGsoT\nFQAAAKD1LDgAStmJnUtxKbsC6IILceGVeCW7AgAazYIDAAAAaD0LDoBShjFciqXsCqAL3oq3\nFmIhuwIAGs2CA6Cg7djOTgDarRe9mZjJrgCAFrDgAChlJ3ZWYzW7Ami3I3Hk1Xg1uwIAWsCC\nA6CUUYyuxtXsCqDF7o/7+9HPrgCAdrDgAChoEIPsBAAAGAsOHACljGL0y/hldgXQVs/H8+Yb\nAHDjPFEBAAAAWs+CA6CUKqrlWM6uAFrm2/HtB+PB7AoAaB8LDoCC6vAZC+xNL3rZCQDQShYc\nAKUMY2jBAezJQiz8fvx+dgUAtJIFB0ApddTrsZ5dAbTG6Tjtp5cA4KZZcACUUke9ERvZFUBr\nHItjDhwAcNMcOABKqaO+FteyK4B2eDFePBJHsisAoMU8UQEAAABaz4IDoJQqqp/GT7MrgBZ4\nMB58IV7IrgCAdrPgAAAAAFrPggOglGEMF2MxuwJoge/H9x+Px7MrAKDdLDgACtqO7ewEoAVm\nYzY7AQBaz4IDoJSd2FmN1ewKoAXejDcnYiK7AgDazYIDoJRRjK7G1ewKoNEOx+ELcSG7AgC6\nwIIDoKBBDLITgEabjMnsBADoCAcOgFJGMfowPsyuABrtbJz9XHwuuwIAusATFQAAAKD1LDgA\nSqmiWo7l7AqguV6JVx6Lx7IrAKAjLDgACqrDZyzwyXrRy04AgE6x4AAopYpqJVayK4Ammoqp\nH8WPsisAoFMsOABKqaNej/XsCqBxZmLmtrgtuwIAusaCA6CUOuqN2MiuABrnRJxw4ACAfefA\nAVBKHfW1uJZdATTLM/GM6wYAlOCJCgAAANB6FhwApVRRXYyL2RVAg0zG5F/FX2VXAEA3WXAA\nAAAArWfBAVDKMIaLsZhdATTI1+PrX4uvZVcAQDdZcAAUtB3b2QlAsomYGMQguwIAus+CA6CU\nndhZjdXsCiDZhbjwbDybXQEA3efAAVBKL3rzMZ9dASQ7HaezEwBgLHiiAlBKHfVWbGVXAMkG\nMZiN2ewKAOg+Cw6AUnZj93Jczq4A0jwQDzwRT2RXAMC4sOAAAAAAWs+CA6CUKqr34r3sCiDH\n0Tj6drydXQEAY8SCA6CgKqrsBOCg9aM/ERPZFQAwdiw4AEqpolqJlewK4KDNx/w345vZFQAw\ndiw4AEqpo16P9ewK4KBNx/SpOJVdAQBjx4IDoJQ66o3YyK4ADtrROOrAAQAHz4EDoJRRjP45\n/jm7Ajhox+JYdgIAjCNPVAAAAIDWs+AAKKWK6mJczK4ADtQ345v3xD3ZFQAwjiw4AAAAgNaz\n4AAoZRjDxVjMrgAOyFzM/UP8Q3YFAIwvCw6AUuqoN2MzuwI4IBMx4etFASCRBQdAKaMYrcVa\ndgVwQOZi7rl4LrsCAMaXAwdAQXMxl50AFLQQC/4zB4CG8EQFoJQ66q3Yyq4ACjoSRw75cxEA\nNIP/JQOUshu7v4hfZFcApTwVT52IE9kVAMC/sOAAAAAAWs+CA6CUYQyXYim7Aiji9Xj98Xg8\nuwIA+IgFB0BBVVTZCUAREzHRj352BQDwEQsOgFKqqFZiJbsCKOIr8ZX74/7sCgDgIxYcAKWM\nYnQ1rmZXAEWcj/NH42h2BQDwEQsOgIIGMchOAIrwPgUAmsaBA6CUUYyuxJXsCmD/DWJwV9yV\nXQEA/A5PVAAAAIDWs+AAKKWK6mJczK4A9seZOPP9+H52BQBwXRYcAAXV4TMWuqMXvewEAOC6\nLDgAShnGcDmWsyuA/XE2zv5J/El2BQBwXRYcAKXUUW/GZnYFsD8GMTgch7MrAIDrsuAAKGUU\no7VYy64A9se5OPdgPJhdAQBclwMHQEFzMZedAOyPk3EyOwEA+DSeqACUUke9FVvZFcD+mI3Z\nQQyyKwCA67LgAChlN3Y/iA+yK4Bb9Ua8cTpOZ1cAAJ/BggMAAABoPQsOgFKGMVyKpewK4Jac\njbN/GX+ZXQEAfDYLDoCCqqiyE4CbNBmT2QkAwB5YcACUshM7P4+fZ1cAN+k78R03DgBoEQsO\ngFJGMboaV7MrgD2bjMl74p7sCgBgbyw4AAryo5LQRof8AwkAWsj/vwFKGcXoSlzJrgA+zVfj\nqyfiRHYFALAPPFEBAAAAWs+CA6CUKqrlWM6uAD7ZnXHnH8YfZlcAAPvGggOgoDp8xkIT9aKX\nnQAA7DMLDoBShjG04IBm+uv467mYy64AAPaTBQdAKXXUm7GZXQH8jkEMDsfh7AoAYP9ZcACU\nMorRWqxlVwC/4/Px+S/EF7IrAID958ABUFAVVXYC8JGX4qWZmMmuAACK8EQFAAAAaD0LDoBS\ndmLnUlzKrgBiKqZ+GD/MrgAAyrLgAAAAAFrPggOglGEMfxI/ya6Asfaj+NGxOJZdAQAcBAsO\ngIK2Yzs7AcZOL3q+SRQAxpAFB0ApO7GzGqvZFTB2pmLqjXgjuwIAOGgOHACl9KO/EAvZFTB2\nTsfp7AQAIIEDB0BBgxhkJ8DYmYiJ7AQAIIEDB0Apoxj9Y/xjdgWMi/vivgfigewKACCNLxkF\nAAAAWs+CA6CUKqrlWM6ugLEwFVP/Lv5ddgUAkMmCA6CgOnzGQlm96GUnAACNYMEBUMowhhYc\nUNTZOPsn8SfZFQBAI1hwAJRSR70e69kV0EGTMXl73J5dAQA0iwUHQCl11BuxkV0BHTQTMw4c\nAMDHOHAAlFJHfS2uZVdAB305vpydAAA0jicqAAAAQOtZcACUUkX1s/hZdgV0ykzMvB6vZ1cA\nAE1kwQEAAAC0ngUHQCnDGC7GYnYFdMfD8fAP4gfZFQBAQ1lwABS0HdvZCdAdUzF1yN9mAIDr\n8K8EgFJ2Ymc1VrMroDsej8fviXuyKwCAhnLgACilF735mM+ugI64O+4+HaezKwCA5vJEBaCU\nOuqt2MqugI6YjdlBDLIrAIDmsuAAKGU3di/H5ewK6Ig/ij/KTgAAGs2CAwAAAGg9Cw6AUqqo\nlmM5uwLarR/9/xT/KbsCAGgBCw6AgqqoshOgxSZioh/97AoAoB0sOABKqaJaiZXsCmixp+Pp\nx+Px7AoAoB0sOABKqaNej/XsCmix83G+F73sCgCgHSw4AEqpo96IjewKaLFzcc6BAwC4QQ4c\nAKXUUV+La9kV0DKPxCPn43x2BQDQPp6oAAAAAK1nwQFQShXVxbiYXQFt8mQ8+UK8kF0BALSS\nBQcAAADQehYcAKUMY7gYi9kV0ALvxDuPxCPZFQBAu1lwABS0HdvZCdB0szGbnQAAdIEFB0Ap\nu7H7v+J/ZVdA030tvua3YAGAW+fAAVDQXMxlJ0CjHYtjrhsAwL7wRAWglDrqrdjKroBGOxSH\njsSR7AoAoAssOABK2Y3dy3E5uwKa66V46Xycz64AADrCggMAAABoPQsOgFKGMVyKpewKaKh3\n4p17497sCgCgOyw4AAqqospOgCaaiIl+9LMrAIBOseAAKKWKaiVWsiugiZ6P5x+Oh7MrAIBO\nseAAKKWOej3WsyugiU7EicNxOLsCAOgUCw6AUuqoN2IjuwKaaCZmHDgAgP3lwAFQyihGv4pf\nZVdA49wdd98Wt2VXAABd44kKAAAA0HoWHAClVFFdjIvZFdA4z8fzT8VT2RUAQNdYcAAAAACt\nZ8EBUMowhsuxnF0BDfJoPPpqvJpdAQB0kwUHQCl11JuxmV0BDTId07Mxm10BAHSTBQdAKaMY\nrcVadgU0yB1xx71xb3YFANBNDhwABc3FXHYCNMi5OJedAAB0licqAKXUUW/FVnYFNMjRODoR\nE9kVAEA3WXAAlLIbu/8j/kd2BTTFH8Yf9qOfXQEAdJYFBwAAANB6FhwApQxjuBRL2RWQZjqm\nfxw/zq4AAMaFBQdAQVVU2QmQ4FAc6kUvuwIAGC8WHAClVFGtxEp2BSR4LV77XHwuuwIAGC8W\nHACljGJ0Na5mV8BBOxWnTsfp7AoAYOxYcAAUNIhBdgIcND8ECwCkcOAAKGUUoytxJbsCijgb\nZ1+IF7IrAAA+4okKAAAA0HoWHAClVFG9H+9nV0ARr8QrvkYUAGgUCw6AgurwGUsH+QlYAKCB\nLDgAShnGcDmWsytg//1Z/NltcVt2BQDA77DgACiljnozNrMrYJ9NxdRczGVXAAB8nAUHQCmj\nGK3FWnYF7LPb4/aH4+HsCgCAj3PgACjIH7rphvmYPx/nsysAAD6NJyoAAABA61lwAJSyEzuX\n4lJ2BdyqC3HhlXgluwIA4DNYcAAAAACtZ8EBUMowhkuxlF0BN2k6pn8cP86uAAC4URYcAAVt\nx3Z2AtyMqZg65K8gAECr+LcLQCk7sbMaq9kVcDOeiCfujruzKwAA9sCCA6CUUYyuxtXsCtiz\nM3HmtrgtuwIAYG8sOAAKGsQgOwH2bCImshMAAPbMgQOglFGMrsSV7Aq4UVMx9e34dnYFAMBN\n8kQFAAAAaD0LDoBSqqiWYzm7Am7I8/H8l+JL2RUAADfPggOgoDp8xtJovehlJwAA7A8LDoBS\nhjG04KDhvhffuzPuzK4AANgHFhwApdRRb8ZmdgVc10zMzMRMdgUAwP6w4AAopY76f8f/zq6A\n6zoX5xw4AIDOcOAAKKWO+lpcy66AT/ZkPHkmzmRXAADsG09UAAAAgNaz4AAoZSd2LsWl7Ar4\nyCvxyoW4kF0BAFCEBQcAAADQehYcAKUMY7gYi9kV8C9mYua/xX/LrgAAKMWCA6Cg7djOTmDc\nzcRML3rZFQAAxVlwAJSyEzursZpdwbj7TnxnMiazKwAAirPgAChlFKOrcTW7gvEyHdN3xp3Z\nFQAACSw4AAoaxCA7gfFirAEAjC0HDoBSRjH6MD7MrmCMfDu+fSyOZVcAAOTwRAUAAABoPQsO\ngFKqqJZjObuCMfK38bdeRQEAY8uCA6CgOnzGckD8FiwAMOYsOABKqaJaiZXsCsbCfMx/M76Z\nXQEAkMmCA6CUOur1WM+uoOPmYu54HM+uAADIZ8EBUEod9UZsZFfQcaMYOXAAAIQDB0A5ddTX\n4lp2Be12f9x/V9yVXQEA0AKeqAAAAACtZ8EBUEoV1fvxfnYFLfZwPPxEPJFdAQDQDhYcAAAA\nQOtZcACUMozhYixmV9BWj8ajb8fb2RUAAK00+IhnAAAgAElEQVRhwQFQ0HZsZyfQVhMxMYhB\ndgUAQGtYcACUshM7q7GaXUFbzcf8c/FcdgUAQGs4cACU0ovefMxnV9Amd8VdZ+JMdgUAQCt5\nogJQSh31VmxlV9AmszHrWQoAwM2x4AAoZTd2L8fl7Apa4514pxe97AoAgLay4AAAAABaz4ID\noJQqqvfivewKGu3tePtoHM2uAADoAgsOgIKqqLITaK5+9CdiIrsCAKAjLDgASqmiWomV7Aqa\n6/a4/Y14I7sCAKAjLDgASqmjXo/17Aqa63gcn4u57AoAgI6w4AAopY56IzayK2iuqZhy4AAA\n2C8OHACl1FH/On6dXUGae+Ke83E+uwIAYFx4ogIAAAC0ngUHQClVVBfjYnYFB+18nH8z3syu\nAAAYOxYcAAAAQOtZcACUMozhYixmV1DQ4Tj8X+O/ZlcAABBhwQFQTh31ZmxmV1CK30ABAGgU\nCw6AUkYxWou17ApKORNnHolHsisAAPgXDhwABfkLf2dMxMTT8XR2BQAA1+WJCkApddRbsZVd\nwf7oRe94HM+uAADguiw4AErZjd3LcTm7ghuyEAuPxqPZFQAA3DwLDgAAAKD1LDgAShnGcCmW\nsiv4NHfFXf86/nV2BQAA+8CCA6CgKqrsBD5BP/oTMZFdAQDAfrLgACilimolVrIr+AT3x/1f\nia9kVwAAsJ8sOABKqaNej/XsinF3OA6fiBPZFQAAFGfBAVBKHfUwhtkV4246prMTAAA4CA4c\nAKWMYnQlrmRXjLuvx9ezEwAAOAieqAAAAACtZ8EBUEoV1cW4mF3Rcc/Gs8/EM9kVAADks+AA\nKKgOn7Fl9aKXnQAAQCNYcACUMozhcixnV3RZL3p/F3+XXQEAQCNYcACUUke9GZvZFZ0yiMHh\nOJxdAQBAE1lwAJQyitFarGVXdMq5OPdgPJhdAQBAEzlwABQ0F3PZCR3xUDx0JI5kVwAA0Fye\nqACUUke9FVvZFR1xJI4ccpQHAOD6/GMRoJTd2P0gPsiu6IjvxnePx/HsCgAAmsuCAwAAAGg9\nCw6AUoYxXIql7IrW+y/xXzxOAQDgM1lwABRURZWd0G796E/ERHYFAAAt4G9iAKXsxM7P4+fZ\nFe12Ls59Mb6YXQEAQAtYcACUMorR1biaXdEy0zF9Z9yZXQEAQPtYcAAUNIhBdkLLTMZkdgIA\nAK3kwAFQyihGV+JKdkXL3B/3fz4+n10BAED7eKICAAAAtJ4FB0ApVVTvxXvZFW3ydrx9NI5m\nVwAA0EoWHAAF1eEz9rP1opedAABA61lwAJQyjOFyLGdXNN18zL8T72RXAADQehYcAKXUUW/G\nZnZFEx2JI34tBQCA/WXBAVDKKEZrsZZd0USPxWOn4lR2BQAAneLAAVBQFVV2Qr6FWJiP+ewK\nAAA6zhMVAAAAoPUsOABK2YmdS3Epu6KgZ+KZR+KR7AoAAIiw4AAAAAA6wIIDoJRhDH8SP8mu\nuFXfie/cEXdkVwAAwGew4AAAAABaz4IDoJjd3fif/zM7AgD2ydxcPPdcdgTAdTlwABTT68WJ\nE9kRALBPZmayCwA+jQMHQDF1HdeuZUcAwE2544548MHsCIA98B0cAAAAQOtZcAAUs7MTly5l\nRwDAp7rrrnj55ewIgH1gwQEAAAC0ngUHQDHDYSwuZkcAwPW99lp88YvZEQD7w4IDoKTt7ewC\nALi+6eno97MjAPaHBQdAMTs7sbqaHQEA1/eNb8TRo9kRAPvDgQOgmH4/FhayIwDgOvp91w2g\nSxw4AEoaDLILAOA6PE4BusWBA6CY0Sg+/DA7AgCu46mnsgsA9pMvGQUAAABaz4IDoJiqiuXl\n7AgAuI633oq7786OANg3FhwAJfmMBaBper3sAoAiLDgAihkOLTgAaJxvfjMeeSQ7AmD/WXAA\nFFPXsb6eHQEAv3H6tJ/3AjrMggOgmLqOjY3sCAD4jePHHTiADnPgACimruPatewIAMbP7/1e\nPPBAdgTAQfNEBQAAAGg9Cw6AYqoqfvaz7AgAxsyzz8bv/V52BEACCw4AAACg9Sw4AIoZDmNx\nMTsCgDHzZ38W99yTHQGQwIIDoKTt7ewCAMbMYBATE9kRAAksOACK2dmJ1dXsCADGzEsvxenT\n2REACRw4AIrp9WJ+PjsCgDFz223ZBQA5PFEBKKauY2srOwKAMXP8ePR62REACSw4AIrZ3Y3L\nl7MjABgnL70UJ05kRwDksOAAAAAAWs+CA6CYqorl5ewIAMbGN74Rjz6aHQGQxoIDoKSqyi4A\nYGxMTES/nx0BkMaCA6CYqoqVlewIAMbGV74S99+fHQGQxoIDoJi6jvX17AgAxsapUzE9nR0B\nkMaCA6CYuo6NjewIAMbGkSMOHMA4c+AAKKau49e/zo4AYDzce28cO5YdAZDJExUAAACg9Sw4\nAIqpqrh4MTsCgPHwgx/EyZPZEQCZLDgAAACA1rPgAChmOIzFxewIAMbDf//vMTmZHQGQyYID\noJi6js3N7AgAxoP3KcDYs+AAKGY0irW17AgAxsO/+lfZBQDJHDgASpqbyy4AYAw8+2x2AUA+\nT1QAiqnr2NrKjgBgDJw4kV0AkM+CA6CY3d24fDk7AoCue+ABBw6AsOAAAAAAOsCCA6CY4TCW\nlrIjAOi67343HnooOwIgnwUHQElVlV0AQHdNTmYXADSIBQdAMVUVKyvZEQB0149+FFNT2REA\nTWHBAVBMXcf6enYEAF10/LhfIgf4GAsOgGLqOobD7AgAumh3N7sAoHEcOACKGY3iypXsCAA6\n5MtfjlOnsiMAGsoTFQAAAKD1LDgAiqmquHgxOwKADvnzP4/p6ewIgIay4AAoyWcsAPul18su\nAGg0Cw6AYobDWF7OjgCgK956K+65JzsCoLksOACKqevY3MyOAKATpqdjdjY7AqDRLDgAihmN\nYm0tOwKATrjjjrj33uwIgEZz4AAoaW4uuwCA9jt3Li5cyI4AaDpPVACKqevY2sqOAKD9pqdj\nZiY7AqDpLDgAitndjQ8+yI4AoOWeeioeeyw7AqAFLDgAAACA1rPgAChmOIylpewIANrsxz+O\n6ensCIB2sOAAKKmqsgsAaK1Dh6LXy44AaA0LDoBiqipWVrIjAGit11+Ps2ezIwBaw4IDoJjR\nKK5ezY4AoJ3m5mJ+PjsCoE0sOABKGgyyCwBop8nJ7AKAlnHgAChmNIorV7IjAGihxx/3OAVg\nrzxRAQAAAFrPggOgmKqK99/PjgCgbZ5+Oh58MDsCoH0sOABK8hkLwF75aViAm2LBAVDMcBjL\ny9kRALTNq6/Go49mRwC0jwUHQDF1HZub2REAtM2xYzExkR0B0D4WHADFjEaxtpYdAUDbPPdc\nzM1lRwC0jwMHQElVlV0AQKtMT7tuANwcT1QAAACA1rPgAChmZycuXcqOAKA9er340z/NjgBo\nKwsOAAAAoPUsOACKGQ5jaSk7AoCW+Ju/iZMnsyMAWsyCA6Ck7e3sAgBaYnY2uwCg3Sw4AIrZ\n2YnV1ewIAFrizTdjYiI7AqDFHDgAiun3Y2EhOwKANjh82HUD4BY5cACUNBhkFwDQBpOT2QUA\nrefAAVDMaBS//GV2BABtcOFCfO5z2REA7eZLRgEAAIDWs+AAKKaqYnk5OwKAxpubi3ffzY4A\naD0LDoCSfMYC8Ol6vewCgI6w4AAoZji04ADg00xPx9/+bXYEQEdYcAAUU9exvp4dAUCDTUz4\nblGA/WLBAVBMXcfGRnYEAA02OenAAbBfHDgAiqnruHYtOwKABjt8OLsAoDs8UQEAAABaz4ID\noJiqip/+NDsCgAb71rfi3LnsCICOsOAAAAAAWs+CA6CY4TAWF7MjAGiwd9+NCxeyIwA6woID\noKTt7ewCABpsZiZ6vewIgI6w4AAoZmcnVlezIwBosNdei+np7AiAjnDgACim34+FhewIAJrq\n0CHXDYB95MABUEyvF2fPZkcA0FT9fnYBQKc4cAAUs7sbly9nRwDQVC+8EMeOZUcAdIcvGQUA\nAABaz4IDoJiqiuXl7AgAGunzn4933smOAOgUCw6AkqoquwCARur3Y2IiOwKgUyw4AIqpqlhZ\nyY4AoJEeeii++MXsCIBOseAAKKauY309OwKARjp6NI4ezY4A6BQLDoBi6jo2NrIjAGikft+B\nA2B/OXAAFFPXce1adgQAjfToo9kFAF3jiQoAAADQehYcAMVUVVy8mB0BQPN85SvxyCPZEQBd\nY8EBAAAAtJ4FB0Axw2EsLmZHANA8r78eL7yQHQHQNRYcACVtb2cXANA809PR72dHAHSNBQdA\nMTs7sbqaHQFA83z1q3HqVHYEQNc4cAAU0+vF/Hx2BADNMzubXQDQQZ6oABRT17G1lR0BQPMc\nPRoTE9kRAF1jwQFQzO5uXL6cHQFA87z9dnYBQAdZcAAAAACtZ8EBUExVxXvvZUcA0DD9fvz5\nn2dHAHSQBQdASVWVXQBAw/T7voADoAQLDoBiqipWVrIjAGiYhx6KL34xOwKggyw4AIqp61hf\nz44AoEkOHYqzZ7MjALrJggOgmLqOjY3sCACaZDBw4AAoxIEDoJjRKH71q+wIABrj6adjfj47\nAqCzPFEBAAAAWs+CA6CYqoqLF7MjAGiGM2fi+9/PjgDoMgsOAAAAoPUsOACKGQ5jcTE7AoBm\nePPNeOaZ7AiALrPgACimrmNzMzsCgGY4fDgGg+wIgC6z4AAoZjSKtbXsCACa4cEH49y57AiA\nLnPgAChpbi67AIBmOHIkuwCg4zxRASimrmNrKzsCgGY4ciQO+eMiQEE+ZAGK2d2NX/wiOwKA\nBpidja9/PTsCoOMsOAAAAIDWs+AAKGY4jKWl7AgAGuCBB+J738uOAOg4Cw6AkqoquwCABpic\nzC4A6D4LDoBiqipWVrIjAGiAP/3TmJjIjgDoOAcOgGImJuJLX8qOACDbsWOuGwAHwIEDoKTB\nILsAgGzepwAcCAcOgGJGo7hyJTsCgDyTk/Haa9kRAOPCl4wCAAAArWfBAVBMVcXFi9kRAOQZ\nDOIv/iI7AmBcWHAAlOQzFmCc9XrZBQBjxIIDoJjhMJaXsyMASDI1Ff/+32dHAIwRCw6AYuo6\nNjezIwBIMj0ds7PZEQBjxIIDoJjRKNbWsiMASHLhQtx3X3YEwBhx4AAoaW4uuwCAJDMz2QUA\n48UTFQAAAKD1LDgAitnZiUuXsiMASPL44/Hkk9kRAGPEggMAAABoPQsOgGKGw1hayo4AIMPE\nRPzDP2RHAIwXCw6Akra3swsAyNDvx/R0dgTAeLHgAChmZydWV7MjAMhw+nS89FJ2BMB4ceAA\nKKbfj4WF7AgAMpw+nV0AMHYcOABKGgyyCwDIMDGRXQAwdhw4AIoZjeLKlewIADI891ycOJEd\nATBefMkoAAAA0HoWHADFVFUsL2dHAJDhrbfi7ruzIwDGiwUHQEk+YwHGU6+XXQAwdiw4AIoZ\nDi04AMbUu+/GkSPZEQDjxYIDoJi6js3N7AgADtz0dMzOZkcAjB0LDoBiRqNYW8uOAODAzc/H\n/fdnRwCMHQcOgJKqKrsAgAN3+HB2AcA48kQFAAAAaD0LDoBidnbi0qXsCAAO1vR0/PEfZ0cA\njCMLDgAAAKD1LDgAihkO4yc/yY4A4GDdf3+8+mp2BMA4suAAKGl7O7sAgIM1NRWH/BERIIEP\nX4BidnZidTU7AoCD9eSTcddd2REA48iCA6CY0SiuXs2OAOAAHT0a589nRwCMKQsOgJIGg+wC\nAA7Q5GR2AcD4cuAAKGY0ig8/zI4A4AC99lp2AcD48kQFAAAAaD0LDoBiqiqWl7MjADgohw7F\nf/gP2REA48uCA6Akn7EA46PXyy4AGGsWHADFDIcWHABj5O2348KF7AiA8WXBAVBMXcf6enYE\nAAfi8OE4cSI7AmCsWXAAFFPXsbGRHQHAgTh1yoEDIJcDB0AxdR3XrmVHAHAgzp3LLgAYd56o\nAAAAAK1nwQFQTFXFz36WHQFAeefPx4svZkcAjDsLDgAAAKD1LDgAihkOY3ExOwKA8v7zf47b\nbsuOABh3FhwAJW1vZxcAUNhgEBMT2REAWHAAlLOzE6ur2REAFPbSS3H6dHYEAA4cAOX0ejE/\nnx0BQEmDgesGQEN4ogJQTF3H1lZ2BAAl9ftx7Fh2BAARFhwABe3uxuXL2REAFPPMM3HvvdkR\nAPwLCw4AAACg9Sw4AIqpqlhezo4AoIwf/SjOnMmOAOAjFhwAJVVVdgEAZUxOZhcA8DssOACK\nqapYWcmOAKCMH/4wpqezIwD4iAUHQDF1Hevr2REAFDA15X0KQNNYcAAUU9exsZEdAUABc3MO\nHABN48ABUExdx69/nR0BQAHz89kFAHycJyoAAABA61lwABRTVXHxYnYEAAX84Adx8mR2BAC/\nw4IDAAAAaD0LDoBihsNYXMyOAGC/nTwZf/d32REAfJwFB0AxdR2bm9kRAOy3qamYm8uOAODj\nLDgAihmNYm0tOwKA/Xb2bCwsZEcA8HEOHAAl+RMfQPccPpxdAMAn8EQFoJi6jq2t7AgA9ttg\nELOz2REAfJwFB0Axu7tx+XJ2BAD77YEH4oknsiMA+DgLDgAAAKD1LDgAihkOY2kpOwKA/fbG\nG/HYY9kRAHycBQdASVWVXQDAfpuYiH4/OwKAj7PgACimqmJlJTsCgP320ktx333ZEQB8nAUH\nQDF1Hevr2REA7Lfz56PXy44A4OMsOACKqevY2MiOAGC/nT+fXQDAJ3DgAChmNIpf/So7AoB9\n9frr5hsAzeSJCgAAANB6FhwAxVRVXLyYHQHA/vnGN3y9KEBjWXAAAAAArWfBAVDMcBjLy9kR\nAOyfd9+NI0eyIwD4ZBYcAMXUdWxuZkcAsE+OHo1D/joI0Fw+owGKGY1ibS07AoB98tRTcfx4\ndgQA1+XAAVDS3Fx2AQD74eRJ1w2AhvNEBaCYuo6trewIAPbD5KSbNUDDWXAAFLO7Gx98kB0B\nwH646654+eXsCAA+jQUHAAAA0HoWHADFDIextJQdAcB++I//0RMVgIaz4AAoqaqyCwC4ZRMT\n0e9nRwDwGSw4AIqpqlhZyY4A4Ja9+GI8+GB2BACfwYIDoJjRKK5ezY4A4JZduBCHD2dHAPAZ\nLDgAShoMsgsAuGXepwC0gQMHQDGjUVy5kh0BwK15/vmYmcmOAOCzeaICAAAAtJ4FB0AxVRXv\nv58dAcAtOHUqfv/3syMAuCEWHAAl+YwFaLVeL7sAgBtlwQFQzHAYy8vZEQDcgldeiccfz44A\n4IZYcAAUU9exuZkdAcBNmZqKubnsCAD2wIIDoJjRKNbWsiMAuCm33x4PP5wdAcAeOHAAlDQ5\nmV0AwE05ejS7AIC98UQFAAAAaD0LDoBidnbi0qXsCABuyle/GnffnR0BwB5YcAAAAACtZ8EB\nUMxwGEtL2REA3JR33ol7782OAGAPLDgAStrezi4A4KZMT0e/nx0BwB5YcAAUs7MTq6vZEQDc\nlFdfjSNHsiMA2AMHDoBi+v1YWMiOAGDvTpxw3QBoHQcOgJIGg+wCAPbukH8kA7SPz26AYkaj\n+OUvsyMA2LsvfSm7AIA98yWjAAAAQOtZcAAUU1WxvJwdAcAeHT8ef/EX2REA7JkFB0BJPmMB\n2qjXyy4AYM8sOACKGQ4tOADa58yZ+Df/JjsCgD2z4AAopq7j2rXsCAD2aGYmjh7NjgBgzyw4\nAIqp6/inf8qOAGCPTp1y4ABoIwcOgGIsOADa6Pjx7AIAboYnKgAAAEDrWXAAFFNV8dOfZkcA\nsEevvhqf/3x2BAB7ZsEBAAAAtJ4FB0Axw2EsLmZHALAXs7M+ugFayoIDoKTt7ewCAPZiaioO\n+RMgQCv5+AYoZmcnVlezIwDYi2eeiTvuyI4A4GY4cAAU0+/HwkJ2BAB7cf58dgEAN8mBA6Ck\nwSC7AIC96PWyCwC4SQ4cAMWMRvHhh9kRANyY2dn4gz/IjgDg5vmSUQAAAKD1LDgAiqmqWF7O\njgDgxjzwQLz2WnYEADfPggOgJJ+xAG3h2zcAWs6CA6CYqoqVlewIAG7MH/1RHD2aHQHAzbPg\nACimrmN9PTsCgBtw6FCcPZsdAcAtseAAKKauY2MjOwKAGzA97cAB0HYOHADF1HVcu5YdAcAN\nWFjILgDgVnmiAgAAALSeBQdAMVUVFy9mRwBwA954Iy5cyI4A4JZYcAAAAACtZ8EBUMxwGIuL\n2REA3IC//uuYn8+OAOCWWHAAlLS9nV0AwA2YmYleLzsCgFtiwQFQzM5OrK5mRwBwA771rTh8\nODsCgFviwAFQTK9n8AzQDof8qxig9TxRASimrmNrKzsCgBtw7Fj0+9kRANwSt2qAYnZ34/Ll\n7AgAbsAf/EHMzmZHAHBLLDgAAACA1rPgACimquK997IjAPgsMzPxwx9mRwBwqyw4AEqqquwC\nAD5Vr+cbRgG6wac5QDFVFSsr2REAfKpTp+LNN7MjANgHFhwAxdR1rK9nRwDwSQaDOH06OwKA\n/WTBAVBMXcfGRnYEAJ/kyBEHDoCOceAAKGY0in/+5+wIAD7JQw9lFwCwzzxRAQAAAFrPggOg\nmKqKixezIwD4JH/1VzE5mR0BwH6y4AAAAABaz4IDoJjhMBYXsyMA+C3Hj8ff/312BABFWHAA\nFFPXsbmZHQHAb5mbi6mp7AgAirDgAChmNIq1tewIAH7LwkKcPZsdAUARDhwAJc3NZRcA8Ftu\nuy27AIBSPFEBKKauY2srOwKA33LsWPT72REAFGHBAVDM7m784hfZEQD8xhe+ECdOZEcAUIoF\nBwAAANB6FhwAxQyHsbSUHQHAb3zta/Hii9kRAJRiwQFQUlVlFwDwGxMTvoADoMMsOACKqapY\nWcmOAOA3XnghFhayIwAoxYIDoJjRKP7P/8mOACAiIqan49y57AgACjLSAwAAAFrPExWAYkaj\nuHIlOwKAiIh4+eXsAgDK8kQFAAAAaD0LDoBiqiouXsyOACDiL/8ypqayIwAoy4IDoCSfsQBN\n0OtlFwBQnAUHQDHDYSwvZ0cAjL177om33sqOAKA4Cw6AYuo6NjezIwDG2/R0zM5mRwBwECw4\nAIoZjWJtLTsCYLzdcUfce292BAAHwYEDoKS5uewCgHF18mTcd192BAAHxxMVgGLqOra2siMA\nxtXkpCszwFix4AAoZnc3PvggOwJgXD36aDz9dHYEAAfHggMAAABoPQsOgGKGw1hayo4AGEvv\nvhunT2dHAHCgLDgASqqq7AKA8XPoUPR62REAHDQLDoBidnbi5z/PjgAYPy+8EOfPZ0cAcNAs\nOACKGY3i6tXsCIAxMzcX8/PZEQAksOAAKGkwyC4AGDOTk9kFAORw4AAoZjSKK1eyIwDGzIUL\ncfZsdgQACTxRAQAAAFrPggOgmKqK5eXsCIAxs7AQ3/pWdgQACSw4AEryGQtwwPxALMC4suAA\nKGY4tOAAOFCzs/E3f5MdAUAOCw6AYuo6NjezIwDGycmT2QUApLHgAChmNIq1tewIgLHR78fL\nL2dHAJDGgQOgpKrKLgAYGw88kF0AQCZPVAAAAIDWs+AAKGZnJy5dyo4AGA9f+1rceWd2BACZ\nLDgAAACA1rPgAChmOIyf/CQ7AmAMfO5z8b3vZUcAkMyCA6Ck7e3sAoAxMBjExER2BADJLDgA\nitnZidXV7AiAMfDlL8ftt2dHAJDMgQOgmH4/FhayIwDGwMmT2QUA5HPgAChpMMguABgDvV52\nAQD5HDgAihmN4h//MTsCoOvOnIm77sqOACCfLxkFAAAAWs+CA6CYqorl5ewIgK574IF47bXs\nCADyWXAAlOQzFuAA+A4OACw4AAoaDi04AIp79NF49dXsCADyWXAAFFPXsb6eHQHQaYNBnD6d\nHQFAI1hwABRT17GxkR0B0GlHjzpwAPD/OHAAFFPXce1adgRApy0sZBcA0BSeqAAAAACtZ8EB\nUExVxc9+lh0B0F0nTsQrr2RHANAUFhwAAABA61lwABQzHMbiYnYEQHd997vx1FPZEQA0hQUH\nQEnb29kFAB01NRWH/K0OgI/4vwJAMTs7sbqaHQHQUU88EXffnR0BQIM4cAAU0+/HvfdmRwB0\n1KlT2QUANIsnKgDF1HVsbWVHAHTU3FxMTmZHANAgFhwAxezuxuXL2REAHfXOO9kFADSLBQcA\nAADQehYcAMVUVSwvZ0cAdNGpU/Fv/212BADNYsEBUFJVZRcAdFG/HxMT2REANIsFB0AxVRUr\nK9kRAF10333x0kvZEQA0iwUHQDF1Hevr2REAXXT4cJw4kR0BQLNYcAAUU9exsZEdAdBFp087\ncADwMQ4cAMXUdVy7lh0B0DlPPhlnzmRHANA4nqj8X/buNsjuur77+Pec3c3mZrNJICQhMSgE\nI2hAlHslVUREblSsXgpFoEq1ar3sOECvEetOa8E4o84647REx7Kh07HDdFTamthaxapTcUB3\n4minpYxaHqRj4oNdO06mPf/d878eYBWBkLv97ff8z3m9HunmhvfMzuxmvvv5nwMAAAA0ngUH\nQDFVFTt3ZkcA9Jdrr43Nm7MjAOhFFhwAAABA41lwABTT6cTERHYEQL9YsiQ+9KHsCAB6lwUH\nQEkHD2YXAPSLdjuWLs2OAKB3WXAAFDM/H9/6VnYEQL8YH48LLsiOAKB3OXAAlDQ2ll0A0C/W\nrMkuAKCneUQFoJi6jtnZ7AiAfrFkSaxYkR0BQO+y4AAoZn4+9uzJjgDoF2edFWefnR0BQO+y\n4AAAAAAaz4IDoJhOJ3bsyI4A6BdXXBEXXZQdAUDvsuAAKKmqsgsA+sLQULTb2REA9DQLDoBi\nqiqmprIjAPrCy18eW7dmRwDQ0yw4AIqp69i3LzsCoC+cfHIMDWVHANDTLDgAiqnr2L8/OwKg\nL6xf78ABwDNz4AAoptuNxx7LjgBouI0bvbYoAEfCIyoAAABA41lwABRTVbFzZ3YEQMNt2xaX\nXpodAUADWHAAAAAAjWfBAVBMpxOTk+84bvAAACAASURBVNkRAI11/vnxildkRwDQGBYcAMXU\ndczMZEcANNb4eAz7aRwAR8r3DIBiut2Yns6OAGisCy6I8fHsCAAaw4EDoKSxsewCgGZ6znNc\nNwA4Kh5RASimrmN2NjsCoJmWL4/R0ewIAJrEggOgmPn5+PKXsyMAmunFL47nPjc7AoAmseAA\nAAAAGs+CA6CYTid27MiOAGigdeviXe/KjgCgYSw4AEqqquwCgAZqt2NoKDsCgIax4AAopqpi\naio7AqCBNm6Ma67JjgCgYRw4AIoZGort27MjABpoxYrsAgCax4EDoCTvcQhwDEZGsgsAaB4H\nDoBiut3Yuzc7AqCBzjknuwCA5vEiowAAAEDjWXAAFFNV8dnPZkcANM3VV8fatdkRADSPBQdA\nSb7GAhytViu7AIBGsuAAKKbTicnJ7AiApnnve2PZsuwIAJrHggOgmLqOmZnsCIBGabVizZrs\nCAAayYIDoJhuN6ansyMAGmVoKC69NDsCgEZy4AAoaWwsuwCgUc44I7sAgKbyiAoAAADQeBYc\nAMXMzcWuXdkRAM0xNhbXXZcdAUBTWXAAAAAAjWfBAVBMpxM7dmRHADTB298eGzdmRwDQbBYc\nACUdPJhdANAES5dGu50dAUCzWXAAFDM3F7t3Z0cANMGrXhWrVmVHANBsDhwAxbTbsW1bdgRA\nz1u50nUDgOPnwAFQ0uhodgFAzxsZyS4AoB84cAAU0+3G3r3ZEQA97+qrswsA6AdeZBQAAABo\nPAsOgGKqKiYnsyMAetjYWLz73dkRAPQJCw6AknyNBTiUViu7AIC+YsEBUEynY8EB8PQ2bYq3\nvCU7AoC+YsEBUExdx8xMdgRAT1q1KoaGsiMA6CsWHADF1HX8+MfZEQA96cwzY/ny7AgA+ooD\nB0AxdR0HDmRHAPSe5z/fdQOABecRFQAAAKDxLDgAipmbi127siMAes+VV8amTdkRAPQbCw4A\nAACg8Sw4AIrpdGJiIjsCoMdcfXVs354dAUAfsuAAKOngwewCgB6zZEkM+xkbAAvPdxeAYubm\nYvfu7AiAHnPRRbF5c3YEAH3IgQOgmHY7tm3LjgDoMatXZxcA0J8cOABKGh3NLgDoMe12dgEA\n/cmBA6CYbjcefDA7AqDHvOEN2QUA9CcvMgoAAAA0ngUHQDFVFZOT2REAPeb226PVyo4AoA9Z\ncACU5GsswBM5bQBQjAUHQDFVFVNT2REAPWN8PK67LjsCgL5lwQFQTF3Hvn3ZEQA9Y8mSWLcu\nOwKAvmXBAVBMXcf+/dkRAD1jbMyBA4ByHDgAiqnrOHAgOwKgZ5x8cnYBAP3MIyoAAABA41lw\nABRTVfHZz2ZHAPSM17wmTjghOwKAvmXBAQAAADSeBQdAMZ1OTExkRwD0hje8Ic4/PzsCgH5m\nwQFQ0sGD2QUAPWB4OJYsyY4AoM9ZcAAUMzcXu3dnRwD0gOc8x3wDgNIcOACKabVi8+bsCIAe\nsGlTdgEA/c8jKgDF1HXMzmZHAPSAZcti6dLsCAD6nAUHQDHz87FnT3YEQA/Yvj1OOSU7AoA+\nZ8EBAAAANJ4FB0AxVRX33JMdAZDtkkviBS/IjgCg/1lwAJRUVdkFAKna7Rgayo4AYCBYcAAU\nU1UxNZUdAZDqzDNj+/bsCAAGggUHQDF1Hfv2ZUcApDrxxFi2LDsCgIFgwQFQTF3H/v3ZEQCp\nxsYcOABYHA4cAMXUdfz859kRAHlGRmL16uwIAAaFR1QAAACAxrPgACimqmLnzuwIgDzr18eb\n3pQdAcCgsOAAAAAAGs+CA6CYTicmJrIjAPKceWbcfHN2BACDwoIDoJi6jpmZ7AiAPCtXxshI\ndgQAg8KCA6CYbjemp7MjAPKcc06sXZsdAcCgcOAAKGlsLLsAIM+wf2oCsHg8ogJQTF3H7Gx2\nBECesTGPqACwaJzVAYqZn489e7IjAJKMjHiPWAAWkwUHAAAA0HgWHADFdDqxY0d2BECSt7wl\ntmzJjgBggFhwAJRUVdkFABlaLa8wCsAi840HoJiqiqmp7AiADOvWxbXXZkcAMFgsOACKqevY\nty87AiDDqlWxcmV2BACDxYIDoJi6jk4nOwIgQ7ebXQDAwHHgACim2429e7MjABbd618frVZ2\nBAADxyMqAAAAQONZcAAUU1Wxc2d2BMDiOumkuO667AgABpEFB0BJvsYCg8bDKQAkseAAKKbT\nicnJ7AiAxXXZZXHeedkRAAwiCw6AYuo6ZmayIwAW16pVMTSUHQHAILLgACim243p6ewIgMV1\n4YWxcmV2BACDyIEDoKSxsewCgEXUbrtuAJDFIyoAxdR1zM5mRwAsolYrVq/OjgBgQFlwABQz\nPx+f+1x2BMBiWbs2rr02OwKAwWXBAQAAADSeBQdAMZ1O7NiRHQGwWM45J173uuwIAAaXBQdA\nSVWVXQBQXqsVw35sBkAy34oAipmbi/vvz44AKG/p0njNa7IjABh0DhwAxbTbsW1bdgRAeWvW\nZBcAgAMHQFGjo9kFAOV5PgWAHuC7EUAx3W7s3ZsdAVDelVdmFwCAFxkFAAAAms+CA6CYqop7\n7smOACjpvPPi3HOzIwAgwoIDoCxfY4H+1mplFwDAL1hwABTT6cTkZHYEQEnXXRfPfnZ2BABE\nWHAAFFTXMTOTHQFQ0vi4t1ABoEf4hgRQTLcb09PZEQAlnXderF6dHQEAEQ4cAGVVVXYBQEkr\nV2YXAMAveEQFAAAAaDwLDoBi5uZi167sCIBiXvOaWL8+OwIAfsGCAwAAAGg8Cw6AYjqduPvu\n7AiAMtasiZtuyo4AgF+x4AAo6eDB7AKAMkZGYmQkOwIAfsWCA6CYubnYvTs7AqCMs86KM87I\njgCAX3HgACim3Y5t27IjAMrYvDm7AAB+jQMHQEmjo9kFAGW029kFAPBrHDgAiul249FHsyMA\nytiwIbsAAH6NFxkFAAAAGs+CA6CYqorJyewIgAV1/vlx6aXZEQDwNCw4AEryNRboJ61WdgEA\nHJIFB0AxnY4FB9A/hobittuyIwDgkCw4AIqp69i3LzsCYIGMjsZJJ2VHAMAhWXAAFFPXsX9/\ndgTAAlm50oEDgF7mwAFQTF3HgQPZEQALYfnyOPfc7AgAeCYeUQEAAAAaz4IDoJiqinvvzY4A\nWAhnnhkvfWl2BAA8EwsOAAAAoPEsOACK6XRiYiI7AuA4LFkSH/pQdgQAHBELDoCSDh7MLgA4\nDkNDMTqaHQEAR8SCA6CYubnYvTs7AuA4bNzopTcAaAoHDoBi2u3Yti07AuA4nHZadgEAHCkH\nDoBiWq3YsCE7AuA4DPu3IgCN4ZsWQDHz87FnT3YEwHF405tiaCg7AoBj8bGPfez2229/+OGH\nzzvvvOyWReLAAVDM8HDccEN2BADAQPi7v/u71772tRHxgQ984M4771y0/+4ZZ5zxyCOPPOmD\n69ev/8lPfvLL//uXf/mXN95446H+hrm5uaFfvyZ/9atf/eQnP/nggw/Ozs6uW7fuxS9+8fve\n976Xv/zlCxr+TH74wx9OTEx89atfnZ2d3bx585vf/OY77rhj+fLlR/43HOGnY2E/aw4cAMVU\nVUxOZkcAHKutW+Paa7MjAI7IT3/607e//e1jY2M///nPF/+/3m63n3S/WLVq1RP/75YtW26+\n+eYn/al//dd/feihhy699NInXTfe//73f+QjHxkdHb3ooovWr1//05/+9J//+Z/POuusRTtw\n/OAHP9i+ffvPfvaza6655rTTTvvmN7951113ffWrX33ggQeWLVt2JH/DEX46Fvyz5sABUMzI\nSLzvfdkRAMeq3c4uADhS73jHO9rt9vve974/+ZM/Wfz/+sjIyK5du57hN1x88cUXX3zxkz54\n1VVXRcTv/u7vPvGDU1NTH/nIRy6++OK//uu/3rRp0+Mf7Ha7MzMzC1n8jG655ZbZ2dmpqanf\n/u3ffvy//pa3vOWv/uqvPv7xj//hH/7hkfwNR/jpWPDPmgMHQDFVFVNT2REAx+p5z4uXvSw7\nAuDwpqam7r///i9+8YuPPvpodsuReuyxx/7hH/7hpJNOev3rX//LD3Y6nTvuuGPFihVf+MIX\n1q9f/8uPt9vtE088cXHCpqenH3rooXPOOefx68bj//WPfvSj991336c+9akPfOADrVbrmf+G\nI/x0lPisOXAAFDM8HFddlR0BcExGR+Okk7IjAA7vP/7jP37/93//rW9969VXX/2JT3ziCP/U\nddddd9999919993vfOc7j7+h2+1++MMf/uEPf7hs2bKzzz77jW984wknnPDMf+TTn/50t9t9\n61vfumTJkl9+8IEHHvjJT35yww03rFq16r777vvBD36wbNmyCy+88BWveMVhzwoL5YEHHoiI\nK6+88okf3LRp09lnn713795///d/f97znvcMf/wIPx3H9lk7LAcOgGLqOvbvz44AOCYrVzpw\nAL2v2+3efPPNq1evnkx94bOqqj7wgQ/88v/eeuutn/70p6+//vpD/f65ubl77rmn1Wq9/e1v\nf+LHH3744Yg48cQTzz777CfuGi6++OInbTrKefwFU596xdi6dethDxxH+Oko91lz4AAopq7j\nwIHsCIBjsmZNdgHA4X384x//xje+8eUvf/lJL+p5WDfffPNFF1300pe+9Pgbbr755vPPP3/b\ntm2rVq360Y9+tHPnzj/7sz+78cYbn/WsZ23fvv1p/8jf/M3f/OQnP3nlK195+umnP/HjBw4c\niIg//dM/Pf3007/2ta+dd955P/7xj2+99dZ//Md/vO666772ta8df+1h/exnP4unvEhqRKxe\nvToiZmdnn+HPHuGn45g/a4flwAFQzNBQ/Pq6DwCAhfL973//gx/84Dvf+c7LL7/8aP/slVde\neeUC/Tvt/e9//y//9wte8IJPfvKTq1atuuuuu3bs2HGoA8enPvWpeMrLi0bE/Px8RLRarfvv\nv/+MM86IiLPOOusLX/jC1q1b/+mf/uk73/nOeeedtyDNx6Cu68fbDvUbjvDTcTyftcNy4AAo\npqpi587sCIBj8rKXxdlnZ0cAHFJd1zfeeOPGjRs/+tGPZrc82S233HLXXXc99NBDT/urP/rR\nj77yla+sX7/+da973ZN+ac2aNRFxxhlnPH7deNyKFSsuv/zye++9d3EOHI+vKh7fcTzRoZYd\njzvCT0fpz5oDB0AxIyPxf/9vdgQAQB+an5//3ve+FxErV6580i/dddddd9111y233PKZz3wm\nI+0XT3P8z//8z9P+6qc//em6rt/2treNjIw86Zcef3mLx//4U//C//7v/1741qd4vOHxV+J4\nosdfE2Tr1q1P+6eO8NNR+rPmwAFQTKcTExPZEQDH5M47Y9g/FIHe1W63b7nllid98F/+5V++\n/e1vn3POOeeee+6hHg9ZBF//+tcjYsuWLU/9paqqpqamnvryoo+77LLLWq3Wv/3bv1VV9cTz\nx/e///2IOPXUU4sl/8orXvGKiPj7v//7D3/4w7/84H/+539+73vf27Rp06EOHEf46Sj9WfN9\nC6CYJUscOIBGWro02u3sCIBn0m63n/qj/k984hPf/va3r7766jvvvPOZ//iXvvSlRx555LLL\nLjvrrLOOJ+Phhx8eHR09+wnP9H3nO9/5vd/7vYi48cYbn/r7v/CFLxw4cOCKK6542mvFpk2b\nXv/613/+85+/6667/uiP/ujxD37xi1984IEH1q5d+8pXvvJ4Uo/Qi1/84gsuuOChhx76i7/4\ni5tuuikiut3uH/zBH3S73Xe+851PfA2OXbt2zc7O/tZv/da6deuO8NNxnJ+1w3LgAChmfj6+\n9a3sCICjd8EFMT6eHQFQ0L333nvffffdfffdx3ng+PrXv3777bdv2bLl1FNPHR8f//GPf7x3\n7966rl/72te+973vfervP9TLi/7SJz/5yenp6T/+4z/+8pe//OIXv/ixxx7bs2fPyMjIZz7z\nmRUrVhxP6pH78z//80suueStb33r5z//+VNPPfWb3/zmd7/73QsvvPDWW2994m+78847f/jD\nH15yySXr1q1bnLDDcuAAKGlsLLsA4CiNjrpuAByhyy677O1vf/u3v/3t6enp//qv/1q9evUr\nX/nKm2666YYbbnjqG448+uijX/va104++eTXvOY1h/oLN27c+PDDD3/oQx/627/92+985zvj\n4+Ove93r7rjjjsV8/5Rt27Z997vf/eAHP/iVr3zlS1/60rOe9aw77rjjjjvuWLZs2aI1HJvW\n4+/1AsDCq+t4xrcKB+hF7XYc4kXyAWiQj33sY7fffvvDDz+c+Oayi8yCA6CY+fnYsyc7AuBo\ntNtx/fXZEQBwLBw4AIoZHo4bbsiOAACAgeDAAVBMpxN3350dAXA0hobiPe/JjgCAY+E1OABK\nqqrsAoAj1mrFsJ9+AfSJb33rW1/+8pff8Y53bNy4MbtlkThwABRTVTE1lR0BcMROOCHe+Mbs\nCAA4Rg4cAMXUdezblx0BcMSWLIl167IjAOAYWSECFFPXsX9/dgTAEVuxwoEDgOZy4AAoptuN\nxx7LjgA4Ms96VpxxRnYEABw7j6gAAAAAjWfBAVBMVcXOndkRAEfm0ktj27bsCAA4dhYcAAAA\nQONZcAAU0+nExER2BMCR+Z3fidNPz44AgGNnwQFQTF3HzEx2BMARGB2NFSuyIwDguFhwABTT\n7cb0dHYEwBHYtCnOPDM7AgCOiwMHQEljY9kFAEdg7drsAgA4Xh5RASimrmN2NjsC4AisWBFL\nlmRHAMBxseAAKGZ+Pr7xjewIgCNw4YWxYUN2BAAcFwsOAAAAoPEsOACK6XRix47sCIDDWbEi\nbrstOwIAjpcFB0BJVZVdAHA4rVYM+6EXAI3nmxlAMVUVU1PZEQCHs3JlXH99dgQAHC8LDoBi\nut145JHsCIBndNJJ3iMWgP5gwQFQ0uhodgHAMxoayi4AgIXhwAFQTLcbe/dmRwA8o3POiTVr\nsiMAYAF4RAUAAABoPAsOgGKqKnbuzI4AeEZvfnOsW5cdAQALwIIDoCRfY4Ee12plFwDAwrDg\nACim04nJyewIgENbuTLe/e7sCABYGBYcAMXUdczMZEcAHEK7HatXZ0cAwIKx4AAoptuN6ens\nCIBDGB2N7duzIwBgwThwAJQ0NpZdAHAIp52WXQAAC8kjKgAAAEDjWXAAFDM3F7t2ZUcAPJ2x\nsbjuuuwIAFhIFhwAAABA41lwABTT6cSOHdkRAE/n9NPjhhuyIwBgIVlwAJR08GB2AcBTtNux\ndGl2BAAsMAsOgGLm5mL37uwIgKdYtSpe9arsCABYYA4cAMW027FtW3YEwFOsWJFdAAALz4ED\noKTR0ewCgKcYGckuAICF58ABUEy3G3v3ZkcAPMXWrXHyydkRALDAvMgoAAAA0HgWHADFVFVM\nTmZHAPy65cvjPe/JjgCAhWfBAVCSr7FAD2q1sgsAYOFZcAAU0+lYcAA9Z3w83vWu7AgAWHgW\nHADF1HXMzGRHADzB6Kj3iAWgX1lwABTT7cb0dHYEwBNs2hRnnpkdAQBFOHAAlFRV2QUA/2vN\nGtcNAPqYR1QAAACAxrPgAChmbi527cqOAPhfGzfGVVdlRwBAKRYcAAAAQONZcAAU421igZ7y\n8pfHhRdmRwBAKRYcACUdPJhdAPC/li/PLgCAgiw4AIqZm4vdu7MjACIiotWKN74xOwIACnLg\nACim3Y5t27IjACIiYv367AIAKMuBA6Ck0dHsAoCIiBgayi4AgLIcOACK6XbjwQezIwAiIuLy\ny7MLAKAsLzIKAAAANJ4FB0AxVeVtYoGesGpV/O7vZkcAQFkWHAAl+RoL9IhWK7sAAMqy4AAo\nptOx4AB6woknxu/8TnYEAJRlwQFQTF3Hvn3ZEQARJ53kTZ0A6HsWHADF1HXs358dARCxerUD\nBwB9z4EDoJi6jgMHsiMAIp773OwCACjOIyoAAABA41lwABRTVfH5z2dHAAPv1FPjgguyIwCg\nOAsOAAAAoPEsOACK6XRiYiI7Ahh4b3tbbN2aHQEAxVlwAJR08GB2ATDwli6Ndjs7AgCKs+AA\nKGZuLnbvzo4ABt6rXx0rV2ZHAEBxDhwAxbRasXlzdgQwwEZH40Uvyo4AgEXiERWAYuo6Zmez\nI4AB1m7HqlXZEQCwSCw4AIqZn489e7IjgAH2whc6cAAwOCw4AAAAgMaz4AAopqpicjI7Ahhg\nt93m/VMAGBwWHAAlVVV2ATCoWq0Y9qMsAAaIb3sAxVRVTE1lRwCDavXqeNObsiMAYPFYcAAU\nU9exb192BDCoxsZi9ersCABYPBYcAMXUdezfnx0BDKq6duAAYKA4cAAUU9fx859nRwCD6txz\nswsAYFF5RAUAAABoPAsOgGKqKnbuzI4ABtLznhevelV2BAAsKgsOAAAAoPEsOACK6XRiYiI7\nAhhIExOxfHl2BAAsKgsOgGLqOmZmsiOAwTM8HOPj2REAsNgsOACK6XZjejo7Ahg8q1bF+edn\nRwDAYnPgAChpbCy7ABg8W7ZkFwBAAo+oABRT1zE7mx0BDJ4VK2LJkuwIAFhsFhwAxczPx549\n2RHA4Hn1q+PEE7MjAGCxWXAAAAAAjWfBAVBMpxM7dmRHAAPm3HPjmmuyIwAggQUHQElVlV0A\nDJh2O4aGsiMAIIEFB0AxVRVTU9kRwIA566y4+OLsCABIYMEBUExdx7592RHAgFm5Mlatyo4A\ngAQWHADF1HXs358dAQyYdtuBA4DB5MABUEy3G489lh0BDJKxsTj33OwIAMjhERUAAACg8Sw4\nAIqpqti5MzsCGCRr18b112dHAEAOCw4AAACg8Sw4AIrpdGJyMjsCGCSve12ccUZ2BADksOAA\nKKauY2YmOwIYJGNjsWRJdgQA5LDgACim243p6ewIYJC88IVx0knZEQCQw4EDoKSxsewCYGCs\nWeO6AcAg84gKQDF1HbOz2RHAwBgZcVQFYJBZcAAUMz8fn/tcdgQwMC64IM4+OzsCANJYcAAA\nAACNZ8EBUEynEzt2ZEcAg2Hr1rj++uwIAMhkwQFQUlVlFwCDYWgo2u3sCADIZMEBUExVxdRU\ndgQwGF760njBC7IjACCTBQdAMd1uPPJIdgQwGE49NZYuzY4AgEwWHAAljY5mFwCDodXKLgCA\nZA4cAMV0u7F3b3YEMABWrIjTTsuOAIBkHlEBAAAAGs+CA6CYqorPfjY7AhgAr351rF+fHQEA\nySw4AEryNRZYBF6AAwAsOAAK6nRicjI7Auh3q1bFO9+ZHQEA+Sw4AIqp65iZyY4A+t3ISKxc\nmR0BAPksOACK6XZjejo7Auh3a9fGOedkRwBAPgcOgJKqKrsA6Hdr1mQXAEBP8IgKAAAA0HgW\nHADFzM3Frl3ZEUBf27IlLr00OwIAeoIFBwAAANB4FhwAxXQ6sWNHdgTQp17ykrj88uwIAOgh\nFhwAJR08mF0A9KOhoRgdzY4AgN5iwQFQzNxc7N6dHQH0ow0bYvv27AgA6C0OHADFtNuxbVt2\nBNCPvDUsADyFAwdASTbkQAnD/gkHAE/muyNAMd1uPPpodgTQj664IrsAAHqOFxkFAAAAGs+C\nA6CYqorJyewIoO+sWBG/93vZEQDQcyw4AEryNRZYcK1WdgEA9CILDoBiOh0LDmCBvfCF8epX\nZ0cAQC+y4AAopq5j377sCKC/rFkTK1ZkRwBAL7LgACimrmP//uwIoL8sXerAAQBPy4EDoJi6\njgMHsiOAPvLsZ8fatdkRANCjPKICAAAANJ4FB0AxVRX33psdAfSR//N/YtWq7AgA6FEWHAAA\nAEDjWXAAFNPpxMREdgTQRz7wgVi5MjsCAHqUBQdASQcPZhcA/aLdjqVLsyMAoHdZcAAUMzcX\nu3dnRwD9Ynw8rrgiOwIAepcDB0Ax7XZs25YdAfQL8w0AeEYOHAAljY5mFwD9YmQkuwAAepoD\nB0Ax3W48+GB2BNAvXvSi7AIA6GleZBQAAABoPAsOgGKqKiYnsyOAfnHbbdFuZ0cAQO+y4AAo\nyddYYEG0WtkFANDrLDgAiqmqmJrKjgD6wm//dixZkh0BAD3NggOgmLqOffuyI4DmW7Ei1qzJ\njgCAXmfBAVBMXcf+/dkRQPOtXevAAQCH5cABUExdx4ED2RFA8z3/+dkFANAAHlEBAAAAGs+C\nA6CYqoqdO7MjgIb7zd+MTZuyIwCgASw4AAAAgMaz4AAoptOJiYnsCKDh3vWuePazsyMAoAEs\nOABKOngwuwBorFYrli3LjgCAxrDgAChmbi52786OABprbCyuvDI7AgAaw4EDoJhWKzZvzo4A\nGuuFL8wuAIAm8YgKQDF1HbOz2RFAY42Px9BQdgQANIYFB0Ax8/OxZ092BNBAW7fG+ednRwBA\nw1hwAAAAAI1nwQFQTFXFPfdkRwANdMEF8aIXZUcAQMNYcACUVFXZBUADDQ9Hq5UdAQANY8EB\nUExVxdRUdgTQQNdeG+vWZUcAQMNYcAAUU9exb192BNA0S5fG2rXZEQDQPBYcAMXUdezfnx0B\nNM2qVQ4cAHAMHDgAiul247HHsiOApvEGsQBwTDyiAgAAADSeBQdAMVUVO3dmRwBN8+Y3e4VR\nADgGFhwAAABA41lwABTT6cTERHYE0CinnBLvfnd2BAA0kgUHQDF1HTMz2RFAo4yMxMqV2REA\n0EgWHADFdLsxPZ0dATTK2rVxzjnZEQDQSA4cACWNjWUXAI1y6qnZBQDQVB5RASimrmN2NjsC\naJSxsRgZyY4AgEay4AAoZn4+vvGN7AigOc49N9asyY4AgKay4AAAAAAaz4IDoJhOJ3bsyI4A\nGuKKK+Kii7IjAKDBLDgASqqq7AKgCYaGot3OjgCAZrPgACimqmJqKjsCaIILLvDusABwnCw4\nAIrpduORR7IjgCbYvNm7SgPAcbLgAChpdDS7AGgCz6cAwHFz4AAoptuNvXuzI4AmuOSSWL48\nOwIAms0jKgAAAEDjWXAAFFNVCwia6QAAIABJREFUsXNndgTQ89785li3LjsCABrPggOgJF9j\ngcNqtbILAKAfWHAAFNPpxORkdgTQ2170onjVq7IjAKAfWHAAFFPXMTOTHQH0tqVLvbwoACwI\nCw6AYrrdmJ7OjgB62ymnxNat2REA0A8cOABKGhvLLgB62Pi46wYALBSPqAAUU9cxO5sdAfSw\n4eFYuTI7AgD6hAUHQDHz8/G5z2VHAD3s1FPjssuyIwCgT1hwAAAAAI1nwQFQTKcTO3ZkRwA9\n7B3viJNPzo4AgD5hwQFQUlVlFwA9aXg4Wq3sCADoKxYcAMXMzcX992dHAD1p+/bYsCE7AgD6\nigUHQDHdbjzySHYE0HvOPDO7AAD6kAUHQEmjo9kFAAAwEBw4AIrpdmPv3uwIoMe0WnHaadkR\nANCHPKICAAAANJ4FB0AxVRWTk9kRQI9pt+O227IjAKAPWXAAlORrLPBU3iAWAAqw4AAoptOx\n4ACebOXKePe7syMAoA9ZcAAUU9cxM5MdAfSYZcti2bLsCADoQxYcAMV0uzE9nR0B9JjTTvMu\nKgBQggMHQElVlV0A9BgvwAEAZXhEBQAAAGg8Cw6AYubmYteu7Aigx9x8c4yMZEcAQB+y4AAA\nAAAaz4IDoJhOJ+6+OzsC6DHveU8MDWVHAEAfsuAAKOngwewCoJe0Wt4jFgAKseAAKGZuLnbv\nzo4AesnSpfGa12RHAEB/cuAAKKbdjm3bsiOAXmK+AQDFOHAAlDQ6ml0A9BLvnwIAxThwABTT\n7caDD2ZHAL3klFNi06bsCADoT15kFAAAAGg8Cw6AYqoqJiezI4Be8rKXxYUXZkcAQH+y4AAo\nyddY4IlarewCAOhbFhwAxXQ6FhzAr2zYEDffnB0BAH3LggOgmLqOffuyI4CesX69d1EBgHIs\nOACKqevYvz87AugZJ57owAEA5ThwABRT13HgQHYE0BvWrIlly7IjAKCfeUQFAAAAaDwLDoBi\nqio+//nsCKAHXHpprFuXHQEAfc6CAwAAAGg8Cw6AYjqdmJjIjgCy3XFHjI9nRwBA/7PgACjp\n4MHsAiDV8HAsWZIdAQADwYIDoJi5udi9OzsCSHXaaXHuudkRADAQHDgAimm1YvPm7Aggz5Yt\ncdJJ2REAMCg8ogJQTF3H7Gx2BJBnxQrPpwDAorHgAChmfj727MmOAPJce60DBwAsGgsOAAAA\noPEsOACKqaqYnMyOAJJs2BA33ZQdAQADxIIDoKSqyi4AkgwPR6uVHQEAA8SCA6CYqoqpqewI\nIMm118a6ddkRADBALDgAiqnr2LcvOwLIMD4e4+PZEQAwWCw4AIqp69i/PzsCyDA05MABAIvM\ngQOgmLqOAweyI4BF99znxsknZ0cAwMDxiAoAAADQeBYcAMVUVezcmR0BLLpbbonly7MjAGDg\nWHAAAAAAjWfBAVBMpxMTE9kRwKL7oz+KpUuzIwBg4FhwAJR08GB2AbC4Wq1Ytiw7AgAGkQUH\nQDHz8/Gtb2VHAItrdDS2b8+OAIBB5MABUNLYWHYBsLie97zsAgAYUB5RASimrmN2NjsCWFxj\nYzEykh0BAIPIggOgmPn52LMnOwJYXJddFhs2ZEcAwCCy4AAAAAAaz4IDoJhOJ3bsyI4AFtH1\n18fWrdkRADCgLDgASqqq7AJgEQ0PR6uVHQEAA8qCA6CYqoqpqewIYBFdc01s3JgdAQADyoID\noJi6jn37siOAxTI2FqtXZ0cAwOCy4AAopq5j//7sCGAROXAAQB4HDoBiut147LHsCGBRnHBC\nnHtudgQADDSPqAAAAACNZ8EBUExVxc6d2RHAotiyJa66KjsCAAaaBQcAAADQeBYcAMV0OjE5\nmR0BLIqrroqzzsqOAICBZsEBUExdx8xMdgSwKE44IbsAAAadBQdAMd1uTE9nRwCL4rLLotXK\njgCAgebAAVDS2Fh2AVBeu+26AQDpPKICUExdx+xsdgRQXqsVq1dnRwDAoLPgAChmfj4+97ns\nCKC8LVvi0kuzIwBg0FlwAAAAAI1nwQFQTKcTO3ZkRwDlXX99bN2aHQEAg86CA6CkqsouAMob\nHvYiowCQzoIDoJiqiqmp7AigvGuuiY0bsyMAYNBZcAAU0+3GI49kRwDlnX56jIxkRwDAoLPg\nAChpdDS7ACjP8ykA0AMcOACK6XZj797sCKCw3/iNGPYPKgDI5xEVAAAAoPH8wAGgmKqKz342\nOwIoadmyeNObsiMAgAgLDoCyfI2FvucFOACgN1hwABTT6cTkZHYEUNILXhDXXJMdAQBEWHAA\nFFTXMTOTHQEU02rFmjXZEQDAL1hwABTT7cb0dHYEUMzQUFx6aXYEAPALDhwAJY2NZRcAxZx4\nYnYBAPArHlEBAAAAGs+CA6CYubnYtSs7AihmyxaPqABA77DgAAAAABrPggOgmE4nduzIjgCK\nee97vYsKAPQOCw6Akg4ezC4Ailm2LFqt7AgA4BcsOACKmZuL3buzI4Birrkmli3LjgAAfsGB\nA6CYdju2bcuOAMrYtMl1AwB6igMHQEmjo9kFQBntdnYBAPBrHDgAiul2Y+/e7AiggFNOidNO\ny44AAH6NFxkFAAAAGs+CA6CYqorJyewIYEHdemsMDWVHAABPw4IDoCRfY6HPeF9YAOhVFhwA\nxXQ6FhzQV9aujVtuyY4AAJ6eBQdAMXUdMzPZEcDCGR6O8fHsCADg6VlwABRT1/HjH2dHAAtn\nbMyBAwB6lgMHQDF1HQcOZEcAC6fbzS4AAA7JIyoAAABA41lwABQzNxe7dmVHAAvnbW+Ldjs7\nAgB4ehYcAAAAQONZcAAU0+nExER2BLBAbropnv/87AgA4JAsOABKOngwuwBYIKOjMTSUHQEA\nHJIFB0Axc3Oxe3d2BLBALrkkTj45OwIAOCQLDoBiut145JHsCOC4rVkTGzZkRwAAh2HBAVDS\n6Gh2AXDchv17CQAawDdsgGK63XjwwewI4Lht2xZr12ZHAACH4REVAAAAoPEsOACKqaqYnMyO\nAI7P9u1x8cXZEQDA4VlwAJTkayw0WquVXQAAHCkLDoBiqiqmprIjgONw5ZWxeXN2BABwRCw4\nAIqp69i3LzsCOCZDQ3HyydkRAMBRsOAAKKauY//+7AjgmCxZ4sABAM3iwAFQTF3HgQPZEcAx\n2bIluwAAODoeUQEAAAAaz4IDoJiqis9+NjsCOCa/+ZuxcmV2BABwFCw4AAAAgMaz4AAoptOJ\niYnsCOCYTEzE8uXZEQDAUbDgACjp4MHsAuDotduxdGl2BABwdCw4AIqZm4vdu7MjgKO3enVc\nfnl2BABwdBw4AIpptWLz5uwI4OiNj2cXAABHzSMqAMXUdczOZkcAR2942FuoAEDjWHAAFDM/\nH3v2ZEcAR2/7dgcOAGgcCw4AAACg8Sw4AIqpqrjnnuwI4CitXh1vfnN2BABw1Cw4AEqqquwC\n4Ci12zE0lB0BABw1Cw6AYqoqpqayI4CjtG1bvOQl2REAwFGz4AAopq5j377sCOAorVkTK1Zk\nRwAAR82CA6CYuo79+7MjgKO0bJkDBwA0kQMHQDF1HT//eXYEcATWrYszz8yOAACOi0dUAAAA\ngMaz4AAopqpi587sCOAI3HRTrFqVHQEAHBcLDgAAAKDxLDgAiul0YmIiOwI4nEsvjSuuyI4A\nAI6XBQdAMXUdMzPZEcDhjI/HsB/5AEDj+XYOUEy3G9PT2RHA4Zx/vhfgAIA+4MABUNLYWHYB\n8IyGhlw3AKA/eEQFoJi6jtnZ7AjgGbVasXp1dgQAsAAsOACKmZ+PPXuyI4BndM45DhwA0B8s\nOAAAAIDGs+AAKKbTiR07siOAQ3vHO+Lkk7MjAICFYcEBUFJVZRcAhzY8HK1WdgQAsDAsOACK\nqaqYmsqOAA7tDW+IE0/MjgAAFoYFB0AxdR379mVHAIcwPBwbNmRHAAALxoIDoJi6jk4nOwI4\nBD/jAYD+4sABUEy3G3v3ZkcAh7BlS2zenB0BACwYj6gAAAAAjWfBAVBMVcXOndkRwCFcdFGc\nf352BACwYCw4AEryNRZ6mfeIBYA+YsEBUEynE5OT2RHAIbziFRYcANBPLDgAiqnrmJnJjgAO\nYeXKGBnJjgAAFowFB0Ax3W5MT2dHAIdwzjmxdm12BACwYBw4AEoaG8suAA5h2L+CAKCveEQF\noJi6jtnZ7AjgEMbGPKICAP3Ezy4Aipmfj899LjsCeIqzz44LLsiOAAAWmAUHAAAA0HgWHADF\ndDqxY0d2BPAUt98ey5dnRwAAC8yCA6CkqsouAJ6g1fLaogDQr3yPByhmbi7uvz87AniCkZG4\n9trsCACgCAcOgGLa7di2LTsCeALv3AwA/cuBA6Ck0dHsAuAJvC8sAPQvBw6AYrrd2Ls3OwL4\nXyedFNu3Z0cAAKV4kVEAAACg8Sw4AIqpqrjnnuwIICIirr021q/PjgAACrLgACjJ11joEa1W\ndgEAUJYFB0AxnU5MTmZHABERcdttMTSUHQEAFGTBAVBMXcfMTHYEEDE0FKtWZUcAAGVZcAAU\n0+3G9HR2BBCxcmVceGF2BABQlgMHQElVlV0ARGzYkF0AABTnERUAAACg8Sw4AIqZm4tdu7Ij\nYOC1WnHLLdkRAEBxFhwAAABA41lwABTT6cTdd2dHwMC77rpYvz47AgAozoIDoKSDB7MLYLAN\nDcXoaHYEALAYLDgAipmbi927syNgsG3YENu3Z0cAAIvBggOgmG43HnkkOwIG2Pr1ccIJ2REA\nwCKx4AAoyTYeEg0NZRcAAIvHgQOgmG43Hn00OwIG0sqV8ZKXZEcAAIvKIyoAAABA41lwABRT\nVTE5mR0BA+Y5z4k3vSk7AgBIYMEBUJKvsbD4Wq3sAgAggQUHQDGdjgUHLLYLL4yXvzw7AgBI\nYMEBUExdx7592REwSFaujFWrsiMAgBwWHADF1HXs358dAYOk3XbgAICB5cABUExdx4ED2REw\nGF7wgjjllOwIACCTR1QAAACAxrPgACimquLee7MjYABccUVs3pwdAQAks+AAAAAAGs+CA6CY\nTicmJrIjYAC85z3xrGdlRwAAySw4AEo6eDC7APrdyEiMjGRHAAD5LDgAipmbi927syOg351x\nRpx1VnYEAJDPgQOgmHY7tm3LjoB+9+xnZxcAAD3BgQOgmFYrNmzIjoB+t2RJdgEA0BMcOACK\nmZ+PPXuyI6BPLV8er399dgQA0EO8yCgAAADQeBYcAMVUVUxOZkdAn3rjG+O007IjAIAeYsEB\nUFJVZRdAP/K+sADAU1hwABRTVTE1lR0B/egtb4nly7MjAIDeYsEBUExdx7592RHQd5YsiXXr\nsiMAgJ5jwQFQTF3H/v3ZEdB3Vqxw4AAAnsqBA6CYuo4DB7IjoO9cckl2AQDQizyiAgAAADSe\nBQdAMVUVO3dmR0AfufzyOOOM7AgAoEdZcAAAAACNZ8EBUEynExMT2RHQfGvWxP/7f9kRAECv\ns+AAKOngwewCaLjh4ViyJDsCAGgACw6AYubn41vfyo6Ahtu4MZ7//OwIAKABHDgAShobyy6A\nhnvOc7ILAIBm8IgKQDF1HbOz2RHQcOPjMTSUHQEANIAFB0Ax8/OxZ092BDTZNde4bgAAR8iC\nAwAAAGg8Cw6AYjqduPvu7AhorKuuiuc+NzsCAGgMCw6AkqoquwAaa3g4Wq3sCACgMSw4AIqp\nqpiayo6Axrriinj2s7MjAIDGsOAAKKauY9++7AhophNOiOXLsyMAgCax4AAopq5j//7sCGim\n5csdOACAo+LAAVBMtxuPPZYdAQ20bVuccEJ2BADQMB5RAQAAABrPggOgmKqKnTuzI6BpLrkk\nXvSi7AgAoHksOAAAAIDGs+AAKKbTiYmJ7AholCVL4kMfyo4AABrJggOgmLqOmZnsCOh5rVas\nWZMdAQA0ngUHQDHdbkxPZ0dAz1uyJH7jN7IjAIDGc+AAKGlsLLsAet7y5dkFAEA/8IgKQDF1\nHbOz2RHQ84aGYnw8OwIAaDwLDoBi5ufjG9/IjoCed8op3hcWADh+FhwAAABA41lwABTT6cSO\nHdkR0JPGxuLWW7MjAIC+YsEBUFJVZRdAT2q1YthPWQCAheTfFgDFVFVMTWVHQE8aH4/rrsuO\nAAD6igUHQDHdbvz/9u48Lsp67//455qBYRFZAhFxQdBUwp1cCIgQzSy5pSwVyWPdnUqLFk09\nlZbyEFuOudydlJNaalZHj93uS7cIWJZklksdO663mfuSQgLeAsP8/rh+zWMaFoeB4eLC1/Mv\n5zvf65rPXPP1Ynjzvb7X4cNaFwE0Sp07i8GgdREAAKBJYQYHALiSh4fWFQCNkqJoXQEAAGhq\nCDgAwGUqKmT/fq2LABoZDw954AGtiwAAAE0Ql6gAAAAAAADdYwYHALhMWZn8/e9aFwE0GsOH\nS2io1kUAAIAmixkcAOBKnGMBFYtuAAAAF2MGBwC4TGmpzJundRFAIxAeLiNGaF0EAABo4pjB\nAQAuY7HI1ataFwFozcdHTCatiwAAAE0fMzgAwGUqKmTvXq2LALTWvbsEB2tdBAAAaPoIOADA\nlXx8tK4AaHDNm0tUlNZFAACAWw6XqAAAAAAAAN1jBgcAuEx5uSxbpnURQINr107uvVfrIgAA\nwC2HGRwAAAAAAED3mMEBAC5TWipvvql1EUCDi4+XAQO0LgIAANxymMEBAK5UUqJ1BUCD8/QU\ng0HrIgAAwC2HGRwA4DLl5bJ5s9ZFAA0uMVGCgrQuAgAA3HIIOADAZQwG6dpV6yKAhuXmRroB\nAAA0QcABAK7k4aF1BUDDMhq1rgAAANyiCDgAwGUqKmT/fq2LABpWcLCEhWldBAAAuBWxyCgA\nAAAAANA9ZnAAgMuUlcm8eVoXATQUHx955hmtiwAAALcuZnAAgCtxjsWtQ1G0rgAAANzSmMEB\nAC5TWsoMDtwq+vSRAQO0LgIAANzSmMEBAC5jscjVq1oXATQILy/x8tK6CAAAcEtjBgcAuIzF\nIidOaF0E0CBCQwk4AACAtgg4AMBlLBa5eFHrIgDXGzxYDAatiwAAALc6LlEBAAAAAAC6xwwO\nAHCZ8nJZtkzrIgAX8/OTRx7RuggAAABmcAAAAAAAAP1jBgcAuAy3icWt4LnnxNtb6yIAAACY\nwQEALlVSonUFQH3z9GRJUQAA0AgxgwMAXKa8XDZv1roIoL4lJkpQkNZFAAAA2GMGBwC4TEWF\nHD6sdRFAverQQUwmrYsAAACoAjM4AMCVPDy0rgCoV4qidQUAAABVI+AAAJepqJD8fK2LAOpV\nq1bi7q51EQAAAFXgEhUAAAAAAKB7zOAAAJcpK+M2sWg6Ro6UsDCtiwAAAKgWMzgAwJU4x6Jp\nYOkNAADQ6DGDAwBcprSUGRxoIkaNYvoGAABo5JjBAQAuY7HImTNaFwHUh1atxGjUuggAAICa\nMIMDAFzGYpELF7QuAqgPLVsScAAAgEaOgAMAXMZikYsXtS4CqA+kGwAAoNHjEhUAAAAAAKB7\nzOAAAJcpK5M1a7QuAqibESO4hQoAANAFZnAAAAAAAADdYwYHALhMaam8/rrWRQB10KOHpKZq\nXQQAAIBDmMEBAK5UUqJ1BYCzTCZx4w8hAABAN/jiAgAuU14umzdrXQTgrDvvlPBwrYsAAABw\nFAEHALiMokjbtloXATjMz08iI7UuAgAAwElcogIALmOxSEGB1kUADnNzk+bNtS4CAADASczg\nAACXMZtlyxatiwAcFhYmcXFaFwEAAOAkZnAAAAAAAADdYwYHALhMWZnMm6d1EYADPD3l+ee1\nLgIAAKBOmMEBAK5UVqZ1BUCNFIV7wQIAgKaB7zQA4DJlZbJ0qdZFADXy8JCxY7UuAgAAoB4w\ngwMAXMZikTNntC4CqJGfH3dOAQAATQMzOADAZSwWuXBB6yKAGnFrWAAA0FQQcACAy1gsUlSk\ndRFANQwGiY/XuggAAIB6wyUqAAAAAABA95jBAQAuU1Ymf/+71kUAfzR4sHTqpHURAAAA9Y8Z\nHAAAAAAAQPeYwQEALlNaKq+/rnURwO+SkmTQIK2LAAAAcBVmcACAy1gscvWq1kUAIn5+YjRq\nXQQAAIBrMYMDAFymokL27tW6CECkf3/x8dG6CAAAANci4AAAV+K3SmiuRQvGIQAAuBVwiQoA\nuIzFIgUFWheBW57JJM2aaV0EAACAyzGDAwBcxmyWLVu0LgK3qvvuk8BArYsAAABoOMzgAAAA\nAAAAuscMDgBwmdJSefNNrYvALePhhyUqSusiAAAANMMMDgBwpbIyrSvALcBoFINB6yIAAAA0\nxgwOAHCZsjJZulTrInALiI6W6GitiwAAANAYMzgAwGUsFjlzRusi0BR5ebGAKAAAgB1mcACA\ny1gsUlqqdRFoitzdta4AAACg0SHgAACXqaiQ/fu1LgJN0aBBWlcAAADQ6HCJCgAAAAAA0D1m\ncACAy5SVyd//rnURaHKeeko8PLQuAgAAoNFhBgcAAAAAANA9ZnAAgMuUlsq8eVoXAf3z8pLn\nn9e6CAAAgMaOGRwA4DIWi1y9qnUR0BVPT/H21roIAAAAXWIGBwC4TEWF7N2rdRHQlfbtpWNH\nrYsAAADQJQIOAHAlHx+tK4AW+vUTRdG6CAAAgFsLl6gAgMtYLFJQoHUR0EJAgNYVAAAA3HII\nOAAAAAAAgO4ZtC4AAAAAAACgrgg4AAAAAACA7hFwAAAAAAAA3SPgAAAAAAAAukfAAQAAAAAA\ndI+AAwAAAAAA6B4BBwAAAAAA0D0CDgAAAAAAoHsEHAAAAAAAQPcIOAAAAAAAgO4RcAAAAAAA\nAN0j4AAAAAAAALpHwAEAAAAAAHSPgAMAAAAAAOgeAQcAAAAAANA9Ag4AAAAAAKB7BBwAAAAA\nAED3CDgAAAAAAIDuEXAAAAAAAADdI+AAAAAAAAC6R8ABAAAAAAB0j4ADAAAAAADoHgEHAAAA\nAADQPQIOAAAAAACgewQcAAAAAABA9wg4AAAAAACA7hFwAAAAAAAA3SPgAAAAAAAAukfAAQAA\nAAAAdI+AAwAAAAAA6B4BBwAAAAAA0D0CDgAAAAAAoHsEHAAAAAAAQPcIOAAAAAAAgO4RcAAA\nAAAAAN0j4AAAAAAAALpHwAEAAAAAAHSPgAMAAAAAAOgeAQcAAAAAANA9Ag4AAAAAAKB7BBwA\nAAAAAED3CDgAAAAAAIDuEXAAAAAAAADdI+AAAAAAAAC6R8ABAAAAAAB0j4ADAAAAAADoHgEH\nAAAAAADQPQIOAAAAAACgewQcAAAAAABA9wg4AAAAAACA7hFwAAAAAAAA3SPgAAAAAAAAukfA\nAQAAAAAAdI+AAwAAAAAA6B4BBwAAAAAA0D0CDgAAAAAAoHsEHAAAAAAAQPcIOAAAAAAAgO4R\ncAAAAAAAAN0j4AAAAAAAALpHwAEAAAAAAHSPgAMAAAAAAOgeAQcAAAAAANA9Ag4AAAAAAKB7\nBBwAAAAAAED3CDgAAAAAAIDuEXAAAAAAAADdI+AAAAAAAAC6R8ABAAAAAAB0j4ADAAAAAADo\nHgEHAAAAAADQPQIOAAAAAACgewQcAAAAAABA9wg4AAAAAACA7hFwAAAAAAAA3SPgAAAAAAAA\nukfAAQAAAAAAdI+AAwAAAAAA6B4BBwAAAAAA0D0CDgAAAAAAoHsEHAAAAAAAQPcIOAAAAAAA\ngO4RcAAAAAAAAN0j4AAAAAAAALpHwAEAAAAAAHSPgAMAAAAAAOgeAQcAAAAAANA9Ag4AAAAA\nAKB7BBwAAAAAAED3CDgAAAAAAIDuEXAAAAAAAADdI+AAAAAAAAC6R8ABAAAAAAB0j4ADAAAA\nAADoHgEHAAAAAADQPQIOAAAAAACgewQcAAAAAABA9wg4AAAAAACA7hFwAAAAAAAA3SPgAAAA\nAAAAukfAAQAAAAAAdM9N6wIAPXnnnXcmT56sdRUAAAAAoKX/+Z//uffee7Wuwh4BB1ALLVu2\nbNGixdatW7UuBDqwc+fOqVOnfvnll1oXAh3Yu3fvU0899e233xoMzKzETRw5cmT06NF5eXnN\nmzfXuhY0dmfOnBk2bNimTZtCQkK0rgWNXUFBwcCBA1etWtWhQweta0FjV1ZWFhMT4+Pjo3Uh\nVSDgAGrBaDS6u7tHR0drXQh04Ny5cwaDgdECRxQXF4tIdHQ0AQduymg0ikjPnj39/f21rgWN\nXUBAgIh069atXbt2WteCxu7y5csicscdd3Tt2lXrWtDYlZaWal1CtfgiBQAAAAAAdI+AAwAA\nAAAA6B4BBwAAAAAA0D0CDgAAAAAAoHsEHAAAAAAAQPcIOAAAAAAAgO4RcAAAAAAAAN0j4AAA\nAAAAALpHwAEAAAAAAHSPgAOoBZPJZDKZtK4C+sBogeNMJpO7u7uiKFoXAh0wmUyKori7u2td\nCHRA/THEDyM4Qv0xxGiBIwwGg5ubW+McLYrFYtG6BkA3ysvLz549265dO60LgQ5UVFT88ssv\n7du317oQ6IDFYvn555/Dw8O1LgT68L//+78RERFaVwF9YLTAcYwWOK7RjhYCDgAAAAAAoHtc\nogIAAAAAAHSPgAMAAAAAAOgeAQcAAAAAANA9Ag4AAAAAAKB7BBwAAAAAAED3CDgAAAAAAIDu\nEXAAAAAAAADdI+AAAAAAAAC6R8ABAAAAAAB0j4ADAAAAAADoHgEHAAAAAADQPQIOAAAAAACg\newQcAAAAAABA9wg4AAAAAACA7hFwAAAAAAAA3SPgAKpw/PjxtLS0kJAQT0/P22+/fdq0aSUl\nJTVvUlRUtGrVqtTU1MjISG9vbz8/v7i4uCVLllRUVDRMzWhIToyQOm4I/eJ8AsfV/RSxceNG\nRVEURZk2bZqLikQjUZfRkpOTk5KS0rJlSw8Pj7Zt2w4bNmzHjh2uLBbac27AWCyWtWvXJiUl\ntWnTxsvLKyIi4pFHHsnPz2+AgqGVNWvWPPfcc7GxsT4+PoqijBo1yvFtG8UXXQuAP/rxxx/9\n/f0VRUlOTn7hhRd69+4Ye3MXAAAgAElEQVQtIv379y8pKalhq3nz5omIyWTq37//I488cvfd\nd7u5uYnIf/zHf5jN5gYrHg3AuRFSlw2hX5xP4Li6nyIuXrzYsmVLHx8fEZk6dapLq4W26jJa\nXn75ZRHx8PBISEgYMWJEYmJiYGAgA6Zpc3rAPPPMMyLi5+f36KOPvvDCC0OGDDEYDIqiLFu2\nrGEqR8OLjo4WEV9f306dOonIyJEjHdywkXzRJeAA7PXt21dEli5dqj40m82pqakiMnPmzBq2\n+uyzzxYuXFhQUGBtOXjwYHBwsIh8+umnLi0YDcy5EVKXDaFfnE/guLqfIlJSUlq1avXaa68R\ncDR5To+WDz/8UERiYmJOnz5tbTSbzZcvX3ZdtdCccwPm+PHjIhIUFHTmzBlr47p160Skbdu2\nLi0YGsrLyzt69GhFRcXGjRtrFXA0ki+6BBzAH3z//fci0rNnT9vG06dPGwyGNm3aVFRU1Gpv\nb775pog8/fTT9VojtOT0CKnfoQVd4HwCx9V9tKi/uG7atEmdAUTA0YQ5PVpu3LgREhLSrFmz\n8+fPu75MNBZOD5jt27eLyP3332/baDab3dzcvLy8XFUuGo1aBRyN54sua3AAf5CbmysiQ4YM\nsW1s3bp19+7dT58+feTIkVrtzc/PT0Q8PDzqsUJoy+kRUr9DC7rA+QSOq+No+fnnn1944YXH\nH3/8gQcecGGVaBzq8pPo/PnzKSkpfn5+q1ateu211954442cnByLxeLyoqEdpwdMly5djEbj\nnj17zp8/b23csmVLeXn54MGDXVcw9KjxfNEl4AD+4PDhwyLSuXNnu3b1IrRa/ee0WCwfffSR\niCQnJ9dfgdCY0yOkHocW9ILzCRxXl9FSUVExduxYf39/de4GmjynR8uePXtEJDAwsHv37qNG\njcrMzJw6derAgQNjY2MvXLjgypKhJacHTOvWrTMyMi5duhQZGfmnP/1pwoQJQ4cOffDBBx94\n4IHFixe7tGboTuP5okvAAfxBYWGh/P6XUlv+/v4iUlBQ4PiuMjIyvvnmm4ceemjgwIH1WCG0\n5fQIqcehBb3gfALH1WW0zJkz58svv/zggw8qb44myenRcvHiRRFZsGCBwWDIy8u7du3aDz/8\nMGjQoPz8/FrdKAH6UpfTy9SpUz/99NOKiooVK1bMnz9/8+bNHTp0SEtLCwoKcl3B0KPG80XX\nrcFeCWhUKioqnn/+eduWiRMnRkREVNdfnb2pKIqD+3/vvfcyMjJ69+69dOnSutQJvajtCKn7\nhtAvzidw3E1Hy48//vjaa6+NGzdu0KBBDVgXGqObjhaz2ax2WLduXZcuXUSkW7dua9eu7dSp\n044dO7777rs777yzwaqF5hz5YZSRkZGRkTF58uTx48cHBwcfOnTolVdeGT169I8//vjGG280\nVKXQsYb/okvAgVtURUXFggULbFtGjRoVERGh5o5qBmmrulSySnPmzJk0aVJ0dHR2dravr289\nlYxGwekRUi9DC/rC+QSOc260WCyWMWPGhIaGzp4929UVovFw+twSEBAgIl26dFHTDVWzZs0G\nDRq0fPlyAo6myukBs23bthkzZqSmpr799ttqS+/evdetW9e5c+e//vWvTz/9dFhYmMuqhs40\nni+6XKKCW5Sbm5vdirtxcXHy+5Vj6lVkto4ePSq/X0VWsxkzZkyaNCkmJiYnJ0f9JoGmxOkR\nUvehBd3hfALHOTdazGbzgQMHTpw40bx5c+V3EyZMEJFZs2YpivLnP//ZxYVDA3X8SaTOGLel\ntvzf//1f/daJRsLpAbN582YRSUxMtG308vLq37+/2Wzev39//dcK3Wo8X3QJOIA/GDBggIh8\n/vnnto1nz549cOBA69atb/qfc+LEiRkZGffcc8+2bdv4m3yT5PQIqePQgh5xPoHjnBstBoPh\niUr69+8vIj179nziiSfi4+MboHg0MKfPLUlJSYqiHDp0qKyszLb9xx9/FJHw8HDX1AuNOT1g\nSktL5felW2ypS9JyVy/YakRfdBvshrSAXvTt21dEli9frj40m81paWkiMnPmTNtuS5cunTdv\n3oULF6zdnnzySREZPHhwSUlJQxeNBuTcCHF8QzQlnE/gOKfPLXbUe6lMnTrVteVCU06Ploce\nekhEpk+fbm3ZuHGjiAQFBRUVFTVI7dCAcwPmk08+EZGQkJBTp05Z+2zYsEFRFG9v74KCggar\nH5pQTw4jR46s8tlG+0VXsXDja+CP/vWvf8XFxV27di05OTk8PHznzp3ff/99v3798vLyvLy8\nrN06dux4/PjxPXv2qBeszp49e8qUKQaDYeTIkSaTyXaH3bp1e+mllxr6bcBlnBshjm+IpoTz\nCRzn9LnFzvz58ydMmDB16tTMzMyGqh0NzenRcvbs2djY2J9//jkmJqZ3794nT57csmWL0Whc\nvXr1sGHDNHo3cDnnBozZbB40aFBeXl6zZs2GDh3asmXLf//739nZ2SKSlZU1btw4zd4PXGnN\nmjUbNmwQkdOnT+fk5LRv3z4hIUFEgoKC3nnnHWu3xvtFtyHTFEAvjh07lpqa2qJFC5PJFBER\n8eqrr1b+s0aHDh1EZM+ePerDv/zlL9X9Lxs8eHCDvwO4lhMjxPEN0cRwPoHjnD632GIGxy3C\n6dFy6dKl5557LiwszN3dPTAw8MEHH6xhOKHJcG7A3LhxY+7cuX379vXx8TEajS1atEhOTs7J\nyWnY2tGgpk6dWuU3kLCwMNtujfaLLjM4AAAAAACA7rHIKAAAAAAA0D0CDgAAAAAAoHsEHAAA\nAAAAQPcIOAAAAAAAgO4RcAAAAAAAAN0j4AAAAAAAALpHwAEAAAAAAHSPgAMAAAAAAOgeAQcA\nAAAAANA9Ag4AAAAAAKB7BBwAAAAAAED3CDgAAAAAAIDuEXAAAAAAAADdI+AAAAAAAAC6R8AB\nAAAAAAB0j4ADAAAAAADoHgEHAAC6d/r0aUVRUlJSamjZv3+/oiiPPfaYBvXpTeWjV52goKD2\n7dvftBsHHwCABkDAAQBAo1ZWVvbee+/Fxsb6+/ubTKZWrVr16dPnhRde+OKLL7QurR4cO3ZM\nUZRRo0Y52N7I6bRsAACaBjetCwAAANW6cePGwIEDv/rqK29v78TExFatWl26dOnIkSPvvvvu\n8ePHExIS1G7BwcE7d+4MDAzUttomg+MJAIAeEXAAANB4LVq06KuvvoqOjt62bdttt91mbT92\n7Ni///1v60OTyRQXF6dFgU0TxxMAAD3iEhUAABqvXbt2ichzzz1nm26ISMeOHZOTk60PHV8z\nQkROnTo1evTooKAgLy+vPn36bNmypXKflStXxsfH+/r6enl5devW7a233rpx44b12U2bNimK\nMmPGDLut/P39O3bsaNeYn58/fPjwkJAQk8kUGhr66KOPHjp0SH3qrbfeuv3220Vk1apVyu8+\n/vjj6tod2WdlRUVFJpMpNjbW2nL9+nVPT09FUVasWGFtXLhwoaIoH374oVRzPCsqKubPnx8Z\nGenp6dm2bdsJEyYUFRXZdrhp2eLYwbezdevWQYMGhYaGenh4tGrVKi4ubvbs2TfdCgCAWxAz\nOAAAaLyCg4NF5NSpU/W1w1OnTvXp06d169YjRoy4ePHiunXrkpOTd+zYER8fb+0zZcqU2bNn\nBwcHP/roo82aNdu8efMrr7zy+eefZ2dnu7u71+rlFi9ePG7cuMDAwKFDhwYHB584cWL16tXr\n1q3Lycnp169fcnKyu7v7pEmT+vfv/+yzz6qbxMbGlpSUVNnuyD4r1+Dj49O3b9/du3dfu3at\nefPmIvL111+reU1OTs6YMWPUbrm5uSKSlJRU3XsZP378okWLwsLC0tPTFUVZs2bNd999Zzab\nrR2qezu1Ovh2Pvroo7Fjx4aEhAwbNiw4OPjSpUsHDx5csmTJ5MmTb3rwAQC45VgAAEBjtWvX\nLqPRaDKZXnzxxZycnKtXr1bZTU1Ahg0bVkPLvn371B/906ZNq6ioUBvVKQzJycnWbl9++aWI\nhIeHX7x4UW0pKysbMmSIiMyaNUtt2bhxo4hMnz7drgw/P78OHTpYH/7000/u7u6DBw8uKSmx\nNh44cMDHx6d79+7qw6NHj4rIyJEj7XZVXbsj+6zstddeE5FNmzapD19++WWj0ZiYmNimTRu1\nxWw2BwYGRkREVHf08vLyRKRHjx5FRUVqS3Fxca9evUQkLCzspmU7ePAru+uuu4xG45kzZ2wb\nr1y5UsMmAADcsrhEBQCAxismJuaTTz5p0aLF/Pnzk5KSAgICwsPDH3/88a+++sq5HbZr1276\n9OmKoqgP09LS/Pz8vv32W2sH9RqN119/vUWLFmqLm5vbnDlzFEVZsmRJrV5r4cKFZWVlr776\nanFx8eXfhYaGJiUl/fDDDydPnnSifuf2qc7LyMnJUR/m5ORER0cPHz789OnTR44cEZH9+/f/\n+uuvNUzfWLZsmYjMmDGjWbNmaou3t3dmZmatir/pwa+S0Wh0c/vDlNuAgIBavS4AALcIAg4A\nABq1kSNHnjx5cseOHZmZmQ8//HBxcfGyZcvi4+OnTJnixN569epl+9uyoiht2rS5evWqtWXv\n3r0ikpiYaLtVZGRkq1atTpw4UVBQ4Phr5efni0hCQkKLP1q/fr2InDt3zon6ndtnTEyMl5eX\nGnAUFhbu3bs3KSlpwIAB8nvqoV6forZUSZ2Ccffdd9s22j28qZse/MpSU1NLS0ujoqLS09M/\n++yz8+fP2z6bkpKSnp5eqxoAAGjCWIMDAIDGzmg0JiQkqDeFtVgs//jHPx5//PHZs2fff//9\n99xzT6125e/vb9fi5uZmu5BEYWGhiISEhNh1a9Wq1dmzZwsLCyvvoTq//vqriGzYsMHLy6vy\ns5GRkY6XXcd9qndF2b59+8WLF/Pz881mc1JSUmRkZGhoaE5Ozvjx43NychRFqSHgKCwsdHNz\ns1vq1cfHxzqhwxE3PfiVpaenBwQELFiwICsra8GCBSISExMze/ZsdWmPAQMG2JUEAMCtjIAD\nAAA9URRl9OjRO3bsWLx4cXZ2dm0Djpvy8/MTkfPnz4eFhdm2q5Mj1GcNBoOIlJeX23YoKysr\nLi4OCgqy21VISEifPn3qtzwn9jlgwIDs7Ozc3Nxdu3Z5eHioAUFiYuLWrVtv3Lixc+fOqKgo\ndUnX6l735MmTV65csQ0UioqK7N6yK6SlpaWlpf3222/5+fnr1q374IMPhgwZcvDgwbZt2z7/\n/PMufWkAAPSFS1QAANAf9W4mNf/x3znqwpk7duywbTx8+PC5c+fCw8PVOQjqGhB293bZt2+f\nXeTRv39/EVm5cmUNL2c0GqWqN1JduyP7rJJ1GY7c3NzY2FhPT0+18cqVK1lZWcXFxTUswCG/\nHxZ1BVYru4c1lF13vr6+gwcPzsrKeumll65du6ZeU8MlKgAA2CLgAACg8VqwYMHatWtLS0tt\nG7/77rtPP/1URGq4vajT/vM//1NEZs6cqV4MIiLl5eUvvfSSxWJ54okn1JZu3bp5enquX7/e\nuiREYWHhxIkT7XaVnp7u5ub2t7/9Tf1t3KqoqGjVqlXqvwMDA0Xkl19+sdu2unZH9lml6Oho\nf3//9evXHzx40JplqP948803pcYFOERk7NixIjJjxozi4mK1paSkRL05iyNlOy07O9suNrp8\n+bKIeHt719dLAADQZHCJCgAAjdeePXuWL1/evHnzvn37tm/fvqys7NixY/n5+RaLZcSIEQ88\n8EC9v+Ldd989ceLEuXPnRkVFPfzww97e3ps3b/7pp5/i4+MnT56s9vHx8Rk/fvy8efN69uyZ\nnJxcWlqanZ0dHR3t6+tru6uuXbu+//77Tz/99MCBA++9995evXqZzeZDhw7l5ua2b99+5MiR\nIuLr69uvX7/du3enpqZ26dLFaDSmpKR07dq1unZH9lklg8GQkJCgrkVqDTjatWvXoUOH48eP\nq6uc1HBYEhMTn3zyycWLF3ft2nX48OGKoqxZsyY0NNRuWY3qynbqoxARSU1NdXNzS0hICAsL\nMxqNu3fvzsvLi4qKGjp0qNP7BACgydL4NrUAAKB6Z86cef/99x966KEuXbo0b97c3d09NDT0\n/vvv//TTTysqKqzd1KtFhg0bVkOLeh+QsWPH2r1Ejx49jEajXePHH3981113+fj4eHh4REVF\nZWZmXr9+3bZDeXn59OnTw8LC3N3dw8LCpk2bduPGDT8/vw4dOtjtat++fWPGjGnbtq3JZAoI\nCIiKiho3blxeXp61w9GjR4cOHRoQEKDeP3XFihU1tzuyzyq9++67IuLr61teXm5tfOqpp0Sk\nb9++tj0rHz2LxWI2m+fOndupUyeTydS6desXX3zx2rVrgYGBYWFhtt2qLLtWB99WVlZWSkpK\nRESEt7e3n59f9+7dMzMzr169qj47bNiwZ599tuZ3DQDArUOxWCxaZSsAAABwWkpKSps2bd57\n7z2tCwEAoFFgDQ4AAAAAAKB7BBwAAAAAAED3CDgAAAAAAIDusQYHAAAAAADQPWZwAAAAAAAA\n3SPgAAAAAAAAukfAAQAAAAAAdI+AAwAAAAAA6B4BBwAAAAAA0D0CDgAAAAAAoHsEHAAAAAAA\nQPcIOAAAAAAAgO4RcAAAAAAAAN0j4AAAAAAAALpHwAEAAAAAAHSPgAMAAAAAAOgeAQcAAAAA\nANA9Ag4AAAAAAKB7BBwAAAAAAED3CDgAAAAAAIDuEXAAAAAAAADdI+AAAAAAAAC6R8ABAAAA\nAAB0j4ADAAAAAADoHgEH6tPp06cVRUlJSamhZf/+/YqiPPbYYxrUpzeVj151goKC2rdvf9Nu\nHHwAAAAATRUBhzNmzZqlKIqiKIcPH9a6FtSPY8eOKYoyatQoB9sbucZW9vHjx9PS0kJCQjw9\nPW+//fZp06aVlJTUvElRUdGqVatSU1MjIyO9vb39/Pzi4uKWLFlSUVHhRDcAAAAATZ6b1gXo\nj8Vi+eCDDxRFsVgsixcvfuedd7SuqBEJDg7euXNnYGCg1oU0EU3jeP7rX/+Kj48vLCwcOnRo\nRETEzp07Z82alZOTk5ub6+XlVd1WS5YsmTBhgslk6t27d7du3S5cuLBr166vv/5648aNa9eu\nNRgMteoGAAAAoMnj23+tbdu27cSJE2PHjm3ZsuXy5ctLS0u1rqgRMZlMcXFxkZGRWhfSRDSN\n4/nEE08UFBR8+OGHGzZsmD9//p49e1JTU7/55ps5c+bUsFXbtm0XLlx48eLF/Pz8f/7zn198\n8cWBAweCg4M3bNiwatWq2nYDAAAA0OQRcNTa4sWLReTJJ59MS0u7fPny2rVrrU/l5+crivLQ\nQw9V3ioyMtLDw+PKlSu2nYcPHx4SEmIymUJDQx999NFDhw5Zn7WulXD8+PFRo0YFBwcbDIZv\nvvlGLSAlJSU8PNzLy8vf3z8hIWH16tV2L2c2m+fMmdOlSxdPT8+2bdu++OKLRUVFVS7TUHMZ\nVdq6deugQYNCQ0M9PDxatWoVFxc3e/Zs9SnH14wQkVOnTo0ePTooKMjLy6tPnz5btmyp3Gfl\nypXx8fG+vr5eXl7dunV76623bty4YX1206ZNiqLMmDHDbit/f/+OHTs6/k7feuut22+/XURW\nrVql/O7jjz+urt2RfVZWVFRkMpliY2OtLdevX/f09FQUZcWKFdbGhQsXKory4YcfSjXHs6Ki\nYv78+ZGRkeqHO2HChKKiItsONy1bHDv49WLv3r3ffvttz549rQt/GAyG2bNnGwyG999/32Kx\nVLfh8OHDx48f7+fnZ2254447JkyYICJffPFFbbsBAAAAaPK4RKV2Lly4sGHDhk6dOt11112+\nvr5z585dtGjRyJEj1WdjYmI6d+68adOmX3/91faygm+//fbQoUPDhw+/7bbb1JbFixePGzcu\nMDBw6NChwcHBJ06cWL169bp163Jycvr162fd8NSpU/369QsKCrrvvvuKi4s9PT1F5Omnn+7b\nt29iYmLLli0vXry4adOmESNGvP3221OmTLFu+NRTT3344Yft27dPT083GAxr1qz5/vvvzWaz\n3dtxsAxbH3300dixY0NCQoYNGxYcHHzp0qWDBw8uWbJk8uTJtTqSp06d6tOnT+vWrUeMGHHx\n4sV169YlJyfv2LEjPj7e2mfKlCmzZ88ODg5+9NFHmzVrtnnz5ldeeeXzzz/Pzs52d3ev1cvV\n/E6Tk5Pd3d0nTZrUv3//Z599Vt0kNja2pKSkynbnjp6Pj0/fvn1379597dq15s2bi8jXX3+t\n5jU5OTljxoxRu+Xm5opIUlJSde9l/PjxixYtCgsLS09PVxRlzZo13333ne2HW93bqdXBr2zU\nqFGrVq3KysoaN25czUfblvp2hgwZYtvYunXr7t2779+//8iRI507d3Z8b2qQ4eHhUS/dAAAA\nADQpFtTGm2++KSJvvPGG+rB3796Kohw9etTa4Y033hCRv/3tb7ZbPfPMMyKyYcMG9eFPP/3k\n7u4+ePDgkpISa58DBw74+Ph0795dfbhv3z71A0pPTy8vL7fd2y+//GL7sLi4+M477/Ty8rpy\n5Yrasn37dhHp0aNHUVGR2lJSUnLnnXeKSFhYmHVDR8qo7K677jIajWfOnLFttL70qVOnRGTY\nsGHWpyq3WN/atGnTKioq1EZ1CkNycrK125dffiki4eHhFy9eVFvKysrU35NnzZqltmzcuFFE\npk+fblekn59fhw4davVOjx49KiIjR46021V17c4dvddee01ENm3apD58+eWXjUZjYmJimzZt\n1Baz2RwYGBgREVHd0cvLy7P7cIuLi3v16mX34VZXtoMHv0pqkJeVlVVzNzt//vOfRWTZsmV2\n7SNGjLD9T+GIioqK/v37i0h2dnbduwEAAABoYrhEpRYsFsuSJUsMBsOf/vQnteWxxx5TG619\nxowZYzAYli9fbm0pLS1duXJlcHCw9Y/YCxcuLCsre/XVV4uLiy//LjQ0NCkp6Ycffjh58qR1\n26CgoLfffttoNNqW0bZtW7WYwsLCCxcu/Pbbbw8++OD169d37typdvjoo49EJCMjo1mzZmqL\nl5dXZmam3dtxvAw7RqPRze0Pc38CAgIcOYC22rVrN336dEVR1IdpaWl+fn7ffvuttYN6jcbr\nr7/eokULtcXNzW3OnDmKotgecEc4/U7rfZ/qvIycnBz1YU5OTnR09PDhw0+fPn3kyBER2b9/\n/6+//lrD9I1ly5aJyIwZM6wfrre3d+UPt2Y3PfhVGjt27Lx582xngjiisLBQfp9SYcvf319E\nCgoKHN9VRkbGN99889BDDw0cOLDu3QAAAAA0MVyiUgu5ubnHjx8fPHhw69at1ZbRo0dPmjRp\n2bJlM2fOVC+aaNOmTVJSUnZ29k8//XTHHXeIyMaNG69cuTJhwgRrKJCfny8iCQkJVb7KuXPn\nwsLC1H/37NnT29vbrsO+fftmzJiRl5d37do12/YzZ85YO4iI3eUGcXFxdvtxvAxbqampu3bt\nioqKGjly5D333BMXFxcSElLlHmrWq1cv25REUZQ2bdqo8w5Ue/fuFZHExETbrSIjI1u1anXi\nxImCggL1N2RHOPdOXbHPmJgYLy8vNeAoLCzcu3fvlClTBgwYICI5OTmdOnVSL+hQW6qkfrh3\n3323baPdw5u66cGv0pAhQ+yuNKkLi8WivrSD/d97772MjIzevXsvXbq07t0AAAAAND0EHLWw\naNEiEbGulSgigYGBycnJ//3f/71+/fqHH35YbXzssceys7OXL1/+9ttvi4g6m2Ps2LHWrX79\n9VcR2bBhQ5X3yLS9ZUZoaKjds3v37o2Li/P09Bw/fnyPHj38/PyMRuP27dvnzJljXX3zt99+\nc3Nzs673oWrWrJn1b/61LcNWenp6QEDAggULsrKyFixYICIxMTGzZ8+u7R/2K8cTbm5utgtJ\nqH/5r5yetGrV6uzZs4WFhY4HHM69U1fsU70ryvbt29W7fpjN5qSkpMjIyNDQ0JycnPHjx+fk\n5CiKUkPAUVhYWPnD9fHxsftwa3bTg1+P1Lkb6qdpq7qZHVWaM2fOpEmToqOjs7OzfX1969gN\nAAAAQJNEwOGoS5curVu3TkRSU1NTU1Ptnl20aJE14HjwwQd9fX0//vjjN95448qVK1u3bu3R\no0ePHj2sndVf6kJCQvr06VPzi1b++/bcuXOvX7++YcMG2+n333//vW0fX1/fkydPXrlyxfbX\n4OLi4uLi4qCgICfKsJOWlpaWlvbbb7/l5+evW7fugw8+GDJkyMGDB9VrZ+qLWt758+ftpkKc\nO3fO+qzBYBCR8vJy2w5lZWX19U5vWp4T+xwwYEB2dnZubu6uXbs8PDzUYCgxMXHr1q03btzY\nuXNnVFRUcHBwDa9b+cMtKiqye8uNh7qG6OHDh+3a1QkjnTp1uukeZsyYkZGRERMTs3Xr1hoC\nEQe7AQAAAGiqWIPDUcuXLy8tLY2Ojn6ikhYtWmzfvv3EiRNqTy8vrxEjRpw9e3b79u2ffPJJ\neXm57fQNEVFXQFy5cqUTZfz888/WPVip1zVY9ezZU0S++uor20a7h3UsQ0R8fX0HDx6clZX1\n0ksvXbt2za6GulMXztyxY4dt4+HDh8+dOxceHq7OQVDX/lBX4rTat2+fXeThyDtVFzqpPIuh\nunanj551GY7c3NzY2Fj1zjhJSUlXrlzJysoqLi6uYQEO+f2wqCuwWtk9rKHshqfORvn8889t\nG8+ePXvgwIHWrVvfNOCYOHFiRkbGPffcs23bthpiCwe7AQAAAGjKtF3jVEfUP0Tv3r278lPT\npk0TkVdffdXaoqYJo0ePVhc7uHDhgm3/H3/80c3Nzd3dPScnx7b92rVrK1euVP+tLrUwduxY\nu9dSbya6Zs0aa8snn3yifpTz5s1TW7Kzs0WkV69excXFasv169f79u0rf7zRhiNlVLZt27ay\nsjLbFvU2Gf/858c/gkAAAASYSURBVD8ttbmLSuW31qNHD6PRaH34xRdfiEiHDh0uX76stpSV\nlT3wwAMikpmZaS3V09PTz8/v3LlzaktBQYE6J8L2LiqOvFP1iom+ffvaVVVdu3NHz2KxmM1m\nf39/deVU6+1g1EVJ1Ykb69evt3aufPTUIMnuLiq9e/e2+3CrK9vBg1+lLVu2zJs374cffqi5\nW2Xq2Fu+fLn60Gw2p6WlicjMmTNtuy1dunTevHnW/yxms/nJJ58UEbtb1dhxsBsAAACAJo9L\nVByyY8eOw4cPd+vWTf1Vzc4TTzwxa9aspUuXZmRkqGs3xsbGduzYcfXq1WVlZcnJyXZXHHTt\n2vX9999/+umnBw4ceO+99/bq1ctsNh86dCg3N7d9+/bqzTirk56e/umnn6ampo4cOTIsLGz/\n/v1btmx55JFHVq9ebe0zcODAsWPHLl++vGvXrsOHD1cUZe3atSEhIf7+/uo1HXUpIzU11c3N\nLSEhISwszGg07t69Oy8vLyoqaujQobU6pDd19913T5w4ce7cuVFRUQ8//LC3t/fmzZt/+umn\n+Pj4yZMnq318fHzGjx8/b968nj17Jicnl5aWZmdnR0dH262/4Mg79fX17dev3+7du1NTU7t0\n6WI0GlNSUrp27Vpdu9MfosFgSEhIWL9+vfw+m0NE2rVr16FDh+PHjxuNxuoWLlUlJiY++eST\nixcvtn64a9asCQ0NtVtWo7qynfoo/r/ly5evWrUqKyurW7dutdrwgw8+iIuLe/zxx9esWRMe\nHr5z587vv/++X79+L730km23zMzM48ePx8XFqf9f5syZs3jxYoPBcNttt40fP962Z7du3azb\nOtgNAAAAQNOndcKiD6NHjxaR//qv/6quw6BBg+SPEytmzpypHuHPPvusyk327ds3ZsyYtm3b\nmkymgICAqKiocePG5eXlWZ+Vqv7SbrFY8vLy4uPjfX19fX19BwwYkJOTs2LFCrGZwWGxWMrL\ny//617/efvvtJpOpdevWzz///JUrV9zc3Hr06FGrMirLyspKSUmJiIjw9vb28/Pr3r17Zmbm\n1atX1WfrcQaH6uOPP77rrrt8fHw8PDyioqIyMzOvX79u26G8vHz69OlhYWHu7u5hYWHTpk27\nceOGn5+f7QwOB9/p0aNHhw4dGhAQoC59smLFiprbnTh6qnfffVdEfH19y8vLrY1PPfWUVJpz\nUfnoWSwWs9k8d+7cTp06qR/uiy++eO3atcDAQNsZHNWVXZcZHGpqk5WVVXO3Kh07diw1NbVF\nixYmkykiIuLVV1+1zkCx6tChg4js2bNHffiXv/ylurPW4MGDrVs52A0AAABAk6dYLJa6pyRo\n/A4cONCzZ89Ro0b94x//0LoWAAAAAADqGYuMNk2XL1+2fVhSUqJe1vHggw9qVBEAAAAAAC7E\nDI6mKT09fceOHffcc09ISMjZs2e3bNly8uTJIUOGbN68ufKtZwEAAAAA0DsWGW2a7rvvviNH\njnz22WdXr151c3Pr3Llzenr6Cy+8QLoBAAAAAGiSmMEBAAAAAAB0jzU4AAAAAACA7hFwAAAA\nAAAA3ft/IriL9IVHlPgAAAAASUVORK5CYII="},"metadata":{"image/png":{"width":720,"height":1500}}}]},{"metadata":{"trusted":false},"cell_type":"code","source":"# Jaccard Plot\nwithr::with_options(repr.plot.width=12, repr.plot.height=12)\nJaccard(FG,Clustering=\"K-means\", K=4, plot = TRUE) # Jaccard ","execution_count":8,"outputs":[{"output_type":"display_data","data":{"text/html":"\n- 0.749
- 0.671
- 0.545
- 0.772
\n","text/markdown":"1. 0.749\n2. 0.671\n3. 0.545\n4. 0.772\n\n\n","text/latex":"\\begin{enumerate*}\n\\item 0.749\n\\item 0.671\n\\item 0.545\n\\item 0.772\n\\end{enumerate*}\n","text/plain":"[1] 0.749 0.671 0.545 0.772"},"metadata":{}},{"output_type":"display_data","data":{"text/plain":"plot without title","image/png":"iVBORw0KGgoAAAANSUhEUgAABaAAAAWgCAIAAAAnwnOfAAAACXBIWXMAABJ0AAASdAHeZh94\nAAAgAElEQVR4nOzdaZCV5Z3/4btZhFZkkwGk0USiLBGwBxFRsIKIUUtAGGNGQzS4TESNFUaM\nEwHLyLCNS2klYskoKoYZMRlbFrdSQR0wqNAgBeOGuNGiImojDoSlm/+LU0PxR+jT3XD69M9c\n16vTz3lO8/WFbz51+n4Kdu3alQAAAAAia5DvAQAAAAAHSuAAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8\ngQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwqsXgaOkpOTaa6/t169f\ns2bNCgoKLrzwwup/du3atSNGjGjfvn3Tpk2PO+648ePHb9myJXdTAQAAgHqoUb4HpJTS5MmT\nS0tLmzdvXlRU9M4771T/g6tXrz7ttNM2bdo0ePDgTp06LVq0aNKkSQsWLFi4cGFhYWHuBgMA\nAAD1Sr34Bsftt9++Zs2a8vLyO+64o0YfvPzyy8vLyx944IF58+bdddddS5cuveiii1555ZWa\n/h4AAAAgtHoROAYMGHDssccWFBTU6FPLly9/7bXXiouLR44cmbnSoEGD2267rUGDBtOnT9+1\na9fBHwoAAADUS/UicNTOwoULU0rnnHPOnheLiop69uxZVlZWoz91AQAAAEILHDjefvvtlFKX\nLl32ut65c+eUksABAAAAfzvqxSGjtbNp06aUUosWLfa63rJly5RSeXl5NX/PypUrd+7cWfU9\ny5cv79WrV803AgAAwHdKo0aNTjjhhHyv2IfAgWN/MqdvVPNEj7Vr1/bu3Ttr4AAAAAAyli5d\n2rt373yv2FvgwJH57kbmexx72t83O/bpBz/4wY4dO6q+Z/r06aNGjdq8eXOzZs1qtRQAAAC+\nC7Zv396kSZPt27fne8g+BD6DI3P6RuYkjj2tWbMm/d9JHAAAAMDfgsCBY+DAgSmlZ555Zs+L\n69evX7lyZVFRkcABAAAAfzsiBY6HHnrorrvu2rBhQ+bHXr169enTZ8WKFQ8//HDmSmVl5Q03\n3FBZWTlq1KhqnsEBAAAAfAfUizM4SkpK5s2bl1IqKytLKb366qsjR45MKbVp0+b222/ffdvE\niRPXrl3bv3//tm3bZq7MmDGjf//+l156aUlJyTHHHLNo0aLS0tKTTz55zJgxefjPAAAAAPKk\nXgSO5cuXz5w5c/ePH3zwwQcffJBS+t73vrdn4Pi27t27l5aW3nTTTc8///zTTz/dsWPHsWPH\njh07trCwMNebAQAAgPqjIPNQVargKSoAAACQ/u8pKi+//PKpp56a7y17i3QGBwAAAMA+CRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwA\nAABAeAIHAAAAEJ7AAQAAAIQncAAAAADh1ZfAsXbt2hEjRrRv375p06bHHXfc+PHjt2zZkvVT\nu3btevzxx88444yOHTsWFhZ26tTpggsuWLJkSR0MBgAAAOqPehE4Vq9e3bt370ceeaRPnz6j\nRo1q3rz5pEmTzjjjjK1bt1b9wV/96lf/8A//UFpaevrpp1955ZVdu3YtKSnp16/fzJkz62Y5\nAAAAUB80yveAlFK6/PLLy8vLH3zwwZEjR6aUKisrf/7znz/yyCN33HHH+PHj9/ep99577557\n7mnTps3KlSs7dOiQuTh37txhw4bddNNNv/jFL+pmPAAAAJB3+f8Gx/Lly1977bXi4uJM3Ugp\nNWjQ4LbbbmvQoMH06dN37dq1vw++//77KaU+ffrsrhsppSFDhjRq1Gjjxo05Xg0AAADUI/kP\nHAsXLkwpnXPOOXteLCoq6tmzZ1lZ2TvvvLO/D3bt2rVhw4ZLly799NNPd1986qmndu7cedZZ\nZ+VuMAAAAFDf5D9wvP322ymlLl267HW9c+fOKaUqAkdRUdEtt9zy+eefd+vW7ZJLLvnnf/7n\nwYMHDx8+/Nxzz73vvvtyuhkAAACoV/J/BsemTZtSSi1atNjresuWLVNK5eXlVXx23LhxnTp1\nGjVq1B//+MfMlS5duowYMaJNmzbV/Ne3b9/+yCOPbNu2rYp7Fi1aVM3fBgAAAORF/gPH/mRO\n3ygoKKjinltuueWWW275zW9+c9VVV7Vt2/att9668cYbf/azn61atWry5MnV+Vc+++yzqVOn\nbt++vYp7vv766917AAAAgHoo/4Ej892NzPc49rS/b3bs9uyzz/7ud7+76KKL/u3f/i1zpVev\nXnPmzOnSpcutt9565ZVXfu9738v6rx911FFvvvlm1fdMnz591KhRVacWAAAAII/yfwZH5vSN\nzEkce1qzZk36v5M49unJJ59MKZ1++ul7XiwsLOzbt29FRcXrr79+8LcCAAAA9VL+A8fAgQNT\nSs8888yeF9evX79y5cqioqIqAkfm70o2bNiw1/XPPvsspdSkSZODvxUAAACol/IfOHr16tWn\nT58VK1Y8/PDDmSuVlZU33HBDZWXlXn8Y8tBDD9111127i8Zpp52WUrr77rvLysp23zN//vxF\nixYdeuihp5xySh3+RwAAAAD5lP8zOFJKM2bM6N+//6WXXlpSUnLMMccsWrSotLT05JNPHjNm\nzJ63TZw4ce3atf3792/btm1K6R//8R/vv//+F154oWvXroMHD27Xrt2bb7753HPPpZTuuOOO\nKg7vAAAAAL5j6kXg6N69e2lp6U033fT8888//fTTHTt2HDt27NixYwsLC6v4VMOGDZ955plp\n06bNnj37ySef3Lp1a+vWrYcMGTJ69OjMn70AAAAAfyMKPP00q8xTVDZv3tysWbN8bwEAAIC8\n2b59e5MmTV5++eVTTz0131v2lv8zOAAAAAAOkMABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQ\nnsABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQnsABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQ\nnsABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQnsABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQ\nnsABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQXqN8DwAAANiHsrKyt956K98rIP8aNmx4yimn\nNG3aNN9D6juBAwAAqI8uu+yy5557Lt8roF544IEHLr300nyvqO8EDgAAoD7asWPHzSn9Lt8z\nIO+6prR9+/Z8rwjAGRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7A\nAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7A\nAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7AAQAAAIQncAAAAADhCRwAAABAeAIHAAAAEJ7A\nAQAAAIQncAAAAADhCRwAAABAeI3yPYAa27Fjx6pVq3bt2pXvIZB/Rx999N/93d/lewUAAJB/\nAkc8//Ef/3HppZfmewXUC2edddYzzzyT7xUAAED++ROVeLZt25bvCVBf+N8BAADIEDgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCEzgAAACA\n8AQOAAAAIDyBAwAAAAhP4AAAAADCa5TvAQDfQRUVFR999NGuXbvyPQTy76ijjmrcuHG+VwAA\n330CB8DBd9ttt9144435XgH1wjXXXHP33XfnewUA8N0ncAAcfN98801K/VOame8hkHe/3bx5\nc743AAB/EwQOgBwpTKlTvjdA3h2e0s58bwAA/iY4ZBQAAAAIT+AAAAAAwhM4AAAAgPAEDgAA\nACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAA\nACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAA\nACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAA\nACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAA\nACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAAACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAEDgAA\nACA8gQMAAAAIT+AAAAAAwhM4AAAAgPAa1eIz69atW7hw4aGHHjp48ODCwsKDvgkAAACgRrJ/\ng+PWW2/t0qXLV199lflx0aJFP/zhD0eOHPnTn/705JNP/vrrr3O8EAAAACCL7IGjpKSkQ4cO\nrVq1yvz4m9/8Zvv27TfeeOMVV1yxatWqe+65J8cLAQAAALLIHjjee++97t27Z15/8sknr776\n6j/90z9Nnjz5vvvuO/3002fPnp3jhQAAAABZZA8c5eXlrVu3zrx++eWXU0pDhw7N/HjSSSd9\n9NFHuRsHAAAAUB3ZA0fr1q0/++yzzOsXX3yxQYMGffv2zfxYUVGxbdu2HK4DAAAAqIbsgaN7\n9+5z585dv379hg0bHn300VNOOaV58+aZt95///327dvneCEAAABAFtkDx69//etPP/306KOP\nPuqoozZu3PirX/0qc33Xrl2vvPLKCSeckOOFAAAAAFk0ynrHkCFDHnzwwfvuuy+l9LOf/ezC\nCy/MXP/v//7vbdu2/fjHP87tQAAAAIBssgeOlNLIkSNHjhy518Uf/ehHGzduPPiLAAAAAGoo\n+5+o7Pbhhx8uWbJk06ZNuVsDAAAAUAvVChyZsza+//3vn3rqqUuXLs1cnD17dvfu3V966aVc\nzgMAAADILnvgePPNNwcNGvTee++dd955e14fPHjwBx988Oc//zln2wAAAACqJfsZHBMnTtyx\nY8eyZcuOPPLIuXPn7r7erFmz008/ffHixbmcBwAAAJBd9m9wLFiwYPjw4T169Pj2W127di0r\nK8vBKgAAAIAayB44vvjii+9///v7fKthw4abN28+yIsAAAAAaih74GjVqtXnn3++z7dWrFhx\n5JFHHuxJAAAAADWTPXD069fvySef3LZt217XFy5c+Nxzzw0YMCAnuwAAAACqLXvguP766z//\n/PPhw4e/8cYbKaWtW7cuXbp0zJgxZ599dqNGja677rrcjwQAAACoSvanqPTr12/atGnXXnvt\n008/nVIaOnRo5nrjxo3vv//+nj175nYgAAAAQDbZA0dKadSoUaeddtq99967ZMmSL774okWL\nFn379r322muPP/74XO8DAAAAyKpagSOldPzxx//hD3/I6RQAAACA2sl+BgcAAABAPSdwAAAA\nAOFl/xOVY489tuob3n333YM0BgAAAKA2sgeOjRs37nXlf//3f3fu3JlSat68eUFBQU52AQAA\nAFRb9sBRXl6+15UdO3asWLFi9OjRbdq0eeyxx3IzDAAAAKC6anMGR+PGjfv06fPkk08uW7Zs\n8uTJB30TAAAAQI3U/pDRVq1aDRo0aObMmQdxDQAAAEAtHNBTVJo0afLxxx8frCkAAAAAtVP7\nwPHpp5/Onz+/qKjooOxYu3btiBEj2rdv37Rp0+OOO278+PFbtmyp5mcXLFgwbNiwdu3aNWnS\n5KijjjrvvPNefPHFg7IKAAAACCH7IaO/+93v9rqyc+fOdevWzZkz5+uvv54wYcKBj1i9evVp\np522adOmwYMHd+rUadGiRZMmTVqwYMHChQsLCwur/uyNN944derUJk2a9O3bt127dp9//vnL\nL7/co0ePAQMGHPgwAAAAIITsgeOWW27Z5/XCwsLrr79+3LhxBz7i8ssvLy8vf/DBB0eOHJlS\nqqys/PnPf/7II4/ccccd48ePr+KDDz744NSpU0855ZQ///nPu79LUllZ+dVXXx34KgAAACCK\n7IFj/vz5e11p0KBBq1atevTo0axZswNfsHz58tdee624uDhTNzK//7bbbnv00UenT58+bty4\ngoKCfX5w+/btY8eOPeywwx5//PF27drtOe+II4448GEAAABAFNkDx+DBg3O6YOHChSmlc845\nZ8+LRUVFPXv2fP311995550uXbrs74OffvrpiBEjWrRo8eijj65evbqwsPDkk08eOHDg/poI\nAAAA8J2UPXDk2ttvv51S+nbF6Ny5c9WBY+nSpSmlI444omfPnmvWrNl9/ZRTTtnrOx0AAADA\nd1v+A8emTZtSSi1atNjresuWLVNK5eXl+/vghg0bUkrTpk079thjX3jhhd69e7///vtjxox5\n7rnnLrzwwhdeeKE6//rmzZtvvfXWHTt2VHHP66+/Xp1fBQAAAOTLvgPHsGHDqv8r5syZc5DG\n/H927dqVUqrij00qKioyN8yZM6dr164ppR49ejz++OOdO3d+8cUXly1b1rt376z/ytatW1eu\nXLl169Yq7vn444937wEAAADqoX0Hjrlz59bZgsx3NzLf49jT/r7ZsVurVq1SSl27ds3UjYzD\nDjvszDPPnDlzZjUDR9u2befNm1f1PdOnTx81apRzPQAAAKDe2nfgWLduXZ0tyByxkTmJY0+Z\nYzU6d+5c9Qczf8myp8yVv/71rwd3JwAAAFBv7TtwdOzYsc4WDBw4MKX0zDPPTJ48effF9evX\nr1y5sqioqIrAccYZZxQUFLz11ls7duxo3Ljx7uurVq1KKR1zzDG5XA0AAADUIw3yPSD16tWr\nT58+K1asePjhhzNXKisrb7jhhsrKyr3+MOShhx666667MmeLppSKioqGDx++cePGSZMm7b7n\niSeeWLhwYZs2bQYNGlSX/xUAAABAHlX3KSpffvnl4sWLP/74423btu311ujRow9wxIwZM/r3\n73/ppZeWlJQcc8wxixYtKi0tPfnkk8eMGbPnbRMnTly7dm3//v3btm2bufKHP/xh+fLlt9xy\ny7PPPturV68PP/zwqaeeaty48f3333/YYYcd4CoAAAAgimoFjilTpkyYMGF/p1oceODo3r17\naWnpTTfd9Pzzzz/99NMdO3YcO3bs2LFjCwsLq/5ghw4dli5dOmHChHnz5i1btqx58+bnnXfe\n2LFjq3O8KAAAAPCdkT1wzJ49e+zYsSeddNKwYcPGjRs3ZsyYVq1aLVy4cOHChRdccMHQoUMP\nyo4f/OAH//mf/1n1Pe++++63L7Zp0+b3v//973//+4MyAwAAAIgo+xkc06ZNa9eu3UsvvXTZ\nZZellAYNGjRu3LgFCxbMmjWrpKSkQ4cOuR8JAAAAUJXsgWPlypWDBw8uLCzMnPdZWVmZuT5i\nxIhzzjlnzwM+AQAAAPIie+DYvn175lDPQw45JKW0adOm3W8VFxeXlpbmbhwAAABAdWQPHO3b\nt9+4cWNKqWXLls2aNVu1atXutz744IPcLQMAAACopuyB44QTTnjjjTdSSgUFBQMGDJg+ffqC\nBQu++eabkpKSP/3pTz179sz9SAAAAICqZA8c55577l/+8peysrKU0s0337xly5ZBgwYdfvjh\n559/fkVFxYQJE3I/EgAAAKAq2QPHL3/5y8rKyo4dO6aUevfuvXjx4hEjRvTr15mrcOcAACAA\nSURBVO/iiy9esmTJgAEDcr4RAAAAoEqNavqBE088cdasWbmYAgAAAFA72b/B8eWXX9bBDgAA\nAIBayx44jjzyyJ/85Cfz58/fuXNnHQwCAAAAqKnsgaNTp06PPfbY0KFDi4qKrrvuupUrV9bB\nLAAAAIDqyx443nzzzVdfffXqq6/euXPnnXfeWVxcXFxcfOedd27YsKEO9gEAAABklT1wpJT6\n9Okzbdq0Tz755L/+67+GDBnyP//zP9ddd11RUdHQoUMfe+yxXE8EAAAAqFq1AkfGIYcccv75\n58+bN2/9+vV33nlnjx495s+f/5Of/CR34wAAAACqowaBY7fWrVt369atW7dujRs3PuiDAAAA\nAGqqUY3ufuONN2bOnDlr1qz169enlI477rhLLrkkN8MAAAAAqqtageOLL7545JFHZs6cuWzZ\nspRS8+bNr7jiipEjR/br1y/H8wAAAACyyx44hg8f/uSTT+7YsaNBgwZnnnnmyJEjhw8fXlhY\nWAfjAAAAAKoje+CYM2dOly5dfvGLX1x88cUdO3asg00AAAAANZI9cCxZsqRv3751MAUAAACg\ndrI/RUXdAAAAAOq52jwmFgAAAKBeETgAAACA8AQOAAAAIDyBAwAAAAhP4AAAAADCyx44vvrq\nqzrYAQAAAFBr2QNHUVHRyJEjlyxZUgdrAAAAAGohe+Do2LHjzJkzTz311BNOOOGee+75+uuv\n62AWAAAAQPVlDxxvv/32ggULfvrTn7711lvXXHNNhw4drrjiiqVLl9bBOAAAAIDqyB44CgoK\nBg4c+Oijj65bt27q1Knt27efMWNGnz59TjzxxH//93//5ptv6mAlAAAAQBVq8BSVtm3b/su/\n/MuaNWueffbZ888/f9WqVVdeeWWHDh2uuuqq1atX524iAAAAQNVq/JjYgoKCzp07d+vWrVWr\nVimlzZs333vvvT179rzooos2bdqUg4UAAAAAWdQgcFRUVMybN+/cc8/t1KnTxIkTmzRpMmHC\nhLKysqeeeupHP/rR7Nmzr7nmmtwNBQAAANifRtW5ad26dTNmzLj//vs//vjjgoKCQYMGXX31\n1UOGDGnYsGFKqaio6Oyzzz7vvPOeeuqpHK8FAAAA2IfsgWPIkCFPP/10RUVF69atr7vuuquu\nuurYY4/d656CgoK+ffvOnz8/NyMBAAAAqpI9cDzxxBMnnXTS1VdffeGFFzZt2nR/t5199tnN\nmzc/qNsAAAAAqiV74Fi2bNmJJ56Y9bZevXr16tXrYEwCAAAAqJnsh4y++eab77///j7fWr16\n9axZsw72JAAAAICayR44Lr744pdffnmfb82ZM+fiiy8+2JMAAAAAaqYGj4n9toqKioKCgoM1\nBQAAAKB2DihwvPHGG61btz5YUwAAAABqZ7+HjF544YW7X0+bNu2JJ57Y892KioqPPvrotdde\nGzp0aA7XAQAAAFTDfgPHo48+uvv1K6+88sorr3z7nr59+95555052QUAAABQbfsNHGvWrMm8\nOO64426//fbzzjtvz3cbNmx4xBFHNG/ePLfrAAAAAKphv4Hj2GOPzbyYMmXK2WefvftHAAAA\ngPpmv4Fjt9/+9rd1sAMAAACg1g7oKSoAAAAA9cG+v8ExbNiwlNKUKVO6deuWeV2FOXPmHPxd\nAAAAANW278Axd+7clNL111+/+zUAAABAvbXvwLFu3bqUUtu2bXe/BgAAAKi39h04OnbsuPt1\nWVlZ06ZNi4uL62oSAAAAQM1kP2T01FNPnThxYh1MAQAAAKid7IHjiCOOOPTQQ+tgCgAAAEDt\nZA8cAwYMeO211yoqKupgDQAAAEAtZA8ckydP3rhx4+jRo7ds2VIHgwAAAABqat+HjO5p0qRJ\nPXv2vPvuu2fPnl1cXNyhQ4eCgoI9b3jooYdytQ4AAACgGrIHjpkzZ2ZebNy48fnnn//2DQIH\nAAAAkF/ZA8eKFSvqYAcAQF169dVXX3zxxXyvgHph6NCh3bp1y/cKgAOVPXAUFxfXwQ4AgLp0\nzz33PPz8w+mH+d4Bebc8bd68eeLEifneAXCgsgcOAIDvprNTmpHvDZB3P873AICDpLqB48sv\nv1y8ePHHH3+8bdu2vd4aPXr0wV4FAAAAUAPVChxTpkyZMGHCX//6132+K3AAAAAA+dUg6x2z\nZ88eO3Zsjx49Jk2alFIaM2bMxIkTBw4cmFK64IIL/vjHP+Z8IwAAAECVsgeOadOmtWvX7qWX\nXrrssstSSoMGDRo3btyCBQtmzZpVUlLSoUOH3I8EAAAAqEr2wLFy5crBgwcXFhYWFBSklCor\nKzPXR4wYcc4552S+1gEAAACQR9kDx/bt29u2bZtSOuSQQ1JKmzZt2v1WcXFxaWlp7sYBAAAA\nVEf2wNG+ffuNGzemlFq2bNmsWbNVq1btfuuDDz7I3TIAAACAasoeOE444YQ33ngjpVRQUDBg\nwIDp06cvWLDgm2++KSkp+dOf/tSzZ8/cjwQAAACoSvbAce655/7lL38pKytLKd18881btmwZ\nNGjQ4Ycffv7551dUVEyYMCH3IwEAAACqkj1w/PKXv6ysrOzYsWNKqXfv3osXLx4xYkS/fv0u\nvvjiJUuWDBgwIOcbAQAAAKrUqKYfOPHEE2fNmpWLKQAAAAC1k/0bHAAAAAD1nMABAAAAhLfv\nP1EZNmxY9X/FnDlzDtIYAAAAgNrYd+CYO3duHe8AAAAAqLV9B45169bV8Q4AAACAWtt34Mg8\nFBYAAAAgBIeMAgAAAOEJHAAAAEB4VT1FZcqUKd26dcv6RBVPUQEAAADyq6qnqFx//fXJE1UA\nAACAeq+qp6i0bds2eaIKAAAAUO9lf4qKJ6oAAAAA9ZxDRgEAAIDw9v0Njr2Ul5dPnz799ddf\nLysr27Fjx17vvvLKKzkYBgAAAFBd2QPHsmXLzjzzzPLy8jpYAwAAAFAL2f9EZfTo0eXl5ePH\nj3/33Xe3bt2641vqYCUAAABAFar1DY7Bgwf/67/+ax2sAQAAAKiF7N/gaN68+dFHH10HUwAA\nAABqJ3vgOOuss5YsWbJr1646WAMAAABQC9kDx9SpUzds2HD99ddv3bq1DgYBAAAA1FT2MziK\niopeeOGFPn36PPjgg8cff3yLFi32uuGJJ57IzTYAAACAaskeON55550BAwZkHhO7ePHi3E8C\nAAAAqJnsgePXv/71J598cuWVV15yySUdOnRo1Cj7RwAAAADqUvZasXjx4kGDBt177711sAYA\nAACgFrIfMtq4ceMuXbrUwRQAAACA2skeOAYMGFBaWloHUwAAAABqJ3vguPXWW999990JEyZU\nVFTUwSAAAACAmsp+BsfEiRN79Ohx8803P/DAA3//93//7cfEPvTQQzmZBgAAAFA92QPHzJkz\nMy8+/PDDDz/88Ns3CBwAAABAfmUPHCtWrKiDHQAAAAC1lj1wFBcX18EO/h979x5cZX3gf/xJ\nRDQUIeIaKZcqXmBVCBFQLsExWNZKubTqeEHqihXZ2PECQp0t6CosaFdlZIbCyoKCiBWwIqKu\nrhpWBLmKLIPihaBBI1aImAiCgCS/P07LshTynEAOJ19+r9dfJ89znsNnxn/a9zznOQAAAMBh\ni3/IKAAAAEAdJ3AAAAAAwTv4V1R++ctfRlH04IMPnnvuuYnX1Zg3b17t7wIAAABI2sEDxwsv\nvBBF0fDhw/e9BgAAAKizDh44Pv/88yiKcnJy9r0GAAAAqLMOHjhatGhx0NcAAAAAdVD8z8T+\nrc8//3zBggUNGjTo06dPVlZWrW8CAAAAqJH4X1F56KGH2rRp88033yT+XLRo0XnnnTdw4MBr\nrrmmc+fO3377bYoXAgAAAMSIDxxz585t1qzZySefnPjzt7/97e7du3/3u98NGjRo7dq1kyZN\nSvFCAAAAgBjxgeOTTz5p27Zt4vWXX365fPnyW2655YEHHpgyZUqPHj1mzZqV4oUAAAAAMeID\nR3l5eZMmTRKv33777SiK+vXrl/jzwgsv/Oyzz1I3DgAAACAZ8YGjSZMmX331VeL1m2++mZmZ\n2aVLl8Sfe/fu3bVrVwrXAQAAACQhPnC0bdv2hRde2LRp0+bNm2fPnt21a9dGjRolTn366adN\nmzZN8UIAAACAGPGB48477/zzn//8k5/8pGXLlmVlZbfddlvieFVV1bJly9q3b5/ihQAAAAAx\n6sW+o2/fvtOmTZsyZUoURddff/11112XOP7WW2/t2rXrsssuS+1AAAAAgDjxgSOKooEDBw4c\nOPCAg5dccklZWVntLwIAAACoofivqAAAAADUcQIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAA\ngidwAAAAAMFL6mdiD/D5558vWLCgQYMGffr0ycrKqvVNAAAAADUSfwfHQw891KZNm2+++Sbx\n56JFi84777yBAwdec801nTt3/vbbb1O8EAAAACBGfOCYO3dus2bNTj755MSfv/3tb3fv3v27\n3/1u0KBBa9eunTRpUooXAgAAAMSIDxyffPJJ27ZtE6+//PLL5cuX33LLLQ888MCUKVN69Ogx\na9asFC8EAAAAiBEfOMrLy5s0aZJ4/fbbb0dR1K9fv8SfF1544WeffZa6cQAAAADJiA8cTZo0\n+eqrrxKv33zzzczMzC5duiT+3Lt3765du1K4DgAAACAJ8YGjbdu2L7zwwqZNmzZv3jx79uyu\nXbs2atQocerTTz9t2rRpihcCAAAAxIgPHHfeeeef//znn/zkJy1btiwrK7vtttsSx6uqqpYt\nW9a+ffsULwQAAACIUS/2HX379p02bdqUKVOiKLr++uuvu+66xPG33npr165dl112WWoHAgAA\nAMSJDxxRFA0cOHDgwIEHHLzkkkvKyspqfxEAAABADcV/RQUAAACgjhM4AAAAgOAd8isq+x4m\nmjB48ODc3NzU7wEAAACosUMGjokTJ+7/Z8+ePQUOAAAAoG46ZODYsmXL/n82atQo9WMAAAAA\nDschA8ff/d3fHc0dAAAAAIfNQ0YBAACA4AkcAAAAQPAO/hWVM844I/mPKCkpqZUpAAAAAIfn\n4IFj+/bt+/+5d+/e8vLyxOsf/ehH3333XeJ1dnb2cccdl9J9AAAAALEO/hWVsv2UlJS0bdu2\nQ4cOL7/88rZt27Zv375t27aXX375ggsuaNu2rds3AAAAgLSLfwbHvffeu2nTpkWLFv385z9v\n2LBhFEUNGzb8+c9/vnjx4k2bNt17772pHwkAAABQnfjA8eyzz1555ZUNGjQ44HiDBg2uvPLK\nP/3pT6kZBgAAAJCs+MCxZcuWqqqqg56qqqrasmVLbU8CAAAAqJn4wHHGGWc899xz+x4sus93\n3333pz/9qVWrVqkZBgAAAJCs+MBRWFhYUlKSn58/b968rVu3RlG0devWefPm5efnb9y48Z/+\n6Z9SPxIAAACgOgf/mdj93XnnnR988MGUKVOuuOKKKIrq1av3ww8/JE4NHjz4jjvuSO1AAAAA\ngDjxgSMzM/M//uM/+vfv/+STT65evbqioqJx48YXXHDBwIEDCwoKUr8QAAAAIEZ84Fi2bNmJ\nJ57Yo0ePHj16HIVBAAAAADUV/wyObt26jRkz5ihMAQAAADg88YHjlFNOadCgwVGYAgAAAHB4\n4gNHQUHBihUr9u7dexTWAAAAAByG+MDxwAMPlJWVDRkyZMeOHUdhEAAAAEBNxT9kdOzYsbm5\nuX/4wx9mzZqVl5fXrFmzjIyM/d8wffr0VK0DAAAASEJ84HjyyScTL8rKyt54442/fYPAAQAA\nAKRXfOBYvXr1UdgBAAAAcNjiA0deXt5R2AEAAABw2OIfMgoAAABQx8XfwZGwdevWxYsXf/HF\nF7t27Trg1JAhQ2p7FQAAAEANJBU4HnzwwdGjR3///fcHPStwAAAAAOkV/xWVWbNmjRgxol27\ndmPHjo2iaNiwYWPGjLn00kujKLr66qufeuqplG8EAAAAqFZ84Jg4ceJpp522cOHCX//611EU\n9ezZc+TIkUVFRTNnzpw7d26zZs1SPxIAAACgOvGBY82aNX369MnKysrIyIiiqLKyMnF8wIAB\nvXr1StzWAQAAAJBG8YFj9+7dOTk5URTVr18/iqKKiop9p/Ly8latWpW6cQAAAADJiA8cTZs2\nLSsri6IoOzu7YcOGa9eu3XeqpKQkdcsAAAAAkhQfONq3b79u3booijIyMgoKCiZPnlxUVLR9\n+/a5c+fOmTMnNzc39SMBAAAAqhMfOHr37r1kyZLS0tIoiu67774dO3b07NnzpJNOuuqqq/bu\n3Tt69OjUjwQAAACoTnzgGDx4cGVlZYsWLaIo6tSp0+LFiwcMGJCfn3/DDTcsXbq0oKAg5RsB\nAAAAqlWvphd07Nhx5syZqZgCAAAAcHji7+AAAAAAqOPiA8ecOXN69OiReAbH/kpLSwsKCp57\n7rnUDAMAAABIVnzgmDJlyrZt2xLP4NhfixYtysvLp0yZkpphAAAAAMmKDxxr167t1KnTQU91\n6tRp7dq1tT0JAAAAoGbiA8fWrVtPOeWUg57KyckpKyur7UkAAAAANRMfOE455ZT169cf9FRx\ncXF2dnZtTwIAAAComfjA0b179/nz53/44YcHHP/ggw/mz5+fn5+fmmEAAAAAyYoPHHfdddee\nPXvy8/MnTJhQXFy8c+fO4uLiCRMmdO/efc+ePcOHDz8KKwEAAACqUS/2HV27dp04ceJtt912\nxx137H/8uOOOmzhxYrdu3VK2DQAAACAp8YEjiqLCwsJu3bpNmjRp+fLl5eXl2dnZXbp0+c1v\nftOuXbtU7wMAAACIlVTgiKIoNzf3scceS+kUAAAAgMMT/wwOAAAAgDouPnDMmTOnR48epaWl\nBxwvLS0tKCh47rnnUjMMAAAAIFnxgWPKlCnbtm1r0aLFAcdbtGhRXl4+ZcqU1AwDAAAASFZ8\n4Fi7dm2nTp0OeqpTp05r166t7UkAAAAANRMfOLZu3XrKKacc9FROTk5ZWVltTwIAAAComfjA\nccopp6xfv/6gp4qLi7Ozs2t7EgAAAEDNxAeO7t27z58//8MPPzzg+AcffDB//vz8/PzUDAMA\nAABIVnzguOuuu/bs2ZOfnz9hwoTi4uKdO3cWFxdPmDChe/fue/bsGT58+FFYCQAAAFCNerHv\n6Nq168SJE2+77bY77rhj/+PHHXfcxIkTu3XrlrJtAAAAAEmJDxxRFBUWFnbr1m3SpEnLly8v\nLy/Pzs7u0qXLb37zm3bt2qV6HwAAAECspAJHFEW5ubmPPfZYSqcAAAAAHJ74Z3AAAAAA1HHJ\n3sGxdevWxYsXf/HFF7t27Trg1JAhQ2p7FQAAAEANJBU4HnzwwdGjR3///fcHPStwAAAAAOkV\n/xWVWbNmjRgxol27dmPHjo2iaNiwYWPGjLn00kujKLr66qufeuqplG8EAAAAqFZ84Jg4ceJp\np522cOHCX//611EU9ezZc+TIkUVFRTNnzpw7d26zZs1SPxIAAACgOvGBY82aNX369MnKysrI\nyIiiqLKyMnF8wIABvXr1StzWAQAAAJBG8YFj9+7dOTk5URTVr18/iqKKiop9p/Ly8latWpW6\ncQAAAADJiA8cTZs2LSsri6IoOzu7YcOGa9eu3XeqpKQkdcsAAAAAkhQfONq3b79u3booijIy\nMgoKCiZPnlxUVLR9+/a5c+fOmTMnNzc39SMBAAAAqhMfOHr37r1kyZLS0tIoiu67774dO3b0\n7NnzpJNOuuqqq/bu3Tt69OjUjwQAAACoTnzgGDx4cGVlZYsWLaIo6tSp0+LFiwcMGJCfn3/D\nDTcsXbq0oKAg5RsBAAAAqlWvphd07Nhx5syZqZgCAAAAcHji7+AAAAAAqOMEDgAAACB4h/yK\nSnZ2djLX169f/9RTT+3SpcuQIUPatWtXe8MAAAAAknXIwFFRUZHkR2zZsmXdunUzZ8588803\nu3btWkvDAAAAAJJ1yK+o7EzOtm3bPvzww7vuumv37t3333//UVwOAAAA8BeHvIPjxBNPTPIj\n2rRpM27cuKKiohUrVtTSKgAAAIAaqLWHjJ577rnl5eW19WkAAAAAyau1wDFjxoydO3fW1qcB\nAAAAJO+QX1GpqeOPP/7444+vrU8DAAAASF6t3cEBAAAAkC4CBwAAABA8gQMAAAAInsABAAAA\nBE/gAAAAAIKXVOBYuHBhv379mjZtesIJJ9T7G6meCAAAAFC9+Dzx0ksv/eIXv6isrGzcuPE5\n55yjaAAAAAB1TXytuP/++zMyMp5++un+/ftnZGQchU0AAAAANRIfON57770rrrji+uuvPwpr\nAAAAAA5D/DM4fvSjH+Xk5ByFKQAAAACHJz5w9OzZc/ny5UdhCgAAAMDhiQ8cDz30UGlp6ahR\no/bu3XsUBgEAAADUVPwzOO67777zzz///vvvnzZtWl5eXnZ29gFvmD59+pHv2LBhw7/8y78U\nFRWVl5e3bNny2muvHTFiRIMGDZL/hBdffLFfv35RFI0cOXLMmDFHPgkAAAAIRXzgePLJJxMv\nNm7cuHHjxr99w5EHjvfee+/iiy+uqKjo06fPmWeeuWjRorFjxxYVFS1YsCArKyuZT9iyZcst\nt9zSsGHD7du3H+EYAAAAIDjxgWP16tWpHnHzzTeXl5dPmzZt4MCBURRVVlb+6le/euaZZ8aN\nG3fPPfck8wmDBw/OzMwcOnTov/7rv6Z2KwAAAFD3xAeOvLy8lC549913V6xYkZeXl6gbURRl\nZmY+/PDDs2fPnjx58siRIzMyMqr/hGnTps2bN++ll15av359SqcCAAAAdVP8Q0ZTbcGCBVEU\n9erVa/+DzZs3z83NLS0t/fjjj6u/vKSk5M4777zpppt69+6dwpUAAABAHRZ/B0fC1q1bFy9e\n/MUXX+zateuAU0OGDDmSBR999FEURW3atDngeOvWrf/nf/7n448//ttT+1RWVt54443Z2dmP\nPvrokWwAAAAAgpZU4HjwwQdHjx79/fffH/TsEQaOioqKKIoaN258wPHEz7WUl5dXc+24cePe\neuut11577W8vT9LmzZsHDRq0c+fOat7zxRdfRFFUVVV1eP8EAAAAkGrxgWPWrFkjRoy48MIL\nf/nLX44cOXLYsGEnn3zyggULFixYcPXVVyd+mTUVEkGhmgdwrF279t577y0sLPyHf/iHw/5X\nsrKy2rdvv2fPnmrec9xxx33wwQexjwIBAAAA0iU+cEycOPG0005buHBhRUXFyJEje/bsefnl\nl48cOfLpp5++8cYbCwsLj3BB4uaLxH0c+zvUnR0JVVVVN9xwQ7NmzR5++OEj+ddPOumk2B9e\nmTx58n/9138dyb8CAAAApFT8Q0bXrFnTp0+frKysxC0MlZWVieMDBgzo1avX2LFjj3BB4hEb\niSdx7C/xkyitW7c+6FV79+5ds2bNp59+etJJJ2X81dChQ6MoGjt2bEZGxqBBg45wGAAAABCK\n+Ds4du/enZOTE0VR/fr1o/97q0VeXt6ECROOcMGll14aRdGrr776wAMP7Du4adOmNWvWNG/e\n/FCBIzMz8+abbz7g4Pvvv79s2bK8vLyOHTtefPHFRzgMAAAACEV84GjatGlZWVkURdnZ2Q0b\nNly7dm3//v0Tp0pKSo58QYcOHS666KIVK1bMmDHjH//xH6MoqqysvPvuuysrKwsLC/d/8sX0\n6dPLy8uvv/76nJyczMzMqVOnHvBR48ePX7ZsWe/evceMGXPkwwAAAIBQxAeO9u3br1u3Loqi\njIyMgoKCyZMn//SnP+3cufNrr702Z86czp07H/mIxx9/vHv37jfddNPcuXNbtWq1aNGiVatW\nde7cediwYfu/bcyYMRs2bOjevXvijhIAAACAhPhncPTu3XvJkiWlpaVRFN133307duzo2bPn\nSSeddNVVV+3du3f06NFHPqJt27arVq269tprlyxZMmnSpG+++WbEiBFFRUVZWVlH/uEAAADA\nMS/+Do7BgwcPHjw48bpTp06LFy9+9NFHS0pKzjzzzNtvv/3CCy+slR1nnXXWH//4x+rfU1xc\nXP0bhgwZMmTIkFrZAwAAAAQkPnAcoGPHjjNnzkzFFAAAAIDDE/8VlX02bty4dOnS/X9FBQAA\nAKAuSCpwLFu2rH379meccUa3bt1WrlyZODhr1qy2bdsuXLgwlfMAAAAA4sUHjg8++KBnz56f\nfPLJL37xi/2P9+nTp6Sk5Nlnn03ZNgAAAICkxD+DY8yYMXv27HnnnXd+/OMfv/DCC/uON2zY\nsEePHosXL07lPAAAAIB48XdwFBUVXXHFFe3atfvbU3//93+f+PlYAAAAgDSKDxxff/31GWec\ncdBTxx133LZt22p5EQAAAEANxQeOk08+ecuWLQc9tXr16h//+Me1PQkAAACgZuIDR35+/ssv\nv7xr164Dji9YsOD1118vKChIyS4AAACApMUHjuHDh2/ZsuWKK65Yt25dFEU7d+5cuXLlsGHD\nLr/88nr16t11112pHwkAAABQnfhfUcnPz584ceLtt9/+yiuvRFHUr1+/xPHjjz9+6tSpubm5\nqR0IAAAAECc+cERRVFhYePHFFz/22GNLly79+uuvGzdu3KVLl9tvv/38889P9T4AAACAWEkF\njiiKzj///AkTJqR0CgAAAMDhiX8GBwAAAEAdd8g7OF566aUkP6JPnz61NAYAAADgcBwycPTt\n2zfJj6iqqqqlMQAAAACHo7pncNSrV++yyy479dRTj9oaAAAAgMNwyMBx3nnnrVu37vXXX+/X\nr9+gQYMuu+yyzEwP7AAAAADqokM2i/fff3/x4sUDBgx49dVXe/Xq1apVq/vvv/+zzz47muMA\nAAAAklHdTRn5+fnTpk3btGnTv//7v5966qmjRo1q1arV5Zdf/uyzz+7evfuoTQQAAACoXvy3\nTho1alRYWPjOO++sXr26sLBw+fLl11xzTYsWLVasWHEU9gEAAADEqsFjxSZS8AAAIABJREFU\nNfLy8iZOnDh37twWLVps2bJl06ZNqZsFAAAAkLzqfkVlf2VlZU899dTjjz/+/vvvH3/88Vdc\ncUX79u1TugwAAAAgSTGBo6qq6o033pg6deq8efN2797dunXrf/u3fxs4cGBOTs7R2QcAAAAQ\n65CBo7S0dNq0aU888URJSUlWVtY111wzaNCgSy655GiOAwAAAEjGIQPH6aefXllZmZeXN2HC\nhF/96lfZ2dlHcxYAAABA8g4ZOCorK+vVq/fNN9888sgjjzzySDUfUVJSUvu7AAAAAJJW3TM4\nfvjhh40bNx61KQAAAACH55CBY+fOnUdzBwAAAMBhO2TgOPHEE4/mDgAAAIDDlpnuAQAAAABH\nSuAAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAA\nBE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAA\nAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEA\nAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsAB\nAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7A\nAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAie\nwAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAI\nnsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAA\nCJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAA\nAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMA\nAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIED\nAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyB\nAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8\ngQMAAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQ\nPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAA\nEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAA\nABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcA\nAAAQPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIH\nAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgC\nBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4\nAgcAAAAQPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAg\neAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAA\nIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAA\nACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4A\nAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQO\nAAAAIHgCBwAAABC8uhI4NmzYMGDAgKZNm5544onnnHPOPffcs2PHjuov2b59++zZs/v373/u\nuec2aNCgcePG3bt3nzp1amVl5dHZDAAAANQR9dI9IIqi6L333rv44osrKir69Olz5plnLlq0\naOzYsUVFRQsWLMjKyjrUVVOnTh06dGj9+vU7dOjQrl27r776asmSJW+//faLL774/PPPZ2bW\nlXYDAAAApFqdqAA333xzeXn5E088MX/+/PHjx69cubJ///7Lli0bN25cNVe1bNly0qRJmzdv\nXrp06Zw5cxYuXLhmzZqcnJz58+fPnj37qI0HAAAA0i79gePdd99dsWJFXl7ewIEDE0cyMzMf\nfvjhzMzMyZMnV1VVHerCq6666tZbb23cuPG+I+edd97QoUOjKFq4cGGKVwMAAAB1SPoDx4IF\nC6Io6tWr1/4HmzdvnpubW1pa+vHHH9fo0xK944QTTqjFhQAAAEAdl/7A8dFHH0VR1KZNmwOO\nt27dOoqiGgWOqqqqGTNmRFHUt2/f2hsIAAAA1HXpf8hoRUVF9Nc7L/aXnZ0dRVF5eXnyHzVq\n1Khly5ZdeeWVPXv2TPKSzz///LLLLtu9e3c17/n222+jKKrmyzIAAABAeqU/cBxKIihkZGQk\n+f4//OEPo0aN6tChw7Rp05L/V0477bR//ud/3rVrVzXveeutt55++unklwAAAABHWfoDR+Le\njcR9HPs71J0dBzVu3Ljhw4d37Njx9ddfb9SoUfL/ev369W+88cbq31NVVfX0008n/5kAAADA\nUZb+Z3Aknr6ReBLH/tavXx/99Ukc1bv//vuHDx/etWvXoqKik08+ORUjAQAAgLos/YHj0ksv\njaLo1Vdf3f/gpk2b1qxZ07x589jAcdddd40aNaqgoOC1115L8nYPAAAA4BiT/sDRoUOHiy66\naPXq1YkfQImiqLKy8u67766srCwsLNz/yRfTp08fP3785s2b971t8ODBjz766M9+9rP//M//\nbNiwYRrWAwAAAHVA+p/BEUXR448/3r1795tuumnu3LmtWrVatGjRqlWrOnfuPGzYsP3fNmbM\nmA0bNnTv3j0nJyeKonHjxk2ZMiUzM7NJkya33nrr/u9s167dAdcCAAAAx7A6ETjatm27atWq\ne++994033njllVdatGgxYsSIESNGZGVlVXPV119/HUVRZWXlM888c8Cpn/3sZwIHAAAA/P+j\nTgSOKIrOOuusP/7xj9W/p7i4eP8/f//73//+979P5SgAAAAgDOl/BgcAAADAERI4AAAAgOAJ\nHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDg\nCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA\n4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAA\ngOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAA\nAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgA\nAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAAwRM4\nAAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMET\nOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAAAADB\nEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AAAAAA\nwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAA\nAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAA\nAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP4AAAAACCJ3AA\nAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidw\nAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIn\ncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP4AAAAACC\nJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAA\ngidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAA\nAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP4AAA\nAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AA\nAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/g\nAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAAAARP\n4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAE\nT+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsABAAAA\nBE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7AAQAA\nAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAiewAEA\nAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABA8gQMAAAAInsAB\nAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcAAAAQPAEDgAAACB4AgcAAAAQPIEDAAAACJ7A\nAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4AAAAgeAIHAAAAEDyBAwAAAAie\nwAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQOAAAAIHgCBwAAABC8uhI4NmzY\nMGDAgKZNm5544onnnHPOPffcs2PHjpReCAAAABwz6kTgeO+99zp16vTMM89cdNFFhYWFjRo1\nGjt27E9/+tOdO3em6EIAAADgWFInAsfNN99cXl7+xBNPzJ8/f/z48StXruzfv/+yZcvGjRuX\nogsBAACAY0n6A8e77767YsWKvLy8gQMHJo5kZmY+/PDDmZmZkydPrqqqqvULAQAAgGNM+gPH\nggULoijq1avX/gebN2+em5tbWlr68ccf1/qFAAAAwDEm/YHjo48+iqKoTZs2Bxxv3bp1FEXV\ndIrDvhAAAAA41lSl29VXXx1F0fPPP3/A8cGDB0dRNGPGjFq/cH/FxcX16tVL738CAAAACMjK\nlStr+v/9j4K6+//tq6qqoijKyMhI6YVnnXXWO++888MPP1T/tnfffbdDhw41XZIie/bsKS4u\nTvcKqBNOO+20Jk2apHvFQezYsWPjxo3pXgF1QvPmzRs1apTuFQfx7bfffvHFF+leAXXC6aef\n3qBBg3SvOIitW7d+9dVX6V4BdcLZZ599/PHHp3vFX9SrV699+/bpXnEQ6Q8cjRs3jqKooqLi\ngOOJI4mztXvhAZL5D9OxY8ckP+3o6NKlS7onADEuvvjidE8AAID/j6T/GRyJh2gkHqixv/Xr\n10d/faBG7V4IAAAAHGPSHzguvfTSKIpeffXV/Q9u2rRpzZo1zZs3r6ZTHPaFAAAAwDEm/YGj\nQ4cOF1100erVq2fMmJE4UllZeffdd1dWVhYWFu7/KI3p06ePHz9+8+bNNb0QAAAAOLZlJB7J\nmV7vvfde9+7dt23b1rdv31atWi1atGjVqlWdO3f+7//+76ysrH1vO/vsszds2LBy5cpOnTrV\n6EIAAADg2Jb+OziiKGrbtu2qVauuvfbaJUuWTJo06ZtvvhkxYkRRUVFspDjsCwEAAIBjSZ24\ngwMAAADgSNSJOzgAAAAAjoTAAQAAAARP4AAAAACCJ3AAAAAAwRM4AAAAgOAJHAAAAEDwBA4A\nAAAgeAIHAAAAEDyBAwAAAAiewAEAAAAET+AAAAAAgidwAAAAAMETOAAAAIDgCRwAAABA8AQO\nAAAAIHgCBwAAABA8gQMAAAAInsABAAAABE/gAAAAAIIncAAAAADBEzgAAACA4AkcUANz5869\n/fbb8/PzGzZsmJGRcd1116V7EfB/bN++ffbs2f379z/33HMbNGjQuHHj7t27T506tbKyMt3T\ngCiKor17944ePbpXr16nn356gwYNmjRpcsEFF4waNWrr1q3pngYc3IsvvpiRkZGRkXHPPfek\newvEyKiqqkr3BghGp06dVq1a1ahRo6ZNm3788cfXXnvtrFmz0j0K+F/jx48fOnRo/fr1O3To\n0LJly6+++mrJkiU//PBDv379nn/++cxMWR/S7Pvvv8/KymratGnr1q1zcnK2b9++atWqLVu2\nNGvWbMmSJaeffnq6BwL/x5YtW9q1a/fdd99t37595MiRY8aMSfciqI7/qQc18Mgjj6xfv768\nvHzcuHHp3gIcRMuWLSdNmrR58+alS5fOmTNn4cKFa9asycnJmT9//uzZs9O9DohOOOGEkpKS\nL7/8cuHChc8+++wrr7xSWlo6YMCATZs2jR07Nt3rgAMNHjw4MzNz6NCh6R4CSRE4oAYKCgrO\nPvvsjIyMdA8BDu6qq6669dZbGzduvO/Ieeedl/ifZQsXLkzfLuAvMjIyDrhNo379+rfccksU\nRevXr0/TKODgpk2bNm/evClTpjRp0iTdWyApAgcAx7hE7zjhhBPSPQQ4uOeeey6Kovbt26d7\nCPC/SkpK7rzzzptuuql3797p3gLJqpfuAQCQQlVVVTNmzIiiqG/fvuneAvyvIUOGfP/99xUV\nFe+8805xcXFubu7IkSPTPQr4i8rKyhtvvDE7O/vRRx9N9xaoAYEDgGPZqFGjli1bduWVV/bs\n2TPdW4D/NXXq1O+++y7x+vLLL58+ffqpp56a3knAPuPGjXvrrbdee+21/b/1CXXf/2vvfkOr\nqv8Ajn/vNv+1OV2uMW/ILBSKOZiKqKgppM5KmVA0h4pKUso0SO1BgjT7R2GmULpEQ62BSpBm\nRZg5RarNB5pOxD0wNJWc5rBS6Y/e7fdgNNbIlfMnd8der0fX7zln3899tPH2nnPdogLAHeud\nd95Zvnz5kCFDNm7cmOxZgL+4cuVKY2PjuXPntm7devz48cLCwkOHDiV7KCCEEI4ePbps2bJ5\n8+ZNmDAh2bPAzRE4ALgzrVy5cuHChUOHDv3yyy8zMzOTPQ7QViwWy83NLSkp+eyzz+rr6+fM\nmZPsiYDQ1NQ0c+bMeDy+YsWKZM8CN03gAOAOVF5evmTJkpEjR+7ZsycrKyvZ4wDtyc/P79u3\nb21t7aVLl5I9C/zXJRKJI0eOnDx5smfPnrE/NX8f2auvvhqLxebOnZvsGeGGPIMDgDvNokWL\nVq1aNW7cuE8++SQjIyPZ4wD/4PLlyxcuXAghpKX50xSSLCUl5amnnmqzeOzYsZqamsLCwqFD\nh44ZMyYpg8G/4bcIAHeOxsbGefPmrV+/vqioaPv27T169Ej2RMBf1NTU9OjRo/U3wjY0NMyd\nOzeRSDz00EM9e/ZM4mxACCElJWXDhg1tFlevXl1TU/PYY4+98sorSZkK/iWBA27CRx99tHPn\nzhDC2bNnQwgHDhyYPXt2CCE7O/vNN99M7mxACGHlypXr169PSUm5++6758+f3/pQQUHB4sWL\nkzUY0Gzfvn0vvPDC/ffff99992VlZdXX1x88ePDXX3/t27fvunXrkj0dANEmcMBNOHTo0ObN\nm1v+eerUqVOnToUQ8vLyBA7oDBoaGkIIjY2NW7ZsaXOoqKhI4ICkKy4uvnjx4r59+44cOXLp\n0qWMjIyCgoJHH3302Wef9bgcAG5RrKmpKdkzAAAAANwS36ICAAAARJ7AAQAAAESewAEAAABE\nnsABAAAARJ7AAQAAAESewAEAAABEnsABAAAARJ7AAQAAAESewAEAAABEnsABAAAARJ7AAQAA\nAESewAEAAABEnsABAAAARJ7AAQAAAESewAEAAABEnsABAAAARJ7AAQAAAESewAEAAABEnsAB\nAAAARJ7AAQAAAESewAEAAABEnsABAAAARJ7AAQAAAESewAEAAABEnsABAAAARJ7AAQAAAESe\nwAEAAABEnsABAAAARJ7AAQAAAESewAEAAABEnsABAAAARJ7AAQAAAESewAEA3F5nz56NxWJT\np05N9iAAwJ1M4AAAbkldXd3ChQsHDRrUq1evrl273nvvvcXFxVu2bEkkErd13xMnTsRisWnT\npt3WXQCAqEhL9gAAQIS99NJLy5cvb2xsHDBgwKRJk9LT08+fP79///6dO3dWVFTs378/2QMC\nAP8VAgcA0EGvvfbaiy++mJubu3nz5okTJ7asX79+vbKysqKiIomzAQD/NW5RAQA64tSpU+Xl\n5V27dv3iiy9a140QQlpa2uzZs/fs2XOjaz/99NNYLFZeXt5mvXfv3gMGDGi98vnnn0+YMCEe\nj3fr1q1v376jR49esWJFCOH1118fOHBgCGHbtm2xP1VWVrZcWF1d/fjjj+fm5nbt2jUej8+Y\nMaOurq7l6OHDh2Ox2OzZs7/77rtp06bl5OSkpKTU1NS0syMA0Mn5BAcA0BEbN268du3arFmz\nCgoK/vaEjIyMW9zi/fffnzVrVm5ubnFxcU5Ozo8//njs2LENGzY8//zzU6ZM6dKly5IlS0aM\nGFFWVtZ8/qhRo5pfrF+/ft68eX369Jk8eXJOTs7Jkyc//PDDHTt27NmzZ/jw4S0//8yZM8OH\nD8/Ozp40adLVq1e7d+/ezo63+F4AgNtN4AAAOuKrr74KIRQVFd2+LdatW5eamnrw4MF4PN6y\neOnSpRBCfn5+t27dlixZkpeXN2PGjNZXHT9+vKysbMKECdu3b+/Ro0fzYm1t7ahRo55++ukj\nR460nFlVVbVgwYLVq1enpqY2r5SVld1oRwCgk3OLCgDQEefOnQsh9OvX77bukpqampb2l/+P\nycrKav+StWvXXrt2benSpVevXr34p3g8/vDDD9fW1n7//fctZ2ZnZ7/xxhstdaPDOwIAnYFP\ncAAAHdHU1BRCiMVit2+L0tLSb775Jj8/v6SkZNy4caNHj87Nzf3Hq6qrq0MIY8eO/duj586d\ny8vLa35dWFh411133fqOAEBnIHAAAB0Rj8fr6upOnz7d8uSL/7sFCxZkZWWtWbOmoqJizZo1\nIYSRI0euWLGi/R0bGhpCCDt37my5P6W1Bx98sOV16/tQbmVHAKAzEDgAgI4YPXp0VVXVrl27\nSktLb/balJSUEML169dbL167du3q1avZ2dmtF6dPnz59+vRffvmlurp6x44d77333iOPPHLs\n2LF2bo3p1atXCCE3N3fYsGHtj/G3Hz/pwI4AQGfgGRwAQEfMmTOnS5cuW7duPXr06N+ecOXK\nlRtd2/xUizNnzrRe/Pbbb9skjxaZmZlFRUUVFRWLFy++fPlyVVVVCKH52RmJRKLNySNGjAgh\nbN269SbezL/bEQDozAQOAKAj+vfvX15e/vvvvxcVFe3evbv1oUQiUVlZOX78+BtdW1BQ0L17\n948//ri+vr555eeff160aFGb03bv3t0meVy8eDGE0PzgjD59+oQQTp8+3eaqBQsWpKWlvf32\n222qxJUrV7Zt29b+m2p/RwCgM3OLCgDQQUuXLr1+/fry5csnTpw4cODAIUOGpKenX7hwobq6\nuqGh4UaP+QwhZGRkzJ8/f9WqVYWFhVOmTPnjjz927949dOjQzMzM1qeVlpampaWNHTs2Ly8v\nNTX1wIEDe/fuzc/Pnzx5cgghMzNz+PDhBw4cKC0tfeCBB1JTU6dOnTpo0KBBgwatW7fumWee\nGT9+/MSJEwcPHpxIJOrq6qqqqvr3719SUtLOO2p/RwCgM4s1PwIdAKBjjh8/vnbt2r17954+\nffq333675557hg0bVlpa+sQTTzTfRXL27Nl+/foVFxfv2LGj5apEIvHyyy9v2rTphx9+iMfj\nM2fOXLZsWU5OTnZ29okTJ5rPeffdd3ft2lVbW1tfX9+lS5e8vLwnn3yyrKysd+/ezSecOHHi\nueee+/rrr3/66aempqYPPvhgxowZzYcOHz781ltv7du37/z58+np6fF4fMyYMc3fjdJ8dPDg\nwbNmzdq0aVPr9/KPOwIAnZbAAQAAAESeZ3AAAAAAkSdwAAAAAJEncAAAAACRJ3AAAAAAkSdw\nAAAAAJEncAAAAACRJ3AAAAAAkSdwAAAAAJEncAAAAACRJ3AAAAAAkSdwAAAAAJEncAAAAACR\nJ3AAAAAAkSdwAAAAAJEncAAAAACRJ3AAAAAAkSdwAAAAAJEncADZI6+AAAAAXklEQVQAAACR\nJ3AAAAAAkSdwAAAAAJEncAAAAACRJ3AAAAAAkSdwAAAAAJEncAAAAACRJ3AAAAAAkSdwAAAA\nAJEncAAAAACRJ3AAAAAAkSdwAAAAAJEncAAAAACR9z/bty2V27D7HAAAAABJRU5ErkJggg=="},"metadata":{"image/png":{"width":720,"height":720}}}]},{"metadata":{},"cell_type":"markdown","source":"#### Plotting the gene expression of a particular gene in a tSNE map"},{"metadata":{"trusted":false},"cell_type":"code","source":"g='ENSG00000171611' #### Plotting the log expression of PTCRA \nplotExptSNE(FG,g)","execution_count":9,"outputs":[{"output_type":"display_data","data":{"text/plain":"Plot with title “ENSG00000171611”","image/png":"iVBORw0KGgoAAAANSUhEUgAABaAAAAWgCAIAAAAnwnOfAAAACXBIWXMAABJ0AAASdAHeZh94\nAAAgAElEQVR4nOzdd2BUVfrw8XOnZSaZ9N4IJCEBAgFCqFIVERGUYtdVWXXdta9t1dVdf4qv\nbV1ddxXdXcsqdsROsYHSayihhkASkpCeTNpk6n3/GBzCzCRMCkkGvp+/Zp577plnAuLMk3Oe\nI8myLAAAAAAAAHyZorcTAAAAAAAA6CoKHAAAAAAAwOdR4AAAAAAAAD6PAgcAAAAAAPB5FDgA\nAAAAAIDPo8ABAAAAAAB8HgUOAAAAAADg8yhwAAAAAAAAn0eBAwAAAAAA+DwKHAAAAAAAwOdR\n4AAAAAAAAD6PAgcAAAAAAPB5FDgAAAAAAIDPo8ABAAAAAAB8HgUOAAAAAADg8yhwAAAAAAAA\nn0eBAwAAAAAA+DwKHAAAz3Jzc6W2hYSEtDVyxYoVreeZPXu2Iz5o0CCXl/jqq68uvfTS+Ph4\njUYTGBg4YMCAiRMn3nnnne+99557PgaD4ZVXXpk9e3ZiYqK/v79Go4mMjBw3bty99977008/\nybLcenBZWdn9998/ePBgf3//wMDAkSNHPvnkkw0NDe7T+sTIr7/++q677powYYK/v7/z57xz\n506XYZdffnk7f2SSJK1cubKjczrt27fv97//fXp6ul6vDwgISEpKuvjii1977bVO5NmJVwcA\nADg9GQAAT/bs2dPO/z6Cg4PbGjl06FCr1eq8eskllzji6enpree//fbbvZnc4b333gsODm4n\nn48//tg5ePPmzWFhYe5jUlJSCgoKWk/rKyOnTJniPiwnJ8flp7RgwYJ2fkRCiBUrVnR0Tofn\nnntOofDwSxGXP1Pv5+zQqwMAAHhD1f4nIQAAhBDR0dH9+/dvHQkMDGxrcG5u7jvvvHPzzTe3\nM+G6deucv/zX6XRjxowJCgoqLS0tKCiorq52Gfzqq6/eeeedzqd6vT4rKys0NLSxsTEvL6+o\nqEgIYbfbHVcNBsP8+fNramqEEEFBQdddd11LS8uSJUssFkt+fv5VV121YcMGx3d1XxkphJAk\nqV+/fqNGjbLb7V9++WVbP9Xs7OyWlhaX4KZNmxw/Uo1GM2zYMGfcyzmFEP/973//9Kc/OR5n\nZWXNmDEjMjLSYDAcPXq0tra29Ujv5/R+JAAAgLd6u8ICAOijWq/LuOOOO7wc6RAXF9fU1OS4\n6nEFx1//+ldHMCAgoLi4uPVs27Zte/bZZ51PDx48qFarHYMlSXrqqaecMzscPXr0ueeec+xS\nkWX5b3/7mzONVatWOYKvvvqqM/j111/71khZlp1vufXmHW/WO5SXl+t0Osf4W2+9tfUlL+c0\nGAzOtTNPPvlk+y/nfZ6dfkcAAABtoQcHAKA7xcbGCiFKS0v//ve/tzPMuTbBz8/P39+/9aVR\no0Y51wsIIf7xj39YLBbH44cffvixxx5zGd+/f/+HHnpo2rRpjqfO5QBRUVEzZsxwPL722msl\nSXI8/vzzz31rpBDC5S177+WXXzYajUIIpVLZ+qfq/ZxLly41GAxCiICAgJCQkOzs7KCgoODg\n4KlTpy5dutRlsPd5dvodAQAAtIUCBwDg9JYuXTruVC+//LLHkffff7/jF/7PP/98RUVFWxNO\nmDDB8aCmpiY5Ofnqq69+4YUXvv/++/r6epeRP/zwg+OBJEn33XffaVN1NqpMTU11BkNCQiIi\nIhyPd+3a5VsjO81gMDj3AV1xxRUpKSmdmGTdunWOB01NTXfffff27dsbGhrq6+t//vnnK664\n4qGHHupikgAAAN2FAgcA4PTKy8s3n6qgoMDjyPDw8EceeUQI0dDQ8MQTT7Q14fTp0xcuXOh4\nXFdX9/HHHz/00EMzZswIDw+fPXv29u3bnSOLi4sdD2JjY51f/oUQer2+9fkgCQkJQgir1eo8\ngsSlKanzaVVVlQ+N7IrXXnvNsfhCCOH4Q+mE0tJS52OVSnXvvfe+8MILzgNxXnjhhR9//LGL\neQIAAHQLChwAgG52zz33JCYmCiH+85//HDx4sK1hb7311tKlSy+66CJnkwghhNVq/fbbb8eP\nH+9cOODk3LvRDrnVYbHyqQfHOp865vGVkZ1mNBqdq2xmz56dmZnZuXnMZrPz8X333ffSSy89\n8MADK1euVCqVjuC7777blTwBAAC6CwUOAMDpuTcZbWuLihBCq9UuWrRICGG1Wh9++OF2pl2w\nYMHKlSsNBsP27dv/9a9/TZ482RG3WCyOGYQQ8fHxjgfHjx9vfcDKk08++cwzzwwdOrT1hGq1\nOigoyPHYZbeL82l4eLgPjey0N99807lF6NFHH+30PCEhIc7HEydOdDxISkpy1LCEEPn5+Z2e\nHAAAoBtR4AAAdL/rr79+xIgRQogvvvhix44d7Q9Wq9VZWVl33HHHmjVrnAWLvLw8x4Pp06c7\nHtjt9ldeecV513333ffwww+np6e7zDZ8+HCXGYQQdXV1zh0fzgG+MrITrFar85SWqVOnjh8/\nvtNTuZws63zsbBPbegEOAABAL6LAAQDofgqF4oUXXnA8Pn78uPuAN95447e//e3q1attNpsz\nWFlZ6fx671w4cPfddzt3QyxatOhvf/tb600T7ubOneuc7bvvvnM8/uCDD5xbP+bNm+dbIzvh\n/fffLywsdDzudPcNhzlz5jgfb9682fGgtLS0qKjI8Tg7O7sr8wMAAHQXyWXrLwAADrm5uc7f\n3kdGRvbr189lwGeffZaUlOQy8u23377pppscj2fOnLlq1Srn+PT09AMHDjgev/zyy3/84x+F\nEAEBAZmZmZGRkY2NjZs2bWpubnYMePTRR59++mnH4xdffPGBBx5wzhMSEpKVlRUUFFRZWblt\n2zaTySSEiI+Pd7QjNRgMGRkZJSUlQoigoKDrr7/eaDQuWbLEcdbs2LFjN2zY4Fh94CsjhRDv\nvvvu7t27hRD79+9fvny5I3jDDTdERkYKIS6++OILLrjA+fORZTkjI2P//v1CiOzs7K1bt3r8\n8/V+zosvvnjlypVCCK1W+/DDD8fHxy9evNixMEer1R44cMDx16BDc3boHQEAAHhFBgDAkz17\n9rT/f5D9+/e7j3z77bedM+zatcv5FV0IkZ6e7rzUTgsPIcTIkSMNBkPrZN544w1/f/92bpk6\ndapz8ObNm8PCwtzHJCcnFxQUtJ7WV0YuWLCgnff+1FNPtR782WefOS999tlnbf35ej9nWVlZ\n640qTlqt1mV+7+fs0DsCAADwBltUAABnSmZm5o033ujx0p133rl27do///nPkydPTk1NDQwM\nVKvVkZGRU6dO/cc//rFx40ZnD06H3/3ud4WFhc8+++wFF1wQHR2t0Wj8/Pzi4uKmTp366KOP\nbtq0afXq1c7BY8aM2bt37x//+Mf09HSdTudYJPLEE0/s3LnTudbAt0Z2yDPPPON4MHjw4K7s\nc3GKjo7evHnz888/n52dHRgYqNFo+vfvf/PNN+fk5MyfP7/r8wMAAHQLtqgAAAAAAACfxwoO\nAAAAAADg8yhwAAAAAAAAn0eBAwAAAAAA+DwKHAAAAAAAwOdR4AAAAAAAAD6PAgcAAAAAAPB5\nFDgAAAAAAIDPo8ABAAAAAAB8HgUOAAAAAADg8yhwAAAAAAAAn0eBAwAAAAAA+DwKHAAAAAAA\nwOdR4AAAAAAAAD6PAgcAAAAAAPB5FDgAAAAAAIDPo8ABAAAAAAB8nqq3E/ANsixfdNFFNTU1\nvZ0IAAAAgLPQtGnTXnjhhd7Ook+46667Nm7c2NtZoEt66++zJMtyz7+qzzGbzX5+fg899FBK\nSkpv5wIAAADgrLJq1aqSkpJNmzb1diJ9QmZm5idKc29ngS65yS+kV/4+s4KjAy677LIJEyb0\ndhYAAAAAziq1tbWff/55b2cB+Dx6cAAAAAAAAJ9HgQMAAAAAAPg8ChwAAAAAAMDnUeAAAAAA\nAAA+jwIHAAAAAADweRQ4AAAAAACAz6PAAQAAAAAAfB4FDgAAAAAA4PMocAAAAAAAAJ+n6u0E\nAAAAAAA4SaFW9nYK8Ems4AAAAAAAAD6PAgcAAAAAAPB5FDgAAAAAAIDPo8ABAAAAAAB8HgUO\nAAAAAADg8yhwAAAAAAAAn0eBAwAAAAAA+DwKHAAAAAAAwOdR4AAAAAAAAD6PAgcAAAAAAPB5\nqt5OAAAAAACAkxQaZW+ngK6x9s7LsoIDAAAAAAD4PAocAAAAAADA51HgAAAAAAAAPo8CBwAA\nAAAA8HkUOAAAAAAAgM+jwAEAAAAAAHweBQ4AAAAAAODzKHAAAAAAAACfR4EDAAAAAAD4PAoc\nAAAAAADA56l6OwEAAAAAAE5SqJW9nQK6xto7L8sKDgAAAAAA4PMocAAAAABCCFFdb6pvMvd2\nFgCATmKLCgAAAM5psiyv2HJsyY+H6xpNQoiYUN3CmYMmDYvp7bwAAB1DgQMAAADntDdXHFy2\n7qjzaVmt8ZkPcwxNQ2aPS+rFrAAAHcUWFQAAAJy7Kg0tn28ocI+/s+qQ2WLr8XQAAJ1HgQMA\nAADnKFmWX/96r2yX3S81m6yHS+t7PiUAQKdR4AAAAMA56pOfj2zcV9HWVZPF3pPJAAC6iAIH\nAAAAzkV2u/x5q9YbLiRJ9IvS92Q+AIAuosABAACAc1F1fUt9s6WtqxOHxoQH+fVkPgCALuIU\nFQAAAJyLFIo2f9UXHxFw97xhPZkMgNYUGmVvp4CuMfbOy1LgAAAAwLkoPMgvJlRXVuvhY/gf\n5gwJ0J7Zz8nbDlXuzq8xmqxJMfoLsuJ1Gj6WA0BX8S8pAAAAzlE3zEh7/uNdLsHM5PCRqRFn\n7kWNJuui93dsPVDpjHzw4+FHrx05dEDYmXtRADgX0IMDAAAA56ipw+MeuWZkRLDW8VShkGaP\nS3r8+ixJOoMv+sY3+1tXN4QQVYaW/3t3e1OL9Qy+KgCcA1jBAQAAgHPXpGExEzKij9c0Nxot\n/aL0/n5n9uOxyWL7cUeJe9zQZF6fWzYjO+GMvjoAnN0ocAAAAOCcplRICREBPfNa1fUtJovN\n46WSqqaeyQEAzlZsUQEAAAB6iK7tFSL+Z7itKQCc9ShwAAAAAD0kVO+XEhfk8VJ2WmQPJwMA\nZxkKHAAAAECHtVhsJqu9Ezf+4dIhKqXrh/DZ45LaKnwAALzEQjgAAADAW7IQR6ub9pU1tFht\nQogAjWpYbFBCiM77GTKTw/9193n//fbAnqM1Zqs9PsL/yikpF42mvSgAdBUFDgAAAMBbe0oN\nhyobnU+bzNZNhTUjrSEpHWlTmhwb9P9uGSPLstlq91Mrz0CaAHAuosABAAAAeKXZYsuranSP\n55YZ+of5KxVSh2aTJInqBuCRUsN/GugMenAAAAAAXqlqNMmyh7jFJtcZLT2eDgDgFBQ4AAAA\nAK/YPVU3HKztXAMA9Ai2qAAAAABeCfRr88NzkNarz9V2u1xa3VxRZ4wN948J9Zc6tqkFANAe\nChwAAACAV8ICNKH+mtpms0s8Plin86Kbxt6C2n9+nnvkeL3j6dABYffMH5oUHeh9AqXVzfml\nBiGktITg6NAOHN0CAOcCChwAAACAVyQhxvcP21xYU910ssYRG6TNTgw57b1Hjtf/6d+bzFa7\nM5J7tOaB1ze98cfJYUF+p73daLK+9uW+77YXy7IshFAopFlj+t02ZzBtSgHAiQIHAAAA4C1/\ntXJaamR5Q0ut0SIJKTxAExGg8ebGj1bnt65uOBiazF+sP/rbiwed9vYXPt61LrfM+dRul7/Z\nVGiy2B68aniH8geAs5ivNhldv379rFmzQkND/f39hwwZ8uKLL7a+ajAYbr/99piYGK1Wm5WV\ntWzZst7KEwAAAGef6EDtoKjA9Ci9l9UNIcTBojqP8f1FdXVN5t1Ha/YX1TWbrB7HFFc2ta5u\nOP2wo7jK0OJlAgBw1vPJFRwff/zxddddN378+KeffjowMPDIkSMVFRXOq3a7/ZJLLtm9e/fT\nTz+dkpLy5ptvXn755cuWLZs7d24v5gwAAIBzmd3jAbNClFQ33frSWsdVP7Xy8kkD5k1Ikk7t\nPppfWu/xXlkW+aX1EcHabs8WAHyR7xU4ysrKbrnllrlz537yyScKhYcVKMuWLVu/fv0777xz\n4403CiEuuuiirKysBx54gAIHAAAAektqfHB5rdE9bmi2aH89nMVksb3/02GzxXb11JTWYzx9\n5v31EgexAMCvfG+LyjvvvNPY2PjMM88oFAq73XUfoxDiiy++0Gq1V199teOpUqn8zW9+k5+f\nv3v37p7NFAAAADjhiinJKqVrMUISkp/G9TeOX24sNFlsrSPpiSGSp0KGUiGlJQZ3b54A4Lt8\nbwXHL7/8kpCQsHPnzjlz5hw6dCg0NHTBggXPP/98SMiJ5tV79+4dOHCgn9/JZtTDhg0TQuTm\n5mZmZrYzc01NTV2d572RFoul+94BAAAAzjlDkkL/8ptRr3ye6+yaER6kNVrt7oULs9VeWNGY\nFn+ychEVort4TOLyzUUuI+dNHBDsdRMQwIcoOB7I53lYi9ADfK/AUVpaWldXt3Dhwscff3zU\nqFGbNm1atGjRnj171q9f79ixUl1dnZyc3PqWsLAwR7z9mTMzM0tKStoZsHHjxgkTJnT5HQAA\nAOBcNG5I9MiBEQeK6irqjPERAYWVzf9dsd/jSPd+HXfOzQjRa5b+fMRxFIufWnn1tJRrzk89\n0zkDgA/xvQKH3W5vbGx86aWX7r33XiHE9OnTJUl67LHHvvvuu5kzZ7Zzo8d1fa3l5OQ0NDR4\nvNTc3Dxs2DC9Xt/ptAEAAAA/tXJ4SviJxxrPv6NWKxX9ok587Cwoa9i4r7y81hgb7n/R6MQr\npqQUlDVIkugfHeiv9b1P8gBwRvneP4vh4eFCiNa1jFmzZj322GM7duxwBMPDw2tqalrf4njq\nWMfRjsjIyMjISI+XGhsbu5g2AAAA0FpKbNDIlPCcfNdVxpeMTdRplEKId1Yd/Hh1vs1+YjnH\ne98dunnWoHkTB/R0ogDgI3yvyaijj0br9qKOx84TVTIyMg4dOtTScvJIcEd70aFDh/ZoogAA\nAEC7/jh/2NThsc6naqViwcQB152fKoRYs7P0gx8PO6sbQgiz1b74q317jtR4mAgA4IsFjgUL\nFgghvvnmG2fkq6++EkKMGzfO8XTevHkmk+nDDz90PLXZbO+9915KSkr7HUYBAACAHhagVd11\nacbiu8576IrMP18z8vV7Jl47LcVx8uuKLcc83rJii2urUQCAg+9tUZk8efLll1/++OOPNzQ0\nZGdnb9y48cUXX5w5c+bUqVMdA+bNmzdhwoS7777bYDAkJye/9dZbubm5y5Yt69WsAQAAAM+i\nQnRRITqXYGl1k8fBxVWe4wAA3ytwCCGWLFny1FNPvfvuu88991xsbOz999//xBNPOK8qFIpv\nv/32kUceeeaZZwwGw+DBg5cuXTp37tzeyxcAAADoGJ2f5w/qAVp1D2cCAL7CJwscfn5+ixYt\nWrRoUVsDQkJCFi9evHjx4p7MCgAAAOgu2WmRBWUeDvjLTovo+WQAwCf4Xg8OAAAA4Kx31bSU\n6FDXfSvJsUGzxyf1Sj4A0PdR4AAAAAB6jiyffowQIjhA8+o9E+eMTwoO0AghwoO0V01Neen2\n8X5q5ZnNDwB8lk9uUQEAAAB8zs+7jn+8Jr+wvEGrUQ4bEHbzxYMSo/TtjA/y19w1b+hd84aa\nLDbqGjinKDT8hfd19l55VQocAAAAwBn3xjf7P/vliOOxxWrfsLd828HKZ383dmj/sNPeS3UD\nALzBFhUAAADgzCosb1i29qhL0Gy1v/rF3l7JBwDOSqzgAAAAANrUYrZ9+vORnMNV9U3mpGj9\nvIkDhg44/ZoLF9sOVsqeem/kl9bX1JvCgvy6I1MAONdR4AAAAAA8qzK03Ld4Y1lNs+NpUUXj\n2j1lv7kw7TcXDuzQPE0t1rYuNRgtFDgAoFuwRQUAAADw7I1v9jurG05Lfsg7cry+Q/PEhvt7\njKuUiqhQbSeT6yyTxeZxOQkA+DpWcAAAAAAe2Ozyxr1l7nFZltfnliXHBnk/1fgh0UH+mvpm\ns0t86vBYnaaHPpDb7PJXGwqWrT1aXmv0UyszU8J+d8mQpOj2jnEBAN/CCg4AAAD0XbIQtc3m\notrmykaTzd6j6w6aWixmq+eTDmsaTB2aSq9TP3Z9Vohe0zo4LDns9ssyOp9fBz3zQc7ir/aV\n1xqFECaLbeuByjtfWXegqK7HEgCAM40VHAAAAOijapstGwuqq5pOLHwI0CjHJIUlhuh65tX1\nWrWfWmmy2NwvRQR1eF/JiNTwtx6cumrrsaNlDf5a1bAB4ROHxkiSsNvlNbuOHyiqNVvtqfFB\nF45KOBOHwu7Kr/5l93GXoMlie+ObfS/dPqHbXw4AegUFDgAAAPRFLRbbdwfLTa3WUDSZbWsO\nV85Ij44O7ImunAqFNDkz9vvtxS5xpUKanBnbiQn1OvWCycmtI1WGlr+8s/VwycmOHh+vzv/r\nDdmp8R3Y/+KNHXlVHuP7CmtbzDatpvtLKgDQ89iiAgAAgL7oYEWjyW2HiCyLPccNPZbDrZcM\nTooOdD6VJGlgUsh91470D9S459YJf/tkV+vqhhCivNa4aMl2q60bJm/NaPZ8jIsst3kJAHwO\nKzgAAADQF1W7teQ8EW/yHD8TQvSaxfdO/GpDYc7hKkkhnT8+KTBQI4QoqDNKQkQF+MUHaaXO\nTl5RZ/S4sKK0unlXfvWotMguJO4qPiLAYzzQXx3sr/F4CehFClYV+TxLr7wqBQ4AAACgTSql\nYv6kAXMn9t9b2WhutbBCFqK8yaRUSLH6E/tlGpota3aVHqtsDAvUZqdFpMYHtz/z8WrXA2id\nSqubR3VL9r+aMCT67RUHmk2u/URmjemnUHS6RAMAfQsFDgAAAPRFEQGa4jqjp3hPNOBwUddi\nNXvaNlLRZIrR+0lCbNhb9vdP9zgPgn1nlXTJ2H53zs2QpDbLBwFadScudVR+af3ir/btOVoj\ny7IkSbJ88iSaqSPibpiR1l0vBAC9jgIHAAAA+qK0qMAD5Q0t1lMWTSglaVhcNzfg9IbR6uEs\nFSGE1S5bbPZqQ8vT7+dYWqVqt8tfbyyMDfe//NSuoq0lxwaGB2mr61tc4mqVYuTA8G5JO6/Y\ncN/ijc6DYBzVjbBAv4vHJo0aGD50QFi3vAoA9BE0GQUAAEAvs8u2Rktdnbm81lTeYKmxyRYh\nhFalmDEoOkp/cr1GoJ9q2sCI1pEeo2h7IYYkSd9tK7Z46jn67aai9uZUSHfNy1C67RC56aL0\n0G56j2+vPOh+zG1NgykxMoDqBoCzDys4AAAA0JusdrPBXCmLE1snbDaLyWYMVIf6Kf1DdOqZ\ng6MNRku9yeqvVob6q9spNJxRerXnlod+KoVaIZVUNXm8WlrdbLPL7iUMpwkZMS/fMeHtlQf3\nF9ZZ7faUuKDrpw8cMyiqW3KWZbH7SLXHS7vyq84fGdctrwIAfQcFDgAAAPSmBkuNs7rxK7nR\nUqdRaCVJIYQI1qmDdd3Wk6JzAv1UQX6qepPriaoJgVohhK6NEx/81Ip2qhsO8REBYwZFRQTr\nAnXqzJSw7qpuCCHssmyxyR4vuS/rAICzAAUOAAAA9BqbbLHJrlUDIYQs2812k59S1/MptSUl\n1L+0wVTRbHK06fRTKhKDdcF+KiFE1sCIFVuOud+SNfA0R73uyKt65oMcw68H33629sjI1Ii/\n3jDKX9sNn9KVCik+wr+40sPqkqTowK7PDwB9DT04AAAA0GvssofWFUIIIQlZ9K1VBgpJSgjS\njowOHhKpHxYVODQq0FHdEEJMGhY7PMW1LWiAVvXbi9PbmbC+2bxoyQ5ndcMh53DV69/s666c\nLzuvv3tQ56eakZ3QXS8BAH0HBQ4AAAD0gvpm8xfrCz788UhbAxSS530fvUuShE6l1ChP+RSt\nUEiLfjv6hhlpIXqNEMJPrZyQEfPqPRP7RenbmWp9bnmj0eIeX51T2l1bSC4d3//qaSkq5clt\nMpEhuv+7MTs8SNst8wNAn8IWFQAAAPS09bllf1+6u6HZIoQYnJI2IM51K4pCUqgVvvQl3E+t\nvH76wOunD2w0WgK0KsmLZqjHqz23JjVZbNX1LXHhAV3PSpLEby8eNCM7cWd+VU29qV+UftyQ\naG0bHUMAwNdR4AAAAECPKqtpfuaDHPOv56q+/nnRIzekhASe/FwqCUmvDpNE9x+YIsvyyq3F\nP+8qragzRoXopg6Pu2h0gjfFCO/pve6H6q9tc2RAq0vV9S3FlU2B/up+UXqVsjPrrxMiAxIi\nu6FcAvQYZRvnFgHto8ABAACAHvX99hJndUMIcay85f5/7p81PnLogKChyaEqhVqnDHTfn2K1\n2b/aULgjr8rQZE6MDLjsvP7piSEdel2rzf7YW1t35FU5nhZXNu3Iq/p59/GnFo5uvYmjx4xO\nj3xrxQH3eHpiSHCARghR02Ba/NW+n3eVOuIRwdrfzxkyOTO2R7MEAN9BDw4AAAD0qFK3rRnG\nFvtnq8v/7608f0V4gCrEvbphaDLf8Y91r3+9b8uBioPH6n7YUXLPqxs+/Olwh173m01FzuqG\n0/ZDlcs3F3X0LXSLlLigSyckuQT91MrbL8sQQlht9kf+s9lZ3RBCVBlaFi3ZsS63rEezBADf\nQYEDAAAAPUqn8byIWKNStLUF480VB46WNbSO2O3yO6sOHTle7/3rrm+jNLAu97j3k3SvOy4b\n+qerRwyICVQpJb1OPSEjZvG9kwb3CxFC/LK7zOUtO7z73aEeTxMAfANbVAAAANCjRqVFfLOp\n0FM80mM3DFkWv+z2UIOQZfmX3ceTY4O8fN26RpPHeG2D53gPkCRxQVb8BVnxNrusVJzy5g8c\nq/V4S0FZg9FsbTHbPl1zJK/EIIRISwi+YkqK4wCXbre3oHbD3rKKupbYMN20kfEDYgLPxKsA\nQLegwAEAAIAeNSEjelRa5PZDla2DAVrVzbMGeRzfYrE2t1g9XqoytHj/uuFB2rOM6L0AACAA\nSURBVMLyRvd4RLDrGS49z6W6IYSQ7R4HCiHEniM1z3yQ0/Trz2RXfvWKLceeXJg9tH9YN6Yk\ny/I/P9/buhT16c9HfnNh2rUXpHbjqwBAN6LAAQAAgB4lSdKTN2V/tvboVxsKqgwtOo1qVFrE\nrbMHx4b5exyvVat0fiqjyUONIyxQK4SQZXntnrKDx+rsskiJC5o6PE6llPJL650dSSdnxur8\nVFOHx7n34BBCTBsR171vsLVGo2XJD3mb91fUNJgSIgMuGdvv4jGJ3pzbkhrveWVKbFjAK8ty\nm06t+DQaLc99uPOdP01zL5R02sqtxS4LbWx2+Z1VBwf1C8kaGNFdrwIA3YgCBwAAAHqaWqW4\nelrK1dNSjGarVq1q//u+JImJQ2O+317sFpcmDYupMrQ88b9th4oNzvhHPx1OjQtes7vUbpcd\nkbdXHXzoqhEzshN2HK5as7O09STnj4y7cFR897wrN1WGlnte3VBZZ3Q8zSs2vFy8Z+vByr/8\nZtRpSxxTR8S9/+Phsppml/iU4TEfrc53H19eazxQVJfRP7Q7EhdCiO+3uf7AHb7bVuxS4Civ\nNa7dc7yi1hgZopuQER0fwZG0AHoHBQ4AAAD0mrYajrq45ZJB+4tqiytPOX7l2gtSByYEP/jG\nptbVDSFEUUVjUcUpW1Fq6k3/9+72tx6c8ui1I6eNiPspp7SyzhgVqjt/RNy4IdFdfxdt+d93\nh5zVDaf1uWUb95VNyIhp/14/tfLZW8e8+OnuPUdqHBF/reqmGemhgW322qiu78CGndMqq3Wt\nrZyIn1pz+WJdwZsrDpgsNsfTt1ce/M2FA685n20sAHoBBQ4AAAD0daF6v8X3Tvp83dHth6rq\nGk39ogLnntd/WHJYaXXTrvxqb2Ywmqw/7ii5cmrK+CHR489kUaO1zfvLPcY37qs4bYFDCBEX\nHvC328bnlxoKyhuDAzTpicFB/prdR9p8v2GBfp3P1Y1ep/bY4kSvUzsf78qvfu2rva2vWm32\nt1ceTIoOnJDRQz9kAHCiwAEAAAAf4KdWXj0t9epppywNKKn0vMrAo2MVTacf1K0ami1txM1e\nziBJIjU+ODU+2BkZkhQaHqR1X6wRGaIb1K/b9qcIIcYMiirwdE7t2MFRzsffbi7yeO+3mwsp\ncADoeRQ4AAAA0EMajZYArdqLDpve0mmV3g/WajowuFtEh/qXVnuoqsS00U7VGyql4oErM5/4\n33bnrhAhhFajfPDK4Spl9/1khbh6WsqGvWUu24Iy+odePCbR+bSkynPNqKSyp2tJOMsoevy/\nVpwdKHAAAADgzGpusS75IW/VtmMNzRadRjU+I+qWWYMjgrVdnzk9ISTQX93WQgkXI3v87I8L\nR8X/77tDLkFJkqZnJXRl2lFpkW8+OOXDnw47mo+kJ4Rcc35KZEg3H3ar16lfvXvi+z8eXrfn\neEVdS1y4/4XZCQsmDVApFc4xbbVQ0fk547JNNttlq0JSKaXu3EEDAO4ocAAAAOAMMlls9y3e\neOR4veOp0Wz9Kac0J6/61Xsmdr3GoVYpfj9nyAsf73KJKxUKm93eOjIqLbLHWm84XTUt5XBp\n/frcMmdEpVT8bvbgto6A9V5UiO6e+cO6OMlp6fxUt8wadMusQW0NyBoY4bEnyKi0SCGE1W5s\ntlba5RPlJ6Wk8VdFKhXdUNgCAI8ocAAAAOAMWrH5mLO64VTbaFryQ969C7rhK/qFoxKiQnRv\nrTiQV2KQZdE/JvCGGWmxYf5vfLN/V36V1SYH+WsuO6//lVOTu3FrjJdUSsVfbxi15UDF5v0V\n1fWmxMiAi0YnJkSePaeozj2v/485JcdOPbMmOlR35dRkm2xqtBwXQnbGbbK50XI8UJOgkNRu\nMwFAN6DAAQAAgDMo53CVx/iOPM/xThieEv6PO8+z2mQhZOcGimduGWO1yQ1Gc6i+l3dGjBkU\nNWZQ1OnH+ZpGo2XNrtLMAWF6raqworG5xarXqSdnxi6cmR7kr2m2lLeubjjIwm6yGXSqnt4r\nBOAcQYEDAAAAZ5DRbPUcN3mOd5pKKQkhuUROW92w2OWCmqbKJrPZateqlbGBfokhOkXPL/bw\nNVsPVr7w8c66xpPHwUzJjPvTNcOdBSarbPJ4o62NOAB0neL0QwAAAIDOSojQe4wnRnqO9ySz\nzb7tWG2xocVktctCGC22IzXNO0vrZdeVBzhFTb1p0Xs7Wlc3hBA/7y5d8kNeb6UEAIIVHAAA\nADijZo1NXL6lyG53rRnMHp/UK/m0Vljb3GK1uwQNLZbjDS1xQdqymua3Vh7cebiq0WhNitYv\nmJx8wch41nYIIVbvLPG4MGf55qIbZ6Q7fkQqyc8sezjdRinRZBTAmcIKDgAAAJxBqfHB91+R\n6a89+Xs1lVK6bvrA80fG9WJWDtVtnC9b3Ww+XFJ/29/XrtlZWtdottrs+aX1z3+08x/L9vRM\nYkaT9UBR3b7C2qaWbt7I0y1Kqpo9xusazU0tJ36kfqpQlx1DQghJKPyUwWc2OQDnMFZwAAAA\n4My6cFTCqIGRa3OPl1Q1RQbrxg6KKq81frIm31+rGto/rH9MYA/ksGFv2daDlbUN5sTIgJlj\nEuMjAoQQVrd1JQ5Wu7z4633uixSWby66eExiemLImcvTZpc/+unwR6vzTRabEEKjUlw+Jfn6\n6QOdvS066uCxum0HK2sbTfERAdNGxIfoNV1PUuen9BhXKCQ/9YlLSkmjV8carZW2k8fE+vmr\nIxUSX0AAnCn8+wIAAIAzLizI77IJ/YUQheWNz3yYk1964uBYhUKaOTrxzrlDVcoztffDZLE9\n9d6OLQcqnJFl647+fs6QOeOTdGqFxea6RUUIoVZKuUdrPc625UDFGS1w/Ofb/cvWHnU+NVvt\nH/x42NBkvmd+h4/UtdvlVz7PXb65yBl57/u8excMm5wZ28UkswZGfPrzEfd4ZnKYWnWyEKNS\n6AI1iTbZbJetCkmtlLqhtgIA7aDAAQAAgB5ittoff3trWc3JDQ52u7x8c1GAVnXrJYPP0It+\n8OPh1tUNIYTFan/1y71DB4TFBmrrWxrdbwlSqeQ2Go02Gs/gnhFDk/nL9QXu8eWbj117fmpk\niK5Ds3229mjr6oYQotFoefbDnanxQXHhAV3Jc1Ra5ISMmA17y1oHtRrl7y4Z4jZWUkp+SqmX\nT+qFz1GoPa8SAtpHDw4AAAD0kI17y1tXN5y+2VRkcWv22V2+317sHrTb5Z92lMQGaeODT+l5\nqZCkgREBcSE6ncbzLwJjw/1d5tmwt2zJD3mfrMnPLajpYqqHjhlsnnbNyLJ84FhdR2dbsaXI\nPWi12b/b5uEH0lGPXZ912+zBjpqLn1o5dnDUq3dPTI0P6vrMANBprOAAAABADymqaPAYN5qs\n5bXGhMguLSvwyG6Xq+tbPF4qq22WhEiL0MfotVXNJpPVrlMro/V+OrVSCHFBVvw3mwpdbtH5\nqVrv7yiubFq0ZMeR4/XOyISMmD9dPVzn18nP2Pa2z6e1d7D+I8tyabXnVqDFlU0dm8sTlVJa\nMDl5weRko9mqVSslTpcB0AdQ4AAAAEAPUavaXHauUZ9+ZbHNLuccrioqb9Tr1EMHhMWdupjC\nI4VC8vdTeTyLJMj/REuIIK0qSOv6qfjWSwaVVjftyKtyRvQ69Z+uHhEWeGK3hdUm/+WdrS7F\ngg17y/71herBq4afNjGPUuKCJEl4rHIMTOjY4ghJkrQaZbOnN+7v9ma7oq2lLgDQ8/j3CAAA\nAD0kMznMYzwu3D/qdA0mjhyvf/6jXc7lEiqltGBS8m8vTj/t2oFxQ6J/3FHiMd7OXTo/1bO3\njt24r3zn4epGoyUpWn/R6MTggJNtMnPyqjwuhfgpp+QPlw7R69TulyxWe2F5Q12TOTFSHx3q\n4f1GBGunZyW476mZnBnbia4ZowZGrt1z3D2enRbZ0akAwCdQ4AAAAEAPGZIUOnFozLrcMpf4\naTuMGk3WR9/cUlNvckasNvnjNfmB/uorp6a0f+9vZ6bvyq+uMpyyUeX8kXGj00//PX/8kOjx\nbdRBjlV66E4qhLDZ5ZKqJveTVn7edfyNb/Y50xg3JPquuRnufUPvnj9Uo1Ys33zM0eVUkqQZ\noxJun+vevPP0Fs5Mzzlc1Wi0tA6OTI2YNCymE7MBQN9HgQMAAAA95+FrR368On/pz0eMZqsQ\nol+U/rY5Q05ba1iz63jr6obTsnVHr5iS0v4ajsgQ3Rt/nLzkh7wtBypqG02JkfpLJyRNz0ro\nwpsQQgi/tk95cL+0YW/50+/vaB3ZtK+8uLJx8b2TXAb7qZX3zB921dSUvBKD3S4GJgR7sxPH\no4TIgMX3TvrPt/u3Hqw0mqwRwdo545Mun5xMvwwAZysKHAAAAOg5GpXiNxcOvO6C1LLa5gCt\nuvWmj3YUlHnuTlpTb6pvNp92kkB/9R8uHfKHSzuzDqItw1PCPfbLCAvy6xeldwku+eGQ+wzF\nlU2rc0pnjkl0vxQT5h8T1sm6RmvRobrHrs8SQhjNVpplADjrcUwsAAAAeppCIcWFB3hZ3RBC\nqFVtfmpVKXvnA21CZMDscUnu8dtmD1EoTlkiYbXZ80vr3UcKITpx+GvnUN0AcC6gwAEAAIC+\nbtgAz91JU+KCArr1TJAOueOyjDvnDnWeqzIgJvD/3TJm2og4l2Ftn/3a3rmwAICOopQLAACA\nvm7MoMjM5PDdR6pbB5UK6eZZg3orJSGEQiFdOiHp0glJtY0mjUrZVqlFrVIkRQd63GWTlhDc\noVc0mq1F5Y2SJPWL0ms1bTYB8ZIsC282+ACAr6DAAQAAgD7nULHhu23Hjtc0RwRpJ2TEjB0c\n9dTC7P99d+irDQVWmyyE6Belv/2yjKyBEb2dqRBChOr92h9w7fmp/++DHJdgdKjugqx4L1/C\napM//OnwJ2vyTRabEEKrUV57QeqVU1JctsN4qabB9NaKA2t3lxnNVp2fatqIuIUz06l0oO9Q\ndLl+h3MTBQ4AAAD0Le+sOvjR6ny7/cT2jRVbjk3OjH34mpG/nzPkllmDiiubAv3V4UHa3k2y\nQ6aOiGsx2/6zfH9D84lDWzOTw/94+TDvW2Ms/mrv1xsLnU9bzLa3VhxsaLac9oRdd4Ym813/\nXF9ZZ3Q8NZqsyzcX7Txc/a+7z9Pr1B2dDQD6DgocAAAA6EN25FV98ONhl+Avu48PSQqdP2mA\nSqnoHxPYK4l10cwxiZMzYw8W1xkazf2i9cmxQd7fW2Vo+WZTkXt82dqjV05N6ejKi0/W5Dur\nG06l1U2fryv4zYUDOzQVAPQpNBkFAAA4m9llm9neaLLVWexNsrD3djqn9+OOEo/xH3YU93Am\n3c5fqxqZGjF1RFyHqhtCiAPH6mRP7UhtdvnQMUNH08g5XO05nlfV0akAoE9hBQcAAMBZy2Qz\ntFhrnHUNhaTyV0WqFP69m1X7Kg2uiwscyms9x88FNlubh63Y7B0uWrWYrR7jxjbiAOArKHAA\nAACcnSz2JqP1lN/J22Vrk6UsUJOokPpuq4Ugf88bLs7lFpgpcZ5XfEjSyUsHj9Wt21NWaTBG\nh/pPGxHXzkaehEh9cWWTezwxSt8t2QJAb6HAAQAAcHYy2TxsXpCFbLLV61ThPZ+Pl8YPif5l\n93H3+ISMmJ5PpnNkWfyyu3RHXpWhydwvSj9rbL+YsC6tmkmIDDhvaMz63DKX+Pkj4yNDdLIs\nXv9635cbCpxtWT9Zk3/99IHXTffcUGP2uH6b9pV7jHclSQDodfTgAAAAODvZZHM7cavNvvVA\n5bK1R3/eVVpTb+rZ1Npz/si4sYOjXIJJ0YFXT0vplXw6ymiyPvTvTU+/n7Niy7ENe8s/Wp1/\ny99+XrX1WBenfeiq4dNbnSkrSdLMMYn3LhgmhPgxp+TzdUed1Q0hhM0u/++7Q9sOVXqcasyg\nqNtmD9aoTn4R0GqUd80bmpncd8teAOANVnAAAACcnSQheezcIAlpX2Ht3z7Z5dynoNUob5yR\ndvGYfh+tzs85XNXQbE6M0i+YlDwitRe+8UqS9ORN2Su2HFu19VhpdXNkiHb8kJgrpyb7qZU9\nn0wnvL3q4K78U7p4mq32fyzLHZYcFhce0OlpdX6qh64ece0FA/NKDApJpMYHx0ecmK2t6smq\nrcXZaZEeLy2YnHze0JiN+8rLa40xYf7nZURHhuhcxsiyXFjeWFrdHBmsHRAbqFLym1EAfR0F\nDgAAgLOTUtLa5Ub3uNms+vObW5paTnaUbDHb3vhm/wc/HW5otjgipdXNm/dXXHtB6k0XpfdQ\nuq3UN1vCAv0uGZeUEBkwuF+oJPV8Cp0ky55PgbHa7KtzStvaM+KiytCy+0h1TYMpLjxgVFpE\n68pOQmRAQqRrlaSsptnjPG3FHWLC/OdNHNDW1SPH619auufgsTrH07jwgLvmZYxqo1wCAH0E\nBQ4AAICzk1YVajU3uxwNq5DUK7bWta5uODmrG04f/pQ/OTO2o2eadtGytUf/990ho+lEhoP6\nhTxw5fB+PtL/0miyuv8YHcq8OwVm6S9H/rfqkMliczyNDtU9cOXw4SntLaXR69Qej5jR6zrZ\nSra6vuWB1zc1Gk++kdLqpsff3vrS7RPSE0M6NycA9AAKHAAAAGcnpaTRa+KN1iqr/cS3X41C\nr1VFHC7Z7eUMsiyvzy3vyQLHyi3HXv96X+vIgaK6h/+z+c0Hpuj8fOCDq1ajVKsUFquHo1u9\nOQXmp5ySf3+zv3WkvNb4l7e3/feBye5bSJzGDIrKL613j48d5NrKxEtfri9sXd1wsNrkj1bn\n//WGUZ2bE+gQpcY3tqShr/GB/08AAACgc5SSRq+Ok4XdLlsVkloSkhBCrezAlo+6xh7tP/rJ\nz/nuwSpDy485JbPHJfVkJp2jUEhjB0WtczvuRAgxfkj0aW9ftvaoe9Boti7fcuzGGWlt3XXF\nlOR1uWXHKk7ZjpSeGHJJ26eiNDRb3vv+0JYDlbUNpoSogDnjki4anSD9uhfoUHGdx7ucO1YA\noG+iVxAAAMBZThIKpaRxVDeEEBn9w7y/NzJEe2aS8sBksTn7nrrwuEKhb7p19uBQvZ9L8JJx\n/TL6h5723sJyDz1ThBBHjze0c5dep/7XXeddMSU5NsxfqZDiIwJunJH24h/Gq1WeP+pX1hlv\ne+mXL9YXlFY3Gc3WvGLD35fuXrQkR/bYk/YMqG00lVY3yz32egDOGazgAAAAOLdcmJ3wxfqj\nLt+lFZJkd/vCqVJKkzNjeywxhSRJkvD4tVfR3Y1GTRbbJ2uOrM8tK69tjg0PmJ4Vf+mE/qqO\nrG1pS2yY/7/vn7zk+7xthyoNTeZ+Ufp5EwdMGe7Vj1GllEyeOni0Vapw0vmpbr1k8K2XDPbm\nVd5eebDK0OISXLvn+Ob95eOGRAsh0hKCd+RVud+YltDVBhwb9pb/+5t9pdXNjpyvnpZyxZRk\nzmcB0F0ocAAAAJxbNCrFC7eN/8+3+3/YUeyoJqTEBS2cmf7WioNHjp9cKKFSSn+4NKMrJ5t2\nlFqlSIkLPlxicL/kzfIHh+YWa3mdMTJY206LzeYW6x9f23C07MSyiMMlhsMlhvV7y567dWy3\nfNkO0Kpmj0+6dnqq+1KO9g1LDt+0r9w9npncgUU3Qgij2fr52oK9BTVmq31AbOCCScnRoSdb\neGw+UOHxrk37KxwFjksn9P9mU5FLGw6VUrrm/JQOpeHi++3FL3y862SSJuvbKw8eq2h86OoR\nXZkWAJwocAAAAJxzQvSaB68afufcjJKq5rBAv7AgPyFE1sDIFVuKcvKq6prMA2ICL53QPym6\np88uuXFG2uNvb3UJJscGTc6MO+29ZTXNi7/at/HXAkHWwIg7LstI9HT8yqe/HHFWN5z2HKlZ\nseXYnPFd6vRhNFnf/T7vy/UFVptdCJEUrb/9soyRqRFe3n7DhWk5eVXOI1QckqL1M7IT2r+x\nodmi16kcTTSKK5se+vcm5xqNXfnVKzYfe/iaEecNjRFCyLJoNHo4Q0cI0dBsdjyICNY+/7tx\nL322O6/4RLEpJsz/rnlDu3KEiizLb6444B7/YUfJ5VOSe/ikHgBnKwocAAAA5wRZllduLV6z\ns7S81hgdqpsyPPbiMYmp8Se/WKqU0pzxSV38ht9FYwdHPXlT9qtf7nWceypJYtqIuNvmDDnt\n5pHaRtO9r26oaTjZEnVHXtW9r2147Z5JrRcvOHhcJSGE2LS/vCtvX5blx9/etvtItTNSWN74\n6H+3PLlw9Oj0SG9mSI0PevEP4//1Re6BojohhCRJF46Kv2XWYD+15xMljCbrkh/yVm491tBs\n8VMrxw2JvvWSQS9+ustlB4rJYnvx093DU8L1OrUkiagQrcdjZWPC/Ftn8q+7zjtyvKG0ujkq\nRDsgNkhzum0y7SuubKqp99ywdnd+DQUOAN2CAgcAAMDZz2qzP/bWVmdjhdLqppzDVb/sPr7o\nt2O6pfFENxo3JHr0oKiSqiZDo7lftN6b01WFEJ+vPdq6uuHQ0Gz5ZE3+XfOGOp7Ksrz1YOWR\n4/Uev94LIeqbPDXA8NqWA5WtqxsONrv85vIDXhY4hBBpCcGv3HlefbO5ut4UF+7vKG1U1BlX\nbjlWXNUU7K/JTo8cOzhKCGGx2u9/fZNzR4/JYvt5V+mOvMqGZg/votFo2XawcuqIOCHEjOyE\n977PcxmgUEgXZMW3jkiSlBIXlBLXPaUHi83D0bkOZqutrUvo47Zv375o0aKcnJzy8nJ/f//0\n9PQ777zz2muv7e28cO6iwAEAAHD2+3ZzkXvbyB15Vd9uLrxsQn/HU0OTWa9TKxW9X+9QKqR+\nUXoR1YFb9hyt8RjffeRE/HhN8/97P6f9g05jwlzXenRIWzkcOV7f1GIN0Hbgg3eQvybI/0Rl\n57ttxf/8PNe5b+XLDQVjB0c9dn3Wd9uK3fuVeKxuOFT+uqzj6mmp+aX1G/aeXMaiVil+P2eI\n98soGo0Wm132svbkEBvur1YpLFYPZY7C8sZnP9wZoFVlJodPzozt7n6yOIMKCwslSbrjjjti\nY2MbGho++OCD6667rqio6OGHH+7t1HCOosABAABw9lu3p6yt+MzRiR+tzv96Q2F9s1mlVIxK\ni7ht9pCEyJ7rLdotrDbPZ446vlHb7PJf39lW4NZ3w8X0LGerC6sQjUJYhPAXwtsfhcdv76e9\n1L7iyqaXP9vt8u427694/4fDJVWez5RtS2jgiY6napXiiRuzN++v2HKgorbBlBCpnzkmwctu\nsmv3lL214kBJVZMQIiJYe8OFaReNTvSmJKHTqGaOTvx6Y6FLXKGQvt9e7Hj89cbCL9eHPbkw\nu50GsehT5s+fP3/+fOfTW265ZciQIf/+978pcKC3UOAAAAA4+9U1mj3Ga+tNj/x3S+6vSw+s\nNvvm/RW7j9T8444J/WMCezDBruofHehxdUZybKAQYveR6varGwqFdOWUFMfWDyFKZfmoEM5O\nnKGSlCaE9vQ5tPETCwv0Cw7o5Df2n3JKPNZuvtt+LDnG84ILhSTsbndoNUqXbTJjB0f9+n69\n9dWGwn99ket8WmVo+fvS3aXVzb+9ON2b22+bM6TFbHOWM4TjcOJTc80tqHn9630PXDm8Q4mh\nj1Cr1TExMRUVno/p6RBFG31ngPZx6DQAAMDZLyLY8/dzpVLKddtY4Ti/88wn1Z0uOy/JvZmI\nUiHNnzRACFFY3uZih6EDwuZNHPDKnef9+i29TJbzWlU3hBC1srxbiNMvwZg6PNZxHo2LeZMG\nSJ3dd9FWu5CaelNsuL/HS4lRgS4rIJQK6c65Qzu0o8SdyWJ7a6WHY1A+/TnfpadpWzQqxYNX\nDX/9j5Pumjf0llmDLjuvv132ULtZs7PU5RwZ9HEmk6mxsbGoqOi5557bsGHDgw8+2NsZ4dzF\nCg4AAICz39QRsdsPVbrH9f6eVxa4N+zo41Ljg/98XdYry3JrG0+0Gg30V99xWcbQAWFCiLZO\nIRFC3H5pRuujZGS5yNMooxCVQkS3n4POT/X/bh7z3Ic7nWfQqpTS/EnJV05J7tibaaWtzRp+\nauWssf2Wby6yua3WuGJK8phBUR/8mLe3sNZssSXHBl01LaXrx5QcLqlvbvFwvqzNLucerXG0\nL/VGcmyQI5n/LvdQLhFCmK32mgZTbJjn8g36oJtvvvn9998XQmg0mn/+85+33HLLaW9Zvnz5\nnj172rpaXFwsRGp3pohzBgUOAACAs9+FWQk5eVU/5ZS2Dp4/Ms7Tb9CFEMJstdvscl9oOOq9\n84bGjEiN2JFXVV7bHBWiG5Ea7uzTmZkcJknC/c0GB2gGxLbeV2IVwvOKCVlukKQTBQ6L1a5u\n48zU5Nig1+6dtONQVWFFQ6BOPSw5PK6NdRZeGjs46vN1Rz3GU+KCHrxq+D8/z236te6gVEhX\nTEmZkZ0ghLj9soyuvK67dlZVdG7BRVu1G0kSgfTg8Cl/+ctffv/735eXly9duvSOO+5obm5+\n4IEH2r9l+/btv/zyS1tXDQbX7rmAlyhwAAAAnP0UCunha0ZOHR63emdpea0xOlQ3bUTcuCHR\n7seFOsRH+PtWdcMhQKuaNCzGPR4fETBnfNJXG1w7XN56yWCv36Yky/KqrcWf/nKkpKpJq1Fm\nJoffOmtQYpTeZZxSIY0eFDl6kLfnwrYva2DEtBFxq3eeUpkK0WtumTVICHH+yPiRAyPW7i4r\nrmqMDNaNHRzVzy0f7zUaLTo/VVs/kMTIAI9FIiFEv+jOtGsZMyjyrRUeFnEMSQqlyahvSUtL\nS0tLE0IsWLDAarU++uijCxcuDA8Pb+eWxx9/vJ2rOl2XzjPCuYwCBwAAwLli3JDocUNO2Wcx\nIzvhkzX57r+BnzM+qQfz6gm3X5qREKl//4c8Q5NZCJEQGXDLrMETMlx2naiE8Bei2f12SQp8\n8dM9q7YeczxtbrFu2leek1f1/G3jBvcLOaOZP3zNiKyBEV9uKCgqbwwNJUGKSwAAIABJREFU\n9BuVFnnjRWmh+hPNPkL1fpdO6NIflsVq/2zt0S/WHa1pMKlVihEp4b+bPTjJrWYRGaIbPyS6\n9eGyDumJIYMSO/MTSI4Nmj9pwLK1p6xPUSoUx6uNlz/x/YDYwGvOT80aGNGJmdGLRo8evXTp\n0oKCgvYLHMAZQoEDAADg3BUdqnvs+qwXP93lPGZFoZAunZA097z+3ty+I69q6c9HCsob9Dp1\nZnLYby5M62InyzNHoZDmntd/7nn9K+uMfhqlc/eKC0lKkuX9buGAg8fUzuqGk8liW/zV3lfu\nPO8M5Ns6Jemi0YkXjU7sltkajZYVW44VlDVoNcphA8ImZ8b+37vbtxw4ceyFxWrferBy95Ga\nF/8wPi0h2OXeB64c/swHOVsPnmzmMrhfyJ+vz+psB1Xx+zlDMvqHfrLmiCOfRqPFZrfXNLQI\nIXblV+/Kr144M/2a8+nF0HfZbDalUtn66fLly5VK5YABA3oxK5zLKHAAAAD0dTa7bLPLmjb6\nPnTR2MFRbz04de2esuLKxhC9X3Z65ADvDoh997tDS344scOlytBSUNbw867jL/5hfFd2SfSA\nyJD2V79HSZIsy0eEcB6sGyFJA7cddN3e4nDwWF2j0eIrWyp2Hq5++v0djjUsQoivNxa++/2h\n4soml2Emi+0/3+5/4bZxLnG9Tv30zWN25VcfOFZns8mp8UGj06M6Xd1wmDQsdtKwWCHEH15e\n60zM6d3vDk0bERdDw9G+av78+cHBwSNGjIiIiCgrK/vkk0+2b9/+yCOPhIWF9XZqOEdR4AAA\nAOi7DpfWv//T4bzSepvNHh2qmzuh/5RhMZ0+c7Qtep364jEdWyBQWN74/o+HXYKGJvNrX+59\n9taxp729os64fHNRYXljcIBmZGr45My47n5PXREtSZFCNAlhEcJfCK0Qotnk4QARIYQsi6YW\na3cVOBqNls/XFRw8Vme3ywMTgudPGtCNK2KaWqytqxsO7tUNhz1Ha6w2u0rpoaY2PCV8eEo3\n7z6orDPml9a7x212eevByrNvw9RZY+7cue++++6qVatqamoCAwOHDx/+/vvvX3vttb2dF85d\nFDgAAAD6qJz86r8t3e08B7Ss1vj6t/sLyhtuujCtdxMTQmzeXy57ajiZc7i6ucXqr23vQ+aP\nO0pe/myPs/HH8s1FX28sevKm7Pbv6lkKIU5ZxhIXHuBxnE6jCg/y65aXPFxiePTNLc69QtsO\nVX69sfDJm7IdJ92elt0ul9U219SbEiL1IXoPZZHN+8vdl0i0M1uL2abXnZFFQ+4ajJa2LtV7\nnTN63sKFCxcuXNjbWQAn9Z3/iwAAAOAU//v+kLO64bRqe8n0kfEJEQFlhpbimmar3R4TrEuK\nCOjEAghZlju9GKStr8qyLDcYLe2UKsprjX9futtitbcO7j5S/dbKA3fOHdq5ZE6rrKb5/R8P\nHy4xCCFS44OvuyC1o7seJg6LeXPFgUa37+HTR8V7XObQUbIsnv1wp7O64dBotDz74c53/jT1\ntC+Rc7jqX1/sPVbReCLboTG3X5YREaxtPaasxvMJuB6FBfr15L6byGCdQiHZ3f62CyHYnwLA\nexQ4AAAA+qKKOmNZrYdvpLIs7zpSs6WwZs+xOmewX3jA/FGJwf5efSM1WWyfrMn/YXtJWa0x\nMlg7cVjM9dMHdvTbbFudLNQqhcflA05rdpW6VDccftxRcsdlGd2++0YIsfVg5ZPvbncuGMkv\nrV+zs/QvN4wand6Bk1yDAzSPXZ/13Ic7axtNzuDYwVG3XjK4W5LML60v+rU80VpFnTH3aO2I\n1Pa2hOQerfnzm1ustpPVgXW5ZUeON7xx3yQ/9ckGkAG6Dnzyv2Rcj+4KCfRXjx0UtXGf6xEt\ngf7qsYOjejIT9BEKjfL0gwA3PbTqDAAAAB3SYnY9utVp97Ha1tUNIURRddNHmws97hlxYbLY\n7lu88b3v847XNMuyXFFnXLb26J2vrKtv7thGgIlDY1p/eXaaNMxz3KnCU9VGCNHUYq1vbnOf\nQqdZbfa/f7rb5Rxck8X29093W20e6iztyBoY8dZDU2+/LGPW2H5XTU15+uYxTy0cre2mr2FV\nhjaXV1S2fcnh3e8Pta5uOJRWN63aWtw6kp0WqVB4qB/FhPmHBZ6yy+ai7MRrL+jps0vumT8s\nOTaodUSvUz9y7UhfaeAKoC9gBQcAAEBfFBmsVSok9y0qQoiaZrM+0LXvQ5nBWFDVNCDyNCeY\nfLupKK/Y4BIsrW7+ePX/Z+++A6SqrgaAn/uml92dne192cp2elm6CIIKgpUYscYWNSbRFBNj\nYqJfjCkao6Kxd42IgIIKKIL0Xrb33nd2ZqeX9+73xyzD7Myb7bON8/uLua/dXWD3vfPOPady\nUMkIoUHSX1yf89zm8+6xg6SowPvXZvV9YICPNBOhgCj8UIOjuFbb2W3xHu/sthTVduUmDa5Y\npkIqHGAD3cFSKX0W8lAp+qnxUVyr5R0vqu1am38xESMmVLFhWfKHvUvDioXMrzfkJUUGfn+2\nqa7NoFKKp6eEpsepBjP3kaEOlLz08MLdJxrOV2ssNseUqMA18xPGbddhhND4hAEOhBBCCKHx\nSCYRLsiK2H++xWNcpZTIfTz1tXVb+w1wnCht5x0/Xto+2NUWl02PyUpUbztYU9uql0tF05JD\nVs2JE/DlCLibmxHxoVf7FQCYmRY2IsUsPLivKPHcpB9H1StTY4PCVLJ2rWeyRpBCnJvcV5FR\nSoHzkbnDcp4pKrdfkZ4WG/TRd5U1LXqpWJAzRX3n6qmxYQoAuHJu/DCmPzIEDFk1J27VIBv6\nIISQCwY4EEIIIYTGqTtWpOmMtrNVGtdIuEq28fLUnYXNvPsLBf0XsDBa+JeBeJfPHIiIYNk9\nVw8uLJIRr1ozP+GLw7Xug0EK8X1rMocwgX55FNp0F6byuWn0CRjyyA25T7x13OZWoEQoID+/\nLqfvJT+EQFJUYGk9TxJHcnSQ92B+VmR+VuTwJ4wQQuMQBjgQQgghhMYpmUT42E3TzlVriuu0\nVgcbH6ZckBkhYMje8jazV4UOCpDgo5Wpu6gQRXEdz8NwTGj/x/ZciNJdJxp2n2xs6jSGq2QL\ncyLXLUgcVPLFQ+uz85JDtvxQXduqD1KIp6eG3royLdj3Gg0XrcH2/p6yguoug9meGBlw49Kk\nfteYpMepItXyFo3JYzxKLR+ThRh9mJEa+vqjS97fU15ar2U5mhar+vHylLjwflJyAODGpUl/\nee+Ux2CQQrwaUyEQQpcYDHAghBBCCI1ruVPUuVN6LVJYnhn55ZlGj91mJqhDvQpzeFs1O+67\n057HOscHMhmOo0+8feJYSZvzY4fOUlTbte9s87P3zpWJB3FjuTg3anFu1MD3B4CaFv0jrxzW\nXyhE2qY1Hytpu3N1+oZlfZXDFDDktz+a9vibx91TVJQy0W9+NK3f1TSjL1Itf/TGvMEetSgn\n6ufX5by+82IX26SowEdvzB1IAYuq5u7qZr1MIkiPU4UEjqOUFoQQGgIMcCCEEEIITTAzE9VS\nkWBXQXO32Q4AEiGzKD18fnLoQI6dlhJy91UZb31d6uohwjDkukVTLpseM5DDd59qcEU3XErr\ntZv3VW1ckTaYL2LQXtxaqPdqs/LON2WLc6Oi+0xdyUwIfuvXSzfvqypr0AFAWmzQ9UuSJln1\nyivnxi/MiSyu1Wr01thQRVZiMG/DFHcdOstzn507XtJTk0UkZG5YknTbyjR/dOpFCKHRgQEO\nhBBCCKFxQW+yF9RoNN3W6FB5zpSQvgtqZMUEZcUEdZvtDpYLVogH9VB6w5Kk+ZkR359tauow\nhgfLFmZHpcQE9n8YAAAcKmjlHT9Y0OrXAIfeZD9f3ek9znL0SFHbtYum9H14kEJ815VT/TO1\n8SJQLp6bET7AnTmOPvH28YrGbteI3cF9+G2FgGE2rkj12NnB0tpWfZfBGhuqiFTLXeM6o624\ntktnssWEKrISgr3/ETpY7uvj9WUNOpalKTGBq+fEj1RXXYQQ4oUBDoQQQgihsbfzaJ37EoO4\ncOUjN+RmJgT3fVSgjL/lar9iwxS3XO75HDsQOiN/5xGt0WezkhHRbbL5aBUCWsM4aoYyUZws\n63CPbrhs+aHqR5clu1dUOVLU+tK2wtaunvYus9LCHro2O0ot33qg5u1dpSaLwzmeFhv0yI15\nUyIDXAc2dRoff/N4Q7vR+XH3Sdi8v/rJ22YNPJqGEEKDNfK9uBBCCCGE0KAcKmx5/rPz7kUi\n6tsMv3/jWGe3ZQxnxctX55Fwlcyv11UHSHyltIQHY+WIQats1vGOGy2Ops6LBVlPlXc8+e5J\nV3QDAE6Utf/61SM7jta+vL3QFd0AgLIG3WOvHzW6jfz1wzOu6IZTu9b89AenWM5HpAohhIYN\nMzgQQgghhEZDh87y2Q/VVU3dErEgMyF4/cJEV/vPT/dVee9vtDh2Hq3zd2ELFwfLna7orG8z\nhARKsqeofdWbXDYtZt9Znia1AyzhMWQyiXBeRsSBghaPcalYgE1Ph4DxvabJfdP7e8q94xGt\nXeb3dpV7H6jptu4+2bBuQSIA1LUZeDvXNnYYC2s0/fa+QYjB1UxoSDDAgRBCCCHkd4cKW5/5\n6LTlQm/XI0WtXx6uffbeuc7qmFXNet6jKpt4FhH4Q1Ft1z/+d9b1vl0qFmxckXbDkiTvPfOz\nItYvnPL5gWr3wcW5UWvmJ/h7kg+uy65vN9a2XvxeSUSCR27IVQ+gd8wly2RxFNd1degskWp5\nVmKwa+1JRjz/6qdgpSQ65GKVjTK+IAUAdBn4VyRVXlj20qox8+4AAM0aEwY4EEJ+ggEOhBBC\nCCH/Mpjt//jfWVd0w6lNa/7Xp+f/cd88APDVr1TAjMZq4i6D1aOLqsXGvrajWKUUr5gZ673/\n/WszF+VE7j7Z0NRpClfJFuZEzs+MGIV5qgMlm36+aMfR2vNVGqPFMSUy4JoFiRHB/l0aM96Y\nrQ6RkHGvkdGH/eeaX95WqNH3BCNiQhW/uD7HGVzISVLPSA09Vd7hccitK9Nc7VcoBV+LSSgl\nwLdRcGEZkVLuszpMgGxS9a9BCI0rGOBACCGEEPKvE6Xt7uEDl3NVnR06S2iQNCsx+GixZ+9V\nAMhK7KfI6Ij45ng97/Q+21/NG+AAgOwp6uwpaj/Pi4dQQK7JT7wmP3H0Lz22KKW7TjR89F1F\nU6dJwJDU2KB7rsro+6/gXFXnXz887b7ApLHD+Pibx1/95eIotRwAntg4842vSnYcreM4Cs5G\nM6unrpoT59qfEEiJDiyu40niCFdJ27Q8ORqusripMUHqAIkrtuIikwjzkjF9AyHkL1hkFCGE\nEELIv9p1PmuFdugsAHDL5akioeddWZRavnpuHN9BI6ymhX+BTHWL/tmPz/79k7NbD9ZY7Szv\nPkNmsbEVjbqmThP11RwFuXnli+J/fnrOWf6T5WhJnfZXrx456FWRxN2n+6q8y2dYbOy2gzXO\nP8ulwofWZ3/6xIp//XT+yw8v+vD3y92jG04bLkvxPrM6QHLfmgzvtKPEyIBl06KdfxYKyEPr\ns733uX9NpkKKb1gRQv6CP18QQgghhPyoRWOqa+WPIACASikGgPQ41d/umfvi54VVzT0lDBZm\nR96/NlMmHo1bNV/rHSile041AMDukw2f7a966s7ZCREBvHu6a2g3nqvq1Blt8eHKuRnh3ic3\nWx1vf1O2/VCN8/E7TCW7b03mohwsFOpTQ7tx64WohAvL0U3bi/KzIoiPcqEDLOwSIBdlJ/rM\nBJmfGfGbDdNe/bLI1Ys3I171yxtyEyICnrxd8PK2QmfMhRBYkhd935pM9zjdguzIF3+28O1v\nSkvqtCxH02KDNq5IG52kJITQJQsDHAghhBBC/vLBnvIPv6uwOzjerSkxQZHqnoKO2YnqTT9f\n1KY1d+gssWGKIMXo1SnITlTvOtHQ9z6tXeanPzj96i8W+XqcBgCOo//dUbztYI0rcSA2TPGb\nDdPS41SufSiFP71z8nTFxdIP7VrzX947+bubpy+98PIfeThT2cGb59KmNTe0G+PCle6D9W2G\nM5WdWoPV5iPppo/+KbyWz4jJz44ordNp9Nb4cEVydJDzBHOmhs9KC2vqNHXprfERSt5/scnR\ngX+5Y/agLocQQsOBAQ6EEEIIIb/YdaLhnV1lvrYqpMJfXp/jPkIIRATLRr9q5vIZMZ8fqK72\nsVDFpaZFX9agc49WePjfvsotP/TqrtLQbvzDW8ff/NVSpayn5OTpig736IbLm1+XLsmLHuSj\n9wTDcXTnsbptB2sbOwxBCsmstLA7VqWrA/tvAeNRntadyepw/ZlSeH1n8ZYfqr1XprjLHHwO\nhUwsnJbCUziDYUhsmCI2TDHYEyKEkJ9gDQ6EEEIIIb/YdqiGd1ylFK+Zn/DGo0tTYoJGd0b8\nRELm2XvnrZwV2298oUVj8rWJUvBeRgEAWoNt75km18ei2i5fZ+7s9lmpZHJ46v1TL2wpqG3V\nO1ja2W355kT9vc/tdy7x6FtsKH8EQcCQGLdNnx+o5q274U4dIFm3IHGQE0cIoQkDAxwIIYQQ\nQn5R12rgHZ8aH/zQ+uyBvLofNUEK8aM35m37y6pNP1/0+C0zfO0WIPe5cMZgtmu6PVtmONW6\nlSBxsPyrdfreNAkcK2k74FUTVGe0vbGzpN9jZ6aFuZYyuVuSF+1KjQGA7T4Cai7TU0L/cd/8\n0Vz9hBBCowyXqCCEEEII+YVEJOBtPiIRCUZ/MgMhFQuSowNjwxRKmci7caxSJuqjQqRYxBAC\nvB1RxMKLX29SVCDv4QFyUZhqtNfmjKbjJe2848dK2iiFvnNnRELmT7fNfOr9Uw3tRtfgrLSw\nh9Znuz46WM5XMsjCnMh1CxKj1PLJ/R1GCCHAAAdCCCGEkJ/kJat/OM/TyHNaMk85g/FDIhI8\nuC772U/OcG6LHRiGPLguu4/QjEQkSItVldZrvTfluX298zMjYkIVjR1Gj32uW5Tk3VJ0MjFY\nPANGTlY762A57ybBHpKiAv/7y8X7zzVXNnVLxYKsRPWM1FD3HQQMEQqIg+WJMKkUktykcf1P\nDiFvzHgNBKNxDgMcCCGEEEJ+cevK9BNlHWa3MpAAkBwduHJWbGm9dv+55tYuc6RavjQvqo9i\nHCxHR//J/7Lp0TGh8nd3lZXUawFgapzq1pVpfZQXdfrJlVMfe/2oxzP29JTQOVPDXR9FQubp\nu2b//ZOzhTU9xTiEAua6RVN+dFnySH8R40sU3xoTAAgNkvYb3XASCpjLpsdcNj2GdyshJGdK\nCG8B17zxHVBDCKERhAEOhBBCCKHhYqnZzuk5ameIWMQECIgUABIilC88uGDT9sLTFZ2UUrGQ\nWTUn7raV6W98VbL1YI0rP2Lz/qoblyTduXqq+wkdLPf5gZodR+qaNSaVQjwnI/yOVenqgNEr\n25Eep3r6rjmDOiQvOeTZe+dt2l5U3qADAIlIsG5B4s2Xp3isv4gOUfzr/vzCWk11s14pE2Um\nBI9+45jRd9n0mI/3VnhnWKyaHTdSl7hzdfojmzS23j2JsxPVi3IiR+oSCCE0zmGAAyGEEEJo\nWMxsq429uDTDymokArVUEAYACRHKZ+6ea7WzXXpruErGMGTvmSaPXqocRz/eWzk1XpWfFeka\n+f0bx11v47sM1m+O1x8rbnvhoQXjPBaQnah+6WcLDWa7zmiLVMt95Z4QAtmJ6uxE9ShPbwzF\nhikeuSHv31vOu/d8XZQTefPylJG6RHqc6vkH8l/eXlRYo6EUJCLB2vyEWy5PZSb12h+EEHKH\nAQ6EEEIIoaGzc93u0Q0nK6sREJmIUTo/SkQCVxeMXScaeM+z60SDK8Cx/1yz91qDLoP17W9K\nf7Nh2ohN3W+UMpF7dw/ktHxGTF5yyK4TDXVthuAAyay00JlpYSN7iZSYoH/dP99iY3VGW7hK\nSvpt/Ds+cBzFKAxCaERggAMhhBBCaOhsnI533M7pXAEOd61d/K0uWjRm15+Pl/J33DjhYxxN\nFKFB0hFM2fBFKhZIxSOf6VPRqPvou4rKpm6RUJCZoNq4Ii00SDqcE2oNtre/KT1S3Nqlt0Wp\n5VfNi1+/MFEoGFBFEoQQ4oUBDoQQQgihoeOow8c4f9cMX6kNAfKL40YL/zmNPjpxTBSUUpYD\noQDf1U88Xx2r//eW867CMbWt+u/PNP/17jmZCT47B/etTWv+2YsHNd1W58emTuNrO4qPl7Y9\n85O5mM2BEBoyDHAghBBCCA0dAf4XzoTw9zicOzW8pI6nl6p7q5HoEP6OG9EhCgCglLZ0mZVS\nkXtMZJwrrde+vrOkpE7LclxCRMAtl6cuyMbKl2OJUihr0Na3G4OV4vQ4Vd9LinRG26bthe5t\ngwHAbHM8t/nca48sGdoE3t1V5opuuJyp6Pz+bJOvTjEIIdQvDHAghCY+jqW1B2jLObBoQRZM\nomeQuHlAMMcVITQaRIySZS3e40LCsz4FAK5dNOWH8y1Vzd3ug+lxqrX5Ca6PK2fFbjlQ7fE8\nCQArZsa8u6vss/3VZpsDAKZEBty/NmtaynhvAnqwoOWp90+xF76cyqbuJ989eevKtFsuTx3b\niV2yalv1//z0nCvQppSJfnLl1Cvnxvva/2RZh3ttVLfzGBrajbFhin6v6F1lw9d6q+Ol7Rjg\nQAgNGQY4EEITHGvnjr8K2rqej1Y91dZBy3ky6yfA8L8+RQihESQWBNs5PUt7vYsWEJlEoOLd\nXyYR/vvB/E/2Vn5/tqm1yxypli+fHnP9kiSx8GJYNjEy4JfX5764tcD9qfKK2XHnq7uOlbS5\nRqpb9I+9fvRPt82amxEO4xXH0Ze2FbJewZoPvy1fNTtumEUc0BAYzPbfvHbUPXvCYLY//9l5\nhVS0JC+K9xCd0TPVwkVrsPYR4LDa2U/2Vu451djaZQoJlC7Kidq4ItWZLeJrHZbBPLHXYSGE\nxhYGOBBCExutPXAxuuEa1FRCwzESP39MpoQQuqQQYBSiBBursXHdlDoYIhIxgRJBMIDPOgIS\nkeDWlWm3rkzr47QrZ8XOSA3dc6qxod2gDpDOywy3O7hfvXrEYzeWo6/vLB7PAY6aVn2HjifD\nxcHS0xUdK2bGjv6ULnG7TzZ4rw0BgI/3VvgKcISpfJYs7WOT1c7+ctPh8oaeKrwdOsvnB6qP\nFre+8NCCQLk4KkRe06L3PiomtP98EIQQ8gUDHAihiY22FfJvaC0ADHAghEYFASIRhEgEI7xU\nJDRIumFZsuvje7vLeXerbTVoDTaVUjyyVx8pJiv/i3rw/Q4fjSAHS788UnuyrF1rsMWGKdbm\nJ1Y0dvPuWdWsZzkq4CvwOTM1NEgh1hltHuNZicERwT4DHDuO1LmiGy5NnaZP9lbefVXGqtlx\nr3xR5LGVYcjKWRjzQgAAIMI8XDQUGOBACE1wNiPvMLUbsQg7QmgysTt4iiA4We0+N425aLWC\nEKCeK1QALpl39XqTfevB6vLGbiFD0uKCrlmQKBOP0h243mT/9X+PVDb1RDRK67V7zzRNiQzk\n3ZkhPpOOZBLhrzdMe/qDUya3mFS4SvbojXl9XL2PKht3X5WxbkFibav+q2P1rnGJSPDANVlJ\nUfzTQwihgcAAB0JoYiNSFTV18o0PsXEdQgiNT/ERAbzjSploPFeyUAdK5kwNP1rc5jEeHSKf\nPu7Low5fQY3mT2+f7Db15D4cKGjZfqj2/+6akxjJ/7c5st76utQV3XDiOFrZzJ/BkR6n6qM/\n6+z0sDcfXfr5geqKpm6JiMlMCL5mQaKkz3fsvroaO6tsMAz5xfW5V8yOO1LU1tltiQ1TLJ8R\nE+57wQtCCA0EBjgQQhNc9HTQVPKNzxj1qSCEkB8tzI58M0jqXc9ibX4C77KC8eORG/L++Pbx\nYrfmuJFq+R82zhQKJnm7K7uD++uHZ1zRDacOneVvH5/Z9PNFozCBfeeaeEYpVcpEHrU8BQzp\nuygMAKgDJXddOXXgV48KURTzdUR2z9zJTAjOTMAXEgihEYMBDoTQxEZiZkNXDW080WswPp9E\nZo/VlBBCyB+kYsFTd87+64dnalt7SjMyDLlqbvzGFf08l445lVL8/AMLDhQ0F9VqbXY2JSZo\n+YwY964xk9X5ak271uw9XtnUXdOi93cSh93B6U38ORR5ySGUwqHCFufHiGDZA9dkzUgNHdkJ\nrJod993pRt7xkb0QQgi5YIADITTBEUJyboSoadB6HkwakKtJ1HRQJ431tBBCaITVtuoPFbam\nxQYlRQcEySVx4YqcKerRWekwfITAopyoRTn8TTomq3YdT3TDqU1r9vffnUjIeGdqOEWHyO++\nKqNDZ6lvN6iUkrgwpVAw8klA01JC7r4q462vSx0s5xxhGHLdoimpMUEvbCmoadUrpMK85JC1\n+YmXQrQLITQ6MMCBEJoMSGgahI73d5gIITRk7+0u+/DbCpbrqdUpFJAfL0+dKNGNS5ZKIfG1\nKUgxGl1vFuVEulfxdBuPAoDQIOmIV2/RGmzv7Co9VdahNdjiwhXXLEh89ReL951rauowhgfL\nFmZHlTfq7nt+v4Pt+Zd8tLht59G6Z++ZN57ryCCEJhAMcCCEEEIIjWuHCls8esQ6WPrOrrKU\nmKC5GeFjNavhOF3RcaK0XaO3xoUpV8yMCRvd0pKUwt4zjXvPNLV2mSOCZcumRS+bFkP6y2Do\n1FvbdOYAmSgqWD7Aoie5SWreHIpwlSw1Jmhokx+UO1dPLartqm01uA9uWJYyNV7lj8s1dZp+\n/tJBraGn5khZg+7vn5xdPiPmNxumOUfatOYXtxa4ohtODe3Gl7cVPnHrTH9MCSF0qcEAB0Jo\nErBTagQiICAHwK7pCKHJhvclvHN8wgU4HCz37Cdnvz9zsfjlR99VPLQ+e+WsWL9e12xz1LUa\nWI5Ghyj++elZV1eXmhb90eK27882//HWmb7CFh3dlo8PVBfV99Q8z1egAAAgAElEQVTLDAmQ\n3LRwSnZ8/6UxZRLhQ+uzn/34jCv1BgBEQuaXN+T20a9kBAUpxC89vOjzA9Uny9o13daECOXa\n/MS8ZH81r3ljZ7EruuHy7anGlbNip6eEAsDBgha7g/M+8Ehxq8XGSsX4GxwhNFwY4EAITWgs\nR2sobQWgQAFAwJA4QmLGelYIITSSWjQm3vGmTuMoz2T4Ptlb6R7dAACrnX1u87mp8ar4cKU/\nrkgp/XRf1QffVpitDgBgGMJx1GOfI0WtXx2ru3pegvfhFhv73BeFXW7P7Z166yvflP7sqsy0\n6MB+r75sWnRChPKDPRXljTqGkIwE1cYVqdEhin4PHCliIXPT0uSblib7+0KUwrGSdt5NR4vb\nnAEO7x5ATg6WdhmsUWq5H+eHELo0YIADofFDR2kjgBFAREgwQBwA1tzqB0dLKe1yG2A5WsMA\nJcS/bwIRQmg0ySX8N2wK6cS7kfvmRIP3IMvRPScb71yd7o8rvrOr7MNvK1wfvaMbTvvPNfMG\nOA6VtnV5ZSVwHN15siEtOnMgE0iKCvzDxsncudzBckIBY3OwVjvLu0O3secbGBzAX5SEYUiQ\nfDSKkiCEJr2J93sRoUmJ0hqAWrePOoBWQqYB4O97nyjoe0c3enC0QUCiMTyEEJo0ZqWHF9dp\nvcdnp0+w9SmU0tYu/sYizRq/ZKOYLI5P91UNZE9Nt5V3vKbN4GNcP/RpTQpVzd1v7CwpqO6y\nOdjYMMVNy1JUSrH3EhUAiArpSc2Ynxnxxs4S1ivGNDM1VD4Bo3XIv0T4TwINBT4AIDQeGNyj\nGxeYKa0cg7lMIJT/phOApcCfzo0QQhPRdYumeC/fSIgIWL8ocSymM3SEELmEv86CQiryxxUr\nmnS8RR+8hQTyd/GglD/jgzpXRl6qzlZ2PvjCweOl7Wabg+Vobavh2Y/P8HZCEQrIsmk9S0dj\nQhV3rp7qsYM6UPLAumy/zxghdGnAwBhCY4/SDh9bOgAowGjUIUMIITRuyaXCFx5c8MG35d+f\nbW7XmsNUsqV5UbdcnioTT7wbudlTwz1qcDj5qVqqj+gEjyV5UbzjcaGKk5Wd3uPxYYpL+dfz\nS9sKHaxn5KiySZ89Jbig+mJypUQkeGh9dmzYxZojNyxJyp6i3ryvqqZVr5AKpyWH3Lg0WSnz\nS3gLIXQJmni/FxGajHjyOQEAgANg8f+pT0Th4/UZQwALlSGEJhW5VHj3VRl3X5XhYKlQMIGf\nrO9clX6mosNjIUN+VsS8jAh/XC4xMoC3qqiHhdmRq2bH8W5aMDXi23PNerPd443DFdMv3YLW\nHTpLTQvPCh1K6YKsyOsXJ58sa9cabHHhitVz4iOCPXsAZ8SrJndREoTQGMIHJ4TGA/6aWwAC\n/E/Kz2ECRkSYQAJBFHQeGwkW4EAITV4TOroBAJFq+Su/WPz2N6XHS9q1BmtMqGJNfsLV8xKI\nf76sIIV49Zy4HUfqPMZTogMj1PLmTlNUiHxpXrSv9A0AUEiFD1+d+d73lbXtPesiA2Si6+Yn\nDKRN7GRlsjp8bTJaHPlZEflZfglXIYRQv/DZCaGxR0gopbXAk40QNgazGToHgAGABVD6DtkM\nD+Wo5iTtPAoOEwABWSQTsZiTidzW+DCExDCE/y0cQgiNK3auy8HpObAzRCxiVEISMNYzGiXq\nAMkvr88FAErBT3ENdz9dmyVkmC+P1LpqW142PfrBddkDXxYRrZb/en1OXYehVWsJkosSw5US\nEX8lkcmqtctc3qgDgNSYoIhgWZhKKhQw3ktUACAmdPQ64CKEkDcMcCA0HigISaK0qneMQ0FI\n0pjNaNDqKK0BcL3SiSIkdcR/wtCmHVRX7PoE5mau9lMSdx2jnEmpEYAhRAmA63gRQuMdBc7s\nqGVpTz8RjlodnF7EqKSC6LGd2GjiOPr92eayBi3L0tTYoMumRwsFfkm+EwmZB9ZlXb8kyXmt\nlJgg95IQA0QIJIQpE8I8S71OehYb+8oXRV8dq3cWW2UYcuWc+HvXZCybFr37pGfH32ClZH4m\n5m4ghMYSBjgQGidiCVFR2ghgAhAREgwQPXHKi9Z6NXxpptRCyPSRvIi52S26cQHlaOt3RHkX\nIfzV7xFCaByysR2u6IaLndMKSYCQuSTyONq05ifeOlHV3O0a+XhvxZO3zYrzahYzUiKCZd7F\nIFC//vnpuX1nLxaF5Tj65ZFai83xwLrsDp3ldMXFKukhgdLf3zIdu70ihMYW/gxCaPxQEpI+\n1nMYAkqpd49bAOgC0AKoRuwyRt6rAFg7wWEAodc9MTWDvZFSAyFyEEbBpfHMgBCaEByUp0Aj\nADhotxAuiR9Wz3x4xj26AQAN7can3j/1yi8WkVFYtYIGplljco9uuHx7uvH2Vel/u2fukaLW\n89Uak9WRFBV4+cyYidjWByE0yeCPIYTQMJncVqZ4aAeQAoxQbgVnH8QmexW1ngfqAGePQFII\nkqlEnDEyM0EIoeGhlP/HJudjfJKpbzMU1Gi8x6tb9MV12syES7d45+g7Wty29WBNfZshSCme\nmRr2o8uSZZKLTwcVjd28R1EKFY3d4SrZvMyIeQNbk9LZbZGIBNgOFiHkbxjgQAj5AwNAKG0C\naAJQEJIyAqkckhAflxKDKLDXCNtJLacvfiQAwIG1CJgAEMYOdxoIITRshAgpZb3HGXJJ3Jg1\ndZp8bWrsMGKAY9S8vK1w68Ea55/btObyBt3eM43P/TQ/NKjnzQTjO5mmj03uHCzderD6o+8q\n9CY7ACRGBtx7dcbMtIlVQx0hNJFcEr9HEUL+JAMQ9k7i8Kgtb6T0HCF5AEHDuQwJSKFCJTgM\nnuOqXCC9rkjt1bxnoLYqggEOhNA4ICSBNtrOM84M6+fkWPn+TNNnP1TXtOgD5KK85JA7V6WH\nqfqqdqHwXaZBIcU3/KOksKbLFd1wae0yv76z+Lc/6imhlR6nIoQ4y4u6YxiSFjeg9xbPbT7n\nXou0pkX/2OvHHr9lxuJcn315EepxifUqQiPFL9WqEUKXEoaQePePfPtQSmuGfR0xE38diNXu\nYyQok0Qs9tyT8wyC9DOOEEL+ZOd0RkeV3l5ssJea2QaO2sSCEAGRe+wmYoKFZOI16fjP5wX/\n9+Hp0nqt1c526Czfnmq8+1/7PepreEiLUwXIeQIZEpEgJSbwVHnHnlONpfVa7+dqNIIOFbb4\nGG91feNDg6RXzuXpvL42P0Ed0H8/+Mqmbu9OKwDw6pfFg/q7pRTatWabg6crLUIIecAMDoTG\nkaZOU3mD1s7SpKiApKjA/g8YLxIJAUprAXgyri/o62Z3oKQRTPIdVF8Glg4QiIk8HmR8r4B8\n5XhfGrnfCKFxxcI227ku558psA6um+UMMmGCXJho57QOqueonSFiEaOaiNGN0nrtF4c9K0Cb\nLI5N24v+fu88X0eJhcz9a7Oe/fiMx3h+VsT9z//gXMsAAOlxqkduyE2MvCSqro4+ndHGO26x\nsRabw1WJ44FrsoIU4s37qpzxBYlIcOPSpJuXpw7kEueqOnnH27Xmpk5jTGj/zXrNVsf7e8q3\nH6q12lmGITlT1PevzZxQN0gIodGGt/sIjQt2B/fKF0U7jtZxXM9LjQXZkT+/LidIIR7biQ1Y\nIiExAHpK6wB0fDuM0Is4IiCBGdDnvQ0RhFNHK88G4YAKoSGEkCfOAcxQbplYanFFN1wocFa2\nVS5MFDEq0ci1mhoTx0p4FtoAwLkqjcXGSsU+M8wvnxEToZK99XVpWYOWo5AcHTgjLfSjbyvc\n9ymt1/72taOvP7pkolSmtNrZpk5TSKAkUD4Bfne7Cm14CJCL3OuMCgXM7Vek37AkuaZFTwgk\nRgQMvBGs1e7ztUcfm1xYjv72taPFdVrnR46jZys7H37x0PMP5CdHY4wDIcQPAxwIjQsvby/c\ncaTOfeRgQYvBbH/2nnkTp1+eCEBNiIFS3gBH/y9qRm4iSWCvBa53zgiREfHU0ZsDQmgSoBxt\nPErrDoJFAwIJCU4mqatB5qPgMR+W8q+MY6mJAks8KxZNPEYLf38rSqnBbO8jwAEAOUnqf/10\nPsdRCiBgyE//fcB7H43e+s3x+usWJ43MdP1Ga7C9tqN4z6lG57KaqfGqB9dlp8WO64oqS/Oi\nP95b6Xqt4nLZtBjvnRVSYVbioIu/xofzZ9+IhExUiOcSLW/7zja5ohsuVjv75tclT985Z7CT\nQQhdIrAGB0JjT2+yf32s3nv8bGVnab3nr/ZxL4o3ckoIzyJefyFCIl8K4lQgYgAAIgJRIlEs\nB9L/gmGEEHKhhZ/Q0u1g7gRKwWGh7YXc0RdA3zyIM/C1SrmwybOggMXGvvlV6W3P7F312523\nPrP3za9KzLbx3jU2Mpj/MVUmFqqUA/qRyzBEwBBKabWPsh3lPjqVjh9WO/voK4d3n2xwFQ0p\nqdM+sumwrx6r44SzoYmgdzeUjHjVHavSh3Na9+Ias9PDovkCGVfMipOJ+3/JeqaSf4XL2YpO\nLM+CEPJlYmdwPPXUU3/4wx8iIiJaWnrVSdLpdI899tiWLVu0Wm1mZubjjz9+7bXXjtUkEepX\nbaue9XqF4lTZ1D01fmIlMIsIyaW0FMB4YURAyBSA8FGdBRERSS5IcoHagUyM3GaE0LhCu6po\n6znPUdZGy3eQGT8Z4EkYwr9UgQDj0RHWbHX8/KVD1S1658cWjenjvZVHitv+/UC++3qB8WZx\nbtSbX5dYbJ5xnOUzYoQCAgBWO3u6vKOp0xSmkuYmhfhed0l8pSsOsB3pGNp1oqGuzTNVx2pn\n391d9ufbZw32bE2dxpI6rcXGJkcHpg+sU8mQrV84ZXpK6BeHa+vbDUEKyaz00JUzY33+TVxg\ntbNfHa0va9ACQFqsavXcOIlIQCnsPdP06b7K2laDXCLMS1bfuXpqTKjiz3fM/r8PTrsXnV02\nLfreNRkDmZ7NxzIWO0s5SgUTJ8EVITSaxu+vzH4VFxc//fTTERGei+o5jrvqqqvOnTv39NNP\nJycnv/HGG9dff/2WLVvWrVs3JvNEqF+M79s3wfi/s+MRQMhMAC2AGUAEoAIYuxADRjcQQkPT\nWcY7TLXVZMAlOYRMIGFbKXgmawiZIIBeP94/P1Djim641LToPz9Qc/PylAFPerSpAyW//dG0\nv39y1mi5mGySlxxy91VTAeBkWftzm8+3ac3OcaVMdM9VGavm8CT0EQLpcarCGs96JQCQkTDo\nlRGj7Hy1hne8wMe4Lw6We+WLoi+PXKzGlZ2ofuzm6WEq/mIZIyIxMuCh9dkD37+mRf+Ht463\ndvX8ne451bh5f9VTd87ecaRu26Ea52C3yfbD+ZZjJe1/v3fe1HjVyw8vPFLcVt3cLRULsqeo\nBx61iQvjL7sbG6aYmHdHCKHRMFGXqHAcd9ddd91+++3Z2Z4/lLds2XLw4MH//Oc/Dz300JVX\nXvm///0vJyfn0UcfHYtpIjQgydGBvhYqZw5+yev4QACCAaIBwsYyuoEQQkPGWvnHKedzkxcC\nAqkw1qPWhoAoJALP1zPHS9t4z3CshH98/MjPinzz10tvvyJ92bToNfMT/rBx5rP3zJNJhA3t\nxj++fcIV3QAAg9n+r83njpfy1yXduCLN+6k1OkSxclas+wjHUQc7vtqFOny0L/WVgODLaztK\nth+qdS+KUVCjue1ve7891Tis+Y0cSunTH5x2RTec2rTmP75z0hXdcLHa2Ze3FwIAw5D8rIgf\nX5563eKkQeWkrJwVy3t3dE1+4mBnjhC6dEzUAMcLL7xQW1v7zDPPeG/aunWrVCrdsGGD86NA\nINi4cWNlZeW5c16JpgiNDxKR4EeX8bygWzEzNj584nUNRAihycBXMVGRHIT910d0ERKlQpQi\nEUSIGJWYUcsEcXJhAvG6ATOY+cttGMz8VTzHlWCl5OblKY/dPP2h9dmLciKdSwe+OFxr43vy\n3/JDNe9JZqSG/um2WRHBMtfI/MyIZ++dKxH1POIW1Gh++fLhNY9/veb3X9/33A8HC1p4zzP6\nEnw0sp0ymG6mZptjxxHPhrsA4GC5v318xrsX75goa9DVtnrmGQFAc6fRexAASuq03Sb+ZrQD\nEaaS/em2We7dXgQMuWlp8pr5CUM+J0Jo0puQS1Sqqqp+//vfv/fee0FBPOWpCwsLU1NTJZKL\npa1ycnIAoKCgIDc3d/RmidBgbFiWIpcK3/mmzHkvKxYy1y9J+vHA+syjQaJAuwGMAGIgQZhg\nghDiRSJyuardhPV8PCPRs2GQi/8JCMRMP71XokLkvI+OA2k2MT7VeK24cfJVTBQA5maEz0xb\nVt9u6NJb48IUYaqLwY59Z5ue/uC062NVc/eT7568/Yr08bB+58o5cVv2V3tXhL1u0ZSBn6Sx\n3cgbD3J6+5vS1XPihIIxfjHpno8zQAazYzhNc2ekhr7xqyWHClrr2w3qAOn0lJA4fPGDEOrT\nhAxw3H333StXrvRVN7SzszMpqVc7MbVa7Rzv+7Rr165tamri3cRxHABotROunwWaMAiBa/IT\nr5obX9dmsDu4+AjlQAqMTya0uZi2lILdRAIjSdI8EPvnnp4aKC0E6rq9FhImFUhsX4cghC5J\ndpFCm7ouuOILxnHxoc6iTmfjFvO/rx+eVbNjjxS1eo+v5qtYMSEIhfxhoL6f0oUCMiUyYErv\nnAiWoy9vK/Le+f095atmx6kDx7hDVphK9uc7Zv3z03MtGpNzRCoW3LYybem06IGfRNDnt0Vv\nste0GFJiBpESMijdJpum2xodqhAL+5qGUja4VwISkcA9/2JoZGLh8hk8nWvR5Cea8I200ZiY\neE9Qr7322okTJ4qKeH7P9a3fotB33HFHezv/ulCLxXL69GmlEmPGyL+EAiZpMBmtk4TDxh14\ngzacdX6iAFDwFZN/B4nO9NjRbHXsPFpX0dQtEjJT41QrZ8U5q/QPmJ1yJwHc38c6KFdMGCGQ\nyGF9CQihScfosFtVyW3T7pV1FIjMHZxQZglKsgXGizjOHwGO/KzIm5enfLy30lWCgWHIhmXJ\n+Vnj4qdTs8b0zjdlRbVdNjs7JSrw5stScpLUfR+Snag+XsJzW5U9pZ8DvVU1dXcZeOqeOFju\nTGXnZdMHEUfwk7zkkNceWXyyrKOxwxgSKMlNChnsg31cmDJALtKbfK5Isvun8khJnfalbYXO\nnvQMQ66YFXvXlVN95VxkJgTzTtIZ+PBeTrVsenTfEROEEBpxEyzA0dHR8atf/eq3v/2tQqFw\n5lM4HA5KqVarFYvFcrkcAEJCQjSaXmWrnR+deRx9WL9+va9NBoPh4YcfFgon2LcLTWJsS4N5\n/05Hcx0TqJbkzpHMXDjWMxo67sxWV3Sjh0XP/fBfwTV/AenF54iSOu2T757s7LY4P359rP6z\nH6r/76457gu2+0Gbe0c3LgzTGoIBDoRQbyzHAQAnlBkjZ7uPOzh/Vbi8/Yr0xblR+842t3aZ\nI4JlS/KixknIu6Ba89jrx6wXSmZq9O0ny9rvW5N5bZ9LMNbMT9hxpM5jUYNMIvzx4BeVmKz8\nBUoAwGQdLzVKJCJBfpZn7diBEwrIbSvTX9xa4GtrXJhiyCf3paRO+8grh+0XlsZwHP3qWH1p\nve4/Dy0Q8QUmJCLBg+uy//bxGfdKqAxDfrY+O0AueuajMzrjxV+yeckh913t+aICIYT8bYI9\nsTc0NOh0ut/97ne/+93v3MeDg4Nvu+22t99+GwCysrI2b95ssVik0p7YubO8qHe/FYQmKNO3\nWw0fvEQdPXd15r3bJXnzgh56kojHOE13KChHKw/zjNsttPYkSV/a88nBPf3BKVd0w6m+zfC3\nj8/86/75A70U5V8QDtQAQD1aNiKELnG+Ej+ZQRbgGJSkqMBxEtRw9/xn561eDUHe+KpkSV5U\nSKDPPAWlTPTcT+e/8kXRD+d7qoFmJQY/uC57CDUUon0XIokNnTzZtWvzEyQi5vnPClivINoV\ns+MGuzxkIN7ZVWb3KvxR1dz97alG3m6+ALBsWnRMqOLdXWXOpI/0ONWtK9PSYoMA4K1fL/36\neH1Ni14hFeUlq+dnRvrz/wpCCPGbYAGOlJSUvXv3uo888sgj1dXVW7ZsiYzseQG7fv36Dz74\n4KOPPrrjjjsAgGXZ9957Lzk5GSuMosnBUV+lf+8F6H33Yz17xLj9feX1d43VrIbOYgA7f9Ey\nqm9z3Rqdrer06EvnVFCtadaYotQDLNgxxFstB8s5WOqrlS9CaFKSCoQmB092gFQwMe6dHCzd\ne6axqlnPEJgar1qYHdnvWl1eTZ3GujaD97jdwZ0oa79iVl8lQsJUsj9snGmyOJo6TWEqaZBi\niMUmw1Sy2elh3v1l48KV/a6UmViumB2Xlxzy9AdnSuu7XIMrZ8XevzbLH5crqNbwjp+v1vgK\ncABAWmzQU3fO9h5XykTXL07yHkcIodE0MX5JuyiVyqVLl7qPBAcHNzY2ug+uX78+Pz//Zz/7\nmU6nS0pKevPNNwsKCrZs2TLKU0WoL7Zu2vADNTYCpUQRRWIXgSR4gIdaDu8BvgRpy4GvJ2SA\nQyQBQoBSvk0X1560anxWbm/uHGiAg5AgShv5NgT5in2cq+p8fWdJeYOOozQ6RHHz8pTLZ8Ti\nKymELgVyocjksFvYXosjBIQEiodbNHEU1LUZnnz3ZL1bYCI9TvXkbbOGUI+z2+hzDUgfm9zJ\npcLhV8d89Ka8P719orjuYrn3mFDFExtnCJjR+Ilsc3CjVksiUi3/z0P556s0lc3dYiGTmRCc\n6KMN7TBRCg4fdT366OeCEELj3AQLcAwEwzA7dux47LHH/vrXv+p0uoyMjM2bN69bt26s54VQ\nD9pdQ0s+hAutB6mxmXacI2k3kuC0gRzOdrTwj2vagXXABHm1eJFQQsKSaVuF9xb3IqN9pOYG\nygectUuiAGoBjB6jhCTz7r7vbPPTH5xyfWzsMP79k7O1rYafXDl1oFdECE1koVK5wW4zOGwO\njhMQRioQBoklfl2iMiI4jv7lvV7RDQAordc++8mZZ+6eO9izhatkvqLQgyiB1B8HyzW0GxmG\nRIcoeKtHByslzz+w4EBBS0ldl93BpcQEXTY92t9tUzV661tflx4ubO022UKDpKtmx920LFki\nEgCAwWw/WNDS1GlSB0pmpobFjnSBjJwktb+TUwiBhIiAKr6uvVP8E1JBCKFRMNGehbzs2bPH\ne1ClUm3atGnTpk2jPx+E+kE5WrEVWFuvog+cg1ZuJzMeBqb/Z3VGwf8ejMgVEy+6AQAAZOYN\ndPc/wdGr/CdJmkfCLsYdpqWESEQC70XgYSrZYNarM0Qwk3IlQNsujMgIMxUIz00kx9FNXxR6\nj2/eX3X1vPjIgS6K6c1ioM1lYNRCUASJTp+gf18IXVKUIrFSNMSFFWOlsKartpVnUcmp8o4W\njWmwP77UgZLpKaGnyjs8xlVK8ez0sKHP8gKWo5v3V334bYXZ6gAApUx068q0a/ITveNIhMCi\nnMhFOaNRE5rjaGuX+RebDmm6e7q3dOgs7+8pP1nW/s/7848Ut/77s/OugppCAbNhWfKtKwf0\nlmJcuXbRlH/876zHoEIq7GN9ykBQCt+fbfp0X1Vdq14pE+Umh9y1euoIhsMQQqgPeHuN0Ogy\nNoHVmWHb+97NbqDddUTFn0rgTjx9vunbrd7jkmn5IzLB0UdCEgRX/p47tYW2loLDCgHhTMbl\nJKVXX5gghfgnV019aWuviIOAIQ9fm80MLj9ZQpg8ACtQExAxgNzX4pSaVr3rvtYdx9EzFZ2r\n5gw6wEGLvucOfQxWU89nVRSz/CckMnWw50EIob41dhj72DSE+Owvb8h97PVj7ikhSpnosZun\nyyQjcBv56hdFWw/WuD4azPaXtxV2G21jFS8orde+vrOkqLbLznLglbdSXKf9eG/Fh99WuC/u\ncLDc+3vKo0LkK2bGjupch23lrNgug/X93eWu9wfRIfJHb8pTBwyrZvmLWwu+OFzr/LNGb/3+\nTNPRorZ/3j9/+CuVEEKoXxjgQGhUUbvP+06w87xw8ybJnSudv9xy+Fv3QSY4VHnjPcOc21gK\njGCW3g8AwNpBwJ/Gck1+4pTIgPd2l1c2dQsYkpUYfNsV6UNNo5UA6efuzWLzzBZx6aNhoS+0\n6iS3981eQ9pm7ot/CG5+BhQDLb+CEEIDIZf6vLuTS4fSiSNcJXv1F4u+OlZfWKOx2rmkqMC1\n+QlDrhjqrkNn2X7hSdjdJ99XXrtoij/6hvTtaHHbn945wXJ8C3Iu+O5UI2/piu2HaidcgAMA\nblqafNm06HNVGo3eGhummJUWxtsgduDKG3RfeP2dmm2OTV8U/vO+gXY9QwihIcMAB0KjiogC\nqK+OpOKBPqsH3fe4OGe2ec82R3MdExQsyZunWHerr6UrE4yP6IZTblLI3+8NGZ2JRIcoCCGU\nb935EHoc0jNf8YzazFzBd8zc64YwPYQQ8iU3KUQkZLzbfwYpxKlDfYUuFDBr5iesmZ8w7Nn1\nUlzXxfFFE+wOrqxBNyM1dGQv1zdK4aVthX1HNwBAb+YvrVrHtyxoQghTyZbPiBny4Tqj7WBB\nS2OHMSRQOmdquHezG6eCao3J4ugj+oYQQiMCf8ogNLqUUSANBUun2xAFICAOJAHxAz0JIbKF\nq2QLV/lhfqiHSilemB3xw3nPkq4xoYrpKYMOstDOev4NHXVDmBtCCPVBpRRvXJH25lclHuP3\nr83yd1XOwXKwPqMJ3gEaf2vsMLZoTP3uppAKXdU33F2arcT3nW3695YCw4Wgz+s7i1Njgnj3\npBQMFjsGONAgiPBfCxoK/HeD0CgjTMp6ruQDcLj6nhIQSEjKemDw/+P48ovrcw1mx+mKi6X1\n4sKVT2ycMZQnBMbHIcyleEOMEPK3DcuS48MV7+4qr23VE0LSYoPuXJ2emzRKGXAD52uZISGQ\nHD0yaYk2B3ewoKWuzRAgE+Ulh/RxWvMAlh8yDFmSF/XRd3Grk04AACAASURBVJXem6anjrtv\nr7/Vtuqf+eiMe86Lg6XurXzdScUCj9IelNI9pxrPVHR2m2xxYcqr58dHh4xwMxqE0CUIH6gQ\nGnUBscz0h2jjQTA2UcoRRRSJXjDw9Slo1Chlor/dM/d4aXthjcbu4JKjAxfnRvP2L+wXiUyl\nNWd4xqMmXtV9hNCEkJ8VmZ8VaXdwhJCh/eAaBYmRAbPTw7wXNSzNiw4NkrqPDG11Q1Ft1zMf\nnXHlZTAMWTU77qH12QK+6tSRajnDEN4lM04SkeCn12RdPiPmWEl7ZVOv7qrO5i+Dnd5E99Wx\net4VPQwhnNcCz8umx7i/HjBaHL9/41hRbZfz49Hitm2Hah5cl716eA1cEEIIAxwIjQWhnCSs\nAF8NPNB4Mjs9bPitEMns9bS+ANgLrwedZVgCw0nW0mGeGSGE+jDMgpGj4Lc/mv7c5nMHCi6u\nB7xseszD12U7/9yhs7y+s/hwUZvZ6ghSiFfNjrv58hSZeEC3r3qT/Ym3TnSbLi4n4Ti682id\nOkDCG4wIkIsW5UTtO9vkMa6UiRZmR8aGKZZNiw5TyQDgH/fNf3dX2VfH6iw2ViggM9PC7luT\neQlmH9S385cdEYkYkYAxuBUryUsOuffqDPd93vyqxBXdcLI7uP98fj43SR0Tesl9JxFCIwgD\nHAj5ja2W2luB2ohABZIUYKT9H9IbNdWAuRGoHcRhJCAdyCT9D2u30IbTVN9GJEqIzCBB0SN1\nYtrVSE9vp521QCkJTSDT1hL12JS4J+FTmGseo/vfoc6iGwRI0ixm0S0gGvS/CoQQmkwC5KIn\nbp1Z2dRd1qBjCEyNVyVE9KQ0tmnND75wQGvoiVDojLZPvq88VdHx/E/zBxK4+f5sk3t0w+WL\nw7UbV6QSwvOK4eFrs7UG69nKi3WyItXyJzbOSOldV0IhFd6/NvO+NZmd3RaVUjJuE2SGr6Ba\ns3l/dV2bXiEVTU8JuWlZisItj0bqo0SCUip65ReLvjpWX9uqV0iF01JCF2RFun+/KaXfnW70\nPtDB0n1nm29enjLSXwdC6BIySZ+XEBpb1Eb134OjrecTAFiKiDIfRAN+uuastPlLaqq5eErN\nESbqapBEjPBUh8PUQTuKwaIDmZqEZ4NkKOulaXMRd+x9sHSD8xtFtpHUxcy064Dv1nNwZ649\nxX3/KnA9ZeqoUUPrzzJL7iaJs4Z55qEhUankpqdA30GNXUQVBdJBt2JBCKHJKjk60Ls6xvt7\nyl3RDZfyBt3Xx+sH0s+lvo0/v0BntOmMdpWSp82tUib6+73zjha3FdZoLHY2KTLwshkxYh/B\nFELAYx3NJPPBnvJ3dpW5PpbWa/ecavzX/fMj1XLnyPTUkB/ON3sfOCM1NEgh3rAs2deZjRaH\n0cJf8aS1q/86rwgh1IfxnriI0ERETSdd0Y0LQzZqOAicZaBnaPvWPboBAGDXck3bgfZfAm10\n0OpvuaPP0/KdtP4gLfuCO/xP2nR80GcxdXEHX3dGNy6cl6Nl39Oy74Y7P9bBHXrfFd3owXHc\noQ+A5e/wN0oCQklkKkY3EEKoX6fKOvjHy/nHPUhE/FWcCQGJqK8b4LkZ4XeunvrTtVmr5sT5\nim5MerWthvf2lHsMdugsL28vcn28YlZcilfPlEC5eOOK1L5PLpMIfeXgBCkkvOMIITRAl+hP\nbYT8iLJgq+Ebt4N9YD1BOSs1lPKMO7qpsXo4UxsptPUcrdoDHHtxiLXRkq2gG1zTU1pzFFie\n/GGu4ofhzrC9Eix6ng1WA22tGObJEUIIjQKzjT+mb/Lx8t9Dno+W3mmxKpkEU5j7caiwhbfe\n6vGSNqu957e/SMj849551y9Ocq5bEQmZRTmRL/5sgSvFwxcBQ+ZMDefdlJ/Fn6lKKS1r0H13\nuulsZafFxvLugxBCgEtUEBp51AKU/1cvZQ0DWndh1wHlfGzq4h8fZY3HeAYpRxuPkaD4gZ+G\ndrfybzB0AscOq4Wqxehzk5U/aRkhhNC4Ehuq4O05Ghs2oCKUM1PD5kwNP1bSK6FSKGA8ql0i\nXt6Lg5xYjupNdklQzy9ouVR4z9UZ91yd0WWwBsnFDF97Gl73XJ1RXNul0VvdB9ctTJwar/Le\nubKp+1+bz5U36Jwf1QGS+9ZkLp02YhW7EEKTCQY4EBppRORzC+FZ8cuD8b3bAM/gZ9TEnx5M\nTR2Dq5wh9JGJygiAGV5+mVLtc5PC9yaEEELjxpXz4r0DHAxDrpw7oEg6IfDErTP/933llh+q\nDWY7ISQjXnX/2sz0OJ5HaOTBV3kRkZAJVPDcigQrB7e0JEotf+2RJe/tKTtd3tFttMdHKNcv\nnMKbvqE12H7z36Pu9WI1euv/fXhaKRfNShtujzOE0OSDAQ6ERhoRgzDcswYHAAABccyAziBS\ngSgI7DrvMxB5/2XVwNwNEuVwAwR9E4rByjNMBIOLv5CIdFp5gG88bZgtdElIAqiiQevZ7Q+C\nIknYlOGceQjsHKe3220sRwiRCJhAkYgZdglVhBAaNVY729ZlDg+W+apq4ScrZ8Y1tBk3769i\nL6yVkEmED67L8i5H6otYyNxyeeotl6d26CwBctEoz39CW5gT+dbXpQ7WM590YXbkSNUlCZCL\nfro2q9/ddhyp5e2G8/F3lRjgQAh5wwAHQiOPKGbR7t1AexezlE4FQfBAzxB2GW3e5rFQhahm\ngDCAtpSCvhMUwSQ8qVcGhN3CnfqCFu8DmwkEQhKXw8y7CQL517gOHgWjhpo0RK4GhZoEp1Bj\nO89eIf3UFfNAYvNIWCpt713GTCgmudcMY6rOUxNmyU+43S+Aye3tnzyIWXI3kFGtPWR0ODQW\nq2sds5VljXZHuEwq8msECiGERkKLxvTKF8WHi1oppYSQ/KyI+9ZkRgTLRufqhMBdV05dPiPm\naElbh9YSHSpfkhutDhxKEcrJ3e7EH6LU8geuyXppW4GDvViJIyEi4L61maM8k/LGbh/j3u+B\n0OSCEUk0JBjgQMgPBGoSdDU1nQFHK1AbCFREmgniQRSnIIokEruB69gHlhagLIiCiXouGAXs\n53+E7gu5IXIVM//HJH4aAADHcl/+nbZfKEHKOmjNabapVLD+cQiKHOZXQ7WN9MTHtLPn5CQk\nkeSugfZCsPa+51BGkJh5gzs1YZgl93NFu2jFfrCZgBGQ8DRm+nUQONw5AwBRxwmufYqW7KUd\nNQCUhCSSjGUgGtV7XJY16yxGCr0eBlhKNVZbhAzvthFC45qm2/rzlw65qiRQSg8WtBTXdW36\n+aLBrkcYjsTIgMTIgFG7HHK5al58TpJ664GamlZ9oFyUlxyyZn6CUDDa0XlfZT0wFRIhxAsD\nHAj5B6MgygXDOoM0iondAJQDygIjgu5WdveTvVqcmrTc3k3Mlb8hYUm0/NDF6IaLzcSd2Mos\nv29Y0zB2ct89D3aza4B21tADrwuWPkCbj9H2QmBtIJSSqJkk6XIQ+Kw/4pNAzORcDTlXg6Ub\nxIphFRb1JpKQnFUjewvEcdTm4KTi/uZprabGUwxnjAJgiaRbmGkQTHGtu7GyLEupAO/OEELj\n2Kf7qzxqQAKAptu6eV/V3VddrNNpMNvr241BClFksHzgNSbRhBAfrvzZtdljO4ep8aoDBS3e\n4xnxPVmxDpZ+f7apqqlbKGAyElTzMiLwtytClzIMcCA0ihzd4OgCRgYiNZCB/e8jjHNJBVf0\nba/ohhPH0YJvyLL7aWMx79G0sYh3fOC40m/doxs97Gau6ggzawMBCnYziPppCDcg0oGuqR4r\nNS36/35ZfK6q0+bgIoJl1y1KWpOfIOC9mzcXUeMJ1ycBtQbbTws4k0508TYRAxwIofHmVHnH\nocKWDp01JlS+Ymbs+apO3t3OVWmcf+g22V7bUbLrRAOlFAAi1fL712bOz+Rv83mJ0HRb391d\ndq6q02xlEyKUNy1Lnp4SOtaTmtiunBu/9WBNh87iPigUkB9fngIAta2GP793sr7tYn+07ET1\nH2+bGcRXCRUhdCnAAAdCo4I1UN0PYKnr+cjISNB8kA2mYkVnPe8wdY47+Gp+AoDdx/jAddT4\nmI8zYYSMTHRjMOrbDOerNXqzPSFcOSs9XCgYjTBBUW3Xr189YnP0VEVp7TK/vL2wqK7rdzdP\n99yVOqjpjPcZAtkygzCZJT3LVTC6gRAaP1iO/v2TM9+dvliYecsP1YEK/qQ8m511HvK714+V\nNVysg9CiMf3x7RN/vn3WvHEc4zBZHTYHp/LP029Vc/ejrxwxmHveRnR2W06Vd9x+RfrNy1P8\ncblLwdHithOl7cnRgUIB06IxOQejQxQPrs/KTlQ7WPrkuyca2ns1hi+o0fzz03N/vn3WWMwX\nITT2MMCBkP9RB+38Ehxu1bA4M+36joAAZEkDPYmvmpTO52QfhTZIcNQg5smLelZQvzBO+cf9\niePoq18Wbz9U46qonxCh/M2G6Skxfs/+ePWLIld0w+X7M01r5iXkJPXuO+voBOrgOweVcJ0m\nQSwASAQCDHAgNNEVNOpO1Go0RptUKIgPkS9OCwuUDn6Z3viw7WCNe3QDAFiOdul5WlcAgLMi\nxqHCFvfohstbX5eOzwBHYb1227G6Vp0FABQS4Yq86CWZESO7pubFzwtd0Q2Xd3eXLcmLiglV\njOCFhsDB0q0Hq7873djcaYoIli/Kibx+SdJ4bitjtbNPvX/qaPHFnnRikWDN/IQVM2MSIgKc\n6ZPnqjo9ohtOR4tbO3QWrCyL0KUJy/gj5H/myl7RjQuo/uQgThLGHwoh4ckAwExdBAxPvJJk\nLhvEJXgFxw5u3J8+/K7i8wPVrugGANS2Gh5/65jJwhtQGDFmq6OkXsu76WS5VzcZ/ugGAAAB\nFgAEhKglvV4eOuoqzF99bPxkk2Xfl9SkH+50EUL+99X55q8Kmtv1VpajRqujuLn77YPVHYZh\nJ82Nkd0nGwa4p4Ah1yxIBIDCmi7eHapb9Garf38mD8Hxio7/7i5rvbDMwWh1bD1W979DNSN4\niW6TrbBW4z3OcdT9KX1M2B3cr1898t8viysau40WR1Vz9zu7yh5+8ZDZNu7+plze313u8X2z\n2dltB6vFQoFrcWhTJ090AwAohcYO/k0IoUkPAxwI+R21+7izcWiAOoCyYG4FcxtQto+TMJnL\nQar0HBVKmNwrAQCCIpnl9/bagWHItCvJ1MXDmjoASVvGU/iTEZD05cM882BRSrcdrPEe13Rb\n951r9uulzVbWV8KKyes+vlUvbdAIWL7EFyIMDhSLouSyiz1iOc7w/gvav9xv3Pyaeddmw7vP\ndf3udtvZwyM4eYTQiGvoMhc0uYWtCQCA1cHtLRnj59gha+3yqrUEAAAJEQFK2cW0FKVM9OiN\neZkJwQDAcT7z+Fjfm8YER+m24zzLPA+Xtbf4+MKHoNto9/WbQmfkz4UZNTuP1RXUeAZfqpq7\nN+/zKk/uB5TSZo2pqdPYx78Zb7xBNwdLvzvd6PooE/tMRZdLMEsdoUsU/udHaOxQoG1Hadth\n4GwAAAIJiVhAwua6em30IlcJVv+KO/wBbSlzDpCQeDL/FtfiFDJlpiB6Kq06QXUtRK4icTkQ\nHD38ORJVDLPoPu7Ex2C8UG1OoWZmbiCqmOGfvB/O1TGkJxagNdp83SPWtvo360GlFCtlIu/E\nYwCIC7sYVNp7pum1HcUdOguASiqiN82z3DjHJHRFh0ThaoXniiHzrk8te7e5j3B6rf6VvwQ/\n9RYTMh5zvBFCAFDdYeAdr9UYHRwVTsBOIoFy/h9xiZHKf9w372RZe4vGHKmW5SWHWO2czcGJ\nhUxSFP/awIhgmXtMZDxo01r0fF8dAJS3dEcGy3g3DVZIoEQoIA6W5xk+YoQuMWRHivhDb0eK\nWzeuGEw5MD4Gs72p06QOkHgvCaEUdh6te/ubUuevb6VMtHFF6roFiaS/RZpWO+vdwcfJVYkD\nAPKSQ3i/5+oASVL0eK9cjhDyEwxwIOR3RBTG/87CzFLjvosfWStt+g4cZhLlY12JKppZ/Ssw\ndFJ9O1GGgDLUswu8REEyloz4nTWJzBCsfpx2VIOpE+RqEpo0lHawg0G1lbR+L5haAAAU0Uz8\ncghMEAl8ZpyJhf5NRmMYcsXsuM/2V3mMK2Wixbk9MYsvj9S+sOX/2TvvwDiqa/+fOzPbq+qq\n9y43Se6WKza4GzC9JXQICYQSkjxeQvi9kOQlIclLAgQIndB7sY1tbNwtd7moF6tr1Vbb68z9\n/bHSarU7q15W0v38pTlT9u5K2pn7ved8z0XPLpsTvXlI0qyjnthoAgAQaJBiuf+VbQe+9g9i\nh912ZLd06+1jNX4CgTC22P0cedxgDA4XxwzaRjoIcLg4m8OllPaUyy3K1nx2mGcxf0mORiUT\nrsmL7TLYX/6m9E8fFLMcpii0MCvyzvWZkWpJW7dvBsQNq1LHffTDxMGbUwcAAM4Av8oRIBEx\nS3KiDl3wzSiUiJilufw+WROG0cK/PGAYXWpJh9724pclnrecHqf6ydWzshLUngPe2Vv59p4K\nz6bJ6nzxy5IOvc27zTAvQoYWCWi7kyezVS7tewIJV4lvXpP29p5Kn2Me3JrL3+OMQCDMAIjA\nQSCMP5I0MJ0Fl6FfkOOwhc+Yo70IRSwCJnBrEnkYkoeN9RAHgxYgTcbEvBRuLcK1O/u2jfXc\npddR2tXyiHkp0cqaFoP/KXNSx/0DuXN9ZofeeqC478k1TCn+xc3z3I3oXCx+Y1eF/1l7Loqu\nX5mcFBMFAj63V45l2/mLa9hW/qY5BAIhGFAFyFAQC2hJELs2urlQ0/XyN6WVTXqOw+Eq8Y2r\nU7csTrzlirQTZW0+tgULMiNWz4sBAJPV+cjzRzxlLByHj5doS+t0T92W9+qO8vJeiyKRgL51\nbdqWJYmjGZ6L5XaeaCip0zldXGqMcsuSxNHng0QoxTSFeAtnokKG1wisy2jXmewaNX+WykNX\n5zZ1mL3vU2Ih/bMb56rlk9yyVBMi4XWEjQodeR80q9312IvHvPMpKhv1P3vp+N8eWpoaowQA\nk9X5/v4q/xM/PVS7fXlKqFI0wMURgoVZEYcutPrvWpzdL73x9nUZCZGKt/dUNLSbaQrSY1V3\nb8yakzLhj0kEAiFoIAIHgTD+IAaFbcbdB8HeW1BKiYGJB3yc52DMYWMd6JvB2IA5F5JFofhC\nEKl5jhwEFlz1mOsGQIhSA5MwNTx3XDZct9c/jC9/i8Jy79mU9avXTvo8pBZkRBSkR4z3uIQM\n9dSt+VuXdJ2r7jBanUkaxep5MZLeEt/6NqMhwPrYhebQpMQAvWwoGgmE2MGThYvEk5zPTCAQ\nBiArSnmkqsPplxeQG6MK8v5IRy9p/+ft055v0Q697fnPL9W2GH+6ffYLjxS+t6/qyCVtR7ct\nNkJ61YL4zYsT3aUEnx+57G/SoTc7Dha3/v3HSy/V6eq1JoVUmJsYMvCsdVBauixPvXrC0xfj\n4PmWTw/VPn1Hwazk0IFPHBiJkF6QFn68wtcTOkotyRxyIUN1i+HNvZW1vRWRBWnht61JC1f2\nK8oIVYief6Tw25MNxdWdFrsrOUqxdWlSMPTyuCI/jlcsWFsw8mrTb4rqvdUNN3Yn+/aeyt/8\noAAAKhr1vAkyLIdL6nWFswbJarl7Y/aF2q5uU79765q8mPz0cJ8jV86NXjk32uHiKIQmpnM8\nYYIIer2YEJwQgYNAmBBoBQrbBC4duLqBEoMgHOt5ljUAAFgOl30Kzp5HSay/jFtPo+wbUVjW\nMF6OM2DHCcC9F2EbwFWNRIsA9bcp5Ry4+RDWlYBDD0I1Cs1F0YVATWbtNDbWAcdXKe2ygqlp\nfkbSH+5d9OKXJe71MZGAvqYw6ZYr0kc+o8AOcLYDZwFaAYLIQTWg2Smhvk1hAWDAJOeB858F\n2fm8lqKCnIKBR0IgECYRhZjZNDt658UW71qV5HDZcr+pV1CBMbz0VYl/IsOOovqtSxNTopV3\nbci6awPPvaa4utM/CADFNZ0IoVlJobOSRiVAePjzh8U+XT/1Zsfv3j37+pOrRtnQdPviRIeL\nO1PT90YSwmU/WJU6xEKGujbT7z44590s/HRVR12b6dkfzPcxs6QptHFRwsZFCaMZ7ZizNFdz\nw6rUjw/WeNt8blyUcGXByBuiBWqjc6nXzXTod8azVR3F1Z0Wmys+Ur42P9a9eBATJn3psRVv\nfltxuqJdZ7QnaBTbliUOMODxLlYlEAhTBSJwEAgTCBMCTIj7RySJDGDM4QCfVUHOics/QYue\nAHqIK2MYO0571I3emAU7ziCRV1MV1s6VvQ7WdgDAAMiuwy2HcXcllX3nZGocbMAmi5i1I4C5\nqWH/enS5weIwmJ3RYdJRFdnaarCpCLieroHAqJFiGQgiR3CluAg5Q1MuvjLvQD58bmTb73aW\nF2Nbv0UwQXaeqIDHsINAIAQP6RpFTIjkYpO+w2SXCOjEMFlqhF+jqyCjVWdp8Vtyd3OuqnOA\nL6tAM1UHn0XCiNHqrBdqeNqsduhtZys7FueMyndZyFA/WJW6elZUjdbodHGxYdLsWPXQxfHP\nj9XZXZzP4R0G277i5s0Lg0vLCMQ9G7OWz47ad7a5pcsSqRavnBPDq9cPHd5bnnc8KUoR6NyU\n6J5ddif7u/+cPVai9ez6z3eVv7w5b25qGACEyEU/3T57NIMkEAgzECJwEAiThCgMKdOxob8z\nFot91Q03LivWVaHw3CFdmesGzOfwz+mBMwDV8wiLtUVudQO8u7ZYtbjtBIpaNqQXGj0uI+4+\nB44OoIQgiUfKWUgcGqiJHBL3PYoppUKPN94IcTRjw4F+EVc37t6Nwq4BSjbci8nEzPoF8V8f\nr/OJp8Wq5gz4BEnHJqt//aL5g385L53CLielUIvXbJOsv9HXPpZAIAQfMiGzKHkqlfrbHQH1\nCFvgXQCQoJGX1PEs1ydqAs5gR4BWxy++AMBYNXNNCJclhA/7Gx4AKpv1vF/KFU08xhZBS2a8\nOjN+eBWvLhbXtxn1Zkd8hNyn1iYlWllUytOcxdO+RBMiWTYr6shF39KYgowIz1/OK9+Ueqsb\nANBlsP+/t0+/8eRqhTS4evEQCISpAhE4CIRJAyVshcaduLukLyJPwqZSvkMBmy8DZQVGjqTx\nwAz4fIYDPiMCtgD0ChwBamRwd9XECBzYVIG1u/oKUoxlWH+Oit0OUg1YtL5HKxJAMpZGG9hy\ngS/qxJYyJB9JecgDW3OcLPftyT5z0NkpoT+/aR41WI4JrYlTPvxb4DhsNSPZWM4WCATCwHTo\nbXVao1wiSIpSjLICYkqgCZUIGIo3HSM+cqDbypbFiXtONfrXtmxbluT52eHiPvq++uD5llad\nNTpUujovZvvyZCZw9yt/BpCtR6tojxpeg9IB4tODIxdbX/iypL23Uc6yWVEPbcv1yBybFid8\nfviy1eHyOev6FSmen5+4YS6FwNv+Y1F25BM3zHX/7GK5Paea/F/XaHEeutASbGU+BAJhqkAE\nDgJh8qBFKPFqpFmGLa2AEJJEAQbc6itwYAGNFBIwX8LmSwCAKSGKXIFCczDXBGACECAUCsjb\nrCvwogfy2hWoGIS18ccBwNSJDe1IFgLKCECjK3Zlzf3UDTf2dty2n8q8gSt7D6wdfXGphkrf\nPqqX88fFkwgNAODiLzUfFCFDPX79nO3Lk0vqdA4XlxKtmJ0cNow8DIoi6gaBMGF0mxwvfnnp\n++JmjAEAVDLh3Ruy1i+Mn+xxjS8SIbM2P3bnCd8mTZoQycKsgarz0uNUv7h53j8+u+RxU5YI\nmXs2ZS3I7NGdrQ7X4y8eq2rq6R5S02KoaTEcvdj6pweWDN0ZIVGjiA6V+hfRiAR0QcbEmZtg\nDAfPt5yt6jBanAka+aZFCeEqcUKEvKSeJ4clIejrkkZMUWnbM2+d9o4cudhapzX+69EV7t9p\npFry27sWPPfR+ebOHtsUuURwz8Ys72IimZj51e0FFY36isZujCEtVpXt1US2y2j310fc+DT0\nIRAIhKFDBA4CYbIRRyCxV26CTANmr/wFikJKab+CBc6BW/diqhYpenMxcBOgBkTl9fxH06GA\nBID9rDqREKiQvi2RGts6fI8BQKIQ/yDotdyRd3DjpZ5jQmJR4e0omqdxLDZdBksLYA4kGqRM\nC/SmsakKWCf4zf+xuRJprqLmPog7LoCpGRACeRwKyx2tnsJDIO1hVC+UFKUYoOqYQCAEAxyH\nn3rtRKVX10y92fGXj88jCq6aPySNg+Vwl8EWohBPuZYND27NNVic3lUDcRGy/74tf9AElpVz\nY/LTI4rK2lo6zZFqyYLMSO+GKZ8duuxRNzyU1nd/fazu2uXJQxwbQvDI9tm/ev2kT47JfZuz\n3Q25JwCLzfXrN06d93iRXoBPDtY8dt2cjfPj/AUOsYBeOy9mYgY28by9h6f3eWO7ed+ZJo8U\nODsl9JXHVxTXdDa2mcNU4jkpoby/qYw4VUacyj/u4886xF1utDqr2eaMDZd5/+naney+s811\nWqNYSM9ODi3IGPcOawQCIQghAgeBEFxQGddyF14HV28ahVjAb8egawWFlyEc7sZcJaKyAQCA\nQYJZ2HG2/wkICeb0m72HzwPeKpXwPN+IzcR+9Qew9E0GsK4J7/gzte2/UHhS32EuK67/HBtr\n+w6TxlCJ14KQz7jOZeRXGDAHrAkEIShiHkTM4ztijBBEgt3XMgMA0IhMRgkEwhTieGmbt7rh\n4e3dlVcWxA+ceNVpsL3yTdnB8y0ulmNotGxW9P2bs4OhD+gQEQvpp+8ouFjbdfFyl8XmSolW\nFs6OGmIhiUIqWJvP31W0qNSvrhAAAI6XaocucABAfnr4S4+ueOPb8tL6boeTTY9V3bo2PTeJ\nT3YfH/69o+x8Tb88PpuDfe6j86/+bOW967Pe/b7KbOvJONCoJfeszwxTTplf/bBwsbgygL1I\nab1u2awog8URFSqlKSRgqPkZEfNHJCXIJYLMeHV5yNpH+AAAIABJREFUQ7f/rvmZAS9YVNr2\n4peXmjstAEBTaMvSxB9emSkVM6X13c++c6atu8+uZUFWxH/fmi8ZTCshEAjTDPI/TyAEGYpY\nav5PccMBbKgHjkVSwLyeGnY/xzXcApDVk5hAxyGRHLvKgNMDAFAhiMkEqt/6CQrJhuhluPUo\n4N4SYkSh6EKkTve9cMl+b3WjB9aFz3yNrvxx32ENX3mrGwAAlmau7hMq/Yc86RKUhO/ND7Zr\n7ECyudjRCNjjq4cBEFBSkA6nHS+BQJiC8E6oAKCt29pltA0wZdWbHQ//86jHksDF4gPFzZcu\nd73wyHK1fJJNIobFrOTQWclj09jVjdHK194bwGDmjw+AO6Nk1CMaCRyHvzvLYwlhd7IHiluu\nX5mSnxZW3qjvMtqjQiTZ8ephOYwMjN3JOl2cXBI8tpoBvUUOX2x1VzkJGerqwuRb16ZJhCOf\nTTywJefnLx939M/ZuWp+fCAz1KOXWn/zZl/hDMvhzw9frm0xPvPD+c+8earL2K/29mRZ+wtf\nljx+/ZwRD49AIExFiMBBIAQfQjlK3eSWBHDjF2Ao5zmGp2SDBXAC9D5kU2okXDzw66DYNSgk\nF+tKwKEHoRqF5oCEJ38Bt1Xzno61XgkgDgM28OWDWFrA0gxS30U/JEvBHQd4HqEkcUBPyIIY\nE4bUV2HjMXC5s44RCGORYgkgr1kK14LZVgAbgATRsUCRZFcCYTqAccDJW+A9AACfHqpt7/YV\nlzv0to8OVN+7KXtMxjZFiVRLGtt5TBOiQidCsB4rDBan1c5vCdHaZQEAuVhQkNbjBqLVWS+3\nGgUMlRqjHE0FzemK9n/vKKtpMWKMo0Klt61NX1cQN+l9tBiaStQoLrca/XcZLT2ilcPFffh9\ndWWj/g/3LhrxgHOTQv75cOErO0rPV3c5XGx0qPT6lakbArvhvLaT54mouLrz3b1VPuqGm31n\nmx7alisWTn8LYQKB4IEIHARCcCON4xU4kMTf8R6N5D9aqkFSzSDHBHrkx31LLtge0JsT2zqR\nn8ABwhAUthR3HukXpMRU5BWDDGYAsA1YPQAArQYkGuxoAIEGhW4D1gisBRhF/+6wGDvPAOdJ\nutZhrhnoOMSQhSACYcqTzmcHAABhSnGYcqCvjuJq/i+6cwHiM4d1BXFnKnlMndYVxE38YEaM\nVMzQFOJtjKL0kjBMVueLX5bsOd3o3hQJ6BtWpd62Ng0Nf5a/53Tjnz4o9my2dln+/GFxQ5vp\n7o2Tn0t40+q0P7x3dtDDzlZ1nChrW5Q98urOpCjFs3ctxBg7XNzAXjAGi6O+zcS7q5TPAhYA\nnC5Oq7MmaqatF+z0BgnIRJUwEsbct49AIIwlSD0bhH7lx4iC8Gi/YOh4/UeH8a+loPBErxEF\nvgkF2IVCF6PY65E0CRg5CEORag6V9EMQjsgqH7PYdhYbvsLmA9h8ABu+wrYLADytEP1HAbQS\nhFH91Q0Ats5L3fAEG4FrGcnwCARCMLEkJypRw2MGfPOaQeaodifLG7c5+OMzhzV5sduWJnlH\nKApdvzJl2ayoAGcEI0KGyk/nvwct9prA//adMx51AwDsTvbtPRVv7akc7su5WPzy1zyN4T86\nWKPV+VWhTjhr8mIeuXb2UKpmfFxLRgZCaFCnW5crYIYVFfg/Vyoi6RsEwsyCCGMEQnBDCamk\nm7F2P9b3PgZJolFkOoh8EkcFiMocryHkrGFL9oOj//MWRaG5G/o2JdFAi3hazyIKyQK2skfS\nBJAmjD4VF1tPgrPeK8CBvRRjJ5KMsJAbBxAyMNuMKD9piUAgTCkYGv3+noV/++TCibI2d0Qi\nYu5Yl751aeLAJyZEyqubfXuFAEBi5ExfH0YIHro6d9W8mAPnm7Vd1ugw6ep5MYFsFAbgTGXH\nxdouq4NNjlKsnBs96Ix3zLl/S05Zw1FPFYabrUsTPe+ltL6bN1fl4wM1N61OHdaAa1sMerPD\nP85x+HxNZzAkv2xanLBybnR5Q7fO5AhXip58uYj3MJ+uN+NHiEKolgu7TTwfWn5mRDGfzpKo\nkUeop1KdFIFAGD1E4CAQgh5GjmK3oOirwKEDRg6MDAAAt2FcB9gEIAAUiqi0PveNMUceSm18\njDvwOuiaeyISJbX0FhSb03cMxaCoVbjpW59TUeQSEAzj0R+3lONTn+POekAUikhCC7ajiKRB\nzuFM/dWNXhzVIM4dUq0Kzzhsw4sTCIQpRbhK/Nu7FtRpjZdbjQqpMD1WpZAOvlK9ZUni/nPN\n/vFty5LGfohTkNykkBF3PLHYXL9796xHcgKAd/ZWPnVr3ghUktGQECl/5bGVb+4uP1vVqTc7\nkjTy7StSVszp07V5++8AgN3J1mlNvM1QAxEoIQiCKSdILhG4m61iDKEKEa/PxYQ1R0cIXbs8\n2d+GQxMiubYwqaPb+tWxfs3RGBr9aFvuxIyNQCAED0TgIBCmCJQQxF5mGSgSoYlraIoiU+nt\nz+C2atC3gSwEaVJB4GsFisILQCDHLfvB3gUAIFCiqBUodBimFbjke+7wW32bjZdwUyl1xQMo\nZX5PyGUBlxVEIf08VtmuQNcDVgfMyBKkhQB8GcIjk0sIBEJQkqhR8NaqBGJWcujj18958asS\nS2+vUImQuW9z9tzUsPEZ4Azi+S8ueasbANDaZXnmrdOv/WzVBDtEhipFj143EXZLMeEyhBCv\n5W18RNDlBCEE25Ylvb7LV1wIVYhWzo2ZsGHcuCrVYmM/PljjYnvSRjLiVE/eNE8koH98dW5G\nnOr9/dXNnRYBjXKTQu/dlJUWOwzJiUAgTA+IwEEgEABbjFzZMdzVghShVGo+CvfzBAUAikZR\nGRCVMcB1kCoTqTKBtQHmgJEObxB2M3f8A7+RcdyRd+ikedjUgBu+BWs7AADFoMhFKGY5UOPV\nUQ/RUdjFs0yHqKlUT04gEMacqxbEL8iKLCpt0+oskWrJouzIAdrKEoaI1e7iTY3p0NuKSrUT\nOX8elMx4/gmzWEgP18kyVCEqnKU5dKHVJ54UpZidMpZNfMeKm1anGizOzw/XenxY4yPlv7hp\nnkw8cbMJhNBdGzI3Loq/dFlntDiTohRzU0Pd1jkIoasWxF+1IN7uZAU0RVGT3YqGQCBMEkTg\nIBBmOlzJUefOl8HW2+Fv33/oxVuY1bcADO3hwGnB+jpwmJAkHNSJgCiePq+Yw/VHcfNZsOpA\nokZRc1FiIVD9FuVwSwW4eHJfwWrgag6B/mhf0xbOhVuPgKUVZdwCAEAHehBEQI80t5lOAq4d\nOE9uCAZAQGmA5pN+CATCTCJUIRqgjSVhBGh1Vs+CvA8NfN1nJ5HMePWCzIiT5e0+8RtWDc+A\nw82j182x2NnTFX1XS4lW/ur2fDooJ+cIofs3Z29alHC+ptNgcSREKhZmRTI0AgCbg23qMCuk\ngshxMLww21w0hbwTeaJCpVGhARdRJt66hUAgBBVE4CAQZjS4o9H5xd+B8yr35Vj26OcoJJqe\nt2bw05uKcNUucNkAAAOAPIrKuQ4U/VUAzsWdfAV0tT2bDhPWN+LW89TCB4D2SsFwWAK+TGsR\niH2ffbGhGgw1SJkClBwE8eBs8D1LmApoxCurNBIsArYBcy2AbYAkiIol6gaBQCAMBYvNVdtq\ntDvZ5GhFiHzwyj6JKODjqDTwrsniqVvzX/q6ZNfJRnd1iVhI37wm7abVqSO4lFwi+P09C89U\ndpTVd9udbFqsalmuJshTD+IiZHERfX3HjBbnqzvLdp5ocH8a8ZHyh7blBmpGozPZa5oNHIbk\nKEW4apAbNMaw72zT23sqmjstCEFylPLuDVkLsiLG5F20dFnOVnbojPa4CNmibM0El0ERCIRx\nJehuGwTCTAGzuPk06OuAdYA8GsUtAoFs8LPGGvbcd/3UDU/8zO5BBQ6sPY/LPu8XMrVyZ1+j\nFj8Gwr73ghuO96kbHvQNuO4wSlndF1EM8NRiAfCtRsEAyFgPyhQAQJKFGInAUe2WWQAoEKUj\n8eyBxz8YCOgERAdsAUMgEAgEHzDGHx6oeXdvldXhAgCKQpsWJdyzMWsACQMANCGSuAhZI1+y\nRqCp8sRT3tD99p7K6mYDQ6PMePWf719sc7JChkqNUQ6ll+oA5KeHB8/bHBYsh3/576IKL+PV\nhjbTU6+eePbuhT7vyMVyb3xb8emhWneqDkWhzYsT7t2UPUC2xZu7y9/9rsr9M8ZQ02J46rUT\nj103Z/2o86fe3lPx3r5qT9JQuEr8xA1zp+ivgEAg+EMNfgiBQBhz7HruxD9w2We45Qxuu4hr\n9nBHn8NdlRM/ENzJU/YMALizafBz6w/xRJ0W3HyyX6SthP/0tkvem0iTBioehwsUmQRinmdH\nBADY1btBI0k+UmxGsuVItgIpNyPxXPL9RiAQCBPMO3srX91R5lY3AIDj8FfH6v7w3rlBT3xw\na65/XcbWpYkT1qFjYHadaHjk+aMnyto6DTatznrwfMvPXylCCOamho1S3fDBxeI9pxuf//zS\nPz+/uOd0o4vlsSANHg5faKnwayvDctjfi/TFL0s+/L5PU+A4/OXRur98dD7QlTsNtg+/r/aP\nv/xN6Si70u4oqn97T6V3SVSH3vabN0+1d/M5ixMIhCkIyeAgECYBXPoZmLT9Qi4rvvgBWvoE\nMBPrV+fXDMUNEg46DAymFv49/ePYGaD2xNF/sY6iqHUPcrv+DiavVvbqaGr1A1zt2+Diu4ik\nfx8ZSgIUaXdPIBAIk4PdyX74fY1//FiJtqpJP3A/iwWZEX97aOnL35SW1nW7WC4mTHrT6rSr\nFsSN22CHgcnqfPHLEo7rpzW4WO5vH1946xerx7CipLnT8vQbJ+u0pt5A3Qf7q//fnfNjwiYh\nwdMfncmuM9qjQ6WefJyLtTreIysaux0uTsj0LDN0mxzfFPF0c99/rvmOKzNiw2VOF/fZ4dpT\nFe06oyMhUrZ1aZLOaOcVd0xWZ2WTPidxSN2IbQ7244M1xdWdRoszUSO/dnlyZrz6y6N1vEfu\nOtlw+7qBbNQJBMJUgQgcBMKE4zDjrgqeuNOMO8uRZu5EjoVKns2VHuWLD9okDwFQADzlLf16\nuAIgsRobePJEkMT3AQWFxtM3PIvLD+GOekAUikxCGYVA0ShyAW4+4Hu+UIlCsgcbJIFAIBAm\niMutRruT76YAUNbQPWjDzsx49XMPLHGx2MVyQeWJcK6605OT4k1bt7WmxTBWjUgxhmf/c8ZL\n3QAAqG8z/fads88/XIgm1ZejrL77H59frGzUAwBCsHpe7H2bs0MVIhfHn0yBMbAsB70CR3Wz\n3kce8lDRqJdLBE/865jnjddpjYcutC7IjOQ9HgBsDv6/MR/auq2Pv3hMq+vJy6hpMRw433L3\nhqz6NiPv8T6fPCEoIH6xhBFBBA4CYcKxdwNf33sAACv/Ysj4Qc9ZxZ7di1v6JYIiiYJeccOg\n5yJVAtbxZJCCKrHfZkwef5VKTD5PkBGi3Ct8HuRQ9HJgbbjtZF8jFYmGSrl6/NrEEggEAmG4\nBLqzDbzLB4ZGDB1csxqjxRlol97sGKtXqWkxVPqVewBAVZP+bFV7UWl7Wb2Ow5AWq7xpdZpM\nzJyuaG/utESoJfnp4aGKwZ1cR0xlo/6Jfx1z9BaGuL0/Kxq7X/zp8uQoJe8p3lkeg/L6rnJ/\nceFkeRvvwQhBhEr0+q7yk+XtOqM9PlK2ZUni8tnR/ke++GWJR91ww3H4tZ1lAoZ2sTxyFem9\nQiBMG4jAQSBMOIKAvc0G2jVO0Izwtqddhz9hz+4FmxkYIZWez6y5HakDLp54QClr8ZnLgPuv\npUjDUXRBv8Oi5kBCLa7vlyeC4haimH6HDfhKCMVfiSIKsLEOWDtIIpAyxSdPZNqBcesF6KwE\nhxlkkSh+EfglvBAIBEJQkRglFzKUg88iISNubNIcJoVIdcCaTU3ImN21mzsDthL79RunHb2p\nMeUN3d+ebBQJKLOtZ5YuFTP3bszetHi8LLHf2lPh/zttbDfvOtlwRV7sf/ZW6ky+Ld6vW5ni\nvZkWq6IoxJvEkRmv+sdnF3lfVy0Xdpt89aP89IinXjvV2tXzWXUabOeqOjcsjH/0un5pp3Yn\nW1SqBT9YDseoxQ1tPMkac1IDtZwnEAhTDCJwEAgTjjgE5BpfDw4AoGgUNhn1n0IJs+Y2Zs1t\n2GJAEvkwhAN1Esq7E5d/AeZ2dwBFzkIZW/o1fwUAQCjnahQ9FzefBasOJGqImovC0oY9TnEY\nEocN+6ypiMuOz76JO6s8AVx3COVcg2LnT+KgCAQCYWAkQuaawuQP/LwhCzIiMuPVkzKkMWFO\nSlikWtLmZ0KZnaD2bpg6SmTigM/kjv6FPy6W8/bItNhc//fphcgQyYLMsWmh6sOly/y5pRdr\ndduWJv3unoV/+qC4psXgDooE9M1r0rYs6ZfIqZIJtyxO/OLoZZ8rrMmLjVRLTFb+BJnsxJD2\nbltVU19Wy7y0MKWUOd3lqwTtPNGwal5MXlpfGxSjxRnInzUtVtmms/rUUqXHqa7ICwrDFwKB\nMHqIwEEgTAJU5jXcuVeB7XdTRylXgngyHwGRlD/XdKBTQlLRokfB1gV2I0gjvLvD+hKSjEKS\nRzW+GQOu/NZb3QAAYJ340icoJBmk/SUelxXMbSCQgiRsuqe0EAiEKcCd6zMpCn18sMbT6uKK\n/NiHtuVO7qhGiYCh/uvWvN+8eco7oSAqVPrkTfPG8FVyEkOkYsZi46meGAqfH6kdJ4EjUNcS\ndzw1RvnCI4XFNZ11WlOIXJibFBqu4kl4uX9LjkTEfHKo5w+DptCWpYl3b8hiaEopFRosPJU+\nceGyp28vOFairWzSMzSVlaCenxFx7dO7eQdz9KLWW+BQyoQMTXnLQB5SopU3rkp94YuSC7Wd\nGIOAoTYuTPjBVRkMPak2JwQCYewgAgeBMBmoE6lFj+Ka3bi7DlgHUkRD0moUkjL4iUEIQiAJ\nA8nMyK2YCDBuPs0T5ljccg6lXtGz6bTgqp245XRPabtYhdK3oMipPYsgEAhTHYpCd67PvLow\n6UJN19ESrcnqYijq4PmWK+fHT+kJZE5iyGs/W/XVsbqqJj1FUTmJ6o2LEsbWtUEspB/YnPOX\njwN2Th2Yutbx8shMjlaU1Xf7x1Njejr4UhTKSwv31hf8YWh014bM7SuSa1oMHIeTo5Ue35CV\nc6O/Oubb2QQhtHJuDEWhZbOils3qaSHPcjhQukd3fzMUIUMtydEcuuDb642hUeGsqNhw2Z8f\nWGx1uHRGuyZE6t+fmEAgTGmIwEEgTBKSEJR7I7mpEnxx2cHpmwjdg7Wr5wfMcefeAEND3y6b\nHl94B2bfiiJnjfsICQQCYUAa28z/+Oyix4Dz21MNnx2u/d3dCyPUU7iTt1wiuHnN8IsrBwRj\nqGrSN3aYQ+SizHjV+oXx0WHSN74td7uNpsep1ubH/9+nQ5I8hILxSuLbvjzl2f+c8QlKxcyG\nhcN2/VDJhP46yA+vyiyt11U1GbyDt61N9/dtoSkUphR3Gmz+V/b3SXlwa051s6G5s68hPUWh\n+zblxIb3pJpKhIwkjMyDCIRpCPnHJhAIhGCCFgLFAMeXpSzoeSzDHWX91I1ecM1uInAQCITJ\nxe5kn333jE97kTqt6bmPzv/h3kWTNaog5HKr8S8fn/ckR6hkwvs2Z68riPvrj5a6/TgpCmGM\nPzlU3dhuHvBKAABzUsYrj3Ll3GidKff1neWeXrkxYdInbpjLW4oyAhRSwd9/XPj18bpT5e3d\nJntchHzr0sScRH5f7TV5MR8dqPEJIoTW5MX6BMNV4pceW/7Z4dpzVZ1GqzNJo7imMDktdtil\nuAQCYcpBBA4CgTDhYCcgGmD6eEaYrM6GdjNDofhIuVg4uqRlRKGITKy9xLMnMqfnJ71vNm8P\n5nZwWiahFw+BMHowdpzc57hQhA06KjJWVLiRSZwM02XCqDld0d5l8G2rAQBnqzo69LaxmhVP\ndYwW589fLvLuP6I3O/70QbFMLFiaq6F6iyYQQk/eOO+/Xj3hXZqBAHz8M5VS4Zhnl3hz9bKk\nFXOiL9R0dhrsseGy/PRwATOWd3CGRlcvS7p6WdKgR96+LqOiUV9c3emJ0BS6Z2NWagyPciES\n0DetTrtp9Th+MgQCIQghAgeBMKVwObhz38Dls9isQ4oIlLkMZa8GaqooBRhsldh6EVgTAAWC\ncCQrAGZcTNFGigNztQDdAByAAlHJAIM45Nud7Nt7Kj89VOs2M5OKmdvXpl+7PAWNovoIZW7G\nujpw9CuoRvGLICSpZwPzW74NsotACFaw3WZ64VfOiuKe7Ypi+9Fdko23STbdNqnjIowErY6/\nyA5j0OqsROBws+tkg393VQB4b1/V0lyNdyQrQf36k6s+2F9d1tDNslxGnHrd/NhPDtYeON/C\ncRghWJAZ+eDWHE3I+Jb/hCpEK+fGjOtLDAWxkP7jfYv3n2s6UeZO95BtWpyYHKWY7HERCIQg\ngggcBMLUwW5mv/w96JrdW9huxh2X0eWz1IbHpoTGgU1FYKvo3eLA2Ya7dyHlGhD6ZpZODliP\nuTMAntoQE2ZbETULUNQAJz330fnvzzV7Ni0210tfl1od7G1r00c+EmkYVfg4rtqNOyrAaQFZ\nJEoqRFFz+w5QBHjKFCkHamRDIAQrtl3v9qkbbjjO+vVbgux8JiUnwEmEIEUuEQTapZQG3DVD\ncLHYbbbq3f3Um+pmPcYY9dfI3dUr3pFf3pL3+A1zW7sskWrJaNMGpxoIwZq8WP+aFAKBQHBD\nBA4CYcrAndvhUTc84KYSXHkEZS6flCENA5fOS93wgLH5BBJeMwnj8QPjEi91ozfGlSI6DID/\nobxOa/JWNzx8sL/6uhUpo3roFMpQzjWBskBQ5Cxcs7fPc9QTT1wJQIxrCVMP+4n9vHHHqe99\nBA6Ww1a7a4Ap9BAxWBzfHK+vbTVKRUxuUsgVebEU6aQwRhSkRwgYyr+3aGy4LC5CPilDmnQw\nxrtPNX50oKapwywRMXNSwjiMeY9ECJXV67U6S3SYNDVGydABVy+EDJUQOUM/T8JMYUwbFRFm\nDkTgIBCmDLi+mD9eVzwFBA5nK3+cNQJrAnrSn9IsgHl77LkAdwHS8O2CsgYdb9zuZKubDblJ\n/B5pg4zD5vq+uLmh3aySCfPSwjLj1TwHUQJq3p1cycd9ZhyUACWvRvFLRvCKBMKkw3V38Me7\n2jw/N7SZXvq69GxVh9PFqeXCbUuTrl+VKhyREcCpivbfv3vWaOkxNdhRVP/5kcvP3rVQLReO\n4GoEH0KVors3ZP3rqxLvoIChfrp99mhq96Y0f/n4wrcne5yhTVbn0UutgZQLCqFHnj/i/jku\nQvbT7bPHzz2UQCAQpiVE4CBMDhgcHNYCWAFoBAoKRZCV58GxBehybzNO7DhGBOZrC9Kzi7+t\n/YSCeWqhewm4iwvsd+H2wB8upyra//xBcZex7xXXL4x/5NrZtP/asjScmn8/6BuwuQ0YCVIl\ngIgUIROmKpRCxel9M5IAACl7VMLqZsOjLxy1OVj3ZrfJ8ebuigu1Xb+/Z9Fw58wmq9Nb3XBT\n2aj/x2cXf3V7/khGT/Dj2uXJqTHK/3xXWdVkEAnonMSQO9dnxkWMWQGd3uxo6jCHKcXjbTwx\nJpTVd3vUDQ8uluPNc7E7Wc/Pje3mp149+cIjhfEzLFPD6eI+OVS793Rja5clXCVZMSfq5ivS\nJEIyZyEQCEOCfFkQJgEOd7G4GsBzF2/nsJahsgIVAhDcIEUYthp4dijGw6cT48566G4GsRKF\nJ4Jo1E9XdIDebIgGOghm5mgA37uAu3ht2wGAoVFy9LDfVKfB9j9vnfH04XOz60RDpFoSwNED\ngSoBqRKG+0IEQrAhmLfMfuAr/7gwr9D9w6s7yjzqhoczlR3HSlqX5g7kkuPP8dI2H3XDzdFL\nrSarc/TFLwQ3c1PD5qaOfeqBVmd94YtLx0q07s20WOWPr54VqKVokHCqop037mK5goyI0717\nJSLGavddCbA72Y8P1jx63RxPxF3aMo1zYZwu7mcvHS+p60mQbO40v7+/+sgl7f89tJT8exII\nhKFABA7CxOPsr24AAGAws/gyjUbhyzgDQOlLcVstTzxj6Ri/kkHLHX4Da6t6NgUSquBqlL1m\nVNcUxgIlBc7iGxclAwqGLyIJICVgf/1IACg00DkZIVx+gvRMve+b2rw4cQTPYfvONvuoG26+\nPl43KstSAiHokW6+w1VezLbWewdFy9YLsgsAAGN8zqsrpDdnKjuGK3Bou/y+hQAAgOVwu95G\nZlDBjMnqfOzFY+3dfV1aqpoMP3+56G8PLQ0kNwcDZht/AiPG8Nh1szkMDe2mUIXof98rrm3l\nWcOo7LUjPVfV+dbuiqpmPQKUEa/64VWZIyuEDHJ2nKj3qBseGtpMH35fc9eGTO+g0eJ8e2/F\nidJ2ndEeGyHbsiRx/YI4NI21HwKBMDSCYV5BmFlwWOejbvTGu2jEAhA/IT9YO244jPW14LKg\nyFjc1tS3C1EobzOKHdMuAy47u+svYPZKF3dauePvUYwYpY9CSUEMUq7BxgPAehXUCOOQbOEw\nLqJv5c59jturweVAIbFo1gYUO3vkQ/IZIMrF+Ez/ghQaUbn835OGNu7gW7ju/C+w8J+ygoPm\nnjQKmkJblibetymb55TBaGznL0HqMtgtNpdUTL6uCdMWJFcp/+t5295PnBeOc3odrYkVLd/s\nSd9wstjdhtkfq53nbjIw8sCNPBRE3Qhuvimq91Y33Nid7H/2Vv76joJJGdJQiAmT8sYlIiZE\nIWZo5C60CTwxRwDw1bG6f3x20RMqru589IWjv7h53vRrJnKirI03XlSq9RY4OvS2h/95pENv\nc29WNen/+vH5k2Vtv7q9wPuTPFfV+Z/vKqubDUIBlZMQcuf6zJlW70MgzEDIEzNhvMFc5Snc\nXAkuB4pMpLILMR3I0QBjcCCYAvW0E4pNxxUaBsWNAAAgAElEQVT/G+y9/eRCAYnDAYUDFoEq\nEqUvReGJY/uCuOZEP3WjF+78Djq5ANcex91NwIhQeAqKnzc85xQmFKm3gqMOu3SAGCTQgGAY\nS6+4tZzb9w/gepbCcHsN3v88NXcLmr1pGGMYACRH9FLM1QHoATgABaIS+etT7Bb2s9+BqQsA\nFJTjl+HHblVdqnGFCRZfm5mbMeKacImI/wuZopCQGIkTpjtIIJJsuEWy4Rb/XUKG0oRItDrf\nmS0AjGCusiAzgqKQv0tOWqwqXDVAqRph8im5zO/rfClAPEhYMSf6tZ1l/nkcVxbEuVvGuslK\nUNe08GRwZCeozTbXv78p89/1/BeXCmdHj8xqN2gx8VWQAYDR2i/++q5yj7rh4fDF1uOl2iU5\nPb7gnxyseenr0p59VvfetmfvXpCXFj7GgyYQCMHEtPpOJAQdVqPrvWfYz5/jTnzJndnF7nrJ\n9frjYOJzkQAAAEQ8OPzAVV/3qRtupEKQmqmVt1FLbh5zdQMAcGc9/w6Dlv3mGe7MR7jmKK7Y\nzx19lfvur+AwD+/qiAZRCpIVIOncYakbAMCdeNejbvQFz38DJv7c9RHBICoVUfmImo+ozEDu\nG/jSPre64SFBYFglqS007B+N412gR665KWHeD8EEwgxkNd8yNYXQqrnRw71UTJjs1ivSfIIi\nAf2Ta2aNcHCE0XG51fjB99UvfHHps8O1nQbfKas3rgDGzoHiQYJKJnzqtnyVrF+PnoVZkfds\nyvKO8DYXl4iY7StSLtZ28RYwGi3OsvqgFndGQFQof8JLdP9EmMCJHj3xLoP9tV3lPntdLPd/\nn1wI0KKXQCBME0gGB2EcYb99GTf2W3PA3W2w8124biuA7+0FgYL8QfrCOrCuEvunSWAWd5ai\nuMIxeAmXHSztIJCBpLeUd4D6VVs/cQp31HCn3qeW3j0GwxgUYzsYtDxxzOGWEpQ+oV1ycWsV\nf7ylcjSXXZgVuSAr4mRZPzs6kYC+d0QFLwTCdIJ3UZfD+HRFx6bFw/bZvX1dRma8+v191TWt\nBqmImZUceuf6zOgA0yrC+IExvL6r7KMDNWxvQs0buyp+dHXOVfPjeY9PjVb6fEP2xIPYgMPN\n/IyI159ctftU42WtUSERzEsNX5Dl6w4eFyH73/sW/98nFzx5HGmxykeunR0TJh1AxTBZA3co\nm5qsmx+3/1yzf9z7rwJj34QODwazw/3DmaoO/yY1ANDcaalvMyVqSKEKgTBtIfNJwrhh1nNV\np/zDuK6a6qY4tU/hNE1TSRMyrCmF0wyY49cbHAFaxg7j4hZcvgM3nezxZJdFoOxtKDwDRaTg\n0v08x/t3KgXAjcXgsIBw3CcG2MmTnd4DbxYJxuCwgmh8BoYDrBYGig8NhOA3d8z/+GDN54cv\n60x2hqby0sPu25RDnsMIMwqXtlX3/ju2ygpKJpPmFai334gEgjOV/H0oTpa3j0DgAICFWZEL\nsyJHN1LCaNlzuvH9/dXeEavD9dePL6REKdPjVP7Hb1qc+MWROv9chutXpo7jKMcIuURw7fLk\ngY/JTlC/8EhhfZtJq7NGhUoSIuVuy8xASQ0Q2OBj6jI/I+KOKzP+s7eS9aoj27o0cV1BnGcT\nIdCoJS18hsGeRI9ApS4AYLQ6xm68BAIh6CACB2G8wHotBMgCRDXtTMFSFpoxtiBgEFJSKA6B\nkPfgGY1ABogGzOeiJxrpgpW1A+vrgbPh6iNg9pozmNvx6ddg/t0oeT66sAvrmnxPFPB9XWAO\nmzvR+AscSBYKCPH/Rcn7rYPhrhbnt2+x1efAaUdyNbNgPbPsakCADa3AOpEqCoSy0Y4mPBEu\nn+MZZMRoK4YEDHXzmrSb16QZLU6pmKH5RCUCYRpj/G5Py6+e5Kw9gqZx907dB+8mvPR6oD4U\nZlvAOQwh+NlRxFMRyXF454kGXoFDEyJ59u4Fz310vqmjR9dWSAX3b85ZkDkevdInCBeLz1Z2\nNLSbVDLhrORQTYgkKUqRFNWv0Xh2gjpRo6jTGn3OzYhT7TvbfKqivdtkj4+Qb1uWONymQsHJ\nbWvTl+Zq9p9tbumyaEIky2ZF+XcCXjc/7q3dFT5BikJX5PeUs2lC+StGEYKokOmmChEIBG+I\nwEEYNwaY9IokCKkZUA/LoZIH7ARzFXbpECUBcQIIw0Z3ueCDFqLQDNxZ6htHNAobftkCZnH1\nN7jlFGAOWA7sfhMGzOGKXWjJT6irHuWK3se1vQk4slCUMAfXHeO9KhJMiC+sSI5iZ+PG875x\niRLF9lXOc20N9n//Euw9qzrY1O38/n1cf5ISs+CwAAAgCmWuouZtA0Y04rFQs9aw5/f0XNAL\nlL95xNf0QRG40QOBMF1hu3UtT//Co264cVyuaf3db2LSbtObeRZdY8NHrVcSJg+PTuFDQ4CW\nUgAwKzn0lcdXnK/pauowh6vEs5NDp3Rn34pG/R/fP1ff1vN+BQx10+q029am+5SKIoR+dXv+\nr18/1dzZ94nFRci6jPb39/eUTHbobWerOrYsSZwebjIp0cqU6IEWcm5clVrdbDhysdUTETDU\nA1tyPGflp4eHKcX+ri55aeHETnjKwLu0RiAMBvm7IYwXKCwWKcKw0c8AkqJRoldrT8yBRQdi\nBdDDzOCwNeCOb8Flgh4/DwTKeSh0xfD6egQ9KG0zNmvB5mVpiSiUtgnEvqsZg4Jrd+PmEz0b\nbACLLX0jcC6QqKhV98PCG7G+FUQypI4Gi46tL+IpwVBqQD5BbuTU4tu4ff/EXV4rfmIltfw+\nb6nCtfcdj7rhhlbLKMoInpkR5nDZPs7USa16cORDkYVQW3/G7fs3dPXmuUgU1LJbUfx0eKwk\nECYL0/f7ODPPjNd06ODGqx8ure/2iSOE1i/gN2sgTAkkIoZXt5IN2BWboan89PD89CnfCMNo\ncT716gnvT8Dp4t7eU6GUCbYtTfI5OCFS/srjK/acbqxo1FMIZSWoz1R27Dvrm2v51bG6VXNj\nZqeEjvfgJx0BQz19R8GJsrai0jad0R4XIbtqQby34ikS0E/dlvc/b53Rmfqa9yVqFE/cMHcy\nxksgECYOInAQxg2EqCt+yH75N+D6VVhQi7YhZTgAgMPClXyDLx8HzgUIoYh0NGc7Ug4tu5I1\n47avgPN+MMJgOAu0HFQFY/YWggGRipr/E9x0FLprscuGZJEodhnINH0HuLrBqQXsAiYUhIEb\nCnDOPnUDwN/ktS/uUTGkaiRV9/wsj0DZ63DJt/2OpWgq/8bhvqGRI1ZSG36Ba0/i9ipgHSgk\nDqUVQv/8Ebamf4oHQjRfkipuLMaddShs5BUlSJNK3/hb3FoJ+jaQhaCo1AkwIiEQpjfOtlb+\nHRy7MpapKUz+4uhlT3tXkYC+b3N2VoKa/xTCVCA/PZy3SqUgYwqXnAydvWcaefWdTw/W+gsc\nACBgqI2LEjYu6tl84ctLvJc9cql1JggcbgY205mVFPrak6u+OV5X2WQQC+jsRPWV8+NJVzIC\nYdpDBA7COEKlL0A3/4Y9+B5urgSORWEx1NLtVOYSAADWyR38Bzb0GmVjjNsq8Pd/oVc9BkPR\nOEyl/dWN3ssYi9E0EzgAgBKg+JUQv9KvlwqHjcfAUtanVgg0SL0KaIXvFQDApgPOq1g9kLmD\nNCxQKg01ewtWx+HS3VjfDLQAhaeiuduQKma472ZUIAqlLEIpi/j3chy4+v1VICENVIBm2B01\nMAqBAwCAolFMFsRkDX4kgUAYArQ6YGIaExr64NawtfmxR0u07d3WuAjZqrkxAzgvEqYEt61N\nP16i7TLavYPpcaoZkphTp+WvxGnpstgcrH/LWG+cLs4SwJim20QcNPuQiZkbVk0BD1oCgTCG\nEIGDML6gmHTmpl8D5oBjge4rlMX1J/rUDQ8uO1e6g1p016CXxc4u/h0uA2AXoBnxh41NJ8DS\n357DqcW6b1H4tQB+s3rU/1GJpgBx/p6dKHnlAK+I4vNQfB5gDlAA1WByoSgUEoW7WvoiA7S8\nDeCASyAQJgt54co2hsEu32mbZM48JiwMANLjVLzekyPDZHXWthhdHJccpVTLicv1JBCuEj//\nSOGrO8qOlmgtNpdKJly/IP6WtWkCJihvMWNNoFQChNCgWQYChlLLhbxaRqR6QoyxCAQCIViZ\nEfNAwuSDKKD7Pa/g9kreA3F71dAuGMhUjAKbESTqaebEwQN2gaWMJ+7qBnsjiPz6JkpCQagA\nR68HO0IgYsDhAk8bNlqAUtei+MWDv3RwqhsAAMDMv9K5+03PJna/Qd50ldB+HxHGuLxB39Bu\nClWIMuPVU9q1jjDhcOBqwFwXgAuQEjFJgEZuYTuTEcTERvzksba//tE7SCuUUU/9ZmxfiOPw\nu/uqPthfbXeyAEBTaMvSxLs3ZIkEA62ZE8aDMKX4yZvmAYDF5pIOaL0x/ZidHPrl0Tr/eHaC\nmqEHv8+uyYv99FCtT5Ci0Oq8ic2snMpwHK5pMWp1lgi1JCVaMZSPnUAgBD8z615CCCJY/tRK\nYIfU8w9J4rGx2D+OjWbuzG+AEVMZa1D6Gu+ckekGawQc4DN06XgEDkAo+Upc/klfgEIgFoA8\nAYXPAqEMhSSPvPVs0MAs2YJ1Wtfp3cBxAAAcZs0srfD9okORaSiyL2e1ttX43IfFFY1696ZC\nKrhnY/aGhTMiR5owWrANO44D52nf2IrZWiTIBzpgWThhAELvuEs8a07X66/YqyooqUySXxB+\n/4+Z8DF2ZHh1Z9lHB2o8myyHPz98WWe0P3Vr/ti+EGHozDR1AwAKZ0dnxteWN/Rzz2VodOf6\nzKGc/oMrMyoa9Rdr+xJaaQrduyk7OYqvTHXG0NZtPVPR0WW0x4bLFmZHSIQB/67KG7r/+vGF\nmhaDezM2XPbwtbPy0qa8eS2BQJhxtxNCsKDUQMsF//BQTUalqSCOA1tjvyCHoUkHAOCycSU7\nUHcDtfie0Y80WBlgnYF/F9LkAS3E1TvB3g0AQDEorhAlrARqGslAFCXYfB+dfwVXfQ4bdSgi\njs5disv24rLvgGMBMABCCXnUwls8OT5Gi/PnLx/3TvQ1Wpx//fi8XMIsnx3YtJVAAAAA7Dzv\npW70hLDzDKKuCJxoRhgIaf58af788bu+yer87PBl//iB4pbb15kSIuXj99LBzKELLSfK2ruM\nttgw2fqF8QN36CSMCTSFfn/Pwtd3lX9TVO92z02OUjx0de6clCH1vJeImOceWLz3TNOp8na9\n2REXIdu8ODFpZqsb7++vemdPpcPV45UerhI/dv2c+Xyeta1dlidfLrLa+xaKmjrM//3ayb//\neFlqDPnjJxCmNkTgIEwOVNJStuqAf74GSl0xtAsgpNmGu0+C8RxwDsCATTZo7MTWvmkqbr6A\nO6pQeNrYjTqYYJRASYGz8OwK3EsFheei8Nwew1FJmK8xx3SBikmlYvoSNFD+tZC1BnfUAucE\ndRxS90vf3XWygbeM+f391UTgIAwCdgDbxhd3AqcFOm7CB0QYnJoWg4v163gNAADlDd0zUOBw\nuLhn3jp1sqzdvXkS2r86VveDqzJvWk2sGccduUTwk2tm3b8lp6nDrJIKQ5XDq25DCK0riFtX\nQL5qAAD2nml6bWe5d6RDb3vmzdOvPL7C35D408O13uqGG6eL+/D76l/ekje+AyUQCOMMKTYj\nTBKyMGrxXSD2kskphsrZhBIWDPUKSIBClqKEH6HIm7jiy7iyxVvd6GGIjh5TEoQUfJ+VOAUE\ngyVYikNAGjld1Q1+pGqUkIeSFvqoGwBQ3WzgPaOm2YCJESlhYLAtYMdlbJ3YoRCGygD/1tyM\n/Jf/YH+VR91ww3L4tZ1lpfXdgU4hjC1ChkqOUgxX3SD48PlhX0cSALA72R1FDf7xyt6iVB/K\nG/jjBAJhCkEyOAiTBtLk0Ff+N265iE1tSKxCkZkgG1Japh/CPqfM/mDWMZ29RiXpCDHYWASs\nCQAAMSCbjWTzJntYUww6QMdchND0t6oljJKBilBIV44gJSVaydDIxfLcNTLi1BM/nknnuzNN\nvPFvjtcfLtVWNRucLI4Pl25ZlJBG6lYIQUygtrt1WqN/EAW4vw/Qe41AIEwViMBBmFQYEYov\nGO3dRKwCgQScPOulSDndSwzEyUicDKwJsAsYJcnJGgFZCeo9pxt54+RBhzAISAKUCjj/FT+K\nmIwGLQqpYPOSxM/9bDiW5kbNTHfGtm4bb/zwpdaQkJ6GozqT/fxl3W2rU6+YSzp0EIIUoYBy\n90Xyj/sHM+JVFy93+ccz42eiyhm8kM5WhBFBpkOEqQ9Fo+RlPHGJCsXMGfbVHGZc9i1X9Dp3\n6m1ccxg4nptl0EHLgVGTf+eRsa4gLibMtzqXptAd6zImZTyEqQUSzPFfKkCCLECSSRkPYSjc\ntynnhlWpnpaQCKGrFsT//Ka5kzuqyUIlC5Bt5CfxfnioVm/mcSwiEIKBQOasvPFrCpNlfo17\nhAx1I7GeIRCmPiSDgzAdoHI2cg4zvnysZxsDKDXUwh8AM7yKVtxewRW9Do4e505cfwqqD9LL\nHgRpyNgOmBA8iIX0n+5f/PwXl45e0roj0aHSh67OnZc2soIpwgyDUiPxKuwsB64LwAVIiQTp\nQJE/nqCGodE9G7OuXZ5c1aR3sVxqjEoTMnMFqSU5mq+P1/nHJWLftVOHi7tY370smz87iePw\n+ZquhnaTSibMTQoJU4rHfqwEQmDuuDLjTEWH1dHPOjQ5SnHVAp6m75oQyR/vW/zXTy5UNfWk\n4MVHyh++ZtbMTOMiEKYZROAgTAjYAawZaPl49U2kaCr/Jpy6HDqqwGEFVTSKmgXUMBPbXHbu\nxFsedQMAAAMYtdyZ96jCH43teAlBRYRa8psfzO/Q2xrbzSEKYVyEPJAxB4HAA5IgIfG+CQ6w\nDcBdb68ANMgEO1QhWphFKongjiszzlR2NHeavYMSiUAs5rlfm6y+vc/cVDcb/vRBcU1Lj2ez\nSEDfujbtptXTtIsZIShJjlL89aElL35ZUlzdCQAMja6cH3/Xhkwhw5/fmh6nev7hZbWtxtYu\na6RanBytJLd+AmF6QAQOwjjD6rHpBDhbejaF8Ui2AOhx6cOHVLGgih3x6VhbBvb+TlQIAAC3\nVYBND2LV6EZHCHbCVeJwFVlyJBCmKCxmy4Fr6O1rg4BKQHQmqd0bFLVc+OKjhe/vqz5R1tZp\nsMWFy1fNi/msqJ734DC+Th8mq/O//n1CZ7J7InYn+9rOcoVEuGlxwniNm0DwIyVa+af7F1vt\nrk6DPSpUytCDCBYIoZRoZQpxzyUQphdE4CCMBgyOOsx2AHYiWg3CVED9S3lZI+7eCdirZNfR\ngF2dSL0FqOBrh2bVBdiBsUWHhiVwcE6sqwJrBwjkSJ0CIiKOEAiTCwZsAbABSIk7xrQEs5eA\na/YOAFeHgUX0rEkb09RBImTuXJ955/pMT6S0WV/m10dTKRXMTuQp2Nx7pslb3fDw0YEaInAQ\nJh6JiImLIBMcAmHmQv7/CSMF27D5ILDdPVvOBrCXI+kSYDR9h1gv9lM33HAWbCtB0rwJG6kX\neKDGnwJfp0kPSBhwF89rdNfiik/B1iOXYEqAElej+BVDvwKBQBhLcBdmSwD3dhBEoYjOBSSb\n1DERxhRs7a9u9MI1ApU2aK0KwZ+7rsx47tOL2u6+9mQyEXP/hiwRX1ODy608bTgBoLnTbHOw\nYiHpg0AYLTYH+9GB6lPl7XqzMy5Ctm1Z0oLMiMkeFIFACFKIwEEYIdh62qNu9IYc2HIcKTb2\nGW042/hPDhQfJzCLtcdx+zlw6EAgR6oMFLsKGF/NAmmygGKAc/mertCAfMh12nYDvvQ2sF6y\nDufEtbtBqECaSdF0CIQZh8nqbGw3hyhEkWoJAgN2nQLg+nbjLuwqQoJCgADNI3hxdoGzEygx\nCCOAIhPmIAMbBtpFBI7AVDcbvj3V0NJpCVOKl+RoFvUaiEYoxb+9PX//hdaqZoOT5RLCZVfM\ni1FI+F20AjkXIBRw10wAYzheqi2t62Y5LjVGtWJO9KAVEwReugz2x1482tzZY5HW3Gk+UdZ2\n7fLkB7bkTO7ACARCcEIEDsKIwA5w8i2XYTu4WkDgSUnleI4BABwgPh5gjqt4B4y95cQOI24/\njfWVVPZdIOjvlS1WUrO2cuc/7RekBVTejcN4tdZT/dQNT7zpKBE4CITxpstg/9fXJd+f6/l2\nStQo/nSvSCXx/8JxYK4eUUNzQGRNuHMf2HrbTFBCpFoICvLvTJjyvLO38p29lRzndi2BHUX1\ny2dH/fKWfPc8nKGpdfNi1s2LGfQ6uUmhXx3j6cOSGa8WBPB3nPboTPZn3jxdUtdX+vrevsqn\n75gfF0Fyx4bNqzvLPOqGh08P1a6YE53DVzM1SjD2b5FMGIhDhw698847Bw8erK+vDwkJWbBg\nwdNPPz1vHvHeJkwaM/TGQxgtnKXXyM1/l5cTO63mP4YJEB8HcOeFPnXDg8OAmw/6H4zSVlIr\nHkaaLBDKQBqK4gvotb9A4cNpim4JkJxibgv4iREIhLHA7mSfeOmYR90AgDqt0eUK4K3DdfPH\nfcAsbvuiT90AAM6BdYdBfwJcHYD5O0oQJhoUyOcItegERgv5NfFwvqbzrd0VHnXDzaELrZ8e\nqmnutHQabEO/1Io50Wmxvr8CmkLeph4zjT+9X+ytbgBAndb0zFunWW7ingQ4Dte0GI6XaGtb\njRhP4SeQwxdb+eMX+OMjg8O4Xm892dR9pL6rqFFX1Wl2shO4GjeV+d///d9jx47dcMMNL7/8\n8sMPP3zy5MlFixYdPnx4ssdFmLmQDA7CiECBU7u9diFJNnY0+B1BIXHWuIyKF0MVbxjrq3gF\nehSeisIfHPnLoQD/UxQ9kP0HgUAYNXtONza2m32Cgf/rhva4b60BZxfPyYaT4CwDQCDJQtJ5\n49UAmzAgNgeLEIgENCAxUHHANfoc8M0J/PcvjgJAWqzqR9tyZiWFTsYwg5TvzjTxxl/bWfbv\nHWUAoAmR3Lspe8Wc6EEvxdDo9/cs/PeOst2nGt0T6bgI2Y+vnpWXFj62Y54qaHXWUxXt/vE6\nrfHS5a45KWETMIaLl7v+/ulFjz1KaozykWtnZyVM3PLSWGF3sla7X+0wAADwWtuODIzhgtZo\n7H0hJ4tbTfYuq3NetFJIk8XgQfjb3/6WltaXEXnddddlZWX98Y9/LCwsnMRREWYyROAgjAhK\nCrQKWF+LdQAKmKi+LUEUUhRi00nAvTchSoLki4HxSil0dmDDWXB2AhKCOAYp8oEaTmH8oLgC\n3P/YYSxPDQNVErSd8w8jVfK4vByBQOil5DJPskZ1M4TyriIHXPPvB3bwzFIAADAHmANEgbUU\nswakvGLo4yQMHdxcjk98itvrACGkSUGLr0cRSQBw6ELLm99WNLSbEEJJGsVdGzIXZuVgEABX\n5y6NdLHw8WHu7e96ZKyqJv2TLx3/w72LJmZuOSVo6+a/CXoyDLQ662/fOfPT7bM3Lhq8E4pK\nJnz8+jkPbslpaDep5aJItWQmJ/k3dfgqrR4a281j+EfIcvh0RXud1iQTM3NSwjz1L3Va0y9f\nOWF3sp4jq5sNP3+l6MWfLo8JG4ZpejAgEtAKKX8eVoRqzFpiac12o5+M4mC5er01LZRUFQ2C\nt7oBACkpKUlJSc3NfJXsw4XP1ZhAGBQicBCGicsKtBgQQpICbD4AmPXeicS5QPW/E4hSkDAW\nHK3AmYGWgyC631Kn6SLuOtBn1WFvwqYSpLl2LGtYxCHAaz8n+v/snWdgG9eVts+ZQa/svYlN\nFNV7b5blJncpjktkJ3FJ7MS72cTe/bzrbDabzSZx4t3YzqbHJXbcLZe4F8mSZfXeSIoUKfZO\nAkQHZuZ8P0iCKAOKBSRY7vPHxpnBzKUAzMx97znvGZOlPExdQE37wdEaFOWUmHf5WJyOwWD4\nEeRyv1/9QlpUxIWtwCmQG2L3yiHM0ryN4GsFZeql92QMBzq7S/r8mYGXdaep/ix35XdeqYt/\n+oOKviBRdXPPo08f/t7WuUtm5ryzz+P1WVxe4UiFp8sGgVn5gkhPf1Dx6++sGue/YsJi0g0p\n7ejpDyquWJI9RHdMnUYxM3vy5QhEHZ064tO1NvKmQM7UdL3waWVVY49CgbNy4r9+5czcVEPI\nPtXNPb946URNf44Gx+H1q3K/fW0px+Hru6sD1Y1eXB7hjT3VD940+Ronb1yQ8c6+UJMXRNyw\n4NLpRUPE4pIvZOuOEGcMQmNj48WLF7dv3x7rgTCmL0zgYAwN0UsXP6fGQ+BzAKfEpGIsvAYN\nV5L7NIgdQALwcaieFZS+4QfVoM6VO6aduveEGpGKDur6HFNujNbAMXE+tR0NT0fHpLHxCOQU\n3Ly7qeZjaj3W56VqyuYKrgVD1G7DDAZDlvx0U6ABRy+nauiZT7T3XCUN5JGhAfk5Q+ysgepU\n+VIW5AADVBOhjQkcUcbjlL58KTRIkvj5sy9duCZ89z++V/bMx+e9A5M6TqMGj0+UxIEPsKyu\n2ydI09b2MoSVpam7wn4v4fQ4vRdbbIWZpnEY0pShMNNk1qusjlDHcQXPzR9C+sY7+2p/89YZ\n/8svz7QcLGv98deXBjZGdXmFR58+3GEdSMORJHpr70WjVrl9c3FFg7zHUEX90LyHJhhfv3Jm\nRb01cPAch3dfXZKfHrWvpaw+DgDj6ZkyNZAk6d5771WpVI888sgld+7o6KitlfEn7mVSG8cw\nYgsTOBgAAOCsIns5CD2gMKK+CPTBHhkkScf/AtZ+q07JR21nqauaW/oA6laO/KSumpAEkD7c\nDSC5o9aIUZ+BOVdRwyeB/V8xeRGmLI7O8cNR6rD4Riy8FlydoDaBImoplAwGYxCuWpr92u4L\n4ZnMxdlFqEgFsgC5AfWA5mEY4mhngCoFvGHmwUp10Ev2HBZtqKkcfDI1FOi2zeA7zgmh5g4S\nkTdsyVql4N3iwJWfCJjA4Wf9/PSdJzjhtncAACAASURBVJoOnGu95J5eQe5OzYiMgue+dV3p\nYy+H1qt+7fKiBJNa9i1+rA7vn94rCwkKIj3xxunnH9mI/ZU/e0+3BKobft7+svaOTUWRLkiT\n9Dpl0Cqf+M6qDw/XHz3f3mXz5KYar1uZW5ARTdFNo+RArmZLo2AlEsOAiB544IGPP/74lVde\nCalbkeWee+55++23x2FgjOkGEzgYRO0fgqMv3Re8beS8APZyTLnevz5JLScG1A0/gouqP8E5\nt438xGKkIlUC0Rk1gQMAU5aguYC6zoC7C5RGjCsGQ1a0Dh4RTgF6tqLLYIwfcQbVf9+97Fev\nnqpt7cvZ1mkU37hq5vr5GQAAmDgin1/ElOupew84zvsDoNIAH2wVpIh+q8Lpjje0K6QfAxe6\nMM5xiHKWD4jAc+hfg02N1+o07LGnD0T88V2LPzhU/+Hh+qYOp16raA7rxAkACh5zUkKLIxiX\n5PJFmUkmzV8+KK9qtEoEOSn67ZuLh+LYeqKqM7y6BADaLK7qZpt/Vu93Dw2hx+ntsnmKs8z+\ny2AgM7MmawERx+E1y3OGYgczMtIM6habjGVbmvESghTDDxHdf//9f/7zn//6179u3bp1KG95\n7bXX7HZ7pK3p6Sz3mTFC2J1+2mMvH1A3/Lgugu0UmPpbWHdFaETSWTkaEzHk9REWExD4aJtg\nqeMxfW2Uj8lgMCYYM7Pjfv9Pa09Xd9W32+ON6rkzEsz6UZsWc1pMvBLiVoOvi7x14L0QmgDC\nx4Eqc7RnYYRgTI60pUUInW/jINoVon/d+qY1zOw5CMSBSaMk0Xee3HuhKdSz6ool2QbtgFsH\nEZyu6bzYYtdpFKW58ZPOsXI8WVCY+NSDqwVREiVSD9kr0R7Z9KHHOSDtDZKIpFRw29bn7z7Z\n5BWCSoA1Kn7rOvYTkMegUhQm6Ku7nVJ/9gsCpBs1aQYmcAwJIrrvvvuefvrp55577vbbbx/i\nu5RKZXx8xOUBWdmawRgKTOCY7pCzUj7uqEC/wCGGLpddIj5EtDMAv5CpUtFkRzF9g8FgTCt4\nDhcUJi4ojHazDN4AvAE12eTQgqtswDxImYbG1QCs6iHKYFohxKWDpTk0nlqoFrOhIaiHF0VO\nve+t4lbwuG1dARM4BoHj8D/uWvKLl06cuTjQF3nTosz7ry/1v2zscPzylZPnavvaFfEc3rhm\nxr3XlHAcm4dERMFzw6pySE2IWNZqcXibOp3pCVpEnJef+OJnMotPeWlGs15l1qt+eveyJ3ac\n9nfOzk01fG/rvMwk1hAkImlGdbxW2e70unwiSGSzeXwOr9ug1qhYlcolIKJ77rnn2WeffeaZ\nZ772ta/FejgMBhM4GJHqRALjugit7PWja3HPGzB+A3Xv6jPj9AcTNozqsAwGgzFWIOoXg7YE\nfO1AXlAkgGJ0l0FGJDieu+q70nv/C7aOgWBcOnfFt38oGP7z+aNVjQO5BiVZcQ6Pry3MkiBO\nr7pqY068XjW/MDE9geUaXILUeO3j9688Vd1Z2WjVqPhZOfGBNgc+QXr06cOBDVBFid7YU61R\n8XddURyL8U5N5ucnpsRp2yyu8E0/+9txACjMNP3DTXMXFiYtnZl8uCKojzXP4b3XzOo7TkHi\nH7+/7kJTT0uXKz1Rl59uGmIrnOmMWsElqhV//qzqvYN1kkQAYNAq79xcfMPqPJZMMAjf//73\nn3766Ztvvlmn073++uu9QZ1Od801MobQDMY4wASOaU+kYhB+QObHjCVUvxek0FQLzFwx8EJw\nk6MJfE7UJoFerpeKLIZSVKeR7Th4O4BTgzoDTQuD+sgyGAzGRIPTgzq6C6FOonoAB4ACMQ4g\ni6WEAAAmZvO3/5zO7ab2i8DxmDIDS9YCr0gD+M2Da74821LZYEXEWTlxy2elVjRYfvLCscD0\nfoNW+chtC0tyJqvpQExAhPkFifMLZBKgDpS1Bqobft7aW3PHpiI2eY4WSgX3b19b9OPnjnTJ\nWUIAQFVjz8N/OPCbf1jz73cu/tunVW/uren17MhNNT5wQ+nCwgHJVcFzM7PjplXj3vo2+7na\nbpdHzEszzi9IHIEq8dO/HT9UPuAqbXf5fvvOWZ8ofWV9fjQHOrXYv38/AOzYsWPHjh3+YGZm\nZkNDQ+wGxZjWMIFjuoO6InLWyMT1AQsyuiScfSuVvwm+fgcy5DB7FWYt731FLYeoficIbgAg\nADTPwPzrQJMwpBEoEzBh06j+BgaDwZgY1Lbanv3ofEW9RZKoMNO8fXPREGYXzUQV/poXog6A\nRsRFAKz2G0ChwnmbwycpHIdr56avnTtgQVeSHfe7f1zz9r7aykYrEBRmmm5cnWfSDdWBhUBy\nCVaRPATEo0rLmzlkD0hB1DTLu1o63EKbxZmRyGofosasnLinH97w/qG6ygbrxVZb+L+8xye+\nvKvqX25d8M2rZ951ZXFzp9OoU0bBb2gyI4j0u3fO+jMvAGBWTty/3LZgWN/M8jpLoLrh58XP\nKm9ak6fgme4sz4EDB2I9BAYjCHb/nvYYZoGrBhzBThzaXDDOCwxgyhyMn0Gtp8HZAWoTJhaD\noS9Ng9qOU837gTuTtYbKXuDm3w8cy8WYQEgSfXCo/kBZa7fNk5Gk37I8R3aZjsFgjIz951r/\n869H/T07DpW3HTnf/oOvzNu8eJC2TZ5AdaMfF1EF4jz5dzAiYNKptl9eNII3iuTt8bVSvyGU\nAB6vaNcrk1VcX4YjEdldglE3re9og8zu2MQv6ug0im3r8gHg4T/ITx3LApxQspKZugRPf1D+\n9/21gZGyOsujTx/+4/fXDf37WV5nkY073EJdmz0/PZqNaRkMxtjBBA4GYvIW0J0nRzn4rKAw\nor4YDLNC2wQAgFKPWSvC30+NX8gc1d1FHWcwZeEYDJgxElwe4ZE/H/Kbw51vsH5+omnruvxv\nXTsrtgNjMGKM4POdOyy2NnAGk6J4AZc45Aq7kMOI9MSO0351oxdJot++fXb17DSdRtGb3xb2\nvvYwdaOXTgARgJnbjQd2XweRGPgBEZDD16FUZ7V0uf/0btnhinaPTzTqlFuW59y2qVCrmo7P\nTrPz5JsdpMZrk80RfTEZoyTkknLJ+PTEK0jvHqgNjze0Ow6Vt62aPdSrur+Fiswm2es0Y6xR\nTseLLWP0sO8NAwAA9MVBNSlDR/SAu0t+k6MZgAkcE4UXP6vyqxt+3thTvbwkJfr9JhiMSYJQ\nfc75wq+kjv5WHbxCs2mbZsudMPzS7cpGa1ePTM281+frqP48S1UNXiso9GgsxJS1wPdNCInk\ny+wBCMADwHwxxxyRfCL1dgQL+tAJpIutXT/47YCvh83pe3nXhZMXOh+/f9U0tJyYX5AY7moJ\nAKtK05j/4thRmGE6UyPzlFWYaR7/wQwCEX18tOHTo41Nnc5ks2bt3LQbVs8Yt59JS5fT7Q1r\nyQcAADXNtqELHIWZ8jkaGhWfncLSZBiMSQPLKpzmSCDZgEbR7XWw5xr2yBM7RAe4L4DzDHga\ne9eHd58K7bbYy+cnm8JiFNTXhsGYopDN4vjDvw+oGwAgCu6PX/Z88fcRHK3HIXMhVXD08ysv\nZsEx8FoACAQ7dZ+Qqp8Foc+sETFS2TwCTOuK+nGDwluV9/P8J9WBrqW9lNVZPjs+TZ3zfrh9\n8Y2r80Ju7m9+WfPzl45HXvlmjIobVueplaGZXDyHE8rzUpLo35898virp05e6Gy3uM7Vdv/h\n3bJ/+u2+SKJD1FEqIk5nBtkUztwZiXPyZPzjbl47I/xTYDAYExYmcExXyEfeU+R4l1yfkfN9\ncu0CsXMkx+FUoEuR32QcpOw8alD3Gap+SSr7nXThb9RxOGYzcxLDu8zEDPtx6niVLLuo5wB1\nf0Adb4Kvo6sntIFiL52BcU8ztb5B9b+jht9Ry8vgqh6nATMYscB7+DNyyXSF8Ox+ewRHS42X\nydK/vNAyO8UZGvXZqH1f/4ukCFpwPEuxHB8QZectBACnL8gX5B+v7JCNT3k0Kl6p4Hr/cQLZ\nebxp/7mWmAxpypOZpP/p3UszkwbSBxJNmke/tqg0V75iKCZ8drzxYFmoN2dFveW13eP0FJEW\nr0sya2Q3zc0fRo4qIvzorsWB7sVKBXfbZYV3bmaNkBmMyQR7fpqeELn3gRRQsCBZyf0lalYD\nP+xqBczaQOdf9R+572Fdl4qJpdEY6iAQXdxB1oq+V95ustdS9zmu4PbxNDelziqq/BB6GoEI\nDKlYdAWmzI7mCXwuaikDewfoEzBlJmiMwaf3hXbVdZ4j+9GgiNBN3R/FG9Nau2U0jkRT/zOB\ns5I6PhzY4G2n9vcwbhWYFkfjz2Aw5JBsINkA1cCZYdybVogtdfKDam8inxeVw0ugyEszFmaa\nqxqtgcElmXbZncle3a9qaBELiYJtnkGFyJ6nxwkelTyqxNBMRkTgXB552TpSfDrw5Rl5IWPv\n6ZahFwIwhsW8/MQ//WDdudru5i5nsllbmhuvUU2sbIIvz7RGiLds3zwS39/hggj3XFPy85dO\nhMTXzk2bNcxG0Wa96ofbF9W12aubetQqfmZWXIKJNbRiMCYZTOCYlghNQepGHxL5ziG/drgH\nw8RSKNpGtR+B19arbmBiKeZdDfLLYlGDus8OqBt+nI3UfhBT14z8uN5m8LURCahIAE3u4FlO\n1HiETr868NrWTMeeg5nX4IwNIx9A4PHrj0tHXwFP/xxJqeUW3owzVoLooJ6D4KkFyQucDvQl\naFjYOz8kxxmZA0mudbNUr+2TETjWzetdqZCoe4/MAKwHUT8L+IlhBOC0kL0dNUYwpgCy7LNJ\njuQkzzEQ+h+LUY3quaDMHc8hoCKCEsrxyI/k5vjIbQv+9S+HWrtd/kicNkLivhRovZGNaCaq\nA7ADKADiEPPY3Xk80SuTbN4WCnJ7Rb0yMSvZUN3cE77/UJpWWB3e8jqLxeHJTNLPzo3HqWJT\nYZErxQKAbvsoal0Zl0LBc/PyE+cNJxlhPLE65L2ELBHiY8FlCzM1Kv5375zrvQKrlfy29fm3\nXVY4sqPlpBhyUgxRHSCDwRg/2CPUdISkCOm1YheANILCJUyagwkl4GojnxN1yaAaF+8r63nZ\nMFnKRyhwkI96doOnvu8VADjMaN4IigiJoJJA5TK1+lT5MWYuBdVo/aiou17a/0xQ0Y3PJR36\nG6fRAp4CqX8SJTnBdow8DZh4A4AEoszjOADcvhpO1JkrG4KWl69flbuoKAkAwNsBYlgiPQCQ\nCJ4G0MV6MdnRJR17lZr6tRtjMrfoFkxj/V8mLyK5vgApILuBPOQ+gsiDYjxK23rhC+fC3vfC\n44qC2cCNREHLTjH8+aH17x+sK6+zSERFmeZZhTz0nJLZVRVyVTEhzhnBGRmD0NXjOXK+vd3i\nSk/ULytJNmgjZvYpUBWnynSJVoE8QMRzKg1v5lG5ZUXOU2+GSsYKnrt6Wc7gp35nX+0zH5Y7\n3ELvy8JM80O3zJsaPSaTTJo6t0xeUnKcfIEAYzqQZNYChC+bQUrcuLbXWTU7bdXstDaLy+0V\nMxL109AJmMFg9MIEjmlJRKMKCq+tHSqcAvQZ43kzIUGmeB4AIFL8kge07ferG32IVrJ+hgk3\nyWejWBvA55KJSwJ1V2Pq3JENY2A8lXtkPykqfw9nJoVGvW3gOg/aYgCU/RB1av6J76x8d3/t\ngbLWzh53VrLhmuU5S2cm94858uLbIJvGB8Et7noCHAEeMbZ2ac/vuI3/gMkjXJwZb0gCdxfw\nGlCxFSEAAPDVB6kb/ZCnDMdR4FAtWOPNKxEulgdFeYXm2q+P+JhqJX/TmhkDr106SU7gwLjR\nXh+mFYIovbn34idHG5o7nclxmjVz0m/bVDB4r9a/76/983vlLm+fxGDWqx68aU5/wpoMiLxO\nEWoueO2K3OZO5469NVJ/S06jTvm9rXMHz+DYebzxN28FySJVjdZH/nToLw+vH0RkmSxsXJDx\n3McyqwuXLcgc/8EwJggbF2TsljEsj+a3wuUVapptLo+Ql2YcKK2VY5xVFQaDMQFhAsd0BDmT\nvIyBeoCJVdg5CKg0yv8VqhGtkpEX3DUycdEG3kZQy6zXkSBv2wkAIEQjLbNHvu8J2boRwgQO\nAPLUo64ElMngC/X6AgBUpSl4vHFN3o1r8mQOqoicdKMYXv1q1KHqA0HqRl9UorMf4IYHYzGi\n4SD5qG43NXwJkg8AQJuEBddgQqwzYmINiTJrfQAAUg+AOH5XIY7X3/9f7vef9+x9D0QBAPjc\nmdqt31bklUTtFNp0TLucWj8HEvwxjF+I8QuidoqpjiBK//zHg/5OmQ3tjpd3Ve090/zkd1dH\n0gsOlbeFZF5YHd6fvXg8K1k/rDQKRLjv2llXLMk6XNHe2ePOStavnZtu1l/CnOXVz2WMFbvt\nno+PNNy8dkb4psnFVzYUnLnYffR8ULPYr24oYO3GpzOrZqfeuCbvrb0XA4Nr56Zdtyo6VYdv\n7b3410/O97Y0QsQrFmd967pZU0AuZDAYYwQTOKYlimzwVYR3h0VlQUyGM0LiSsFyLjyMcSMy\nNxVtABESW0SrbBj1yZHSXVAnI0AMm0hWqZHyZCQfAKBxCXV9GPq3KJNBkzfYuRRG0OSAO8xz\nURkPmoyhDHbsoM6L8vEOOUFqgkFlr1BnQIKAq4PO/BVKb8eksbbgnbRQ5G/4GIBavXbrt7U3\n3iN2NHPGeNRFP8UGExahIZ96ysDTBUojGotAGzGPgBHOR4cb/OqGn4Z2x8u7LtxzTQkRHCpv\nO99gBaDirLhlJSmI8PaXF8OPI0r0zr7a720ddu5MXpoxL8146f0AAECSqKbFJrspxIB2kqJS\ncP9997LPTzbtO9vaaXVnJuuvWpo9O28CdfRgxIQHrp+9Zk7ap8caGzscKXHatXPTomU6+8ae\n6j+8W+Z/SUQfHalv6XY+dt+KqeJsw2AwogwTOKYlqELNSvIcDcgS50BZDMoJ1Fb9kqC5GJIW\nU0dQxxA0FWLS0hEdLvJSQKTmDrpETCykzqrQuDEd4qKxapFSCO2VYVHC+AgLZb1ZGKoMTLiK\nevaB0NvgEEE3Ew1LL2mtgomXU/t74A3wQleYMenq2DeTHqyiamJjvRikbvRDNR9Oc4ED+Xjy\nyW3gTGNtTiwPr+BTs8fw+Ko4TFo5hsef0hwsl0lJA4CDZW03rs77rxeOnasdSAgqzY3/4fZF\nta3yzWtqW+WlhyiCCByCKHdx4kdk7DIBQYSNCzI2Loix9s2YaIyFDaoo0Ys7w56yAE5e6DxV\n3Tm/gOUNMRgMGZjAMV3h4lF7GYhtfT0a+STAseyUIYngsYPWFN3FWcy8EswzoesUeS2oNIF5\nJsaN1HiSNwGvBzHcvwNBFfExDufdSseeA2uAc4chlVu4HaKxrMAVbxBr9oPTEhRVG7lZl5Pn\nWPjuqOvPq1dlYNI2EB0guUEx5O6bvB7TvgLOKvK0AoioSgFdcWxmm8FgfBbVh/+9AIY04cRu\n6mxGUyJfvBDN0ciaiSpkvSi/wdUFnh5QTwXHwRGizAZvRbgNB6qZcSwjFLtTVgwDm8v73y8e\nD1Q3AOBcbfd/v3hcpZSXEtTKMb+gIWJJTtzZizJFWKUszYHBGCZNHQ5bhCtARb2FCRxTn7G/\naDOmJEzgmM5wwKcBP8aN6+0d0okd1HwWJBEUKpyxkpuzBVRRE1PQkAeGvKioJmhYTtadoVFt\nCfCR/SnUJm7Fd6ntDFjqgSQwZWL6/Kh1MFXp+U0/kE7soIYTQASAmF7KLdwKxhToEcB+eiCF\nAZUYtwaUwXd6Xg/8cDu5IOiKUDceXeuHDuavgoqdA71yAQBAsrp8lSfJua/vtVKt2nyHcs0N\nMRjfIEhieEx0eNzn26Qzv+YS09ULV6qKp2fvDB61a2XaxI6jwyhjspCWoD1zUSaeYFCHl64A\nwOnqrrVz0xvaZdymx6fL5p1XFP/rnw+JUlAWR06KYdMiZsPJYAyPQRI1pQmfxMlgMGIFEzgY\nY4m9Q/zkMfD29x8VvFS5W2qr5DY/DPzEc4dS52L81WQ/DL5OAAJej7r5oL2UHyQips6FUfdM\nkUcXz626G0QvOLpAFw8Kdd85TStBWwyeOhIdqDCDpgD4MM1IdJDjFPg6ADhQpaB+PnCX8Mab\noKgN3Ibv0qG/UXdfpowkKr11jSAFNtD1eN9/GuNTFLMnUiGAPiUk4K5o7dlTST4RoBoA7G/+\nVbdxi/nefx5ZU9LJDadD7RqQ7CD1AKqBG3KqEWOasXlJ1qfHGsPjJbnxVU3yXbEXFiWevNDZ\n4wzymUpP0N2wOm/o5xUlqm7qae5ypsRpCzJMSsVQf6QLC5N+8s2lv3nzbFNnn8iyfn76t68r\nVQ35CAwGo5eMRJ1eo/B3XA6kOCvy4hODwZjesAdKxhginX1/QN3oh6xNVL0Pi9bHZEiXQJmG\n8dcBiUACcOpYj6YfXgWmsEQbZSIoEyOmrnjqqesT8PsceBrIUYaJW0KzPGKLJFLNSeqoB7UO\nM2diskyrml4wLgs3/zN1XQRbG2jM4sFdIMn0KRD2vTuhBA5MLCFNPLj7ktWFLqd1V0XIqpNz\n13uKzFz9tbfFYoAAggckXyyb13IG4Fjr3KmDT5B2Hm+sbLQqFXxJtnndvHQcdb3ewsKkr185\n8/lPzgfmRFyzPGfFrJR399fKviU1Tvfkg6t+9865Q+XtRMRzuH5+xn1bZuk1Q33mKauz/Pr1\nU3670PQE3T/cPGdxcfLg7/KzpDj56YfXN3U6u22e7BRDnGFyissMRqxR8NwtGwqe+bAiJF6a\nG7+wMBEA9p1tfX13dW2bzaBRzitI/PqVxYM3kWUwGNMBJnAwxhBqOy8fb62YoAJHL8hPBO+J\nkUMCde+CEBdHyUWWXZi8LZonkiSxs4XT6tEw7IUU6qgX332K2vv7tiDHzd3Ib/4mcBH+5REx\ncQYkzgAAqfNF+eG01cvGYwan5ObcKZW/BvYmAHBXtMjm1Dp3vjf+Age1V9C5v1NPEwCAxsQV\nbcbclVGrrmKMlAPnWl/cWVXTbFMr+dl58d+8emZu6lD7d4w35CbhIlAPgAK5RFBk17Q4fvzc\nUX/aAgC8vqfmx3ctSTCNViy+fVPhytmpO481NnU6U+K1q+ekzslLcLgFtZL3+EILwdRKvjQv\nXq9R/OQbSz0+sd3iTkvQKvhhfLdbupz/748HXd6BRePmLucPnznyxHdW6bWKV3ZdqGrqUfJc\naW78VzcWROoay3GYlazPSh5unSCDwQji1o2FRPDSzir/j33t3PQHb5qDiH98t+z1PX2rHTan\nr7nL+eWZlv+5f+XQ2x4xGIwpCRM4GGOJENqJtg8xQnxwiMBnAU4DCu1oBjX18TSB5JKJ+zpB\n6AZFFIzuyOd1f/SK+9PXyOMGAD4tR/eVbytLlwz1/YJXfOMX1NMRcERJOvUZaPT8+tsv/XaF\n3IyCAJQTb5lUl8wtup+6q8DRKu59CUBGghFaG4AoKsa0Q4Qaj0nHXvC/AnePdPoNtLVyc28e\ntzEwwnn+k8rnP+kThT0+cf+51sMV7T+9e+nCwglnoAtiC3mPA/RKAERio+C9+JPnpabOoJS9\ninrLL189+bN7lo3+hDPSjHdfUxIY0WsUd11Z/MeA/pG9fP3KYn+mhlrJj0BieHPvxUB1oxdB\nlP7vnbOVDVaf0Fccd662+6Mj9T+/Z3nRoKnylQ3WDw/XN3c6E0zq5bNS184dY98rxmTmeFXH\ni59VXWjqUSv50ty4b1xVwjQyRLh9U+GWFTmVjVaXR8hPN2Um6QGgurnHr274sbt8//f22V9+\na0UsRspgMCYKTOBgjCFoSqMOmVICmYKLwZEEattH7QdB8gEAaFIw83I05I1+hFMTScZdrw/R\nERWBw/GX//ae3Od/KbbU2Z76V8O3/0M1f9WQBlh1JEjd8MdPfMKvuQX4S1yX+Pw5Yvnh0CgC\nnz82TiijBTG+COKLuIQvAE6Fb+a0+vFUN4Ak6dw7Aa/7Tk0Xv4QZa8AQ6hvCGB9au10vfhba\nFloQpafePPOXhzaM5xfk0pA3QN2A3q/QmRpbQ7vMKI+eb2/tdqXGj4kqvW1dfmq89pkPK3ot\nRbOS9V+/cua6eemjPGxlg1U2Xl5rkSgoCcvm9D3+2qnf/9PaSId6/pPzf/usSupP3fr4SMPy\nWSn/vn3x0B09GNOH13ZX/+m9PsHO7vJ9cbrlQFnbT7+5bEHhRKotHUdsTt/J6s4OqzszUT+v\nIGFJcI3YwTL5BtKnqrucbkE35Ho0BoMx9WC/f8YYgoVrZQQOjsf81cM6DtW+QT0XBl6726j6\nZcjbhqbCUY9xKsLJziUIACNsGh5C1ZlAdcOP640/DlHgoI4G+Q1eF/V0YPwl9C/FsquEQx9L\nHUG+g6g1KC/76lDOHitU85c5P38/PK5eMK5rTWRvA7esNSNR5wVkAkeMOHq+XZSrYGpodzR1\nOnpXLCcKYkuAutFHk0xf1P5NnY4xEjgAYO3c9LVz03s9CIdusXEJIshJIepGL9XNPQ3tDtll\n9tPVXc9/EipaHSxre31P9W2XsZsXI4gOqzvcacInSE/sOP30wxNM4hwXPjpc/4d3y+yuvmLb\ntATd97fNC9R6IrWPJSKby8cEDgZjOsPWEBhjCOYu5eZsCXJVUOm4FV9H8zBW2Mh+MUjd6ItK\n1BzW0jUqCG5wdgDJNPicNKgz5BxSERRxUTEZ9VWdlo2L7U3iuU+o7B2q/Jg6q4K2kUQdR6Sq\n56RzT0qVz4JHfuEFYEhlJqjSaL71M8WSzX25HhzHFy/W3P8YlzChc7+1Kzaq54ZW8XCmOONX\n7x3XcQxSIDay2rFRQiK4K8ixnxwHwHMeYDL/9EaBbJuAXvyP+BMEIpkKOF3kH65OPeYzDb1G\nETV1A2Bmdtyw9u/sccvGPzsu0/wFAHZGiDOmM8cqOwRRCo83djga2u3h8anNofK2x187FXjp\na+ly/vCZw81dA0VwKRFkU7WSC3iFmQAAIABJREFUjzdOGJN4BoMRC5jAyRhbcPbVfM4SajlH\nLgsakjFrPqiGuRRpj+Ac6e4AwQGK6C1s2pqk838Hy0UAAOQxYwkWXAHKsParEx9UoXkdWT4D\nCnhaQiXGbYjO8X0Rp8FS2buoVwMAXABImY3zbwdeCSRK1S+DrbZvXdRnB4V8EQ3Gp6EhYShD\nQL1ZffN31TfcL1nbOWNCqCwieMjSDD43xmeCZsKYjSEX/y+POd5/zfnp22J7M2cwqRetMt56\nHx8/rg4LqE8G5IK+G37GP31DtJDtc5D6Ht/JA+AuQ8NlwE+YT228SEuQf1hHxLSEiXUVQlSF\nZzLMzyelAn1hKk2CUV2YOcm6Od60Ju/DQ3UhkpOCR0GUyeAAgEhdG9qtcl5IAK3d8nHGdGYQ\nHdM2ZhKnIDlF8gKggtPwOIFEgTf21IQHPT7x7/tq77t2Vu/LtXPT/vJ+ebjN8Pr56awlM4Mx\nzWECB2PsMSajcf3I8ysp4sImSNFb7LU1SUd+3+fxAQAkUuNBstZxS78Tsa/HREabj8oEsh0H\noQOAA1UKGhYBHx0xiE/Pk42jguO1A0IDtZ2Fivew9EbqPA722sCsb4zTY6qZWkML3bkN24c5\nFD48a4PKdkrH3u7rT4wcFq/llm4F5YQwpkWF0nD97YbrbyefD5XK2AxCqcXMRdRwJDRuSMGk\n4vEdCpH9C7+60YdoI8cXaLpmfEcSe5bOTEkwqrtsnpD4ytKUSH06YgafAj4ECJrtx+vhaxu1\nz3wSOnX/zo2zeW6SpdenxGl/+a0Vv37j9Pl+M46cFMN91876xcsnwrPiCzNNkWwgzRHSWuIM\nE2gmyZggRCrjQoS0+OhLnBL5nEKbIA0kHyk5g89rbmhzxhnU6Ym62P5sa1pk6yihunkgnmjS\n/POt83/1yqlAS+BZOXHfvq50zMfHYDAmNkzgYEx4NMnycV4LSkO0TkLVnwyoG37szdRyDDOW\nUkc91ZwkpxXj07mZy0Ed63p4r4s6GkD0YXIOaCL8IyjiMH7jWJxcOX8Fl5gqdbaGxFUzkiH4\nkYgaj2DJtdBT2ecAEgA3J4cS3NKFBvC6AACTsvmNd2LeaF1C6czH0uHXAl5LVLFbsndwV3xv\nlEeOLjFTNwAAgJu7VRLc1HJmYDymDG7xneOt5QkdIMq5OQpdIHYDHwU33Ngj+chyGtxtgBzo\nMtBUGqkXr0bFP7p90X89fyxQ4yjOMn9v67zxGuuQQT0qZ5KvPOh3jcrbNi3Jy3Q9/0nlxZYe\nDnFmTtzdV5eU5k7Kz7Ew0/zUg6vr2hzNnY6UeG1eqpHj8AdfmfezF08Erhib9aqHbpkf6SAr\nZ6d9ekymGmXV7NQxGTRjMrO4OFlW4lxclDz6RsvhOHwtIg0kY1pswrMfnN93uk8+yEjUPXDD\n7GUlMbNkitTXOSS+dm76rJz49w7W1TTbTHrl/PzEyxZm4DQ0LJm6oHISLjEyJgBM4GAMAZLA\n3QMaU6RH8zEFzTNJaQSfLTSetDiK46FuuW4vANBdI1aUSUc/9Kf0i1+8zF/1La5gcbROPTyI\npINviwfeBJ8HAIDjucVX82tuke+cOjagUm38zn/Zn/6Z2ND/j8ahKidBWxJmgSF6wdNDglPG\ntY9HLMxQbvlPsrajWgvaaJQkSKJ08r3wMDWepbYLmFIQhVNMDRRqbuk3qasaui6S6EVTBqbN\nicGve7B2P/apIHC4W6T6NweuXd0nqPMwl70NlPLf9jl5CU8/vOGDQ3U1LTaVgp8zI37jgon6\nsK4oQi6efFVANgAe+ERUzATUrCw1rSxNFUTiELjJlrgRAiLmphpyUwcU5FWz0/70g3Wv7a4+\n32BVK/lZOXG3bCgw6iKKlWvmpK2dm/7F6ebAYE6K4Y5NRWM4bsbkRKPi/+2ORT954ajFPqA7\n5KUZv/+V6EucguQKVDd8Av34mbrG9oFIU6fzR88e+endyxYVxaZH9ey8hN0nm8Ljc2aEFrEm\nmTV3XTHOuYcMBmOiwwQOxqC4LNLpt6npJEgicDxmLeTm3AAa07iOgVNy+V+Vat8Bd78zJSIm\nLsLUNdE7B0WqdqGueulIcEdSZ4/4zhP4zcfRHCG1ZCwR974iHXhr4LUkSoffBXs3f+2D4zkM\nPj3X/MhvfecOi401qDXwagfXfTTCrmpUmsgVmu4BAKg0AyLGRW2NiHpa+ypTwmmvBiZwBIMJ\n+ZCQH8s5KEZOY5FxyZ1skCjVvx2qzLrbqel9zI3Y7kenUWxdlz/mY4sKXBKq5Sc/Cn5ySxuD\nkJage/CmOUPcGREe/dqiT481fHCovrHdkWjWrCxNvWVDvpqtSTLkmJuf8KeH1n9wsO5Ck02j\n4mfnxm9ekjUWpSKB6gYA7D1tDVQ3+vaR6LmPKmIlcNyxqfBgWavbG/RglhqvvW5lbkzGw2Aw\nJhdM4GBExmMTP/8fcPXnkEsi1R0ROy7wlz08bKPQUaJJ4Yq/SbZqcLcDr0F9Nmiie9NF0KeA\nvTl8A7XJzMxB8EpnPudXfyWqYxgCHqd0WCZDQSr7klu1FRMyxnUwHKecs1w5ZzkAgLVe2i8n\ncJiyQKWH+NnQE9orEQAgfqjzhKEi18Sxf5OcpyYjtihSAJVAYaVhqAZFFNr9xBZy1INPpgCH\nHLXos4JykvluTkDI7RCPfiK11aFSzeWU8nPXwMTLdkGEzYuzNi/OivVAGBMdm89n9fokolVL\n09dxmfEqlVYxVkIYBudUVtbJtwGqaLCKEsXEjCMvzfj4t1c++eaZinpLb2T1nLT7ry+NYrMk\nBoMxhWFXCkZEpPOfDagbfpzdUuUubva14z0a5NBUCKbCsTp89ioqeyM0yquoMULpSqdM8uRY\nQ+21IEZo/N5UOd4CRyDmbMxcQo3BvpWcgpt1PQBgXCnY66jzWOBGTJiHCaN13AgBTSmgVPcV\n74SQyJZ9Jh6oRN0ScuwHgAA3B0T9MoDJv8Tts0TaQl4LDipwEIEo0RTOgxg9Us1p72uPk6Pf\ncfDwR9yBd1V3/BvqZRMMXUA2AAA0Ach3PGEwYkiXx2v3DdzcBUlqd7sT1Wq9ckye0hVckKGp\nXHdaAAAiIgr1zxo3irLMTz24uqvH0251ZSbpDdpYGlcxGIzJBRM4GJFpl1t1HyQ+RBwW8eDb\n1FINAJiWzy27Hg2xL7bHjCXgtVHNTpD67bg1cVj6FTr5nwBymoIqFin0g2UoRN40LuCcbRCX\nQzWfg7MbOB4T8nHmFjCm923NugriZoHlLHgsoIqDuFlonBH9QfBKnH0Fnfj7QIQAEDClENNn\nRv90jNGjLkDeRK6TIHQCICgSUbtgCqRvAADwES8RyIfNsSWRqvZRe3VVJz19IaWsk/eJUm6q\n4fZNRWvnhlnbMDxO76uPkzOoz4LUWOl79w+qrz4cvKtIUgVQU3/PFwTMQq4YgHWRZEwUBEkK\nVDf8dHu9OqViLNQFDpUq3uQV+35BeWnyF6vcVGMks89xI8GkHguPVQaDMbVhAsd0hYiqD1Nr\nFQgeTMjGmWtBGXoLoQjJAiCG1moO47R1Z4U3f9XbOAMAqLFCOv254uaHMTv2bb0wbyOmLaSu\nKvDaQZeESTOBU3I5c6SqsIaaAJgb5eyDoYBJ2cDxsnYhmBbrun3kMHsFZq8A0QccH+5YiYZc\nMIx5GgW34DoJiE5/1JfqgoC5i7hVX4vVGlQMcNRR1xnwdoPKBOYSNE14O0NFMhovj/Ugog/q\ncggVMl2uFQbQBPvOOLqkj58gS9Nhe+qP61eIQAACAFxo6vnJ80fvuLyIWeiFIFYcDVE3+uLl\nB8llR+2ALShJZ4ECywwJqJ4kEbnZYz9MBmNIuCNkUEhEPklScWMiMegUSTwq3UI3gbRugemt\nLzq7baEXq1s3MuMqBoMxKWECx7TE1SN99AS11fS+IgA48T63+QFMDSoAQWMq2eQcKIwjXVGU\nRPH93/rVjT68LvH93yrufWK8W1TKoonDjCWBAW7trVLd2ZAxY1YJV7JyfEcGAABaIzd/k3T8\n45AwFizC5Jxon4yksv1SzWly2TAxk1+0GU1D8z3hY5pHisgtvAFKNlBHba94B+ZptABOjR9T\nR4Ae13Ua4mZhzg0x6X90CSQP+LoACJSJ8pai5AOhGyQ38CZQxI37+EaNQo8pa6l1V1AQOUy/\nIkRuk/Y+S5YmIvxty3wxTIl7eWfVVUuzU+O1wOiHLHI3JgCQJLK2BwgczmB1w//+ZoBCALYs\nzJgQEERMwKQxzM1ENR+n5uMkEswq/MW96b969eT5hr6qZJ1G8Y2rZl62MHPMzs5gMBhjCBM4\npiPSF8/51Y0+nBbpk9/yt/48sNso5q+hplPhb8eCtSM7LzVVkq1TJt7TQU2VmFUyssOOKZiU\npdj+U3HX81R7GkQBNHpuwWZ+xU2xmjHyG+8EXiEd+8ifx8HNXstv+kaUT+N1+V59TKo94w+I\nh95TXHMfP2ddlE80RmjNmB391noTHLKeD1I3eoOWMtBnY9IS2bfEBpLIehCsR/uyG1AB5sVo\nXh70m3JXkf0wSP3Wd6oMNK4CPhq9hMcRTFwK6mTq+BLcbQAc6jIwZR1oUoN2clqoqRwAGrz6\nFp8u/CCiRMcqO65elj0+Y54caCK6XKNmIH2jz3dDBgKyATKBgzEhUEbO0RhkU7TgUAEAeWnG\npx5cfa7W0tBuN+tVs3Ljzfrx6z3PYDAY0YUJHNMPt51qT8jEnRaqP40zFvsDmDKTW3iLdPpt\nEPpdGxUabt5NmDiyagiJHJFd9xzdY11CQI5a6CmH3v4FphLUD7VcAhMyFFv/BSQRXHbQh1kD\n+pxU8xl1V4PPifpUyFmDiWOZT84r+I13ckuupdZqEAVMnYFxqZd+1zARdr0YqG4AAPg8wru/\n57JKotjVlRFlLGdlw2Q5O6EEDuraDbaTAa8FsBwk0Y2JG/sinhrq+SLoPd4msnyECTcCTrJ7\nFhry0JA3yA5k7+jNonNRxNQnhztCteB0hS9c4OM4kEIT+7nUXIyLQfduBmM0aHheyXG+sO+z\nXqHgxrExECLOzoufnRd7TzQGg8EYJZPsYZExesjWEdGQsqc9JIAzVvNpc6jlLLm6UZeAabNB\nI+tRH/l04JGoTiILgIjatki7oX4I91RrC7VXg+CDhCxMGV5pKDV/RN0Bc6rukxA3DzOuGsYh\nOF5G3XB2SEf/AF5731k8PdBVCXkbsODKYQ1vuKAxAY0JY3V0IvH0Hpm46JPO7eNX3ThW52WM\nDvLJuBIAAHgjxGOC6ACbTF4Y2E5B3DLg9QBAjpMyO4g2cFeDdqq5UWB/y+1UpRNBPlU9I3F8\n23JPeDAhXbF2q7D7taCoQqm89r7g/SK1qkHA4d3IGIwxJVmj6XC7vQEah06hSFCzHAoGg8EY\nCUzgmHagKnItt1puk9aMM1aNbBGBwC1IZ3oN8wCAMpLAaARbaNowmpIwY1ArRFGQDr5E5/cC\n9d3+MX0Wt/YbMBRZBICs54LUjd6g5RTos9E8Kqs5Ov+uX90YCNbuxtR5YEgfzZFjidsBHqfs\nloil74wJAPI6eeVSIVP4EDM8rRFm8QSeFtAVAIkgdMu+lYQOhKkmcEBcGhiTwdZu5j0rjM37\nbaHXjZQ47ZKZLCshFOVlt3GZhcKeN6TWWlSpuZxS5eV3YFKIZYAGMBOoMfTNmAPApo6MCYSC\nwzSd1iWKPlFCBDXHq2LdvoTBmBBMBHs+xiSECRzTD3MKmFKgJyyZguMwM8rG8hLV+9WN3lPA\n1VfAW++ANyDjWqXhr3lg8EuYdOgVqgjKKaDmMmnnb7lr/xWGksBpPRcxPhqBQ/JRl1zHXCJq\nL0N5gYOkC8eoqRJEAZNzuZKVwE+836BaG6lXC2gnmQnC9MI8E3pkvpBonlAtcgfxzItxq+MY\ngdyq7dInT4Ik/GP6CYugLnMNJGelxGl/uH2RSsGmOjLwM5fyM5cOvg9yJSQpgeoAesVxDjAP\nudj0nHJ5hFd3V5+p6XK4fTPSTFvXzchPZ4kkjAG0PK/l2XSOwWAwRsvEm1wxxhzk1myXPvx1\nyAyWW3gdGIfWJmPISGQNDWVnwzfuwsNnobkdADC9kFt+PRoGrbbwuej83vAwdVyklgpMv7Q1\naaTUffL1jKq81ef2Z5SEbXLIBJ09wtv/Qw3l/oB04E3+hu9jUtZoRhF9OJ4rWCBVHpXZUjyB\nrBxGi+QEAOAmUnbD6MD4uWAto54LQVFdBiYvj9GI5FBFTkZQpQAAIA+KeNkkDlRE+QI1QcCM\nWfyNP5KOvmluu/B40dF90qxzqtlupbkw07R5cZZaySY8o4FDrgggD8gOgICGWD32NHU6Hvr9\ngQ5rn29uVWPPzuON371xzpYVUW+AxWAwGAzGtIYJHNMRzJrN3fwjOvQ6tVSB6MWEbFx4LeYt\n7NssiXRhL7VVgtcJ5nSuaD0YR+wrKZcFYDDgZVfzONRUc7K2ghTanr2PrgYYgsABCh145OL8\n6DovKnXAK0GU8//TyHS1FD/8Q6C6AQDU1SS+9bjim7+aaDl4isvv8jWeJ2dQMRG/6Aouc0oU\nCHgqyXWyr0MHp0PtAlDHZkU3yiDijFug6xR1nQZvN6hMaC7BpKUTq0eswgT6EnCUh8YNJaDo\nW81G/QKy7grdgTeBZnjOO5MJcxp32f29/7sOYJI0K5pEKAFjbJ341Jtn/epGL6JEv33n7LKS\n5OQ41gOYwWAwGIyowQSOaQomZOFV3wMAICm4O6NN+vwpsjb1vWw7L17Yyy2+FfNXjuQsoCFw\nyW3RDOMog0z+h1bfgcZCctTJxUc3Xed4TJlLzcfC4gpMmRsSI3u3dCFsTwDqbqa6s5g3sXqa\nYkK68r7/Ffe8IlWfIlcPl5TNL9vCla6K9biiALlOgCugQYzkJMc+JBdoolyfFSMQE+ZjwvxY\nD2MwMOly4pRgO9Nfk4JgnIMJ6wf2UOehaR3ZD4PUf/VQZaJxFeDE0gEZjCFid/mOVXaEx32C\ntP9c2/WrhtrSi8FgMBgMxiVhAse0J3h1Vzr+xoC60RcSpaOv8KnFoE8c7rE5TBGpVuaUOIyU\nEIzLALUePHJFH6mDWpP6jxC/kHrOg7MhKKrLwoSFEd4x5LEVbSF7K9gCTOw4BZbcJJPBYWmJ\nZDFAXc0TTeAAANSbFVffd+n9JheSG1znAAggqDiJXKdQXQwYsU8nI5qgAhM3gXkpeNsAAFQp\n/tyNATQFqM4FoQskNyjigGdWBYwJgShRc6dTqeBS44eRdmGxeylC87Ium1s2zmAwGAwGY2Qw\ngYMRgOijhhMycUmghhM4c9Nwj8dhOoFbosDWGzyP+TjMDA5u0Y3S/r+FhLFgBcZnyr4jFOS5\n3Fup+zhZy8BnBaUZzbMwfmEUUveVOm7pA9R8FLougOACfQpmrQCtnBI03OY1jLFAaAeQQtQN\nAAASQegA5aRtfDMZUZhkdI1AUAHKERfHMRhRRhDp9T3VL+2scnkEAEg0ae7dUnLZwiHdg+IM\nKkSU1TgSTcO5GzLC6OxxP/vh+WNVHRa7JyfFcOPqvCuWZOFQ3McZDAaDMUVhAgcjAI8jktsF\nObtH9rzA4wwOk4msBF4EDYdJAMNeJ8eSDZxSLR15A5xWAABeiXOv4uZdM5xDcJiwGBMWD/fU\nQzpyxlLIuJSZf1I26OPAYQndwPGYMzWKIyYFcqYwl97EiCmSG1wVJHQDp0RlGmjyZSQqBmOM\nefLN0x8eqve/7Oxx//ylE3aXMJQCE4NWubgo6cj59pC4SsGtLE2N8kCnEw3tju/9374ep7f3\n5YWmnsdfO3W8qvP/3bYgtgObSkgSNXY42iyu9ERdeoKeaUcMBmPiwwQORgBqHSAn2xkENSNP\nEUcwIBpGMSwAACxYyeevAFs7CV6MSwNusn11OZ6/7C7x70+EhlfciMZh1/4wRghvjrxJxheW\nEXu89WTdA9Q3gSHXeXCdQ/Nm4Ni6N2P8aGh3BKobfp79qOLqZdnKIfTxffDmOQ/9/kC7ZcCU\niufwOzfOSTKzb/LI+dN7ZX51w8/O441XLs1aWDg1my6NM2cvdj+543RNS5/jeGlu/D9unTsj\njTWMZzAYE5rJNktkjCm8CtNmUfPZ0DhymDmoSYTXSp4uVBpAnTiGLRsQwZQSi8UDosr9VHMM\n7B1gTMaiVQMdZ4YDV7ISDXHi7heppRpIwoQMbtVWrmQqOHdOGvh4UKSC0BoaV2UBN1oNjhF9\nJE+gutGHr4PsB9C0ITZDYkxLztR0ycbtLl9Ni604K7Jy2k96gu7PD617fXfN6ZpOu0vITzdu\nW5efxyaKo0CS6HBFaFJML4fK25nAMXpqWmz/708HPb6B9MZztd0P/X7/H7+/Tra06nR11+GK\ndovDk5Wkv3xxVoJRHa2RON2CTsMmLAwGY6iw6wUjCG7RNnFnA7isQcHZV4MpTf4NXgs1fEi2\naui10FTHY+aVaJwSTTd7EQXpoyepob/vRkcd1RzFopXcxntGkCePWbMUd/wEJBEkERSqKA91\nYiGBu5qEDgBEZRKoJ0pZAdV0gqEH4wcykqirB5NYF4MJibcuVN3oxV0LRt8YmsKSj3qOgOsC\nCHZQGEFXhMZFgOx2OX3xiTKJjX2bhIibQtCqFNs3FwEMyRubcUk8gihE+FzsTrn27Yxh8squ\nqkB1oxeb0/fm3ov3XFMSGBRE6Vevntx5fMCi/sWdVd+7ee6GBRmjGUC7xfWn98sPlrW5PIJZ\nr7p6Wc7tmwo1KtZOazoxSCNFBiMy7IltiuLpIXsLAKAhHdTDWSMyJPNX/ZtU9hG0VpLXgeZ0\nLNmEyREeyDwWqep5EGwBkS6qeRUL7gB99miGP3Ggs58NqBv9MarcT9nzsHD5CA/K8VPwki16\npaqd0HyK3FbUxkGSDlMMgAgA5K4A51k0XQZ8jLMkqLuWLuwHAIgzglEPRGBzkNUOiT5c/4+x\nHRsjHBLtEbZIIDpAMTZVRZKX2t8AX3ffS8EKPUfIdRFTbmYax7QlL1X+NspzmJMyIZK/nG7h\nYqtNIspLNRq006IhlFalMOtVVoeMBpqeqBv/8Uw9yuussvGyuu6QyMu7LgSqGwDgdAuPvXKy\nONuckagf2dlbu13ffXKv//O1Orwv76o6XtXxvw+sVPBjlinMYDCmBOxxbcoh+ajyA2o82Gul\nQchj9kosuHIYphUqHTf/psF3oc5zVPcpiFZQhkzUEUii1r2Yf9sIxj4BoerDYTHsjY9c4Jh6\n+Fzil0+Cra/0g3wu6AHoNOCsgl6NA0QL2b7AuKtjOUgAaCnr/S9ZbGAZEOaoswZ8blCyYviJ\nBaJKvrUmwKfHOxYUa8bCv4DsJwbUDT++DrCfBuNoG0szxoJD5W1/+7SyutmmVHCzcuK+eXVJ\nQUaU+wrPmRFfnGU+3xA637tiSZZRF2M1QZLopZ1VL++60LvYrlRwW9fO2L65eCjOIJOdK5Zk\nvba7OiSoVHAbF44qcWDyQkAOn9MtekRJ4jlOy2t0Si2ONH1SitDbONyoTdahRhClT4823nlF\n8cjO/tePz4erVxX1lo+PNFyzPGdkx2QwGNOEqX//m25Q2Q5q2D9w/yGR6vbS+XeGcQQggTwC\neQjkkz+p9TCdfxXcXcDJ3zXJ0TjMUU9cyCm/ggHOsCnQNEaq+syvbvghq53aAwrXhXYQw5rI\njC8kuCNtgYibGLFDnSVb2VTRonzstYq7f7n73QO10T+pu042TBHijNjy8q4Ljz59uKzO4vGJ\ndpfvcEX7d5/ce7CsLbpnQcT/uGvJ/IIgQ+jLF2U+cEPse2D96f3y5z4+7y8l8AnSy7suPPXW\nmcHfNTW484ri5bOCmklrVYqHbpmfnjAdMziIqNNtsfucgiQSkCCJNp+jy20hiCQUX4KiTHmh\nsCjYdEYQqd3qkt2zqdMxslMDwLHKjmHFGQwGww/L4JhauLqo5WR4mJqO4IzLQX3pRS2XaHUL\nfbdDBNQozFreHDTHkASq+6z/uJEOM8K76QSB7NXgqAfJDeok1BrJ3imzk4413RiAWsOMaXvp\ntkFKwJRAtMW2WQnqk+S/mgr18Cq5GOMDbwb9PHAEXdNcPvzNpwYAcHmFJ3ecyU42hEw7R4vk\nGV6cETs6rO6/fnw+JChK9NSbZ5aVbMSoNrRMMmt++a0Vp6u7LjT1KBXcrNy4/PQo54mMgB6n\n9629NeHxjw7X37GpKDVeO/5DGk/USv4n31h6sKztaGV7j8OblWy4amn2tG1M4xBcgiSEBH2S\n4BTcesVIvglfWV+w/1yrIAbdNrUqxU1r8gIjCh7VSt7tlWm1rteMPL/J5Q39W3pxuuXjDAaD\n4YcJHFMKsjXJiwtEZGvCSwkcTqHLLfYMvAnIJViIRJ0iYP7gaBlY65YiCBnaCI6kEx/JSw1v\nkz3gedEogJxNO+YvjcLp3B3kbARJQG3K5HYt8Trl475gpzeMsa8qZs6HM38PT9bAnGVT0BVl\nSoD6RaBIJucpj6ujxwmnG1XPfqFv7Rn4sN7ZVxtNgUP0gVuQvzEqYj+bncqIbnK2gOBGbTJo\nhvqBHq/qkLWZbLO4Lrbax6KZ5dz8hLn5CVE/7IipbLCKcjdiIiiv657yAkcvy2elhORxTE88\norwI6xG9IxM4SnLifnTnkqfePNPW3944J8XwT9vmpQUnyAiitLAwaf+5sPZkAMtKRv65ZCXp\nw4vCACArZYSmHgwGY/ow+QSOL7744oUXXtizZ09dXV18fPzSpUt/9KMfLViwIHAfq9X6yCOP\n7Nixw2KxlJaWPvroozfffHOsBjxR8HaBpwNU8YDyEzkiMVDd8OMWbRo+jut/F0kBU1ZRApA5\nGqZO1tan1LorSN0AgDQ9dBuh2xYYw4LlWDA6Aw4SqeEj6j4JRNArShlyuZzrQDkpJ1GoSyCP\nTWaDJqBLHKpBEeu+fWojt/wu6fAL4B3Im8XUEm7O9TEcFOMSqLNRnf2Pz+ypaZH5jtW1RTIi\nHT7WVvGD/0W9gAtl+kB/hsNEAAAgAElEQVShflbUTsQIhtoOUePnIHoAgADQXIS5W0B1aXnC\nEXktd5r00Yi0yhC+yeUR9pxqrmuzm/WqRUVJhZmXbm3LmFxEtMwI98wYMstnpSwoXF9Rb2mz\nuDMSdcVZcQp+IDHqTE3Xn98vr6i3iBJxiCEDWFmaOhqB45rlOecbTocEeQ6vXsYMOBgMxiWY\nfALHL37xi7q6ultuuaW4uLixsfHJJ59cvnz5Z599tmbNmt4dJEnasmXLqVOnfvrTnxYUFPzl\nL3/Ztm3bjh07brzxxtiOfBxAUyYBAlFo3ToCde0hy15Q6DBlPcbNDX+vINuOsW+TR4V9gj1q\nA5L8icDtAxUPXL+Zi0KPGZvQWDDavyQmkEjWc6FBDrE0E7xZ0NIK9i4wJWPRKsxfMtpTNX1K\nXSeCQvZaqeZ1rugbENW06vEBsxZTt5wbQlL8wD6GpZHEtfEEU0v5Kx6l2oPU0wwqHSYXYVrs\nq+gZl0QdoTVgFFsGSrv+DD1t1ANg1uGM1IBfIgemxaBhvYTHBGo7QnUfBUWslVT5Ild6L+Al\nbMIiWS0gTpc+GgXpJkQkuZltUYCEcayy45evnOzs6Ute4zi8amn2gzfN4SMYaTEmIzzHi6JM\nnQgfducVJHL4REGSVDxnUCoGf+hQK/l5+TJJVXvPtPzXC8ekfiGtV91QK3mvIGUk6q5flXv9\nqrzRPM5cvSynscPxxhc1/lPoNIp/uGnOWGRmMRiMKcbkEzh+/etfFxYW+l9u27atpKTkscce\n8wscO3bs+PLLL5999tm77roLAK688spFixY99NBD00HgAE08pi+k5mOhcZ26T4MQnNT0AZCA\n8eHtAAYxzgjYpDJi4mzq7PdckAjcAnAI2gTMvxZ1mcCNSxmCz00XjlB3C+rMmDMH4tOjcEzB\nAZLcoh8CZmTgwtujcIpeRA91npCJu1rIUYuGvKidaLzA3FXYXUcNRwJCHOTMwPg4AARFIuoW\ngjJ6KcSClawHwNsMkg+UCWhcBNoZw3i7SodFG9lz/eRiQUFieZ2MSe3CwiilBVlbqe1C7/9S\neQM0d0FaPGhV4PRi4XVoikZJGkMOatkrE3W1kaUC4y+RNbOwKCklTuvPn/ezpDh5mhgxJJjU\nVyzJ+uhwaA+LtXPTs5L7Mvm7bJ4f//WoyzOQ7SJJ9P7BupQ47e2bCoExVdDyaq8os1KlVQT9\nFjpdvha7R+wXxdQ8l2XS6JU8ALR0uw6fb2+3upJMmsVFSZmRO7wS0e/eOSeFZRB5BemP/7Qu\nNy0KvZMR4d4tsy5fnHWovK3T6s5I0q+fnx5vUF/6nQwGY9oz+QSOQHUDAPLz8/Py8pqaBvpv\nv/XWWxqN5tZbb+19yfP89u3bH3744VOnTs2bN29cxxoLsOQmUOqofj+QCACAADoNGIPKL6lt\nL8bND1kc4yP7I4RswoLrQBKou2IgpM/iiraBeixTXj0t5CgHwQq8EewK6eNXwdENvdILx+Pi\nLdzqW2QbLgyDQaQZPqr3VE9X36cTjqsNJqHAAchxC2+n7KXUfBJcVtAnYdYSNGf2S2NRFRO8\nzdT+d6D+h3VvK3V+AMbFaGZde6cyW9fl7zzeFDKVTY3Xbls/HG0rMiFewmR1grXPWQZTI1jM\nMEaPzw5eueo2AHA0waUEDpWC++H2RT/+69EO64CxTmGm6QdfmR/FMU5wHrxpjlrJvXugrne2\niYhXLM564IZS/w47jzcGqht+/r6/lgkcUwmtQuOTBKcQdJHUK3VqfuDZxuoRGm1BLlQeUbpo\ncRUn6j8+2vD63hq/qc2OfRevX5F786o82XPVtzvaw4RFACCi0xc7oyJw9DIjzchSNhgMxnCZ\nfAJHCI2NjRcvXty+fbs/cvbs2aKiIrV6YEY6d+5cADhz5sx0EDiAU2DRFsxdT/Zm6qkE+6mB\n+hE/ogu83aAOyjnkUKHi9F4ptKeXitPxGOyDzWuw5Da01ZGtAUgEfTrGFUR5EhsMde8F61EA\nqe8sBFgcRye6+6bPkkiH3yFDAs7fPKrT8BrQpoOrKfxvQYNMTf7IGcTPcgIUcYwYTCrCpKKQ\nWNTPQt1fDKgbfmzHQD8TFKy1zZTFrFc9+d3Vf/mg/POTTT5BUiq49fPS77lmlkkXpZQxdeQn\ncg17vI4JQ7p6zMyOe/rhDR8err/Q1KNScLP/P3vvHdjGdaV9n3Nn0EGCJNh7FUmJqlavlmzL\ntlwi95LiEpdUp22Sze7m3S/7ZpNsNvtm12kbJ7Ed24lLFLnJRZKt4qLeGyWKvXcSJDowc74/\nWAEM2EkA5P39JZwZzFyRIObe557znOzYaxensvCuvCCi94/VHTjb1GZxJMboNi9N3XpN+oR7\nvqhF9rXtJfdem1dWbyGi/DSTX+VOfZtyq86OHqfd6dVrI34eyBkkWm3UiRqn5JJkWWCCVtCo\nmM/vt82ukOIhEV1usrxysMInKNPrh6ozEwzLCxIC36IomQ0cCrJ/w+FwODNFZD/YZFl+/PHH\n1Wr1D37wg8FgR0dHbq7PcjQuLq4vPvLVfvWrXzU0NCgecrvdAOByRU6bQLUR4wqAOskepIZZ\nyXTKoIpHL7qkIdM+jWDUi0Ec46MyMWpGrJ7slWDpq30YmP8hYFY8dFqpZqjBiXx2rzBJgQOA\nJV8nV7/it37G2CWgTZrklX3QxIOgA0lh9wONkdxLZQaQbOBpVzpA4KwFIxc4IhlnLzl70JgA\norJmERet+e59i79zz6LOXldclGZqF7FozgCjGQJ7Qqs0mFo0hTfi+KAygtoEboVeCWBIHeM1\ntGph+7rsKRzUtHK+quNfnz9pdfSXQ9a32U5dbf/obNOPHl4x3L5xvCTG6BJjlDtlBPOpYQzV\nqgiW1EPI4UstH51rau12pMTptyxNW1YQav/sYaiYSsWCNmd1epUNR9usyi5s+881KQocqWaD\nwFCxg09GAu9ywpk6eIc7zoSIYIGDiL7yla/s2bPn1Vdf9atbUWTU7ZEzZ87U1tYqHvJ6vRBZ\nAkcfmiDLcqYCTWxgGAENYrxWMEnkBgAB1f65G6GAbKXKB7LMMEzggK5GIHlUU7pR0KWyvIep\n5SDZaoE8oI5D80qMKZnUNQNBhimbqP79/pfUL91gbAloea+7EZGD/g2S7ArrTVtOcKi9kk6+\nRt31AACAmLOKLd4eLKWCMZwWewVEtvEh+f2nQfbRN9maB0DD5+vTCKZuoOpd/lF9CsbMC8Vw\nppeDZ5v+/S8BJlkAx6+0vXes9rY102JkuzQ/fufHVYHxRblxk5FUphW3V65tsUqynJFgDKsc\nE0mmn/719Efnmvpenq/s3HOiftuqzG/cuTAi/MGDjTFYRkZzl8JODABE6VUbF6XsP9PoF0+J\n018zT0EQ4XA4nJkkjB4b44KIvvzlL//xj3984YUX7rrrruGHzGZzZ2fn8Ejfy748jhH405/+\nFOyQ1WqNioqKjo6wFp5ozCJtEjj9m5Nj3DWAQX/1AqrCQdcYwqtcoY16jc/egaCarLrRhzoO\nM+5AACBp+gpG0LwMmIaaPgSPFRCAiZi4BhNnvr0uTWtt0dQjGAGZYv4RihH258npgzqq5P3/\nA/JgVjNR1RG5s5Zt/R6wGX1CYXqJcPeP5BOvU2slSF6Mz8JrbsfEyOwJFTlg/FIgooZ9MOAd\ngLHzMfOmqfk+Dye8Ev32zYvBjn5yvnmaBI6VRYkrChOOX2kbHtSohMe3hWPnYyJ489PqF/aW\n9SW5iAK7a0PO57fOU4th8Xl492jtoLoxPLisIH7joqkwO59mDCqhx62gZVisypsHek3QL+Gn\n7lzYa/ecKBv6XKUnGH74+WtU4fGb4nA4c5mIFDiI6Iknnnj22Wf//Oc/P/igf2+LBQsW7Nix\nw+l0arX9u3znzp0DgJKSqd6HjwCQZd5Fje+RtWowgHHLMXFDSEc1Tpjybi35bjhgxmjNPq0t\n1FUNkguMyWguGL0b6zTbYWDsAoxdAO5ukD2gMc/obN7dTD3HwNMGAKAyY9QK0KRN171kSTr+\nrnTpEPR0oCmBLdwoLLth4v9ZpgZtLjjKA+Ja0GZPbqCc0EDndw1TNwaClkaqPoa5E5f85NY6\nz6G3qbUOtAYht0S1+hYQx6DbxqSw678y4ZtyJgYmLENzCdhbSXKgLhHU0+lXHToqGi1dQZaR\nANDZO/EU0U8uNPd3mjAbblyRkZ/mo/Yiwr8+tHzHwcrXP6my2NyigItyzU/eNj88vRtf3lf+\n/O4hC3OvJL96oKLN4vjHBwJbv4WA/af9cxb62He6MSIEjiSjurfL69dWWCOwaFF5wrMoO+jW\noEEr/uSxlaeutl+s7nS6pbzU6I2LUkSBqxscDif0RJ7AQUSPPfbY888//9xzz33uc58LPOGO\nO+74y1/+8vLLLz/yyCMAIEnSiy++mJeXNyccRgMRjZh5DzpbyNkKTIW6VFBFxEY3AdlAtgPT\noz6HHNUKpzR3Df1bpcF19wa9mCxR6ZtUf2xw559M6WzRA2AIg0RK9YzbRjjKqGv/0Et3C3Xs\nQtMGMMwP/p6J4nF5/vIjueFq3yuydskNZfLlI6oH/mXCdZUYu5GkXnAPy0tiWjRvBca7x0Uk\n1F6pHG+rmLDA4T36vmvXMyD16ybS5ePeY3u0j/0Yo0fJ4+OEDKYGY/qg6kyWKmj4iGzNwEQ0\npmPmFtCFwdf15LAH92UEgIQJFV55vPL/fenUkUuD34dtbx+p+cIN8/zao6hF9uB1+Q9el2+x\nuQ1aVdhWprg80iv7A/RrgH2nGx/Ykp+VFHpFpr3HqRiva7PuP9MYF6WZl2HSqcN3aq0ThbwY\nfUOv0zFgxhGjEVOitFlLtZ9camlo9/GjTYrRbVsxii/YsoL4sLIg4XA4HIhEgePb3/72s88+\ne+edd+r1+h07dvQF9Xr9tm3b+v59xx13rF279qmnnrJYLLm5uc8+++yFCxd27twZuiGHAdok\nnFqbzGlF7ibPOZAHbOdU0aBJBlezzzkekao6AQCQYXoxbvo8xgd9DFPZe1R3xCdkqZdPPsfW\nf3uGc+B9cHZS86dkbwEmoCEdU9aBqB/9XZOEJLIcUgj3HEZd/kiNcieEdPzdQXVjELn6gnT6\nQ+GarRO8KNNi4p1gLydXI5AHVWYwzOfqRqRCFJi+0Y/smeAlu1pcu/4wqG70X6y9wf3OHzUP\nfG9i1+TMJNR0hKrfH3rZWUpdZVj8IJoiu1wo1TySmcvmpWM1VR3O3w5WDlM3AABkmZ7ffWVR\nXlyJ0t67yTDFX/JTS2VTr9Ot/IVwqaY7HAQOk0Hd3KnQN7qu1frTv54GgFij5ku3z9+8ZCK/\nzZlBrxIK4gweiTyyrBGZgAgAKib864NL3zhc8+mlFovNHa1Xry5KuHNtjsHXAKW6ufev+8rL\nGyyiwIoyYz53fUEwa1sOh8MJIZEncBw+fBgAdu7cOVyzSEtLq6/vM6gDxtg777zzgx/84Kc/\n/anFYikuLt6xY8f27dtDM1zOeCEbuQ4DDNvpoh4wRaMnh2wV4LWAGI36fDBdI+QLYO0EXVSw\nngv9yF5/daMPezu1Xcak0BQuUeclqnoDqH8mR9Z6aj/DCj8H+mnOcfW0Kvt0kpesVZ6LlXJr\nHUbFigVLhIwChdPGiVx2PEj82MQFDgAABH0B6qdghJwQg4imFOpWaF+FpgmWTXkvHgZJYavc\ne+moxuMGVVgv8GYZFpv70wvNDe02c7R2ZVFi+ljaK3isVPuBf5AkqtyFS5+KMM8gX5JidcsK\n4k9dVegDtXlJ2vXLJvKB//C0cuu3D081KAocYY4kK/f4GPnQTLKuJPlKXfcIJ3RZXT/962mj\nVrWiKKxzjlQCqgSfPEqdWnxgU94Dm/LcXlnR8WTf6cb/fPXMYOeU6ube/acb//2LKxblmmdi\nxBwOhzNmIk/gOHJEabHqS0xMzO9+97vf/e53MzCeWYgs0+WDVHsW7D1gSsTia2eySyJ5K33U\njT5QIr0OTf5+KxA1hseqowsk5f5n0NsMIRE4JCfVvDOobgwG5aq32IInp/fWcpAfBYBr1/+6\nT/VnW7jYi+oVN2jv+iqwSdXTkr1nXHHOHAQLt9DRF/2jKt2E61PIEqQjuOQlWzfG8EZFM8SB\nM41Pv35hsB/qH98tvW9z/kNbR+mNQpZKkL0K/sfOLrC3gT6yf33fvW/x/3n+xNX6oba4Oo34\nxZsKb59om9u2buUmF61Bml+EOdlJUaKAXkmh+Wh+alg4s9yxPvtoacvF6q6RT3t5f3mYCxwj\noKhu2JzeX71+3q8vrMsj/b+/nXvue9eO2qaQw+FwZpLIEzg404vbIb/7C2odMCVtr6aKY7jo\nRrb6vpHeJbvAcZm8XcBUqEoEbR7ARBfGcpB5Q7D4qIxQhBKi+hTqqQJJqY7X0QrODtBO52ZI\n8FYjUtOwfUVZdh/djeZkzZZ7JnM3jI6nLv8mPgCApkid+XGmHMxexZy98oV3h4TIqES26vOg\nnWA6OhqD+NowAfUR4UA0G6hu7v2PV84MXw55JfrLB1dTzfobrkkf6Z3evu/GwPUSDbZZiVzM\n0dpff33dwbNNl+u6PV65IM103bK0yXSdMBnUTrfCjyXMS1GCYdSpbl6Z+fbhGr/44jxzUeaM\n+1UpoVEJv/jSmrcP13x8vqml0+H2St1WhW2D8gZLYDCiOVvRbnMqZMY1dtgrm3rzUvlXK4fD\nCSO4wMHxQT719pC6MQCd202Zi4PmcbgbqefAYOEDOcrAfgljtgKbWGWmwtbNpNDFgC4WHAr6\nCMblTvG9xojHGuwIeXpxegWOWFAn+Th0AgCA3N4jNfv/iDyH35ukwMFKNsg1Cm0RhZKI6uPD\nmWaw6Hohazm1lYOjB6KTMKlowh60ACDMXwV7X4KAhHZh3jJQT8THkTMB3j1W67fZ28euwzWj\nCByaYOtYBG1YLHEnCSJeuyT12inyaFgzP+mNT6sD42tLkqfk+jPPl2+fzxB3HakZ/PysL0m+\nf0u+xeYOE9VGYLh9Xfb2ddkA8OOXTgV2jQUAjORaKkUUdZyBQxNvAMThcDjTARc4OD5QpbJp\nAlUeUxY4yD1c3ejH20m9h9B03URGgDEASvULOOH0VMR52+jsX/yjyQshJnOi15wcqqBb0xj8\n0FSBsddRx/vg7RyMkIMcu04GKktyVyt43aNYnIyIsHgL1ZZK5w/6BJffxIpWTfianNmJLgYz\nl0/JlVhCuvqGz7l3vzA8iCaz5rbHp+T6nLFQ32pTjNe1KccHQVMOqaPB7f8UQFPubG0fOxk+\ne33BibK2et+f6oaFKesWRKrAIQrsq9sX3LUx50qdxeWRyhste07Uf3KhGQDSEwxP3jp/VXEY\nlSkVZcYoChxFmdFOySGgIDLV7BA74oN3+UngPqMcDifM4AIHxxd7kLxKaxt17AZ3E8geUJkx\n+hrQZgEAuOqVTStddSC7JtDbAsVckuoB/HZfGYoT98/HlMUgqqn0LbB3AAAIKszZhLlbJnzB\nSYLROSTqFNKt9cnTW5/ShxCFiXeBvZw8rQCEqgTXsQNyp1JSiagCQTWpeyGKt3+NlayXS49Q\ndyvGJrGSDSxzGvrRcjjDUG26i+Us8H7yptxSC1qDkLdItelO1Ex/l6I5D9kqwdkMQIuSbSev\nAgXIplr1aLk5TMXm3S1fec0n002XgPmfmeKxzgpMBvVvv7Hh1QMVR0tb2i3OtHjDtlWZ1y9L\nj3RLhOQ4fXKc/id/PX3gTONgsL7N9sPnjv/zZ5duWhwuPUpuXpm58+OqdotPzanA8NaN8T3u\nbgAQUIhSmdRCxPf5WpxnjovWdPb4z/fy00yZicaQDIkzJ5hEOidnLsMFDo4vehNYFS362sBR\n0f9PdzO1v4PRKyF6OcjBqi0IZOtEmneyKNSsIvc5oIEtKTSgehGwSaU2YEIxJhSDqwe8LtCb\nASflnTlZBA1m30aVO0EeVtEq6lnO7TM1Agb6eQj9bn9ifrf7k12BJ4kFi2EqpsksdwnLXTL5\n63A4Y0fILBIenDl35Miiod12tqLDYnNnJBpXFSVOxgNiCK9VbnobnP3L0XsLYVGs7scfpLbb\nfETSZQXxo18qKpMt/Rq1nARbEzAVGNMxcQkgn+Yqo1ULD22dN6p7a8RRVm8Zrm4M8sw7lzcu\nSg0TBcegFX/xpTVP7zw/2BwnMVbz0C0ZhVn9a36JJIu7K1ZjFtnkdgtCjUYlfP/+Jf/2wsnh\nThzmaO33718cwlGNgMXmfnlf+cXqLrdHykmJum9zfk5y6NsMczicmYELHBwfMHc5ndutcCDF\nv/iZek6gfh5g8PoFnGi5O4tH7bUgdwPZAfXAYiZuWeqHJhrCYx8FYwpxwZep+RDZm4GJaEjH\nlLUghMYgQFywSpy31Ft22meEGp1228NTfi+ylEFvBXh6QWPGuMWgHcNqh8PhTBFE9Kf3ruz8\nuHKwUUWqWf+9+5fMz4qd5JXl5ncG1Y0+ihId/3Rd47ffyhqMmAzqz98wtnW4oMXUdZMcEiei\nOV+p3A6prdvR1GlPNYdLQlaqWf+zx1c1ddrr22xavcccC6Lgo74QkN1ri1LpZXAhiAy1Edrt\neGl+/HPf27zz46qrDRZRwOLM2DvWZ+s04biOqGjs+d4zR3rt/S2cqpp7D55t+ubdC29cnhHa\ngXE4nJkhHL+YOFOKB6gLwAWgA4wbVSlgy26XG69Qe/XwIGYnojnQIlsGZy3osgFYQEUJgBgH\ngmESw2bA4gDiJnGFsEcTg1nbwmKag6h/9P+4Dr7u/uRt6u0CUS3OW6K97YssIW0q70Jeqt5J\nPeUDr8up/TimbMYE7sfB4cwQOz+ueu1AxfBIY4f9h88df/a7107KwdHdAY76wPD8JMf8FO+l\nJlElstXFiY/fUpwUy8v1OWPC7Q2YVwwe8kjBDoWKlDh9Spy+y9XukT1+hxjKiG32gdZpCCq1\nkCJO3FYslMQY1Y/eXBjqUYzOL3ecG1Q3+pBk+s0bF1cVJcUYw8KqlsPhTCtc4JjVUCPJZQCD\n3/I6ZPMBR1QN1Dq2/Z/p0n6qPQu2bohJxoKlIJ5RPll2gWBEw1KynfSJo4BRa6Zg/JwZQ1Rp\nrrtXc9295LChRgds6kt4qOXwMHWjLyRT4z40ZII+Zcpvx+FwAlHsuNFr93x4quHODTkTu2Zn\nj6u0tHRNkAfLLx8r6oJsk0EtsLCQczmRQlaSsrODRiWkhE36xqggkFa0IAwpMgQel1SLQpaA\n422tKgM4AbRTltM6OCSi+jZbU6c9KVaXkWBkEf6n2trtKKtXsJNzuqXjV1pH6eLE4XBmBVzg\nmL1QO8l+HTodJJ9GYS3AiHtoTMCS67Hk+v6Xsosazyp3bxWjAQAMi1AVT/az4OkEVIE6CQ3L\nQOC1jhEJ6iaTdzMS1HVeMUxdF5ALHBzO9ON0Sy1dAd7GAABQ09I7sWvuO93wPzsvFCdY1twc\n5AymijOER2UgJ6JYUZiYFm9oaPdvu7NtVaZGFaaGLCJT+2VwiMw1XN0YxC236oSxCxwuogqA\n9oGZmBkxH2Bqalqv1lv+Z+f5QUUgNyX6qTtLJl+zFkK6eoO2rR3hEIfDmU1wgWPWQlSjFJZJ\nrkM2HjcypgFdNjiqAuK6/kYqAKBORXW4uJpzwhSPUvdfAPAEadzD4XCmFFFgiEiBrU0A1OJE\nVoz1bbZfvHbWK9HlVp3Ty7RiQE0BqlA3pZVuU0pnj+uFvWVnKzrsTm9mkvHeTXkrihJCPShO\nPyqR/d9HVvzkr6fLG4aeEVuXpz+2LXzNg/WiwSk5iIb+EBC9imfK5ACgsZlxeIhOAwxfmXcQ\n9SAuB5hstUVzp/27zxyxD/MNrWzq+f4zR3/zjfWR2xvFHB1U+hmh2S2Hw5lNcIFj9kLB+puM\ne6cOYzaRZAV321CI6dC8FRgvZRwRksBWSu42AEB1IhiK5nQjAEEHXv+9uP44h8OZfkQBF2TF\nXqjuDDy0KG8iDar3nqzvMyu1u9nzxxO+tKbF7wQ0r5tIL60Zoaq59zu/O2x19O+3d1ldZys6\nHtiS/8hNEWAxMEdITzD85ql1J660VzT16DViSU5sbsp4yzpmFAGFWHVcr8cymMfBJt2yjaje\nV93ow0NUi5g/yYvv/LhquLrRh8sjvXag4h/uDdP2KKMSb9KW5MRdqPL/ojNoxRWFiSEZEofD\nmWG4wDGLCfZYHf/jVtBj4t1gLyNXE5AHVfFgmB+209ZwwdNFbbvA2933igCg5xQm3AqqCM78\nnAwYXUCdSmYuptnW3ZDDCVse3Vb4vd8f9Uo+qRYlOXHrS5ImcLXGjiHJ8o0LsS29qodXtGbG\nuAEANXFoXo/GgkkOePr47ZsXB9WNQV7ZX7FlaWpWEi+xDBcQcUVRQgRl1ohMFauJl8gryZLA\nBKIel2wPPG08vVS6g8SnIPnxcp3yxS/XBrtpeNFlddW32uKiNSlx+uHWId++e9H3njnSbnEO\nRtQi+/Y9i6P0kd2sl8PhjBEucMxeMAaoWSGM/g1fx3g50BeifrZsbckSNZ4ESz0AgCkD05ZN\neW4Fdbw/qG704+2mjt2YfP/U3mjiWNuosxYkD8RmYMy0p5FjyiayVoPb52eCMfMxOnyXQBzO\nZHC4vOHWQ7EkO+6/vrzmt29evFLXDQBqkd2+NvvzNxQgTsRW0KD1WS0crjEerjHqRNLrhJd/\nGMyTIyywOb3nKhUyWYjoSGkrFzgihUs1Xa9/UlXTYjUZ1Evz4+/amBMm9hwCioIgAgBhDMqt\nBP5SmoqNPZUgWDeZoF1mxo5SvRoAAAF09rreOV5b3WJliLnJUbeszIjWh1HSbrvF+bu3Ln58\nvn+WmxZv+Nr2BdfM6xfC0hMMf/qHTX//uOpidZfLI+WmRN29MTc5LmK8aTkcziQJr7kXZwpB\nzCFqA393Ky2gYhtwL5AFwAmgA4yZco/u8MLWJp96HmwDFTd1R6jqILvmEdBPJElbGXcruNuV\n4m3gbgV1qJMkZdhkn30AACAASURBVK98agdVHoKBUmFMW8iWPwja6ZzWiwY274vU+in1VIDH\nCpo4NC/DuIXTeEcOJxT02N1/3l22/0yj1eEx6lSbl6Q+fGNh+OwcFmfG/Orr66wOj8XmTo7T\nT6a5yTXzEt49WusXdHhxZW7y5MY47VgdHkUvEgDosblneDCcifHXD8uf331l8OXZio49J+p+\n8aU1YeWzgMC0Qo5bbpDINhAR1GxcbWINAIoVx1PgCF6QZrqilMSREKP73rPHXB6pzyfkcn33\ngfNN371rUX5qWJQIuTzSd39/ZLgBbUO77V+ePf6zx1ctHqi202nEz13Pt084nDkKFzhmL2hE\ntpzoMtBAHiMmIitU+KVTM8lXAAZndXpk8wEjuZLCZaHqD8hSDV4HGJIwfQOaB23JSD7z0pC6\n0YetVT7zElv71JhTRkfDG8RQEwC8lpALHPKpHVTxyfAINZyX3X9iW74xZT8BRQQNpmzBlC3T\neAsOJ6RYHZ6nfvVpY4d98OXbh2tOlrX/+ql1Rl24aBwAYNSpJj+e9SVJKwoTjl/x+To1GdSP\n3hzuuX4xRrVKZB6vwh54Uizf5o0Aqpp7/7ynzC/Y2GF/5p3Sf3pwaUiGFAyGGq2QK5NTBheC\nwFCP49lDQkwlag3sZIc4Bc7ud27I2Xuy3uXx2QlTiazT5uoP9s8IyO7y/v690p8/unJi2V5T\ny96T9YHtdSSZXthb9l95a0IyJM50wcIiJ4sTcczqjXoORiNbicImZKtQuBbZYoW+YtRJ8vlh\n6gYA2Ek+DaDcTTACsDbKJ56mltPg7AKvEyw1dPElqtrTf9TSAL1NCu/qaYCeRoW4rNDgbXRG\nMChhod5cctup8lBgmNrKqaN6xkfD4cwqdnxUOahuDNLYYdv5cUAjqhnA6wRLLdjbgaYgmz0Q\nRPzRwyueuLU4JU6PCNF69fXL0n73zQ0pYZ8KrlEJGxYqdKfWqoX1JeGefsIBgE/ONyvm4By6\n0NxnfBtuMNSKaBLQOC51AwAAohGLfLemRIDC0lrYc6L+0MXmLuvEW5+mJxh++tjKjGENU1LN\n+i9uK3Z4/GY+CADNXY7q1mDu9TNKaY2yRUhpTXewohsOhzOn4BkccwE1YNDKSaJqpbBEcj2y\niMzuk8vfAcn/eU91H2PSEtAnkqMj2BvJ3oHR/VYU1N1IJ/5OzVdBcqMpGRfejHkrx5HdoEkG\npgY5INWZaUCjMKueSainKehqp7sBzDkzOxxOZEAOm/3tl9ynP5W724XENM2Gm3VbPsO3VgI5\ndVWpNg3g1NX2L2ydQT9dr4PK36fG4/1F9toYLLgVExfYnN63DlWXN/QwBoUZMbetyZqkZ4Eo\n4N0bc+/emOvxyioxkrZMvnz7/Po2a1n9kFOjVi38w72L46K5f3Y4cuhi8+mrHb0OT2ai8aYV\nGV29yqt6t1e2OjwxxjByi5gKEhFjAboAHADaxg7tz1+5eKmmq++YRiV89vqC+zfnTezSJTlx\nv//WxvIGS2OHLTlOX5BmOnqlNdjJ3VY3TMSPeIqRZGUZQyYionDIMeFwOKGFCxxzHgrWNXbc\n3WTDAq8DevxrwgEAgKjzCuoTQQzelFTVf4iar8i7/xvk/t5p1NVAH/0R26vYqjH7g6IKYzdS\nxwf+4diNgKH+oxuha92kG9pxZiVyT7flJ1+T2vpTn7z1ld6Xf+M+e9j0rf8Axj8zPjhcyjlf\ndpd/L8ZphEg+82ew1AxFnN10/qWGlO3f2dE7uN978GzTG59U/+SxlZnD9m8nTGSpGwBgMqif\n/tq6vafqz1V0Wh2e7OSo29ZkhZV9A6cPh9v7oz+fHC4dvnawYmWhcl8VjUoIH7+bKUUFkAgA\nLo/0/WcOtnQN5di6PNKz713WaYTPrM2e2KVFAYsyY4oy+x3oTQZlecgrySeutJ0ua09L0G8o\nSdFrQzaZyUuN3ne6ITCemxLNJmEqxOFwZg2hXmtxQk+EzUpHweMILFUddggwNptELXid/kdV\nOozJ7vsnHXl5UN0YhEr30bwNGDvmbiOGYhRNZDkC7jYAAHUCmtaEPH0DADAmDUQ1eBWM9DA+\nd+bHwwl/HLteGlQ3BvFcOuU68oFm7daQDClsSYs31LQoqMPp8VPgCDhGqOOyj7oxAFbu7bL6\n2BO0djt+9vKZ335j/UwNLbxgDG9cnnHjckXjbU648Oy7V/wSo+xO76cXWxlDOWAnf/3C5Mn4\n5oY/B882DVc3BnntQOXta7IVcxcIyCN1S+QCAIFp1cw0cjpqYXqMSa+22IdPEqjX6nHYPX87\nUNH3+rn3rnznnsWhat9744r01w5UWAL8gCecxsLhcGYZs2txy5kAQay8J9pNNtRoooAFke20\nsQAAghqLbg08iEW3g6ACALB3U5fCzgAQQeOlcQ4mFRPvxPQnMf1JTLwzHNQNAABBzebfFBjG\n7JUQzYvPQ4bF5q5rtYZn9bj73JEg8aMzPJLw55ZVmcrx1crxacGimMUGKTqbSe3frrK8wVLd\nHJn5epw5gCzTB6fqA+Mer7SyKMFvuz4z0fjErcUzNbTQUNmkbGHe1u3osSvsW0jksnqqHFKr\nW7a4ZYvD29LrqZZppFZBapE9fnOReljxmt0u2e2e4Q+nzl7Xv714UlFqmQGi9eqfPb4qP22o\npYtRp/rGnQs3LgqPWRaHwwk1PINjrjPQTdbPlEETpJts2MNUGL+AWs/6xwUNxs/v+yemr0R9\nvHx1N/TUAyCY0lnBjRA74D3hCUjuGIDcjtmxMYTFNzCVVj6/C9x2AABBhYVb2IKbQz2uOcrp\n8vbfvnmpb9tfJbLt67I/d0OBTh1GX86yTdlYTrYG7xY0V1lRlPD4LcXP774y2KFDLbKHbyq8\nZt4MbnUGt0YWUEFBa+lyZCdPZ4toDmei9Do8NqdyeVeCSffrr697/ZPq2lZrtF61rCD+9rXZ\nEVcqNV5YcIMJxdQVu7dRJh9ZUya33dtkVGWNcJfFOXH/8ciKt4/WVrX0MsQL5QrWQi6PtOdE\n/edvCI1ZW15q9K+/vv5SbVdDuy3WqCnOjJ2lpUkcDmcihNEcmhMaMArZcqLSITMONCMrBojU\nRwXm3UKODugdtucjaLDoHlANSxGPy2Wrvtxvv+c3XTDEgiCCpDCjwugQt3edOhDzNwq566in\nGWQJo5NBnGWWbBHDscut//r8iUHLNI9X/tvByvKGnp89vip8jNKE+CRvrcImv5DAt8sUuGdT\n7rqS5E8vNLd02ZNi9esXJs90V5Eo5d9Lp0vd5VL4Sw9Wcj/bIBncjeDpBkEHmlRgwf2YOGGD\nTiMKDBVNJaP0qvw003fvWzzzowohxVmxivGsJGNg42eJHIrJGhI5JXIJOJKfbny09pEb5gEA\nEd30j+8pnlMb0qYqjGFJdlxJdlwIx8DhcMITLnBwANCEuBrAAeQE1AOEpYe8p4HcV0GyAtOi\nmAKaIsAg5v8qPVv6JLWcBUs1eB1gSMKUlaBWctFTXEGKGsxZSeUBjVS1UZg5uyZSTMCYMVuK\ncKaHP717OXDufrq8/cSVtlCVNweiWXejt7ZcMT7zg4kIUs36ezaFzNEGE0uo8gNwdPrF36rP\nCFwmxpu0BenKhYqzCncrdX0Inv7GE4AqNK0C46KQjokzOmqRLcmPP1nWFnhoZVEiADS02z69\n0NzS5UiK1a0rSU6bQbObkLBuQVJhRsyVOv8+qQ/fWBh4sl/uht+hkQWOQRBRLTKXf+NYAACt\nmjfS4nA44QgXODiD6ADDdEeL7MfA3W9tBXIvedvAU4PG6yDo4xkxaQkkLZnY7diq+2VbFzWV\nDoX0Jnbtk4NtVjicKcHq8FQFsT84X9URPgKHbst2b81V16E9QyFBNNz1RVX+gtANihMcpmJL\nHpEv7RiyGmUiZl+bGVvIKs8N92UUBfzWXYtmtykjAIDsoPa3QR7WWJQ81P0JMi3oZ7B3L2dC\nfOm24m/9ttvq8Fmrb1uVOT8r9tUDFX/eXeaV+svB/ryn7OEbC0OoLc4AjOGPH13xh3dK956s\n70tCTYzRfen2+etKFCy0EIIKEBhsi0iJJfnmo6UKvWOX5seP/SKhwuH21jRb7S5vdlIUbwLN\n4cwRuMDBmSm8LrK2oSYKdOPcLfQ2D6kbg0gWcp5H3fKxXqSvK0ow/1E/1Dp207ep7iy1XAWX\nHeMyMH8NqHj7QM4U45HkYIfc3qCHQgBjUV/8vnbtVveZQ3JXO0tM1a67UUiZQddMznjRx7Pl\nT4KllqytoNKhKRM00dfnQHZS1At7r5Y3WASGhRkxD22dlzEVPWJDAhFdbehpaLeZozWFGTEa\nVfAFm63UR90YvELvGeQCR9iTlRT1zLc3Pvf+ldPl7RabOysp6q4NOVuWph0tbf3Tu5eHn+nx\nyn94pzQ7OWpFkCayswOTQf0P9y5+4tbi2lZrtF6dFm8IplEKTIfAyN9kDRAEEccxpXnkpsJz\nFZ0Ot0/p7oLs2GsXh3uV4hufVr+wp6xPHUPErcvTn7y1OLCWh8PhzDK4wMGZfly98vk3qPZk\nXwNXNKXhkrvRPNY9FvLUKR/w1MEYBA7qukK1+8DRBgCgT8SM6zB2TJ5YmLEYM2ZXTQonzIgx\nqGOM6m7rUI10sannmoTuGLWnIJbAnalcWhUiVMVLVcVLRz+PEy4gmLLQ5GMlmJ9m+reHx6wL\nhzHlDT2//Pu5q/WWvpdx0Zqv3L4gWA8F8nQoX8XTCUAjt8zkhAPxJm2g18Z7x5S7Bb17tHZ2\nCxx9ROvVo9pPIDCtmOjwNvvFdWLSuD72uSnRT3993TO7Lp0ub/dKZNCKt63JeuC6fBbeyV87\nP67637eHmt8R0e7jdc2d9p8/sTp8LK44o8B4GRRnInCBgzNWyGah2lKydaM5lWUtGPrScXRQ\n+0VwdYMmBuPng843ZVHySB/9Cnpbhq5jaaCPf802fgPjRjLxHkJp522k+PAxNx2m6t1Dr23N\ndPkvkHsLJq0Y0605w6DGs1S2j3qaQG3A+Dy24FbQRWYv4bABEbevy3l+9xUAEJC+saB8a9rA\nX4qrST58EovvxMSFoRwihxN+dFld3//DkV77UM1CZ4/rp389bdSplhUo5swH6ayByNWNyKWh\n3R4kbpvhkYQzamYSVGqn1C7JTgAUmFYrxAvjSd/oIyvJ+O9fXOmVZIvNbY6OgIRWSaa/fqhg\nHXW2ouNcZcfiPPPMD4nD4cwYXODgjAnp2Lvegy+Du7+FKsanqW77GqbmU93HVP0BUL/7FFV/\niNnXYcbGwTdS7THobfHfJJMlKn0X1315TPcO5nU/qge+10G1+wLDVPMBxi8GYfoaB8zCLUH5\n3OtUfqD/hddFtZ1S43m26Sk0cZvSSXH/5rxeh+eNT6ruzqobUjf68Drp4msYlQo6PhXjcIbY\ndbh2uLrRhyTTK/vLFQUO1KSQ/YrChdQKtgWcSEGvUd7a1Wt8ZrbdVveVuu5ehycr0Tgn/HQD\nEFBnEDOm5FKiwCJC3QCApg57j12hgwwAXK7r5gIHhzO74QIHZ3SkCx979z43PELtDe6Xf6y6\n/ymo2u1zKklUtQcMSRjXb+hNHZUACut9aq8c491RlUWuMoUD6uyR30i9dSArWYhLLrLWo2mK\nfciopVL66BVqKgfJi4lZwtq7MWeqK1y8TrC1gqgFvTloE5lpgCwNQ+rGsMHQ2b/jxqdmbBiz\nEsbwyVuLb12daTr/q4BCaQDZS02nMPeGEIyMwwlXyhssQeI9ym/Qz4Pes+Dt8gkiQ9PKqR4a\nZ+a4Zl5Caa1/M5G++OC//3aw8sW9ZU53/x7Molzzt+9ZlGqe2bbNnFBApNBaeNRDHA5ndsAF\nDs7oSEffVog6bfLxt5hiBWjTcRgQOEBWaC0GAEDSWDMdxHjQLgTnhT4Lj4FgAmpG6+AgK4v3\nAADS6OUt40IuPyG9+cvB/yw1XvXu+Klw7efYilun5gZeF1XspoajQDIAgDoKC27G5GkzRCAJ\nrGfJWQWSFYQosMqAPj/+/rPaK8Hj4M1lAECiXrfcKpMTARnq1SyZjScHOM2skynI2szePjVD\n5HBmCzjeAnoUMeEzZPkU7Ff7I6o4jNkI6nC3SOSMwJ0bcvafafQrSElPMNy5Iafv328eqv7D\nO6XDj56r7PinPx575jsb1WKQqqW5QWVTz/mqTqvdk5Uctbo4SRRmW84pAKSYDUadyq/5Th/z\n0nl1LYczy+ECB2dUiFqVrbzkjlYWp6BwkKNj8GmJ0akEpwPPwejUsddxoLYEVCnkqgC5B1CL\nqlRQ54z6dtSaKYiGgrop7W1Gsrz32UApR/r4VTZ/AxgmnxNLdP4l6hxWTerupYuvgSxh6jT4\nBZKb2t+CQVs+2QkawKIsulwToHEQFzgAwCO3ueV+FzcCkKjXIVm1QpaAUWO9BDJgKpCUJDmR\nt7XjcHwoyow5dNHfNxEAirOCr1sEPcbdADEbwdsNgh6EMf9tcsIVo0719NfWvbCnbP+Zxh67\nO1qv3rI09Qtb5xm0IgAQwav7A/qvATR22D4613T9sjlaXOmV6NdvXHj/eN1gu+iMROMPHliS\nnzbbindEAe/ZlPvc+/61acWZMUvzeX0KhzPL4QIHZ1QQmKCYiIFCkM+PMLQkw+zVcHUfeBz+\n52TngqcWVBljlTkEM+rH+UwyJIMxHawN/vHoLNBNpcU6tdWStVPhgOSRay+w4nWTvX5XlY+6\nMRiv3Isp18BUu4GT9SwENB1AkwESYqnVN8dbUIFmrq8TCDxuuSUw7JIa9GLR2K+D5nnUekHh\nwGAyFIfDAQCAW1ZlvvlpdUePc3hQFPCz143WIYtpQJ00jSPjzCxRetVXty/46vYFDrdXp/aZ\nkPTY3e0Wp+K7yhssc1bgeGHPlXeP+mxZ1bVaf/jciWe/u0mnmW0rgvs35xPBy/vKXZ7+Gez6\nkuSv31ky7hQwDocTacy2rzPOdMDSC+Xq84FxTJ8HoLCN5tOHVRvN1n1JPvkX6G3tjwgC5mWi\n2Uv2IyCUon4dsHH1wiRwVZK3BWQXCDGoLQj+dmTz7pGvvAq2pqGYMY0V3DWe240Bl7KX+yiH\nxo6lJsjFe8DZBbpRGsWNG2eQ28VFga/AgRnXgDA3+8nLYL1IzjqQ7LLKwHQxssr/Q0jgkck5\n9kIVzNtKnRXg9ZEC0TwPE+dPzZA5nNlClF71n0+ufvr182fK+6XYVLPh63csmJ8VG9qBcUKF\nn7oBACz4IlYI7+amE6PN6qrssFvdXq3IEoya/HhD4E9AkmnXEYWE3I4e58fnm7cuT5+Rkc4c\niPDgdfm3rM682mCxObx5qdHpCYZQD4rD4cwEXODgjI6w6T65rhQk7/AgxqeL6x+Sz/8JHL4e\nATozpvvkLGBctnDdP1LbVer4FEQXxkSDZqCDiWQh+yE03jCUx2FtJ0s9IMPYTIUupOSm3g/B\nO5Bf4Gkg12U0rAZ1jvLQNTFs4ePUeRlsjQAAxjSMK5r6FiemxGBHMDqOyg9STyMwEc05mL4M\ncPylvxRoPjmGQxNGVt71ApWPloHmXLZw+9TfPfyRndT6Brjb+l4xF2it4InO8xj9beoJghjQ\nKKJPYKueovL3qeMKeJ2gjcH01ZixfvZ15OFwJk96guHnT6xu7LA3tNvM0ZqspKhZuWrlAIDT\nLb2yv/zY5bbOHmdqvOHW1Zmbl6SNugcfpVelJxjq2xRaxibG6v/77+ermnp0GrEkJ+6ujTmB\n+khkcaGpp6Kj/39qdUG7zV3f7ViXY9b4Wo109jgVPSkAoKald9pHGSJMBvXyeVOZtMvhcMKf\nyP5O58wMLL1Q9eAPvbv/NGDGgaxkvXjdF0AbxZY+STX7qPUseOyg0mPCIsy+DsSAXWsmYEI6\naKMAjP4LNqkbpE4QzOBxyGf/TrXH++PIMHcdW7gd2NCnlOwnh9SN/pBEtiMoJgILIswjQ/N8\nME/jNjhGx2NWCdX41xdgfJJ85U1w9Bv+U+UnePUAW/sEaKPHd4OoVOW4qAXdNOxYCkaQFCaF\nGFeI8wuppwlUOkwowPSlc3PtTd2HBtWNQVQ9FZI2ThZ9PoQMx9mKWBuDJfcjAEieuZoaw+GM\ng1SzfnhHjMqmnk8vtLR2O1LN+k2LU1LNfLc24um2ur/120ODTqKdva4LVZ1HS1t/8ODoHtsP\nbZ3373/xtwBLidP//u1LXql/b+DU1fb3jtX955OrI7e1SofdPahuDNLr8l5u7V2c6uOsoVYF\nbb6mCX4oQvFKcm2r1emWMhKMUXr+POVw5hZc4OCMCZY5X/34f1FPO1i70ZwKmoGpgKjDvFsw\n7xaQ3CCMuJyT+/YHlJbEUi8IZvn4i9R8cShIMlV8LMsSW3rf4CXArVQ9QRK4a0FbPN7/lA/u\nBvB0AAqgSgTVuMV+YdtXpL//nFqrByNoisdE46C60T/S7jr51Cts7RPjujia55ExCaz+Rg+Y\nuX46msWibh65A00lAA3FYM6ci5KGH4ONGHwRHK1yVH8mEQGIGIUw0UkVVzc4nPFABH967/KO\njyoHrRNf+uDqozcV3rVxituBc2aYF/eW+fVJAYD9Zxo3L0ldPX8UO5VNi1MB4H/fLu2za0GE\n9SXJR0vbBtWNPtq6HU/vPP+zx1dN6cBnjqYgViONFudwgcNLpNGKWclRNc0KyRqL82aV7+ae\nE/V/fLe02+oGAMbwllWZj95c1Oc+y+Fw5gL8r50zDjA6HqKD9B8ZWd0AGOnDhiL1NPmoGwNQ\n9RGYv63fyVJ2AXkDzwEAkq0TX3hLvdS9HzytQxFtLpo2AI5jkYnGOPELP5FLP6WGMpAlTMzG\nhAT52PMKQ22+BM6e8SVxoMAWP0ylO6hzwBOeCZi5EbO3jOMiY8dQDO4WcJQNHwEYF4M2c1pu\nF1nI7mDth9E71HtYQJ1GmG31zBxO2LL/TMNrB3xaZni88u93lRakmxblzqqV21zj0EUFtR0A\nDl1qGVXgAIBNi1PXlSTXtlqtDk9mYtShi80fn1cwDjtd3m6xuU2GcebczTiVTT3Vzb16jTgv\nIyYuqt/N3SUpV6q6JVkmYoheok6Xx+GVAWD7lrxfv3JWkn06oq1dkDSbBI53j9b+99+HbONk\nmd4+XFPfZvvZ46u4u2jkweZ0R2fOhOECx5zFDSACzOAXhxgHqAIKrP8UQEyA7nPK7yKZLI2Y\nWAgAgGoAhIBWpQCAY7ZyDLwBde0Fr28PFGcloYCmTeO7EjI2fwPM39B/3fIDQe9o68DxVqlo\nY3DpY9hTT9ZmELVoygDN9DV1Q4zdDPoCclSB1AtiNOrmgTqoz8jcgqkARUWhjYlxIotFYAz1\nIprmZv0OhxMS3jtWpxh//1gdFzgiGotNWVC2WJXjgYgCy03pf+B29LgUzyGCzl5XOAscbd2O\nX+44f6KsvzpSoxLu35z34HUFiKAVlSdyGpExRAJocbi9A4pGSb75O59f9sqestqmXgDQa8W7\nN+bee23ezPwvZgAiemFvWWD8dHn7ucqO2aTjcDicEeACx1xDBqoluQbADYCAMYjzAMe52J4g\nAmqXkOO4XxS1CwA1I/Y6HTiEAqhSwNOocIJ6orvl7mZ/daMPRzlErQamUTg0RlS6YEdQPdFa\n3+h0jJ6pvABNOmp4DkIAskxiGnoUSqUEw0KBcRmIwwkBTR3K/aoCqxs4kUVijLZR6ZebFBv0\nCTsCg1kPfiBCrHESj/tpxivRPz97vHpYaYnLI/15T5kosvuuzUsz6cqVPudpJh0AWD2S1zdf\noyAr5oePr5S8soEwMUY3y5IamrscnUFkrEs1XVzg4HDmCFzgmFuQfB5osBaDgLqIjiG7BnBG\nuuupc5DpyXkOpG4AACEaNSWgSgMAjM1Wzs5gAsYONadA/XLq2Q3k+/TSzgdhouP3dgU5QODt\nBvXoGbDBwMRCYALIAX00DGaI4svgyEPubLP97ffuU5+gXjA9spHF+KpU0dfwJBcOJ1Tog1TX\nG7TcziayuW5Z2ot7FWyPtixNm8DVVhUnqkXm9vrXdCzKNccYZyJ940RZ2/vH6ho7bOZo7Zr5\nSTetyGBj6P5z7HJLtZJxxt8OVN6zMTdGpypKjLrc6nNCXxAA3LJyAYsgsiTjhFNfwxdZVkjy\n7UMKfojD4cwyuMAROUgd5K0EuRdQg0ICqPIAxmkwSV3D1I2hKFEZ4kzZa4lJaLwBQAYiH4NM\nYzxmLh9qoTIAFmz2SYUQojHmNrKfA28zkBsEE2rn90kkE2QEk85J+nfqYljxzfLFXT5BJrAl\n9/LihYhD7u7o/vFXZUsHAFCPp/t3H2rXzlMXpYmpqaA2Y9Ri0Pr3iOVwODPG8nkJiivAFUX9\njtFOyeuWJBlIxQS9qOJfwZHCfZvzL9d1H7881LiKMXz4xsKizIAu8mMg3qT9ymcW/Or1C8PX\nunFRmm/cuXAKxjoaT++8sOtIf/ZfeUPP0dLWD042/OSxlVr1KJONisYexXiP3d3S7UiJ0xcm\nGhOjNFUdNqvbqxGExChNdqy+PzVjtizqT11tP3i2saXLkRyn37Q4ZWm+sh9cUqzeqFMpdsMt\nSJu+wl4OhxNecIEjMiD3ZfBcHnoptYG3FrUbAMeRVEmkVIsBANQD4IEJd3yYCCxwjc+W3idr\no6n8QH/Wg6hmhVtx3nX+56EWDSunbCCq5CAD1IAqbpLXxsIbmCmVLu8mSyMwFZpzcMGtaArS\n85UTxjjefblP3eiD3JLjQKnjQKl++8P6224N4cA4HA4A3Lc575MLzc2dPrUM+WmmW1ZnyUSd\nLodTGvLN6XW74rQ6NZttfTFnJWqR/fujKz8613S0tLWz15UWr9+2KnPQU2MCbFuVWZQZ87eD\nlVVNvXqNWJITd++1uUbdtM9/jl1uHVQ3BrlQ3fnqgYqHts7re9nYYa9t6TUZNTnJUcNVDxa8\njGTwUKxOFZuuIPqoBQbegExSAHXkeDcS0f/bcX738SGfnXeP1m5blfmNOxcG/mBEAe/emPv8\n7it+8bzU2ywq7wAAIABJREFU6OWF426Qx+FwIhQucEQCcjd4/L+sQbaS+wJqrhnPhRQecsMO\nhTqVV1Cxktuh8HqyNAETMDoFxOmviRVNoC8Ge+lQhCEAQ30hkAw42RkAJi/A5AUAxLM2Ihr3\n5TOKcU/pabjt8zM8GA6H44fJoP7NU+tf2FP28fmmzl5XQozuhmvS7t+crxaZn7oBAF6SO5z2\nZL0R+ddyhLBxUcrGRSlTdbXclOjv379kqq42Rg6ebQoSb3xo67y2bsfTr184WtqfYxtjVD9x\n6/zrl/VnpwZLV4k3aRNjRvEiMYiCxe2VyD+RwzRa2kj48MGphuHqRh/vHq1dlGveslRhx+iB\nLXkeSf7bgYrBWqTl8xK+fc8iYQzVQBwOZ3bABY4IgLyNylmG3kbQLBv7yhnREPCM60MFEDb2\nWio9xs+ooTdGrwHBRLbTQG5gqr4fJzkvg7sWjatAPSWlB/yxGuG4HIphcjtneCAczmzBRXIV\nQF/uvQlZ9iQfQ1F61Ve3L/jq9gUer6wa6CtBRA6vQrK6ROTwevViqGV9zpyhs1fZ+bKjx+X2\nyt//w9H6tiGj0G6r++evnFEJbNPiFABYVpCwMDfufKV/Eu5DW+eNahHKEJJ06g6XZ7CbLEOM\nVYt6MWIEjn2nA63lAQA+PF2vKHAg4kNb5926OvNybbfDLWUnReWnzYyVPofDCRciJkVtTkPK\nz0UASbFdZVAwCUDJRgvT5vYKnIGhBONuBUHj82OQ7dRzELwdQd/HmTMIKZlB4lkzPBIOZzZA\nnSR9ClQHZAGyANWSdAgomOXz+FAN65rppSCqPoCXlM0XOZzpIFgDF3O05uDZxuHqxiAvfdDf\n7hQRfvTQ8ptXZuCAnhFjVH/n3kU3rhjTBoyKYbJOnaJXx2tVSTp1ml5jVEWMugEAbd3KGwxl\ntZagf94A5mjtupLk65elcXWDw5mD8AyOSACDOF2jCDiu36CIbAnJ5wGGPS0wBdnsaYE+JtwO\nurSf2msAERNzsfhaENXkvAQQON+VyXEJozaEYJCccEK7+TPu88f8o4zpNt8WiuFwOBGNTPLF\ngJJJL8kXUFg/tWr7CDnpCkZQHM60sXFRyt6T9YrxK3Xdim+pabE63VKfGYdRp/rW3Ysevbmo\npqVXr1FlJhnV4vh2KNWMqSNzTzPaoNzgxmJ37zpSc9savs3A4XD8icxvuzkGiqnKcz4hSHyk\na5lQWIusBDAbWQGylchKgn4MnD1Ud4rK9lPjBZAUsnwjEWqrkl77J/nYDqo8ThXH5MOvSK/9\nM3Q1Bu0X6w3izMqZS6gXrzbc/xVUD23BoU5vfPgfxNz5IRwVhxORkAVAsbbLCWSZ2lsJyMQg\nZooagW/wcGaOVcWJ21b5ZwLOz4q9f3P+CO8i3/Jkk0G9KNecnxY9XnUjolm7ICnYobcOVc/g\nQDgcTsTAH/CRADOhuojcpb7BKFQvmNjlAFNGrdukqwflc2+Dd2AaaoxnKz6LiQUTumPYIMvy\nh78Hu+8c2toh73sGr1uh/JZJ+4xyZge6G+7SLFvvPn9M6mgRElLUS9ay6NhQD4rDiUSCFV2O\nfGiCxKi17U67X9CoUqsip4sEZ3bwzbsWrl2Q9P6xuro2W4JJu7YkadvKTMZwXnoMgH+DFQDI\nTDTq1NM+SyeCjh5njFEtCmH6F3H72uxX91dYbO7AQ3VtNiLCUWe0nMiFt7viTAgucEQIqkIU\nEslTCXIvoBqFRFDlTl8CDtWdlk/9zSdkbZc//l/h5n8BfQQv6qilHHpaFeIdtWBbrdxyV0yc\n7lFxIgVmTtJey2tSOAAA0NeYg2cBTIQRzESn3u5aK4iJOoPF7XJLXgIQGYtSaQzcXpQTClYW\nJa4s8p9UbFqc8tcPyxs7/G04Pnv99G4p2ZzeF/aUvXes1umWBIbL5sV/+bYF6QmGab3pBFCL\nbFVR0p6T/o1U+g5xdYPD4QTCJ2eRA4sdZ1NYX2QPWevA1Q0aExoyQFCuaeyDruxTiHpdcsUn\nbGEkL/Cs7cGOoCcGtBZ/P1dUo25iaTKc6cdjo7qPqKceyIvGVMzYANq4UI+JM/vxXDzmfPdF\nqbEaAIT0PN2tD4mFS33OkBpIbgFwAeqRZQDjH0tfMAZAq1SlogM0TccN1UxI0OoBgIACW8NS\nVz1VHQZrG+hiMLUE0xZNxxg4nGBoVMJ/PLHqf/5+/kRZW18kWq9+bFvR5iUKLUKmCo9X/u7v\nD5c39LUxAkmm45fbLlV/8h+Pr3a4vZJM2clRwYxRZ54lBWZFgWNJfvzMD4bD4YQ/XOCYE1BP\nBVW/A+7+0gxSRWHmTRhbFPR8i3LDduhWbtYVMWiMQQ/p4jGmmKxHwNPSH1ElomEVCMHfwgkh\nvfXy+ecHS6iot4FaTmPxfWguDu24OLMb14E3HDt/P/hSqrli/c0/6R/4pnrNjX0B8pwAebD1\nUhdJDSDkohj0y3ZOgsgWkHzG12dURLZguvt5KagbF9+TL74HAx1VqPIQpi1ia7/I86I5M0lS\nrO4nj62sbbXWtlqj9aqCNJNOM73z870n6wfVjUFsTu83fvOpJBMAMIa3rs58bFtxn8tpaNm8\nJPXtQ9WltT5urDq1+MhNhaEaEofDCWe4wDG78LiouYJs3RibjIk50Je552ilq68CDZtKenqp\n8u9Y9DAY0pSvE2xuF+H52JhSCGo9uP3rsUFvwoQcYAKabgTZDpIVBCMwfSjGyBkLJF/ZOWQQ\n04fspbLXcWXeyNlJHM6EIbvVuev5wLjjjT+orrkW1RryVg5TNwaQKoElADPPwAgjBoxDYR3J\n1QAWAASIRpY9HfUpI0Nt5fKFd/yDDefoyodYvHWGBzM9eAicABpU7BDPCTMyE42ZiTO0p3Km\nIuCbCgAA+tQNAJBleutQTVev64efn0Tu8BQhMPzZ46te+uDq24drnG6JMVySZ/7y7fOzkqJC\nPTQOhxOORPZ6lTMcufRTaf8LYOtP08CUfOHGJzAhk1qO+agbfZBMLUcw9y7FS2FCHjWcU4qP\nZPcdAag0bP3n5P1/HNyvAwBgjG14aEjTYXoubYQ7tlawK3ipgMdO3ZVo5rvlnGnBW3mR3Aou\nmOSwSTWXxYLFIDcCUGAaAskNyAUOfzTIQrz7StXHFeNy9TEhwgUOAodEVUT9W/QIUQLLQdAD\nQGevS6cWpjtBgBPmuD0B00IlPj7fXN3cm50ceh1BpxEfv6X4sW3F7RZHjFGjmkt9ZDgcznjh\nT7hZAlWelnb9yifSVO597ceqR/+LHC3Kb7G3BMsGxpJt1HwZJF/P6qgkzF0zJaMNIZi/msWk\n0Mk3qa0KkGFiLi7fjrFBMlk4YQm5rUAEMgEBMIDh3RDcvaEbF2eWQwHNOPwPkVOxyIJqT1Dd\nKbbqHtCGnYFfOCLbyXkepFYgNzATauaByr+/5tTgCNId3B7Z3cEJ3F75IoB3WKTX6bm063D0\na/tr+rpR5KdFP3HL/CX5XHebo2QkGAGUJ4d+XKnrDgeBow9ESIjRhXoUHA4n3OECxyxBOvqm\nQtTeI5/bB6YgOnfwBqgYk862fJNOvkqdfa3LELOWs8XbZ0fyP8Zn4Y1PhXoUnIkiuaHqI3B4\nhiIig8FeeupwmYdxIgQ3gGqM1g9CYlAllCWmAwCgBkhJBLE76MJRubGM3fP/gTgbvkWnEamb\nbPuABhbnUifZj4C6HXXLpv5e6iB6kyayv0ZkahqubvTxPzu6950eWtCWN/T84I9Hf/j5ZWsX\nJM/s6EIBecFTR1IPMh2IKSBE9u93SrhpZcbOT6o8Xnn0UzkcDifS4ALHLIFaqoLEK1lqCVnr\nAw+hIX2EC2JcJt7wXXBZyWFBYzyI4WKmzZk8Vc29Zys6emzurCTj6vlJGlXoLcTGDl34G7WW\n+oS8MpAXNCKoDBiTG6JxcSILD1ElQDOABMAAEhDzALQjv0fIKBDS86T6Cr+4mFciJGUAALBk\nkCoV3llVBwDUWU+lB3HhDVPyH5itkPP0kLoxiLsc1NkgTHE/GkxbSDUKVSqYtnBqbzTDEPkn\nslU0ePed9q+ukmT637dL18xPnuV9Nj2NZDsMsgMACACAgW4B6haHeFShJi3e8C+fW/bLHee6\nre6RzyxIn5beRhwOhzN9cIFj1sMwaTV1nAOP3WeTUtBgyrrR360x4gidR8aC2w6CCgTVWM8n\nD/ScIVcTkBfUCRi9dK71MaHuRmirBpIgPhvjMqb24rJMv33r4q4jtfKAkVhynP779y9ZkB07\ntTeaLpwWalJwhwFJBhCw8M7ZkWTEmWYkolMAtoGXMkALURfiilF8LhENj/6z7Q//JjVVD8aE\n9Dz9F77Xf1zMJ7kdyKc3AV2totr+/lNUX8oFjpEgL3iDNPP2Nk9M4Dhd3v7q/orqll6tSlyY\nG/fQ1nnxpn4lCzOWYvpiqj/r84boJLZg2wRuFE6Q3+uzlR7F85o77S1d9uS42Ws7JdvI+pGv\nZCaD4zwwPWgKQjYqf8gjt3upUyYPgiiwKDVLwonMzyWiRqIuAg+iDiEZcaQ/mTXzkxZ+99pP\nLzY3ttvN0dqyesueE/6tWNcuSMpNiR7/SDgcDieUcIFjloAp+VR7USGeWgDqKFb0kFzzLvTW\n9EeN6SzzZtBM55qWZKr6VL6yFxwWQIaxGbhwO5pH2133dFHLTvAO7D45aqn3HCZsA13ONA41\nfPC45MMvUcWRwQBmLWXrvjBSd9tx8vK+8rcO1QyPNHfa//X5E89+b1O0PgKkAbI2B87d+8Cs\nGzCOd4zjjIWGYerGIG6iasRRPkIsPiXq+792n/pIqrsKyMTMeaol64e5wIioXgtSjdzwCWrV\n1GOFK+VU3TD0fu8om6VzHfIE+wMnck0gz+CvH5Y/v/vKwCtXY4ft4/NNv3hydX5a36Y0srWP\nUfURqjhE1jbUxUDaQlZ0Q6SXESHqiXw+4S638k8VAFxjM5uMUMhVHpgQRADgvILhInCQU6qS\nBn5fBB6v3CnJvToxD2HMO0MAAG5JPg8w0DqdnARdCCkMR5p3GXWqG5f376NIMsVFaf7+cWVf\n3Qoi3rgi/cu3zR///4jD4XBCDBc4ZgnC2ru89ZdB9pmpYHQ8W7QZAEAbzwq/AO5ecHeB2gTq\naU84lE+/StUDC3WSqbOGPvoVW/1FTCkZ4V3UvndI3ei/kIfadmP6o8Aie8Y5FuRP/0xVPvnS\nVHNa9rrZ1m9OyfWJ4O3DNYHxHrt7/5nGz6zNnpK7TC8YvJomwsvmOTMGURBrSQgW94UJ6uWb\nYfnmYIdByIFjL8ot5QoH47if8YgwDaCoUKICgGzcOm9jh/3FvWV+QbvT+/TrF57+2kACIyLm\nrMGciPfPHg7DFJnah0tFmUnK35walTCb0zcAQOoJjCEASJYZH4oyXuqWyF9vJfC45VYNG8fX\nhUw1g+rG0HWoidCMMKYpn8Dw0ZsL79iQXdHY4/HKeanRidzOkxNyRpj1cTjB4W2WZgmYMV+8\n4x/QlDgUyV4o3PdDUA97PqmjwJg5A+oGWRqG1I2hqCyfez3Y7hwAgLcXXI0KcdkJDoVl+WzD\n1kVVJwLD/z977x3Yxnmlez9nBh0gwN4pkVQh1Xu3JFuWi+zYiVvsJI7TnLsp9jrZTXY3u/d+\n27N3s7k3ud5N793xJo57kavcJKvLahTVKPYOEr3NnO8PdnCGYgFAkHx//9g4M5h5SUKD933e\nc57DTae5W8NCZTJ3CEW7vRpNLgHUt/kScotkQ65SSFqyLEmUWZ7q0QhmKnpb1gnbyqaVuzWi\nskFapieLCPqQtBumkAHGCdfrHaxpV1SNb5ya+h69J+HsgGAzUBVhaFdgQ5WjIEuj/OqmDaUz\ny4NpwuitjtJm1aSo2p2/9OJ6MGu3/tGL65HlMK9fnLdlaYFQNwQCwcxFZHDMHqhyjeHBldzZ\nAH8PsoopM//q70kSHee14/5O+Lth1+lLp+ivsZXZ3/uTe5p01R93I7LHcoQdJ0aDRATWusmM\nmeMaLFR5HV94OS5M87bCIozQEo0SRKQLkhmm7PRZDyQCG9CjE08MtHir5O1UDz0FZcD7wOaS\nrv0UMqfasYIRAUKAiWAeZ/OXmQVZVrHqQ6x9IMAgE1k3gq5iATsab0DbeAKANxDJzpjNztlE\nmQZazexhhAgWoznjnz4V+Ppvjl4ZpmVft7r4s7cumcZBpgAyFHBYy/fXmC69Yxh6fUwmpLfy\n6L45A4iyOIFAMOcQAsfsQpIpv3y6BwHEdDfHWBlZR62GEe0EAGMOJP3tAtkGVhD2wpwxuxZa\nw5D0f64xDk0Es1FeMi/rzBWNPPzVC3VUp/SDFu6Gyc7nX0I0CACyiRbspood0z2u2YUS4s43\n2XOqX3ST7ZS3kzJmyVqIqIS5ZbSeSJQAGXHoautulxdt4YZT7OumzEKqWAPTlAQURljhy8z9\n0gzBKlMF0ezx/2vpDrx0qKGp059pz7tpdd6CghA4SrILpopJqBsA8rO03yVLlOuaC7vTElHm\n4BduRWHG9760Y/+ZtrpWr8Ukr6zMrirLnM7RpQZzBcK1iHWNCJKBrKunaUDxSGRStHYdiCYk\nwBFgBEYpegxM7DoCgUAwGxACh2BqKEEONiDqhdFJtvn9ThkZBdonSwayDSykWWXvYXiPgRUA\nIAmOlTDlIjLKQp8lbjjBHb8DKyCJcqqpcg8sM6Trx7ihnHLIBiijNmGIKH9Bou7y4C3Vf/XD\n92LKiC2j9YvzNlRNX77PhCGat5XKNsPfCVZhz0uUAJRG+N3s6yR7NhzZ07FLz2rznxBq5sF7\nK35ufR7M5JwdhnMZREuYa4fteUpEFUBegu/jzKNl1yXo76co6mkethnLCMb4rIGWEWZDn6nn\n36v/7lOnI7H+R9NT72LrssK/+9hGo2HyhbRblhbYLQZ/KP6h2hef/FhnLAaZtq8o3L4iXZIX\nUoJEGbs5eALh82AFIBgLyLYecrqIOwbKiqJLMz6h6xDlMY8q8iUQ5U56bAKBQDBDmYvf8YJE\nwZ7T3PEG1AHXboOd8m8g+wIqXAprJoI94BGrM5q3AYb+zQTufQe+k8OupcJ7HOZ5iHmgDsuo\nVJnbA4icHDxN7TxDnnppzRdgnj1blwBgstKKPXz8mbgwVV8H+2SaI2qyvCL7W1/Y8r2nz5yt\ndzPDajbcub3ivusW0IxLdScJjhkkygzBHRfQfZljYcospaIV8epMT4u6/zfc2t/3gfIqaMv9\nlKPlSpC8EfrrEGrGKGWFu96ZLQIH0N9AsYM5SGQBcoC03tJXuY01Us1Z4UYDVU/DgBJKXav3\n0T+dUkf6Zbx7uvWx1y98/IbFk76s02b66/tW/+/HjgeGaRyVRc6H7hjL61ow2yAj2dbDtg5q\nAGQGpde8VyKrSSqOqK0YVqtikLKN0sTSKiWap7KXMbyelySaNzsEUIFAIJgQ6fWgF8wgOFDP\nbS+OCMX83PI0lX0c5lxpy2fVg7+Ab7COGlS8Ulp5Z/8LNQjfKY2Lhuup4E72nUG4BRyFKR9e\nBZH3hp9CACI+bniTFn4gwT/SdCOtuY0tDvXoU4gEAMBollbeSstvSuxdqsoyv/3FraGI4gtG\nc5yWmSdtzFyiIfXQL7m1v50zA3AWShs/Rc6BDdVAj/L8NxAeKpLnjsv8wn/It/8vOFOo5oRa\ntOMxD2I+GGbNdNkElMyUzz9D26KI2TcLjDhePdqkarmB7j3cOBWBA8DmpQU//cq1T++vq2v1\nWkyGlZXZN20ok6WZ/ysTTBiCZJ/uMWhjlHIMlBHjHpXDREYDOSWaRDmbLEkrmdsYbnAUZCUq\nIqTpjywQCARJRQgcgsnSc1QjyCr3HqP8GyizVN79N9x0nHubyWhBTiXlDquziHToGmqqPsrZ\nNfSq5SeaZ3HPpaROUbm5Bu2XwCpy5tG8FakqEyBaskuu2glPG6sqZRZqdwxJBBaTbDHNusqO\n9EY98YdBdaMfT6t64Efy7q/1/aHV068MVzf6iYbU91+QrvlEqoYJ6JrejX1IkFT0+k/p96Wa\nObS6g5rx9p6gqrI0NT0i22n+5E1VU7mCQJBsiExGSoCKTVRAKNCcsDBzXZuvpSuQ67JUFGZM\npfhLIBAI0hwhcAgmCY82y+iLhzv7v1slmcrWUdm6CV535Hxd0bHBV/UMw6dMyKe+9gNuHFqI\nUv4Caffn4EiVDackI7NY7DDONqIBbjiiEfd1cvs5KlwGAO0XNd/KHdrxyRPq5kAz1CjZimAb\nVZBv1plnyzYYMhI8EsF4sQMa9sCzY4c2w2rUjNstximqGwKBAMDFZs+3/3jyXEO/RXFRtu3h\nO5evX5xo1yGBQCBID4SCK5gsuu3lx/GhMubq5kSYRiyuyKa91iJbsr6Y1X0/Ha5uAOD2i+or\n350dO6WC6YL9XWCd9Adv28BJOieoE+oXOCZqjK88p576Ll/6E9c9q575EZ9/DFH/8FPIvgCm\n7NEfd8raOCv7ks4IJMrX3JCQqTj1g0k4m5ZoP+f14oJEo+o3GZ3BRGLqqXr3G6daj1/uDoRn\n4Q84Tro8oa/+4MCgugGgpTvw//3sUE29ZrdsgUAgmPGIDA7BJCFrCUe6teLjaLUo22BfAv+Z\n+LhtEQyuEVcr3sjtxzUWfiWbJzLYcePr4ivHR4e5/TK3X6b8yqTcVDAHINmke2zwUHYZOi5p\nvDc7YSaj3PASd4woLuPe83zxD1L1sBIYkqXiu7h9LweuDEQMlL2JsiaYjSVIHASTQapW1IuM\nwWoOg0zlROnSDGIqbKzO376i6K2TI8xfsp3mT9881dISjkYiR99SmurI6jBWrzJUzHhD1sTC\n3KNwPSMAgGCWqFSiWbKrf6ah57dvXur29Tett1sM92wt37holvx0E+Kpd674gvHJsDGFH3v9\n4j98QjzVBQLBLEQIHIJJQlmb2FsLNTwiasigzLXjenvmdiYj/CcHxAuCfSllbos/L6OUqu7i\nC88iNjCtl01UcRNlLZri+DXhnlbdY+5mCIFDMGkc+f2theIgorx+G0Vp6S7lwrvxZVmSTMtv\nTMwYlBB3auh38NXD1wjHMGnS6KSSuyncxpEuSGayFEKeDaUQMxqCwyCtZHiZQwQTkWPWfIMT\n4X/ev+aFg7nPHrhS3+bLdlo2VOU9cONil11fFhwHsUtnvT/8V6VjSDcxb9nt+ORXyKBdETPX\nULld4SFFlRFW+CIjJFPZNI4qIbT2BL//0rnhDdH9odgv37jospmqSlxjvHFWUtuonamhFxcI\n0ghJuMUJJsMsmR4JpgGjSyr7CHe8xoH6vgA5FlLudZDH122RZMrchozViHaCGaZcyNrdGSh/\nFWUt4q4ahNywuCi7CqakGQEY9OfTYxwSCK4KkbTyDvW9n8WHK7cjYyAPP7NY2v2Q+u6v4B0w\nuLFlSls+SnkViRlDsEOvCoaDbeQYlXtlLiBzQWJuPTasINQDsyt5rrqzBSI4iWZRh2xV6Zu/\nEtEtm+bdsilxyUpBv+c//6fqGbGEC+9/RXJm2T/8uUTdZSajKlyvEeVmiQoIM/v7bt+p1uHq\nRh+qyq++3zIHBQ49WNTdCgSCWYqYTQqmgCmHSu4hNYSoF0YXpIlPiWT7uHaGjTYqHFdiyBSh\n3PkwWREZZekvyVQkfPgThBJVDj6vXjzGPjdlFcprb5AWrZ/uMaUCKlktbX+ITz7FvY1ghjVT\nqr6RyreOOKd4qXzHP3HbBXg74Mih/IUwmid/S1bgO82RdgBkyscY5Qzjsc5JBmEPX3qRO0+B\nVZBE2YtpwS2wZE/PYAQpI+xVa57n1tOI+GHLkuZvocqdiZW3wkfejFM3+gi9+Zz9rgchz/XJ\nDyOg47vBzB6i3FQPKKE0dQd04n7N+OxmcWnm0fMarvBVZbOhwE0gEAhGM9e/4+c0igcxNyQj\n5BxIU1hESRaYLfFBZm46zu4rUGKUWUrz1qfX3mzIw72NUCLkLIZjmI+dwSRtuEt959dxp9Pq\nW2BLzrZPoIk9ZxH1wOgi1xJYZ4Nl4FiE/JFf/T2395s7cFezeuGovGa34ZY/m95xpQbKW0S7\nvgIlAiUKk460JxupeAmwZKo3i3ZzxzOIefpesf8s5AxIRqganYnIMR1J6dGAevwHCPf2v2SV\nu2rY0yCt/SLMsyhJQRBH0K289W2Evf0vA93q2eeo/Zy05XMJFNqU1gbNOAcDirtTzh3VPGiu\noWdpDEy9G7QnEJGIHDrNcVKArNN8xyClr7O+JxB54WDDlVav1WxYUZG9c1URUWJMnT+4bf6z\nB+JtOAwyfWTXgrgzz1xxH6nt9PgjJXn269eUZNhEMZdAIJiRpNOaU5Ay1CD73kNkID2VDGRb\nAeuKhF0/1Kvu/zF31/e1XGAAtS9Lmz9DzjRYvbOqnnuRL74x2GiWilZJK++Cqb9Ahpbtkmwu\n9eAf0NsGAPYsaf2HqOqapIyl9RXuHnJ85O4jlL2eCncl417JhVXuqkGgDZKRXOXI0DWajb31\n+KC6MYhy7BWperNUuSrJo0wbZBPG8BxNENz50qC60Y/iRYYTvV1xZ1LOKlimYcOWm/YPqRuD\nRP3csI8W3pb68QhSg3ruxSF1YwDuusBNx6g0YZaHZNItlqTRivwchCx6ncGIxldnOgpmvHyk\n8Zd7a9t7ggBK8+yf2VO9bfmEtaRAOPb8kcaaxl5/KFqcbbthdXF16cRyDRYUZpxrGvVsARYU\npmmj68O1Hf/222PeQL8G8cz+K0++U/dPn1rvtCXgmyLHafmPP9v8rT+8X9vY/zspyrY9dMfy\n4RkcMYW/9Yf3Xz7SOBj59cvn//LDK7csTUmVokAgECQUIXDMQZg9ryE2bJHDMfYfIxCsyxNy\nA/Xwb9hdP6KhpK9Tfe9n8vV/M+12Qeq5F/j8q8Mj3HJCDXukbQ8NtsCkinVyxTqE/WAVlmTN\nh7j3zHB1oz/YfRi2EnLOqHKYQLt69vfw9/c6ZYDyVlLVHZA0Nn/Umvc0r6HWHJhDAoc+HKhH\nuB28T4zvAAAgAElEQVSSkSzFME/B8D/SjqhGTjKZFZRs57bD/a69koEKtlDx9snfaApw72Xt\neM9l0Y12FsPtNXrxBAocxiVr8NTPR8cNZQukDJGZD4JJoiyV3aPidoK2H9ZV+cXec7999cLg\ny8YO/z/+8sjDdyy/bcv88V+kozf0jSdO9gYifS+7vOGTV9y3ri/74ET8Wa5dXvTO2fbBi/Rh\nMck3rx1Hl7eU4w1Ev/6bY3EZFmeuuL/z5OmvfXRNQm6xoNj5nw9vu9zqbe4K5LkslUVOo2FE\nMsvvXrswXN0A4AlEvv6bYz/56s78zEkKXgKBQDBdCIFj7hFpHqFuDMCB02RdCkw5gTPQze3n\nNOLedu66ONgwIklwZw1ffg2+VkgGcs2jhTfDMWz7SInwpbc03tV9mbsuUc7IdE1zkttG9J7W\niZ/CDBI41Jh66tcIjWgYzB3vw2ChRbePPp39GrtqANg/5+3coz1q6/MI9Xd8YICcyyh/N2hS\nT+lY/A75IOQqocIdCHWBY7DkTWftmKppAaAfF8wOYmGd+CjzoylgXLTcvOHa8KE3RkRlg/3e\nzyfwLjMamRYwzjMPPZMJdllaBExGYOzyhB5/4+Lo+E9eqLlxfanZON69jd+/fbk3EI4bw/NH\nGtYtzCnNGe+XssNi+MqHlj/+zuWTV/oVnMqCjPu2V+S70jF5593TraPbuAJ462TLl8IrrObE\nPKKJqLLIWVmkXf33wkENx9lwVHn1aNNHdi1MyAAEAoEgZQiBY+6hpW4AAIeh+CBPtfSd/Rr7\nxv34OpBMgYPrXueLe/tfqFHuOsfdF2jVA5TTf1P2dUCJaL+5txE58fWoSYWjHu14pHcGbV9z\nd22cutEfbztKlTePLsQgRxb3dow+nzJykjK+mQKravOTiIz4t8me0yAj5V8/mQuO4aojWUAS\nrFNID0kQZMtnb6NG3J4/OpgIGGhm7gIigJWoBMgER6H0AoDsAomC81RA9hz2tGgcsCe4Tirj\ns18zlC8Ovvi46u2BJBkrl9jv/byhcsruNrMHg4GWMHoZPoZKsEuUNTl1A8Cpy+6YolH0EgjF\naht6V1SOyzk4qqin692jx8CME5e7xy9wAMjJMH/+5mp/ONbRG8p2mJ1pbCfR6taW9mIKd/SG\n5uVPMqFm/ISjSmdvSPNQY8dctGUVCAQzHSFwzEGSvHw26O+QGJOZ6Bjq5UuvxgdZ4XNP0dav\n9P/UY1l2pVxVkHV+UePss5smBDTUCgBQYwi5YY8v35WWbVPefXL06dKybQkf2tVRY/A1criX\nzC44Sqcxl4EDV+LUjf645yTlbp9MfyJzISQL1FFzVskMc9Gkxph4qHgTtx/XMDss3pKoW3T2\nho5d6OzyhEtyzesXt1hMg4ktHuZWhI0ItQIKAJAM8xKyLEtAFptgTKhsE58e9RAgico2JfhO\nssF6873Wm+9VPW6y2Mg0BS/t2QuRi5AAC+1wVNE7FNI/FEcwHFNUbWsQT0Ajx+Gq2M0Ge/IF\ngimSoe/GmhqbT5NBMsjS6Ma6AOwWsUwQCAQzD/HkmnsYdXbJJAvkBPhNUGYpzBmjPeQgGShv\n0dSvrwe7L4C1ZlHBbvg7YM8HQI58GC2Iau1UZE2gSDghkGMBB5o04hkzKh1U1p9+aXlwGLbd\nxQ01asOIInx5251SWaq3Vbn3Il96FiE3+nxwzVlU+QHKTFAWD4fBMUjj3m/UUjcAgBVE3TBP\n3OaNDJS1k7v2YoSRIFHWzknWvCSDjBKqvpsvPIvoQE9H2UyVeygrMX+FP7x56Rcv1Q6uu3Jd\n9OW7jesXD6TKh7wIB4HB35CC0CnmCFkTZgMh0IQqriFfG1/ZPxSSTdKKO8mZLOlNcmYl6cqC\nQcr0dYTx5yDYLUajQYrGNFba2Y5x6VOqys1dgVZ3oDjHVpRtT1AfkuSyviqPniXmeGVncakr\na3w/9RQhojWLcg7VaGxXrFs8/bl+AoFAMFHSZqYrSBnGIhjzEW2PC5NtlUYWQ8zLnW9zoA5q\nCMYsylxDrpVXSXYgSVp9t/rez+LC0tI9ME9cQImGuWYfd9ZDlim/khZfo2tTGtNOsASGlXZL\nBmnhbvXss/FDLlhKqRc4stextxbB1hFRazFlJ8ZULDWQq0J7u82SBavWosJkMX78n5QTr6sX\nj8LrpuxCae0NUml1ckc5mkAb1/xuhNdD2M3nfkcrPgvb1EzjIw3sPwLFAwBkJtsKWJdcPT+I\n9AvUJ61H2BeT0cU97yHSBgCmfMrcDFN6WeJT3krKWsTdtQh2w5JJWYsG+xlNkTeON//w2bPD\nI529/E+/jP7gL6SibIKq9qsbGPnHCZ+HeRmkdCzUnz2QJK28h8s2cttphDzkyKOSdbAK48+Z\nTXWZa3Gpa7BPxyCblxYUZI03LVGWaP2C3P3n4ucnBpnWLbx6GeOZK+5Hnzh1qaW//HPJvMxH\n7lqh5zqRAphx9HzH+SaPLFF1WaZenc68fMeHd1b+fqSDidkoP3xHYnzfx8ODtyw5ddkdDI/w\nP9q0JH9jdZIKBgUCgSCJCIFjLkLO69h3GOGBb1MykX0NLKOMLSPdasPvhrLcI13c/goC9VR0\nlQ6OVLJa2vklPv0Mu+vBKjmLaMkeKprwVzW3X1L3/hcC/faTXPMW3t8r7/kyMrSSUKx6Jb40\n/BAtvE6SDGrti/15HCTR/C3Skg9MdGwJQDJK5R/jrkPsqUGkB6ZMci6hnPVjrXXTEEcRFazl\ntvh2MFS5R3dVTySv3iWvns5uuNxyQMPJUo1x835a+KHJXzd0gX3vDrtNmP2HoXjIsXns95G1\nVFsnku0wjat2XRtTAeVrWL2mFwYr5Se+gc6T79SNDoaj/MJ7sU/vMULRS3dnKF2QShI+HkEc\nlDU/9bKyIHkQ0fqqvPNNvcMTEbIzzH9x98Q60N9zTXljl7+hc8j6wSDTR3csyHVeRXa80ub7\n6x++N7xS5mx9z1e+f+AHX94uy9K+E83NXYGsDPP6xXmLSxNQknNVOntDX//NsVN1Qx5VG6ry\n/uYjazSrTj5zS/WS+ZmPvX6xrtVrMxtWVOZ8ek9VUbYtBePso6Iw43tfuuaHz9YcO98ZjMTy\nM60f3FZ+xzUVMyIFRjCbme7ei4IZihA45iRkpoxtsK9BrAeSEXKW5i4xd745uoaffbUI1JGt\n/Cp3yKmgHX8OVsE8yceTElNf+d6gutFPT4v6xo+l2/5G447ZC9nsQjh++4hyq2AanjlCVLlD\nnr+Zva2IhclZDFOSu6WMAcmUu5lyr7L6TXNo8YdgL+CGff2FBvbCxFUZMDce5/ZzCPvgLJDK\nt8KeGC9S9mt5HALsb5nCdI45EC/0AECoFtYlkMecUpvzKGMpe8/EhSl3xzS4w8wK6tq0+8jU\ntfUtv7QFpasdEggE2py54h7eI7aPbm/49ePNd1xTMf7rOCzGv7tn1Vtn2moae3yhWHG2bdfK\nooJxdCp9/I2Lo31AfMHot584WdPQO5ib8NvXLuzZUPaF25dSMtfuzPjnXx05Wz9iAnPoXMc3\nHz/xj59cr/mWrcsKty4r1DyUGopz7P/wiXXMCEcVi0msKgUCwQxGCBxzGMkG0xj7A8yBOu0j\n/su4msDRD0mTXp1xSw18WraLLbXwdmokcUhGaeXH1Pd/jfCw7iQZJbTkTo2ryybKnDfJkSUJ\nNYyoG7INhmnLp50kJFHpNirdhogXknEsl9kJEQur+3/EHef7X7acVC7sk1bdTeUJ0YN0jCSn\nMuWN9WiYevYRbbuKwAFQwY0wZbP7INQIABhdlLuTHEm0rZndmAxSQDsOYEzvGHkKKTMCwVxl\n72GNjkh98QkJHAAkiXYuL9y5fGKr/XMN2r3GD5/rlOWhB7uq8nPv1Zfm2T+4tXxC158QtY09\ncepGH/vPtLW5g+Ov2UkSMZUvdwd6glEGMi2G8mybSe7/TiSCUDcEAsFMRwgcAh04pu3ZCUAN\np2IAXt12s+xpJ80qFWeZtOUvufkwfC2QDHDNp4JVU1qypgbFz91vwn+u/6Uxh3Kug6V0Wsd0\nNTiKQB1iPZBtsMyDIQPAyEyZqaKefm5I3ehDiarHH5dzF8Ix1XaSlFHC/maAh+VHMEDkmMqv\nXb9TAMcAQIlB1n/kkkzZmyh7I6I9ICMM6e78nxjUMHftZ/8FxLwwZpJzGWWuS0iV1oqKnLdO\nauTpLK+QAECSYbIgMkqQMpVDSl1auEAwa2jT6XXa2q2pNCYedZRJ5wA8Og/uxUMNSRU46tt9\nuofafNMrcPSGoocaesIDTq4dvnCdO7ihLDNLv5mLQCAQzCyEwCHQgYyQ7VC0WqAbU+IGZ9av\nHDHrr/1kE5VtTcZwkgXHuPUPiLqHItEubnuCCu+GuXj6hjUmwXruehmxgRIAMlDmJrg2JPQe\nzA2HNMKqwo1HqPqmKV6dirZyx0kow9e3BNlMxVPoVis7AdIocGCOnqwJv/kdtb2RjCa5Ypnl\nA5+SCvXcBwjGOdPxQQmqDb9BdKCsLNLNnW+x/7JUcg9oqr1aH7hx0eFzHcHICKeVefnmPRuz\ngShgI+sKxmVELg38yQimBWRdO8X7CgRzE4fO8jjDNvEu15NiYYmrsUNrxqKVR9rUmVzZxWzU\nVWnNpulsRM3A8WZPeGSfmqiiHmvqvW5BbvrvBwkEAsF4mM7nrCBtYc9lbnkH7MDo9vUkU0Yq\nOnpS8RIYtCZGjhzKKUvBAFKE78wIdaMPVtm9X+vsseCeJj67Vz36OJ97DUHtZN0EEPNw+9ND\n6gYAjrH7Hfhq9N8zcaJhRLQnoBzo1oxPDEuWtOwTcAzzknSUSMs+CcsUxAUywVI5Ohx+7Wzw\n8Z+qrVegKhwOxmoO+771iHLx1ORvNFvg7veG1I1Bgo3sScAvZ35Bxre+uGVlZb9piyzRTevL\nvvm5HRbTWqJNRCtAeWTbSM4PkP0asl9DztvItmGGWfwKBGnDpiXa7TY268QTzj07Kg1y/JxW\nkkiSNFbtNktyt/eWl2cbZI37Ws2GqrLpbBjkCUV94VEG20AwqnQHI6kfj0AgECQDkcEhGEks\nwJeeYM/loYgswWZG3xRBMlHBjTCmwoEcFoe06R71nd+MCEqStP2BGVB1Mm44rOl2ydCO615G\nPfEkn3sdrKJvP/rUc9Lae6gi8fal7DvVX3ARF/ceI0d131jYfR7+NsgWcs2DfVKuabIRkkGj\n0QmQMFNYe5G04kGE3Bx2kzkLlqyp23mSfROzgnDdYER1S5HDp+PPU2LBJ7/v+Mv/muLtZjoc\nuKR9IHAZrpVTv35lkfObn9vsD8U6e0PFOTajQUvQlxyQ5kY1kECQTHatLn7lSOPR8yNqSwuz\nbR/bnSIjoUWlrn/8xPpH/3RysFimNM++c1VxXPvVPtYtmmqd49hkO8337Fzwu9fiXVc/dVPV\nGMkdKSAUVSdxSCAQCGYWQuAQjIAvPz1C3QCgqIgYKL8KphxyLoOcup4jtOx6KbuUjzzFnfWQ\nZCpcSOvvoOw0NadgRfE888fgkfeUnh5TeaXrrvtM5ePoJMKaUwoCq5qVw9rXuHSAa14dEYqF\n1UO/lTJLKCvR2S4RnQSKSBcABNrVs4/D39o/MJKoYA0tun0CG+NRHze9xt2nYZYR1BA4JtFv\nWB+CJZssiTOVJANl7IB1KaIdQAxytnL6ILQqw9WWOvZ0k3Nu+1kq2m4+rIQSKGHaLQa7RUgY\nAkFykST6189sfOqduhcPNTR1+vNcli3LCj92/UK90pVksKE67ydfvfZcfU9bT7Ao21ZV5pKI\nzlxxn7w84mvLZTd9/IbFyR7MJ2+qKsy2/XJvbZcnBKAw2/aZPVU7V01z5alJU+e92iGBQCCY\nWQiBQzCMSC/3nteIh73kWAl7icahJENFVfSBv0r9fSeK0tvT/OX/Ea7tL9MIHjvkeeaPuY/8\nteuOe8d+I5lyedBedDim3PEnFPCld7SiKl96l9ZdZQATRk+qIBlqTD31K4SGVdywyq1HYLBS\n5c3jungsqNb8DOEeAJRl53AU6ggBiCq2UY5GGUh6YciFoX97kMPaxnsAOOib6wKH0QVFoxCJ\nUuPyIxAIEoos0Z3bK+7cPrGeKcPp9Ucef+NiTUNPLKYuLHF9+NoFE/XjNBmkFZXZK4ZF/vlT\nG/741qUXDjZ09obsFuPG6rxP3VSV60pQty99iLBnY9mejWXdnrAsk8ueIi+Sscm0GC0GORSL\nrz42yVK2MBkVCASzBSFwCIbgkH7jkmAnTYfAMVPo+u63BtWNPjgW6/x//25bv9lYpmcnCQBw\nLEPvkdHtRcm1fvx3Z2+79gG9+BQgS4m2ImMp5a6aEerGANxykMp3Q7r604bbD/apGwBgkKg4\ni3v9CEbBEmWW0oKdVLpmSqNPOVJOkfYBg5GyUlSaPgQz/G0cdJMlC478qRt5ThFyLueQRikW\nOZelfjACgWB6OdfQ87UfH/QFo30vz9b37D3c+D/vX6vn7jFOTAbpI9ct/Mh1C8NRZVrKQ7Kd\n5tTfVA8irCx2Hm7oGd50hggripyyllmJQCAQzESEwCEYgiSjXps1SELa10dVfa+9NDrMsZjv\ntb1Zn/jsWO+VbVRwB3fuRbSrPyKZKHMb7BNJoDVatC05jUnYpHIshfcEIl0jKmjISJlbuOV9\n7bcoEYR7YL16zXN8eZRElOVAFmArlJaO+WucCkoYZICUlImvYdkmcrjYF2+laVyzk0xJ30Ic\nQW+DevqP8DSjz6Ulo1Badhcyx1Tfkgy5ViDczr0nhoUkytkOa5qWoQkEgiTBjP/4/YlBdaOP\ncFT55uMnfvW1XRZTAp7P02t+Me34gtEuT7g4x5ZnN+1ckHO+w98TijKzy2JclGd3mMRyQJCW\nJGduJpj1iCeaYBi2YshmjcJ4kihj3mQvqiJcx7FuQCJjLkyTvk76ong9alC75UesvfXq7zcX\nUPHHEG5CtBuyHeYSyBNLyqWCar70zuiSFipMQr8bMlDhPex+G94zgAoAlhLKvham3LFyNMaR\nvgFA21UUgJoEd3dmbjnMda8j6AZJcJZKC29J+IKfLDbbJ/428Kt/Z083ALKa5FyHXFpuuu7W\nxN7oKgS61IM/gDLs1+htVQ/9UNr6COwpTyQZgih/NzmXsu8CYj4YXZSxBKa5XbZzdVQoXpAR\nkm26RyIQJIy6Nm99u290vNcfOXGxa4pJHHOc842933nq9JkrbgCSRDetL/30nupVxc7pHpdA\nIBAkCyFwCIYhGahkF9e/EBemomtgnJRLn9LLnjeg9G9fcxAw5JHzWkgTW8CnOZLdQUYTRzUW\n4XJ2zrguQRIsZbBM0hBUWrZHaT6FUO9wjYOy51PFlsld8Gr3s1DObmRfh1gvZDuk/vxbclVo\nZwBZsmEel6sCWXM5oFWzYMmb9GD14NpnuHGgFy+r6K1Xj/6QVt5PuQlWheSKZY6//kH08KsU\nqJEzQyAGwJce47YSqeKDsIzvEzI1uO7NEepGH0qUL++j5fekYABjYSkmyzQb780MOMbhUwhf\n6BcWJQdZ18CgUwMlEMwouj3xdZqDdOkfElyVC029f/G9/eFov+mGqvILBxtq6nv+68+v0W4s\nJRAIBDMf8XQTjIDy19OCe4YWXSYXld9OxTs1TlVi7G5BVLsPAgCA2bNvUN3oJ9bBXi1HzJkM\nGQz2bTs0DkiSffuuVIzAliXf+NdUvglGKwBYMmjpTdJ1jyQ3tY9kGLMH1Q0AcJZRnkaLk/E6\njAKUt077QP4EHEnGhb+dmw7EB1nl2mf6CjgSC5mtxoX5clawT90YGEOTev63UKP670sY3Nug\nHe/Rjif69goiHYh2geOd7cZLzAfvCe5+E57jiHkSOrgZAwfeRri2X90AoPrY/zaizdM6KIEg\nMWRl6BpV5DhTW803u/jF3tpBdWOQy63el480Tst4BAKBIAWIDA5BPJRVTVnVUEJgFQaNLGj2\nudU3fq2eOwBVARFVrJZ3fYKyCuPPi7ZD6Rn9dkSbofggp33jxlA3N77JviaAyV5MpTvGcJHI\nfegroTOn4gpSsu7/tHlxdfIHCgCwuqRNDwBALATDtE0Hqepu2Au44e3+QidbPi3YQ1mLxvt+\nRxmVf4Ab9g6lG0hGKr2enAnunMLui5rdWxF0I9gNa+KzKrhtv0Y03MPdZyh3lcYhVUEsikT5\ndOiKNolXc0ZeXoXnCPccBEcBQDJT5mY4V4+/QxAAeN/n7rf6rsAA3G9T1lY41yZlwGlLrBWx\n0Z7BzKH3ySjyXwQznopCZ1m+o2FUlYrLblq1IBVpbrOVuBa5g7x/qfuWTbOwZFggEAggBA6B\nLrLOyirkV37zv9gz0G+FmS8di7VcMDzwb+Qcuf5XvLoXVz1pLnBwzwU+99igJQQHO7nrNC2+\nh7K1BQtDYfG8Xz3h/tVPAocPqL1uY/mCzHs/blu/OYVDHhzKtG52SQaadx3NuxZBNwwWGCds\nE0C5a8i1iHtqEe6B2UWuRTAloVR4dL3GILExkpImSyyIiE7eQbAVGCFwcHsdv/Vbbj4HJYaM\nHGndrbRy9xSTcchVonoaR4sK5EqunSd374N3mIeoGubufVBDlDnu4qlQI3e9NvKiMe56k4zZ\nsJYnapzpD8c6tA+oHnAYlEZtGgSCSUCEr3541d/+5OBwn1GzUf7Le1YmxGF0psOMM62eMy29\nPYGow2xYlO9YU5Y1nr4n0ZiqE59sPp1AIBCkPULgEEwM9eiLQ+rGIEGveuBP8o0j+1yQ/qeL\n0rsnCyt88el4w0tW+NIzlLlAr6GMZHfkfO6RHDySihGmOwTrFKwijQ7KS/L+vE0nGYfkKY1c\nj7Easo44xI1n1T/9bygDnz1vl/rGL6n1gnTzF6d0//Id1HwUyshyGMlAFddO5bJXIeaFV6ux\nTu9hONeOKG7Sh70nNaIE9r5Pc0ngGKpMGQ2rE0qIEQjSk7J8x907Kt840ewNRM1Gae2ivA9f\nW1mYLcx0wYyn32+61Onve+kLx1o9oXNt3nvWlZnkq1Sazy/IuNAU38YLQHmhMBkVCASzFuHB\nIZgYasMZzTg3nI0PGQtBozdeGJIZhiRnnEZ8iE3BlszXpL3fHvWzp37ylxWkCe46XHkPMUZM\nhaIOL9OgwlVJSYGRzbDq+KSO7E/Eb/56SN0YDNa8yy21UxqAPU9a/yAc+cMjtP4zcBRM6bJj\nE27RKIFhgBWE28Z7kahbJ66ddz0pGNEWBM8idHGsvLNphSRXW6/0m7eM33ja/JPXTCfrBx6t\nZIYkHAoEM57jF7o+/Y03fv7SubpWb5cn1NwVaOjwZdhM0z2utKCmzTOobgzS7g0fvnL1x+Ad\n15SPDlrNhj0bJ2lqLhAIBOmPyOAQTBBFxxMxNirnX7KQbSX7j42MEtk3JEtZY5WbDvLlVxHx\nAYA9nxbuodwJu2BwVLvnKwBE4ycZgpkF173JNc8Oew2oDIMEIsqpoqrbk3RfKrmOLzweH7WX\nkGvx0MuQj9vrtN9/5RSKFmsfGidZFdK2L8PTzIEusuUgozj57eW1kg5I/5Ameg2GaXwrHzUG\nXzuUMOz5MNk1TlB62fsWYoPrBIJ1CdnXTcwlJPk8dwTff9YaHnj6Pr7feOPK2JdvDcu2xek2\n1OTA8LVyuIdMTjg0pXPBDMYXjP7Lr496AiNmEScudn33qdNfvVfLomiOcaFDo4EugAvtvq2V\nutZgfdywrrTbG/71y+cHrUYLsqxf+fCqXNdYwmhMYYM8Fx4sAoFgdiIEDsHEoJxSbtLaTM7V\n2g2wriDZxYETiPUABEMO2dfAOMqONEFw7bNDjT8B+Nv5xC+w9B4qmli9A5mcetaLZBZZnTOZ\noJtr47sgA4Axg1bcR9kLkndnyqzCwg9z/UuI9GULE+WuptLrQcMmkfo9iTgaTMBkk2S4ysiV\nqo07o35zX9NVJuVDWOYhpNUoxHr1n4KbjvC55/rlTiIqWU9Vt47wheEY974CdbhqyQieYTKS\nLY2WVReaPI8+eSbOFXfv+4by4ty7d6XKxnga8TWrtU/C14y+jCBrDi28nbKS+K9VkGLePd0W\np2708caJ5ofvWC48OIIRbb+MwKj2KJrce+2CXauLj1/s6vaGS3Pt66vyzEbtXykzv3io8Y9v\nXmrq9FvNhlULch68pbokV0saFggEgjRGCByCiSGtuVE9tQ9q/NeqvFanFahpHpnmgRUQJbck\nKtil0fgT4AvPU+HqMU0QRuEogjUXwVFWI+ZMZIiszjFRIwi2sxohSx6MGdM9mni489zojy4A\nBHvJrr8aTxCUWUWuxQh3IxaENVfDx9eeCZMVkaDGe7NmYKcMUw6s5QjWxccd1eP3GCbnGvad\nRWxkDbkhg5xX6RzMTYf55FDKDDOj8RD7O6SNnx8SlSL1I9WNAYI1sK1Mn8yIl480slbTn5eO\nKXfvSpdBJotwj/r+T0eUHAa7+NQvac2fwTED/1EItGju0k6NjMbUjp5gWX5aW5KngAyL9lzd\naR6vnVlepvWGdVe3lP4///3+3sP97WN9weg7p1oPn+v4P5/fsrjUNc4bJRuFwwoHmVWZzAZJ\nKC/pQldX19e//vXDhw8fO3bM6/X+6le/uv/++6d7UII5jRA4BBOD8svlDzysvPwTBAeK1U1W\needHqWLMDU+9jOJoJ/ceQqQDJMNcSM6NMExyVczuy9qNPyN++NqQUTSRi5G06E717G9GFKQY\nrNKiu0RqdDwRP3s7yJYFq4s7D3Prm1BCABigrOVUfAMM1uke4jAi2om+ABDxIwXpOUSw6BvQ\nSDIt28nHXoyPWzNo0aakjitJUN4e7noF/vNDIccSytk1gUtIZiq6j3vegb8WagRkhH0RZW2D\nPPbnivn83hEj6fuPu447z1Fef9YDx7T6WAPgMNQA0mb23NKtXTTXorMsnE1w0wENQyVWuH4f\nLf3IdIxIkHjsFt2Fut2a3pbkKaG6wFnTqmEPVF2UyF2EM1fcg+rGIOGo8v2nz/zfL4y774CG\nLJIAACAASURBVFUS4WCsLaIOid0yWWyGImmc5YqCZNLS0vLzn/987dq1N9xwwxNPPJHISye9\nllYwOxECx2yF+cpx7rgEVaHsMqrcoFvKPnGkqs3S/BXq5eNwt8KZK1Wsgj1zMhfy13DXq0PV\n+NEu9tdS/u0wl0zmavqNP9l9EMo8slfAOO5x2oul1Q9z6wF4mwCGo5iKNsMg7NyH4e9WD/2e\nG/ubgNL8+RhZ2cPuUxzpkRZ+PH12wmHR+QAQwZIWO1TStvtUn5vPvzcUysiR9nwR5gl+9tRe\njjWDAyA7GUogTVM2jWSmvFvh6kSkDZBgLoBx4k1qZCvl7EbObigByOP7PYQ8COmIF731GBA4\nxkzsSqNJlUNn/9Zhm/1rP/Y2acd9zWnzWBFMlfVVeT96bpRPObCwxJmdkawWyJ29of1n2po6\n/QVZ1k1LCopz0vf7vSLXvm5e1pH6EY7LC/Mdq0szm7v8V9p8TpupsjjDaprSNO9IrXYv6tNX\nugOhmE3nKZQygrH24eoGAIVD/lhThrE8jeYYc5WlS5d2dXUBeOWVVxIscAgEk0IIHLORoEd9\n7bvcfrHvFQN4/3l51+eRmbiEXotdWrJtSldQw+zeN9JrkMAx7nqVih+YzAXt+dpxIg6eR/gC\nd8qUvYWyx70TbrBQ6bWTGUkKUX0epbNVzi2UHKk1B4kElJf+A4HB+RbBFtNYE/ob2VtHGRUp\nHZs+lL+UDZbRG8KUWz3CmmEaMRilW/+cm86h8QyH/ZRTRos3wzixKT5HTiN6YbCDCUdryVQN\n49Q8SqeCKXcCphtjME51A9Do3jJ0ZFjTHGOB9nmyM61ak2yozn/lqMY6f2O1zkNvGlG6OHQS\nihtgyFlkXg5Dkoq/9P/EgplGRWHGh64pf/LtuuFBk0H64geXJ+mOLxxs+P7TZ4KR/pZVP36+\n5oEbF997bfoau+xYlLcwz3Gm1eMORJ0Ww8I8h8tk+IdfHDlwpr8jldNmevCW6pun0BvFH4pv\n4NUHMwLhaRY4GGqcutGHypGo6jdKc72IadqRJNGUU5BeCIFjFqK+9bNBdaOf3lbl9e/LH/wH\npM8zKNQIVSvnItaLaCeME14OUVYl2wvgH9V+0maGRADACne9DVMWOaZvpZc4Yi313l/+v8jp\nI30vTcvWZTzwJUNRiixCuHbfMHUDMMsw6Ox4B5qRNgIHjDZacS+//7sR+T72PFp25/SNSQMq\nqUJJ1ST3pGKNiJ4fGVI5coakTMjptx5OEmYnTA7tiiTnsAQxYxGMxYjGm5iSfV0yBzdhdq4s\neulQw9HzI1yBcpyWB25Is0dZ9AoHhiUfxTo49jpZ18NUOelLUkYJ917Wil/dUEAwg/j8bcuq\nyzJ///rFhg6f2SivqMx58Jbqeclx3zhzxf2tP7w/PBKNqT95vqYsz7512YAJetTHoU6SzbDk\nJTABdioUZ1qLM/tL86Ix9fPffqu+fegR5wlE/u8f3jcapOvXTioHFijSyWGxWQyZjmTl0YwT\nlaN6mqbKutm7AoFgzpIWT21BIvF3c9MpjXhPC7fVUlHaWO6rGk6K/ShBTCLzmiRp1cfVk7/D\n8JRmmwnZIwrpued4cgUOfz27zyDqgdFJWctgT4rioHS3u//5IdXnGYxETh9x/8tD2f/8Yzk7\n6WaZAOIVtJmzmUoFy2j7V/nKO/A2w2BGVgWVbZkhRZ5RcBhkGfu5zbF6nfgVmjsCB0lUsZPP\nPRcfdxRS/pIRJzqv5cAJBM/2Z5PJTrJvgGmSK4QkIUn0L5/e+OQ7l59/r765K5CdYd60JP+T\nN1W57MkrPlcQvcKqG2CSMmEoB11ttsAKB49phEPHyVgGmmQ1DZVs4dbD8VlXJFPZjsldUJCe\nEGHXmpJda0pS0KD0uQPaD8ln99dvXVaIWIAb9nLXSfR9sxnsVHo95aZRWyUAb51sGa5uDPLr\nV85PWuDYubL4Zy+eC4zK47hxXem0t4ylsYpQRH3KDOaRRx559tln9Y6Gw7qt5QSCsRECx2yD\nPe26xzxtSB+BY4w2CuPusBCPNUfa8EXuqoG3hb2nIfkx2mM80jXJi48DbnyRu472/S9A3HWU\nctdRyU0Jv1HguceGqxt9qN7ewAu/z/jYQwm/nQY8shdJJIaYCoNWfpBtQvauKcHioqpbpnsQ\nE8LPag24u/8V5ZO0GNBx2WQd40nNdiGzF6rYATXGl16DEu2P5CyiFffE+wSTgezrYF8DxQMy\nQUqPSqVRGGS6e0fl3TsqmZkoyRN61ceh/YMfJEYjohfIshnSmB5GShc0t1I5BqUThsk+B8wu\naeWnB9vEAv1tYkULlUnz7um2x9+4WNfmdViMyyuyP3VzVUFWGllBp2At3dih7Tbd0OEDs3r+\nd/APy+qK+bnuaRAoJ400jtpGjXoNAE2dfl8w6piUM2umw/R3H1v7jceO9/qH/iFvWpL/mVum\nf94okUkig8oaRTSGdH1oC8bDfffdt2zZMr2jDz2UkgmtYDYiBI5Zh1G/dNyYRpMYWEoh26CM\n6g5gyocxa/KXJaLcJchdguZe9l/SOEFKli0fu08NqBsY3FLgziOwl1Hm0sTeK3peK0kHiNae\nTOyNdMksQcsITzju8FDRqPWPrTh9DDhmLEFWDgLDJnbczkovyVugnemk8wmf7Bb6VWCV6w9w\nRy0ifjjyqXwbOYsRC3H7CQTaYbAis5IyJ1+hMAWIFlxPZZu4px6xEDkKRhSnxCNBkdlbDzVC\n9kLY0jfVJenqBsDhw/EyGYc4fIis14/V6puj+oemlkPuKJbWfh7+Ng65yeyCvQDoZfUY2Acy\nAtkkVeh+7AUj+fHzNY+/0Z9/FwjFXjvWtP902zc/t3lR2vQBTQFWs/bU12IycO/5EerGANy0\nL60EjiSxoSrvZ3917d7DjXVtXqfNtGpBzoaqVOSEjgeLnBeItcQFjZJTpmkunxFMhS1btmzZ\notuj55FHHknlYASzCSFwzDYouwyWDIRGdRSTDFRYNR0j0oEMlHMjd74AdVgGmuygnBsSc33r\nPGgJHGSbn5jrj8atLTrAfRKJFjhY0TEDi2nHE460eKdSu29wbxwAOr0MUHEuBvZYyFVNpTeJ\n9NEpwurlEepGP2FW60nSssSTC6BqNBAhuSDxg4sG1P3f596BzoJdF7n+PSrfCl/tkP9F/RvI\nXUrV9yRPWxwLk4Pyr/avj1W+8io3vgM1hr7kq5wltOiDMM1J4zrVq/n5geqH0g15DHckC3d3\nIhCAopDRxJmZZBsoD0yABSDBXkj2QgCsXgQPPNs5BHhZaSF5AyA2cq/ClTbff++L/1oMRmLf\neer0t7+4dVqGNC2sXpgb52jTx9pFufBrd+1BpBdRH4zp8kxYrCNIleTaJ5e+MYjDarxze4K3\nJRSVT1zsaujwZTnMy8uzs52TkSSMktNmkEJKR5/pBkEyy9lmeeJtuQQCwRxACByzDkmWNt2n\n7vtRfHj1B2Ab+EaMdiHcBjBMBYnpbjA5LGVU9HH2HkekHSSTuRAZqxK1z0yulew5GV+QItso\ne3NCrj8ajsbXjPTHI70JX+IbSitjDRryjWFeqkzgM/Kk6x5SD/wKvoFposkmLbqDyldzsA1K\nmCz5ME8hE0cwCOu0O4VbM0rGhaw0QR2Zgy25YEx8Ko169rkhdaMPVvnyO3CYSR7a6ufOM7j8\nMi1I07IgvvIq1+8bEek6y1G/tPqzc1Ge0ytxAsCjEu4GiXrVmt8h3P+Z5EgEfh8ys5CXD9mF\nBC5C2Ace7TkaYfUcSWsSdpdxEI4qZuOM8O4Z4mBNO7OGYdLZercnEHHakmfpkl58cOv8l480\nNoz0sMh1We7btQA9rbpv0/rVTRfbVxT99tULo2047t+9aFrGMwa1jb3/8fsTV9r6d90sJvnj\nNyy+Z+dk0vqMksMoOZhjDFWiufJxFQgEk0AIHLMQqtwo2TL5yBPceQWsUmYRrb6dytcCgBrh\nrtfgrxk6276IcnZD0hLUw40caQVHyZAD64L42vWEIFspUzc5bUpIRqn0Pu56h72noUZBMtkX\nUO5OGDKScjsAsk4FkCHx+4q2m+4OHXwdykgjDNlgu+nuhN9LDyqskm/7e247B087bFlUuBgm\nOwBylKdsDBqEfeq51+BuABGyy6Wq69KrMmsyqBOLk5Es13K0BrEmcBBkg6GMTIs1mvhOFeYm\nDVNJgBFVII+oZeC2o1R5MyhtujipMb78FnfUIuIDaVWze+q55/IYxTXdnvDFZo/KvKDYmetK\no56yU2WMZYP+IW58bVDd6I8A6HGTs4BytyRUJ2rX9jTmLkBJwoc8Hk8g8ouXavedaPEEIpkO\n0/VrS+/fvcg+rR00x48vqF1GxAxvIDp3BA6r2fDtL2z9+UvnXj3WFAjFzEZ5+4rCz9xSneUw\nc7RQ+z1GR1qldBkN0r89uPHRP51672y/7ZrTZvrMLdWTdhhNEr3+yNd+/J43MPTBC0WUHz13\n1mk33rR+khbsRIa5JzzPAJ5++ulIJHLy5EkAhw4dslgsAO68807RQVYwLcyMb2XBRKHCxXTr\n30BVwAx56K/MXS/DP7KFpP88c4zyPzgiyBHufgXhhv5XAHxHKWv3JLq3JgeGyldveStbKP96\nyr8eMR9kW7IXV+RaxP4Gjbgz8TsqxsrqzC/+vecX31Z7+40nJVe285NfNpantm2kbKTi5Ugb\nsz/uOK++/SNEBvaZW84oF9+SdnyBslLUPTcpkAOs2XJIX6ojA5mWw7S8r94iWQOLheN7Wwwy\neqszFkLUD1PS5MUJEfYq734XvjYAJEtw6MgTvmZoCRzRmPqTF2qefrcupjAASaJbN837Hx9Y\nMuP287WRMkEW8Ki/LBkg5ei9id01cZH+j10kE5IzgaNjXacPBqLJFjh6/ZGH//Od1u7+J0yP\nL/LHNy8dPtf+6MPbrKYZMJvSMxM1GqRZJdKNgwyb8eE7lj98x3K3L5xpNw1a21BmFVvzEOyI\nO5+Krkm3fK68TOs/f2pDY4e/vt3ntBkXFDv1vEWmkb2HG4erG4P8Yd/lSQscgvTkgQce6O3t\n3y149NFHH330UQDBYLBP6RAIUkzaPQ0FiSSu+WXME69u9BG4jGg3jENZxNzz9qC6MfDeXu5+\nifLvvXqzwGTCnXV8+AnuvARFoexSWv0BKhuH75chFRsvlLOee84hMLKC115KueuScTvz+h25\ny9ZFzh5XOlrkvCLT0jVkmdsl6KqiHvjlkLrRR8irHviFvOfv0m1uOn6I5jHHz7YBiWg8s8Nk\n/tSyGbIJipZ/pIaWSJDTZX9YPfNMn7oBjJl2riOJfuep08+/N9RmUlX5mf1XvMHo3340pSUS\nSUMi8yoOHYxLlCDTSt36QTUKVcdGNKpf1TIpiCw6fzEJSPoH7L/3XRpUNwa50uZ78u26j+xa\nmOy7T51tywt/+NzZ0X1Ad6wsmiXy3MTJcoxMXyVZWvQRrnuOPQOt0CUTFe+g/A2pH9t4KM2z\nl+bZr37eNFHXOsoPDgBQ3+5LQT9gQSrp6dErp50aIgFEMCmEwDGXGKNDaqRzSOBQIwhd1DhH\n8SHcAMu0NcXgK8fUN74PtT8znzvr+JX/ktbdQSvTo7ZfMkgL7+eOQ9xzBpFemFyUtZRyNySl\ntAcAQFa7ee22JF18xsEdFxHQsqXwtLK7cQYncVA2SctYrQUG98HMJC0BTXe+NBEVLuemoxqH\nDKO+WZzzIKeH1z0zt7w//BXppLlQhsZnptsbfuGgRqLWG8ebP3nT4uKc9F1pTAC5iKzXceQM\nVDfAkLLIVAVJ30dDMkK2QNFK50l4zg7lAxc06rOoYKwOLwni6PnRUiMAHKntnBECh8tu+tpH\n1vz7Y8eH16pUz8v8wu26bRpnBCcvdf/+jYuXW702s7y8IvvjuxdPzsayH5OLFn+Ugm0c7IBs\nJXtRMupM5wo6CoYsxVUxCgQCQSIRAsdcYowajeGHFC9Yp7w/lhyBdjywqh747aC6MYh67Bl5\n4VbYRjUonRZIpvzNlJ8sH1PBWAS1TTcBINCDmStwAKBikvPA3UAIsIKy0+TRLS29TXHXIdA9\nPEhZxfFtOCSjlD4Oo7HQiKwTBociZI3f/KfsKjg1PjMXmzyaNo0Azjf2zhKBA4DkJMsEnmOU\nvYw7joyKEmUnfOVsJama1ZoRGgdlkJSKHmGBkKIdD+u3yE0zNi3J/+lXr33uwJXLrV6H1biy\nMvu61cUp6D2cPB5/4+KPnx+qkLrS5tt3ouWbn9tcWTS12ihrAVmT0HlqjtHdG9aMLyrNnNGf\nOoFAkOakxSxZkCLMBSAZPHqKRjAXDXul38ckQS1OJgG7mxDQbF4Y45YaWiA0hTmPWX+v2DLd\nyQ4JwAhKv9m2xSXv/Kp68XW013DETxkFVLGDchdyy2FuehtBN2QjuSqo8mbY8qZ7rAMYLPGV\nNZEYg8hi7N9sJImKNlDFjdpvF3NyLah0Fwea4W/pf82ARFRyPZKxRKQSkrNYbQB8gJEoG1SS\nmj9MSZ6tuUujy0xJ7kx6wmQ6TB9Lv14bk6O1O/Dzl87FBX3B6KNPnJpTjW/TE1XlmgbtjYeq\nMu02twKBQJAQhMAxl5AscK5D78H4uHMN5GEbjwYnZCeU0U1PCebSpA5wLCKaPotXOzTWBf3s\nbYPBQhkF8WYlgvTB26GeeBadVwBC7nxp1a3I0F4qU/5CmOyIjFp+2LIoe37SxzlnMZilqptR\ndfPwGBVvpOKNUKOQDGknCRBR0UpuPDwiGIlyTJHW3Q+Li+wFMOpmpC8qcUkSqWp8EgcRqsrS\nI49sWpAtUvWnufM4PBc56iNrPuWtg02nIUUCsKUmZSOOWzbOO1SjUaVy66Z5qR+MAMB7Ne19\nXr9xnLnidvvC8f4agtTSG4j4Rxm+9KGXJSwQCAQJQQgccwvK2gLZyj0HoIYBQDKRayNc8S6Y\n5NrK3Xvj65zty2GYthk8OfMB0m4Q6Myf2LWiQfXU01y3v99f0JwhrfggzUtTC7G5DDeeVF/7\nHpSB9G93o3LpoHT9F6hkucbZskna8FF1/0+hDstRko3SxvtT1p2UO5uUK2cQ9FF+mbxwzVwX\nzqRpS/gaG2npbUpPPXztI4KLb6bC1Vd9b6bDdNvm+U+9WxcX3722tDB7bhfqk0R5a5G3Ns0E\nrUSybXnhJ25c/JtXzw8uqk0G6dN7qlcv1O0vI0gqHr+Ouy3g8UdmoMARBvyABDimZX7e3hP8\n2YvnTl7q9oWi5QUZH752wdZlk0/CspoMRKRZ02e3itWHQCBIIuIRM9cgONdQxipE3YAKY7a2\nBaZlPuXezp4DiLaDVRic5FgD2zTsmA1hy6SSZdx0Kj6ekUdFExoYq/t/xJ3DXFTDXvXwryVW\naf6mqQ9TkDCUmPr2L4bUjf5gVH37F/I9/wZJ49lFpavkm/9WPf0iu+uJJGSXS8tuhj0law9V\nje79eey9FwblFSm/zHjXl6TCaTPlFehizpB3foUv7eOOWkR8yCikiu2UVa557vELXc8frG/p\nCmRnmDctyd+zsezPbltqsxj+8OalaEwFYJDp9q3ln95TndIfQTBNfGz3oh0ri9453dbuDhZm\nW7evLCqa48LWtJKfqd34VpIoxzmzmlPGmC8CrQMvZaL5QEqto8439n7lBweC4f6cizNX3P/w\ni8MfvnbBg7dM8uFmMckrKrLfv6Rhb7+haoL7UgKBQDARhMAxJyEJpoFVHyvcXYtAOwxWcpXD\nNvCtYyqg3A+CVUCZRuuN4UjbP6m+9l8ItLHZQKEoB8Jw5Ei7Pq+51tWD286NUDcGUM88L8/f\nmHbp9HMY7riIYK/GgUAPt1+iwsXab8sokDZ/IqkD0yT21h9j+58dHlHbGyK//lfLw/8Js/YU\nXDCdSAZaeD0tvH7ss7739Jk/vX158OX+M20vH2n8twc3fermqrt2VFxq9qrMC4qdLnu6dMAV\npICyfMd9+Q4AUP1Q/VABaSZpHMzc6g6GI0ppnt0ww1tZbF5aYDUbBtfkQ/El+Q5rWsxbxgnz\naWC4y5jCfImIgdRVP33nqdOjf5P/ve/SDetK5hdMsh3S525b8hff2x+KjLB+u2Fd6bLyrEmO\nUiAQCMaBEDjmNr4WteZxBPqLipkkKtpIC24ZSuknKQW998YJxzqwOAdRY78VINmkeR+CfYK2\nIN112vFgD4K9sM7hKvp0I+jVPRQabRAzrTDH3ntOI+ztVk6/K6+9yipakJ4cPd85XN3o43Sd\n+7HXL3zypiqnzTQ7CxNiYQTdsGbCMLM2wFOL0s2BQ1AG+gcZCsi6HvLU2nakhFePNv3o+bPd\nnjAAs1G+Z2flR3YtNBrS5Vt+orjspq/eu+objx0fvoSel+94+A6tMsb0pXekutEPcz1Rqd4c\n7FKL551Tbe3uYHGubcfKopLcKbVw8gaiZ+s1DEGZ+WBNx6QFjoUlru9/eftPnq85dqErEI6V\n5Nrv2l6xZ+NMbmomEAhmAkLgmMMoYfXULxEZtoxklZsPwGij+buu8t5QK/svIOaDMYsyqmGc\noCF20Mu173JPK6xOmrecCsfh6B5o5MYnRzhTcUBt/KO04DMwTMDBnjVdPPpQtVsApgXeLvXI\nc+ishySjcKG07laYk7tnyO5W9fhe7m4hi4PmL5eW7UCKm7rZ9cUmW3pt/rC/l/3amovaXj+3\nfThmMPtONGvG3zje8smbprVeL0kEurnm/2fvvQPjuK6z73PubF8Au1hUohcSIECCvYtNYlOn\nClUsq1mW5Vh2ZCuxk1d5lbh89hsncewkjixHliWrRM0y1UlJJCWSYu8EwQISvXdgsb3MPd8f\nqLuYQd0FFuT8/uKe2Zm5BBY79z73nOd8SC0Xel5hfD4W3A7GiGl/EzmI3WTfAzRor9vfTPbd\nGH0zsIhWhT46XP3b9wbKPD0+8fXdVxraHf/nawuncFQTZPXc5Nk/Wv/+warKxm6DTjU323LL\n8oxpkplCwJuIbMicMm8QAZwAEjOcl3ZeemdfRb/b8Wu7rnzjxvx71uWMeyg2l0+m//VwRiej\nISXO+I8PLQYAv8inye9FQUFh2hNKgWP//v2/+tWvGhoa5s+f/8wzz8ycObP/0I4dO5544om6\nuroQ3k5hglBrSYC60R9vOIIZ6+V9GYla9pD17MDrjiMYvw7NI/vz9b6//Dj/8iXw9Ha7oOPv\n4ew17PrHhndkpPajEr7booc6TmHi2lHeGgDQlCr9ENcYI23Z3A9VnOI7fgv+vklGTYlY8iW7\n6xmMD9c2CC/eI+7+U4//BQHAha/42d2qbf8HtBPaIxoTGJ8D0fFgaws+EJOICRFmbKGSz4VW\nKcULEQNx6mwGjwvjU0E9svtgh80jGW/vdod6ZBGA186PPgeegScCtZXS0d+xVd8HnZLXFgB5\nzgeoG31R8paibv5UjGgEqptt9W2OaIPm5SEdVQHgi9MN967PzZkxDdJP5Ig36cbtEzFlkJ3E\nM0B2ACBQjcmS+cszDW99GVBp6xf5Hz65OCvVNO60srgYrVrFenyFgpgRF5rdFEXdUFBQmDRC\nJnCcPHly48aNAJCVlfXqq6++/fbbr7zyyt13391z1Ol01tfXh+peCqHBKdHuDgDA5wSfAzTS\nGYlkLR6sbgAAkJ9av0BdMuiGNAV0tPNLu6GzFgQ1xGWx2ZvAbee7ngcxYHZIl74icxIuvn2Y\nwZKrWfqAq0k6LgMmz8GYZOoOPovlbZi0Xhtjw+vmn//PgLrRg9NKn/8eH/hFOG5InU3irpeC\n8lmo4Yq47w1h87fCcUdpGGNrH+e7f9uvhQEAaI1s7Tcj7TeFOiNLyuTN1UMPsaw5kz8ehaGI\nJfvFPa+TvRMAQFAJS29Wrb13eJnDLNOCwRI97VozjAxV7h+sbvTidVDFXiy8YypGFMH4JUwT\nAQD8Q9TYqaau1fGf28+dLZcZcB/nKjumtcAxDeEkngLqS9yQbZoqAEiIC58eq5V896fHa8Yt\ncGjVwtp5M/acCp6oG3Sq6+aGr9+zgsJIXOPd6BTGS8jWCT/72c+Sk5NLS0svX75cWVm5Zs2a\n++6774033gjV9RVCzzDenPKHqPucZJi6g/ubUN1ZcefPqewraq+ilit0cZe442f83KdB6kYP\n/PzeUY15KGMtm2ACW/VXmDgow1xQs4KbMG+kqpxxIPqodC8//Ao//AqV7g1uCDI6qKYE3HaJ\neEsVdI1N3BntHUuPSFbr8IuHJrl5PSbmCnf/HOfdjCmFmJwPCTlojOX7/8j3/I5aJJxipxDV\n5oeHPoZZ7gIhNxJ3dK81xNO7/R/8tlfdAADRLx750Pf+fwx/1tqiGdLxeTMAwCu11Tl9oc5g\nt5Hh49c2cnWO8vWPU4HT7f+7F46MqG4AgP/q+jBPA6h9QN0AAOKSz1bEDMlZekO7Y2gQAOrb\n5EpdRsWTt88pzAzIYzXqVM98baFioqygoDDtCFkGx4kTJ55++uns7GwASEtL++STT5588smH\nH36YiL7+9a+H6i4KocSUJR03JoNKvu+DT6qxBQD4Ai2yfG5+/H+Dl/QeOzQWS59uawfRD4Ls\nBxL1M8h2ReKAPkV2qHIYYtnqJ6mrDrobQaVDSyboQr95RV0NfO/vwN67p0flh+DibuH674JJ\netUki0PC96v3mvZONId+a4VsMhNirwvcDtCP02xsnGij2OI7oatR3Plv4LIRAgBQdwvVnGZL\nt+HcLZM6GHmEmQu1D//Yt/OPvXkcKrVqxa3qdfdMtnGJwlCIi/veGhrml0/w+sssVaYdD8DS\n2Qm3rMj45EjN4GDOjOiWTufdP/nc5vQlxepvWZFx95qc6evROICcAxGX0KOvdQQzcAnRGYTI\nquX5/ERdm3VU5VQ5Kddc+kaL3VPT6XJ6/Tq1MCNGly7TbjZc0BCFQvQCUw9SyQXEDLkWKkad\nGsAlFZ/QlD7aoP7Nkyu/PNNQXNFhd/myk6NvXpERK5PIFlacbn9Ni12jZhmJUUphDD3nLAAA\nIABJREFUi4KCwjgImcDR0dERHx/f/5Ix9vzzzwPAww8/zDnX65VGiREHxuaCZRZ1BKoGyDDn\nxuFOY1oQpeZMLOApSM2XwCuxmUCiGwQG4pDNCpUahOHy0DB+BdkrgAJn4YIBYxcNN9phLmhO\nA/MYO7CMHiI68GK/utFzQ7C18QMvslueHVvaiUHWwBXlD00IOQlDUIFmav6Q+ZE3wG0L+rHx\nk+8JmYsgOlJMEFn2XO2TvyFnNzhtaElW8iojBOpoIoe0LEu1l0Be4ACA799VtKIgaeexmrpW\nR7xJV5gZ++Gh6orG3lKO5k7XSztLT15u+5dvLWdseitZGJ1E3RImWRg9RkH2GgC1s8lXH5yv\ngQJqI8t69nKdRGOOocxKM83PuRr7Aclzpt5a3dk3P3H5GrvddV2u5ZmxLJx6tEe0e7mDk5+h\nSoNsSFIEAfcCIQpzAQ0A0cPMzxfnxVc0SthaL82f6NMQEW9YmHrDwtQJXmfcuL3inz4r/fBQ\nlV8kADAZNY/dNFvpuqKgoDBWQqaMpqenX7kSsFRGxOeff/6xxx579NFH33zzzVDdSCF0IBY+\ngBnrB9ytjMlY9A2MnTncOYYs6QPGQN9Hl3wjTybxqcOMohGW/foZmLENNJaBiCGVZd0PqvA2\nExkf1FFNXUNaMCBQZz11SFfPyoEZcyUbpmBcGljGnr0yCtjMxdLxnIXDpNiEEa+TmiS88YCL\nVCuTEDR1oCEG41MVdSOCGKY70igaJy0vSPzJI0te/OG6X35reUO7o9sZ3FDgbHn73rONExzj\nlIMZ10lY2yDDzNVTMZzIRpWAxuuADVJ7WRQa1wGb3Oy2ULBgZtxPH1ky3eW5MdHY7R5QN/po\nsXvKZeo+QgHZfM0Of5uPu0Ty+bjLIXI7BS/aCQBAC5gCEDv87uN91+emDDH+nJkac+vKzJAO\newr4f2+c3v5VZY+6AQBWh/c37xa/f7BqSgeloKAw/QjZcmXNmjWffPLJL34R4HqIiC+88ALn\n/KWXXgrVjRRCCVNj1kbMvAHcnaA2DFeZ0gdaVpKjAvyBdnT6NIwuDIzIprxi8iyqvRgQ0hrY\nyntHvrUxE3MfA08b+WyojQ0QOyINu3zZs60VLNJ5p9JoDWzDN/mnvwtYjGkNuPnb4x+eJF4X\ntdcDIlpS2JKb+YkdAUeNZnb9QyG+4yhx20Gmfx25uiN6Ys79ZK0EdztoYjAmE9ST14NGoR+M\nTQa1FnwSLVEwcWzrgZOXpV0kT15uvWFhWNTGycOUhvO/ThfeA29f8YXagAVbITZrKkcVsajT\nUZUMYhtwB7BoEOIBI07TzEs37x7iGQkAOTOi71yT7fXxnBkxc7IitHdY+KiXKdup73LPih9D\ny/nR4xHtPh5cUeIjoxvMOhzIskEAZAWjSfCMMWj++6nVr35+eX9xY4fNk2DWb1yU+rUbZmrV\nEfchHBOltV1HLkjYyb+26/KtKzJVQkQ/7RUUFCKKkAkcjzzySHNzc1lZ2eDusACAiC+++GJM\nTMzhw4dDdS+FEIMM9KPOUFUZWcZD1H6QHGXgd4DahDFFGLskaPcPk/JBrQdf8EMdTSls04+o\n+HN+bjfY2kCjx4witup+iI6H0YAMdImoSxztaEeJ6KOmi2BrAW0UJuaBccLSiUY+r2TsnVYx\nb4UQn8GPfwit1SCoYMYstvR2MIau3puL4uHt/NhHvb1a1FphxR3CnT/kpz6ltjrUR2NmkbDq\nLtCFZfI3MvoYYAy4lAebMXJn52StpIoPwd1roUKCDjM3YtKSqR1VLx6X/8gHVHORPE6WmCGs\nuH2sS/3phEotLLlJPPx+UBiTsljO2CxgHW5pQwqHezz+wZEGJhdh3ExqLwNXB+jNGDcL1JGY\nHzcSBNBI1ATgAtAiJgKkj92MehSgGlQRXb+zZUnau/sqWrqCn8KP3TR72exQP0OnD26/dN6W\nXHzieLm096cfZgB6gZwACBiDQj7gaOceUXr1k1vnPLl1js/Pw+EB5PaLl5vtnS6vTiWkmvVp\nk+JRcqFa2nHM5vTVtdqzkqdfhpSCgsJUETKBY+3atWvXrpU8hIi/+c1vQnUjhXHi7abWYnC1\ngdoI5ployh75FDkEPSZuRNgIxGUbdqr1bOkD/MgrAR51GgMufwgEFS68WVh4M/i9oJp6d25q\nK+dHXxvwy2AqVrgF59w0kWtiQi5oDBIuJBojJuSO54qWFLblryTiHieVHSdrCxpNmFEEseOZ\ncIu7X+Zndw+89nnEr95mS25W3fvseIYactQ6TJtHNWeC4yoNZiycigGNAk8XXXoD+KB1r+im\nio9BHYWW2VM3LAAA6mj0vf5jsvVOJcXmKvH8QdWWx4RFm6d2YOFDtf5+EH3iiU/706BY1lzV\nbd8dayVRaryhulnCXTI1/mrJzVHrMbloqgcxEYioGKA/gc5D1A3QgrgQYHpvbo8DvVb1r99e\n/p9/KTld1vt0i4vRfef2wmtE3Wizuj86XF3dbDPq1PNyLJsWpzGGbVa33yctZOhU4fqEcJK+\nIwcVqtYC+AHYWArGyS3a/NzNgQuo1gkxISw276G6w/nl5RZPX2+dM3VdWXHGDfmJqjCXMolc\ntg/RMIcUFBQUhjIVFfUKkw61FlPlxyD2VY83HIT4Ipx5x0SzaqXUDWoppzMfU0cNMBXGZoPe\nAM4OYCqIz2EFm0A7KAsgSN3wdFH9F2SrBtENugRMXomWORMa3mhwd/P9vw/INOF+XvIJ08Vg\n7nXjv6xKw5bexw++HBBExpbdD4Ja5pwxQxWn+O4/gLMbesp3mYBLbmOr7hnbRWztvHjP0Dg/\n9Zmw/A4wRIS7Plv5ddHaBNZBbXEFFVv10DAOrOPE3U0tpeDqAmM8Js0G9Ti3raj5BHAfAAVt\nHVPj4SkXOPyfvtivbvTCRf/nf2IzF2PMVeo1yATVpkeFpTdR/RXyuDApc5jmKcNw07KM3390\nISgoMNy8RPHAixAaB6kb/XQD1AJk9b4SHSB2g2AAIWYimR0Ot7+2xa4SWHqiMWJLA1LijP/y\nxPK6VkdNiz0uRpuVHB2xQw0t+4sbf/XOWbe3V1nYdbLutd1XPF6/1eEDgJgo7arFqbmZAVmQ\nKSZdmAbDUBClluesdwI2hnk4J9Hmaxapdy7nB7dHtBtVFq0QsuwGh1fcXdrsDxxxVbvjeHXH\nyuzwPiByZbr5aNVCWoIRAFq7XB4fnxFnEK4l1xgFBYVxoAgc1wCuNir/AEgcvNyitnOgj8e0\ndaG9FZXu44deH3jp6ACmYhu/h6kjSRXORn7pVeDe/pdUsR3stZgxbEuXCUOVR4bW0QAAv/yl\nMBGBAwCzl7OYJDrzIbVXAwDGZ+L82zEuayLXDKCrmX/yXwGNeLlIx94nUwLOWT/6y1BjmbTD\nBRepqRxzwpkiQR7gLmBRgCN9ERnMwtYf06UvqekyeJ0Qm8bmbAx5/xSqPMjPfQD+PrMGbTRb\neA+mjK2KoRdnCwBILJ+cEgXGYcRnJ2cLMhUYkkDQAgC47LzqnMQ7RR+/fFxYEt4/t6kFzUlo\nTprIFe64Lqumxb7j6EDjWL1W9dSdczOTpqh6SyEQImmTFKJWxCwQbWQ7DN4+WwqVGaNWgmbM\nnbY9PvG1XVe2f1XpFzkAROnVD2/K23pdVsS2hE5LMPasD68ROro9g9WNHlo6Bx703XbPp/sq\n1q1In5vX+xCxGDS5ceH6EWmYcagHR098rJdyip396kYf5PB3qJhOwNDsnZS32v1Sekxps21F\ndlxYP+MLcuPy082ltcHdf+5cnXX0YssLH1/sqbfSa1T3XZ977/ocpYOsgoKCHIrAcfVDbcV9\n3VUDN5NbTodY4PDY+bE/Bwe5nx98VbjnlzDs7I/XfDagbgyM8DjGzwdDGOucySrT/sDWPFwB\nzujAuCzc8NRErjAMvOTLAHWjDzq7a0wCh5x/JwAQ5+GazYgd5DwBYt9eqyYLdQuBDbuBJqhw\nziacsylMI6KmC/z0OwEhj40fe4Vd/0M0jd0/Uu6TM2lOhKKHqndRyykgTgAgaDBtHaasIqdV\n1rHVLl3/PJlQZy1d3gvdTaAxYGIe5q0PYcbTxGEMf3B30Y1L049eaum0eVLjjTcsTImLCdfG\nr8LYCX6IDMTJS507YLAbgr+LrJ+h+WZQj00q/dU7xfvODjTJsrt8v/vwvNvnv//64RqQKUwa\nB883Bakbkhw93bi0MDlKp5oRo8uKNYRPn9IKUT7u8vKALi0aZhx72gX5RMlWL+TjTkEITT5j\nt4yjkMfPPT5RF84MIET86SNLfv1u8bFLPTsEoBLwjuuykyyGn79+qv9tLq//T5+V1rXa/+7+\nBeEbjIKCwrRGETiuAdwy6xZPF5AYwhUXNZYO7H4PxtFBHbUYJ986RPSAXbp5KlnLMZwCh2zf\nU2TDKzJTT+eQNrQAAEAdErb5w4CJWXJHMEnu0MQQO8m+GwaXJXsryd+O0TdNYScCKt8vEeUi\nVRzAhSO3+AkmOgM6Lg0NY8wkeXnSlb9Q5+WB16KXqncB92P8EkCU1DgweorbElHpF/zs+0C9\nhd/UdBEqDwnX/wD0oS5EmhizM8yzM0Jn8asQSnQAkh3KdeC6BEO9HomT8yyaNo7+BlVNtsHq\nRj9v7Cm7c3X2NVIAEuEMTtYYBrfHn27QFEzK33KUOsHLjV7RwcnPUKURDONI3+DECaTlaU7S\n/sfjQCOTFoEI6vBnTFhitD9/bGlZfXdFY7dOI+Snm+NNuvv/v91D37n7VP22dTk5MyKiilYh\njIzRKktBoQclv+saoCc1XSKuGWE9ST7wNoKrAnzyTU8H45FvIz/MIQAQpWSRHvyjmqmMG0yQ\n3nPDhJlhMd4PISqZX6tcXAaMTWZ5y4fG2ZzVGB2Wgltyl0Cw6RoCt4G3PBy3GyXULZPLIxcf\nFkxaDLohHV6YOuRFYdLYG6izdGiY6g+CRsOypYpuVGqWtzTsAxuG7ubB6kYvtlZ+5i9TNCAF\nAO4GX/uQv9bIBVG6BAkxmXyt0ufIxWW4WBOcP9+D2ytWNtokDylMMjHG0ZqXe2UMR8OBhhmi\n1AkxmhlR6oRxqBsAwJDJTUswdHsD6bHSvZNSTPpJc76YmRqzeUna2nkzkmL1tS12q0M6M+tc\nZcfkjEdBQWHaoWRwXAPEzoLmE0PDaJ413FmuK2Q9DP2Fo5pkNK8F1bBdOaNk18M4fBdYtRGY\nOqDlRD/aEPQBJXuXb88bvOIs2btYfJpq+U3Cwg09CRqYsQQv76POmoATmArn3T7x+4YVTC+k\n0kMS8Yy5Y72UcNNfgUbHS/b1XQLZvBuE6x+e4Ahl8UuvKMjfgtrxWD+GBrlSiPGVSAhaNucb\nVPUZtZ/vjUSlsuybQRVFZbvB1giCGmKzMG1pOJJWyFEvPQ/mXnC2qm563Pfaj6l7kGrJBNWW\nx6c2g4PqzgSrG73xYuB+YMqjanLxNlHXV+Dt+VNFMBagacUIRWSThtdGtjrwu9GYBFFB5WMJ\nAGkAdYHBJIAUgIvSV5P61A0DVxo9RDzLZie+uOMSyVdf9oCI6YnTyz0HNUwv2XRWw0LW0XmG\nSTcrIepKa0C7KLXAwu0wKofPL/sXOswhBQWFa5ywzBr37t375ptvVldXu93uoHg4bqcwPBib\nB7H5wTu6KgNmyOfluquo84uAiLeJ2j/GhPuAye6NYHI+GC3gCNbUMXHmCH6QKKBlLrWdDo4L\nWrQUDHfiKKCORs8fniFnb94yb6r0fvA7oaJYs+1vAACYwNZ/j5/7iCoO9bSQREsmLroHLZNU\nSjBusGANFu+hlsqBEAHoDGzltjFfS6MXbvoOW3EntVQCICbloDmsfQTlJiVTOVnB+Jlkl1Be\nMH68dfWaGMy7B8XbwN0BmhhQR1HbZTr6woCpbcMpqj7IljwOulCXYAw3syc0J2m+/R/i0Y94\nzUXyOFliprD8VkyY4j4g5JasLADgfvA4Iq1K5SrH20StHwxK3CBwXCBvCyZu6zWXIZqiCj6i\n6r1Uu79HDScANOdi3h2Ds6UQ8wASiZoAXAA6xASAeAAAVRx4pAoh1WNbtuXINHpQCSwrOWSd\nLBQmQmZS1NduyH1jT9nwb1s3f4YlemwJj1OOQWXx+7xBBSl6lVnA0SatjIbr8xNnmHTF9Var\n26cRWKpZvzzLEqObGjuk1HijSmA9hr5BZCt/cQoKCjKEXuB47rnnvve971kslry8PK12mj08\nrlIQ8++D5uPUdBzcHaDSo3kWZmwAjeyzgexDtAYAEJ3gvARR82TvI6jY+if4F78D16C1Skwi\nW/vYyENM3wSeTrJVDbqaDrPvANVEjc19n7/Wr270I547wBfewHIXAABoDGzxfbBwG9hbQRcD\nmpDthIQXQcXu/gd+5C9U8iX4PMAEzJjL1j0EseO0LMHYZIwdc0OB8SCYwC/V7ECYSl8Dlr9J\nbDgL3sDNMWMc5q6Z0HUFHRhTAAD8bjr7RnDLHnsznd+Oi78xoVsMAY0p0goHU4MhAQBAoxPW\n3BNRha2oi5EZswq011ADiEiArMckylJ8bbzxoOvjPf6K8+T1CDOydJvuVS9YPakDq/2KqgN6\nWlNXOZW8yhZ/LzATyowY/GWC+nxyXoDgDhSABvknmhSz083zc+POlgeXbd66MsOoU/KMIoVH\nt+TPybS8s6+8qslm1KnzM0z1rY7Lddb+N6wsTPrBXUVTOMLxwVBl0qS4/VYfdxNwAdU6IUYV\n6tQqBChIjilIjuFEbKrNyAw61U3L0j86XB0Uz5kRs2DmsKnBCgoK1zChfx7/+7//+7Zt215/\n/XVF3YggkGHyckxePtrOID6ZZnu+1uGfdZiYK9z1cyrdR23VIAiYkIt5a2SNPAcjaDH/Iei8\nBLZKED2gT8D4haCasNZAJJadkjwiXj7VK3D0wASImZTlfQjRGti6h2Ddg2DvBH3MqH7OEQBq\n82mowIEq1ExpDwJjnLD+aX72PWq+CECADNMWsqKtoNaH5PLUegl8EqnF1HYJvQ7QhHQNH52G\nphyyVgSFccaKYTKwphZMmw8lnwytF8DUIqU+ZbLxSvvO+M596ivp/ToVa684XvqFbvP9ulsf\nmaRREae6gxJxZyu1X8L4kTqRMwOat5DtAPj7XLeZHqOWg2ZsPZIQ4dkHF/3X9nNfnWvqiQgM\nb1uV+cQtE002VAgtS2cnLJ09kDpKBCevtJbVd6sELMyMLcwcVfWrX+TvHajac6q+sd2ZGKtb\nUzTjvutzB1vJukXuFTkBaASmn5SupQhMr4oNzWNpJKZc3ejh27cVen38sxMDGVhzsyx//7UF\nk+YJoqCgMO0I/cSxubn561//uqJuRCgT63s6Kt9NjR6LbhzfYwdjZ0Ps7HGdKoPfBz5peypy\n2yXj0xCEqCnufzE21Bmod5P7LPTn2bIoNCyHkFQRkwjEgY0rmTYqkV33bfB7wdUJxrgQr6td\n0t6EQATuzhALHACYdw9U7qS2s4P/Zqm7Ch2NYBxdjg/3gLcTBAOoJ8WmPiaZzbudn30/IBgV\nzxbePRl3Dx9eJ3U3AlNjTDKoIlRdCoRkbSmGPD3cu9/RLN/EEsbeR3kceLvBJ2NWbW+EEQUO\nAFDHo+V28LWB2A3MAOoEwPF8UZiMmn98aHFVk62isVslsPx0c9IkrTcVxg8iLMlLWJI3hpbA\nfpH/3f8cLanqrbqtbrZXN185UNL0H0+uMuhUIlGb2+ceVDqhFViCTi1EhigwIj2WMZGsEZTW\ndp260tbR7UlNMD5+y+y71mZfrO50e8XclJh5OXHT5MesoKAwNYRe4FiyZEl5+VR2Q1AIAepE\n8DYNDaMm0JrB2cGvfAHWRhBUGJeDM9ePtYVH2FFrMDqWbBKNclmstN++wmSgzUN1OvhbgFzA\nokGVHAKvTVsNr94N9nogDjoLpq3DhKLxtMJRaSA6DJ+NYTJB1GEowVDpceZWcjSBq2UgaKvl\nJS+xuY+NoHH4HdS8l6x99qjaeDZjExjCbtKBszeyxFlU+gVYG0EbhUl5mH8DCNNCFJBC9PJz\nH9OVfT3mPqDWsTk3Y/71kd6eCRDUFsnOWdQ6xCeFc9+lU9rJETiG+YoYw1qHgToR1CHwGMpK\njlZMN65udhyt7Vc3+qlqsr2zr/zRLfntgeoGAHhE3ur2Jesj/SvrxOXWVz67XN7QDQAzU2O+\ncWP+wgir9eCcfvt+yc5jtf2evq9+fvnpbfNuXp4xtQNTUFCYLoRe4Pj1r3+9bdu2BQsWbNiw\nIeQXV5gcMHoRte8IjgrRYMjvf0X1Z/iJ10HsbX1CzZeg8pCw5rsQFVZ/yjEjLLjB/9WQTpOC\nSpi3diqGo9AH04MmZE6u1HGRLv95YOfZ3U5l28HZjJmbQnWLCYIJ+cSE3rXuYKJngH6kZGl3\nJ3WUg9cGhgSMyx9lYxdqOxegbvTAfVTzBRZ8Xf40P69+Czzt1L8Q97Tx6rdZ5v1gSBvNfScC\nWjJxZYgdSaYKfuRVqjsz8Nrn5me2M9GHhVumblCjAqPmB5tMA5DH7ymWMOkkVyhT4c6Wt+88\nVtPQ7oyP0S0vTNy8OA37xQtNFOjM4JbKhIpRlj0KoefopWbJ+JELLQ9uynNJ2V56RO7lXMMm\no1ZlfHxwqOq598/3v7xU0/X3Lxz9m23zblw2xT7Tg/nLV5WfHAnobWd3+X755unclHUpcdPE\nJU1BQWFKCb3AsWjRom3btm3cuDEqKiouLsCfvKqqKuS3UwgL2nS0bCHrQRDtAxHz6oGEXq+T\nn3yzX93oxdXFT73F1j41wsXddtAaJlwsM1rU6++llhqx9PhASKPT3PpttMhvYnc384u7oasB\nVFpMnIkFGyIuM0UhAKLKnUPz6qnxMCYtGdxhYSrRmXHWjVT6SUBQULM5dw1/HlXuoaq9wP1A\nAAikM2PhPRibM/IdrVXSF+yuGma/m6wXwNMOQWkGxKn1AGbeP/JNFQAAgLrqA9SNPvjFz4T8\n6yM9LcU4G7mLuo8PFJEJ0c7tn5PTM/hdPRKYEB+y9I3ffXj+/QNV/S8PlDTtPln/88eW9lke\nIGZupNJ3g08zZWPslNr3KFyl2JxSresBbC6fT74lsI+TJlL1DYfb/8cdl4bG/+fjC9cvTBns\nLTK17DhaMzTo8/NdJ+se2Tx1veQVFBSmD6EXOJ599tlf/epXc+fOnTt3ruLEMY3RZaE2Hfwd\nILpAZQZVQB0+NV8Av3voSdRWDm6rdNtLn4cf/4CK94DbDoIas+azNQ+AOfx1Iiq15oFnxCun\neNlpcnULKXqM1QA/TxUNELcQTcGWH1RxlB95DXjvzJ4aL0DZAWHj0yN0ulWYQlxt4JVqMkqc\nrBWoWzzpA5IGs9eBKQ3KvyBbAwgajM3CWVtAP5x/CtUfp4rdfecDAIC7i86+iiv/BrQjWWME\nthIcFBf7FqdSOOukT3LWR3hlRWTRXiUd93upqwHjssZxSbKVgb0SRAdoLGieB5pwdh2KXoiG\nWeBpANEFahNo0zG+Fmq/GvwWBGAxsao5S0Nyw+OlrYPVjR7Olre/s7fioU2zeu+YtAAQqeJT\n8Np6AjhjCWZvifiqH4VpSVKs4VKNRMZQskU/TFFUJH8Wz1d1uL1DsggBHG7/herOCClUIaKG\ndglDbgCoa71qrNMUFBTCS+gFjhdeeOHJJ5987rnnQn5lhckGBVAngGQ6vMsqFQUAIJcVhwoc\nXOTb/5kar/S+FH1UfkKsuyDc/7NxtzUdE8KsRUJOAS97Hdyt4AYAIG8X2CrBMg/Tbx14n6ub\nH/vffnWjF0cHP/YG2/D9SRjntYXXBu4O0JpAO7GlmuiRPcSlLWanCrTkgiV3kO2nCJ4rJHYC\nEAoW0OQEGQ1Q3SGJq4geajiB2TeMcDN9jyQ3RMvQJww3CZcymCQAJD6cLKIQTOAGr0YFfg6c\nA0j/hEe6mJ9qPyD7gLkVtZ/A5A0YO3+CoxwOIQoMA5ulhnu/5+hq91de6I+wGIvhsX9AbWj8\nNfeflW7dsu9sQ7/AAQCYOB8T5oKzDUQ3GBJBpbh7XuW0Wd3vHagsb+jWa1WFmbG3r8qctESD\nTYtT951tGBrfvDhNwxgG/5EDACCAdlJ6qYwPp1tG9R720CSDiDqN4PJIjMegVTpqXYNESmKR\nwvQi9F8WLpdr8+bNIb+sQmShlTVXQ6lDVHp4QN3ox+Pkh95ht0yScEBNB8DdGhzsKAbTbIzp\nzXCm+mLwSyyJqfESeOygjQr7KK8RXG105SPq6luwRaexWbdD1Hhz3XUWQCa9btTFSQQjBLGb\nHPuA925JEVSA5xIa14LQrw8SOIaYaPTgkC4OHwwmLqSGQ0PVH5yxfLjTdAkwRL3Enriibowe\ncxoAAENMicVkMwgMCMjugtouNI35c06thwarGwAA5KemXWhIA+0kfcLRGBP1g1/5zhzwV5wn\nj0tIydas3BIqdQMA2rslUgIBoNU6JI4CGBWL6GuCQ+ebf/nm6f6kg4MlTR8frv6XJ5YnWybD\niGHZ7MQHNsx868tyPqgg5ZYVGZuXpCGCWavqHLIIN2lUkdxFJSVe1tA6Vf7Q5LN4VvyBEgmf\n+8V9TXBare6/7K+oaLKpGOanm+9ekx2lH1frNAUFhauU0Asca9asOXPmzNatW0N+ZYWxQZwq\nDlL1EXK0g96EM4pY/qZQeUlgcgEIGhCDtQCMzQSDhOUB1ZRIj1EmHg7IWip9wFoKfQKHfGYK\nkdOKisARErw2fuYPAR0fbXX87B/Zou+AflwpsioDWgqo/XxwXGdB0yi8KoIQfVR9jDrrQFCj\nJQvTF46lR8MYIOfBfnWjF24n5yGMvrFPSkBgAkhZ2Y2qha0mBvPvp/IPwNOXZc1UmLoGExcN\ncxKai6jtKIiuIDkD45aNfEeFPjAuCxPzwGJHU99KDAGj9VBoALED1GPTOAY62gREOVnPY+Ik\nmiUjqheuUS9cE45rx0ZLP5viYpRC12sUu8v3q3fOBpVUNHY4f/PuuX95Ylgwyuu0AAAgAElE\nQVSVNnQ8uiV/9dzkL8801Lc5k2L1a4qS52b3FhXGqFUCYpfX7+cEACpEk1YVpYro3eZZqaaZ\nqaay+uB5TmFmbES1BPrGjflnytvtrgAPlIUz49cUJQPAsdLWHtmLgADgTHn7p8drf/HY0pwZ\nk9LRXEFBYToQeoHjueee27p1a1pa2h133BFkMqoweRDnB39PLX1Lep+LupvEhrPCuqdBE4qt\nD200m383P/12wLa5xoCLZGwIfTJFBD7P5OW9+6WrOsE/aKWtk3tAIuqVZ2dooPrDAepGD6KH\navdj3giOm3Jgzq3gd5G1YiCki2P5945KCBg8tq46fuAP4OjtkUkAWJrJ1nxb/oMxXsQOEKX6\nQYhdIHaA0PvNieYcapcS5kZjMgqApmxc8F3qvAKuNtBEoSln5GogwcAy7uENO3qsRgEAmAYT\nVqOpcDR3VOiHLdlE7Z8NCRN1foUz7hvDhYiDzyZ9yCdbKjjtWF2UvOukhP/LmqJQ1DD6bMA0\nIChayXTi2KWWoCVuD2fK2zpsHouMIhZyZqaaZqZK2YoBGFWCUSVwIgKI5MSNfhDh2QcX/fhP\nJ6qbB75ScmbEPPPAwikc1VDSE6N+9/3Vf/jk4onSNpfXb4nR3r4ya9u6HER0e8XfvFvcI3th\n39TR6vD++5+Ln3tq9ZSOWkFBIYIIvcCRm5sLAI8//vjjjz8edIhI1ndaIbRQzYkBdaMfWwsv\n/ZwV3RGSW2DWChabQZd3g7UeBA3EZbP8zbJFHJItSxAxLoHO7wS1HhJmYmyYu5Spo8HbKRUf\nWLtiahEI6uDuMACYNAt0EbS/Ma2hbgmDdAAga834Z4gqPRY+DF1XoLsWuA+MyRg/N8jPYmS4\nyA++2K9u9I6qo4ofe52tfXLcQ5O51xCJZ/ChfoEjdxN1VgAP/EBGp2DygtHeiKkxbozahH4G\ny/kGOWvA0wEqIxrSQBVB2cvTBfK3SR/wNAL5AUf98EUGTCPtJiNcPQ4UKwuTNi9J+/xEgMaR\nn26+//rc8V+U/NRyhFqP9hZq6ZNZ6kYwKj1lpwcS1UkAAEAEbVb3pAkcI8Kmg7TRT0qc4fdP\nr9l7puFynRUR8tLM6+fPYCzi/gvJFsM/PrSYCNxev36Q9cbZinarQ+LLsLLJVttiT09U0mwV\nFBQAwiFw/PjHPw75NceK1Wp95plntm/f3tXVVVhY+Oyzz9511zh3hqcp1CSV0gxAjechRAIH\nAKApBZc+PJp3ssJ14skdAcKBWgCzkVQeKtkBAIAMs5azJfcDC1eGJ8YWUvNBqficgRcGM1ty\nLz/2ZkBmii6GLX8gTKO6FpEzWRyH+WIgaJ4F5lkjv08GarkM9qGLUqTGi+DsAkNIm1agVKPQ\nnmSmwYeiU9mSv+KlH4K1GgCACZiyFHM3j1m7GfPwGBqzwJgV3rtc3XB5074xCRwAGD2TrBck\nDkRNYPEfefzw3vmr5iTtPFZb3+aIN+lWFCTdvipLJYx/6UXV75P18sBrVxMvfwOz78XosVeu\nKUw65ijZbsrDHFIYEYHhhkWpGxalTvVARgYR9IHGou3dsp7ibd0eReBQUFDoIfQCx09+8pOQ\nX3NMcM5vueWW4uLiX/ziF7m5uX/84x+3bdu2ffv2O+4I2cJ+GuCVKcfwyu8bhxVzErv5e3zX\nH8BtBwBgAJZoGLxpQJwqD3Otkc3v+zVxD3AXCNGhWsth4ipw1JO9KiCYtBqMAZkjOGsNi8+i\n87ugqx5UGkiaxebcGJq6HgUAAMCoZN4tkayBUZPRT2c47MEetH0QOVoxtAKHEA+oAQrciepR\nN4TAhsTRKWzJX4HfDV4b6C0T+nMgETyl5G8CcgGLQW0eqBSzxnCBaguBVAWeYASmG9ulEteS\nowb8AY4taCrAqOyJjjLCWDUnedWc5NBcy14ToG70QJwa9mC+InBMA5bmJWpUzOsPFr5npZkS\nzVdP7pLCmLDIa1uWaEX2UlBQ6OUqbLm0ffv2gwcP/ulPf3rkkUcAYMuWLYsWLfrhD394bQkc\nUk6fAIAGyzgv6G6h7kvgs4E6Bk0FoB2zGSTmLhFSZ9OVo9TZCNxBzeeGvofKD0LRbSB2UPdh\n8PX0j2BgyMOopcAmnI/K1Jj7AHSWQHcZ+WyojcO4BWCQcPvD2HRc/dhEb6cgA6asxKZTwfvb\nyDDtuikaUR9q+UnzMIfGBwqoX0zOI0GtBlG/WFrCUOlANbZVcTDkIftuELt7X4rd5KsD7WzU\nR1b19dVDVD50HQYenGaP0fPGfCl1DMv9BrUeJHsF+J2gtWDsIjTPDc04r1LIUS19wN0KfodS\ndRX5WGK0376t8LkPzg9uYmLUqZ6+u2gKRxWxcIJ2l9fmFf2c1AKatKpYnTriKk8mBhGkJBgN\nOpXD7cNA5Tg9MSojUakjVlBQ6CVkAofdbhcEQa/X2+12ufdERU1G8tj777+v0+nuv7/X7VIQ\nhIceeuhHP/pRcXHxvHljn1lOTzBjGVUflYhnDt8KgQAIILiLO7Xso7Zj/Ssxaj+K8SsxYezL\nUV0UFm1AAF78IUgJHOBzga2S3F8B9Runc3BeIm8Lxt0BGIL28hg7F2LnXmVP/WmGIQHnPEhX\nPgB3nyWKJgpn3gYx/bXxRJ1nqbMYvJ2gjsKomZiwEljYN2cwMQ+YAFwMPqA397b2JBKbanhb\nAzPFCylZoJpYXzpNFgrR5CoGsQMAQLCgrghUQdKhD8APoJu4ES+5zg6oG/14LoE6fchNFUIB\n02HS7dT6Kfj7f+wI0fPAPK5+NIIekzeO9CHwA7ChX+DXKMOUCA1zSCGSuG1lZl6a6a0vy8sb\nuvUaYU6W5cGNsyxKY50hcKIqq9vT13JL9JPb77V5xYwY3VUz2zl2ufWt/ZWddo9Go3K6xcF7\nAwat6m/uLppWXigKCgrhJWQCR3R09Jw5c0pKSqKjZTXUyTEZPX/+/KxZs7TagUdgUVERAJSU\nlFxDAkfCTFZ4M7/46WBfA8xYgjkyLtOeerIeBV8rAIA6HmOWga63cIO6L1FboFZCnFoPgj4Z\nx1sBjoJa7qNA7guD1I0+/B3gvEStTrA1g96EyQUQpTTomcZg7Exc8hRZq8HdAVozmjIHNTgg\nqt1OtvLeVx4PedrJVsqyHwQhzIVCehObczM/91HgWBlbfC8gExurXG/91l/Za4XALEn6e55U\nz5lY81QhDqOulznWQXQFoKemTABIR8ya0NrVVysZJl8NKgJHmNCmYOrD4CwnXwcyHejSQROe\nHzWvJ14O5ARAwBgU8gHHm6x31SCXZijoQK3s9E4b8tPNP3548bhPr2627TpZ39juiDPpVhUm\nL5h5dc4c2l0+T2BDcQJw+kSr22/WXQ2Z2gcvNL/4eW/FmVanilczh8OrYiw5Vl+QYb5vfa5c\nn2kFBYVrk5B98f3mN7+Jj4/v+Ueorjk+2tvbc3ICKmwtFktPfPgTS0pKmpqaJA+53W6Ybl1g\ncPYWNqOIao6DvQX0sZhShIn50m91XKDOvQMvvc3U9hHGrgPjHACALqlUi574uC3uEvMAPpEY\nsykFoEPyDF7+KRX3FVQLalawkfwErZVABAlZrGgz6BRzqWkFU2PszKFh6i4dUDf68XZR6yFM\n3hjuQWHhFmZKofM7yVoPKGBcFs7fipZMsnXZ/+vvyTGQAcE7mh1/+FnUX/9SlRuOSoE2onOD\ndqhEgCoiO+K4JVoe7PfRD0m3Kgg7HgfVnANrM0THY/pcMEg3Ypz2oAqM+WHdWSTxEvCq/ldA\nVvIfQ2E+sKk2tRklfic5asHvAK0FjRkhydQDADTnU9OX4LURAWJPChQCAMYtDNUtFCKct74s\ne+Wzy2Jfhcv7B6o2LU7723vmRWDTkAli8wZvC/X8D22+6SFwWB3e3SfralsdMQbNkvz4eTkB\nOhQRvHuwanCEMcYE5vb627rdta2O0tquFYWKn5SCgsIAIfvi+8EPfhD0j0gDR0pfu/322ysr\nK4d5w4gSSaSBphQs2jrCm8hH1kMS4a5DqJ8FTENeq/R53q5xzxEwIRfTFlDdmcAowwV3Ah2W\nGeegpGKvlx//YGD1V1ciXviS3fh9TJJYMCtMM4aqGwAAQLaySRA4AABTizC1CLgIyKDvS8Nz\n4JPB6kYvXPR8/rbqO6EXOIjKgrw5AACgDaATQNpeZyQYoBZIyn8ep8Cuj8qO8X2vgNvW+1qj\nZ6vuxzlyySwK8pATuITZBImXkCVPvLIp3FDbCWra19vGFYB08Sz9VklrpDHjtSE3kKuj90fA\nEDQqjF+IyetCcHGFiOdcRcdLO0uDgrtO1s1KM91xXdZUjCiMiDLbbyKfBttyB0qafv3nYrur\nt8veW1+WrV+Q8nf3zVcJvUJkW7e7a1BrWM6po9MlihwAOn2eTpvndFnbzcszvrd1ztCLK0x3\nOE0DhU4hArkK9zHi4uI6OgKyAHpe9uRxDENFRQXJYLPZAKAnReVqw9sMXGprl3zgbQKAQbUD\ngUzM9ZOtfJQV3dbv3YiWDHb99zG5AFQyvSpsroF/+/zBqz+Pg3/xAvCJ9hlVmHpE19jiYYIJ\nMEgSFWuvSL7LXzOkTUMI8AJId0Ei6hr/VTUZUlFETeb4rzkuqLWa73p+QN0AAK+L732Zaksm\neSRXA9QhpYUBgAdI1g8rQqCuC1T/eb+6AQDgbuMVb4F/wt2+/E5e8jLZ6gYinMDL0aKkb1wr\n7DpVJx0/KR2f1qhlclLUQmg+7S6P/8iF5vcPVh292OLyhNLCprnT9cs3TverGz3sPdPwv3vK\n+l/6A6tv7HavKAZP9nYcrSmukM7/VVBQuAYJsTBmt9tfffXV/fv3NzQ0IGJKSsq6deseeugh\no3HyHMvnzJnz7rvvut1una636UBxcTEAzJ2reM5LIalu9B7yAABG5ZCrcehBjJ5Ypz2mwsIt\nQuEWcHWBeqBDBBrnknV/8Jv9IjW29f5bJJDclLC1UUs5Js+a0KgUphy1TKmCXHxqCUvZ2jA6\n3fglPNTNI397r6HpQHAuCJNt1kAluyWcXAHo7OeYrnxLjxWJn+QoDkUE1HJEIiq6qf0MJk2o\npxI1HQffEH2Hi1S3H/Pvo85aqj4B9lYwWjBtPiYoT42rkOYOaU28qUNaPp7WmLRql18iQc+k\nDcEk/0BJ03+/V9Jh672+JUb71J1Fq+aEpiTki9P1QzsBA8DOYzWPbM7r+XeCSafTCO6+Mhy3\nV1phOXyhaV7ONe89pKCgAAChzeA4duzYzJkzv/vd77799tunTp06ceLEW2+99Z3vfGfWrFkn\nTpwI4Y2G58477/R4PG+++WbPS1EUX3vttdzc3GvHYXRsqOTXjSozAGDcUtD21kMOLOZ0iRi7\nKDQD0JsD+l/q8zB6SUCnTB/QmTLw9Av88ktKu6LfT3vQLJ1lOrVNMYV06SWQKlPG12ZCaOWk\nZ8QJKMWowejNqF8C6lQQ4kCThdGbQTcVP9WOeskwdVyFO6thR/YjgYBhNuWdOO5WmXjLRK9s\nl/4skb2On/uI7/o3Kt1D9cV0eS//4j/58TfCo1QqhAaH219S1XG2vN3m9I387j6iDdJdrqL1\nYW/INfnE6lRDtYx4vTpKLdV0fCyU1nb94vVT/eoGAHR0e37++smyeuni5bHS0CatN3V0e1x9\nQoZKYBvm95atEQDJ1N102eW36xQUFK4xQpbB0dHRcfvtt3POX3zxxa1bt/ZUc7S2tr733nv/\n8A//sHXr1gsXLphMk7EHe+edd65ateqpp56yWq05OTkvvfRSSUnJ9u3bJ+HW0xJ1HGgSwTtk\nQqlJAE0CAADTsOyHqPUwdV9CXzdoTBhTgPErgE2sR+YwGBegbiZ4G4C7QDBRbQ11DRLIhvFS\nMcSEa0jTGq+Dqg5SdyOoNBibhenLgE100hNG9CmYtJ5a9hPx/t80xuSjZckUDkq7+hbP/g+D\nbTiYoN18XxjuhgBpAFVD4jqAhIleWTsLtVO9Xy3IfHVMsO3utQnGARqBhtR0sGSAiF/IIZPo\nmdUTnyBygoXopQufBb+34hDFZWHOqoneVGHCENHnJ+r2nK5vbHcmxRquK0rqdvje3Vfh8YkA\noBLYnauzHtmSr1GN/AlZXpC4v1gi+XRFYWLoxx0BpERpTVqVzSv6ONcwZtKqdKP4KQ2l2+lV\nC0zfJ5e8d6ByqJGHX6T3DlT96L75Ex00gFEvvQxRCUyrGpio3LUy0+fnu882cE6MIRdpqL9Q\ngnkK/KQUFBQik5AJHH/84x+tVuvJkycLCwv7gwkJCU888cSqVauWLFny0ksvPf3006G63TAw\nxj755JNnnnnmn//5n61Wa0FBwbvvvnvHHXdMwq2nKWjZTO07wddjoUoACOpYtGweeAfTYNI6\nTJpEbzYhCvS92YmYk4qX91Fn364vQ0CpNA59DCbJr9zsbbzsINhaQBeNqXMx5VrJhKfWUn7y\nVfD1JutS7XGs/Iqt+DboIrLiw2MDZBi3DKNyqKsEvB2gioLoWRiVPbXjwmhz1F//i+ut//RX\nXeqJMEuiftuT4WmhAojZRD6AhkEf9CjEQoAIVqZGDabOpvqLwVECTC2YiuFMdxCFhSSeCXDc\nwHgUpoHfHhrTyCZl7G1Mn+iljcnQVdb7OBuMKK2PU+VRReCYckRO//Ty8eOlvXk9zZ2u4ooA\nZ3e/yP+8r6LL7h3N0nrDwtQ9p+pPXWkbHEyNNz6w4ao1IzeqBeN4UzaIaOex2td3X2mzugEg\nOzn68ZsLls5OqGi0Sb6/vGGI6/a4WJyXsP0riS+BxXnxg5vdMIZfW5ezYUFKeWP3jqO1J0uD\n9+QQcd28adI3SkFBIfyETODYuXPnvffeO1jd6Gfu3Ln33HPPjh07JkfgAACz2fz8888///zz\nk3O7aY8qBpPuAWcZeVsACDWJoJ8VQU5sgppt+ht+5gMqOwTcD4CYlEUtdcAH1WEyFVv3DRCk\nP8905St+4h0Qe7NbqXQvpi9gqx+Xe//Vg8/NT73er270QLYmfvYdtvxbUzUoKYhqjvLST8Hd\nDQBgsLCCWzFl/RQPKhAhJSvq6V+LjVW8rZGZ4lhKNqrDt0OOiPkAaQBdAH4AI0Bc5HfEGA1E\ntNtbuAB2WyBw3qyPZotum6JBTXMwClXXAW8m6kZkgLGAcSOfFQFg0hqy1wQncWjjMHai9aSY\nvJSajgfYlwIAMnLJCBzOadYi7arks+O1/erGMOw+Vff1jbNS4kaowGIMf/HNZR8eqvrseF19\nmyPBrLtuTvIDG2bqQ2FLcfXxPx9fHCw0VDbZ/u9Lx35033yVIP0nE6pOu0vzE66bm3ywpGlw\n0KhTfesWCb070aRLNOkWZFv+6ZUT56s6BwbD8NEt+bkpSg6vgoJCLyH7or9w4cLXvvY1uaOr\nV6/+6U9/Gqp7KYQBBoY8NORN9TBk0BjZsgdgyX3g6AB9DKi00N3KT31IrZVAhInZbOFtYJK2\nvKKuen7sTaAAFyuqPcPPf8rm3Topo58yqOUCeCX6EVDrJfDaQfTz0t3QWQMAYMlgeZtAP1Ja\nB/mp/Th1l4LXChozmgrQsjjAMGVc8Es76Mqe/nuAs4OffJV5bJi9ZoJXDjGIQkq2kDJp6SRG\ngDGbbnjPn7J//Ja/toJFxWhmz4+6+1EWHSnZOkTwk1dOHr7QHK+6/nHzmTWGWoZEgGJqkXb9\ngxA9PZblEQkCS0ZInuphjBFjGsv9Gq/7rN+MA82FmLIR2IRnJhoTK3iIl38Arr4FszoKs2+C\n8/sla1dQM3k+6ApyBK1y5SCC0tquEQUOABAY3rk6+87VPd/YnKie6JzIPQhaxATElAjvJGh3\n+fadbaxttcdGaxfPSpiZGq4FfGOH870DVUPjL3x8sSAjtqxeIlkjhKY1zz646KND1e8frGrs\ncBp1qsV5CY/fPDspVrbeRK9V/eu3lu853XDicmuX3ZOWEHXzsoycGdGhG5GCgsK0J2QCR2dn\nZ0KCbH14UlJSUOtWBYUxwwSI7vuMxSSw9d8czUlUeTRI3eiNlx+Cq13gAJdMV1EiXneGSj4A\nf58pV2e1WH2MrfzWcA0FuJdXvTHg/+duJncz2a6wzPulNQ7uH9VCxd1NZV8Oeo0AAAT80g4h\nYzkIEe8jEEnY//In25//2PNvsa3ZV3XFdWh33E/+W5Uy2b1gJfnqXOPhC80A0ObX/7Jt5a9x\nWbLK0eI3bJyR+5R5ui3OFUKCMYPlfwu8VvDbQRsHgm7kU0ZJdBqb/1dgryd3B2hiMCoVBC2k\ndFDjeYk3pxSF7L4K46XLMVqTSJHzulYHYzjDosdhbLkCzjhH0Cv3EziJqpHaGSuKWI3j8IXm\nX/+52Nr3M3mZld66IuO7W+eM7v87Noor2knKtsbq8IpcunVXp02iacv4EBjesTrrjtVZPj9X\nj843BBE3LkrduCg1VGNQUFC4ygiZwOH1elUq2aupVCqPJ2TfhgoKY8Auk3vs6ATiEVSJM2rI\n1gTWekBEUxpEDeuXppbd46JLnw2oGz34Pfzk/wpb/knuZ0LtJyS6GzjrqfMMWhYPRLifyvfy\n6sPg6gS1HpMKWcGtw1h+UEeFhAKFAH4PddViXK7ciQpB+OurbX95OSjIu7u6X/5Py//99ZQM\nKYijFwM+P14SanwxAHDkYvNTcK3Y4ihIoDGBJgx5RihAdAZGZwwEclZhzQlqLQ94lymFzd4Y\n+rsrjJH4GN0VGFVvjv/aXtLTNNRk1Hzzptk3LhvBtIWosV/dGAiCnagRMRIXyc2drl+8fmpw\n/1TO6cND1ckWw7a1OSG/nccr24Dc5ZXuNm0dtRo1ekapbigoKCiMSChrEc+ePavTSW+/nD17\nNoQ3Uohc3HbqrAMgjE0DXWRkDGpkFvka/fRTN3xufu7PVH+67zVi+hJWdLdcmgMm5gMTgAdP\nUNAQR9aWoQZ84OykjiqMk54/kb1cMg62MugXOIjzIy9Qe1nfgF1Ud1JsuSSseRoMMg3qRfnO\nf6LS9W0MuE8eBKndNk/JSXI5UD/1SfjdTulfaHcY5soKChIwga1/iq7spapjZG8FgwXTF7KC\nTUqmWCRw/YKUngyvEXH3rbqtDu+v3y22uXz3rBtu2U8knUFM1BlRAofd5atvc1iitZ+fqBus\nbvTz8eGacAgc6YnSTwdETI03llRK/PTiTaFLtlJQUFAINaEUOJ599tkQXk1hmsH9/PSHdH4X\niH4AACZgwQ1s8Z2yLSEnC0wtoitfScYnfzAThJ9+nZovDAoQ1R7nXGSLHpQ+QR/L8m/iFz8O\nCAoaSF0K1k+kPSvdfdW23A/uDlAbQB3VGxFdUicAiQPGfdRwekDd6Mfr4Jd2skVflx5ktLR5\nCgBgdASULXCRyg9SWzmIPjCnsplrI0W5GwLvlqtI4tzWLUSAwCFXVp1kGbmcXkEhNDAB8zdg\n/oapHodCMOvmp5wua9t5rHZwMDXe2NTh7OlUioiSlRSv7bp828pMnUbWDYrAL3NEXl6fXNqs\n7t9/dKG/r22UXnri1NjhEDkJoXL47GNeTlxmUnR1c3DDlHXzZ2xanPrZ8dqhp6yfnxLaMSgo\nSMKviuZxCpNPyASOl18OTo1WuKbgR96i0n2DXot0fhf3ONiab0zdoAAAMK0IMxZSzemAqN7E\nFk63zsH25kB1oxeqPw0Ft4LeLHkSzryBmdPpym7qbgBBg5ZsNvtmcnbKOoTpTCC6qeZLaj7R\n293AkIjZN2NMFqpN5JVYQqN6ILecWkolr0qtl+RuiOZ0jM2kzurgePIc0MfKnTVJODrEfc+B\nrW9Tsb5YvLyXrXoMkyOxoakQL12vhCo1M031TxIAADYuSvvwUPAvGgA2LU6b/MFMe3xuurSH\n2quBixCXyWbfANqokc+KGMrqra98fvlSTRcRzEozPbw5ryBD+ktM4RoBEZ7eNm/tvBlfnG5o\nbHcmxurWzU9ZWZjUZnWX1VtFTs2drt9/JPEQdHvFsnrr3GyZJEEARC2RlECPEZGG4PL4//b5\nw40dzv6I3SWtvGhUQsjVDQAQGP700cU/f/3UYD/R5QWJ37+ryKhT3bc+9+29AfmbhZmxD2y8\narvtKigoXAWETOB49NFHQ3UphemH00qX9w8NU9lhWHg7RE1tcwRka56g8oN0eR9Zm0BvwtS5\nbN6t4VoMuO3U1QAqDZpTQBXKtGeyNsgdoe4GlBE4AADjZ2F8gHUo6kygN0tYkBosGJvOL7wC\ntkE7Ns4WuvAqFDwEpjngkFidgnmQe4JfxmrH75YbHgCyJY/wk69Rx0CPOkyczRbIdmWaNPiJ\nNwbUjR58Ln7kFeGWn4A6ImbGg9EtW2f73+fJF1zuoVu2FrURMdrZGeYnbi14aeclvzigsK0p\nSg5H0vXVDXXW8i9+C66+1UjDebF0H1v/HUycHquO/cWN/++N05z3fgxOXm49Xdb2t/fMU6Qu\nhcV5CYvzAhzr4026noIIyVSCHnyirIsEACAkEkip8zCsidVksfNY7WB1YxgWzYoP0xhS4oz/\n/derj1xsLm/oVquEggzz/Nzemds3b569vCDx4yM1De0OS7R22ezEG5emszDoLAoKCgqhQukH\nrjBqRAe4a0G0gcoEukxg2v4j1F4NUomjAERtlTi8wEE+8LcD94HKDEJ4kv8RceZqnLk6LBfv\nx+fmR9+li3t7LTO1Rrb0Liy8fkLX5D6q2U+tZ8HdBSAvl4zVTIQJbMmD/PAfAvQIlY4teZA6\nLweoGz0Qp5rdrOhb4GqgzjODb4xxSzF6kHpilJ5+oVG2xRIAgM7MrvsetZZCVz0gQmxmRHiL\nurupSSohxWOnxguYsWjSBzQCgiXB9MTfWV/418EahyojJ+bR70/hqILYtjZnaX7i5ydq61od\ncTG6lYVJS2cP+9lQkIDo4MsD6kYPXgc/+JKw9Wch6LEaZnx+/tv3SvrVjR44p999cH713GS9\nNtLHrzBVZCVLzxAQUe5Q3xsSEGxEjYEnpSBGRGvq81Wdo3mbQad67CtJSBkAACAASURBVKbZ\n4RsGY7hqTvKqORKVoXOzLcMkyCgoKChEGspMQmF02M9R50GgvrRJQY+x14NhwqtQ1wWynwbq\nW49pszB6BTDZ/ueRDN/1O6orGXjtcfADrzHux7mbxntFHz/zAtj75mTcBSjVfZ4JaB7BQ34o\nmDBL2Pwsv7wHOmsAAGIzWN4G0MVQ1afSJ9jrgftxxmY0FVB3Kfi6QW1CUyHoZwSMJX2ZWLFv\naFcUzFgx4ogwYTYkhHH2NlbI2Sn14wYAAGeENr3Wr9miyS9yfPoXf20FGqO1hQsNG24DIbK+\n5zOTor51SyTW+EwXqLOeuqTyuRwd1FKGyeP9I/I4yNGFpsRwGyeV1nZJtmBwuP3nKjuWzY6I\nTXWFCCQvzTw32zLU83LDwpTYKK3kKf0wzCFMIGoD8ABoERMQIqWkS64Vq1pgRr2qy+7VqNji\nvIRv3VKQljD1VkoKCgoKkU9kTXwVIhRXFXXsDYiILmr/FNX3gToefDYwMIzRk9MDwb7fiHFZ\n8pe9SLajARFPFYl2tNw6pL3HGOEi1RdDdyOodJAwEy0Zw7zXX1PmqygFAHXObFXGOCUbai4L\nUDf6B3LyQ6HwBmDjMUmi+iMD6gYAMAS1CrzBZmmYux4045r06GLYvDuDg1zOjA2ARAAVGNLR\nIK+nRCexBV/j594dnBuCmSsxe814Rjil4DBFTNrInWUKiSkxD//1VI9CIZw45fd7HaPaCg6C\nWqto/2vUdAUAABnOXs1W3gv6mPGObwRsMv4CAGBzRorpo0KEYHf5Wq3uJLPeoFMhwj8+tOhX\nb589Xtra/4YbFqY8ddeoLMMRohEj0SI6e0bMofMS7WPy082/fnKl3eUzaFVKSYiCgoLC6FEE\nDoWRIZtUl1/iZD0NXh91lQAAFKShSNTQDg1d/fvemLsCouVKRokcUpf1t4GnDrRjTkkYuG5X\nHT/8MnQPTBcwaxlb+sDQzG3usHW/9G+e4wPuIbrl18d844doGPvytVmmharHQdYmjB1PIzrq\nvBwUQb0GGJJH7E2RUGlZ3mbMWT+Oi8tikNk+1ZpBGGGLrAdMWyzEz6L6k+RoA20MJhWgeTiB\nKXL5/9l778C4yivv/5x7p89Io96r1WxZsuXeGwZj0zGQACFASGWTTc8Wfrtv3s0uu/lls2Q3\nCQlJKNkEAqH3bhsb27jJVZZsWcXqvUzR1Hvvef9QG83cK42kmVF7Pn8x57bHYsrzfJ9zvscY\nj+Y0GesTToUpxTMxIAYDAGC8Pj6Tb/FDXQ3Sq/8GwnBKBUlUdUjsqOXv+pfQugiNoNRMBwBS\n41k/HcYQzV0Dj79xsbx6SMtYtyTp4VuWpsUbHv3y2sqGvtpWK8/h4qyYRanhUuIixo3rsl47\nXO9w+e8u3LltESh3VGEwGAyGEkzgYADYW6mvBjx2MCRiYgmoAmafXoUe8n2XwOvjTM4jZiYQ\nILT2Asdh0XZuzZ2KDxUdICm0HRW6ccoCh+CRDj3hZ59JV09IWhNXttfvXOvvHnWfPeYbcR0/\nQKIY8+2fTP7Bim1JAus1gkXWmFOrRnMSFn0eADEqOeT19piwjJoPgnfAP546YY2JD7pozNsx\n2/ebPAPSxXeooxJcVjAmcLmbMG+Ln5sJrrmHPvnV6NoPAAC45beB3gyzEnLawNqD5kTQBSHS\nkQtEC4AEfAzgnKwLW5hgXBYY42Ag4GtZa8TkArkrxoNOvub3DgcA6G2hS59iSVh6qeamROen\nR/v2axgkK8m0OJM1UplDSACTtH8Kms5+53cfP2p1jL4zj1d1Xmm2/Oa7W+KitMXZscXZs6Iz\nVEhIMOsefWjtf710rrlr6JfXpFd/5YbFG5cq9lBnMBgMxjgwgWNhQxLVvEXt5SOLcLr6MRbt\nxbiiMaehXIWFKIFXpmUGZibj0nsxLhsMMzBVpZbzMs1BAKj2CCy7xVcOENqa/NSNQdynDomd\nrXzSJHu8xyskKai1aJax7AoG1MWSb4nKSNyQgOaw9RpQ6bgl90lXXgXncA4w8pi+CVPXheuJ\nM4KzXzzwGLgsQy9tHdL5V7Gjktv4DcBRZQbjc/nd/yRVvEPdtSB6MSYdi6/HhFlggBoAdTUJ\nHzwlNVwcfMkVrFLt+hLGKM2PJXJfBE8NwOAHH0Gdi9pSQPaLMBdAjlv/RemT34DoU9DB8dy6\nL4AqqDQrX6hVobVz66UwCRyI8Mi9K//p6ROtPaOdI5Ji9I98YQXLw58LOInqAPoARAAdYjpA\n+nSrSgN46ZM6X3VjkF6b+5VDdfPSwWdpTuzvv7/1fF1vc9dAfLS2dFFctCEs+VMMBoOxEGDT\n2QUNNR+mtpNjQt4BqnwB13wHtD7yhC4d7Bb/axW7sgmYkAa6idQN3gCcXjaJA1VTb4RG1naF\nQbnB0Qem0X4NQnOd0k2EprrJChyYtgSTFlGn/z1x6Y6pO/alrILuSpl48qqgLvc4qL8VOBWa\nU0E9mWWPMZVb/g3qrwNXN6j0GJ0N2vmzVzaIVPnuqLoxDHVcopazmLFiTNQYx637YuRGNiWo\nv8Pzp38G12jejXSl3NtWp/7qz9Egk79NrnPg9X2vEnjriNyon0yeDmPmwNQl/M0/ls6/Q931\nQCLG53ClN4A5deIrAxEVbC+EMNphZCQaf/+Dbe+faLrc1C8RFWaYb1iXpVVPxauIEVnsRGcB\nxOGXLqJaAAvi0tA+5kKAk+j48YghiFTXZu3ocyTF6PPSolV8yHJYVDy3siAhfI1gXR7xzaNX\nLzdZJKK8tOjbNuWw4hcGgzFfYQLHgsZf3RhE8lLHWczaPhLA6DXkqANpbMUErwOYzgwY0bic\nbAE5FOoE0E49PQFVGsVaEX7MfgiO01pCNfnPBSJ3/belQ89Qw7CxCIeQHgdRzWStwuip7Dhh\nXBFkX0ONQ31nCQCRw4zNmFgywZWiVzrzBl3cN+QYqtZxy2/Ckl2+uQkTPZvH2AKASae7+0K9\nV8B6FUQPGJMxaTlwk5xLkQSCB9S66YxB8d7tcsoRALVf9Bc45gLi0dd91Y1ByN4nnnhHtf0e\n/7PJDd56mbsILSDZgJuNJnwMGUwJ3MYHpn8bjMsIVGYBAOKnboQUDBoVd8vGbIDssD6FEVqI\n6nzUjRG6AXoBQtlGVFDYQfH6G5lHlIqrvf/zyoWGDvvgy4xE43f2li7PmxW9ZsenocP2yFMn\nu/qHtpSOVLS/ceTqvzy4ej5V+jAYDMYITOBYwJAILgXLfUfXmJeqaEy5i3oPgasBAAAQjIWc\nNldqfE3mWk4D2uAmOvolCED20/5tYqeT7JqksCaPTvbzTVDnFQPPg+g/V0OVWp03JQtJfTQu\nW0xxVnS4geMgSg8aHkiglvdQnwHqqawbMfsaTFhKXefB1Y/aGEwsAdPEm7TS4f+lOp/2NF6X\ndOplFFzcilunMIapILqp8gXquzISoIZPuCWfh+jglkyWdunEi9R2CUQvGMxc8bW49NoQu414\n5f1fFOOzG6mpSjZOTZdkoqJF0TJG7GMCx0IDl19PH/3WP6rWcsXbZ2A0jFkNAchUgAIAUR9i\nKAWOnJSoxk57YHwGLUWbuwb+8Q8n3F7RN/L/PXXi19/enJMy2782f/r82RF1YxDLgOc//nLm\nmb/bHsIkFAYj5EjEFqqMqcDeNwsY5IBTybcFDeyXoYrBpFuAvCDYQGUecuUwZIKjyf+uCevk\nPTtk0S9BXT4IPSB5QRUD/HRnCRifi1mrqLF8bJTjyu7wO5Mzxxquv9Px7l/94oYb7uZMU5pC\nkUSWKjRqwTj2r0cCWS9h/Jqp3BMAjMlovG4S51vax6gbI6O48AGUXB+mhAj/Z9W87atuAAC4\n+6XKv3BrvueXRyNzbU+j9O7PRrvMOizSqVew4wp37bdCWeZtjAebTFs+MIYrPTi8iArNfWXj\nzOWA4QMWbuAc/dLxV0atRqMSuJ1fhag5sC/NiCxSoDba0CG9cMBb31anVrUtyYq9Z2d+XJRi\nReSAS6hrtboFMTclKj56vB+j2zbnHKloF6Uxj1PxeNumnGmMf1q8fKjOV90YxCNIj718/j+/\nvn5WFVgNjnNkSFfbbbWt/p6+ANDR56yo7yvLZ590BoMx32ACx0IGMWYR9fr3IgUAiM1XuEIN\n6tFdGi7zdmrfR5aLw6/VmLABEyZZxo9qUE/RhlMWbt39FJspVX0IHgcAYGwWrtiLiTL/oqjP\nfZ2Pibe/9r/kHAAANJhMtz9o2OXfbCVYRCdIAc0IBvH62z2ED+pS8BYRvdTTiCmFY4K2Duqu\nB8EN5lRMKgjN2lf0UOd5mbjHRj2XMGnZ+FfTyZdH1Y2RYNN5aq7AjNIQDA8AALjsdVLFm/5R\n5DB7bageEUkwKVvq6wj8n4dJct63XAwAyidx8KHcg2XMFbBsD5+/jlqqYKAfYlIwqzRMDWKD\nocfq+uP71Wdqui0Dnqxk0+2bc3euSENoJbIAiIhRgBls6jJD8AAagNGfuX2nhcdedglDq35P\ndbPl49PNP/3quqKAbjhE9NdP6v6y74rLIwIAIu5Zm/m1G5cYdPL/K0ty4v7u7rLHX784YjUa\nY9L87e2lBRkz1sHqSov87/ilxv6v/Pzg/31gdV7azDesPV7V+cz7l+vbbQCQlWS8f1fRltKU\nzn7FzMSOfgcAEzgYDMZ8g80SFjSYez1ZGkAcs57E2HxMCM4zgtdh+o2YtJlcXcCpUZcMfMCe\njCh6G2vEzlY+PlmVU4Cq8JtacTwuvpZffC04LaDWgkp5m4jjDLs/Z7hur9DeDACqlEzgp7EJ\nw2sBkIBkRILAP0v4IOWGtb5rWkmUzr5CNYdHGuhgfC63/gEwTSaFQXSTsw1EF2rjQTds4Oru\nBwos0gYAAGf3BDeUJOpQ6ulQGUKBAwt2oLWNGn08aHg1t2wvmtND9YhIwq+9Ubp8IiCq4tfs\nkTkbNaDJA0+Nf1ydBZwpLONjzH5McVi0aaYHAY2d9u/95qjNMeTudKXZ8sz758pyq+Ojh3KR\niNoBGpBbBsi8A2aEFIDGwf+yOehXr7uFsV/2Ay7hsZfO/+77W/0ue/bjK3/+yKdokejd441d\nFuejDylqyjvK0lYXJp6p6e7sdybH6lcWJBoV1JDIMI7839Hn/Nc/n37yh9tUfEQT5AjII1oE\nyQ0AKk67r9zy3y9XjBxt6LD/65/Lv3rjkmWLFJXrGOOk+y4xGAzG7IcJHAsbYzK38mGqe5/6\nakASQGPCtPWYsXlyO/lqM6rlN1W8NRetT/9caB5yNOST0qIf+J6mdKrFGkEieocal+iD2+rh\nVar0nBA8F1Voygb7VZkjJoWMmHCg1LCW4zB2dPUuXXiTrhzyPU499eKnT/DX/yNwQak81F1O\n7QdBdMGgAWpULmbcABrzeEUo44hNg4gekBQ85OR6Ek8d5LjV91H2euqoBGc/mpIwey0Y5mr+\nApdVrLr12+KHz5DTNhjBqFjVnq9hovybAbXLCNTgqR72C0TQ5KF2IvNaBiPM/OGdqhF1Y5Dv\n7eVH1I1hPCRdQH4TwCwqClggIOYQuQA6AeD0FcHpltHT69ttzV0DGYnGkYjbK750UCa18OSl\nrstN/YHpHiNEGdRbl02pN1AYKMqMqW5WTMZs7RmoqO+NZLmHSJ4Bb5tEQ58Or2RfmicsStfV\ntYzxg//fDy4/+8jOBLOu2+Lyu4NJrx5H+2AwGIy5CxM4Fjz6BFx6H5IEomfi9edkEDtb+372\nQ3I5fSP9v3gk9v88rs4pHOdCf4jo6md09TOyd4EuGlNLuKJdMl4Sgpvq91H7GfDYQW3ApBJc\ntAs0Rrk7hgtM3knO5wbX/KPBuFWgD2UNzgRjiMvE9BJqqfCPF20D7fD+vOilmk9lLra2U1sl\npk+cKEG956jlA9+IZKvHuhe4oq+ANgYMif4+tQAAiDF5E9xXrQODGRwyk0g0J084qsmCifmy\ntUtzEb5kC5+/Urp6gSzdGJPE5S4DzTgfZ0RtMWjyQbIAEfBmwIjs43l6yVoNnn7QmDGqALRz\n0/GEER4Ekcqrx3xvxEfjynxZB0Q3UA9gUmQGxvABEZcApBH19dm6BpWOQPpsbl+Bo7HDPliZ\nEsilcQWOWcUdW3M/Lm9xehQMjwBaewYiKXA4vB0j6sYgZqPqO3elff+Xdb4taDyCVNXY9707\nl/34j6d8e9NwHH7rthK9lq0CGAzGPISZJzMAAAC50KobAOD48BVfdWMQEryOd56fxF2IpM/+\nIJ35K/U1gtcJtg6q3ifu/xm4bWNOEz1S+RPU+Cl47AAAXge1nJBO/ho8/r0zw4s2nst7CGOW\ngdoMnAb0aZhxK6bsjOgYALjtX8VF63xec1i8k1v7udHIQM+ooaAflpZgHkEdR/wiCADuHuqv\nAgDMuzHQaBbT1oExGQQbWMupdz9ZToAnUAQBrmibzPPUOlw0SW+XBYjOyC1ez6+7iStaO666\nMQxqgE8EVVJk1A3qPi7VPkOdh6j/PHV+KtX9kbr830WMhYzLIwjimIyA5BhU7m099seFJPC0\ng6Ma3K1AiktQRogwI+YkxCh2xYo3R7AqM1KkxRv//6+tS09Q3DUx6cNfgTuMSG6RAn7EEVLj\nNQUZBr+w0y2sKUr8/fe3bi9LS40zJMfqNy5Nefzbm69ZkRah4TIYDEZkYdotI1x4r8rZlwJ4\n6+VNFmShlrPUftE/OtAjVb3Pld3lc9pxsLf7n+bqp4ZPsODG4B8XAlQmTNs9w30qNAZu21eg\n7CbqbQLkMTEHjGPTUMdpuRpMN1bRBR75ZoHgbIfYEozNxxVfp7r3ydoIkgj6eMzaiskrwH6B\n+g6PLD/IcgKilmPsFt8b4LIb0d5DV3yWvvpobstDYJgxbznG9CF7PXUeHBuSqOsI6JIwSqG1\nM2OBYdSpTXq13TlaomJ1KhSsAQD4LCY9bdT7CQjDXc95E8ZuA112eIbJGGJVQUKUQe1XUgQA\nRZkxafFj1thZySatmg9sQQIAhRlzI31jkMVZMb///tZ7Hv3YOuD/r9aq+eV5kUvfkJRVvMRY\n1aXGMZHMRBMAZCQaH7l3RbgHxmAwGLMBJnAwwoby1lvwyKgbg/G2CvAROKDnivxpvVcWbk9M\ncwqaFUpjjPGgjwGnnEiROFEVSZCY0nDZQ0gSkAicGgDA0069n4w9icB2FtTxYCoejXEct/lB\nKtoGbVXktGJMGuauAY0+NKNizBT9co11AKDvHDCBgwEAAIiwa3XGq5/Wj0RauqGlm9ITAr/F\nOcDhxaRgoa63gXwWnKKdet7DxL2gYTUsYcSgU/3oc8sffe6Mr3IRF6394eeW+52pVfN3bM39\nyz5/Y+NVhYlLsuaSwAEAahX33TuW/duzp6WxLWy/vGex2Ri53kOobEBjc4wRkooyY/LT2fYA\ng8FYWDCBgxEu1LmLvZdlVjXqvOLAoCIeRzBxEhUcKAV/Vy0GAAAit+wW6fif/MMZZRifO/Hl\nvA60ceDulTlkGJvyihzgUB0c2atkb0YDF9Hk/5bAxFxIzJ114pTXAZI0amXCCBpSSPkhT9+s\n+7/M8Nqp/ROyXwXBAdoETFyDsSFrYDQ+D15f1NhhPzXsxEEET7xN//IAx+GYxSRy+QBDdVVk\nvzBG3RiKimQ7g/HXh3/IC5r1xclP/2jbS4fq6lptKh6X5sTesWWRbOfXB3YVqnjurwdqR9SQ\n61ZlPHzLZCYDs4bNJSm//Namp9+7dKmpXxQpLy36/l2FKwsi5ygkEQ0InEOIAwAOvVregUN2\n0eB0S1UNo7Oj/HTzP923MhSbTQwGgzGXYAIHI1wYdt/pPPQuOey+QVRrjDfdO5m7KPQCHFtz\ngfp4sjQGnoUG5mIoD+as5VQa6eyrMNALAMCrsegarnh3sJcnb6HGNwbbp4xGdUloXqx4jaDg\nP+9VqHaZTVDrOaniLbB3AQDozFzxHsxZP7lmQ1N4aG8dXT0K9k7QRmHSYszeGGSDm9kIp7C3\nybMmhbMMV7dU82cQhx0unO3U+BbYGzDzpgg8XKfh//0ra49e7Civ7rI5vZmJpj1rM3mVRFIN\nUD+ABBiFmAvo8/3vlbHyAUCFOCPEJMbo/+aWpROehoj3XVtw84bs2lar2ysuSo1Ojp3DeXmF\nGeaffnUdABDROD4x4cAjSZ1Ol0QEoAEAII2XDHq+X4VuAIgzpj5yb/Slxn4iKsiI2VySHOHh\nMRgMxmyACRwLEkszdVwAZz/oYzC1DKLC0oaNj0uK+8dfWJ/5L2/dpcGIKi076kvfV2UuCv4m\nmLWG6g4HxrmstWNOS1tD7Wdkrk9bKxNkAAAAZpTxGWUw0AuiB0yJk1o8Y+xSAIla94MwZOOK\n5sWYvmskX0PuGgUDNm62L3Gp7rB09qXR1y6LdPoFtHdzJTeH76FS1TtUs290DJ1V2HSS2/Aw\nqCO1KpBEkERQhSbpGk255GiSiRuDyBhiRBBq2zeqbowEe89j3DIwKrSgDjUblyZvXDqmaxJy\ny5RPR5LXGtm6btZhNmoimekQASIvH/S43BKNSWgiQpdgjtE69KoYHrUbik0bikPfdCx4BJHe\nOFL/8emW1m5HUqxuU0nK3TvydZo5q84zZhSJ2DuHMRWYwLHQILr0DjUcBhoyb6P6g5h3Debv\nmsQ9bG1U+wFZm0ASwJSKi3ZinHwVvSq7IO7HvxFaG8XOVj4+WZWRC9zkGvdgXA5Xept08S2Q\nRstKMWMF5m8fc15sLhbeTDXvgTTsvIUc5uzA5HGmxQwA8M+FCR6MLUXzEnB1keBAXQJoJqjy\nRX02OetkDugitGqaIqJXuvh2YJiu7Ie8LaAPSwE59V71VTeGgpZm6fL7XMnt4XjimAd11tGR\nF6ijBkQRzIncyptxydZpWupg3EqyVIK7e+QhAAiaOIxnEuRsgiSy1csfsdZgpASOyaFJRner\nbDziQ2EwwotXkrySjPOuBBzPJfABzcsijyBKf/+H4xfqhipYGzrsDR01Ryra//ubm4xytUsM\nBoMRDtjXzcKC2s7R1UNjQxLVfAzmTExcEtQduqvo/LMj+ghYGujM01BwA2Ztkb8AOVV6jio9\nZ8pjxoIdfPISqfEE2DpBb8bUEkyWGSpmbsSEJdR5AZy9oIvBxCVgZBPcySFePileOEyWLoxJ\n5Eu38oWrJriAU4EhNdiFr3EJ2KvAM7bZDW9C87SXuCSRpR2cFoxOnrJeo3jv/mbw+m9oDz20\nuxYzJ/oTTe2hbWfl461nIcwCBzWel975xaieaOmUDjyF3Q3c1vundV9Ow+XeR11HyVIJgh1U\nURhdhImbwlqiQj0XoaOcnN2gMWFMAaZvUqyUYQwieUa/2/0QZ6mfEZqW0UAlSGNtmFCFUStn\naEQMRrgQx+ZujDkkkbLxaOR4/2TTiLoxQkOH/cVPar+0u2hGhsRgMBYgTOBYYDSfVIwHI3CQ\nRJffCJwBU+2HmFIGmqhpj0+B6BSu5JaJT9PHYvbWcI1hfiNJnld+IVYMN2dtvCSe/5Rfvk1z\n+7dD0g0HAAB5TL6drOVgrwTRDpweDIvQvAH4adVcUOtF6eQLYBuqt8e0Ym7tvWAKXRa06JnK\noWnissrH3TYgabw6oGkjffqsb7bUIFSxj0p2Ylz6tG7NaTB5AySlA1kBeQAzYBh/gKjmdeoa\n1ok8VrK3UncFV/IQqI3he+hcYqCFbLUgDIA2DmNLQGUEAOB1wOsDS1QAADQKdkgzDm/ExNuo\n7+CoeKqOx9itoA6x1slgzDic8s8xPzu8No5XdcrGj1V2MIGDwWBEDCZwLCzIKdf5AoCcvUH9\nNg50gEvOKlISqLcWU8qmMzbGDCKc2T+qbgwjnjsoLlrOl20P2WNQheZ1YF4HJEIokmmp84p0\n4HFfxY1aK8UP/4u/+ceg1k3//gCAUckACCC3bxal0IV3+mgVtEKtKazqBti6ob9dJk4ETRUw\nTYEDuokuAojDjgm9RC2IKwBCrzhQf82oujGCq4eaDuCimwAASKLqQ9RSAS4rRCVh4VZMLgz5\nMGYpJFHzu9Q72uKKOg5jxh6MKQYAjCulrhP+l6Bq8OgsRR2PSXtBsIBgAVUUqGKYAQdjXqLh\nOBXHCQFVKhyijg/nT0PQ2BwBLY0G4075OIPBYISDWfGFyIgcCg6FqApuF92rnKUsyG36MeYI\nYoWMkysAiBc+DcvzQlQqTOffkcmod/RRTeiGrY/BtJLAMMZmYVxOyJ7id/M0ea0QU8OrIdI4\nH/BxDgWFSFQFIAL4pgR5iCqnd1sFei/Jhqm3CgDA45De+6l08gVqraDeRmo4JX30mHT61bCM\nZOzzQbSB0CPT1jSCUNdxX3UDAEB0U+Nbg42fMWUbRo31geY0mHXTsMmOYob8zKMygy4LVLFM\n3WDMY+K1Gr88DgSI02pmSbeUlDj5yWRKrCHCI2EwGAsZlsGxsMDExWRpljmQFJQBB+iVs5T1\nLB94DkPWnknFZwnUc1U+3lWPwb2jg4Fbda8k/ok6qkYiGJfDrX0wZMU7AWBcLubtoNoDY4LR\nadziPWF64tAjohKAV4EoyByLmWa6Si+A7KreBuAACPXcV3AExggAvQ4Aks6/Tb3+XaWp8kPK\nWIZJ+SEeyQjuBrKfAHGwbTYH+iI0rVJsLRROqEfO4YVE6ruAKduAU+Oiu8FSDfarIDhBF49x\nZeB10Zk/U28tiG4wJWPuNkwtYzoCgxF5tDyfatBbPB6PKBGAhuPMGrVqkvbtSjjdwrvHG+va\nbGoVtzgz5rrVGTw3+jFv6LD/6cPq6uZ+IshPj/7idYV5adF+d7h2Vcb+MzKmv9etzgjJCBkM\nBiMYmMCxsMCcrdR+AewdY6LRGZi5IajrdTEYl0e9tQHxWIzNC8kIGTMCGs3U3SJzwBSWLiEh\nQ8kQMbT7zGoDt+kb1FUDfVdBkiAmA1OWhHt1xxXfTEmL6epRsHeALhqTlmDO5km18p0Ejlay\nN4LoBF0CFq6nqoB0HlMcZk8zecQ97qFQCxya0Wm31aOq6TfY+Ah36AAAIABJREFUParsaGd2\nogYAqaFc9iJqOBUugcNVT9ZPAIa6xwBI4KwisR9jdoflceNB4OmXP+IeLWBEcyGYh2t2+hul\nk78DcVifsrbSueehrwGLbwvrQBkMhiw8Ypw29PbMVY39P/lTeY91KFnv3eONr3xa/+9fXpMY\noweAzyo7/vXP5YI49Nva2e88VtX593eX7ShL873J6sLE+64t+Mv+Gkka/RXeszZz9xomcDAY\njMjBBI4FhkrHbfhbqt1H7efA2Q/6WExbibnbgQ92IxGL76KzfwS7T6G+1syV3gscey/NYfji\n9VKDTLEAXxyc8jVDYFwWddbIxkP/rMR8SAzb9r7sExMKMEG+AXPIIJGa3qW+C6ORlCgUS6i6\nYjQSnchd/01QT3M+PU77ktB3NsGEEmo7RgSv1KQ+V5XmFIe2N9flcN/Jc8Yqebg65QyGQgEN\nDEsqvrKYpw08LaCZprPJZEHgNSDK6U0KHW2kS2+OqhvDUONRzFgL0WmylzAYvhDRR+UtRyra\nuyzO1DjjrtUZ65YkzfSgGGPwCNKjz54eUTcGaeiw/eeL5372tfWCKP3PKxdG1I1BJIl+9VrF\n+uIkvWbM9O/+XYWbSlL2n2lp6XYkxeq2lKSWLmIZvgwGI6KwRenCg9dg4R4snGquu9bMrf1b\naj8LlkaQBIhKw7TVwLPmi3Mb1Zrd4uVTUt2Yynw+v0y16tqZGpIv1F1PFe9RXzPwakzM45bf\nDIZYAMCl18sIHLoozN88A6Ocg1D7oTHqBgB4bZCq54q+S2314HVjQibmrwPV9D/gcQAqgMDi\nF2M4TEbBlIFZO187UPlkxZhtw+NXpX96+tTjWdHyWobeHPqRAIDkBNEme4S8nRhpgQPQlEsW\nOY8SP+uNQbwO6Pcv5xmEui8jEzgYE+H2iv/8zMmzNUPVjjUt1k8vtO1anfGDu5bPDteIWY1X\nkNSqSJjlna3p7uyXcVI7W9PT2e/s7Hf22mRUUbvTW1HXt2Zxol88Ly06sHqFwWAwIgYTOBiT\nBzlMXQmpK2d6HIwpIXjI0g6iF2NSQTNcGsCrtPf/WCj/SKw4Qv2dGJusKt3Mr9gZPpuJ4KHq\ng9LJv45Uo5C1Q2wo5679LibkYnopt+khqfwlcA0tIDEuCzd8EXRh61g8nyCSt2MQnWAEbt0d\nIX2YCrGIqHJs9ZAKQ+iV4kfa5hdrnYHGH3Vt1vbiJSnOY4FXYPaq8AxFqZBqnBqrMIKp28ne\n4NcLFqMWoXmxzNkCM5ZmTIvXDtePqBsjfHiqed2SpC2lqTMypNmP2ys+v7/mo/KWrn6n2ajZ\nXJry4PVFZmMYd5I6+hQ/zm09jgGXnDETAABYHGFrl85gMBhThQkcDMbCgahyv3TmTfA4AAA4\nHpdcw628bWh/HlG1epdq9a4wPNYNQg+QC7hoUMVPzr3CaZXKX/ZfBwpuOvYs3vTPAIC5a/mM\nZdRzFRz9YE7BuOzZIMrMDUSH3yp3FFc4zGWTEU1EDQA2AA7AjJgTjvqUQbotrr4B+WYlB2nl\n3bEt1NfkG8Ql12FSeAqCOANwOpBklAJUzUTmtjaOK/oytR4gWy2ILtCYMX4VJq5RODkaeHVg\niQoAgD4+rMNkzA8OnW+TjR8818YEDlk8gvSD335W3TyUZWYZ8LxzrPHEpa5ff3tTrCn01huD\nmPSKdcpRBvU4R5NjguvBx2BMCYktVBlTgr1vGIygsbZQXz14XRCVjEnFoep1GjHo/HtS+Wuj\nryWRLn4kDfRyO74Rxqe6q8l1frQvJh+HhvXAB1sLQG0XZRdX1N8Cti6ISgQAUOswRW7zeS7i\nbCFXC0he0CaiMR8wnMnJnPyclQBQ4dC0MSIWh+fO/ozTNNHL6bhd/0DVn1BLBTgtEJ2Chdsw\npSiMYzEsJXuAsykfDdrQm8UEhToas29FAJCECeyTOBWmllHzyYA76DGlNGzjY8wfeq3yBsN+\ndg+MEd470TiibozQ1e98fl/N39y6NEwPLcuP16g4j+CfU5Ycq89NiQLA3JSo+nb/Uru0eGNx\njnJzPQaDwZghmMDBYASBJNDFV6nl1EiAjEnc8nvnksee6JXOv+sfJKCr5dTbhHGZYXmop56c\nY9d1Yi8NHMCoG4NtkOm2Kx0htx2j/Et/5zCSlzreI/uVkQBp4riUm0GbEK4nchowpIHDv6Uf\nAkBUbrgeGinio3UJZl23RWYRtSQrBjgeF+/ExTsjNBpDKZAAjgujuUjqJIzeMvMiaRDm0Lj4\nZnD2UY+P2Y3GiMvuAXWoe98w5iOxUVpZ+4b4aF3kBzMnOHW5SzZ+UiEeEmJN2of2LH7irTFe\n4yoev723dFAs/sd7Vzzy1Anfb9S4KO0jX1jh20eWwWAwZglM4GAwJoaq3/dVNwAABjql089w\nW340VwxWqa8FvAGzzMGZSWcdhEfgIHeVTFRygqcetIUyhwIxKOXwIxrn1cYRdR3wVTcAADy9\nUtvrXPaDgOH6oubSdkq1fwESfYNoLkJTdpieGDEQ4f7rCh97+bxfvDg7dk1R5HUxRONK0BWB\ntxPIDapYUCdHfAxTRaXDNV+DjovQWwuCE6JSMX01UzcYQbJ1WWptq0zfoq3LWH2KPE63KBsf\ncMnX3IWKvVtyF6VG//nj6tpWq5rnirNjH9xdlJsyZGiVkxL19I+2v/VZw+WmfiIoyDDfsiHb\noGOLCAaDMRth300MxkRIIjUfl4m7LNRRgWlzxGyVSPlQmJwOJRDl+26S2Bfkpg+mLQWtKTCP\nA1MKQR8zveHNJiQP2WTa9ILXQgNX0RS29rTGTK7gAan1YxhoBpJAZcTEtZi4NlyPiyy712YC\nwFPvXbIMeAAAEXauSP/6zcXjVK+EF94I/FxNjcHkpZAcnvR491Vy1YBoA86I2izQF03Opocx\nu9m7Jff0le5ztWNsfa5dmc4MOJRIjTecr5NxQUpPCEPDqbGU5ceX5Sv2htdp+Lu2yfVaYjAY\njFkGEzgYkUcCoZbELiA3cCZU5QA3uwsNXBYQ5KuIwd4R2aFMHYxJBV4FopwXenyYjAAQAMd2\nzfA9FBxqHbfpS9KnfwCvCwCAQzQZwRzPrbwzVKOcFXgtfmkUo3h6AMImcACAPoXLuw9IBNEF\nqrBPoCPM7rWZO1akXW23DbiE7GQTy4qfbZD1ILjqhl/1k6cFXHUYsyt8WUuMCKNV8z/72rr3\nTzYfrmjr7HOlJxh3rU7fuDRlpsc1e9mzNvODk02B8RvWzZBlD4PBYMw12ByCEVnIQ+4jQHYa\nXOOKdhLbQbUI1eGyzgoBvPLHZI7UpwAAqHW4eAdd/Gg0QgAImFKISWHak0Hg40CU2YlC1SR8\nJTBtKX/LT6TLB1Bqh1QdqBEAyH0AKANN64CbF2vycUw9w+X3ORbk55+6MYhWzRdlzqNkn9mH\nxS3Y3IJIpFNxCXrNJGryXfU+6sYw3g5wVICxbPIDISAKry8vY0og4p61mXvWhsfpad5RnB37\nzduWPvnOJbd3SPXmOLxjS+61KzNmdmChotfqtjo86QlGtYp9WhkMRlhgAgcjopBwCcgOfjv4\nQh3wqcDNRMfEiSHgrWAwg0Om2gLjw7m1Hmq41XdIiFS5DyQRAAABc1ZyG74YvoRw1JeS/aB/\nEgdvBk3O5G6kj+aKV5L1wJigp5ksVoy5eeadGqePOgZU0SDIVKqjIbhdO6+DuqvB0QP6OEwo\nBM38VCvmNk4bVR+jvjY0xkD2MkzKmekBTReRqK7PYXWP5oW12925MYZobVBTC3JfVYrjpAQO\ndzv1HQZ3O4AE6niMWQeGufTNzGD4cevGnHWLkz4519bSPWDSqfvs7stNlr//w7GijJi7ti+K\nNsydnZWxnKnpfvz1i42ddgDgObx5Q/YD1xcZmZEHg8EINexrhRFZxDbZMIltOAsFDslGzmMg\nWSEnGaqsfjYWmLIMYuaUFyPHc2vuguJrqbseRAHjMiAmzF1gVKlo3ErOcpCGTTTUWahfCTDp\nfRtynJOJilZw14NuPixmMHE7tb3pHzQvA83E2S7UdoYq3wCvY+ilWo+Lb8b01aEfJWOq0JXj\n0sdPgXsABgW/oy/h8uu4bV+EmTIECQXNVpevugEAgkR1fY6SpChVMHkcklM+LirEZXHUUOc7\noyqqp4s638aYDRCzbhI3YTBmGSlxhrt35JVXd/3kT6ednqFP2dmanvdOND365TVzMSvtVHXX\nPz99UpSGPqqiRK8fuVrTYv2vh9fPmC8Sg8GYp7D0MEYkISCPwhGZbo4zDQ2pGwAYG4tLl4Bx\neFdcrcfCPbjsnpkc3ZQxxmL2Sly0NuzqxiDqNIy+CaNuRNNONN+Oxk3A6Sd/FwmEXtkDJISx\nc14kQVMBZnwOtMPNNVRGTLwGk66d+Mr+Rjr/1xF1AwDA66SKl6E3IPmfMVP0tUnv/2ZQ3RiC\nJDr7AZ37cObGNF2IoNcp09ZBJOoLst0D79+KhbweslnI0irV/opaXwOPTIGb/yh6Pgk0+iHL\ncRAUO0wzGHMCt1f8z7+eG1E3BrE6PD/767lxTMNnLU+/d3lE3Rih4mrv0YtzxsuMwWDMFVgG\nByOSIKAWSM6wE30WvS47DfRiVCJoprASDh1i96C6MYQ5CstKQBRBEDH+tjEDZkwAAh8NEB2e\nm8/BiZ4CqM/ErPtA8gIJwAf7BqPGozJ9cEiixiMYxxzvZwVS5SFZf1+6sB/Lro/8eEKCV5Ik\nhWWWWwiqMRNqc8lVP/ra5QC3CwbdgchDA3XkaODS7wS9svWApwdEOSGDJHA1gqk4mGEwGLOT\nC/W9vTaZ+VJTp72uzZqXFqaf1LDg8og1LfJd1S7U924qYaazDHkkmvs1yIyZgAkcjMjCp4FQ\nHxhGPg0AqKeJjjxL7VeGgrmruA13gyk+oiMcQZKbN/M88DxItuDXn3MXsafLU1/DmaI0OXnI\nO8l6GjzdwGlAm4bRqyJkfjkEB6pYEPoCD0zKr3RuwKkBJvG3JYVWPmTrYFm/s4W+dtkw9bUN\nmf3OQXjlrPJgfUa12aAvBGc1AIAkknsoj2/0YhKlrv1c1v2Kd5A8AAp/QkkhW5DBmCP0WBUz\nW7strrklcAiioujpDU4PZTAYjOBhAgcjoqCqiKQ+kPrHBNVFwMVAf5v05n+A1zUyXaX6crHr\nKn/nv4DGP5M5Iozz6ZjnHxzRaul9/Oe2j94BIiCIvq40/t6Vo0sIZyPZKzD5TlBHrgwY9aVk\nO+Qf5YygXfBJCpzCu3Gc7j+MCKPRysfVujmqbgAAz6FRww94ZNobm4MzGQUAjNoE2hxyXSFb\no/wZ7i4QBhRb/KjNAAp/wgh+OzEY4SDOpNjWOi5K4StltmLUqRPMum6LjGSzKHUuKTUMBmNO\nwGbAjMiCatRuBqGRpE4gN6ARVTnAxQKAdOZt8A7++PlMV+09dHE/rrhpKs+SRGr8jHpqweuE\nqBQudwsYJpMMokoE4AAC9hZQC3zops6SKHU2k7UH41O5uJRZ4TgoSR2PfMd18RwAAAEfq4+7\na7n/EkKwU88+TLkj9E8nCbwO0Jj849ocJIEGykdLnNTJaNoAuNC/xDAul/ob5OJ5kR8MQxbM\nKqXKT2Xi2aWRH0wIyYzWV/cM+BWqJBo0BvVkkoo16ahJB+9Rcij46UhuAAWBgzeCIRccAVmB\nKjPoWFPSeUt1s+WNI1ebu+xRBs3KgoRbNmar+HnoKFe6KM5s1FgG/HOR0uIN+emzQhQQJJdI\nbgDgUaviFOUYAECEvVtyf/92lV88Llq7oywidmAMBmMhsdDXBoyZAEGVjeDff2SkMsU/3laN\nKyb/ELdN+uy3ZBvODO+qFq8e5ZZ/HjNWBT1MHWiKwFPll/6M2mWhcucVa8953nhC6m4dfMnn\nLtXc+jdc0gz3uneWHxtSNwAAwbA8EzVyyxVXE4jOUJbqOHvoyrvUUw2SACodpq/F3GuA99mn\n0uWjNhuEHpBcwJtBFRuyR88KSKw4LJ3+kHrbwBTLLSpTbd4bjA0N5mylltPgto55o2pMmLst\nDGMk6qsHexdoDBibC9oAHYohBxZtwAv7qOXymKjWyG383AyNKDQY1XxxgqnZ5rJ5BEkinYpP\nNmni9VPqYamUcIE8qKLGuQ7jryPxLXD79OdSmTHppvnQPXr+4JWok8AJwCOYOZxWx7QXDtT8\n8YNqadiu8sSlzg9ONv3s6+vNxrnaPFUJnYb//l3LHn32tMeniEOvVf3w88tnvO0IgeTwdnql\nUeNkFWcwqpJQ+XN3x5ZFdqf3xU/qRspVclOi/u7uMgNrE8tgMEIN+1phzBrkTPgAACSF+LhI\nFa+OqhvD95HOv8gn5IPOHORNUFsMXBR4qkCyASDwMagtAT5pCuORGWFTteuPP/H9V4v1F11P\nPqL/zq/QGOwIw4Gr6qLvS5VZeY0tDoRM4HB0SSd/A8Jw/qrgooZD1FfPrf76mIUKqkE9P93I\nvK//Urp4eOjFgEXsuCpVfaZ+8NGJ3wwaE7f+m3Tpbei8CCQBIiYW4+KbQRviLT6yttHZ58nS\nPPRapeWKduOiMMgo8w/kuNv/QTr5Fl34GBxWUGsxezm39V6ITpzpkU0XrYrLizXAtK1E0JRH\n3foxDWIJAAGjFk9g98MbMPXz4KghdxtIAmqTwFjEErtmDxL1iVQLMPJL1yFRtIorApiKAlXX\nZn3m/WoamzRU32576r1L379z2bQHO+vYUJz8+x9se35/TU2LheNwSVbMPdfkx0ePlysRGRze\nDq/k8I0IkmNA6DCpFdMxEOHB64t2r8m8UN9rc3gzk0wrCxKC9ethMBiMycAmAYzZAsZlkKNf\n5kDc5DONRQ+1V8jFvdR2HnO3TOJW6kxUZwIJgDi1CZkSnk9eCtR0yG4Rjr2n3nl3CB80aca2\n5BAtTqUTgVfIG5/CM+s+HlU3RrA2UftZTA066WbOItWcHlU3hqH+DvHgC6obvj7x9fpYXPFF\nFL3g7AN9LPBh8H/1uqRjT4DbNhoR3NLFNzi1HjPXhv5x8w+VhttwB2y4A9wO0OhnRTFaSJnu\nv4fTcqm3SO1vg3dg6F4IaMjCxGuCutyQj4b8aQ6BEQa8ItUAjDFqIbCK1MDjVOyTDp1vI7ne\nPQfPtX3vjmXz7lMFAJAWb/jBXbNLu5HI66duDCJITpE8PCqm0vTbPU6PuHVZqnZSVWwMBoMx\nSZjAwZgt4LJd1BygSvBqbumOSd/LbQdJxvoOAMjZP5UpUBj2A6Vm+ZIcsak6cHlKA1Zyu7i4\n0CSPjI+2cInvy4FzzXEeUaZKRZcZwvoU6q2RP9BbAwtB4Kg+qRgPRuAYhFeDKVzvEGo+NUbd\nGEaq2c8zgWNSaGfEMnkuoM/gsh8i60XwdAGnBX0GGpmPzNxGoh4/dWM43s1jzhSKPXuscm3m\nAZxuweEWjKzYISKIpNiiSEngqGzo+/XrFTUtVgBAxD1rMx/aUxRtmG9VRQwGY5bAfgwYswXM\nKOG2fUk69iK4h6s6TXHc1gfBPPmSBI0BEEFunwdnj2uA3PAA/BMovBc+c77+lNTVAgBoMOl2\n3a3dfhtwYdz9MKzbrC1Y7L5yafCl2O/oee54/P0bkPeRhngjxge3sxokosKESSk+D3D2S3WH\nwdoOWiP1NMmeQg7rLGkjStZW+QP2ThC9YckZYSxAOA3GTMFyiTFLIZDXIwAkAi/CpFuBKHUP\n0WtUBi1LChjl4tW+d483tnQPxEfr1i5J3LUqI4S2Haj8kyR7qLKh74dPHBux3iCid483Xm7q\n/9XfbpqX7rAMBmPGYQIHY4oIIlU19LX2DiRE6xZnxYZk5wSLtvDZK6i1Cgb6IDoJ05eAaqIJ\nkOAGyevfdEOlw4QC6qoOeACHKSUAgyICzawLHZeWJ1aXy8QzRhOtPcc/cjz32MhLctidrz8p\ndjYb7v7OBHe391JfC2j0GJcB6knW63Jcyn/8quuxf3McPTh0sxPNupVRpk2Z4OkGTgPadDSv\nAi6kbeoMiWBvk4/PR6ipXDrzAgjD8o3dInsamhNng7oBAIAK01BExUMMxjQhooZjVH+UBrpR\nFw2pJVzRrol/FBizBlSeZI5zaBw2l6Q8v18m3W9zacqM+27KQMKM2ME89e6lv35SO/Ly0wtt\nH51q+beH1uhk/cInD88N9rcO3KRBFcrMN/74weURdWOE2lbr/jOtu1bPsKs6g8GYlzCBgzEV\nqhr7H3vpXEOHffCl2ah5+Jal16wIRa8vnQkXrQnmROqqpsq3yNoKQKCN4gquw5yNI2strmSv\neOTX4LH7XsIV7QbeRW3Pg6cLiEAThzEbYIYqt9Xb9oo1Z/1KaVBvUq+7YeiFJDnffDrwQs/R\n97Xbb+dTsuTv67JLR/9CNceHJh8aA7fmdly6c1Jj4+MTUh79b2/jVU9dNWeK0hQs4c2h64wr\nB2aso0uv+0c5FaatDutzZwZnv3T6eRC9IwHObJB6BwJP5Eq3RnBY44FxOdRwVCYekxXWfCLG\nwoVIOvYktQ3VLZLHAdZ2sfksv/17rH3PXAExBkgmPQ0hamqeVgUZ5i9cW/Dcx2MKPDMSjV+5\ncfEUhxgOyEPuShCagDyAWlBnoqYYMEJpbudqe3zVjUHO1/W8cKDmweuLQvIIBE6ninUJvX5x\nHR8T2EWFCCrq+2Tvc6amp6HDdvRiR7fFlZFovH5N5s0bspntKIPBmD5M4FgYkAiOavL2AvKo\nTQWdf4vWSdFtcf3jk8cdrlGDTMuA56fPn4kxaVYWJEx7rEFBrWel8j+NvnbbpIpX0dbGLbtr\nKGJK4nf8vVT9IfTUkteJ0amYtwO1bmp7ZXRH3NNDnW9j7CYwByWphBY+t0R3z9+53/o9WXsG\nI1xqrnbvtzB6qIWe2NFINjnXVQCh5gKfkkmtl6GvBVQaTC6AmBQAACLpg/+hDp/JjcchHXmO\nA5isxgEA6qwcdVZOUKcKdhpoAK8dNDFoygVu0oW1mL4WnL3UeHi0QkdtwCV7wRChd1QkoeYz\nvuoGAIBOzSVFS10238IlLq9MtXFvpAenAKatwNpP/AtVkMOiPTM0IsY8h1rOjqgbowx0S1Xv\nc2V3zsSIGJMGwchhikQ+Hc0IAHmey5nyPR/YVbgyP+H1I1cbO20xJu3KgoS9W3JnkWklechx\nAKThzRVyg6eGhA407IiMxvHJOflywgNnWkMlcACAjo/lQOUSeyQSAQCR1/FxWl6mdRcRiZJ8\nQe7RinanZ2gmWdtq/c0bF49XdT760BqOaRyMYSSaNR9txpyCCRwLAE8X9X4AgnXwFdkAtOkY\nv3vKJQZvfdbgq26M8OIntRESOIiki2/KhBs+o9wtGDXs2aExciW3+xwWqen3gfn+1H8MTUuB\nnwHnP37pekPhCrH5Cll6uIQ0Li0POJ9sf6+i/QQ6+qQ3/oM6hjN1kcMl27mN91Br1Rh1Yxjp\n9Ft88Y4wlRJQ72nqPATS0GhJZcK03WiarEM+Yv4eTF1FPZfBbQNDAiaVgHp+2jGSU2Y7i4s3\ncSYtmZeTtQdNMVzeCm7JhsiPTRGO59Z/Q6p8g5pPDyUHGeO5kr2YWDjTI2PMT+Q7YQ3FmcAx\nZ+AxByFKolYCJwDPcWYOM6fgvuFL6aK40kVxoRphaCHPlVF1YwTJBt4a0CyRuyLEKPmwdlkC\n+pRNDw0fpeGjJBIAgFOuxOE4zEoyNXTIeFSPqBsjlFd3HTjbunNlemiHymAwFhpM4JjvkEA9\n74E49ufW3UL9hzDuuqndsrbVKhsf9MeOBANd4JJPbYDuKxClYErq6QBJ7oefRHC1gLEgZMOb\nFGotn1sie4RLTAdeFdhKFhC5zjPk8Fkkk0SV+yWVBjUKbU2cVrB1Q3ToW2yQ7Qq1fzwmJNip\n6TXM+xJoJj8BNSahMRKdYmYWVBvk97N0WtWuL4FqthrLa03cii/A0tvI3olaExjimfsGI4x4\nZPpQAgB4ZIq5GLMZDuM5jJ/pUUQKsTMwRgAgdGBEBI4Yk/wviJI/6zQZR9oY4Y4tuY+9fN4v\nqGAED8eqOpnAwWAwpgmbns53XI3+6sYgjlr51X4QKGUPcpF6N5Ffer8v4xySlPtxKPc8m0FQ\nb9Sslakr0WRlokMmBYAqDyg1xw0f1HNKLipS79kIj2QukSovaWFS4exVN0bQGDEuF4yJTN1g\nhBeDvEKKCnFGWOnqdx6uaD9wtnXEe2sGOVPT/dzHV55899L+M62B7pVKCKKk1LsslJDMJAQB\ngEb3Ki419r98qO65j68cvdiuVL4xZTaXyO/xbC6dfEO6ELF7beZDe4p8y4gyEo06haoim2M2\nzscYDMbcgmVwzHcE+e4MABIIVtBMpUXFkqyYY5UdgfHi7AjNO9EYDxwvv5iPSla8TB0rHycA\nlcKhmUZ/x8PgdnlOHxyJ8FmFuvXr6eJHMmcLbjKY5W+kM4EpPNVD7h6FeFdYHjcvwJgMLNhB\nVw6MiWqM3LLZ4rjBYMw4mLWG6g7LxddGfjALGUGkp9679MaRekEcWopvKU399t4Ss3EG1FiH\nS3j0L6dPXhr9fXn2Y+M/3bdyUaqM+8MgRHDgbMvz+2ubu+waFb80J/YrNy4e5/zpwplAkinH\nAM4EAG6v+PMXzx/0scnITYl65Asrs5ND5pu7dnHS7rWZ758Y4+26KDX6vmsnl6YqiNJrh6+e\nuNTZY3WlJxhvWJe1oVh5fjURd+/I37ki/Xxdb7/dnZFoWlWY8K1fHqlrk0n7TYmbn6WpDAYj\nkjCBY74zjt3j5J0gB7l5Q/ZbnzV0j63nVKu4+66NVDsSlQ4zVlPjYKMQn3QSYyImKlupq8yg\nywSXn6M7gTYRdKkTPNFtp74GcA9AdArGZk63c+dAF9V8RJYmIAmj0zH/OoiSHwBqtIYH/0F7\nzR1C3UXyuvn0PPWSVXThQ6UdHy5tsZSYQ11X/eNlN4YrwYbjQTZrhGPfLePBld5GCXl05QBZ\n20Gjx8QirngPaKNmelwMxmwB43K4kluki2+PGg8DYHpUh3g7AAAgAElEQVQZ5m+fuUEtRJ58\nt+rVT+t9I59eaLM6PD/72vrIN2b95WsVvuoGADR3DfzL/5Y/+cNtapX8b9zv3q4cGb/TI5yq\n7jpb2/3vX15Xlh+WqhlU55Ig0/Ic1YsA4LdvVh4cawJa32778R9P/uEHiuOfAt+/c9n6Jcnv\nHG9o6XLEm7XrFifdvjl3Uve3O70/fOLYiADR3DVwvKpzz9rM7925bMqjSozR+9ae7Fqd8cRb\nlYGnscaxDAZj+rBFyHxHmyHfrlwVDSqF3f5ARDc1H6L+GvAOgD7emLLm59/Y8PjrFScvD80z\nclKivn17SX560DecNlzJ7ZLXSW2jVZ0Yncqtun/8jpWYuJs63wK3j6O7OgETbxpfsKAr+6Wq\n90EYqujB+EXcqnvBNJXkFwCgrkt05n9H0k/I2UedlbjsbkwtU7qEzyrgs3z2XlIU9mGMsWBO\n4XZ/Vzr8Z6ovHwqqtdzKW3HZrqmNdkLQkEkWmTkKGDLD9MR5A6aWYmrpTI+CwZi9YOFOLqWY\nGo6DvQt0ZkwrweTimR7UfETykO0UuBpAsgNvRkMRGEsHa9AcLuGtzxoCrzhX21PV2FecHdHk\nR7vTe1CuRUhbr+NUdZdsfkFjp/21w1f9goJIj79R8YcfbAvHIEGVitoSclcCScMzCw61S0GV\n5PQIH5U3B17R2uM4eblz49JQlpBsXJq8cenUEy6e/fhKYHrFeyeaNpemrima4uTHj1s35Vxp\nsew73TISUfH4pd2LI/ymYjAY8xImcMx3VGaIWg42P0MEDmO2BHsHj1WqeBrcw6aeXjtZG1IT\nyx798m3dFldL90BijC4l1hDptl68hlv9IPXWQ289iV6MTsWUkolNAXgjpn4eHLXkbgci1CaC\noXD8q6jusHThjTGRnjrxyG/5a/8B+MmnwJBEF1/xL64hiSpfw6TiIG+ISYswd9WohDEMt/ZO\nQAR9NHfdN8HSQb3NoNFjQjZojZMeZ9Bgwgay1fj7m2hiMVZRr2EwGIwgwehULL1tpkcxw3gE\nScVhuH5kJSd1vQbicFWF0EfWY+BuxPgbAbiGTrtXkDe5qG21Rngt2t7rUHKsaOq0ywoc5dVd\nJGe80dBh7+x3JsUo2HJPE00RqtJBaCHJgZwBVOmD9SltPQ6lP2ZDh33j0rCMZWocvtCuEG8L\nlcDBc/j3d5ftWp3x2cWObosrPcF43eqMrKSQleowGIyFDBM45j9o3gjqBLKWg9APyIEmGc0b\nQRNsrwpq3DeqbowEu85CYmmCOS/BrAv1eCcBxuVCXO4kJ30Ihnw0BFtNI1Xvk4kO9FDzGcxe\nN7knA4ClGVxyriheJ/XWjVdfMxZux1el2DQ69/6QqaopnttwN+auGj3DnIzmqe/eTAJtPJd7\nH7Xvo4HBXT5EczEmb59yARSDwWDMLM1dA3/Zd6W21crzWJgRc9+1BTPyS0cE+860PL+/prlr\nQMVjcXbsV25YXJQZE+Kn2E6PqhsjuFvBcQUMReOIKlzEC1R0GsUpq14rf0i2pf2Eh0IAZwJN\nkd8fSKtgqwkAGmwEgQdVznQLYENEv13ehL7XNkVzeiVW5CesyA+PQRiDwVjAMIFjYWAoREMh\nkAiIk22dQ32X5Q/0XgZzXgjGNpvxOsDRK3uE+lswe9L3I69yg8NJ9T5UabjVt8PKW8DSAWot\nmGa0A582AbM/j5IHvDbQmCGIpnEMBoMxO/n0Qtt//OXsSG+OmhbrgTOtj355TUlupLu3+JpH\neAU6V9vz3ceP/uTBNWsWh2YLfQh3k2yYXI1oKMpJidJrVE6PjBawJDvEUsuEpCcYk2L0nf1O\nvzgiKi2S0xLkExhVPJccG570DWXS4uXHDwBl2TZynwOhGXUbAcartI0M8dG6tl6ZPs2J5kj/\n0RgMBmMKsD5/CwnkJ/1/nCQQXPKHvDI/fvMO5T/XlDavUKc8I9RPfrLI8RCbNsPqxgicBrTx\nTN1gMCIBeUBsB6EBxB4ZiyXGVHF6hP9+5YJf51GnR/j5i+dkKx3CR3PXQKB5hCjR429UhHgg\nSg3jyQ0AWjV/z06ZnYyty1LD2IhEAUR4+BYZE5bbNuVkJMoLGeuXJMWatIHx7ctTlZI+wgci\nfO2mJYHx61dIi5IJAEDsAW9NhEflBxHsP9Oq5Ei6vWwiR3ZlLAOewxXt7xxrPFfbE/LmuAwG\ng+ELW40wxgU50ESDR6aVF4yzVp83qHUQnQLWgGJUAozPncoNo1IgKhVsARbr+jiMzZnKDRkM\nxkLDW0feSqDhTXXOjNqVwEXO43neYu8+W15tc3gDj7T2OGpbbfnpkVvSn77SLSuptPY42nod\nafGha6XJR4Ekt43BDzV1+vz2fJ1a9aePqu1OLwCoeLx1U+4Duwo/Km9+5VB9Y6fdqFOV5Sd8\n5YbFEciJ2FSS8tjDG37/TlV1s0WSKDlWf+/Ogt1rFPtu6LWq/3P/qkefO+3b9215Xvw3bysJ\n91Bl2bos9adfXfe7tyrr220AYDbA3VvEW9eOCmoktKC6aEbGBgAeQfrxH0+VV8t3eb97R96y\nRVPcUHn98NVnPrjsdA99a+WmRP3wc8sLMti3FmMCiNhClTEV2PuGMQGYuIxaDgdEOUxYEA0g\nuOIbpGNP+wUxNhPTptYsDbnl90innhrjxKExcsvvBZz5rFQGgzHbEZrIc35MRLKQ6yjqrwVU\nz9CY5hZEA03g6QFej/p0UEcBAEiCdOZVqj3c15UGIP/T1qfgShAmZKtCBgmteQQaisgis6BF\nw5AnFCLctjlnz7rMxg67R5Cyk00mvfoXL59/78RQbYtlwHPwXOuJS52PPbwhL23SGlC3xdXa\nMxAfrUuNC8qtvCQ37pff2uQVJLdXNOknfs8vzYl96kfbPi5vqWuzGrSqktw4WTvSiLGyIOF3\n399q7//U5eqJD+wMTmF5m52r7Tl0vm3QV/WaFelLc+TdYV8+WCerbqzIj//irsKSnClWae07\n3fKbNy/6RurbbY88deLJH24zG5ldF4PBCD1M4GBMAGZsA3sbWWp9Qjxm7wLDTE4RIgamLefW\nfUk6/xo4B51WEbNWcaW3T9yxZQxE7TXQ1wZaA6YUcFt+RA1HwdIEIEF0OmZtAjWra2UwGBND\n3ityUTcIjaCe76ZI08fdLbW+C86hpDxCFSasw8RN0plXqOZTAIjXKC4vEyPrM5oWL19zwfPY\nK4pNVmeaSceHpK+KsRi83eC4NBpBDqPWgWbMT7xWzY/st1c19o+oGyM43cITb1X+59fXB//k\ntl7Hr18b7TefnWz629tLgswRUKs4pTKKQPQa1c0bFE2z3F5RzXMR7gRn1OuMsuIMhvhtRkT/\n82rFu8cbRyJvfdZw2+ach29eGlhoe+CsTBdeAEiM0U9Z3QCAlw7WBQYtA54PTzXftW3RlG/L\nYDAYSjCBgzERnBqL74OeSuivAY8d9PGYvAr00zA5EwWx9hz1tKLRzGUXo3m2G2hjehmfWkr2\nTvA4MCoJtIF7LuPS3ybt+wO1DxfWqjTc6ltx1U2zxCydwWDMHQikgIYXgwekfvaFMgGSR2p8\nCbw+f0ASqOsISEB1RwcDpaaeOLWr1+u/yMxNicpOnuQ3//RYuzgxwazzLawYpLQgkVdznQMe\ni0tYnGBShWBZjhizDQyF5GoA0Q58NBoKQTVeCeqpy52y8fN1vS6PqNMElY1od3p/8NvPfP+B\nDR32f/jDiV/8zYaQd4qRhYjeO9H04ie1bb0ONc+V5MZ99cYlU8hAmRqoyiChRTYe2gd9fLrF\nV90Y5PXDV0tz47aU+htq9FjlPdcC34fBI0l0tUP+W6u2Va78mcFgMKYNEzgYwYAYvxTiQ9Cl\nXWq67HntV9QzvEug1qi3fU61Ze/07xxeOB6jp+StJXjEN/8TbN2+EenYS5xGh6XXhWp0U8fT\nDZ52IAHUCaAL8bwqElgaqf8qiG4wJmPiUuB4cLSTox04FRrSQBfprgezApKo5Rz1NwMCxmZj\naunUDHEZcw1mGT4BZL00Rt0YRmo7BpI4+N9aTvphzvl/q1vpEEdnRzEmzd/dXRbhj5FWzf/f\nB1b/65/LO/pGm27kZ8bces1Qno5blNrt7ozo6Wz4EwgdIFqB04EqAaODTb6wO+VrZIhowOUN\nUuB4+1hj4LJZEKXn9tX85MHVQY5kOvz3KxdG8lA8wv9j77wD4yjP/P88M7Ndq1XvvdmW3I27\njXHBxjaEHggECAkpRwIkOe4S8sulXJLL9SRH6hHgCBACxpSADQFs3HsvsizZ6r2tpO27M/P8\n/tBK2jKjukVlPv/APtNea8vM+32f5/uIZ6o6n3j28M8fW7agMCK+3Ww6cLnA1/kHU0Keh7Xn\njISMAgCfnG4KFjjiYtT9TisBqFXsgQstWjVbnGWSNG0dBkRkEEUpL2SOVe5NCgoKYUEROBQA\nAMBjJXsbEI/6NFCHy/aJrD3uV35KTp9+qB6355NX0GBiF2+UPUzgqbsZADAhA9iRPrG8i7qq\nwdYFhgRMLARubHfikENVx/zUjQHEM7vYeZuimcRBPHV/CjaftGRNOiZuAS6iq5TjR/RQ+ZvU\nNmRGQLoEjEsmu/eBlQAxeRFmbwFmJv3K2brEY89Tr/eJlgAwPpdZ8SXQKV5u0wMENgGELokN\n7IyU88aEU9o6EXi/jmCLYrueKzv4dltelT2WNaWWzs6/a23+aLweQk5JlumPT6/79Gzzmdou\nQMzNiJ1d4Pcu97o8WTBegUMwk/04iINL6Cxq54BGokdJMOkJ0hanei0XN+rZb0W9WTJ+pU46\nHlquNfUGV9nwgvjbdy//4ds3RmAAAICaRcBlEF8Pog1Qh1wGcNkhv4pkV1oA8BXOBlldlvb6\nvuvB8WPlbcfK2wBAzTH33lT48M3FOGrBDxFKc+MvVEv8apVNoOxFQUFBYRhm0qP/9IO3kqsb\nOR2oE8doCeEDidT0KbUdBxKgf1qYtBCzbwY29NKAcO5TP3VjAP7oe9ICB4niyfeFo2+D2wEA\noNaxq+5mbtgutyJNLZfEc2+AY8C/U2tiFt2L6dE0Q6WuwEcoL9ZucNpAGxPZ4QxB5gN+6gYA\nuFqo831Mu39K1M5Q1Qe+6gYAgKObnGYwagc+HkQdZwAQc7dFY4BRgcTjLwyqG96QuU48+Sfm\nxieiNSaF0IKq2SQcAauVenqQ50mnx6REUJnCMTWabsgYOaNWTZwG+CH3jQSV80tZFQDAbv0+\nxKZFaHhSaFTsLcuy8wsTrFKeo+PvtUlush3w97MUyHkJUQPqkTMIbpyf/uKHV4NtUG9enDV6\nWxC5wUemgejpKomFBwCoabV097kSYiO1NMKmIhteO7NYvRpA4rlL0t3zcxuKTld1XmvqDd7U\nj5sXX/2kCgEe3lwy+jE8srnkO88d4wW/dzY31bhxceboT6KgoKAwepSk1qkJb6XGd8XK31Ld\nX8TrL4rX/kB9leM7EzV8TK1H+tWN/gB1nqWad0M1Ul/E9sBCUG+8o2EwQ9gXYd8rwv4/e9UN\nAHA7hH2vCAdfkzwJ9TSKx18YUjcAwNkrHn+RzNIXjRDDCE9M9L59ogtsVyTi7k5wNkZ8NGNH\n9FDLKYk4EfCiX6DzLPDS61fTDzI3UI/E20ed18DSFvnxKIQFJgGq++jseaipo4YmqKyi0xfQ\nkaXczUdGLzOb0qcwpVuCw5i7NLrqxiAaVvrN1XDj7b3lrpPs1kGuq6M5OiFW88wDCwOyWpaU\nJH9p2+zRD0HO7WKCvXjL68y/f6/8Ry+d/sP7V6429MjtNtivNBi7/KapyAqZljGSrWT0Wu5/\nvrHqy9vnzM1PSEvQl2RJm6HsPFDj9r/VDs+8goR/+dLynBTvig4irF+Y8W9fWa4etU2sgoKC\nwphQMjimICSIda+Dyyffz2Ohxncg+y40Fo3tVLyDOiQmitRzFR3toEuZ2ECDkCswYTkJIcDW\nI57+MHhf8eQu9oZbQR/4DETX9kmoJKJA1/bj0ofGNdwQgOnFdO4DiXhCFqils3wjAd8DJPN0\n4ukC7aRfCnb2giBRJwwAIIoAPg/9JIKzA2JyIjOuKGOVdv4DALK2o3FGtD2a9tC1PdR00S/k\nsotnX2Nu/EdQRe8nZSqAxiLSZYDDv08EMphyI8YUMCqtePF9cNsBABgOZ29kSm+JyjiD0chk\nRSTpx1k4Q6LMEr1oBRJ8U126+1x7zzU1d9rjjZpls5MH7T9XlKa++I83/e1kQ12b1ahXLSxM\nlJtIy3Hritx3D9faglre3nfTOE0oiOB3f738zuHawcjbh2ruXpv/5e1zgnfOTJJuUqNRsclx\nEW2XE27uXJN3+FJrgNYzNy9h+wrp2yLHMveuK+hvbrLzQHVlo4RI5HDzjR3WgvQxSFELixKf\n+/t1bWZ7t8WVkxITlZovBQWFmYMicEw9qO+Kn7oxGO84NFaBgxztchNdsjVjqAUONmeOcGZP\ncJzJmRNcdSK2XJMemyhQ63UsWBQY75Vubwa90UxJwPzFmFpAbYE90nDFPVEZzwDy634yWdyT\nC3ZMz0YzZo2Ik38uj7YZjUJoIKKGoxJxj51azmHOqogPaGqBTM691L6fzOeh3/JQHYepaxE9\n0HsS05PZ3B+QrRdEHmPTJsNXhhfENw/UvLm/us/uXrsk85Y1eYOtTBEg2aBJ1ElUGYwOuV9F\n9L0X7znT9D9vXxpMdnh1T9W2ZTlP3FnW779gMqg/O14xAgCSTNqfP7b8v3ZcqBtosWEyqP/u\nM6VLSsbZo+3Tc82+6gYAiCLt2F89Jzd+zdzATJxVZWkmw5VemzsgvmlJpkY1FW6Co0ajYn/x\n+MqdB2v2n29pM9vTEvQbFmbcvjp/NAafwxhtjN6Dw+cQSEvQp8kYuCgoKCiEEEXgmILYW6Tj\nznYgHnAM7ymClLH10MYQw86/kTn2vtha6x/lVBsfkNhbkCha6YcEXmJwjMxDSWRm7Lwd3BbQ\nxgPj/8SJDHPr0+KRv1DFQSACADAmMWsexPzFkRiVHKoEYLQgSjV+04xcEyt2tbo+fk2ovwqi\nyGYVaW6+n0mNbIqExgT6JLBLFVEHpLwyatBPiiTzCICJBcCqJHJbOC0m5EVhQAohh3eAW6Kc\nHmC4/B2FIVgNpm/G1JvA1Q2sFjytZD5AoqvfkBcYNcathZhRGW1GgJ++cvbI5db+/z94uunK\n9e5FpSmblmSmxOnitSr9BObhyCWR+5rEBjZxUPuoa7P+5xvnfR0xRJHeP1aXnWK4c03+uC/t\ny+ycuN9/a+2VOnNTpy3JpJ2dE2/Qjv+h9JPT0osZH59qDBY4DFruR4/c8LNXz/h2clk+J+Wr\nt06Wdz+EcCxz302F40iNmZ0jXaISo1NlJ0unwCgoKChMBhSBYyoiL0oQjU2X0KcBsj4GHENg\nTBjMn1hO/ciP+U9e5s/s6Z/tM+n5qu1fZrIk3KowRXbOjCm5EtGEfJC020gqGPd4R4W1WazZ\nBVavsyMmz8fczaDycQ/VxjAbHoPVn6PuZtTFQGyKrBYTMZDBuFXUvZcCdKyYUlAlABE1n4aO\nq+S2oCEZs1dA7NCHga88a3/hJ8B7F77EzmbPxSO6B55WLVgT0X9B8Xa68HJgjo+aA9bvb4sZ\na2dQFxW1nindJl4MNNBh5t8B7LhXehUmE4xKVpdW3uLRw6hBlwauFur6ZOCPiQAAopu696DK\nNBqdN9ycv941qG7009nj+PhIXU1dz2+fmvCPrSob2EoQuvzvAAxqhzy5PzrVIOn3uft4Q6gE\nDgBgGZybnzA3PwTdNFrNdsm4ZLsQACjLi3/hH27ae7apts2i13DzCxIXFydNfBjTidLc+BtK\nkk9VBrYfemBjESfjC6OgEFrEYTKOFRTkmTGP/tMJrUzliCYRmDGWNbIaTF1OrUcCwphQBtpQ\n3umF5mrP6b1iZwtjSuLmrdJt/ZLY3YoGE8ZIrw8AAManM0VLxGunA+LMrOVokvgLMCUbhIaT\n3grqQdR6pli+Ae3EsbWIl18EcWjNnDoukLWZmf/VwPdCY8D04jCOZKzElCGjgZ5DwFsAABg1\nxt4AxkXAu8TTz4O5tn8v6q6mxpNYvAUL1gMAiILzjV8NqhteBN658zfc7BtQE7nSZUyajYu/\nLFbtBksjEIE6BnPWAitQ+zEQeQAAToeZ6zF5ScSGNBnA4g2MIZnKd5GlFQAxNh3n3oapEiXo\nClMSVgVxOdBTJ7EpcYwGTDMesl6QlIrIcgHDLXCQQG3HqOsSuHpAE4cJpZi6MkCKPXddohAV\nAK419VodnglbGCAabiTnBXDXeP8IbCxqFwM3VB7S3CWtFzR1yuQQRRu5diFGKacSIuIF0qrZ\nbctnhkPTePn+5xc/t/vK7uMNRAQABi338OaSO1aHTOFSUFBQCAeKwDH1QFMpdR4FjyUwnrR8\nPGfL3ACMiloPe6eFyGDyEswKpSjg+ujPrk/+AqIIAATgPrJLtWS97r5vjdhGhN32dfjoObFi\nqOacmbOa3fyY9N66OPbGp8TzO6jDm3mLSYW48LOgjw/NP0MKatjnq254cXRS+xlMG8/bEVH0\nRagvAsEKxANn6l/Ko2sfD6obXkikqg8xsRhMWULjNbFHojCE7Bah+hI354aIjHuAuDxm6eMg\neEBwgXrAnj1jDTg7AVnQJk4NP5FQgxnzMGOet1BlbGYlClMAZtZ28eT/en+uB8CUUkwaQ9dG\nBQAAj1km3h3e64q8ePUlsDUDABCAo52a2slcwcz+gq8s7nLLFmk63UIIPBpRjbobQLsIRAug\nBhhdwHa9Rvr5cCJVJGFl+ZyU8jqJ93TFHL8VkarG3j/uriivM3sEMSvJ8LkNRRsWZY7dUGKm\noNdyT90175HNs6pb+nQaLi8tRqeepB8ABQUFhUGU36kpCKNmcu8Tmz8Ae9NARIMpa9E0dzxn\nQ8SMGzF1GdnbgATUpwIXytJKvuq866M/D10NAAA8pz9lc2apV986wsEaPXvbU8zKu6jlOiBi\nWiEmZQ23f2was/YJcPaRrQsNCaA1TXj4I0AWmR60ffUw+QWOftgY31fUck5iHyJqOYemLLLK\neO8DkFW2IV94YVV+03hGBfr06IxkUsGqAJwkXgPoAyAAIzL5AIq729QnPo9Z8XWx4n0w1wCJ\noDZg3lrMuzHaw5qCyAmgYRZGqeOUV90AnwIReyu1Hcf0odqT7JSYwCMBACBGp4o3hs4AFVlg\npfMol5QkfyzlajFuE9Bwc+ea/IMXW6419fkGZ+fE3bYqb/DlyasdP3jx5GDpTX279d/+cu5a\nc99Xb1XS3IYjLkat1O8oKChMIRSBY2qiTmDyHgRHK7k7gTWgLh3YiVUHsFo0ShlbTBjP6b3S\n8VN7RhY4AAAAk7IxaSyNS7WxqB1D97IJIWVfAgAB66tTBhLBFZgZ5MXZCwAYJ/toO8wmhShA\nZhLPAgx+Pi0ktCIzH1B5m6Y+sZnMsq+CKADvGMxdUhgzmkxwtUrFh5XRJ05PlWSYeip9BY61\n89Je/LCixxrY5uPWFbksE4l8g5sWpH9wov68f6WMyaB+9JZZEbj6ONCq2V88vurN/dX7zre0\ndtvTE/UbFmXcvbZA5eM8/dt3Lwcbi7x1sGbbsmw5RUlBQUFBYcqhCBxTGV0a6obtEOFsI8t1\n4C2gikPTHFBFatrvA5mlvf1FmfgUQ58CloA1LgJAMKRGZzwA0NsqXvgIzE3AaTC9BOdtBm7U\n7oPIgEoHHqnSa00MALDpeUxajtgamLfCxCVzBWUTGrbCMJAI7j5QGUZtskMkXvZRN/oRSSxH\nds1wTYIVphAMO/XUDZEHVy9o4qJvtAyAsYvIdhUEq1+UNWBseFtckSDteQn+8Rid6sdfWPqv\nfz7b0j30g7z5hqyHN0eoFolh8F++tGzH/updx+s7ehwxOtXyOSlf2jo7yRQ5r6WxolGxD24q\nfnCTtN1VS7dd0kCEiE5Xdc5kgYMXaDRdYxUUFBSmCorAMW2htk+p69SgiRp1HsHU9Ri/MNLj\n0BslwygTjzSCB1hu3D1xMX0lWXYAgFfXAABAYNWYGh1vS6o8LB54aTB/hBovQcUB9tZ/BONo\nk0sxdS41npDYkFzqPvKh5+JR0cUTcih4YGAVEfVG3YP/AKzyYxIGeAfV76WOsyDyAIimAsy/\nBXQjZWGQBUByEuUG6gFMDMNAFRSGxdlDVbuooxxIBGQwuQyLt0eginA4GB2m3Uvmg2CvBhAB\nEPQFGLcW2PBWcqE6juxtkvGAyJycuOeeXnfoYmtdmyVGp1pYmFicFdG/mIpjHthY9MDGIpdH\n0EygK+0kweGSzay0O6dm0uXEsDv5V/dUfXquubPXmWTSrluQ8flNxZPWY0VBQUFh9Cg/ZNMT\n6rlAXSf9QqKHWj5GbSroIupQoCpdxl8M7NICAFxZVC0qSKS6I+L1/WDvBlaFiYVM6WfAOOa0\nC0wsgzwrNewBYSCRWBOPRbeDOrIP7oKbzFXQ1yyefx8YAt/eqZZO8eCfmG3fHuWZsOQW6q4G\nu5+TKGYts73ye766fDBCAGxSGpuRw2YXq9fePlG5igRw9oAm1i9DwdlFjm5UG0GfPDPtQkHk\nxcv/B0NzIaLe63Txj8y8x0bSOALT2n1whWx4CgqjxG0RT/12qPyNRGq/SL11zLInopyEwsZg\n0lYgAQQLsMYI/c4kzoeeqxLxpAXBMTXHbFiUEfYh+WOxe/adb25ot8bFaJaUJM3KjpsG6gYA\npMbrORZ5QaJ1TlZyKK3HpgR2J//Ubw7XtXkzmDp7nTsPVJ+oaH/2G6v1isahoKAwxVF+xaYn\nZL4gGaaeCxhhgWPJes/Z/XzlWd8gk5qt2fDZSA4jAPHsn6npjPeF4Kb2K0LXNWbl4xg/ZiMS\nTF+OiaXUVwfuPtAloqkwoNtfuKHuKqp6G1x9AICpRkiOoY4+MA8l4lLTZXDZQDO6Bzh1DLP6\nW1SznzorwGVBQzLkrHKdOu2rbvQjdLZq7/47VTiU4OYAACAASURBVNmyCY3ebaVrH1LrOSAB\nEDGhBEtuBQSx+n3oq4X+BCRtPOZvx7gZ1wWTOs5C4EovgeCihk+xZPivzzAehKGzJ1TwgWpO\nUeUB6mtHnQlzFmDZzVO7hY27FTxdgCpQpwI3UbmW6g5KmPu4+qj+IBZtneDJQwCywMk2LA/9\n1eJnQ9pKajsGNDTTxpSlmDAum/BQc7S87b93XOi1eUXS//vb1a3Lsp+8a15kjD/CikHL3Tg/\nfe/Z5oB4kkm7bLZE7/npiiiSxeF5/2jdoLoxSEO7defB6oduVloyKSgoTG0UgWOa4pZpaeGW\naYwnex4H2PsgNmn8JdMMq3/sR+6jH3hOfCx2tWBckmruKvWGe1AdtTpe6q4eUjcGETx0+V1c\n8+R4zqg2YlKUnk0dXVT+qp+nKYOYaiJeBMtAkQIR2XtwlAIHALAqLNqERZsGA56zv5Tc0XP2\nwIQEDsElnvo9OAZM7Iio6yqdrAOd1q8c3Wmmitew7BEw5oz/WhGGiGy9aDDBRNoP9tYGhRAA\nqLd2hJOiEcAAEFxtrgEMY9dkSfjyE669O8SWWlBruYIyzdZHmKR0ABDqK527X+EbriEim1ui\n3f4wm5Ef4bGFCBL3P0/Vx70vrF3UUY3XjzPbvwPqKdi2RrBQ915wDbToAoSYuWhaDThCV+9h\noJ4a6bi5ZvCT3Nptv1Rr7rO5c1NjFhYlTYPp9DBg1iaML6XuS+DqAU0cxpdCTJidTUdHm9nx\nL6+edXn87Hs+ONGQnqi/f/1klJjbexwaFWsyjNZn6ht3zO22uM5dG3JOTYnT/dNDi7Xq6ZCi\nMiIt3fY/7rpy/Eq7mxcZma/YyasdisChoKAw1VEEjmkKqwZByiqSGe36LbXX0L4/UXMlAACr\nwnkbmJX3gGZcz+sMq159a3DPFGqsoLZqAMS0AswMgTE7tddS5TGwdIIxCUuWY4r0fInapdKD\nAchcB7wTuMnroBYMtZyU7tiSYBgSOABRN6E1WLGvWzreKx0fJdR4fEjdGIR3gt0NGv/VbxKo\n8QDO+fxELhcZyNrr+uBPnnP7yeVEtZabv1q7/QtoHJesINeLZxQ9epCZS+IZAA8M2cNwyMwF\nGP80dRy4/vaK6+O/DLxweM4f8pSfMHz1p0J7i/3P/w2i2D888eIxz+WThi9+TzV/VSSHFxKo\n/jxVHx8y4ekP9jSL595jlt0XvXGND5E6d4PH91tJYL1IyKJpAm+N4JG5mgcAiOClj66+sa+a\nF7zFdQXpsV+5bXZxZpxRN5WzYIbHkIGGSNeejMieM00B6kY/u47VTyqBQxTp3SO1r35yrc/u\nBoD0BP1j2+esnTes5zoAAMToVP/+lRXHr7Rfqul2uPmC9NiNizOnRwHOiDR12p789WGL3ftl\nFIO6yfRjm5F2JAoKCtMMReCYnqAhn9xnJTbEyK6RUncFdF4gpxk1JmKT6MNXgB94KhU8dO5v\nYksVc9+PQuN+77Dwu35NNeeHBpy/kNv+ddCN38pBPPw6nXofyPuITKd34ZJtzJrPSewqyNkQ\nEPDuqSVwBJUweEE1N/jwghmzQDuhQnc0xpNDwnyeiU2YyGnBXC0dJ1EiZm2a/Eu65LDZnn1a\n7GrxvnQ7Paf2CNcuGL71KzSMvYeRPhnMUmKcfhStXjEW2dUk1gL0IRBALDK5Ea5PETtbXHve\nCIx63M6dv+Hb2/rVDZ+9BfvrvzaVLZtybrVUfxZAwqeY6s7ClBM4nI3+6sYA1ksQuwxwnG8N\nGlLIJmWraUgFgDcPVP95zzXfeHVL3/97/lRqiiE1TnffjQWLC2eMLa7YQWILkANQj0wGMJH+\nh0s2GQGANrPDzYtqLqLy6DD89q+X/3qkbvBlS7f9Jy+f/ubd87YtH1WW3/I5KcvnzKCalH5e\n/rhyUN0YhsykGWdHojCZEWmKPRIoTBImy+1KIbRg8kpQBYkFunSMmye1O1HVTrr6F+oqB1sL\ndVdAxyEsTAH/DEZqq6arR0MyPP79//FVNwCAas7xu3497hPStZN08q9+E2MS6dT7VCXREAQN\nMi1FVLr+ZqhTCbkpx6C8oYtl1jw8wYuoF62VjKtk4qNFlHnYIkkpQ3q5aVLhPvjuoLoxiNjT\n4drx71R/BkSJpdFhwJTFkn4umDZKg14VMsXILEHmBmRKIu++wV89Hahi9MebayX1MrKY+Trp\n7KpJjTOwjt2Loy+y4wgFkuoGABAPfO+4z4pZK6WiDGatJIK3D0kUsAiCaLd72nudz75Xfuiy\ntIw7vSDynCPPSRAaQewCoYE8x4m/FOFByFVqqDhGxU6Wx8XmLtt7RwNblQPA8x9UDCYBDdJh\nc19qs5xq6rnY1tfc56QpcBsJF2eqOkfeCWDrsuxwj0RBQUEh3EyWO5ZCiOFimPxHMH4BsDoA\nAJURk1cxufdLusRTx0XqvBgYjdNDSmBdAzUGOk2OA+pqotqgywFQzXnqDnT/8tJdTSefE/f8\nSNz3Mzr7MtjaA48tPyB9Lak4ZiwElUSaBmYvm0ideXSIk0nJ4VlMKcAFW9nP/gziRk7cHR7N\npnu5/DkBQfWqraq5E2uFY5BpWyP1JoyQzk0i8L1Aw3QPiQR81Tnp+PXL4oH/FXb9hMyNYzid\nNgFL7gO1EQbVHYbDnA2YPH/CI40E5JCZ+cvPMcg2BUUBvUz9kWFi+U1RYZgfwIn8NsbnY+k9\nfslxnBbn3A1xuVaHp7PXKXkQz3snq28cqpFLp58+CA0gBt3+hHoQAwXTsLKkRFr9X1ycNBE3\nodBysaabpIQKi91T3eLnZXulw3q109rj9Dh5sdfJV5vtF9r6hJkqcjjdIyjsLIMPby5ZWTrm\ndnIKCgoKkw0l82f6wukxfQumbwHiR0gt7rosHU80Qqu/WalcKfVYoC7ZOR51NmJC4DyW6g7T\nlXe9LzxAzovUUY5LvoiJxUM79QZKHt5jJePqGOaGR8Uzr/ga+2P6AmbO9lH+EyYPmHYDNZ8A\nh//KDKth1nxtVIUMo7yKRhfz1H+6jnzguXiUejqZlCz1yi2q0qUTPW3mcmo6ART01KUx9JtH\n+OzKQKZMtojopL6T4Kjw5u+o09G0GlRRyml3y1Q/9U/PelvFfb9lb/sRcKO1xMP4Ylz4DTJX\noqML1EaMKwRN5No9TBAmXk7AYgEkMjsAgEmcqBgXebBgGV3dHxxnCqPaCXt8qGV6bLH6CfYZ\nwfQlmDiLuq6Cwwy6BEycBWoDAHDcyPNmi8PT1G3PntaZ8xSsbvTHhWZkItf4bGVp2g0lyacq\nO3yDei332LbZERvDiLg90r8eAOD2MRDpsLm77IGSt8XFN/U6c+J04RrcJCYrOeZak0Qe1ty8\n+IwkQ1qCfu289NzUqZbEqqCgoCCFInDMAESR+uqAd6AxDbQSD6nkkV5oRRUXsNKBCSFwekf5\n7onIBW1yW6lyd2BQFOjSTrzxO0MtKrTSz75yrUMwqZjd8Aw1nSFLK6j0mFSEiZPIQW0MsGpm\nwWNU8yG1X/DO8OPymcJbQ6hueGEYzZrtmjUh1YBiUnHu5+jq2+AeKFhgVVi4BRMKxer3wDrQ\nx0FtxLytaJLKVSGBut4Dj4/XqbuFOt/BpDuionEwyZlC0/XgOGoHPti2bmq6gLk3jOGkrAaT\nJCvLoo2zl+ztqDKAIUWylIYrW4Z6I9kD+4Oq56/01F8X25v8fTmBzSpk0/PCOODwgGkluGA7\nnd/lF8woxXm3RGtI40edAroCcASa42DscgmXkTGfPAbTlwTEdGquKDP2WpNE5o5GM/Sh8vCy\nc9ppAkmnscjGwwMi/PgLN+w8WP3O4druPpdGxS4pSfry9jmTypdBbhLOMJidMrQpWN0YjM9M\ngePWFTm/3BmYPMuxzLfvXZCVPIneXwUFBYWJowgc0xyqOype2QWe/o4qiJmLmbl39C+dDYKq\nGMmUTfL4m2mrdVi2buJDwoxiYDkQgpy6WRVmFAfEqOuadNqIoxusbWD0rvdi3kJqkqjex4JF\nsuPgtJi7atJk3U4AdQzOugeL7wBnN6hjRzBJdfVR7afUWw8kYEw65q0HQzS91jClDBMKqbMC\n7J2gjcPEEtDEAgAz7zGwtZKzC1VGiMkARkYUc1T6qRv9EE+WU5iwJcxjl0C1cqvnnERVlCpx\n6BtHPS2YG8ExhQNXH1W+R+2XoL/cRBOLxdsxNbBwBrUG3UPfdbzyb76FJ2x2sfaux9U9nbY/\n/FDsHXJ8YBJS9Y98Z0JddaMHs/gOyl5AlQehrw10JsxZiAVLQ6AIRANM2ER9p8B63ptaxRrQ\ntAr0gb/MwyMSOHiBF4ljUMeN0PL10Vtm/+DFk4J/EYpazWq13oJKlsG0+Ok+I0U1UHDjMwIc\nbbZXqFBxzP3ri+5fX+Rw8Vo1i5PvKzk3L6EwI/Z6c6AotmFhhm+/WI+UARAAuKd9uZMMW5fl\ntHbbd+yvHvyuGfWqb949T1E3FBQUph+KwDGdoboj4oU3fQPUdFq0dzKrn/SbSCSWSjdrMPsY\nAcYmM5u/CoZQpMfrjMzyO8QjbwaE2ZV3SjT78DgCIwOQxzb4b8CFW7DqOLXX+u6AyTm4MAqz\n3OjAcKAfSarorRfPvjDYRIasbdR2EcvuxdQFYR/eMHBaTFsYFEUwpKNhhNxscsmYtsjFwwxX\nOE971+Ou918g98C6K4Pq9Fg2dkh1wlHXp0xSRF48+7yfD46rjy69BshiSlnAvlzxgpjv/MF9\n/G9icy1qtGx+mWrxTYDIxpiM33/OfXi3UF8FDMPmzlKv2oqqKfyXweR8TJbtUTWVQA5NKyB2\nKfBmQBVwsWNVamweocPh4QcmUSyDyTpVjHwnzqWzkn/2pWW/efdyQ7sVABDAYFAbfb4yq0tT\n9Zpp/qyCTCqJPcFhZKJmiKCbrH9zhsEfPXLDz/98trzOPBhcOy/tibvm+u6mlnFF1Uwat9QI\ngwhf3Dp705KsExXtXX2urCTD2vlpsfop/KuroKCgIMckvYEphAAisfIjibC5jtqvYGrpYAST\n54O5kvydODCukJnzNWq8ArYeSMjE/IWjNw4YEXb13WiMFw69AbZeAABDHLvms8z89RK76mQl\nFdT5ePipNMxnfyieeo8qjoClE4xJOGsVs/S2EI556kPilZ2BLXJJoIp3MHFWFJrj2q+T7Qrw\nvcDGoL4QYsrGs+Id7N/hJSg/KFKoV23jypbxl48LJ3YiuVmTllH7/8ymzYrS0EIDtV8MdvkF\nAKr5JFjgAADUGzXr75GIa3SaDXeHfnwzAdEJJAKrD+MlkAWVTLepYXEJYqvN7btELojUZnOr\njJphJpaLi5Oef3pdZ6+zvsP6/qnGmtahsqbls5IfvKlwHCOZYrB5ILaDaCYAHKzdYpKAVVpa\nSJAar/vF46vOVHVca+5TsUxpbvzsnMBHhWSDpsMmUaWSbJjRTwU5KTE5KYrRhoKCwjRHETim\nL84ecEq3JKCeel+BAwCx5F7oKoWOC+Qyo8YEiWWYvAAAccINOGRAZv5GZv5G6usEAIyVfZLG\nxGLSmsAZ5IyVUAg6/+YFnJpZcTesUKZMMtg6JOelwDup+xqmzJXYFDao6yOwDSQNebrJWQ+2\nq5hy+whuuMGo4sEp0WMSuIg0sBDdwPcAowlY5WZMSepV2yknW9z/e7/WxQCYvxwT8yIxtvDR\n2yAdt7aC4AF5hx0FCRx91FwO1i6ITcHMMlCPpFnYqsh8yNuxlTVg/CqIkRCVokiPiw8uACCA\nHhefOtJacZJJm2TSLipKulxnru+wqTmmKCM2b6ZMxlhUrQChFsRWIAegHpkMYLOnaKFTCOEF\nsabF0tnnzEg05KTEDOaeIsKSkuQlJbJuUwk6VYZR22xxBgfDOmAFBQUFhaijCBwzEqk2aZhY\nBollEX6YGkba8MJwzMLPi2deArePE6ohhZn32bAObBJB5Dm7X6gtJ4+bTctTLd+M2vGs3JJb\npmcnALgDbSDDi/3akLoxiKsZ+s6CaWydWVA/m6wXgALzNdAQZldO0UW9R8F2xdvylItF0xrQ\n5fmNIWs+s+mbdPpN6q4HANAYmLItOHtjeAcWCYapYJ+hxe3jgyoPiid2DFXhaY3MygcwT96A\ntu8cde8beinYqPNj8PRg/OqwjnNMuAXpz4BLJh4MAszNjZ+bK9N8dzqDwOYjOy0KnULEyYqO\nX79zqaXb605Smhv/zbvn5aUZR3l4QYI+Ua9qs7mdHkHNMUl6ddI0rcggcgMyqDzSKygoKACA\nInBMZ7Qm0MSAS2Jai3FTKus1Lpe58R+p4Rj0NgHLQVwuZi4FRraiezpBFrP9+R8Ljdf6X3oA\nXPt26h9+hs0vHf7AYFATKzvD0JjGP8SxQ/ZrcnEco8ABrBETbqGefSAMfM6RReMS0JdMbIzD\nQ9T5PrjbhgJ8H3XtxsStoPObnGBqCW77HnicwDuHKbaaYhgzpeP6ZGAnMHkQeGCnwf1IBPd1\n8tQDWQH1wGWgepZkXhI1l4uH/+QXclrE/c8xxhRMzJE4MfHUc0Qi3nsaYhcCO9ltAmd6HkIQ\nokgt3XYPL2YlG7iZ6goxPJdrzT986STvI42V15n/4Q/H/vj0OtOoy0xMWpVJO43TysgjdnnE\ndgIBABDVaiaNw4je0BUUFBQmIdPggVJBBmSYok3i5Xd8QgSAaMr0r0+ZCnBazL8p2oOIAo4d\nzw6qG/2QxWx/+V9jvvu/qB5jnq0+CYyZYGkKjKtjMCGyLXIF29jiw6PJxJT7wFkPfA+wetBk\nARvmnHZHjZ+6MQD1HUed1OqrSguq6ZMUjakLqHYvOMyB8XwpD52RII/LvXeH59Re0dyOxnjV\n/NWaLQ+ifrQrtJMMkeyHQOjwvqI+cPcR34z6mwADp1hUvlfqBCJVfIqrH5HY5GoDUbLtpQjO\nJjCER9HzuIXOZtTqmfjR9lrScYxLkOheoeVm7hyeF+hybXdTpy3BqCnNi4/Vq3cfr3/xw6u9\nNjcAaNXs5zYU3buukGOjrgKJIFgBGWAMk0GSem3vNT4o8afX5v7rkbqHbh5bW5/pikts5sVu\ngP6HOyByu4R6YDI5JiJFmgoK4UekGbGcqRByFIFjOoMFaxkSxMqPgO+3lkRMm8vMuwdw5j5r\nTiHI2stfOSkR7+vmK06r5o85L50pvVc89zy4fApSWDWW3juhhfdxICdAcOMVJpADXcG4hzNW\nyN0qvcHTDaILGE3ERhIdWBWz6EvilbfAXO2NcFos3IJp8i2Z5eDd9t98Z1DCI4vZffh9/spJ\nw5P/jTFTcBHSUzekbgwi9pH7KmoCPW6oR6bRjzlIgvQeIO+bS1KNtCcGOe2uD19xH9kFogAA\nTFK69vavcnPky2cGiNNwFrcg+FdBsojxk7UlR7ipqO/5rx3n69q8KWYxOtXCosRDF4d+Q5xu\n4cUPr7aZHd+8O8yFdcMhgvMKucq9HzPUom4BqCP3oypJZWNwW5nh4pOHivqemlaLXsPNyY1L\niQtXh2ORXF51A/z0KLfYyjHxk0GiUlBQUIgWM/SZY8aAWLiezVlOPY3AO9CYDjGjXYgbEeq7\nDn21wDtAl4SJC4AL1118xiKa2yXdUgCAumTm2MMTk8qs+HtqOAS99SDyYMzAnLWgiZ3QKMcO\n6ovIXikVnyKLciSxQD3AMJumEbpEZvGXwdJCtjZQGzA2e3xdeNzHPgxIUAIAsbvN9ekO7W2P\nhWKgEYX4FukNfAsECRyybqxycZX8kqwqccSxjQ0i+wv/LFRfGgyInS32F3+if+R7XNny4Q/l\nGMyM0XQ43A7e+13QcUyyTs0xM3G61d3n+t7zJ6yOIQXK6vD4qhuDfHCi/rM3FWQkRqfUiOwn\nwV3t89pJ9uNIHtBEs+WTzN1PNj4ZaDM7/vON8+evd/W/5Fjm7rX5j94yiwnD518kO5CEjkEg\niORkUHkkU1BQmLkoAscMQKXH5JAmMIseur6TeqsGA9RyCPPvQFNkKx2mO6iTzWhA/XiTHTgN\n5kfb57K/KazVry0xaLPBuDBKAxobqEqSfsBmY4CZSc+UxnQ0pk/kBPzVM9LxitMwBQUOIMkS\nEuk4phVLJ3HIdRHmjKAvAHt1YFyTDpoQ97riK075qhteRMH5wUsxIwkcAKBmMTNGIxB5BFKx\nyOJMlDb62X2i3lfdGAYiuFxrjo7AIfb5qRsDkPMiqosAo5YfXpJtOlkRlBIFMCt7kqZ38QJ9\n/4UTg9k6AMAL4uv7rqtVbDhqaghEuSwNmiFSu4KCgoIMSqmCwpihxr2+6gYAAO+g6p3gke/T\noTA8QiO5T5LrALlPgeBd32OS0pnkQENHAgCWY2ctjvQIQwombMDkz4C+GNTJoCvAxE2YckcU\nn6QlcHdS9zFq+xt1Hwe3v9+EvlCyygYnp0AjCtEegSzksMvEx+XGEnUYmfZGjMSslZm/DbRB\nnyJDAlMqqz9i0uaATj2gycDk7WMb5CgQasol42JrPdlH23GJRdRyzExWNwCgulm6U7skHj5K\nk1K+UzpOHhCjWQzyufVFbFDig8mgvnVlblTGMyInKtp91Y1B3j5Y6el+iyy7ySnR82vcMCiX\nN4fymxQUFBRmBEoGh8IYIaKu8xJxwU3mckxZFvEBTXVEcp8EoX3gZS8JLcBmoXoRAGjvetz+\nxx+CMPRIhACam+9n4pKjMdSQostF3SR9TqWuw2Q+MViKQt1HMXE1xg90eEEVJt9G3XvAPfCu\nIYfGxRAzP5xjEqm3FWxdYEhAU/rINjokUu0RseYg2LuA02JyCVN6G+i8rTep+xo0HiV7B6hi\nMLEYc9YAE4VGA0xSOl9bHjwDZpIzIj+YiYNcLnkaJOIqqa4ohgR2+zPiidep8SIQATKYt4RZ\nei9o5NfwGS2m3gHOJnC1ABCoUyA83yAS5Odgw2xSCIIdi29oYUakqwW9DFNzN1w5XtiZm5/w\nw4eX/Pqdy+093lbKs7LjvnXPvPiYSepzVN0irWdZndDWK2QmWMFVQZ4mjNkIGALfKxb1DGpF\ncgbEOYxDkF4taO9x1LZa1BxbkGGMnaYdcxUUFBRAETgUxgxvB8ElvcnZLR1XGAa+1kfdGEBo\nBCEF2EyueEHMt//HufsloaYcPG4mI1+z6T5uzhh7qQoeMjeBy45x6WCID9XApytkraTuY/4h\ngToPgCYF9QPzSS4eU+4GVyvw3cBoQZ0W1j6d1FlDx18lc2P/S4zPxGUPYvJwFoDi6T9RywXv\nC4+dms8JHRXs6qfAmEpVu6n+4MCOHdRTQy1nmSVfAXWYu88EoV52s+fUHqn4lgiPJDRwqaCe\nA+6K/kQrL6o8UEn11gGA2BRm0xPAu8DaDcYkWfeNALSZoJXp1Bsi2PQ8yTga4zFmunQ7jgil\nufH7z8s4s/gzvyC+JD3SX0AvrFzFBwIbJc1lgBWlqYuKk6pbLJ29joxEQ0G6EUfKCSKCPrt7\n9H1kQ0hwvonPpoH/Ey3kqkBtSNRw1DK5TrFBpKFUOA5NGlZCILY6PH9478pHpxuJCAA0Kvb+\n9YUPbCwa8e+poKCgMBVRBA6FMcKqAdDvCX5ok5IVOWZIaJa0CSOhGdlMAGBSc/SP/tP4z191\nVDz+Bth7+19i4TJm1QOgi/Jjayjx2MjWBIIDtclgCMXKf+9F6XjfRdD7LpgjaNJBM5IJRW+7\neGQHtVSC24nJObj0dswJ8pscHku7+MkvBxohAQCQuYn2/Ird9j2ITZU8gjoqhtSNQTxO8cp7\nzKzNVH8w8CNn76Drf8M5d49tYBOGLZirvf3Lzl3/B/yATwHDqG+8Q7VkPB1nJwOoKQVVBnka\nQLQCo0MuE9iRkq04DcRNyMok5HDzVzMfviL2BNofaNbdAcp0aCzcsiz7nUO1Ld1+pVgcyywp\nSTp+xatrl2i6v5lRnocd4v/+L8SnMzd8BotXRLQFBpcMXJJEoYq6ADD6uRIaFTsnJw5gZGWt\nu8/1/AcVhy62Oty8QcttWJT5hS2zjPrI5aaV5kqvHyQZKdXk88jkaYbQCByAqNaxhQJZRXIA\nMCzq5bxFf/bq2dOVQ99ol0d46aNKXqRHNoenw7SCgoJCVFEEDoUxwqjAmAOWuuAtisnoeCCn\n1JMsQlDeqSzOPnJa0JgSvAJM146Jnz7nF7l+QuxpZe78ATDTwX+HWo9Q8wEQPdAvucXkMPm3\ngUa+38RozumRLjsnd89Y5xzUXCm+9XPgvR6T1FBODeXM6vtw6WdGfxLxyie+6oYX3iWWf8ys\n+Lz0dduvSsc7rlJ8OkhNnqj9UuQFDgBQr72dm7PMc/6g2NnMxCVz81ayGVFuTjla+D7qOw3u\nDkAG1KkYuwRYPQAAE4eaqZ3mgGqt/ss/drz230M9blhOc9Nd6nV3RXVcUw+dmvuvv1v523cv\nH7rkdVbKSzM+ccfceQUJdW3WigZzfFfF4uq9KApeU8juJvGj32FnA7Pys5EcJ+rXkP0Y8D7t\nXdT5qFsSyTFMkG6L6xvPHurs9d43bU7+vaN15653/fqJ1bpItSheUJi4pCTZV0fo5wvr3X7C\noJwb8XhhMYbF4dJ/Kup7gkcFAG/ur75/faFGNZncrxQUFBRCgSJwzGxIJOt1cHYAo0Z9FuhG\n5cbPZN8sVrzUP6scBJMWQExWeEY5rUENkEMqPnI6DLVX0cm/kLkJAAAZLF7DLLwD1ENOh+Lp\ndyWO6qqn2tNYMMY6l8kHtZ+kRv8CB2u9WPlnpuxrwEzgl42RWbR0WMS9vyCXDWPTcNZ6TCoc\neYR7XxhUNwYRj77JzloFsUmjHU9nrXS8q0b2EF5GHRMFcMm4HvJOED3RcuLQbIzodC4EOGqp\n84Mhv0BXC9nKMfn2kDc0iRZMao7hqV/w1y+IrfWoN7L5pUx8yFqMRx1RpHC07ZQkyaT9wcNL\nem3uhg5rYqw2LV7fP9fNTY3JTTUIL/8y2AaYzu6G0nVgks7PCguMDmPWA98JghmQATZJvm5l\nkvLGvuuD6sYgDe3Wd4/U3r8+cksvP3hofx4TrwAAIABJREFU8Yt/u/rekTpBJABIjKHHNro3\nzPV3rmHGWYt0+FLrzoM1De3WGJ1qUVHSQ5uLR2lHUtnYKxl3eYTaVsus7KktyCooKCgEowgc\nMxhXh9j4Hri8iakEgKYyzNgCONKnQp/OlH2FGvdSXw2ILtAkYupyTF4U9gFPR5DNICmnepQq\no/WF2irFPb8aejgmkSoPiF11zC3f8TpQumzQ2yZ9bHt1iAUOIr69hW9r5ZJTubRRWGCG5Jqt\nRySiLjOZyzFx/AnAaMgjl8TfjVoaoaMXAKivlRrPMXO3Y9nW4U7U10mdEpaTIApUdx7nTbhZ\nr3SvWgAA1CcRSFU+aYyglTFhUemjom5MSYinrk8CuyGIbur+BNOlc2qmJIhc0QIoWhAY99io\n6ShYm4HhwJiFGSuAnRpuhQ4X/8onVQcutLT3OFLidDctyHhgU5FOHYmnIJNBbTIEZZb1tEKf\nxLo6kEgNlzGSAkc/XBJwoxZeo42HF1Xc0I3mbJV0L5gzVZ2RFDh0Gu7xz5Q9umVWXZvVoOpN\n1x5ng26GqJYx5RmW3/718juHavv/v9fmbuq0HbjQ8suvr8pKjkZrYQWFIHp7e5955pm33nqr\np6entLT0+9///l13KUl/ClFDEThmKsSL9TvB47ecS72XgdVg2qaRD9ckYOE9CAAkTK7unlMO\nLh+EdhD9H87YDOqw0eXnoLcV9HGYtxBnrQmofqfzf5VY+uuqo7pTmLcMAIDkZ8DDbBo77ppr\nXc/+3HXZ21tHU1Ka+NT31EWzQ3gJCXg7uGWSEeytMBGBI+4GslwF/0IVcrihw+9y4uUPmKwF\naJLVocgp3zXZMZaGygk50F0vEU+U7aCBWYuh8m8g+s7ACQAxZwWmzqf6A8EfAEwNmscqyOFs\nBFEq68pjBncHqKd+hyN5qOc6lb82lCLUWU7Nx5h5j4J+sv+rbU7+qV8frm/3fvXazI7X910/\nUdH+y2+siozGEQy55esQ3YEfMJFEJiLC8SSnucv+/O4rZ6912Zx8RqL+rrX525fnMAw63NL9\nsB2uKPTJ1mm42f2+IS47OS/5qdHqQlDnjfWEVxt6BtWNQfrs7t+9d/lnXxy5dV1JlnQ+jkbF\n5qUZxzoYBYVgRFHcvn37hQsXfvaznxUWFj7//PP33HPPW2+9dccdd0z0zKRMVBXGg3K/nKGQ\n5XqAuuGN91wcW592Rd2YKAxqVqB6AbDJgEZgU1G9hE5fF3f+lCoOUksVXT8p7nlOfOfn4PEx\nYiCROqolT0ftA2Xz2hiISZTcB5PzQjV6vr219emvDKobAOCqLG99+iueJqk5eQgZ5ll/gtMA\nVsvkPIhxi72NUbgYaO+Dqy0g+osCJEKjVL/kwVHEJsk6MprGMBtkSjcBF7RCzqqY0ptlj9HF\nM4s/DypftznEjIVMyWYwZmDBzYF/ImMmFm4e/ZBmOoJ9PJumAaKHKt4MLIBy9YlX34zSgMbA\nWwerB9WNQWpaLe8ero3GcAAAMDZZ9scqzlvrxIuC2dXbau9od3S1OTotHhsNk7s13bne3Pd3\nvzh48GKr1eEhoqZO27NvX/r5a+cAIFsmkUEuHiE0czBmM2hmgyobNLPQsAF1S8bhIDtoSRvA\nmcpONz9yK9/ZOXE3lEjcdD57U4FiwKEQEt56663Dhw8/++yzTzzxxLZt295444158+Y9/fTT\n0R6XwsxFEThmKq4u6bjoAbd0uaZC2EBgc1C9ArU3oXoZtfXRmfcD9qDmCvHMe0OvRRFI5rFG\nGNKnmEW3SuwQl475N0x4zF763npVtAYqZaLD3rfjT6G6hDSsFrQy2dSGCXvBMFpMXs8UfI0p\n+iaT8SA1doEo8dcmp0wKST/aGMyXqtvSGb1xImq9TJUfUdUe6qiUPU9sGrPhSYj1MXeITWU2\nPAGm4VpvYPp8dv0zTOltmLMcizYyKx9nljwMDAsAmLeeueHvMH0xmHIwaQ7Oup1Z+jhwSguk\nUTNMS+BwdguOOtRTDW6LxAZLEzikCwQmDyevShWDAJyokI5HAm0MFkr9FBsTMWceAPAi3+U0\nu4QBl2Iim8duds7EG7TLI+zYX/3MH4873IELMPvPN5+71rV9RY7kgbeulM10ixCsCbXzUb8S\ntQvGXQFksXsk44JIVof0pgD+3+cXb1maPdgUVqNiv7Bl1uc3FY9vPAoKAbzzzjtarfb+++/v\nf8my7EMPPXT9+vULF4IauikoRAQl82emMowLY+RL8a3t1NsMrArjskE7jTqYjguqOiYdrzwG\ny+/xvmA5MKaARWJVB+Mzh/5/zjpG8Iin3h5MeMasMubGLwAbsi++64p0U1Vnedjvapi5jq7v\nDIwaMjBuVuiuwYJaD6wKBImHSNSN4M3GbPyS2NdJnT7JLNoYZus3QK0De7d4+iXqGTLpwOQS\nZvFDoJaYIWNKEXvrP5G5ASydYEzC+KxRuahqYrBwvfRaYWwWlt6rNPwcJ9pMYHUgBFWpqOJB\nPWUsDMaDS3ZqTa5e1E3qf7vNKZ2ZaBvd/DBMMOseFe191FwxFDImMVuf7E/akszXcIseJ+/S\nctFv4Boxmjpt333ueJtZqi4MAABOVXY8tm32l7bN/tNHlZ6BjAatmv3abaVyrVunFinx0s1f\ndWrOZBiVA45By/39vfMfvrm4ptWi4piiDFMkG+gqTHsuX75cXFys0Qz9Ls2bNw8ALl26NH9+\naJoiKyiMCUXgmKGgPkc6z1UdB6oISgxum3huBzWe9b5kOCxez5Rui4xL5STFJt2mFGzdvq+Y\nORvFE68F7qM1YsEK3wDO3cQWr6SOWnBZIT4TE0Ld6UYqtWG4eOjA+FIoBGr4eNCMAxMXYPYm\n2cKQcV6GwayFVHdSIp69cIRjDXHMAz+lKwep6Sp4nJici/M2gs4IROLJF6mvyXdf6qgUz7/O\nLP2i9KkYFhPzIDFvvP8MhdCBHCZs8uuiAgCMBhPli4amByrZ/BSU3zRJyEjUNwSVqABARlJU\nR641MHc+QzVnqbUKPC5MysGSVYMlaW4pXRUAXKJbCzNI4PivHReGUTcAwOb0AMB9NxWunZd+\nrLytzexIT9SvmZuWZJomiWk3zk9/6W9Xg6tR1i/KYMfSDyg5TpccJ62VKChMhK6uroICvxbv\nCQkJ/fHhD3zrrbdOnDght5Xnx1Iyr6DggyJwzFR0aWgqpd5yvyAymLo+goMg8djz1Hl9KCDy\ndPVjEYApk6qtmEyQINj2f+KqrECNRjt3gX7pypCdWicjMOn9fMKwZB3jsoqXPhxKLjClMase\nlUgB0Bgwqyxkw/NHXVjiqiyXiBeFLo1CHowvxbjZ4OwEwQnaJOD0Ix8zdpiFdwnmBuhr9bkw\nwyy8C4yjaHPAsFh2E5bd5Bsjc22AuuGNt14GZw9oQ9G0z22jzjpwWcCUjgnZI1R9OzrJ1gxE\naEgH/STtBkpOOxChbnJMpHV5mP55spwBVzsgA5pUjF0CzDSfOWBcAbEaEFyBG3SJYIh4y48x\nsmVptqSRwS1LsyM/GH8Q8xdj/uLgDXJ2GxRSl+hJTkeP41JN9/D7ZA6oVP22o+EfVKRJjdd9\n+975v3jzossz5Jlalhf/5e1zojgqBYURwZEWnCorK0+fPi23VQz/UpnCdEUROGYumLENtMnU\neRwEJwCANgVT16NhoGCVtwLvAHVc+CpWqLPaT90YjF/bD7M3T+bWg+7a6tZ/+nt3zdDg9ctW\npf343xljCJJfsOAGKt8nFQ9s7IrztrMFK6m9CpxWMKVi2px+k4VIEnvnA9Y9u8nt9hsYx5nu\neShCI0AGdGGek2uN7JZnqGo/tV0Ftw1i07BkPcZljnygHFZpxzgAIks7jixwiMC3gmAFRgNs\nMjCBsg5d/VQ891fwDNQlpRQzKx8Co9RfSXBTzS7qONevgBAQJs3Hgu3ATqKVT8/5w873XxLa\nGgCASc7Ubn9IvXhdtAcFwBkxfhIMI5JwOiy6lSrfHjQAIgBkVEzJnePwTYwwa+amPbCx6PVP\nrwsDbsEsgw9uKl4+Z5IqegDAMSwf1CoLALjRVKhNFzp65XvNAACATs2tXzhCV/VpwIZFmXPz\nE3Ydq69rs8Qa1AsLk9YvzAhttuJYcboFrVrxKFUAAEhMTOzu9hMi+1/253EMw3e/+93vfve7\nclt1umm+bKAQPmbQbVIhEGQwcTkmLgePBVg1MAMpr/YGseUTcPVbryHGz8eUdWGZ8PQ2SgQJ\nQHCTpQ3jor6wJg3xfMszT3ka6nyD9hNH2v/jJ2n//B8TPz/mzsfSmwI0DkzMZpZKddsyJGD+\n8olfdNyocgtS/vmXXb/6Gd/iTUngUtISn/hu2NvERhiGxVkbcNaG0JyNlRcNgxumBCB0kf0E\niINejyxqS0EztI5H14+IJ1/3PYLaq4Q9v2Jv/WHwyen6u9R12Wd2itR5EUQPzrp/NP+OCODa\n947jrT8MvhQ7muz/969iV6v25vuiOKoZC6YuQkMa1e8jawsgw5hyMGc9aKeGzcEXtsxatyDj\nwIWWtm57WoJ+3YL03NRJ3SNTz+n63IFlNQiom0mWwHExw/0kxurV/3DfgsTYGfEHSYnTPXpL\nJFIjh6fH6n7xw4pDl1otdk9cjPrmJVmf31Ss0yiziRlNWVnZm2++6XQ6tVrvl7HfXnTu3LlR\nHZfCzEX5SVIAUPk85NkbxLrXfTp0EJnPk6ONyf98GHwxpE6Ifv+ZhDhOnwhQN/qx7vtY6DGz\ncSF41mfWf5HyFtLlvdTThoY4zF2IC7aE0Bk0tOgWLct8boer/ALf1sylpGvmzEfNDKoPHweY\nWAjISPTBUenQNKxJCrnIdgDItzJfIOdFRA2oveWv4qUPJA60dlHtSSxa7Rd09VDXZYmLdFeg\nswu00j2GIwm5HM73XwqOOz/8s2bVVjTMdEPi6BCTjqWfm7w/0MOSn2bMT5vUooYvek4nkGDz\nDNlPMMiY1EZ2JnlUZSQa8tOMNa2B7XtYBh/eXHLritzommXanfyn55rr2i0mvXphUVJZ3tQQ\n+8aN2er6xv8c7ujxfiZ7rO4d+6tPV3b+6hurlI6zM5k777zz1Vdffe211x599FEAEATh5Zdf\nLiwsVBxGFaLFJJ0yKYQSwQ7ORhCswJlAlwM43NOA2H5QYt7lbKW+CjSVhnhgCTL92zgt+jbF\nnGR4GmqlN4iip6E2JAIHAMhVZUcNjxOAQCWdLogqtXZByFrPAgA5bWJrHQAwqTmoi5HeSXBT\n9afUegGcPaBLwIwlmLc28kU640FrwqINVPVJQJgpvW2E9ijuGn91wwu5KrFf4OBdYJFue0nd\n9Qh+AgfZ2+SuQ7ZWnAQCh1BbQW6pBHWPm68uV81bIbFJQWGyQ+BqIL4LAFGVBOogTVN0gK2C\nPD3AaI3aHJ021SV6RBI5ZLWsZsSa9unHt+6Z/53njjtcPh3QGXzqrnm3LItymufpyo7/eP18\nt8XrSvPSR5UbFmU+/dn5HDttFai/7L0+qG4MUt3S997RuntuLJA8RGEmcOedd65aterJJ5/s\n7e0tKCh44YUXLl269NZbb0V7XAozF0XgmO5YLpL5EIgDtnBcDCZuAl2ezN4EdgnvQwAAeyOE\nWuDA+BxMK6PWwDVkzFlFjVdBb8KE9Ek4X0X5mkDUhcXkMrpQywWxYrfXNkKfwMzailmLx5xi\nI/Cuw7v5qgtk7WXTc9XrbmdTZZ5NBcH96eueA28D7wYA4FSqNXeoN94fmMDicYjHfwvWgSm6\ntY0qd1P7ZWbZ1ybhZyYYZvZWMiSLVz8EhxkA0JiGc7Zj6ghesCTI9OkU+wBEAGa49yV4XiS9\nDkwAOEnaGJE7yM9yaNMIlfkKCpMRwUq9nwLf2f+KAECViqYNwAwUWdivU/eewVs29Z1i9cWG\nxJsBp8DPWpiYnRP3/NPrXvmkqrzO7HILRZmx968vKs4yjXxkOOm2uP755TO+sgsA7D3blBKn\n++LWMBaSmK2uli57Yqw2VaZ3bFg5UyUtoJ+u7FQEjpkMwzC7du165plnfv7zn/f29s6ZM+fN\nN9+84w6pwmoFhYigCBzTGkcNde3xi/BWan8fMx4ElVSiAYkgbdtOfg0RQwez7BHx0ntUc7g/\nbYRIBTYUP/Z2P8X4NHbTFzFvcmW46RYvA4YJboPKJSZrCopDeSUi/tQn/KXD1NuJCWncovXc\nvNUjHxVSqOageOntodf2bvHsq4y9C0s2j+Eklh7rb54Rmmv7X/LXL7mO/k1399c0a7YH7+ze\n9UfPsd1Dr3mPZ98Osvdp7njcf2D7h9SNQXrqqPE45qwa/diiB2L2UjZ7KXjsgAyMsqhedpIz\nIG1waozLpB4JmRKTA58+MSaTkAUKcDFEQAZjQt1OeFywqbLDkBXIFBQmMb7qBgAAAXjaqO8A\nxm0GAOAt1PVR4N3WXkVcHMYF5Su5LMCqgZsR9YBJJu03754X7VH4se9cc4C60c8HJ+ofvaUk\nHIk2zV3237x76WSFV2IozjI9eefcWdmh6Lo1auwuCddbALA7pVsaK8wc4uLifve73/3ud78L\n7WlFmLnarsJEmBTLdAphgvrOSkV5slyQPgBZUEs6HiNowmM1z2mYhfew23/KrP06s+br0GGn\ntoahkZpb+Z3/Ro0VYbn0eFGlZ8bd93BwPOnJfwAmdF8o3u18/vuut38tVJ0V2xuEipOu1/7d\n9dp/QCQbBApusWJ3sOQlVn0MrkDru2FwvPPcoLoxcGbesfP3YkdzwJ5kMXtOfBh8Bv7kR9Tr\n102dOq5IX6xjcn1aRkalH626AYBskvQGLmnIvWbBbRIHxmdizpKgo/SYLlHlgWnLQD0pfAqY\nlCyuZGFglIDLL2UzldVChamGp8NP3YCB5kXuJhAsAAD2Sum1BJtPniMJVHtA3Ptj8dOfiJ/8\nk3jkV9Qt0YxMIdw0ddok4702d5899LP9Prv72787MqhuAEBVY+/Tvz9W1xboThJWMpOk01Sz\nkmUqSRUUFBSigSJwTGvcXTLxTuk4ACZKOSmwOtTnU+UHdPI5OvkcVX4Abulb+zhRGzC5hOqr\nyBLU7l4UhKM7Q3ktACBhggkpSY9/K+V7P1FlZAEAMIxmVmnmL5+L2XhLaIYHAACew+8J1ZcC\ngvzFQ/z5/SG8yvCQuR54l0TRgyhQd/VozyLwnnOHJOPuoLjYdC04NQYAgEhsrPKLeALLgL07\neuyjHdhURJ0DbHDuFYPaIaNyzF7IrP0y6IeW9TB3CbPhKcnKHczdhLk3AzuwCMyoMWcj5o4h\nPSfc6B/+R67Qz4adzZulf/QZiYobBYVJjtAnFUUAAL4HAIjvkTnQDqK3FTdd3EEV7w/dgvua\n6ORz1C7hFqwQVuT6hiBiOJqnvnu4rrsvsGTP5RFe3XMt5Ncahm3LcqTjy6XjCgoKClFBKVGZ\n1sgV0stX82L8QhAc1HF0SALQJDIxi8Qjz8Lg1LGrihqOMUu+CHEyLqHjgpql79Ny8fHAt5Dr\nMoh9AASMETWlwI0rFR8xdtvtsdtuF60WVKnD0TSEv3RELs4tvCnkl5NGcI9nkz+izUIe6Z2p\nJ0hok89PoQDvW30COCUmA6gboen6FIdBwzpyXgB3jbeajI3//+ydd2Ac1bX/z7kz27Wr3nt3\nlyX3bgPGYFOMqSmGkBCSkPqSkITfSx7v91JIeC+/8PJeAiGhQxIIOCFUG4ONjQu25SZZtmxJ\nVu9dq+1zz+8Pyasts6q7ap7PX94zM3eupdXu3O8953tQVwjemR2YvkRILaCeJrD3Y3gC6IbJ\nYUZMWoMJK8DaBgCgiwE2lS0J/GGmyLBvPe48XyzVlANxIS1PNX+5om4ozEgw8BPXgPk3Bvgq\nQTZoP9xTT40nfY8SpwtvY9wIDj4KwaUoN+a1/TK5Mwszo0LRT+R8TZdsvKxaPh4iNi5Oqm7p\ne3VfpcQHv6w1KuHL2+bO+vYxCgoKMwtF4JjVaJLBJZOxj9rkYS7CmFUYvoAsdeCygCYa9an8\nk/8Cn41xp5Wf+Qtb/wMZDUVyUlcTMgEiEsZm9+jfvWX4+FhxVpHNo2aH95H1U9T0g3r8fmAs\nLFSZ/GSW38qjXr8kl5CBYfEBjw1zyGcQnQEEESSZlBk0+i68WVIWIMrIHIhCco5XIGmJfBZJ\ncjCbuUwyvKPZ9v6fpbpLACik5Wq3fIZF+7UTQjXqloK2ELgZmDbgioiJGDlqlwomgiFx/PMO\nNYiqeUtV8ybym+0g6gJwAugQEwHGq0g6+qi3Dlw2NMSBMXk6d7MOFcQB8Wr8jwcFVbx8f2hU\ngSoGAFCXRn1yhaXatIF824DVKJYOsHaBTllkTh5FuTFrFiQcKm32DGpUwoM3zQ3F7VyyuY2B\n46HjC1vyNy1OOlLW0tplS4zWr1+UOCV2pwoKCgrDoAgcsxmMWE7WKndq6yCiCYwFI1ypMg41\nhe2uAYvcotraCd21EJkxFOESnXiLH/8nOO0AANowtuoOLLhulE/DGJ8BFz+Vi2eO5vIRIIns\npYMdIjzD9jJUZQRcJU4daIykHplKIgyfxOadhmiMnUN+rhYYmY4Ro818QZVaNW+Zs+SI3wFU\nLfJ1A8XwGHHxRtepfT5xsWA9Rnq5wGDKUuitp1qPYZFh9rUYkzfKiU03nGXH+5/5GVzJdpEa\nLzuK9xu+9BP5hT0KIExxE4EZAicqAxgq1iOqQ8wDGK1CN3Rd7X6qPQDcCQPJM+EZLG8HzPKM\noSGo7QKV76aeBmACRqTinG0YGcwMvqsCpkX9Yuo/6fM9hGFLB5M7tGmgzwaLt4rB1BhxxV5a\nCmzuMMwhhdDwr58r+ufh6jcOXm7rtmpUQkF29IM3zU2LC4kbRVai6XSFTNFxTtIUfBGkxxvT\n46eFT5OCgoKCLIrAMatRRWHCXdS5D2xXuioYcjFyAzD16McgW4DOlABk6/F8SuP7nqeSj4Ze\n28x83/PMZsYVt43mRmzRtdKJd8Hq65glrLh19LMNCO8EcspJLRykDhCTgnALAAAgq9l+8B1X\nXSWqNWLWPM2q68fXtVRctNbh4zoxEF+4bsJzHAOs6LO8+EVqH6oSwsh0tuS+MW3h6nY8KNVe\n5N4uodobPickZfifrNn+EKq1zuO7B804EMVl12u2fcnvRMR5t2FiIbWUgLUL9DGYVAjGaZyG\nMDwup/XPvwGfWh6nw/rn36j+7wu+LXIB+izO5k5LXKQu3DCGv+WrEKJqT3UDAAA4UTmiCWAM\nu45Uf4iqvTtS9VTz0hfYkm8O1g7MaqjqY37uzcEX3EXtFXTof1jR5zHJzwJWYXgMBSgYof/U\noB+HGImGItAM+RdgzA3QV0J9Z8DVC0wN2jSMWA2iafBwWAC3b0E1U9M3yAVkAdQNFumEGJtD\nevfT2oqGHoGxvNTwG5alqsTxW9GJAu5Yl7ljXabNIWlULBSdU9zcsjrjnaO1dqdXExOB4Z0b\nFLtlBQUFBV9m/5PZ1Y46BhPuBG4DVx+I4WOSNgZAdVhAXwSNx05FbxuV+G68AwA//k+h8EZQ\nj6JPhN4k3vWv0vt/oJbL7oiwaSdmFY5xynL49sL0PBS0jS/XpbN9T/+U+noGlv/2w7ttH/3d\n+I1fsMgAzS8Co1p1k1RxRrp4pdyaABDEomsmu1OsOoyteojaLkB3HRCH8FSMnzvWBHUWnWD8\nP3+w733NWX6aLH1CQppm0w4xN0D3X5VafetXVRtu5w2VBCQk52BEbMChIzPQM4doxuK6fJ73\nypRS895O1+XzYs5Qf8TGDsvv3zx37ELrwMtFWdHf2D4/I2HabKZJNgAcci2devx6CQMAEFEL\nYsaoByGqlzPKtXZQ+zmMGykhbqbj6Ofn3/ENEuelu4SEheMTcK9qtFmozQJyAKDcqp6BsQCN\nBUCSv1sWxs4lXSRYfT8rMGU5CNPLPWdkyEqOEnBd6aUlxKJ6ETAjcAvZSkBqBXIAM6EmD1TB\nyRWqaOj5t+dPtPfYBl7uPlG36+Dln39pWVK0YYIja9VCRUPPix9cqmjoERjmpUTctyUvuKkc\nSdH6n31x2W9eP9vYMVgvHG5Qf+2W+QXZk5jUqaCgoDBDUASOqwOmHZXEIEtEOmhMYPezf9eY\nMCLD/YoaL4F/Q1EAcDmo9TKmjKoqFeMyxJ2/oOZK6mzEsEhMzAF1kGo7WeBHDRacxSHZbeY/\n/YLMPZ7Lf6mxuv/l/2f85i+unESukx/y8uPU24FRCULRdUJOgC1QQdTe92+uMx+7zh2hrlYW\nnSgWbhLmLBvn3Drr6fS70FkPggric1jhNtCZRr7sChg7B2LnjO/WgyPoDNqb79fKNDANcH5E\nrDCMrjHrCGS5AgDUN7SY6TLb/+V3h7vMQ176Z6s6vvvkkd99e21ilHz3vkmDesqo9SA4ewAA\n1JEYtx5N43e3CRIcIJAbrm0MwzjM4AjQF9ncDLNd4KCOSuBybafsZuqpVwpVxgmOtNkg6wUu\nqFjR/fzMK2Bucde5YFIh5m8L+gSDCBF9dLrxdEVHn8WREhu2dUVaUpRI1gNAHs2wpDayHUD1\nUrIcHtp1kLrI8imo21A3UXMlSbL/7OVit7oxQEN7/y//cvq335jotsHekw3/9doZfsV3s6XL\neqSs+dF7l66YGyDjZlwUZEf/8XsbSi53NrT3x0XoFmRGGbTKM7yCgoKCDMqHo8JIMAEX3kkn\nX/B6xmUiLrzTa++OB06RkDOYDAgiJuZgYs7IZ44JFgZCHEitvnEhEoTgVNE7y05wOQdQ5/li\n3tPJwqPAYbO//DNeUzZ4oKlKOndYXHaD6qYH5UdEFBdvnHjPFCr/hB98fqj9attl6dJhtu1h\njFH6uk0XMDxgjg9GDB36+8HLnurGAGar89V9ld+5fSFMHdR2mNo8chwcXVT/JsRfI992evJg\nAAxA1oRvLN99gdpRDX9oGuCUOCfQTCAJHyBgS2YAAFfgQwohwpjA1nyHWs9jXxOIGozMhPBR\n2wlPFkSw73TDwZLmli5rbLiuqdOZBzJgAAAgAElEQVRS3z4oER4rb3vraM2vvxiZHev35iEn\n2YtlciodVaDOBGG8qQpSJ9nPlFZ1N3bIiEoXartrW80Tybaw2Fy/+0epW90YwCXRE2+UvPTI\nNaIQzLoVlciKcmOKcsecE6qgoKBwVaEIHAp+OLvA0QiSDVSRoM0AZBiTj2u/T5V7qbceANCU\ngtnXgd5bF4gJ8IyFiIEOTS6oW07WIyB5FOQLEahdGazxeaefejIAEe9oZuFRzkP/GFI3ruA6\n/j7LWyLkLQnWNHyx9vJDr4CP0bq9nw48hzseDdVNFcaImJHPohN4R7NPnMUkiulDuTOll+U7\nApZeDtBbh7ugv5WcVjTEgmYMOTtjQ7JSu5+JLAC1HcTIReMoiwsq0QBtg/8cUFoFEQAQx7JY\nUhlAFw1WGYc/CJ+mKmGn1VnbY7W7OAAIDJON2gSjZpwrLUNg9U1/FaVZTSNQwPgFEL9gquch\nj0vi//5CsbuMrqqplzGvt57TxS1W38+6K/gKuAOQswnHJ3BIHWQ5AMBbegLWUjV1WCYicJRc\n7uy3yezidPTaLjX0zE0bpku3L5zT5ea+5k5LTLg2K9E0EX8QBQUFhasZReBQ8ISo+xCYS4f2\nPMUIjLoO1HGgj8KFdw3zfIxxGZg6n+rO+cbnrgXDGL7gQwhqUL8RXM0kdQJwFKJATAxiv0PU\nB3xCQoMJAKRzh2WPSucOhU7goPpScMk8MlJ7DfS1g1HZCJoeMEF/78P9f/h3sgyZ7KLeqL/3\nB8CGnnEDdgqUZOLUfJoq3gV7HwxYuMQXYN5NoA6+wz9Z6uU9briTrI1oyAj6HUcPYiZRN3U2\nQGMN2K0AABodJC3E6LE5MmLGdXT+Vd9oeDpGTceuPa39jstdQ429JU61PVabS8qMHE8dE0Zl\nQlgcmH0FXIzJAYNS/x8SiOB8bVd9W79Rr5qbFhkRNpO8hN86UuNWNwDAR90YQAi0cqcA38gU\nqNZsBMh+buB5Jkwb0EzMqJ+QfUmvJeDcevvHMO2L9T1PvHG2omGwHDgpWv/N2xYsyVM0RIWr\nGk7KQlVhPCjvGwUPeovBfNYr4uqm9ncw4bPARnYNZFu/yff+kSqL3RGct45d88WgT3NCiAko\nJoRiYNXcIhBFcPnu5AjxKUJcMgBAn/z2O8kVtgQNq595ivu+1h5UBI5pg5g5z/STP9k/2uWq\nvQgAYnq+ZtNtA9KYm8wE44VaGbeOzETf7AxqOUPnXvWNWFrZsm8Ev6pC1qBh8NCUt67UQQuH\neo9Wx3YrXD5GzjBMGEPhPcYuBECqeveKGxFi4hLM3BJEhTRYEEFdj0zlSGu/I8Go0Ylj9wRF\nxpbex48/B/1DjavRlMwKPzuReSq4sUsOi8vq4pKATCOo27ukX7921v2XrlOLO6/PvWP9jGmW\n8fGZphHPqWnFeaNtNQ4AgMO4aA0HuXM2F6ZxnQqsfh9IUUZNXsqE9mDiIgI6hcVH+h5q7Ojf\nc6K+qcMSZdIunxNbmDP4Fdzabf3h00c9M0EaOyw/ee7EE19fnZcyxR3BiYATCXJClYKCgsL0\nRBE4FIag/lKZKLeCtQIM80e+XmdkN3+XWquh9TIwAROyISo56JOctrCIGP2tX7S88bRXVFTp\nP/ttGOgeFxYBtn7/C9EYyvZ++oCDY+BDI+BygjjTHPtnAmgwaW/+wjAn3Lom44PiBp98DYHh\n7eszfc6kqr0y1/c1Uds5jAuyWwdqogLtjaJmqnf4JTs1HvQPU+MBjF0CwhislzF2AcbMA2sH\nuCygjwMxSP7Hwcbiklxc/hfSZ5fGKnCcrugovtjW3e9Ii73r2rzWCHsjMIaR6ZhUOM39R2YK\nvY4+i2vQ+VIiqddm+9HTJZ29Qzv/Vofr6bfP6zXi1hXTtB7Kh84++TITT94uFrYUcoZ+b1QW\nB1K9bxAFUI2v0JXc6ahhWvjKFtcTb3s99AoMv7VjwQRtMuZnRCVG6Zs6LT7xnORwn/5Wbx6u\nfvrt807X4JTeOFB1TWHyw3cXCAzfPFTtX+fikvir+yp/srNoItObCOdru59970J5XbdLoqxE\n487NecG1TVVQUFAIEYrAoXAFbgPJ9xsaAACInJ2j//7HuAyIywjWpGYW2uvuEFKybe/92VVX\niWq1mD1fd8v9QvzgRpUwf7Xr47/5XyUsCGHnV0xdAGodOHx3dDEhF8LG6K7qclLx27x0H/R1\ngDYMMwvZ2rvBEDJ1hgj6O0FjANV4GwDNOrISTY/eu+S/d5W4ewGEG9QP3Tp/QYb3r9JlA0u7\nzPUA0FMLwRY4QBsP+mSwNPiE0ZAB6uA4+I4b6q+XTzDhLuqrw4jcsQ2HDKa96wTRwKKR/LNL\nOAXM0vfH6eK/+uvpA2eHduP/rBX/5Y416xckBmGWCgAA4JAcbnVjgKMlHZ7qhpvX9lfOFIEj\nyqhp9ljtExGi71uxuhWb+xckGS8BXfnvowrVC0FIInKCy6O7M6pQtwzY+PREBiwM+KC/6Y2F\nrrQY/vIB8WIjCoI4Jy36C1vys5Mmak4kCvh/Plf46PMnPJWd+EjdI5/xapF2qb7n92+Wkfff\n4EenGrKTTHduyLpY3yM7+MX6gA22Qs2Bs02/+PMpt3nqxfqenzx3/IGtc+7amD1VU1JQUFAY\nJYrAcRVDBI5ucvaiOgLU4fJN6QAAEFB5n4wW1ZxC1ZxCmQNEkmAgUYvej7PikuuEvFB2mtAY\n2IYv8n1/BJfHQ7Mhkq2/f2zjcInveowaywdf2sx0/qBUc0a456dgCnadi8vBT71NJR8MuIdg\nXDauvgfjlIcqAIAVc+OeeXjD6YqO5k5LXISuIDs6TOeXTUPyVh0AAGNZ4o4elnIrr3/TS+Mw\npGHyTaG419iQAtfITH35TEjQigICkFztjF41hvSNVz685KluAEC/zfX4X0/npYQnTHVP4lmD\nTfLVMmqaZbcZoLHDYrW7dJoZ8F28bmFCWc1QPSaXiIm+CsetqzOS47OB0kFqBbIA6kCIHWid\ni4YN4GwgVwuQAwUTqLMAx69xozqHbKfdL+en8sc+5wBUo2ET4MiFt6MkPzXi2Yc3/vNIzaX6\nHsZwblrEtpVpGu8/tw9O1pPcx++eE3V3bph29UcSp9+/eY775YK9sOfi5qUpkWFB+9EpKCgo\nhIIZ8GWpEBL663j9brC1wsBOX1gGptwA6jhwyLQCQVRR+7vg7ALBgLoMCFukJCePDZer57//\nj7OsGADEMLWoVTERQRehufPrQv5y35O5i5qOUnsJ2LpAE44x8zFpDbDxl4Rg5hIhJp2f3Q2d\ndSCoMCEPF24ea1oElR8eUjfcWHr5kdfZlq+Oe26yt+Lv/zc1nh963VpJ//wV2/Z9TJyOho6T\nj04trpoXP9wZKj1oI8Em5/liCk3VmBjGMj5H5iqwNgMiaBMxLCMkNxojqIsJWD6jm+kGNBKB\nFUEA0Homa4gMYwzqNj93Q71KMI5lefz+sTr/oMPFPzrV+Nlrg93Ge9JwtpD9MkhmEAyoSQdV\n0tROh/tpkcM4Hci6dU5Dblmdcby87eSlwSQyApAknhprUIlCr8WZFmfYvjpj5cAnGIogyv0K\nVMmoCtInlSobuB0c5UPW6cyA2mVBVDcG0GvFezYNp8I3d8r3VG7qsABAXkr4mUqZPk35qVPj\n0V7Z2CtbauR08TMVHRsXT/EfjoKCgsLwKALHVYm1hVf+BWgoc5vM1VTxEsu8mbr2+G7/CmHU\nfaUHpLOTbHXQfwHjdkx198eZhHX/PwfUDQBwmR0u88Daw8yaW4V871O5k597AcxXipCtbVS3\nnzovsPlfBGECP3BjDFvzufFfDkA1Z8cUn8CNzniqG4NwF336N9z+r8G91ywGMzbShb/7RvUx\nQTfg8LppWBaETbOtSG0MGjOor9o3bkwH3RQVk0sOMrcCMgyLBzZ2y08AAIlTHVEjwIB6o2aY\ngThUO5MRoeMEHUPNHcioVmVH6Ue/PrY7pUBOCg3tMkZCMwIyfwq2KyqtE8h2CTTZaFw9hU6x\nzG+rIDslDD5t8T8zJ9mkGUsCzhSiEtljDyzfU1x/4ExTa7c1IUp/XVHyhoIpWxKjZh6o0kFq\nA7IDM4IYDzAFP0mjf6rdQFyvBoDb1ma+d6zO7G2CqhLZ8KJJ6LDINb4dQLYnroKCgsK0QhE4\nrkao5ZCnujGIq5/6GjDmVur5BBxtAAAogjYD+v327R1t1HMMI9dOxlxnBY7ig3IV8WA/cUC7\nwSuNn1pODKkbbvqbqekIpmwI5RxHwi6/+wQO+YTqcUNNfu+3gXhrFbgcIM5gWU1qqXN8/KbU\nVIMGo5hboFm7DYRQfQJj8nLgTqr6wN0kGKOycc7twK66z3zMuo2q3oC+2qFQWCrL2jEFU+Eu\nfvEDqtw3aAui0rH8G0FjosojZG5DXTgmLcCc9SOqHpzKiTzTcxycLjIgxEHJhiHmROkTwzRm\nh4sT6NVC+BhLG9QiU4nM7YboiUE3M99C9pohdWMoWAmqeNBOWUKKTtRYXF4frcvmRb1zqKnW\nr1Dlvut9tPBpDSJuWZq6Zen4nEFDADMAM0ztFJbPif2g2O/LHWDAtjMmXPv4gyueeKPEbcaR\nHGP41o4FOck+LVS4RC2czAASgk5g8Qgh8ahKjA5YhpYUo1SoKSgoTHdm5pOKwsQgPzvAQfob\nIH4dxt0J3A7cBqKJOvfLn2mtAEXgGDW8t0t2j1CmQWzXRdkRqOviFAsckQlwWSaMEcF2HHTJ\nOiMQAIDknIYCBzVXUV0Z2S0Yk8Jyl4Egv01n/+Qd6xtPgTQoLDrPHnEcfi/sG4+FrocOpq7B\nhELqawCnBQ3xEBaS7sgzAFUYy7+PeirB0ggAoE/E8KlZ0PIzr1H9iaHXTisv3QVOCZwSAFBv\nM7WUY/Vxtulbw1SQEfR6qxtXBqcaAb1yUgxqwaAe5041Ihblxnx6XqZicfmcIGS+lF7uLKvp\nckmUmWhcOTfO34Qy6JC9KlAcQyFwEKeGE9BZBS4bhMVj2mrQyjT7VDFVmMpgdg4lxYgC/vi+\nBa9+0PDRqYYBx4b4SN1Dt85XulfMdNYvStxTXH/8QptnMDZCd+/mwdLLnOTw//nmmsvNfc2d\nlthwXWaiURS8EnwI7E7pIoE7tcosSe0iyxAw+M2q4iN1Rbkx7jojNymxhoWZU90bS0FBQWEk\nFIHj6iRQWfqVONMA0wAASAGykQPFFeRgEdFSs0xBO4v07chA3hakQ1zZ5aO+GrA0AxPRkAT6\nyWtngPM20Knd/u6VuHBTkG8UmSj37kTQh4Nmmm0cSU5p9x/5uQPuAI9MEG76Jib4JhXz9iZP\ndWPw6qYa69//qL/3ByGcoUqPUWNsFDJLwfBsCJ9Sn1pzq5e64UYlgEtyf/RSVy0ve58VbA84\nDvUFOOAAsEHwtnO/vG3uueoun5z5dQsTluaNvY+M1AuOJuA2EMMtPPGXr5YcLRuqwshJDv/x\n5wuTokOwwW7t5mV7oLMaiIOeMD0adH4/Hx6C7zJHPz/xJ+i9spHQWkY1h3Dh3ZggUyAWptJr\nBLXFZZW4xJBpBHWCXvvDe2K/evO8ulZzuEGdFGMYxphDYaaAiP/xhWVvHal579Paurb+mHDt\nyrlxOzfnGfUqz3OyEk1ZifKNXVy81kPdGIBcvIYJJoTgN25/+O6Cf3v+xCWP9i6JUfqf7Fwy\nwa66CgoKCpOAInBcjaAunpxmmQM6P+dCIcBDpxAW5DnNajTLNzkvnJaN+0RQE0H9Tf5noiYS\nHL388t/dmfYEgFHzMePmkfxHiSqPUdVRMneiIQozl2HuqnHUnGNMKtv8IN/3PDjdXf0YLroW\nF20e61Aj3Ch7BRS/CTbf9ydbcN0UlsrLIh181VPdAADqanbt+k/VA78BtVdTQ+fpT3zUjQEc\nZw7pJVfoClWmDgJbPbi6gGlAnQCizN711QZ11QQ8hujZ3YbqT0NggYMoQANggMDK9XhIiwt7\n6l/WPfvehRPl7WabMylav31N5k0rx9yplMzFYCl1Wzw6bGpzrwE81mMVDT3//kLxk99ZF9xl\nPLVV8gNPglsy7gJqbMSFORjrnTM1gQ4dAW99/s0hdWMAyUElr2JkBmiM/uermBiu9o2HG9Th\nmVPcZVkhuAgMt6/J2L4mY1xXS5xkW8lyTt0CBr99dbRJ+7/fXPPxmabztd1OF89NCb+2KFkt\nKgbzCgoKM4DZ92CtMApiV0LfZd/deKbGaN9+pajPJXOpzAh6ZVt4DGjX3eg8V2wv9loMa1Zc\nq1nlpw7EFkCnn8UmAMQV8IrXwOKlfVDnOUABM28NeGPO+Ue/p7ozg+d3N1JDKV4+xq775jjc\nDXHeOiFtAZUfpu5mMERgZiHGh8BRUhvGtnyb7/sj9F5Jj0eG86/BghuDf6+JwCV+9iOZeH83\nv3iMLfCqJ+I9Mvb4AABOBzf3sPDZlfHr7KTOveC8sg5HBmGLMHw8stqsgqTAx9BLm7D2BhyD\nOgjktGkAAAEgyI0h4iJ0P/pMIQC4JBrntq21DCxePsThWsd/3Ob60jNRXZahlVJ1c9/Zqo7C\nnOD1tSHix14Cn4Q4IjpfjVHh4JH5j5pg+0RITmopkYs7qKUU01YF+XYKVwcEAdtaEzlC9OGK\niBsXJyk9UxSmEE4zw1xZYbqhCBxXIxiWDmm3UsMecF3JztVEs9RtoPZLjNSmgKkQek95BTUJ\nGL5sMiY6a2CC8aFH1ScOOI7vlzpahJgEzYpr1IVr/E/EqDmQvJYaD3vKT5i4AjXhZJHJ7KDO\nEkzdDKJ87QZVHHKrG0PBhnNU/jHOvWY8/5GwSFyyLdTrVIzPFu78KdWcpq5G1BohKR8jQ9Pc\ndAKQuQvs8gar1OHrcYPGAK3+BBH1Mju6MxhyUvvbIHkswolD32lADZiWTN20pgGmwIsE7q01\n6wMmvBAETN9ATAII1ebquJPSySIj1xo0fPMC22vHvD61alrMQRQ4qLsezHI/K6eTunox5srf\noxgL2rnBuukg9j7gAcQsq1znZgWFUYCBH9cRR1ufQkSfnm+91NArMMxPDV8yjnIzBQUFhZmA\nInBcpWDEXDRlU38DOPtQEwH6FPDrV3flzLWgzSRzKbi6QdCjLhPC5l/tm7HjQrN0vWbpeq8Q\nl6T6Ct7exCJihNQ8UKkBANOuw+h51F4Ktk7QRGL0PDCmUlux/KBEYG0Ho3zeOFXLX0XVxeMU\nOCYNQYVZy6bzmwxVAXfL/Q+pFq6yvfuS7zoWQDV/GapGbZsquajkA6o4RuZONMVi/hqctxFC\n7844NqxVXurGFch8Fk1FV/PnBkakYUwOtVeAT0clv04lmOabSTcEXdnF9e3KxBhOm44VboiD\nJJ+NkhbtW7GlDW4PVFsgpxIAlxaYDpgBNRmgmxN8VUil803JcaOe4kYeCjMZkaGRy1jwIEMZ\nAZ2I9hTXHzjb1NplS4zWX1uYNCct8mcvnyyv63afU5Ad/ePPF4Ubpp11t4KCgsIEUQSOqxim\nRmPmqM7UJqM2SFvoXALuAjHIqdQzEanmgvVvv+XNg54aLCJGu+Mhcd5yAABDEhq893sx8NP/\nMI0/A+S6k6VbNj4y/d10/hPqagSdCdMXYeq8cY4zC9AZMTaN2mr9j2D6Ap+IkJiuvfHztnde\n9AyyiBjdjq+M9nZOG//HL6l1sBME9XdR00WsOsG2fXcc1UYBkSSQnKAevysBOQMU43ArSJaA\nnj5XB2zJffzMa9R8pX4BEQzx0Or1FsLYHDY3sK8NqgcXzt5KEYJ2OopHiADM7b7hiVNC7xNx\nUXZQC7V0AbNgMHotRoXyg0ulg6hM6PRr2oIMY4OdLaJwNSGydKdU7l2rgiJLRfBVKJwu/pPn\njrt7oNS09B0tazHqxD6rl7B4prLj8VdP//yLy0M7bwUFBYVJRxE4FCYJ6rxMZW9Rdy0QB30U\ny70OU1fI7z9LLupuAqcdIxNBMztXRLyzxfL0j8k+VCLOu9stL/zC8NAvhfQ5/udjWJq8f6Cg\nBb2fNawbXTiATPcW1I+nNSmVH+EfPgOOK/1cTryF+avY9V+djR6Zo0LYuNP1xi990tFZ/ipg\njH/0HPS2QVgU5izDjAIA0G75jJiz0L5vl9RUi/owVd5izea7UDvavjB0+n23ujEUrC2hCwdx\n3saJ/194/UXnnhd5fTlIEkbGi+t2iEXXjSc9ZBglbphDwYP3dDhPfzKQFSUuWCHET6e8BrWB\nLbufehuhpwGYgBFpYIih1otUeRjMbaALx6QFmLlqmB87QgxBm0w8BBaDwQBBHQ8Omdq6M7Ve\nS7Lb1mYkRQezRxJGJIEpAXqbfQ/owjEu5AZSbO52fuxJcFq9ppR9DYQprV5DjtnqfOfT2srG\nXp1amJceed2SlGncg4ZA6gXeD8wAgmlEjRJBqxYWuHgTgZlAQtCJmIAo84fz5uFq/w6vPurG\nAMcvtDV3WhKihvvr67e5tGrB88fokuiD4vryum4iykkO37IsVTEfVVBQmFbMvJXJwYMHX375\n5QMHDtTW1kZGRi5btuzRRx9dvHix5zk9PT2PPPLIrl27uru7582b9+Mf/3jHjh1TNWEFAKDm\nUn7i+SFfCUsnP/Ma9jayBb6/F7rwCT/yKlh6AACQ4YJr2Mo7fXpSzAIch97yVDcGkVz2fa/r\nv/BjmQu0URi7xL9QBVOuGWbdiJnLqEHGIxazxm6h0tPC9/g2OqXyIzwyka28fcyjzQowY6H4\nmUelfS9RUwUQgc4oLLsZbL381X93n0Ol+3DOarbla4BMzF4gZvsmd4wSunwyUHziAod06aTj\nz4+5lRrqanH+80lqvqza9uBYh0K1bJdfAFUksOC3q/DBcWS3ddcfyH5lYfn2C9rr79be+PlQ\n33dMoCnJ048D4/IwLm+012IUQgJRs3cwHHGaugCiYQk53/MxWDW7Iqu7IgHMABBl1Hzuutxx\nNGcZ8c5sxb3849+Bw6MLrKhhy3dSTz21lYPDjIY4TFkSkrIRYwJb+z2q+IA6KsFlQ2MCZG7A\nmPxhriCC/Wcaz1Z19NtcGfHGrSvSIsKUwoExc6ay42cvn+zpdwy8fO9Y3d8/qf75l5ZFm0L+\n4TNmpA6yHAfpii2LEIG6ZSCOaEMjiCxlxLE/KfGT9gJT22r2ETh6+h0VDT09/c7LTb0fnmpo\n77GJAluYGfWVm+dmJZoaOyw/ee54Xau7FLHubx9X/ccXlmYkzC4/KQUFhZnMzBM4fvWrX9XW\n1t511115eXkNDQ2//e1vV6xY8eGHH65du3bgBM75tm3bzp49+/Of/zw7O/uZZ5654447du3a\ntX17wMZ7CiGG+Ll/+DZtAaDqQ5CxBsKGEhDo3D6+/znP66hkL+9uZrf8YHImOmnwBr8E5oF4\nfWWgSzDtBtBEUtNBkOwAAGoTplyHUfOHuQvmrMKGUrp83CuYXoi568Y6YTp/SLbRKZXuh6tV\n4AAATMoTP/dTcNrBYQVDBF0+xd/8L59z6MJhSszDggn106VAngKBO26MHud7z/jbIrqO7xaW\n3cDixrjy1KaBJgWcTaBSD0pvXAKnE8ND3jxCqim3vPpbL6MTyWV97xUWn6ou2hD4uhkGw2yC\nKIJWIBugGiEacRrnBahiMWIrmT8FZysAAAqgm2M0FD79PZXZ6nRKPDIsVOWKGJUmbHuUX9gL\nndXAOUSmsrxN/OJ7VD8oExMAXNzNFt+DCQuDf3uNCeffPsrMgT6L8yfPHS+rGbIgff1A1Q/u\nLlg5L3B2noIfVrvr568MqRsDVDX1PvFGyU/vn2a26LyPzB8BeXylSt3Uvw+NNwALgkzQ2Wcf\n/ck69dBCgAj+9nHly3sv2Rxe3wguiZ+qaP/m/xx6/MGVT71V5qFuAAA0d1p+9vLJp7+7nk3f\nZBkFBYWri5kncDzxxBM5OTnul3fcccecOXMef/xxt8Cxa9euQ4cOPf/88/fddx8AbNmypaio\n6Pvf/74icEwZ/R1g6ZSJE1HbJXQLHET82C6Zs+pKqfECJskUbsxkAjwHDFMUgAwTVmHCSrB3\nAwoyLW/kRmMbH6TMZVT1KfS1gTEGM5ZhZmALw8BQT4v8AXMnuJwgjtbFfXai0oBKAwB0/hPZ\n43ThkwkKHBgWRX1y9hbGiTaeoO5W6pApIgAiXnl2zAIHABrnksVDdmEMVNpAjX7GCnW1SGc+\nos5G1Iez7EKWO9SZxX74fX8bVwRwHHp3NgkcAIAYiRA5DT035FHFYOQ2IAdwGwhhblPPMF3o\nPzTUerboFvcrXr7brW4M4rTyky8Lm34EuvFU7QWLp94q81Q3AMBsdf7yr6df+OEmxQBy9Bwv\nb+s2O/zjxy60dZsd0yojhmwXvNSNwairsqakvjfTqFflpYRP5A8kyqhp7pRv8uWDXivmpQ4Z\n1uw6WPWndy8EOtnp4v/z95KqJhm1vbbVfL62e37GVP4dKSgoKLiZeQKHp7oBAFlZWRkZGY2N\nje7IP/7xD61We8899wy8FARh586dDz/88NmzZxctWjSpc1UAAACSAm8muDwO9bUNVqb403QJ\nZpfAIaTluSp8G7gCgJA+XBozAAAgaMb2DIHphZheOKZLZFAHWKCKahBn3sdIqOiTE/IAqDdg\nd89RgnmrqemSbHyCI5PDr1TKjcMa8FDg4cjqV01DEvUfwfBbxzyaN9LJPa4PngfXoMeeVLyb\n5S5V7fjugL7GW+tlr+It8vFZDAE4OUcAcfrsqKIahCleYVLtUZmo5KD6Ysy9btKnM4jdKX18\nptE/brG5Pilp3hb84p1ZS0uX/OcVEbV0WaaVwAGSr1rd3M2eeFd3qroLoAsAwnSqL2zJv2V1\n+viG31CQ6COZBeKBrXM0VxoYcU6v7g+YQzrA5eaA/YkaO/oVgUNBQWGaMONXJg0NDdXV1Tt3\n7nRHzp07l5ubq9EMJb4uXLgQAEpLSxWBY0pAfTQwwT8HHgDA6JFc7bf7OoRfeYv7APS2k7UX\nIxJmlh2peu3NjqPvk8X7WcZOzqoAACAASURBVEFUq6+5a4pmNAKYsYjO7JGNT8f2DVOFLkw2\njLpRpNsMC87fhI0X6dIRr2DBFswsmuDILCIORJVbNfAaP3rszg7O5qFWpp5IfSB1gyDTznCU\nUFut6/1nfD4K+KUTrkO7xA13AwBqAjj1aGebg88wEECvw9HrcA44oTDEcLXKqLq6E6wGkJxg\nkxXQkfrbpvAjrLPP7vBrFTxA0+g24RUGGCblwaif1n8Cdhc88hd9Y9eQT6fZ6vzff5Q2dvY3\ntlvq28zRJu3S/NgdazNVo/PyvHlVRvHF9mMXWj2D1xQmL8uLefGDSwPvq/T4sPtvmLN6/lAZ\nVFuPTTYFxhOS91gCADBOQk6WgoKCwuiY2QIH5/zLX/6yWq1+5JFH3MGOjo6srCzP06Kiogbi\nw4+2aNGi+vrh9vpGHGG2wR3UVQrWVhDUYEhF03jN50UtJhdR3XHfuD4K4zza5hljQK0Hh9wj\nXYzMPgbVneMHXoTugex6xPxVbPVnQB+wO+C0Ak1Rhod+aX39d1J12UCExafpbn9ISBpd495J\nBzMXY/YSqvRO8NaGsTX3TNGMpiOYvYSq5NxAs8dTFuQ9NGPXf43yV1HlCehrB1Mc5q/BpBHz\nfUaBWissXCed+sj3hqZoIX/s06bA+SDcBhPooyKVHJQVOvnZ/bDhbgAQ8wudZX4fMgCq/Ilq\nQDOINqvVJg39lDhRl91BBCb1Vb/2YCIwEbiMkRCIU+lAadSpEOXXjdN8WT7dWJIXIwroknx/\nlGlxYUnR02z/Q4gashcFOHBe5aluuNl14PLAP+rb+s9Uduwtbvj111aa9COnoogC/vT+ZftO\nNxw429zSZUmK1l9blLx6fgIAXLskxWx1MkS91vf5fzTpXmE6lcDQx+gEAHQacWFWUDs9KygA\nAABN5LlB4SpmWgsckiT19Q1tcZtMJsaGvgOI6KGHHtqzZ8+rr77qU7ciC47U8vD555/v7JTP\nMLfZbDfffPOAUHK10F/Ha/4BTvfP/yiEZWDG7SCMxxOOLdjBHf3UUjYUMsSypfcB83gHCiJb\nvIUf+7vPtRiThmm+JnBUX8bf/i+PrBCi8sO8rZbd9R8zpWspS0g3fP1XvKOJtzeyiDgWlwJs\nOjdaQ7bt23R2Lz/zAXS3gEaPGYvZmrvAqDzTDIFz1+HFo1RT4hWMy2BLtgVn/PTFmL545PPG\niPrGLzn6e6SLQ+oVRsar7/r+gLHI2GCBFxLDHBoF1CPTHhUAqLcDuARMUK/Z6vj0A6nxstc9\nw6M1W2a/Bsc5vX20dt+Zhn/ZWeB/tMfhMKpU4+j5O6tAxNg8r+8g95G4qax/DNOp5qVHnqv2\nLShAxBVzprF97PQjLkJ37/X5z77nZSGhFtm3d4TARHZioGYOOavd3YUqmke1hKtp6Xtxz8Vv\nbB9VKy5EuKYw+ZrCZP9DgVJdYsK10SZtR29gkRpg64q03GTTY385zbmXkPSVm+Ya/BQTBQUF\nhaliWn8elZSUFBYWer5csGDwk52Ivva1r/3pT3968cUXb7/dq4lDdHS0j04x8HJEeaKoKOBG\nn9lshlFIJFMIdV6CzgpwWsAQhwlFE219J9l59S5w9XvGyFwNjXsw9ebxDChq2PIHqP0SdF4m\nlx3DkzBxMTDfL3Vcuh25RKfedTfswNQF7JoH/M+kY2/417xQZz1dOoJzxtwiZMpAZDFJLGaa\ntnj0hQm4eIuweMvAenKqZzN5UE+786O/8JoysFswLk1ce5uQG+Czggls+w+oZB+VHaDeVgyL\nxtzlWHjjdDdh1ejUn/tXXnVWqikDh43FpQkL141zzmI8MB1wv2J4MQaEUXQHcFp4+V5oqyCX\nDU2JmHcNRg5mb2GA8h/Q6AbejajWhH37P23vv+L4dC9Z+lCjUy1arb3lfmaa/cL0r/929oPi\n+tUFCbJHCcDBJY1wFf3NyoJzb6LOKnB6rd8wYX4oBI4+i/PlvZdOXGzrMTtSYg23rE7ftDg5\n0BPE12+d/72njlrtXtkld6zPVPpujpV7NmXnpYS/8uGlqsZejVpYkBH1hS35KbHTLH0DAAQT\nGjaS5TjwXhhd6sQAB882j1LgGAeIuHNz7hNvlAQ6YdPipPuuz1OJLCFK/9z75eV13ZxDTorp\nvuvzFinpGwoKCtOJaS1w5ObmHjx40P3SXXhCRA8++OCzzz77wgsvfPazn/W5av78+a+//rrN\nZtNqB/NOz549CwBucWS2wZ1U+ldqG9qYosv7cN4dGDtv3ENS7yUfdWMw3lWGyVuAjdOsC2Ny\nISZ3uK9yRLbiDph/DbVUgtMO0SkYmyE3P6IW+Tar1HRpJgkcM5SrSd3gjZX25/8N7IMrdqo+\n56g+J66/Q3Wt7yfPIMhw0bW46NrJm2KQYFmLWNaEXYpQRMMaMh8A8shhZno0jMIMta9VOvBb\nsA9mjVFfCzWeZQtvxZyNAMByiqTi3f4XCblDdTSoM+hue1B324Nk6UNd2HA9iWYR5XXdHxTX\nA8AwjqKBC+enKySBrQFc3SDoQZsCLAhVJGhMENZ/j5e9TW3l4LKBPoplrsOMtYMuQmQDqQ3I\nBmgAMR4mkBfd2m399v8edu+El9U4ymq6ii+2P3y3TH4NAOQkhz/93fXPvV9+tqrDbHVmJhjv\n3Ji9doG8XKUwPEW5MUW5E+0tNRmIcWi6EaQukMz5WXY4JuMh7U93v52IQrfZtnVFGgA8+155\nr8UBAIgwNy0iM9EUG65bnBM9L33QQzQ/NeKXX14RojkoKCgoTJxpLXAYDAZ381c3RPTAAw88\n//zzzz333Oc//3n/q2677bZXXnnlL3/5y/333w8AkiS99NJL2dnZs9VhlCo/8FQ3AABcVjr3\nV1z1PdCM15DCEcB/myRw9IA2dpzDjpKwKAwbdtOVeEBHUtkS62kP9fe6Pn6N154nu43Fp4lr\nb2MpeSG+pyRRN5ENUcXQhDCVVegTwtFPXbXg6AdjAkakBH145zt/dKsbblyf7BIXrcfY4N9u\nNqBKwPBbyH4BXJ2ADMVY0OQDjvxdw8+87lY3BiHOS/8pJC4EQzTLKWLzVvOyw57H0RgpbJJR\nmlB/FW19n7w02KOnvsUc6Bz1tC5/88PeRO17wHnla4hpMHItGINRaKCPZkvvAwCQHF5dXZwV\n5DgPcCUr0KFDTREI4/yme+bdC/55/h8U119blBxo7R0fqfvRZ4JffaYwvWEgRIMQva6A/3V/\n8zANStxEm7ShTiXeuiLt2qLk6ua+PqszM8EYbZqxzwYKCgpXMdNa4JDlu9/97rPPPrtjxw69\nXv/6668PBPV6/datWwf+fdttt61evfpb3/pWT09PVlbWs88+W1paumvXrqmbckghaiqWCUtO\naj6D6evHOSoLXHsvTINvOyZgVBJ1NvgfQTk70mkOb6lxPPcTsg4uTqTOJqn8uGrLF8SVN41w\npd1CzRVg7oKIBEzMGX1iBaduF68hcAIM7O2igAkikynWnebQpX38/HvuZsMYm8eK7gFD0HJl\nydLL6y/KHOBcunRSVASOQDAt6sa4WnNaqU32Ry1RUynmbAAA1fbvSFmLpeLd1NGAhnCWUySs\nuxN1V5GWIYvVPrgsr27sK6/uzs/wbVUTphJZKBZFDhtVHKfuJtQaIW0BxqQGZ1jJTC1/B+6R\nAcTt1PEhCjrQj2y2NVo81Q1XPTlKvY6SlWxHUX8tYID22MNy9HxrgHjLzEguUJhcRIE99sCK\n37157mDJgGk6mPSqXotMO6qNBZNR0KpRCfmpEQBwsb7n8LkWl8RzksIXZs3+Wj8FBYVZw8wT\nOI4cOQIAu3bt8tQskpOT3Q1QGGPvvPPOI4888thjj/X09MydO/f111/fvn371Ew31Lhs4AzQ\nSc46/p4vaMwkQALyfSjWxoJqWiwnsHArffhH36jOhHN8U36mP863/uBWNwbh3LnnRWHuSgwP\n+DRMZQf5gZfBNnghxqTi5gcxPivQ+UMXgt3JqwA8U2BIoiYkjYAz6eGbqj7hJf/wirRdlA49\nJVz7AxCC5HlhDbgl7tviV2GC2M2BOhCSvW/wgwhRKNgkFGyaxGnNADwtBp56/dy9N+cX5g/9\nIYepxEjNeJyhh4dqS/juP0B/FwxqpAwLNrMNOydeFkR9JV7qhjveU4xBFDg8R3bKFjxK5KxG\n9ZgrPV0S93HTcNPr13tCQWGAKJPmJzuL2rqt1S3mcIM6I8H4l48qXtnrVbcyNy1i5+bxNrMb\nI1a769d/O3vgbJM7sjgn+kf3FEaZgv9hoqCgoBB0Zp7AcfTo0RHPiYiIePLJJ5988slJmM8U\nw9SATLZ74oRa32ljMWYptHv3XEQBk68f/5hBBeesY/Z+/ukut2McRqfitQ+CZvrZiQ0Lmbt5\n3QWZA5JLunhCXHaD/FVVJ/mep7wi7XW065fCvY+DwXf/1ndg3u6tblyJU+uMEjiIl3/gFwMw\nt1LjGUydcFtWAABAYxQIotvy1utQ5EQbHEhlR1xH36GORjSEs+wCccOdqJ1h795gojEG/Chr\nKCNDEmaEpturrY+f+wg66kBUYXwuzl3v1dppJrBmQcIf3zk/0Lixz+L83aulKfFhGUnGgqzo\nbctSxVAUp5i7+FtPeLl1EqfTu8kYjRNvGORoDxCXb6MTBKhXPs4DxIdFFFiUSdPZa/c/FB85\nnnwQhauH2AhdbIRu4N/3XZ+3LD/2naO19W3mKJN2WX7sDctSh/HZCS7/7/UST3UDAE5XdPzH\nS8W/eWj11WFtpKCgMLOZYU9yCr4wASOzqVPOnio6fyIDY/Jm0CdS62GwdwCq0JCCSdeAdhp1\nrcOCG4TcVdR0Eay9EJmESfmAM6rOHAAAyBL4Gbq/J+BVJ96Sidr7qeQjXLljhDuCfBM4Ir/O\nF9MZhwWs3b7BAa/A7vpgCRyg1grzVkoln/jeR6sX5k7IYs35z9+7ivcO/JvM3bylRio9pHng\nsWFydqYEunySit+iznoQ1JiUx1bdBRGJIbmTSosJ86ip1P8INVZQwyVMK2Cbvhpc9YHqz/EP\nnwL7oKEyVXwK5/YKW78HYQGrnKi/htoOga0VUEB9EsauD7kn0UgYtOL//cLSX7xyqrV78E+4\nvsWcERt2Q1FKSNQNADp/0KcXyQD8zAfCxAWOQB/jGDpv4yDf8bqilNf2V/oEBYayPTsVFPzh\nnI5daL3c3JebHH7b2oyc5PH6qY2Lzl77gbON/vGymq7yuu45aSNsoigoKChMOYrAMePB3K1U\n/JTbhmAwmLAYI0euVhhh5MgFGLkASArlk+XE0Idj9rKpnACX6NLH1FhC1h4Mi8Gs1ZgyNusB\nNEYBomxyPpoC16e01QaI14xicyXQmmdmyUPD/EeDucGk2voAdTTxxqHlCmr1qh3fRr1p3GPy\n6lK3uuGGejuce19S3/4v4x426PBP36ATbw6+cNqpqliqOcNu+SEmTUg8DQRbfKfU1wxmzw18\nAotz4K+Das9Q2Ye4YEvQ7ue08Y/+4FY3Bulu5h8/x7Z9X/YK6jhOLfuGXvZVkvkypu7AsIl+\n2E6QeemRzzy8Yf/pxsvNfQatqiA7KqRdG6lLZvEDANDbBi7nBJsioyaJ+uUke03I3AdYNEhN\n/mFk4/wZ7tycW9nYW3xxKOVEJbKv3TIvPT5Aq2MFBQ9qWsyP/flUVdPQ5sd1RcnfuWORWpyk\n7+ia1r4A9YJQ3dynCBwKCgrTH0XgmPmEJbAV36aK3dR5CVw20Mdg6hpMCt6yf9qqG1OO08b3\n/Td11Q28or5WairDjOVsxb2jHwN1YUJOoXTppO8BjY7lB/4lBtqYHUUOC0MjJxl/FobjX7FP\nAWo9hMWCWSZrHaMzgngf1Js0Dz4undkv1ZSBtZ8lpAtLt2DYhJ7wpAvHA8aJpktz055WKvZL\nFJJc9PEL+JlfhOSOugjh2h9R5QF+cR9YeoBzsDuBDz1oU9WxIAocVFfitrDxijecB0s36P1+\nxa5+aj3gdzanpj2Y+5XgymrjQKMStiwLks3niKgC1D8KIggTfqgIWwC9p8Hlnb+GAkasnOjI\nAUD1XLK2DrVQGYAZQTVOy2qNSnjsgeUHS5pOlLd19tnT4403Lk9NjrmKC9AURo3Dxf/tueNN\nnV7eantPNoTpVA/dOn9y5qAKrKQMc0hBQUFh+qAIHLMCbSQuuAcBgPhMLNOYBNq6rftONzZ2\nWKJNmhVz4/NSgpDwyc/vdqsbbqj6GKUsxuQx9CRW3fxV/vyj1OmxhahSq2/9OhoCKg6YmEvV\nZ2TiSSM7kAkYLWErkefzEwGIM66LCpt7Iz/+ok8QI9MwMRjtJL0GRWHxJmFx0LwtyRrAoNRh\nA5cTVGr5o0Gkr4mazoCtG7QRmFgARpmqE6orkXXEoM566GsHY2hKaQQV5l2LlSepv0Xm1v2d\nwbyXOdBoROYO9BM4qL8GSJI53dkLtrZpVb4XajB1Hp3xc8ABwJR5QZDnmAoT7qSuj8Gdx6GO\nxehNoImf6MgB72hC3XpynAVpQPlFEFNRPR9gQuL+uoWJ6xaGpp5LYfZy7EKrj7oxwHvH6r60\ndY5GNRkbTrnJ4Tq1aHX4mk8h4oJMpZeKwqTidAXIJlJQGBZF4JhdKOqGHO8dq3vyn+dsjsHF\nySsfVty8Mv2hW+dNsJ881Z8OFB+TwIHhMdqvP+E69h6vOU92C0tIF1fejBHDFfbjituo7pyv\n+aUpFhdcM5obqlm+izdK1AbAAZChSWRpCNOgAfBYwNQlDICX/ANsA6m8iKlL2KLt0/+vIFDx\nERrCJ0HdoEu7qWqfW7ygy/sxaxPm+mVG+NRueI5gt2BImylp5UdHbVCTjLQB6wVQdgKSvHkN\nAJBkmx5ZN5MEZi/DlLlUf94rKqpxzd3BuYEYhrHbINoOzi4QDCAG790mOamnBfURvr99Fo7a\ndUBOICswwwSlDQWFcVPbIt+6y+6Umjst6fGT0cZOoxLuvT73D2+f94lvX5MRH6mbhAkoKCgo\nTBBF4FCY5VQ09DzxRgl5VJRyTm8erk6LD7t51TgzkAeRy28HgCvr7bEgqsTVt8DqW0Z5Oibm\nstt+xD96DjobBiNZRWzjfaAepUghiCxVhFQCB4JqyrPrxw2mLhGSC6i3GRwWNCVAcBfAIUNY\nuM71yS7gvvkRQsGGUN+aWkqp8kPvEKfKD8GUgvHe+c+mABIbE9AYQn8HAMC0QqouljmQXhjM\nu6TMl22Rg5HJYJJLx1AHrEvCwIdmJ4hs+w/4sTfpzB6wW4AJmDIX138eY4JaI8M0oEkI2mg2\nMz/2BpUfHJD2MCYd13wO4737zqIKMEgdphUUxoVaFVCgV09K+sYAt6/PMhnUz75X3tFrA4Aw\nnerz1+VuX5MxaRNQUFBQmAiKwKEwy9l9vJ7k/LLeO1Y3UYFDFw5Ouc4juslY7WDKXGHnL6Gn\njcydGJk4YndY+UEg9NUQoYaJGJEy1ZMYGywuVXXjl5zvP+e5umYZC1Sb7gn5vRtOBIgfB2+B\nA9MXg84IftU0mFkU6mbMmLUca4qp5pRXMDqdLbwxmLfRR7AVd/LDf/EKimpcf5/8rAxppDKB\n01e+REM6qGaGshZMRDVbfSesvhP6u0BrDIL1RiCcfdRzERzdoDahKXcYmWk4uIu/85/UMVRR\nSO019Nbj7OYfYnx20KaqoDBhCgLYA8dH6hImt83w5iUpm5ektHRZJU6JUboJZrwqKCgoTCaK\nwKEwy2nslM+0b2gPmIE/SljaEl76jn8c04LUo3REkEFEPEaErDRdIWSIy28Usha5Tuym9kYw\nhAvZi4WFayfBXpQsMv6yA3Hfe6t1bMs3+O7fgXVoSY/xWWzjF0I3vSu3QXbN16jiKFUcpt4W\nNERheiHOu3YMPWKddnLY0DCC1Q4u2MxiMujU29RRC6Ia43PZ0u0B7UVQZCm38rq/g8sjdUsT\ni0lbRzurWYkhMnRjU8dpatwL3DH4snEfJqzHuDG7jVLFp57qxiDcRcd34U0PT3yeCgrBIjcl\n/JrC5I9ONfjEH7xp3pQoDEpNioKCwkxEETgUZjl6jfyb3KCd6Jsf51yHbRXUUu4VzL8GE+ZM\ncGSFoEIA0jT8rMOYZNUNX5zsu6oCPK3KxTF5rvC5x6lsP3XUg0qNifmYu3Kymrwg5qzCnFVj\nvYzXlrn2vkhNVQCEhnBh1XZh2Y3AAqZ2Y0Iu3jjq1ry6RJbzAHWdBXsroAC6JAyfP/09X2Yq\n/fVU/65XhFzU9BFoo9GUCwBAHCQniJoRR6Kmi/Lx5ksA5K7Rs9pdf/6w4pPS5rZua2K04fql\nKbetzRQFZeNaYVL5/l2LMhONr+6rNFudAJAeb/zKzXOX5g1ny6WgoKCg4Mm0e+hXmMkQ9JeR\ntQokMwjhaMgH3dRn/y7Nj/34TJN/fFn+hB8XBBXb+A2qPk6NJWDpBmMsZq3C2JH7mChMEtRH\nUjlQJwAH1CDLAJYOcFUvRzEmn7qqZeKxAVQ5jR4Lt86UFR6/eML5xn8BH7QTpv4e194XqKVa\nvOUbQbsHU2P0ZKVoXd1Qh1/z7AHaT5Kko5NvUOslkFygj2TzNmP+xuGUJu7rtHLlHhJwAoYA\n0GtxfOd3h+vbBjP7alr6/vjO+aPnW3715ZWyGgfv6eQdzSwmkZlCmMOicBUiCuzujdl3bchu\n7bbqNaJRr/jCKCgoKIwNReBQCBLkora3wHFFSnB2ka0a9LkYtTmodyG6dJiaL4LdAlEpbO5G\n0I+QhX5tYcr7x+rOVXd5BqNMmp2b84IxIcSM5ZixfOQTJSc1lVJfM6rDIDYXTcEzz1OQhbrJ\ndQzgipEn2UkqB+pGIZhGlTMOzFhHzWegrxk8d66NiZi+diqnFSRce19wqxtupJKPheVbMSEr\n0EVAfQBOQANAaL1FFMYE2eX7+PKOWvj04JB/jaWLn3gN2y+ztV8KNBRGJst2GsSIJGCDssir\n+yrd6oabkqrOPSfqtq5I8wxKDZf7//o/rorSgZeqOYX6u78hJIzZYLWj19bQ3h9uUCfHhCl5\nIgo+ICrlIQoKCgrjRBE4FIJE3+khdcON5RLoMkGXI3fB2LGZ+Xu/obbLgy8vF0sle9jGBzBj\nuCWrKOCvHlz5t/2V7x2ra+22mvTqVfPj778hP8o4cmJzsKCOKn7iZejvAAACAGSYtW5GNDSd\nuZBUPqRuuOEtwDoA5VzcuERdjWDuwPB4iEgAQCAJcNZ1ixTUbOU3qPIjaj6N1m7QRWBCAWZf\nC8KMt5ul3nbqapY9xKtLBVmBg+qJVwA4B19iDLK5MNP6Jc9WkKnkVYn6ZvLrfUPVxylvA8bJ\nf9dg/lo4/S44LL7xhde7//3p+VbZaz893+opcPD2pt5ff5esQ1KI88Kp3l9/N/zHT7Hw0XYX\nau+x/f7Nc5+UDr5dE6L0X791/oq5cr17FBQUFBQUFMaIInDMCqzN1HWK7B0oGsCQjpGLJ3/l\nTNZK+bilCoMkcPDDrwypGwM4rHz/n4S7HwOdXBcD4tR6jLovis6+z6ZFfnbJYqdxjloM0k/G\nbiZHHxpiRzY+tJv54T+C0+PZmjhVfkxaI+YHNb1FYQgJqFv2APF2FHzXIdRwnn/8PHQNKnQY\nlwDL5qBJDUwH6izULZhVzSMFNebdgHk3eOZwzAac9rEdogbi570j7SSdRGHlVV7HNF0IywBz\njW+Qc+r31SkGoKbzgQQO0JnYjd/h+5+BnpbBiKhmhTfhnHXuUwb8Dvzps3jFrXte81Q3Bm9t\n7rHtfUN/+4MB/ide2J3SD54+6pkt0txp+fcXTvzsi8uWKD4LCgoKCgoKE0YROKY3JAGy4Rch\n1H6UWg8OZAYQAPRdou4Sln43CJO7D8nlGqYCAJ9os5JBXHaqKpaJO6xUfQrnbvCNS3Ze/hJY\nBpesZOuE3kpV9ELI3D7BiVBnFZXsot5GAABkmL6Kzdka0LsRgOqKvdSNK/Cqg4IicIQKaeCv\nIcAhL6itmr/1n54dW6m1GfZ24NbVoAWwnSNnA5puAJx9n5azSN0AwPBYENXgcsgcikn2DxK/\n7B8E6AdqBVQqyKYejFlKXSXgU6iChoB/2i7bcKPF5wh3/JTqS6GnBXRGTJwDYVGeJyRE6Tv7\nZISwxGiv3pyuqjL5m1eeG+bunnx4ssG/Fkbi9NIHlxSBQ0FBQUFBYeLMvkf22UL/Beo+Bs4u\nQAE0CRi5FjRyz9y2Fre64RPExMldPDM9SHIba0JYcMa39AQyiqO+Nv+FGjUdcqsbQ8GOEoiY\ng5Hj73JCHZX86FNDdf7EqfoQ765ja78VKGuG+lpk42DtAad1GGVEYQKoANQAcmtd9PVZoJNv\ng1/GO9idVF6LBbkAAFI32C+Cdl5IZqoQLES1ULBJKt7tE8bwWJbrbwvqBJDXZIl6URE4pgOC\nhuXcR837qesccAcwFYbnY8JGqfxRcMr87kY2NhJETF8c6ODmJSllNV3+8euXpni99v+sGD7u\nx4U6+eSyC3XdnBNjs0p2VFBQUJgITsmv1lhBYRQoibjTEer6hNreJ2cnAAG5wFZPTa+BRWa/\nkXrLZbezqPe8fzCkoD5A8XOwDDg0hkAbzqiV0VCo+4L8OIHio4MuvOvvYkjdtdR4JuA1QsDq\nBmq5QHWnwNw+kSkpyIHAUuTiIrBEnxC1VMmP0dEzdI6zMVgzmw1YeqCryf8PYcoRr7uXzVvj\nGcHoZNVdPwRxxjuMXKWIOky5kS38Ppv/bbbw+5h2C6hNmLNG5kyNAdOLJnKrrStSfcxEBYb3\n35BfkO1V0SakyLvVCqmjbRnGuXwGChEEOKKgoKCgoKAwBpQMjumHqwd6isF3Nc+pcx/qM31P\ndvbJDyLZgDuBTaJxQFgB2BvAVucVNMwHnd+cx4fGgAk51HzJN44M0wpkzncGKI1xmsc/B+Ky\nLTYBgDorMVne6xRjc6liv2+Uc5CIH3pm4BTMWM6K7lCyOYIICjkENuAewgRqUFgE4L/WDbCq\n8AyTfH3+1QZVn+H/LuheHgAAIABJREFUn737Dozjuu7F/z13ZvtiUYkOEgAJAqwixSKKoqhC\nFYvqzYrk2JZb3JJYyc8p7yV5qU5e8nOKW1xip7hJcmJbVq9Wp0ixiJ0EG3rvwBbs7syc9wcW\nZXdngQWwi8b7+Ut7pl1Ci8XOmXvPefNHkWIlqpWu3Cu2372A0geq1XLvY8b2vdx4hkMBkb9C\nVG+HMK0UawEcppM4iKZozCRF8XVzfxNYh6eYPMXpuoo6PvFKbLrb8PdzwyFgpIwMw5ktdn0K\n1lk1wSGix+7fcNOVJW8ebztT3wegvChjeb6bGTThj7H9xvtDR96Jze6pqv2Ge5O80KrizJfR\nHB+vKMyQvVQkSZIkafZkgmPhCTSa33Fpgwj3wZIdFVQTfKUT1vHshq5BG4YtRUtFEiGF8u6E\nv5YDddC9UD3krIF9ecxeBnca3MEIECxEWQqVAslmYWjnR/jZv0co6p6ENu1FVuwzeQCwZiBg\ntiTbalaONElsgBPcDCd+mk1F6yh/NXeeGw/pRvSjOub6A0bIL6797MzHJsUSpGyEWA7uYQ4R\nuSGKTD/xaFk5D/WYnCB3wltFZKRljMNd3HcSwT5YMpC5mtwr0nKVFOELh4xn/3n8tRbi958y\nuhrE3V+ev0GZECWrUTJ1E2gSlWzE1U2gDFByzSyCnTx0BuEBqBnkroLDdMbQkmZoxsmnuHE/\nODKFmAo3iCs+PMtEw9QUi7j201x9PTpqOeinrCIq3wY1NV2x7Fb1/TOdbb1+AOdaBl4+1Lyu\nPPsvPr410xXJ4qkVNe5P/4n/iW8ag5HiICI7z/XIY0pJsqn8m7aUPPHGhd7B2HofD9+Y7BwQ\nSZIkSZImIRMcC49hUjgg0SbyrOae9+N3JE81AO66xO//jLvrYBhwZooNt1HNDRDpXJfkrCZn\ndaKNOp83OHInyQgydxjcq4r1hKS+m1LecuXBvzEO/oLbahH0U24pXbGXlm803zlnHbe8YbIh\nZ30y1zInVLjy4DNZUUIZZkmW0Y3i6s8Yta/whTehBQGK714KgFtP8EArZabtEejlibJAWZM/\nFaXNt3P90dgUlcVCq8fTc2SrSvaKepi7TsPbBZubclfBmZdoR+7cx+1vjd0covsgcjZS6e1R\nz4tjGDr0MCzz08fUeOfx+CDXfcDNZ6h0zdyPZ7aomASzcX5Cm9h8EjXJ1F7lnn3cu38sE839\nRyhzA+XfvMTqtk7OOPlLbnhvYoTbTxhaQFz9hTm4OuWvQv6q1P64w5rxlz881NEXlUM/Vd/3\nz/9z4i8+vmUsYt28y7Jmi3bhuN7driwrVldtINs0fiXdDsvff+aqr/7seO1oMQ63w/Kp22qu\nu0J++EuSJElSCsgEx8JjyUqwgWCJmzvtKKbcq7jnQFTQmkP5u7nllPHq12GM3j75B4wDT1B3\ng9j9yRQPODnM/WPZjQnCBjcqlPTdoytbXP+pZHakgqsx1MiDl6KDO8gTt4Kah9moAw8BBMoi\npXySSSWicrdx4hexUauLyuKrGE6gWMXa27F2LwID3NdivP1t8936miATHHOOCleJvV8y3vwh\nhiKpK8rKwPa1cI7ctCjk3ARLQTKn4t6LfPxJDEfuW5gUqriOVt9qct/rbeS2N+IOPw5HEeVt\nid0Z4O5GfvdxbquFriEjV2zaS+tvTLD4InIEeuvY2wGLg7JWwJGdeM/k+PrR326+qeUMZpTg\n6B4Yfu2DluYuX06GbXtN/rryWQ9yuqiElEKwFwiB3EBSa8TYX8+978UGB07AVkiZ5vnWJSjk\n48YD8WHuvsD9jZQVO3dvUThyvjsmuzHivdMdfd5gtns8EU92h2X9VTNeArqiIOPrv72ztmmg\nsdObnWGrKcvKcC6hRtSSJEmSNK9kgmPhcayAmgEtrriGqwoi7jERh6ngOrjL0XuEQ71QHOSu\noNxtINV4/2fj2Y2x3S++x2tvpLzy2Q4yNMCd+zjQDhA5iyl/JyxTLIExYFKgHoDB/WlZdyxU\nWv0R9JxA/zkOD5Ith/I2IyPuazd3sXZ0vHUo97DRROo2kPmSBCq/RoR8xvnXxlu6uPPF5kdg\ncZruH3M0HFkY6pxsB2k+0IpNykfWcWc9vN3ILKDsTGitbPhIuGFdASW59SnBQT7yn9AmzDxn\nnS/9GvZMWn51zL7cd9z0HNx3PD7Bwa21xtN/P96mYajHePtH1HlR3PQ585H4uowTT6K/MXK4\nUKjielp1y2RzQ6akJ6xCwro2g/O+crj5m788FQhF/lGP//rCrVvLHntggzLSRYLDoLm55VMw\n3aIbg+aNQnno9OWT4OCh9vHJRzEGWrE4ExytPeaVm5i5rcc/McExe0RUszyrZnmi5xmSJEmS\nJM2QTHAsPKRS/p3c+Sy0wfGgvZRy94y/ZI0HDmLoJHQfhA3OCiq+mSY2ZPUPoD9B34fW05hd\ngoMHL3LDz8fu8Nnfyr3HReVDcJVNelyiKhX6SKW42QwpEcrdgNwNiU+ts3YibmAh1k+QujPR\nKWn1rUrZdu65iKAX7nxaVj3pg/S447PLIBSzmh1EuSkqyCrNgGKhoipgdDKRJXe670huORSV\n3RiLN7wbn+BAaCB+z0Rxfucn8U0ouXYfr7+JCuO6FOlh49APEOgdjxg6X3wNio0qr088/Km4\nc2BzImjSCppyJ/nFZ64/js56ECG/glZsGIk2dAz9438fj2kn8dKhpvJC5/2bB9h7EroPZIG9\njDJ3Qp1F3Zw04ImfzBOFE/w/XZIo8YdeWldBppPTnvAbkdMmvyxJkiRJ0uIg/2YvSNZ8KvkY\nfLUc6gKpZCuGc8LCCja4/ecItkVeGkF4z3KgkYoehjr6qFlPWMiDtdCscgmscdOz4/MXImMI\nGY3PiJrPT/qI2PzxF8E6bzMXjG7A7AfFg2AfKHGpPEc2lU66JmUSFgdV38hnXokJU/l2ZCyb\n4TmlhcCbYG6Orwusx94QKgkW7cfHh33cVW++c9NJuLKMU6+jtwWODCpZS6t3cMeJqOzGKK5/\niyqum/kkDqHQFTfz+7+KjXuW0UqTNTUA4O0znv86t46X16WyteK234HT8/Kh5vhmmUSocb7L\nA6NP0TmMwCUOtlD+fVCTWL3CHD6xT2+sRTiklKy0XHk91LTMASFhNy81HD/DbumizGIoVtM/\nNJRdPufDSY1NK/MUQXrcOzM/y7GiIM1VuiVJkiRJShGZ4FioSIV7nfm9iO/MeHZjBAO6n/vf\no7xbIhFnNlSb6fNkyiyczbjY2wTNbB5vqB+BdjgT1toUlGdwa3yDGEqyZ0FamPx8JmxKVy8A\nseFOVu3GmZeghQBAKLT6BrH+9jRdTpojIsHHqVCga3zqNe6sB0D5FbTueniqMFAbvy954urR\nhM2aAQEAuOOCceTZyLsI4NNv0snXqGaD+d4hL4KDsM+8B6rYcb8RDPDxV8e7ZixbIW77YoI2\nsRyT3QDATaeNF/9V3PfHbT0mM0GuXeVfWxj32WIEeeAA5X5o8rHxUJ//P/5Gbxz/kQZf+5nj\n0T9Risqn+mdNn6sSvovxYXJfTl0wFKuousk4+3xMmMq2wT2PH+mzUpDt+I0bV/3k1ahm5Iqg\nL96zjmazvEuSJEmSpDkkExyLRKAeoS5AwF7EgYbYrSNfvSbGFZWqruEzv45d/OHMouWbZzUS\n0+wGAIA17yTfAQkOhSp0rseEJiKCchSax7Kak6ypNr1nSxEStPZWpeo6HmyDoVFmCazJlPCQ\nFraclWg2aWkEe4H+wz/CYNfIKz71Jo68oNz1+3CXs7c+ak9bDhXErY1yZcFiN01zcNPJmDo7\n3HYOnsRv3UQpmCQJRdzwcVxxM7fWIuhDbhmt2AAyX4/AXY0x2Y1IvPEE+tpcdpO5FZvLTOo7\nAsBw05RDCzz5tYnZDQBGT1vgv77i/oNvQ0nxnznyrOehMwg0R0WtuZS9LdlT6Fro6DtGax1s\nDrVirVq1KCt3UNUeodqN2hcQDgCAUGnl9WL1LVMdt6B9/JbVFYUZP3n1fGOnVxG0ZkX2p26r\nkZUyJEmSJGkRkQmOBU8b5K4XoqZsKAlK/XNUFUCx7QFjeJDrDo2HMpaJ638LltlVSlMTzmsg\ndYpJvILyiTIN7gSGAYtAFtF0vjiygeFeHu4new4c2SlY2CJyoVvGm0SOITco/ROSLXZZdGMp\noaIruPHdsdKeEYoFdQ1j2Y2IgQ7jxX8Vj/w1ug9z73GEemHxUGY1FVwDEZeeEAqtvY6PvRQb\nt9gQNiv50VpP+WbvXtcyWFMxKSmnmHKSSEr2JqgBBHBv69bq4pcOxaYtHFbzlR/gMGAACSs7\nGAPdWu1hk3h3m3bxhLp6dildAPowD11CaADWTHKXQ3WKkge5/wMeOoVQPywZ5K6i7KsgkloR\no7dc8v3H3xmd4/kRy4Ydzo/9IdmS6uGykBBV7FKKlvPQBXCIHMVwrwEt+i8VuzcW7d5YFNYM\nRZAQcuKGJEnSvAnrCapZS9KkFv13kaWOufNZhKLX9usJnnOq0ckCxSKu/yyv3YP2cxwKUHYJ\nlW+BMttF6eRezhY3wt7YDbYcOE0Xv0TdmRBsCk1eizSBwUbj/NPwtWNklYtnuai6C65ZLbcB\nVFLWsX584qQSQCUlwSR/gAdbuX4ffJ2wuim/hkq3zaozhbSUkBBbP80XXuHG92BoACG7nPI2\nGae+Gb8vdzVwVxPlb6dl26c8sdjxoOHr4wsTpoe4cyi/gi+Z3NVjyIvV69AfO8+LVt82rX/N\nbE2SSLVYd28sfPHgssPnovI+PX4bYDZBTM2cJLsBgLvbwebJEaOrBbNLcHD/aW55GVpkTQ0r\ndiq5mbI3UPYWyk5QfGSSs4VDvn/7S6M36iM9fGJ/4OffcT7ye7MZ5zzgMPe9iuHRZj1DDfAf\np+w9sCZcqLiIWNTFWip1tvQA9x9BsBOkwF5ImZuTzNxJkiRJ0gIhExwL23Argp1JzlSgDJPb\ncspfhfxVqbwFJ4XK7uT6n8OYUF5OsYvld0ZPqWCdO3SjkxECFEGZqiilGa/78Hcax/8DxoSp\nFoONxrEfiC2/A9vsOiyIQqIMNi6BBwEByiKlMtHSFb74unHmubECBNx6lBr2ix2/BfUyKi4o\nTUa1U82dVH07Av2wuqDa+Nz+hDsPdiK/PLnTWqlqJ/d3Y6ADFgsVrBQ3fso49hJMExxCEZs/\nzhdf5ab9kfeqI5vW3EX562byL5opKqqCYjFpLmuxUWEViP76E9t++U7dc/sb2noDHqdlx5qC\nm3cVw/dzcGyDIXJNNXJ7wrkPZJ/d4i9/Kzc+HdUPVR/mpmfJmglXbCdU9l6Crwl6APY8ylxv\nWkdWO3MoJrsxInTw1477Pjvb0c4tHnhvLLsRofu492XKf9hkIpK0GLC/kduegTG6IM57nvuP\nipL7Yc2d13FJkiRJ0jTIBMfCFu5NmN0Qlgk3/ATPZpglOFKPA7ANUMUG9LZwcBjkIOdyyt8R\ns3QlbJwzeGj0lW5wb1gftChraLKyF4mv2fRWVHZjhBbgln1UOUX1wamRa5IpG+NjGGwxzjwb\n86CY++qN2hfFuntmOwZpKSEBZ07kv22Jb1ltyS6DMl79Pp98fewlD/QYPa10/ccYcT1NAFq+\nATY3rb2HqvfC2wmLA46ceZhn5MgQV91j7PvvmLC4+kFY7QBUhR68rvLB6yo1nVVldHi2m7nv\ndRgTlt641iLjiskvpRRVkCeHB+N6xyiqWrVpNv8I7j4Uld2IRJm7DtHEBIcR5uZfsffS+C5d\n71HJHeSOXYOmdzTDlK4ZPe1KSaX51gWIdQTOm8SNYQxfgrNmzgckzRpr3PH8eHZjhDZktL8g\nlv/mPI1JkiRJkqZNJjgWtsTrmWnZXoT7ODxAqguOcljnpHC90cvB/YAOBVi2jACAyFIVk90w\nuHdCdiOCoWlGi0XM5Bs8D5rfFfBg05zdt3HLB6bT4Ln5CGSCQ0qAiqthcyIY1zTE7qaiuIYp\nZrjh+MTsRiTY04yLh2njzXw8ut+wI0Nc80jkvxUrMktnNOrUoO13C0+ese9/IiVIMgvENR+m\n1TtidhvPbgBwVJKtGP7zrPWRcMK+PKlPNiHs93w28MO/iwnbbn6YPDmmRySJh7sTxLuipqt1\nvDExuwEAeoCbn6aqz0CJynCRLeFsL7Iuqolgug+smW5hbUAu21uM2J+gRVqwA8PtsM9yQagk\nSZIkzRGZ4FjY7CUAxbdWhbDBvhyOijn+HsmhY0DMBHLm8ElSCkHjc5INHjQ93OCBGV95mvE0\nGE4w+JAXenj2xU2kpcliE9d/zHjpO1FBEuKGR6Em9Z7hCwcTxZVPf4NL1vDRF7mvFXY3la4T\n2++FY3aLtlKJqGaXUrMLw14AsCc3Y0XY4d4w3U82y8ZrxO98dfj5/9QbzsHQlcIVtlseVtdf\nPd0Rxw0mcevfMWzwwCmTfYwgD9ZSdlQFELXKfDaKyC0UeYuqdEXiugwk16csUlrsY4kxRt9B\nUXTnXI5FkiRJkmZMJjgWNtUDz2YMHokJU/YukGJ6RBqxFxxXWxQAdBhdUErGd4xNgoyZYTFk\nchdzoMc0PrMTzoQ1we2Zah/Jbhg9beGDrxndLeTOVtdsVauvTHgqZoT6AAGb7D649NGaa0VW\nEb/339xxCSAqrKSdH6aCpKcy+c3ThfAPAKDKrVS5NUUjTZskUxuzo6yocX3+/8IwwEaqWsOS\nq4x9JtPHyLVi/IXujypINFGoL3aQRStsu24PvvNcVFQojvs+u8jKFQsHLHkIm81wsc2ojLQ0\n7xI1aAMQaJnDcUiSJEnSrMgEx0JHOdfCksX9+6H7AcCSRdm74Fw12TGsQeuFEYCSATUV7VQj\np03wJT5uU6JCGzMrwAGASndx9+nY6oOKlUpm/YQ2+TEUrudLb5rEizYACO17bvjp70OLFAoJ\nvfO05Ypdjke+HHujxcyd73PrW9CHAUB1UekNlDfrNpbSwkZFq+i+/zXDg93ZCeJTL77Q6s4E\n33jKaG8kt0etusJ24/1knV2X6IVPiMlbrkwLLdvOfSdim0apLsqfsNBmkgkLwuSn7XjgC6K4\nIvjKk0ZfF4RQyqoc93xaXbk+VWOeM5S5k3uei/1Ydq6BJW+eRiTNCjmXMwmTojNAbGEOSZIk\nSVrAZIJj4SNkbKSMjdB9IAViqnXaoUb2vg9jdM2/mkvunVAT3CNNbyCJL01RT34UkafrHfHr\nRxSxbIaXziihdR/h808j2B+JOPLE6nvgmLvS7pS7ksp3cf07UVFnrlhzh956afip78KI+l4Y\nPvaOKK607fnwxCA3v8IdB8Zfaz6ufxaanwqvSePQpcWMVl/Nx14xidfsnPzA4ZefGH72v8Ze\nauePhw684n7sqyIzLb81rIVJiKi1G0uA6hKrPs6tr/JA7UiAPKuo+GZYMsb3EVY4S+A3ecRN\nbrN5OkLYdt1u23U7+4dgsZFl0S7osBZR3n08uB+hNrAO1UPuTXBWz/ewpJkSNjhK4G8aC/DY\n4xF1LiZhSZIkSVJKyATH4qG4pt4n3M6DbzJ4fM6G1sMDL1P23VNnRqZETogcGHGtCsgGJSpz\nQbBbREXYaJhYsEOhZQrNvBIq5aymbV/iwWYE+8mRg4zSuV+kIzbcx/nVXPcOezvJloH8GrHy\nBqi28OEnY7IbI8IHX41KcIQGufP9+N249S1atg3Kor3PkdKJSqpp+z38/lPRwRqx7e5JjtLb\n6oef/1FM0OhpD/zie65PzHQuSQLBg2/4n/6R3t4IISwVNc4HPmNZtZDmI4QCxrHnuOkYAoPk\nyafq3VS1axrrQayZVH4/GWGE+mHNNJ2vIQpuNBqeiOn0RFkb4ZisrAY5MybZOjUjCNaS+ruQ\nPpYcyt0LMFifpCS2tFhQ9g6ekOAY+yUht8xbSZIkSYuG/EaypLD/BCZmNyLRIA/XknOKVovJ\nIOsmDu4HT2gJQRayXgnE5hoE5ViVDIN7mYcBi6BMQbP+Ii4slBXbdnGOUcE6KlgXEzR6O013\nNno7wDx2K8XeZtM+LDA09rdSRnkqByotIWLng1yxiU/8Gn1tcGVTxSZau3vyW/TwsX3mSbcT\n70HXUlWiAoD/qf/0PzOaSTGM8PmTA3//exmf+zPblt2pusSsBAb0Z/8O3kgFH+6u5+56ajou\n9nxhemv3hAX2xBPQHEWi8lHueIP9jTDCsGZT7nbKisvy6AHueo/9TTCCZMuj3O1wzqjNTaCO\ne99GuBcAFAdlXgXPFSlbijgTJLMbSwM5lyN7C/cdjoraiykntvmRJEmSJC1Y8kvJ0qLFTa+I\nxE0qdM4Euch+PbR6NvoAA+QhtQKUqOKGRaGC+fzWPVfIYT59l5zuqLvQBF0VAcBIVJZVkgCA\niqqSbCs7godiy1tGaGH2D1FGKtasAUZvl/+5n8ZFDd9Pv2nbvAsiZbUwZsz44Omx7MYYbjzK\n9UeofEsqr2TNprJ7CQDr5pPLgj1Gw+PQItlhDvXz0AXK301507x19J7hrpfGP1f1APe+gXAv\n5d4488HPN+4/j873ebgbqos8FVS0C8pSLxazUFHe9XCtxMBxDvWR6oJrJWVumNf0mSRJl6+w\nPsPuBNJlTiY4pOlSoK5M+GUn0Mf9dQj74VpGOavmodXLCA5C7wcAJStR/iWFLGu3hd9/OT6u\nrtk28SU58hN1tSXHTKuTSJIZSlBogyzWRCsjuKcJ3U1QVMqvgCepN2T47AemuTmjv0drqVPL\nViY/4DThpuOJ4ilOcIxJ8KHH7a+OZTfGg13vkKca1uTzTcx9b5vcbA4dh2cTLFPXnV2AuPGl\n8bV7oUH2t3HvSVHzCVgXTsPjyws5yuAokykNSZIkaZGSCY6lxZKHkFmtO8pEfx0sLjhz05Z0\nYL74Cje+NXbDw658se7DyCiB7kWwDYYfajbspSnscWDG4MAJBM+OtqQVsFWTY+MML8oa9x1i\n30VoXliyKHMDZayN30tdt0Ndd5V26sDEIGXm2j/00aj9nIXIWIGhhpjDKWed/CovpZblimuG\nn/9R/CoVy6ZdJutTAoPG6//OdaPtqIVC6/eInb8x5UoWHo69Yx/fFEi4aU4FfQniph2v08YI\nsq/RJM4GD12g3G0mm0yFeiLttOINNy/KBIe32aQyUWiQm1+jynvnY0CSJEmSJC1uMsGxpJBj\nA4faRu/tRxlkHHkKmgYA9ixafSctM7lLnyVufJfrX48K+TqNo/8pNuxh37HxVoKWHMrZA0u6\nJiyw/zBCFyYEDATPMIfIuX3a59KHjeYnEBqd3655OdAM70UqujN2TyLnx/8ktP/F8P4X9M5m\n4clR12yz3fqR+EflYuX9XPcrHrg4fmj2Wlpx+7THJi1euqad2m901JPVIcrXKivWpOMiSkGZ\n4+5PB371/Yk5DqWgzHHPp+P2ZeOFr3PbufGAofPxlw0isesjk19F5JeYbyBSCopnMu6Uc+di\noN0sPretTLVAfFep0U3TyQRNssxtkk0LGPfXJorThCYekiRJkiRJSZIJjqXFkk+eG9j3PvSh\nSGRY44bGSHYDwHA/H/8RrvgY5aX4toqb3o0PkpPYe2RiszmEe7nrWSp8BCINK0eMAEIXTeKh\ni7Cvh3BO62Tcu388uzEW9J6D9xy5V8fuLYR1517rzr1TnFR1UdUj5G1mfxtIkKsYzsn6LMQK\nh4zORvYPirwSyi6YxoHSwmC0Xgo+8f8b3a1jEXXDNbYHvgRL6n8dbDfcq1ZtDL75K729UTgz\n1OpNtt13QbXE7MZtF6KyG2Pxk69h+32wOuI3jbHWbFbyS/TO2Flj1k0709SMdrrEqquNw7+M\njRLRqqvndByqCyTAZmuJLdPppWLJBkRsCjuyaRFO30Di/I4Rhh6WvaUkSZIkSZoumeBYcqwl\nZL0bWj8MP/e1cO2v4p8c8sVXUpzg0IIY7jeJ54xU34x+CmcE4D8H94ZUDmCE3pvwManeM+0E\nh++C+QbvBcQnOKbFXUruaXdP0I+/HX7pP9gb+Tkr1dssd/wWeRbEbaSUlHBw+Edf4YHuiTHt\nxLvk9Fjv/lw6LqiUrnR+5Pen2Kk7ds1UhK5xbwsVrprsWFXN+OJfDv3rn+sd4zkOS9V696P/\n33SHmia0/hbqbuCGI+MhoYit91Ne+ZyOQ1goYxUPxiWSSCXPdD5MhA3uanjPxMYtObAvn8X4\nDOhdMLwgC5RczL7jVfISrc5T7DK7IUmSJEnSDMgEx6IQAvsAFeRKrpaEgJoD5GDgA/Mbfm8b\n9DCU2Ge5MycUgGKvJQgW8zcYh3sWwcxjPZAgPg/FBfTT74V+/s9RkdqDRk+r/fP/FP9MXlqY\ntDMHY7IbI8JHXrPu/UQ6JnEkRSQuyjPJplFqaUXWX/0geODXetNFqKpl5Trrpp2Tt7CdU0IV\nN36em09w0zH4+5FZKFZdg6zpTJtKESq8iYM9CE6YFEYKFd8K1bwHU8Lz5NzIRhi+C+N5Y2s+\nLdsLmmltI6Ofg4dhjE76g4BlFVlTv4zRFGWv5ba34/tnU24aMuCXJ8MXSf2LTCiZ8z0aSZIk\nSUo7meBY4MJs1ILbRl9aSawGJf3t3HRGdERKGy8JFZ5SDDZFXx0J11DP+Lv45JTEzQiU6c/f\nVjPil6gAgDoPBUG115+MD3J3i37yHWXTDXM/HmkGuNukADAAhENGX6fIL5vb4YxKNEfD6qDc\npOYZkWqxX3NrKoeUalS6gUrn+4ZZdYvKR7nvKPxN0IdhW0Y5W2DNmvZ5hIXy70CwDcOtYB3W\nPDgrZl6rgsM8/B44OCFkIHwOZINlTprgOJZR2a3c9HLUXytXCZXIj7XZ0zlwFKFL488eLGXk\n2AKSU2MkSZKkpUwmOBYyZuMIeHBCJMTGSRJINsfhTlDnz5kHJcWPi2nlrXz036O+pDJDA8ym\nF5A1PQUIhRPWSoQuxcatFRDTnnRNGWu45x2TDZ60VIWcTDhkdJq1YACMlvMywbFoWBP+0pHV\nPpcDibp0bhldU4T2AAAgAElEQVStuoovHIiJi613p3KSlwSAFMrZgpzZtqfl4YDWOgDhUYuX\nU+I3VVK05ujsxuglwhdobhIcAOVvI08Fdx7CcDdUJzwrKW+jLC86exz4IPavYbiJOUSu6+Zp\nRJIkSZI0F2SCYwHj7ujsxmjYuEhKUgkOKryC638dXx2DVqT++w3lrMTmT/G5Z+BtBwBSqPQq\nKljPvS/Fzhax5sNZmfIBRIbh2MqkInh+9JkVwVZF9k0zOVX2Vgy3sm/iF0SinB3kmPsn7QkK\ni0y6RVpolMqNpnGRW0xZ6eorlAyx5zOGJ4+PvQR9pNeSS1z1AK3fM49DkkyxFvb+8oe+Zx7n\nUBAAOVwZ9z/quv3DM54Tx2Z/YgCAA+AwaK4yXPY8Wv6hObrWZYKDCNWZxLUO6H2TzXaUJEmS\npEVOJjgWLuaBBFsCQAhIYpapYhWbP2mc/jkGRusIKjZaeTMVb03VICei7Eq66ksIeRHywZk3\nsoCflDu5/y2E+0Z2gWstZe5IrpLIzAahkGMLbGuh9wEMJQdisjYQU5yq+F54z8N3EdoQLNnk\nWQ97YUqHmxyLjZaVcldz/BZRPEdPWaXZE8WV6pY92uHXoqOK9Y741q1zS7WKqx/C1ru5txWK\nStnFUOSfhoVo8Af/5H/92bGXHPAN/vhbht+b8eEZv4USfxSnaSGhNBeCrB1LXHK7XyY4JEmS\npCVMfotdyCZ5Op/0g3vnMrH1sxhsZl8nLC7ylMGa5gr5VjesE8rm2Uqo4GHoPhgBqFmgOXnL\nCcfM8xrRyF0Fd1VKTjUblt0PhH7+LzFByilUNlw7L+ORZsZ27xdFUUX4zZ/zUB+IRMkq695P\nKuVzVNBxChY7FaRratW80Jsv6g21rGtKSaW6cn1armFoEHP0Z1TvbPO/8Vx83PfMT913PEzO\nmXywk8hlmLXWFtnA1CVmpQVJYz4MDCXeQS7/kSRpcdB0OVFZmgmZ4Fi4iDLiSsuPsCU1fWPC\nmeApI888lTAcobigzGHrwSVH2bjbEg5pr/yQA96RiKjcaL3r87DIcnGLilAsO++07LyTfQNk\ntc9b55SljoOBwBNfDx1+Yyyirt7k/OiXRWaK2ioHB7juZe49By0AexYVbaeSnenOdITOn4rv\nNgKAw+FwXa113ZUzOalaBC0Xekw1ZZqzLipSGjQDw1BUk9ZmI1TZXFySJElaymSCYwGjfMAF\n+GLDYhY186VFS91yk7p+p9F6kf1DlFciClbM94ikmSPXnPdrDAX0429yZwOsDlFWI6q3LeGP\nkcCT35iY3QCgnTvq//e/dT/21RS0sA30GEe/i/Bou+jhfq57GX0XaeOj6f2RGnqiLTxSPGUm\niGw7OHwW4bpIsSThIetGKHkzPaE0z5j7AYCIrQ4KxTU1t5ZDZMz9qCRJkiRpzsgEx0ImSLmS\njVPg3tGIQqISNK9zMaR5ZHOKivnudrmoscGDHQj0k3sZ3LlL+A4/htF4Wvvlv7B3pBQO9APP\niOVrLQ/+IexLa16VEYKhc2A4JrsxQqs7rTXUquU1s7wI1786nt0YC/ZfRNdJWpbGX0/LigRr\n5UhYViTo9ZsMspB1A6zrYHhBNtDczCoaArwAAR7AOSdXvHxEqnqT3QkihPzjE39sa8i+bt7G\nJUmSJElzQiY4Fjg7iS3gke+CFpBnmotTJEmK4M5zxuEnMdgx8pKWVYmtD8EzH1Vj51gwoP38\nH9kf1S/DaDwdfukHlrt/d74GlVo8cIkbXoGvDQCE01K5LHypM356vtFaj9knOPrOm2/oO490\nJjjU5ZW2TTuCR/fHxJ3X3yYyc2Z9egHhmfVJkhFmrgUmLoopJKpKY+Xpy44LGG2dZnPAaoeh\nAwyRT0LmxyVJkqSlT36lWAwoA1QEypPZDUmaGe5tMN781lh2AwB3nddf/xqC3nkc1dzQzx+K\nyW6MME7vQzAw9+NJOe4+wad/GMluADD8ju0V9s1ma7jUKfueMrReBBuh9cb2tx7bQQ+ZH6oF\nkx3xTGX9zp87rp7QvpeEc8+dnk/8XuquYEDrQ7AF+iQlKmeF+VR0dgNAO3OCnJE0fUQlUXPT\niKCoUCwkls/foCRJkiRp7sgZHJIkLX18+iWTEgbDQ3zhLVq3dz5GNIf6Oszjhs4DXZS/yG97\n2OCGl+PD1tWFofMdxtDweIhIXTnp/PxwFw/tgza6JFDNooydsBRE70Sw5yDQbXK4I+11K4TL\nnfWlv3Df//Fw3TkoqqWyWi0sTdnZgy3c9xa0yDom2MsoazfU1BaLGQJM2593AJXAlOknKRlu\novXMtcBYJk4hWgXI2qKSJEnSZUEmOCRJWvq4t8E83lO/9Otw2BLXOFgCNTgCXQiZTTcgKPkZ\nExMctl13iNzEK5L0Ie5/CRwej2j93P8SZd8JNTvqxEVb+dKLcZdTqHDzDIY/A2pphVpakeKT\nhjq4+xnwhEkrw03c9UsEK3igA0JQdgWVbAbNctZnbM3sUQz4gKzZnVwas4woG+gBAoAdyJHT\nPyVJkqTLh0xwSJJ0GUjQcjlhfAkRlVeAKP5fSsuWk2fxP9Q1wom2CHcG0AWAHC77rQ/brr9n\nktOw/1RUdiMS1dl/gjy7J8aoZCcCPdx2cDyk2Gj13XMwgyN9ePBwVHZjhO7nrre4vQ8A1++j\ni2+Iqz4N+2zmdEySTlz6mca5pQIFU+8lSZIkSUuOTHBIkrT0UXYpt5022ZC99HsSUV6Jsv0O\n/cAzUVFFVT/0qckP5K4GXDzMQz2UVUCrdyAzP42jnDF7DkiY3JwD9vses93rga6J3CKIqaYe\naGarTgCE4+IkqOpuKtzCvecQGoIjj/KvgNU9g7EvIKEE65ic9rH/5MEW4+jjYsfnZnJ+1hDs\nYmMADtPNCrDIf4CSJEmSJC0MMsEhSdLSR2tu5vazsbfBFoeo2p3giAUvHAq/85R2ej8P9oqc\nQnXLHnXLTSDzx+DqTR8TxSu1fU9xVxMsVlG2Rr3xN2nZJMkdNt5+nI+8MPITYwAHfiF2PUyb\nbk3Lv2U2VCflruPuE7FxRx5lVRIp5kf5B4yDT6G1lsPDlFtKV94Bu/mOCWWUUsas619oXh48\nhVAPVBecK8hZPtsTzpwBsMk0iuh3FHedQ6APjuzY3SbF3vPc9Tq0IQBYVk6ZsVNdiFYACf5P\nSZIkSZIkTYdMcEiStPTRsiqx81PGkf9GINJAkbJKaNsjcCzKZf887Bv+7v8yOiKFRfShPr3h\njHb6gP2jf5IoxyHWXmNdew0MHWLCnaS3D3YX1Nj1+XzmHT78XFRICxtv/FDkl1NxdQr/IaPX\n40TDTgZV3gE9yH3nxkPOfLH6w0iQ3eDuRuOpv0XQH3k51M31R8Vtt5vPIbCka+EJD53lzldg\njFaC7DsE9yoqvB00H3+XLcsQbDaJ+4djAuzroekkONhfz+3PjucWuxugB5FdNFrOw0JUARTN\nZMySJEnSkhbWTduZSdIUZIJDkqTLApVeoRSt4Z4GBPqRkU/ZZbOumDhvwm/8z1h2Y4x+9qB2\n7C1103WTHTmS3dBCxsFn+IMXEAqAiIqq6LqPUkHl2F588g3To/nkGylMcPBgt/b6T/nSMR72\nUU6hsu12ZXPCSSiTUWxU8wgG6uBthh6Cq4hyaib5n8tv/udYdmOM8e4b4tadgBYVJYWcG6Y9\nnmSE+7njRXBUZx/2XkDvfsrdlZYrTooyNnN8gkM3uDuqwTBZFPZdRKgF9jzKrkmUQpqIew9E\nzZxi5t429HVQ/k7ybAAcsvqGJEmSJEkpJBMcUlqZzXmWpPmiWCm/ar4HkQL6mfcTxA9MkeAA\nADae/iduHF3Twcyt5/hnfynu/99jyQseSFCRob99JsM1HUR3c+i//hTDvtGXLdoL3zMaTlnu\nfWxmJ6TMCmQm0VvEP8DtF0zigz705iA/PN4nVfFQxs6YFiqpwkNnY7IbkfjAiXlJcMBeRjm3\n8MA70EcntgRCaOxAeDTjQyCPC04rut4fKVfLthxReS9cxVOcOWj2XmIDvk54Evf3kSRJkiRJ\nmhGZ4JDSgA1u3s8t++HvgdVFudW08hZYMyJbwwHj3MvoPs8hL7kLqHI3Fayb1+FK0iLD/kHz\nuG9g6mPrjo5nN8boGr/9OD30F5GXVifQF7tPJJ4a2ms/GstujDFOv2ts3iPK0zNpAgDAw96E\nmwZDouZuaH3QhyDcUHOSmuNj6HpnqzHYqywrFtnLkh1HuN88rvthhCEsyZ4nhZyryLECoQ7o\nPqiZaDpj+McL01KGA87opUzBXuPCk2L9F6DYJjvt0u9TJEmSJEnSAiITHFLKMR//EXefjbwK\nDnHrIe4+K7Z+AY5s+Hv1d7+O4cjtGQ8Pcvd5qtwt1k3WwVGSZsIIIdgDocKak8xc+kWEMvPY\nZ5LjoKypG51ws1k3GYDbLkALjdTjoBUbuLfF5PzlG6c30ISDMIxLx0y3GBc+SGuCg1xZibqu\nUEYuIKDmQk22e2747BH/49/UOyOLOyzrt7se+m2RVzi6XWP2AhrIQXBFHSkSFDUlBWL+/i6T\nBbZI5VRaWSgyivjcSzzYCqHC5TDJVYS93HeG8jZNdk57AQJm1T3ssu6GJEmSJEmpJxMcUopx\n1+nx7MaYkJcvvkTrf8M4/fRYdmP8kEtvcfFmyl4xR0OUljwjzF3vcu/hyCoAxUH5uyn7ivke\nVsqom64LtV4yi18/9cHhUIINPJbgENvu1i8ewmBUh1QqqKQNN057rOZjCMIwWaABAMHYaR0p\nZnPRiiu4/oPYuNVBFZundSbt4qmhb/4p9PGyHeGT7w+2fjnzz75Hdidzm8GNkaIeDKJMQasw\n1qzFVY7+w/HnJGfFwlnWR/k1lF8DZoQHjeNfN99pOEF73bGT5Ozg1l/EZpRUN2WmMY0lSZIk\nSdJla7HW2JMWrp5a0zD31IIN7kjw9LjjZDrHJF1euOVZ7nl/vMaBHuC2l7jX5H5ykbJcfae6\nfmdUSAjL9Q8qq6ZO4lBOgqIJrizYR2cZOD3KI1+hjTfB6QGAjFzafo944E+hzHzpROvA8KGm\nvrcvdR9t6R80LDRy5vjhZafuwb7u59793PYMd77Mg6fHJiCI6x9FzFUsNrHnt2DPiD5cCx47\n4Hv+Z4F3X9V7u+JPH3j+JxOzGyOM3s7gO88zdxp8aWLJUuYB3TgFRO7zyVlO7rhyrYqd8hZe\n32KiyZbMiNgWPLFHO1dQ4R1QJ/xsHaWi5EGISRe2SNKo7oHh2qZ+byA83wORJEmSFgc5g0Oa\nPn0QwVbofqhZsC+PbWqoBRMcFUQ4ACP2fiAi3Y9tpWQMD/BAG1QreYphSTCFfuEbbueh8/Fh\n7nqXsjctkbUqimJ75I/Uswe10wd4oFvkFKpb9ojSpOqnUvVO7P8F4kpRiCtujswdYGP4/TfD\ndWdhGJa199i374bVMZvBGszv1vU09wfGIrWd3huqdmUeez52V9Uq1qemxCb7LnH78zBGP44G\nTvDAEVF8PxQHXNnKQ1/hU69zWy1CAeSWiY23wJ0z8fDwhTP93/6K1hJpVUNWm/u+j7vv+ejE\nfbS6M6aX1urOWNi03OkwczdRZBkRFd2OgeXcfwThPggHucopdxdU01618011wpGPQGf8FvKU\nT3k0uavIVYFgF2s+smbDmuwKIOkyd6ax/xu/PHmhJVJa6NoNhZ+/a11e5qL92yRJkiTNCZng\nkKaHhw7Ce3R8vrGSQdk3wDrhcagjx/RA2HNgccJiR3g4fiM509KqQEpWOGCceIobDoAZAFSb\nWHMbVV2/cGbLJ4/9reYb9GEEe2CfukrFYqHUbFNqtk37MEeGuOv3jRe/jcHRWQkkaOMe2nYX\nAKOvu/+f/3e4/tzY7r5nfpL52FfUorIZj/N0+9DE7AYAg/nNwuv3+jrVC4fGozan5Y7Pkydv\nxhcapweishsjhju46zUqvAMAFJU23kwbbzY92hga6P2/Xza844vpOBQceuJ7IiPLuefOCfuZ\nFPIAAEMHTD7lADC8hLF3IFHmRspMUVmTNKOym/n84zErTSh7LdzLkztehb1o8X2aSPPnQsvA\nH3znvZA2/pZ7+0T7+ZbB7/7etQ6b/O4qSZIkJST/SEjT4TuFoSNREX2Ie16k/IegRNorUOFm\nbngrvoYfFW8BERVfyQ37ojYwoChUPGmZusuK5sVwM3QfLFlwrIidIJMWbOz/d+4av6eFFjRO\nPCVYp9U3pf/qKZfgtnOKTZcRKq5WPvYPfPEQ97aS3Y2ytZQXuU0d+M7fhOrPTbwR1doaB77x\nf3L/5gcQM1zSWN9rMj9LE2rtNZ/dtP0249Ix+Acpr1RsvJ5cmTO7RAz2XYjNbozEvefJCE25\nqiLw5gsTsxtjfM89MTHBoZSt1C6YrK1Tlq+a5ngXAfJUUvXHjaaX4WsFGKqDCndSwVXzPS5p\nyfrxq+cnZjdGtPf6nz/QeP/uynkZkiRJkrQoyASHNA3sM6uUwSEEauEerc/nyqc193Ptr6CP\n1zKkwk20YjcAseYOY7CF+xrGD1cUsf4+uJJur7i0DRzm/vfAowt5VA/l3Qz7zB+eJ4N76qKy\nG6OM2leVVTdALLY1HbYEczRIvUzmxnN/p95yEcxKcSXlFJrvpFqpemfME3W9vTl05mj8Y3at\nuS584ZRl9QyrQvpC5vVEvUFNrNwoKtIwhSFs3kYXbEDzwppgltnY0U0mBVwBaG1NHA6TJVKQ\nwnHrQ0NxCQ5yeWy77gDqGP74MxAy4oOLhrtUrPkkjDD0ICwLcimNtCjp0NuZvUQWUA5E1kj0\nVL1Zp2rgZH3f/QuvUo0kSZK0cMgEh5Q8A1q/6QYO9028KaKiKylnJbcdga8LtgzkVlP26PMW\ni11c87vcfIi7ahHyIyNflF8jsxsR3jPc93ZURBvkzqep+GNRJfpSrq/RPB4OsLeDPAlqUi5U\n5CpjRxECbbHxnCsnq5W4NIRDwed/oB18ObJ6gkjdcpP1jk+TNalV61qnSWvYyKb25hknOKyq\nGA6b5DhsatqqXCuJ/72TbBpFaoL3CREp4/k+y/qrXI/+of9/vsveSI0ApbTS9dEvi4ws5uXM\nsc2kCE6iVCzAGaEF2N8BNsiZP6fpBmFZ+r9H0pwx+lg7Cg5grAiwUkLqBkBouvmEu0RxSZKW\nnnDcNC5JSoZMcEhTYYNbD6D3LAcHiXzscZIrruJgfOFGWyaV32B+QiIq20Zl068dsNTxYFzr\nSgBGmL0nKevqdF6YE29K42XThkTZfdz6InsvjgUo+0rKX/pP/YJP/av2wetjL5lZO/QKggHb\nw3+QzOHC4ZrBpimVeOwXenzxE0NKMmdVu3QS5KxgvGny9rUXjS2mm4S1ZqP/tadN4tUbYtbp\n2K66ybrxaq3+rNHfoxSUquU1IzsQ5QpUG1wHRCayEeUKqkxNURs2uPktbn0XRhgAk6CCbbTi\nJpl3kBabMGuHwdGNq/UWho3Umooiz8m63vhjVhabN2CSJEmSpBEywSFNSg8ax/8dQ5HnugzA\n54fHhfyoOd5kW2QP+ReosMmXOQAI9aT3utkJlsCodspYnCU5VRctv5+GO3i4E8JCjiJYUlPc\nYSHjgR7t6BsTIyM309qJdyy3/KbInbr9qlpRLVwewxe7voMsVkvN1A1oE9lQnNk2NOyPXqhS\nluUozUpXggPWHMq5inv3RwWFReTvid3T0PnMO9xxEQAVVFLNLiiqY+ce33NPhuuje/EoasZD\nn4m/FDlcljVbTOKUp1AOww/WiJzAFIU/kseNr3LrhEpGbHD7AYR9tPqBVF1CkuaC3h6b3YjE\nm6BWP7C7Mj7B4bCpd+xYMRdjkyRJkhattM0QlpYEbnxrLLsxHhz0sW94/OmoNR+OlXM9siUp\nUQfTNHc2pbyVlGfyf1CsvhFiMedA7QWUtYE8NZdDdgOA0XYp0WQco9W8qEQMUi0ZH/lifNz9\nwKdExsx/hg6Lctuawpr8jAybalFEjtO6fXnONZWpW6xhhnKvoeL74CiFsEH1UMZaseJR2Aqi\ndupv13/8x8Yr3+Pjr/Hx14xX/k3/8R+jrw2KmvMn/+y88Q5Q5E+kurwy90//xTrtLI8guImy\nUpjdgDbMbQfiw9xzEoGu+LgkLVjMidrDh8HBnesKHrt/g9sxPi+pJM/1lU9uk21iJUmSpMkt\n5rsXKf24+5RpnLx+uOwgBc41lLFNZspSw14Mf118mOwlab4wiR2/aXzwX9xSHwkoCq3eTNWL\nsYXKZYwS/xpSsisj7LtuFbkF3v/5vlZXy8yWskrXvY/aNu+c5dCsithcmrW5NGuW55kWclWQ\nqyLxdjae/wZ6WqNWjfS1Gc9/Q3zkKyIjM/O3/sjz8S9p7c3Ck6Vkpzcdkzz2t4PNi7ayt4Uc\ns65npIUglMVXWlhalBK/zUgBsPeq5bs2FB6/1Ns9MFy2zLWxMteSvsI9kiRJ0lIhExzSpMJe\n87hSQPn3Q8mY7J4qJXTN+OBlo+EEhn2UUyS23Eb55em94vyhrKs50DTeQmWENRfutWm+MrNa\nS1vX05pKHhyAolJWJqxW5vNENWm+tJQyomQVhIiUF52ISCmrTv481jWbcv7sm9B1ZiNhuc3F\njzsbuLM+viYGdzVwRx0VVAIgm92yYoH1fJ2kYs7sTstn3zYOP4OBTigKFayknQ+P/BAkKU1I\n5LJ+3mxDJhD55PE4rbvWJ2gFJUmSJElmZC5cmpQ1QfMOWxbUzLRnN/yD2g//WH/9h3zpA249\nZ5x8U/vh/zYOPpvei84jaz4VfRi20VoJBsG9jgoeAKU5Ecm9YC8AuJxUVET5y2C1AgC3ANrk\nh0oLB7kzLTtuj4+r22+lzOn3x1WUJZzdAIDBzoSb+jvmcBzTQ67CRB+85Jp5LSTj7R8bv/4+\nBjoAhq5xa63xi7/mxuMzPqEkTU3kQMTXBhKkrpmHwUiSJElLhUxwSJOhZQkaQ+bPsGHktOhv\n/Ji7m6NCbOhv/pR7mhMcsfiN5DjCW/hgl/Hrk8azvzJe/Tf0x3Y8TbUE83RggP1pvrSUSta9\nn7DueRiW0YoPqtVyw4O2O0xKY0qwJm6nYp95y5i0Ux1UaNKCinLWwDnTksC9LXzy1digoRtv\n/3iGJ5Sk5JBlE6k1INvIK4gcsl4NEali3t7rf/KNi1/7xYmfvnbhUlts8WNJkiRJMiWXqEiT\nobJreaAO/XXg8f6GVHI1ZVel/dpsGOdMaumBDa49QDtL0z6AeWLse3z8ZiPk54ajevNJcfsf\nUGH6fuaTJDplDnRREYplz2+ou+4yOhphGKKwnOxTd0W9PFFRFWxOBONSeFYHFa+ejxEli1bc\nAmHh1vdGi3EQ5W+m8ltnfEJuPmW+8qW/HYNd8My6rockJURQKkmpBIdA6sS/OE+9W/+D588G\nw5GKMz965dyD11V+8ja5alKSJEmagkxwSJMSFrHxk9x+BD1nOTRI9lwUbaGsOemZEvQjHDTd\nwt4E7VQXP+5t4VOvxUZ1jff9hO77i7RdNlHpRytoAT/KlhIgm1NZLm8D4nj7uP0itBCWLafc\nUlhs4rqPGi9/N2Yvcd1HYVnYbRpIoeU3UdEO9rWDdXIVwjq7PkHh4URbODycbH1aSZoNiuo0\ndLK+919/FVXjXDf4idcvlhdm3Lg53VW3JUmSpMVNJjikKREVbkHhlrn+mmt1QLVCC5lscs1p\nL4Y51XwSbCC28iFzdyP8A3Cmp90pZYAKwe2xYbEqbiSStAgZurHvZ/zBi9AjNWVo5Vax51O0\ndrfILOB9P+POOjBTQQVd/SCVpnj9f1gzXjrcXNs8ENaM5fnuvdvLslyp6BprcVNWigqgZiYo\n4qioJKdvSPPhhQNN5vH3m2SCQ5IkSZqcTHBIC5VQxKqtxtl9sXEisfqq+RjQXOCQ3yynQAAQ\n8o8nOAwNQklh9oHEOjac4HpgpAeHjcRqkKxdL02HFtbqzug9HSInX61YQ5bZ3caHfLA4UlLJ\n2Nj3Mz4UVZyYLx4yAoPiw/+HSqrpwT+LLNBIupluchgc7Ojnv/rpB+29kYUwB891vXCw6ffv\n37B5Ze74bmhmbgUCgA3IJaqc4z/NVL4Jrmz4+mLjVVcv9Mks0hLV1mNe/qm5yzfHI5EkaR6F\n9bjGcJKUBJngkBYuccNvcvtFju5ooOx8gJYtn68hpRt58s2bQCoq3DkAc/sRbnoLgV4IlTJX\nUOWH4EpJGkKQWAlUgP0gBXCk4pzSZSR89ojvJ18zuiMFcUVugevh37WsMymHOQU9yHW/5pb3\noQ1DqJRbTVV74ciZ+ci0EB99OT7Mree4+WxkvkZqUxuGn4ePIdwCGN/6pae9N+rvrD+off2p\nk9/67WucNhVg5mPA2Jq7ANDM3E20FUjBLI+QZnQODDttSo7bNtl+qlXsfcx46ZsY7BqL0fKN\nYvdHZz8GSZoBh10xjbvs8lurJEmSNAX5p2Lp8vXypfd4sAOOTCpaS0WLr+8auXPUR//BOPis\n0XACgSHKLRVbbqPSSYsLDHZwbxMsdspdAXuCHrcLGJVvht2N4di2JrTyKqg2Pv8rbjsYCRlh\n7rvAR74tNj6KzIoJ+zLC3dAHIZywLJtmi1kBcs9m/NLlSW+tH/rWn0ELj0WMno6h7/x55h99\nQymdTskeQzeOfB+Do22SDI27TnHfJbH9i3BMv9MtAIB7W81XugHoakCqF6TA8LP3FXAQQK9P\nnGox+QUc9IePXuzZubYA6JyQ3RgzzFxHVD2bUfiD2rOHm9+r7TKYASzz2O/fsWJNacI1bpRf\noTz8d3x+P/c0w2Kl4hoqWz+bAZjTB6H3AYCaDeFJ/fmlpWLr6mUHz3aZxKvlmilJkiRpCjLB\nsTTxxX3GwSfGvtbz6VdoxVZxzScgzJ+KLFwWm9h5v9h5/9R7Dg8Z7/2E6w9HXioWsfE2uuKO\nVE87TzOrU9z0BeO17yAw3hKPimvEzkfgbRvPboxh3bjwnNjy25GX4W4efAfh7shL4STPDtgr\n52Dg0n1mGQ8AACAASURBVOVs+Ne/mJjdiNC04Vd/7nr0D5M/D7cfGc9ujJ8nwJdepXUPzXBw\nkyxyScOHAwfPjGQ3AHQPJbx018AwAOaeBNu7gegEh6+J+88gNABrFmWvhXOyMgQG83dePtfQ\nNZ4n7Roc/t6r5z5zU9Xa0sQFjFQrrdmdro9LDnPgEEL14xFrBTm2TjMDe9kZ8IX2nepo7/Xn\nZdq3VS8rzLlcmiLtvWr5K4ebL7REtYYtyHY8fGOK6s5IkiRJS5f8brEUDbQZ+38Mjlq3xg2H\nOKuYNuydr0GlF7Px2re48+J4RA8bHzwtQLTpjvkb1kxQcY3y0N/xuXe4txkWBxWtpvLNAHHH\nIfMDfO0IeWF1wwhw3wswJrSeMfzc/wbl2GCVVdmkNNKbLpjGtcbz0ztR70XTMPdemPGNN+UU\nm7eDBVCUhtbLWufYf3rsCRcPZzpHVqDEZYVGzzLxBTe/yD1Hxl/2HKK8bVR8U6KTn2jom5jd\nGGEY/Oyh5skSHOnE/gMIN2Fiv/FQHbNOrmvmZTyLwutHW7/xy5PeQORNYlXFR29Z/dD1c9LF\nbL7ZLMo/fv7qx1+78PLh5t7BoMdpvXZj4aO3VmempECvJEmStKTJBMcSZFzaH5PdiMQv7lPm\nL8ERDOv17UP93tCKAnfKH0Nx+9mo7MYo4+TLyoYPQVls73Org9bfHHtHp5s3zR3d5Gb/majs\nRoTB3mOUIxMcUjpRgqlh050ypidYSzLJm39Kiiq232u8/ZPxCAMEWrmFCtIxuWk8N1GYZZTl\naE29sZ8/VlVsihQZTVTsZjzOfScnZjcAgJm73oerlDLN1+td6ojNboxo6fUHw7rNMufz+Iwh\nhEeaYkR/qoUbYWyCkL2oTVxoGfz7J44axnhRppBm/OD5s8W5zms3FM3jwOaMw6p+8raaT95W\nMz9vWkmSJGnRWmw3flIyvN3mcV8P2EhJV4LpevVIy789e6bPG7lLuXptwe/cuz4vM3X1+bsb\nzePhAA92UPaSuL1PVINAWGDzABhfmRJDSxCXpBRRK2q0hlqTeOU0K1w48yY+458Qn9XCe9qy\nV6gWY99/I+gDAEWhjTeJaz5ssmvYy30nEOyG6kJGJbnLp30x4YERGHv1uT3+v/plRlAb/ycJ\nokdvWZ3ltgIgKmSOW5IDEE24g+07YX6h3hNIkODQ2bxUMQDdSLgpjfT+xJv6ZILD1HMHGgyz\n/1lP72u4TBIcY2R2Q5IkSZoWmeBYiqwJ5kekqOfidL1xtPUfnjg6MfLe6Y6Wbt+3H7vWoqZo\nPJOspV9cNTgSo9w1bHEhHNskjwo2QVhG/jPRoWkdmCTZb3ogeOA1DkRNHCC703Hzg9M6DxVv\n46Z9YD02Xnr1LEdIV9ysrLuOe1ughSinBHaTYrrcd5KbX4QxOoukcz+y1tLyOxPOTzG9kHUl\na+ONn6qLtK99dOBnB5xnOzJCGpUXZDywq7yqZKzYp4doNfOF0fbMIwqB0vFRhQZML8ShgUS/\n2MXZ5hNDslxWh21e/uhP8hEkP53MNXWa90NtTDA9R5IkSZKkETLBsQRR8Xo+/7ZJvCQNVfGT\n8ONXTdbhN3Z63zzedtOVs55b4e9ibzvbEtyB2FzkKZjtJRYI1S7WPmKcfRLBCfVHc1ZT5W2R\n/7bmc7DB5EBLfvIXYe5k9IBDIDshnyh7doOWLgsit8Dz+1/1/fRrWt2ZkYi6otr5yO+KZcVJ\nniHc0hw8d5YcDnvFHdT0CsKjJTNI0PJrqXhLCkapWim/IuHWYC83PRtbuqj/NGy5VHjtNK5i\nKSX7Rh4+BUTSNHke5Yt3rYNlRYIDSolymNuBAGAlygOif+kU82wFqQl7OW+pzH3xg9YBf+x6\nnxs3FM1POkHJBQiIn49AUGfYHGfJs1vN/6glikuSJEmSNEImOJYgKttIpRu5+XhU1O4Rm+6Z\n+8EEQlpjp/kTp9rG/lklOEI+rn2KO09GXjqsCMR+oRdX3L74GsdMInOF2PoYdx6Drx2KHVkV\nlD2hpLyjBr5TMGKKKQpyb07u7IbBp5lHHxezl9FNKBAkq9ZLU1NKKz1/8C9Gd5ve067kFoq8\norHJU8z8+tHWk3V9wbBeXphx2/Yyt8MydqA+0N/51b8deum5kZdkteU++vHsD92EQC9sHsqt\nhmsaGboZ476TpqWLuPfY9BIcAGw1ZCmD1saGj0QGLCUg26QHOIkSVgOhzCr2t5is3PEkrJBq\nsyifu3X1T9661NwT+TSwKOKmjUW7185Ttlc4YKtC8Fxs3FYNSt1CxZRgHaEeGCFYcxOllubG\nFStz3z/bGR/fXJU394ORJEmSpEVEJjiWJBLXfY7PvcXn3+LBDtg9VLxObLoLds88jCXxim+e\nZFsS5zWO/wgD4xMWKDeD+3zwD0fOanWKzXfR2j2zuMSCpFipaJv5JmGjnNt58F2EWiMRNZM8\nO5OcwcHcOp7dGA92MLKJ5uMp6zzVi5Fmjkj8P/beOzCus8r7P+fe6VXSqHer2bJky3JvcRzb\niUOqU0hCAimULASWJUBggXd3+b1LWdjlZQlLLwmEJCwkjhNId5pL3Kvkrmb1rhlNn7n3nt8f\nkqbekVVGI438fP6x7rntscqd+3yfc74nIzcia8Pq8P3LU0cutAVdGP76QeO3HlheM+KySdT1\nz19yHQv2Pyaft//Xv5b8mP65LyZq3AAA4IvhE+EfBhInVaUCAMDpQVUWl3QJTF9F1vPg7g6L\n6vMwfbysltxU3Vduq2rotncPuQ0aRUmWMWVW20+gdjmghrxngQQAAFSgugo0k7RomWHIfpb6\nPgBxRBVCNFVh+rXAz44Ec8u6wjeOtLWFLw+Y9ar7tzLFmcFgMBiM8WACxzwFOVy4GRdunu1x\ngFatyM/Qt/fJlBNXTKNhIQ01h6obAAAcosUAFjMuuhdUekzJA6XMqin5vL4Db4htDcBxfGGF\neu0NoFBGH5asKMyYdhOIdhBswOtAkQIwUY2ASN6LlKAfIbECh+si2Y+DYAVUgCoXzetAySpl\nkpWf7KwLVTcAwOrwfedPx//w9et0GoX79MlQdWMUgqHn/2B55FHUJHBuycdIsuCUk1Y34gun\n5MofpN5DZD0HPiuoUzGlCjNWXXFUHGJFjqkiZzxdu6Pf2dbrSDNpirONqng5IsmDoKlC9UKQ\nhgEAOPMsf1ejoOEz1PNGaICG68k3yBXcNytGIVqV4sePrXvqjQu7j3V4/aKCx7WVWY/eUpmZ\nMpt5JQwGg5FI/OJsGGMzkh8mcCQtk1zfpv7z1PIeOLqBU6C5CMu2gz5B6cr3by2PMBkFgLx0\n/eZlE63Pl8HeEWOHiOYMMGTL7+tocv7629JQ3+j2wbe87+00fPb/chnzos1KAN4IvHGyJxHE\n6NBJvkS+3pN1LzjqRvPxyQeeFvK2YfptoL66GgfMD+wu/4GzPdFxm9N38Fzvltpc7/kzMqch\nkMfjbW7UVFbN+BADGEuh/5jMWIyliRtDLFCBWRswa0McL9k54PzJi/UnGkZlzXSz5vO3V22o\nln9yxg1UAJ82s7eYKjTwoUzU00nOFtTHtm6ZSUw61T/dueSLd1QPDHtTDGoFzwxZGQwGg8G4\nMkzgSDZEv3TkVanuPbL1oj4Fy1byG+8B7RVms9T8DjXtHruCj/rP0eBFrHkY0xKR7LpteZ7P\nL/7u9fN2l38ksrw8/fG7l05vwXDytvyS6Hzqe0F1YyTW1+n8ww+MX31yGiOZKiSCtw2EYeA0\noM6ZgiQRXxBUBH65HQnMbPcPgKMOIn6EJJJ1L2bJNfVkzG36bG7ZVpcA0D3oAhi//9HMjCnW\n3UxlYK4gW7hPBK/FnOsSOo6E4PIIX/3lwX6bJxDpt3m+86fj3/nkqhUV0+rIm6wIDhCGo8ME\ngJ5OmCWBYwREjGdL9bnKqcaBd050dA24MlO0G5dkr5stvxgGg8FgJD9M4EgqJFH4y3ep/fzI\nFjmG6OTb1HhM8YnvgT52uYfHSs3vRl+KLryM676cmGnETWsKN9fkXuqwDbt8hZmGoqzpTubR\nVCA/bVJoQS/vwSY0n5N6ZfI+xNZLYlcLn1M8zSFNDl8PWd8DYczzAnk01IIxHq0ipgqihUim\nkghh2p52zj7qOEHOftCaMXsJphTGPNLTJh/394PoAj5G/2PGXEWviVn/ZdQpAUCzeInsXk6n\nV5ck2msAi+6EgePUfwx8g8Dr0FiCOZtBOcvK40zwxpG2UHVjBFGiZ3c3XKUCRwxDKIy9a77i\n9Yst3faBYW9Bhr4gU6abctwhgv/ZVf+3A8Ga07ePtW9amvON+2t5jiWtMBgMBmPSMIEjmZDO\n7AmoGwHIPijuf4G/4dOxzqLBS7LdAcDVD64B0CXIkl2nUYzaCsaFlCJMK6fByAa0uGBLrNJu\naVAmVX50V393QgUOyU2Dr4EUUhJCItmPIqcF/eLEDSMcxDwkK8FweDBzmg6j1PSBdO5VkITR\nzYb3sGg9t+SOUGWN2k5Rw16y96GKAzNgvkWm/Ip8AEzgSDKyUrVFWcbLPfaIOM/hyoUZAKCp\nXqpff43zw8i21mkPfRpV43cemQGQw/SVmL4SiMZLLUl+LrZH2gmPcKHdOt//6zFQGIDXjdmL\nhqO+ilIJ9tZ1/eKVswHxa1mZ5Ut3Lcm16Gf0pvvqu0PVjRH2nO5aWmK5bX2s5soMBoPBYMSE\nNSlIJqj5VIx4pMNFGELkSl3ILvf0RjSb4JIHMG918GVcocGKW7FwY8zjtTEXo1Cf2EVa14Uw\ndWMMctYldBiRcBxXzWEpYgqCDjGNw0oOY7ainAg02CydeTmgbgAAkEQt+6j1UCAgHfij9MEv\nqKMehnuov4sau+hYAwhi2IWQBz4Ra4mMuPPFO6qji9Hu31qWkzYqV+V897/Md3wUuNFjOJ0+\n4x+/kvbwZxI6ygjm/xRfPiuBrq5khVAQU1fLhFXpqJ8DJixx5dC53m//4dhnfrTnG789/MqH\nl8WxIrJD53r//Znjoak9JxsGnvjVIZdHiHGl+PD+SXlHrXdPxHLaYjAYDAZjPFgGRzJBXrn1\npdjxUTRypm4EwKH8rmRBocZFd2DJ9eToRl4Fhmzgx3OLUJRWo1pL3khNB40piqKFMznQSEgY\nkt8hWGe7NyoiZiNkx6tuidoOy8dbD2HhWgCgjnpqjPT2I4cHWnqxLMRVVFcBeDU8rIh6zoD1\nMgheMOVi7vLxf6WTgiUlab98fNNTb5yvax70+MSSHNN915WuDSmw5/SGrG982/LoF3wNF1Gt\nUZdXcHomZs0s5Xnmd090Rscr8s3zX9uJAaauABJo8BDQqBUR6oow6wZADkgE0Q68ca51fpkC\n/++F028cHq0EvNxjP3axb/fx9h88ukarUjz/bkP08X1W95tH2+7YOIMuJH1R1VIj9FqTeAGG\nwWAwGLPI1TBnmD9gSpbsAhumjGd9j5ZyUhvBG54ljoDplaDSA3lA7AVyA+qBzwKMR89U6iUa\nAhAQDYC5ADPZh1VlmKBVKmr12jv/wfX8f4dFOU730c8Dn+A/hBhvycjNt6Vj54BsmMbi1HZC\n/oB+a1DgUOejOZ79I+Yofpd0/GkYagkEqPFdbtnHYRzLkiQhP0P/L5+4gr+MwpKusCSoXI6x\nfVXBC3uaB4YjJ5b3b02078mcAtPWoHkJebpB8qLKAupMEIap/zVwNY34jYKuFFOvSd5ssoNn\newLqRoDzrdY/v9v48PaFMQuX2uTj8cKsl5dxUwwJL1JjMBgMxryACRzJBLdks3Rqd3QaMbd0\nXJ9/XsVVPyDVPxumcZjycdEd4G8i/1mgsQRUVKOqBhTT6N4KfpJOAY1mKBABQDNyS2AKPg5E\n0vkDUtsF8Hswo4hfdh2otNMYGACAat12LqvA8/qfxNaLwPGKooWamx/kCxLuZajOIdc5ufHl\nJLp1hCzCMHh7AERQpoNqenNOZYwfWSDululcAADgl0C/GDgVqnNBUzytMSQJVP9iqLoBAOCx\nSief4a75GvAzKRFOE/KDOAQkAG8CLlknfkmM6KeeBrD3gcGCmaWgnFC7DYNW+cNH1/z4hbr6\nlsGRSJpR/dlbF69elDmpmxP4ERRz4qkVL3gd6ktGvxad1PPXEWMOh5c72GLoHh5KM766YsUN\n2ZbU2RzkVNlb1y0b33O66+HtCU1jDGVDVfahc73RcbdHuP+77xi0yqUlaR/fVpFiSPp0NgaD\nwWAkBiZwJBOYXcpv+6T43jMgBB0cuGXXczXbrnBmShG37ivUeRTsXcArwVyEWTUg9ZDndNhh\n5CXvEeQ2A2ee2ghJuhBQN8bwk1SH/IbIPA4SwdlPPgfq0kETeTtyDQt/+YHUEWzZKB7Ypbjz\ny1zBoqkNLICiZLHh89+b5kWmi6YUlHXgD2tYC8ijcdUsDWgMEmloHwyfAhhzpdWVoWUr8FOU\nljBzEXXLGItgVuXY9WN0/9GlYermqd00KfG7qPeMTNxjo/7zmCXfamT28V4kb31QIVXmo3Y5\n4PxvaTlHoM6z0v4/gmMsT0pn5tbcj8XLJ3JuQabhR59b19w93NHvTDWqy3LNGtVE6y8IRL/U\n45eGAKSRJ5eKy+ES2U86IdDw8RF1Y3+z4X/2ZAy7R78/qr0H79+28J5rS8Y9ey4yaPfKx4e9\niFCRbz57WaZ8cmHBFN8HJsj1K/P31ncdOd8XEe8YcAJAv83T0m1//2TXjz63riiLSagMBoPB\nuDJM4EgyuGXX44JldG4/DXaCMY0rW4k5E0tA4NVYEJbnT/4mueOI/M2oXjal0YlAsp1K/EB9\ngMHEEOo7R2dfBvcgjCT+Zlbh4ttBE5zrCq//OlTdAAByDAk7f6T63E9BlfzTJ+TQcjPZD4Pr\n/GiDG2UGmjeAapbt+mloLwyHG9a6GkhyY/ZHp3ZBLFiN7UdosCUsqkvjykclOSxeRZciO2iM\nxKd2x2TFPSjf6ghilvnMPt6L5An/bfG3k+RCw9Z5taQ/Z7F2Sbv/B0R/MOKySR/8itN9DTMn\n5IuJCCU5ppIc0yRvLHnEJokC5S0k0rBHdGr5MpxnGoe3AwAuD6p++HaWXwr+TvtEePrNC9mp\n2k1Lc8Y5ew5iMckXfVjMagD42Jayf3nqSMSujBTt9pUFMzoqnsPvPLLqzSPtu493dA44zXpV\nc5ddCs9UHXb5fvZy/Q8fXTujI2EwGAzG/IAJHMkHmjNw7Y44XEiK7N04CsWIXxlvcOU/4pLk\nCphLUP9FOv6H0Okc9Z4hRw+34fHRVHy3Q7oQ+ZoFAOSwSo3Hucr1Ux3eXIJTo/kaMG0A0Qac\nFrg5oNpIPrDLtXHxdIC3C9RTepXneG7dY9T4vtR2GFwDoDFj9hJu4XZQjnbQwKwKrLqRzrwR\nehJmVXBVN07ldsmLIvYvwMSKDhIOkVeuzEocBKEbFEk28UtGpHPvhKkbo1GJzryFmZ+bufv6\npaEQdWMUAtEn9ar5/Jm77yxAIgC8ec4kSDLeSH87eDnpBI5rl+a+dbQ9Or65JhcA1lRmfuuB\n5b/425nB4dFEj5pSyz/duUSnmfEXRUS8cXXBjasLAOCvHzT9plPm2XKqcdDpEfQzPxgGgzF3\nEMQYaz8Mxriwj4qrGORj9AqcslF8zF8nDPEupYa3ZRarXf3UeQwL1gIA2fpirWbTkGyGSNKC\nHCjmTC23f2DkhV4GX+8UBQ4A4BRYvo0v3wZEshaqXO0Oyqumhn1g7wWtGfOWYMm6+Wa2ekV0\nFtBZwBWVrIEcWqbVqXemkFxA8unuIA4xgSMRDMrMVAGABiJdJOOLSI5JxZMYZRr4B9us8mkp\nbb3J9/9dtSjjtvVFr3x4OTRYvSDt3utGU36urclZuzizsXPY6vDmpeuLshLbQB0AAKwO+QcL\nEQ27fEzgYDAYDMYVYR8VVzFcOkgyr2jIT9lXUgVoAoq2jUSAMZNRIrDJv5eDtRUK1gIAaHQx\n76DWT3VsjCsyTnvaeHSuja1ZYGYZZl7V7RsAEBfvoGNPR2hMuGAz6CZv0Mu4GuBiKNGx4nEj\n1nrafFtnQ+NScjWoFPLrACplUraM/cKO6vVV2a8fbu3od1pMmvVV2TeuyseQh7NayS8umk3Z\nPTNV3vJJwXNpRtZXhcFgMBhXhgkcVy+oqiCxE8gXHtWDYuod7xEXER2NfNPFQsCANxhBjLyR\nQNYGpmShJZcGOqOuznElNVMeG+MKqCzAqUDyyeyacvoGY8Jg+kJc9wW68BpZL4MkgCELS7dg\n9lz9hed0gGr5JA4+LeGjuSrJKIGui9FuJxM04JgyHGpkkzU4nHeTT3UeWrYtzTtxsFlGWK8p\nSVblcXl5+vLyuduSeUNV9m9fPe/1R6YTblySrU5OUYnBYDAYCSYeC7OMJAV1qNkEfPaYIyCC\nogC11wBOoyclmpFfB5gJoATgAE3ILUWuIuQADowxZsumvMCXihs/DXyk+sZvuANTZ9mGcwYQ\nY6b6JxhUoHm1TFxfMd1msYwJYsrDVZ/hrv937vrvchsen5vqBhF1DbrqmoZcJFc7w1tAMf/+\nSOci3OJtoI2qIFBquKU3TffSJIDojLVTgamyJrIKLlkn/OOhr/zIltsK0yM/E41a5QNbr/Kk\ns5ki3ax5/O6lEVpGSY7psduqZmtIDAaDwUguWAbH1Q1nQM1aABEkN3C6OAleOuTGm5hhyWY6\n+WxkVG3CvJXBcRUvUX3qB8K7z0rtF8Dvw8xCxYY7uYVy0+/kRRwgbx2IgwAEqEbVQlCVzrLm\naF6JnJKGPgTJCwCAPJiWYcq66V6WJBo+B55eAA602WiqYF02xgVnvspgipxpGfrpS/VNXaNl\naF+8Je0jNcMcBNrEFqK2FgjA0UcuK5qyQDvZDh2MCaMz8zd9TTrwHHWNOjJixgJc93EwZU79\nmt5usu4Fbw8AAadBUy0YawHDfhs51Kj5Ap/YQRBYY0cVl6nAGC2fkxy1xvDDz177x7cu7D7e\n4RMknsMV5emP3lKZnRa7lJIxPbbU5lYVp768v+Vyj12nUS4rtdy4uoDn2KcGg8FgMCYEEzgY\nAMADl7j28phdA9VeuvAq+N2jIXMBt+QeUIZV3mJGofLebwAAkAQ471KNhB5y7w9W65CHvKdB\nGkLNbIs4xho0VIF/EEgEpQW4afd99A1KbS+BN2ifSdocruAOUCTuV44RFxo6bF//9UGfECxA\ne/Lv3p0HTf/zWJVOA8CZgdNR13np4HNg7Ro5AAtrubX3gZ4VrcwM5mzuxi+Dy0r2PtSngWF6\nORSeNup7JWjwLHnIegC8PZhxc8SBCjTzCoMoDUvgQ1DwaORQdbpp4Ll3Gho6hlVKrrIw5eHt\nCwsyDQAAnm5yXgbBDWoLmhbG4ZGScEw65Rd2VD92W1X/sCfVoFYqZuHzqN/maeiwSUSlueas\nGC4VU4UAOom6ANwAaoAMxMJpeI1PbyhE3UNup1soyNQ/ekvlrIyBwWAwGMkOEzgYswDmr8as\nJWRrA68dDZlgyh+va8b8UzcAyHs63IsEAQD8baAsm30LA1SAahqLwGGQ1PZyqLoBAODuoo5X\nsejeON1iXkH9DdRxElxDoE/HghWYWjjbIwry3DsNoerGCO393l2HnPdvLQMA6mmQ3vpvkILF\n89R6Qhxq52//N1DOO4OGuYMuBXVxyJ4g636Z9lXuJvC0gyay/ysCr+CCVpQv7Wv+xStnA5t7\n67oPnO397idXLNMfJ+vp4C1692Lezagvmv5oEw/HYWZKfJWFCeETpN++eu5vBy6LEgEAIn5k\ndcFnb12sUcVFgyCi0wADY02u/AAOol7EFYl/P9xb1/2rv53ttboBQMHjbeuLH7qhQqtmr6kM\nBoPBmBzsk4MxSyi1mF4RFhnulY7vot5GELyYVoA1N2POolka3AxDHpCie80AAIDYO/sCRxxx\ndYK3LzpMzsvoGwLVnGmROxcgkk78L7UcCAYa92D5Fq761lkcVChnW4fk45dH43Tyb6Hqxij2\nPrq0DxdvndGxMaaL5AWfzJ8qAJC3HaMEjlCGHN7fvXY+IiiIUnP9WzULw72iBQe1vYRlnwFF\nmG0ngQiAyEzB5PjpS/VvHgm2/iWi1w61Oj3Ctx6ojcflewAGILLJlZOoBTGhJiPvnuj8j+dP\nBDYFkXbubb7c4/jep1ZfbU3DGQwGgzFN2PsEY05A3RfFXd+m5iPgHASvk7rOS2/8iOreHO8c\nTzcNHqP+A2RvkFl4nMtQ1CQwsIeEWLuSEfLJT4nH33V1QpcPUfOB8JBEF3dT95lZGlEkkiTf\n/ygQp75G2QOoVz7OmEOM8+SR/OOfeuJSf3RqDwBsKpBTTCQf2YK5Hn7J5hQaHP4LDv95p9Ao\nSPYJjvcqYWDY89ZRmcbqH5zq7ByIaQQ7cYj6Y+yJFZ8RiOCpNyI1MgA4drHvVONAdJzBYDAY\njHFgGRyMOYF04E8gRr5GS8d38SWrZAr4SaDON8kWnPiRJoPLuw3USWLjz2kBFbIzCuTmlylj\n7Hp7TMJS/BmFWo/IWq/S5cOYPSfaB5Tmmo9dlJmyluWN/dJKMXTG2IoeY67A64BTj7oLh4PK\nK+SU2d0yCoheJabpYigjvsGRf71ir08KTqQl8rrFNg3kKDmW2zVKY+cwkbyweKndlmuR6V87\nSfxjxSmR8WlfeRIMDHt6htyyu+pbBpeVJcknO4PBiDd+ManWLxlzBpbBwZgD2PsCroRhSAJ1\nnI0OU/d7oeoGAICnT2p7cbxFyLkFB8pimTBqQJGb6LHMJKgvAJRTUXkNaGN0C75aIdeg/I5Y\n8YTz0WtLooNateLWdaOWCmiRdwxBS1J6LlxlIBiqZcK8FnRy/YBDyJJzpvAKXCy9a6QTOYHg\nk2QW571ib7g/0VUNxi7PGGfXZNDEuIwmHhefKELsOcw4uxgMBoPBkIUJHIzZh7yxU229jsiI\n5CdrncyRPivZm+I5rJkE1dWgLAgLcQbUrpeXA5IXXouZGwNbgVkLZm+J6D3JQFWMrpOq6S/S\nO2ed+gAAIABJREFUxofl5enfuL82xRBMvSnINHz/06szxua3uGS7zGlqA1Zck5gRMqYDmteC\nPrxvhcKMGbdcse9JbXl6ujlyPixI2GyPYX1qKAYAUXLJChkEokjyi/lXIeV5Zk6uPWqRxb88\nu4kG3yLbPvDK1LBMEMSsGPHsKV9zCmSkaA1apeyu0tz5ldXIYDAYjJlnfs2mGMkJGiwAKL9q\nZ0yPjPhtMTM1fElUrMujZjUoy0DsJ/IhZwZl3iQFRz9IwwASoBEwoattkwItq0GVSn37wduP\nAKDJwsxNSdpGYWbJqQarzEQFc+TW1WeJ65blrl6UefbyUL/Nk2vRVRWnKfjg7AsLa7mND0lH\nXoAxyRJT8/Gah0FjnKXxMiYDcmjZBsYa8LSR5ENVGmhLJyJEqpX8Nx+o/b9/PGZ1+ALBgkyD\nJSUVyBp5E8MCNJQAAEHMlflxdiUeAp9EPQAeAAWCmcOEmkCnGFS3rS/ata8lNPjRFfZPbhzm\nhB4QAADIeQa0pZiy5codx0QfAAIfKiWkARQDXA7//M0CGM9WNu7wHN6xccEzb1+MiOdn6NdW\nykswDAaDwWDEggkcjDmAxoj5VdReHxnXmjBvSWRwnByHpEt/4NOAT5t8njGB2ERCA8CItQEC\nn4+KSvk/Z/KT9ywIvUAe4EyoKgdloqtg0FiOxnIgEQDnZdPfuMCVb5E668jWERrE9FIsXjdb\nQ5JFr1GsWpgRay+Wb+QLa6mvCVw2MGViZhlw7CeeVKgyQJUx2YdSdXHa75/Y/Oqh1oYOm0rB\nLy5KvaHCzncfkjnUsGDkXw5jdg7mYK40FZaoX6QmAAmAABCgRwKzAhcmMvv1H25ZbNAo//J+\n44iT65I8/6evsUUe5G4EZQYYamJdhHrq6dKb4OgFBNBnYcWNmLl4ZBdiCUAGUReAG0CNmAEw\nC54XD2wt8/rFnXubBHFUallclPq1+2qUCvYAYTAYDMbkSLYJIWOewm14SHrrv2koZHanNnDX\nfgaUUW+6qhRQmsAv02YV9fIWAPMMEhpAvBQaALGNyIPKVZGHSi5yvgPSWL635CahB9TlqIlL\nf8FJIvrBbQWNGZQyFfsMUKi5zY/TxXeo6zTwLjSawbwAi7YBl2y1PGo95kfpkoz5jkGrvHdz\naWCT2nbKGmnQ8AVMWwkAPGp51IjkCdsLoOSMHMpXKyQYAq9IjWOpDaOaD5FNhDYeE5eDxnP4\n4A0Vd16zoL5l8OiFvg0FDbKHkesCxhA4qGUPnf/72AaAo5uOPw2Vt2PRhrFDjIjxT7Macnif\neevSycZ+p1soyDTcvWnB2sUx0zE4Dj9906Jb1xWdaRlyevwLso1VxWmsQSyDwWAwpgATOBhz\nA10Kd9u/UsN+6m0Avw8tBbjwWlDLuw9g1hZq3xUZNFeB5mrIZRVBbBpdTQxF6gPJClxY0Tt5\nTgXVjQDeS6AsBD6Ba3TuQTr3CvWO+sWipRwX7wB9zCyAqxdeicUVYOoEcaTEo426n8PUayKd\nERiMOQ8JUfZJI/iDcQ1f4BHbQx03FKjX8HPFaJmoX7ZwUqK+RAocI9Q3D/7nX07ZXf5VO6LS\nN0YQY3TY9bvpkkzDdbr4OuatBMVMJcu09jq+/PMDw67RqqUhh/d008BHry35zM3jPc2yUrVZ\nqUwBZzAYDMa0YAIHY87AcVhxzUT8CNFUAUX3UPe74O0HAOA1mL52ZFUwCSABJDugGrgYppJX\nON0BIMr2EwWyAYQKHAT+Tvlr+DswYQKHzyEd/Bl4gy/fNHCJDv6M2/Al0MTwILxq8fdT/+tA\nIQYEkpcGdiNvAE1B7NMYjDkH8nr5ViiKoGzNoVKnWCBIdpE8iMihVoEzYKlLEjXsp54L4PeA\nOYdbeJ1M63HZ80CmaS4AAAgAQiJfn/ptnu89d8LjEwHA7o5RssHJOzGRtSW6BTsAgOgjWyta\nrtAiZ8r84pWzAXUjwAt7mrfU5jHfUAaDwWDMKEzgYCQlqC/G0k+C6AXJB8oksTAkL3lOg69l\ndFWQM6J2OShmLOuEhDGTDpmRzNRNo2/VsjdU3RjF76Km93DxHQkbRlJA9rowdSMYP4lM4GAk\nF6aF4GiMDqNpUUREwRkVML1nuGAl2xHw9QEiqDLRtBoUYxf0DEvv/CRY/NhRL154n1v3IBZH\nFfTJEOsFCQESWjj2/qnOEXUDAA41a7dWumQO0sRIKhEjVYYgwkx9EHj94omG/ug4ER0828ME\nDgaDwWDMKMy9iZHM8OqkUTdAIucH4GsO5jxLdnLuAaF3cpdBQ+x3ayP4O8FzAXztQD5AJcQq\nZZ9a8sgkIHD10OB5cHTQkHzvXhpsnuExJCH+GG2AYsUZjLkKplShqSIyqC/CtHgbALmbqOs5\ncJ4H/wD4+sFxlrr+BJ7RhkTSkb+EWTsBgOiXDj4D7sj2LtFwmBojnhJVIjizdPYHFY09l7SH\nmqOSNXgTGuVzGFEX2xJ4xuoEHW6/JMln8Nii0joYDAaDwYgvLIODwUgI/jYQo1+piTx1aNg6\nmQvxwC8AMcpnDk00/D6IY/XtnAb1q0BZBL5oRzpE5Uy6sbp6pMa/gaMdAAgIgAcFD0JULokU\no9fv1UzMlpzJ5jMKBPZ6sp8EvxU4LWiLMHU98DNQfcCYuyDm74Dhc2A9S34rKk1gWoQp1XGW\nBkiggXeBxMjg4G7MfQgEP7WdlDlL8FHrSVy4efxrIxg5zJQoQoNWcAk34NBrgq9qRPDtv6Xf\nVOX8yBJHXqqo0ZhQW4TG5YAq+ZNNOZBSSNbWyO97WgkYrpw/KJEgksCjgptMkzKzXqVW8l6/\nTAphdupMy+sMBoPBuNphAgeDkQhIkMnXBQAQBwGkSeVSoaIckCOhMViBwmWB/UKYZCB5yL4P\nTVuIHwIxdP2fQ20tcDOW9uJ3SGf+AEJgvREBJNCrwekBIaz4Ao05MzWG5EWdG1h5DkOTl/Ch\nTAvqew2cY41+RAc4zpC7CbPvBSVzXbm6QFMlmCpnMNvB2ynjowwAgh18PeBTxBJSyTk4kVHx\nWIJgkqibwI2gQDTzWACQ6CYvKxdm/O/7wXofSYK/1+n/XqdfXp7+H59Zc6WzkVv2cenEM2Br\nC8ZSCrma+8c/TSSfUxgQJO9Ii1wFqvVKCx9LRglHwXOba3LfPNoWEVcr+U1L2ZOfwZhvDAwM\nfO973zt69OiJEyfsdvszzzzz8Y9/PC5X9osydbsMxhVhAgdjRonu9nHVEusZTbJG/eOCwJch\nVwg0DCABGslZJ/ceT+S5gKYt4LtMQjeQFzgTqstmUN0AoO4jIepGCGplWL03clh8ZTfZqw00\n1pDjbDANZwROjaYkMdAdwd0SVDcCiG4a2oeZt8zGgBjzF1HuaTO6yw3qbEAEknnAosYwwTtw\nmM5h+tRGFy9qSi3X1uR+cCrMNFqrVjx6y8T6K2lSuLVfoL6zYG0DRDAXYGbl+B/NEgnDvm4a\n/dhCABDIO+zrNqtyJ5jK8Q+3Vl7utZ9vDeYtqpX8lz+6NN0sb4bKYDCSl66urqeffnr58uXX\nX3/9zp07Z3s4DAYTOBgzgeQj+1FwNYDoAIURtOVoWhHTD+LqAPkUeRmDM06xAAFVEHjtFgfl\njxGGABBUxagqnsotpoBTvm8LKBQQaEmg1GHl7ZCaqCElEZwGs+4m615wja3WagowdRMoksmT\nj9wt8jtixRlJSEe/88U9zS3ddrWKry5OvXPTAq1qNl4n+Ng6BW8ApRazFlL3+chdyGF+zYyO\nK+5842PLlpak7dzb3Dng0msUtWWWT91UmWuZcLkHImZWQWbVBA93izaKEuUJJLdo0ysm1IHL\noFX+5PPrdx/vONkwYHf7i7MMt6wrykxhLWAZjHnI4sWLBwYGAGD37t1M4GDMBZjAwYg3kpd6\nXwRhbN1GsIP9OHkuY+adV7XGoSwCz9no9iWojuwpMCViVLjgnHERVmqx9i5wD4ImFS2loGRl\n2DFQGDH9JpB8IA4DbwROPdsDmjxSjNYMJAAJMJlKfsbc5N0THT/662n/WNHZsYt9rx1u+89/\nWJNrSbjNijoHeENk0hMAKNNAlQEA3Kp7xbd+BN6wA7ilN4NpxtpXzQwch7euK7p1XZFfkJSK\nGX+wizH+ioVYf91yIOL1K/KvX5Efp0ExGIw5CsfNmbdNBgMAmMDBiDvkOBVUNwL4B8BRD8Z4\n++fPJOS8DANHyNsPvAb1hZi+Hvhp5NaiCvWbyH0kaDWKPKqrQLUgDmNVZoK/WyauyIzDxSeF\nPg+GosoTANCQj1nViR5M8sKpgJvlrPipozDLx3k9UzfmAUMO73+/WOcPt9Tps7r/+8W6Hz5S\nQ4PN4LaBPh0tJcDNvDku8ph+A/W9GiarcVq0XD/6tTmHv/XfpLpXoeci+VyYkielLXIdPiS+\n/BJqdIrSJZrt96Fu7rbiaum2v7inuaXHrlMrqhek3X1tgjJlJls2yWAwGAzG3IG9bjLijadV\nNkyey5g8Agf17qX+AyNfgn+YPL1kO8cV3w8q+caBE4JPRcP1IPSCZAdUA58OXFS+rs8Fgg90\nk/NiRM0i8lyKNNtDJeqWTH20UwKzV1L34UgbDuQxjzluXC2gfhHZDgNFmc4YJ5oez5jLHD7X\n6/HJdMew2OqEN15Bv3N025jFLf8YWuIh4I6POg+y7xHt+9A3BACSyijqCxSKkJcbjZFbdd/I\nl953X3T//v8F9oitl3xH3zV88Yd8VsGMj3PyvHao9acv1Ytj/VZPNPS/caTtvz67NidNd+RC\n38v7W9r7HEadauXCjHs2l8RX+FBwKlGU6eeq4CZkMjpxBFF6aV/LntNdvUPurDTt1uV5t6wt\n4jlm3cVgXBX09PS0t8t5qwMAgCQxh1HGFGECByPexEphlWTeluYo3j7qPzi2MfamJTip+x0s\nvHt6l0ZQZAHIZEdTZ7107EWwdQEAqPTckhtx0daJ1phwGjTfSM7D4OsYjSjS0bAW+IR7NygN\nXNVDUtPfwT7in0+gTsOSm8AYOn8g6jtBfcfA0w9KA5rLMPdaULCilfmCMgUtN9DAO0D+YFBX\nhuYrtntgJAEDwzJP+JWpfV8trYeQHzjYe6T9v+Sv/wZow+TaEcdPjOcEVvRCOxnzAILNhvxS\nBwLPc2FuEdJAt/tvT0ecTHar+68/M3zhP+I4oLjQZ3X/7OUzAXUjEPzpzvrCLMPOvc2joQHX\nhTbrO8c7fvzYOospbv6dGt7kFZ1RmRyoietniscnPvGrgxfaRrMahxze863WfXXd3//0agXP\nMt4ZjLmIKIp2uz2waTKZplOf8tnPfnbXrl3xGBeDEQYTOBjxRmEGwSYXTxqjRLI3yKbokrMF\nJT9w8XcSoZYj0r7fBbd9TunYi2jt4tY9ONFL8EY0bQXJA6IdeD1ws6cX6LK46k+Cq488g6gy\ngS4TuLDnDDW+QENjtn9eK/UepaHzXOUnQRWjtEEGAnABKACS0KLiasCwCDX54DhD/iHgdagt\nBG3xbI+JER9SjTJ/dHdnyyXuCR6paR9XNdo650zL0NNvXrjYZiOgslzzQ9srakonZFc5PoI0\nRGHKyih+6uEh7Pr+uoMgynSNFS6dJpcDdRPtq5IYDpztjagDGuHYpf6jF/sigt2Drt+9dv5r\n9y2L1915VBmVmU5hQKLR7xiHCr1ivDaxwy7fwLA3J02nUU20NOmlfc0BdSPAqcaB1w+33bqu\naGojZzAYM0pdXV1tbW3oZnX11AuQX3jhheHh4Vh7c3Nzp3xlxlUOEzgYcQb1lSRXpYL6xYkf\nzBSRbXQKACSB5I2/wEEkHX9RJtz4IVVuxZS86F0x4TTAzYUmfAi6TNTJOICQ9WJQ3Qjgd1DH\ne7hgxwSuLJLUBNQ61nZXh1wFYMZ0x8uIOwoDpKxhiebzj9WLMlQKzhc+916gj/GGahvNKdt9\nvOOHfz4ZCNe3DD7xq4NfumvJTWsKpzkeArd8nLwAUqgBMznklHcAICKHda4JHIN2j2yc5Lre\nAsD++h6ieKbGKDltiirPL3klEjhUKDg1xugse6nd9j+76s+1WgEAEa5blvfoLZVpckJYBB+e\n6YkVZwIHgzE3KS8v37t3b2CzpKRkOlfjeT41dRql3wxGDJjAwYg32lIwrgD78WASBHJoXAWa\n6b7IJg5ljGQTVAA/A13u7L3girJlHaHnIkxK4Jj72GQsSAGArJcm8mZO0mmg/pCAi6STyNUA\nJtxOlcG4KrGYNI/dXhXqDQEANG4jJ69f/PnLZ6J3/urv5zbX5Oo0M/ceEvZQwZQYxr0cj6a0\nGRvDFEkzxpKqUTbB0O0TPH4h3hakqLySYt7YOfzlXxzw+kdtWYjg3RMdF9qsv/jSNVdM5bC7\n5AtXbc5J9GphMBiJRK/Xb9y4cbZHwWBcASZwMOIPmteArozcjSDYQWFCbRkok0mgRdNC6t0D\nFGmkh6aFgPHvC0ByXm6jCMljXDJBRPllSRA9cMX1RxoKVzfGwtJF5JnAwWAkiJvWFC4qTPnL\n+43NXXaNiq9ekGZI6YAeGQljxGT0fKvV4ZapInF7hfqWwdWLpvXHy6EeQOaxwKEuQuBQLlnr\neenX5I98qCqrV6NmznkArV2c+au/c9FVKjlp2q5BmRxDo06ZmAYrETzz9qWAuhGgo9/52qHW\nO6+5gsVsRoq2c0Dm/5KZMud+HAwGY3xeeeUVn89XV1cHAEeOHNFoNABw5513sg6yjFmBCRyM\nmUFpQWUc6qtnB6UJc7ZT15thGocmC7O3zMTd0JABvEK2OBxS5l39oSpGgxh1ygSyq4dixN0A\nXubHwWAkjJIc0z9/LFiGTVaD1H8RxHAVQ5eKCzYAgNMj93ADAACHO+auCcJjKoe9EkUUqqCC\ny4k4kjNbtPd90fX8T0AIjpPLzNN+9PPTHMNMkJmi/dxti3+2K8xnNN2s+fyO6n99+ogkRSZx\nXLdsdj4szrQMysbrmwevKHBsrc071TgQHd+2Yn7lLTIYVwEPPvigzTZaBvjkk08++eSTAOB2\nu0eUjinjF1nTasZUYAIHgyEDplSjLo8GT4C3H3gN6IswZclEe5pMFqUGS9bRpb2RcVMW5lTO\nyB1nD7QsoZ4DEFVGjpaaK54bq/gcAMYsORgMxiyAKQXcxsfo5F/J1jkaya7iau4CpRYAciwx\nF+RzY++a+M1VfJlfbBdpVABFUCn5fB5lKg1Vq7Yqiiu9H7wsdjajVq8oW6LaeAsq49z6NF7c\nsrZocVHqi3uam7qGDVrlkgVpH722RKtWfPqmRb997XyoxrGwIOWRGxfNyiD9ovyzNxAXJXrz\nSFtd86DbKxRlGW9bXxTo9rJ9Vf6ZlqE3j7aFnrhjY/HG6uwZHTODwYg7VmuMUmsGYzZgAgeD\nEQNV6gylbETDrbxH8rup5WgggqkF3DWfjug/Mh/QZmLhTdT2JkjBlVtMXYQ5G654KqI+hsSh\nAJgL1qrzCBJg+Di5mkCwgzIVDYvBkDwmwYzZAC0luPVr4BwktxWNmaA2BnYtyDYuLEiJ7pdR\nkmNaWDDx3kmxbw0KFV9MkE/kQVAgqiGGHSYAcBm52rs/N/2bJoaSHNMT90aKv3dvKllenv7K\nh5dbex2pBvWKivRNS3Kee+fS/vqefps7x6K7YWXBHRuLE9NptTjLePayTG7dgmwTAAzavd/8\n7eGmrlEP2g/P9Oza3/L1+2rWV2UDACJ+5Z6l19Xm7jnd1Wd1Z6XqtizPrS6ec34oDAaDwUgu\n5t30iTGPEQWQRFDOx0oEXslt/DRVboPeRhC8kJqPedUzlTAy22DGcjQtoP5T4BkApR7MZWgu\nm9iZmQBaiG6agIXjzGcYk0byUvdfwTfmayA6ydMOribMvJl9nxnjgqC3oF6mOPGbD9T+y++P\ntPY6ApG8dP23HqjF+LX9QFAgzq1OKDNHSY7pS3ctGfl62OX7wk/3dw44RzYv9zh+8+q5g+d6\nfvCZtQp+xv9g79q04OwzkQKHRsXfvLYQAH6260xA3RjB7RV++L+n/vD1NLN+NHFmeXn68vIY\n/q8MBoPBYEweJnAwkgCppV5471nqbgZJwrRsxaZ7uKoN82+uhZZisBTP9igSgjoV8zZP/jQO\n+VqS6oECb8wImI/ctLqUMSIg25GguhHA1QDOi6BfOBsjYiQ9OWm6Xz6+6Z0T7RdabRLRwoKU\nbcvzlIr5qeEmmOffbQyoGwHqmgbfPtb+kdUF0ccLotTW7xy0e7NStHnp+ml+jl6zJOex26ue\nev2C2zealJeVqv3qPTVZqVq3Vzhwtjv6FJdH2F/fPf0OwQwGg8FgyMIEDkZMqP0c9DQBImSX\nYu6sTWzE+r3Cy08GRzXY5d/1E76/XXHtfbM1JMbsoUduNdAggANAAZgKwPz2442rUTZMrkZk\nAgdjqih43L6yYPtKmSk3YzocPt8rGz90rida4DjVPPjH3Zd6baN5cGU5pk9uX1iQrp/OAHZs\nKN60NOd040D/sCcvXb+8PF2t5AFgYNgrxDAI7LVGJeIxGAwGgxEnmMDBkMNpld74ObUF2/5h\n8TLuxs+BJuEJwJIo7v5DdFj8cBdfez2akrZRS7yh/kvUcwY8NtCnc/mrwDCP26YioAWA/ehn\nDDHG3CNWnMFgzB4Ol0wLXgCwR7Xmvdhh+/FLdaFtWRq6hr/3vyf/45FVZt20nFbTjOrNUW1c\njDolYrSjNACAaUq367W6X9rX0tw1rFMrqopTb11frGJJQAwGg8GIggkcDBmk135KHedDI9Ry\nUnrzl9ztX03wSKjnMjltMjskUbp8hl+yKcHjmYuQJJ14njqOBQJi4/tc5S1Ycu0M3MsDQi9I\nbuAMoMgCZA+Q+YjCCD6PfJzBYMwxstO0Qw5vdDwnLTK77e+HW8Wo/rIOt3/3iY67NlyhpesU\nMOtVlYWp0RakPIdrKictwe+t6/rhn095/aO92/fVd//9YOsPHl2TmaKNw1gZDAaDMY9g4jcj\nEuq7HKFujMabT4BNPhX2SlcUqG+/1Pg76fyPpcanqP8gkDjRU/1yE60RfFHryZIodTQIdful\n9ksgyC9qJTPyub7UtCdU3QAAkETp7Cs0dDnO9/c1kP01ch0kzyly7SfHGyDI1Fczkh35OhQC\n1M9OH0oGgzEO16/Ml43fEBVv6rbLHhkrPn3+8Y5qvSZSB//E9RV5kyyKsTl9//WX0wF1Y4SO\nfudPdtZNd4gMBoPBmHewBVhGFIMdsfbQQDuaJ7nwIvmklmfB0ze66e2j3j6yN3DF901k/R9T\ncwBQdm6PlrzQTbH1vO+ln0k9rSObnCVHteNzfGlkg71JIDqBCBSzbcvv6JXOvUqDTSB40JiN\npVswrzZ0P7UfkTmLiNqPYGpR3Ibh7yD38bCI5CLXfjRsB262v0WM+GKqBW9XpBNHyirQMlNA\nRtzot3lUSm5qpQqMUG5eU9jQMfzaodZAhOfwoe0Ll5ZE1fHJi+QzSGmu6TdfufaPb1883Tjo\n8vpLckz3bC6dQs+UQ+d63V4hOn70Qv+wy8d+ixgMBoMRChM4GFHwsd8VFJN+jaCBo0F1I4C7\nk4ZOYtpKAADBR211YOsBnRlzK8GQFnogGlO5ihXSxaMRF8D0fK6wMrApDXR5fv9voXn10kCX\n5w//rv3cf3I5k8+8dZwj634QHAAAvA5T1oFxyaQvEg9osFk68AuQRl/syNZBx59BaytXdXvw\nGNeg/MnOGPGpjcR3SS4qkq8RNTIqEtmHyOfl0rIgfm0gGQkCecy8FVwN5GoEwQHKFDQsBnXO\nbA+LMR8QJXp5f8tz7zQMu3wAkJ+hf/TmyrWLs2Z7XEkMIn7priXbluftP9PTZ3XnWnRbl+cV\nZckUlC3INp5sGoiOl2RPtPrsVOPAxXYbEZXnm2vLxtcpaKTTWbpZ8+W7l07w+qE0dNheO9TW\nOeBMNar9fkn+HkT9Ng8TOBgMBoMRChM4GJFgbjlwPEhRVSQKFWaXTfZq5JDvyAD2RkhbSR1n\npQ+eAsfYK5dCxS2/DZfdFHbbWx4TXvyRdDnE8TQ9X3n3V4HjAxFh/ysyrgGC37/3JfU9X57c\niG1HaWhfcFN00cA7INgxdf3krhMPqP6lgLoRDDbvocLVaBybcCp1IMgUYIMqru1FRDknFLm4\n/9R+967fSgPdAIA6g+aG+9Sbd4T+sBhzCxLB7wSlATC8YlFXhrpJ/70zGLFo6ra/V9d14tJA\n/7BHxNHMvPY+578+ffSJe2uuXyFfZ8GYINUL0qoXpI1/zK1rCutaBiNsOAxa5bbaPAAYGPZc\nbLd5/GJhpqE0xxRxrs3p+/5zJ45fCjaQrim1fOP+2jSjOvxAAqGVhCYgJ6ASuAxUVgJO2ibj\n2d2Xntl9SYpyDIkmRa++4jEMBiNJ8Qvy4iaDMT5M4GBEoTPjylvp8K6IMLfuLlBpJn01Ud5E\ng0Q3OgakN38aNjkXfNLhFzhdClYE1QTUGpUf/7bUcFxqvwCiwGUv4CrXRUyYxQ55GUVqb5jc\naCUfWQ/KxIePgqkG+Gn10ps0XjvZ2mXiRNB7HsYEDsyqpJYPo4/CrMXxHAxy8unN4bNi34E3\nXM//JLBJLod712/F3nbdff8Uz8Ew4oJviDp2k70RSALkMbUac64DBWu7y4g/Lx9sfeVQq0QE\nAFqNQqtR6HTK/gEXSQQAv3n13JbaPJ5j2V4zS0We+fEd1U+/c6nfNvq5XJpj+tQNFSat8k+7\nL+36sCUwl6gtS//HHVUWU/AT/z+ePxmqbgDAqcaB7z174r8+uzY0SL7jIHYGNkDsIKkX1RsA\nJ2FRfPby0B/eujiRIxcXpaaZmMDBYDAYjDCYwMGQgVt/N5nSpQMvgNMKAGBI4zbcg5XXTOFS\nqDSRL9JEHQBQlSKd3yObekD1b4cKHKNDKluOWiU1H4C2PdLAacyrwZJ1wdl1rDqIydZH+HqA\nZAp9gSTwdkHYgrYIwI2k4M4UQkyDVfK7AzfmKraLvechvFAFMxdhbm30iVOHTwepLTrQXG0/\nAAAgAElEQVSMfEZwQxTcrzwVfYzvwJvq6+7kswriOR7GNPFZpYtPg+gezSMnkQZPkbONq/gk\ncCzfmxFPmrrtuw5GeB6TSsmbjGqbzQMAVofvco+9JCprgBF3akosPyxMbe1zDDm8mSnagnQ9\nIj7/XsNf9zSFHnaiof/f/3T8R59dN6I6tfc5j12MKjUFON000NQ1HPzBSX1BdSMA+cl/DlWr\nJz7I905EXUQOg1b5xTurJ35ZBoPBYFwlMIGDIQti9XV89XVgHwCOA33q1K+UUg1OuXYe5mo4\n96bsGTQU7XJK0qE/Ucuh0S1bJ3WdxZbD3ObPjziG8PllUqtM5xcuv3xyo42qB5HZJXWS1Ajk\nBEBAM/ILAafx/RkHjRk4heyQUB9S/Kw28pu+Il18i3rqwW1DQyYWrsHiDfE1v0BNFQldkeoP\nZwBVaWBL7G4l57DMyURiQx0TOOYU1LMfRDdESHTeQeo/jplrY5zEYEyFQzJzYwQArVZpG0sl\n8MUwWWDEHaWCC61AEUTplQMyn9HN3fYTDf0rKzIAoLXXEetqrT2OgMBBYow+a2JfwJJjIvTZ\n5MX9kf6yjZ3Deo2yekHqA9vKUw0sfYPBYDAYkTCBgzEuxigb9kmC5ipwd9FgaAMOxPQ1aCwj\n/l35c3hlRIDaTwXVjUCwv5HO78aqmwBAueF24di75HWFHaFUKa+9a3LDVcX+/6osAEDiBZCa\nA0MAspJwCPllwGVP7kYTgVdh7jJqjzRYBZUes8NNT5Varup2CHEejT+cCfVbyHMChJG5CoKy\nADU1Ya1wBD/EeI0lv28Gx8aYPOSI0UXY0QJM4GDEFatD/s+f53CkRZaCx/yMxBYAMsboGXK7\nPPLKfnOXfUTgUCs52QMAQK0KKReVzX8EAJAAxIm/cJr1ke8AI6QY1N9+aOUEL8JgMBiMq5aY\nH1pJwXe+8x1EzM6OnFvabLbHHnssOztbo9EsX758586dszK8OEMkNJ31HXrbf/YIOYfBdpla\n91Lzu9R/Dijhzd8mA2Zv44ofwLSVaCxHyypuwScwcxMAYE6F/PE5CyMi1HZC9shAHFMzNZ/+\ndy43mE3AZRZoHvn/uMxJZg0oTKCT67qiyQdVBpAbpBaZYUjnZqj/Hld9B1pKw0IqA7fiQVBO\n2rMtDvApqL8OTTvQsB1Nd6BuLXBhw+Ay8oDjZRfp+Jz4NaxlxIUYUxEaJ4mJwZgSJp1S9gEp\nSqMfXVuX5xu08nNaxkwzjvUJN7ZrUWGqWiljFK1UcIuLggmMyMVw8EH1pJbT1lfJLxhsqGbd\ndhgMBoNxZZI4g+PcuXPf/e53s7IiP/AkSbr55ptPnz793e9+t7S09He/+93dd9+9c+fOHTt2\nzMo444LY2ex69sdi22irTlQp1YuyVAtG6xTImMtVfwx0k+4tnzh0eajLi4hhxQY88w4Ntoct\n+StUuPKOyNM9coUPAOQOtvDg8sq0n/+R1NtKQ71oTueyioCbin6H6dup7w1wtwRDmgLMuBEA\ngAbkhQzyAjkmZaI2UZRabv1j1FVHA40geNCUiwWrZ0fdCICqWI2EUWdQrd7qO/hWRJzPK1GU\ny7SSZcwiqE4nv0zaOWrm8GOEkZysKEvffVLGVcHjFgBg09Kcz99elfBBMUbJStWmGtRDDhk/\nrEUFKSNf6DWKT1xf/tvXIutA799SZtaHfBzw+eC/CBBVbcQXTmpIayozty3P2308rFg1P0P/\n4A3yiyIMBoPBYISSrAKHJEmf+tSnHn744UuXLtXX14fu2rlz5/79+59++umHHnoIALZv3758\n+fKvfvWryStwkMvu+Nk3yW4NRnx+z+l2VCmUeSkAAPZO6fQz3JovAiZVM05eyd3ydenwi3Rx\nL0gSAGB2Oa5/AC1RaRdas+wFUJsSvo1cVhFkTS9ZgNNg1g7wdoK3G4hAnQWaQP/CqNa5QWZu\n3RsxZynmLJ2x608RsnVS/as0eBkkCdMKseomTCvU3v0Y+Ly+4x8EDlMUL9I99PWpiU2MGSR9\nBThaIoPIYfryWRgMY16zKN+8tSb3nVNhGodBrdhek1tbZllYkBLrREYCQMSPbSn7+StnIuK1\nZelVxcHsjHs2l6abNU+9caFnyA0A6WbNw9sXRjb3RS2qlpP/FJA/GORzUBkmTBBd2SHqa/ct\nW1OZ9eqhy+19TotJs6Yy86PXlshmkTAYDAaDEUGyChxPPvnk5cuXX3/99bvuijRZ2LVrl0aj\nue+++0Y2eZ7/xCc+8cQTT5w+fXrp0jk3S5wIvkNvh6obwfjFnlGBAwCcvTTUhGmT9NScCUii\nvuNgayDBieo0zFwJhijBgkSyNoO7H5QGbu2dsPEBsPWCPgVU8gmuWLCcLkdZUQBg4Yq4D38U\ndS6oc6PuF6uDJgJeXQXk1HFa2v9boNGVOuqsp66z3OoHsHiN7uF/Vm+9S2g+Bz4vn1+qWFgb\nX7tTRlxA80LI3UrdHwRdbHkt5t8ImsxZHRdjfvLx60qXFqe+e7qre8ht0imXFKdtX56nUoTp\nnkQk0XgVE4zpIYHYA2QHUAJnAS7oM7p9Zb6Cxz++fXHELYXncPuqgk9si3yd2FKbt6U2z+rw\nSURpxhjunnwOcmkgtpNkB1Qjnw5csNNWXdPg029daGgfBoTyPPPDN1ZUF6fFGu61NTnX1uRM\n5z/MYDAYjKuTpBQ4mpqavvWtbz3zzDNms8zC/pkzZ8rLy9Xq4KfvkiVLAKC+vj5JBQ6xvUk+\nbneHLYU4e2HWBQ7RK118BpxdI1vk7KTBeszZiHnXBY+xd0gXXgTXqN068Sos2oL5G8e5KuYt\nxZIN1LQ/LJhZgQu3xHn844MWQB2QKzLOZQFcTZ01JVE6+ueAujEKSdLxF/j8GlBo+IJyvmC2\nfxUZVwIz1qC5kuyN4B8GVSqay4Gf1eonxrxm6YK0pQvkZ7Pn2qzPvtvQ2DksERRlGu69tmRF\nOSuViiuSjXzHgQJVaQiKAlQuCRixba3Nu3ZpTke/0+0TCzIMek3Ml8MUw5U+7FANitJomer1\nw20/fuF0YPN008CXf37giXtrItNAGAwGg8GYHkkpcHzmM5+54YYb7rzzTtm9AwMDJSUloZG0\ntLSR+PiXffzxxzs6ohuUAgAIggAATqdzKsOdPrHS+xHD1sa52Tdpo873A+pGMNi1D83lYMgH\nAPA7pbqnQXAHd4s+anoDlHrMqh3nytyqj1F+DTUfAEcf6FIxrwaL1yQ8NYBDfjmJJ4BCfhPQ\ngvzVVUBOQ63yrih+N/U2YG51wkfEmCoqE1rG+7tjMGaaffXdP34pWGfa2DX8vT+ffHBb+e3r\nmDNxvBDIdxgotPcqgdBKoEblokBIwXNFWTPgJAUAAG6v8Ku/n42O/+KVs5uW5rDaEwaDwWDE\nkTktcIiiaLfbA5smk4njuN/85jdHjx49e1bmk3J88EqT4fT0dJcranEeAMYEDm6WfAQUxYt8\nh96WiaeMVkwQACJiakn0MQmGBuV/LjR0Bg35AEA9x8PUjcAB7fvGFzgAAHMWY87i6Q9yWqAB\nFRtA6iayA3DIpQBmXPmseYY3ttLnmyURkMFgJCGCKP3uzYvR8effb7yuJseku5oy42YOsStc\n3QjEW0C5UK6vd/ypbxmSbUbrcPvPXh6qLWMJOwwGQwZBjDItZjAmwJwWOOrq6mpra0M3s7Oz\nn3jiiX/+53/W6/VWqxUABEEgIqvVqlKpdDodAFgslsHBwdDrjGyO5HGMw7e+9a1YuxwOx+9/\n/3utdnbyt5Wrt3Hv7ZR6w7NLENWLRstTEQDz182JLipyfRkAAHxjQpWzR/4AZy+QBJgUVpQc\ncLlXdZm4Pvaf0ji7GAwGI5ymbvuwy0dRk2y/IJ25bF1XyRxh4gBJMT6XyQ/kAUzEi43L44+1\n6/VDrWolH9pulsFgMBiM6TCnBY7y8vK9e/cGNktKSi5evGiz2b75zW9+85vfDD0yNTX1oYce\nevrppwGgqqrqhRde8Hg8Go1mZO/p06cBoLo6WTPnUaky/OMP3C/+0n9y30iES0vX1BTxJhEA\nQGXABVsxb/VsDjGA0iCvcSgNo1/E6vOC3NyzoiTwD4IwDLwBVJZArTIDzbmYmk9D7ZE7DBmY\nXjobI2IwGEmJ2ytAjBQC2QV/xlQYr71agmpDstNiWXTD+6e63j/Vdf2K/MfvXqrg59prAIPB\nYDCSjzktcOj1+o0bw7wny8rK3nvvvdDIV77ylebm5p07d2ZnZ49E7rjjjmefffb5559/5JFH\nAEAUxWeeeaa0tDRJHUZH4MwW/Se/JQ0PSb3taEzhM3KB40HwgCSAynDl80cQOkhoAckOqAE+\nC1UVcX+5wdRF1CvX7iRtrLTEVAjdx2TONBeGveW6hkESwTB7Szq+Xhp8F3x9o5vKVEy7DtR5\nszaeqeF3UccBcHYDx4MxH3PXxMuohVv7iLjn5+AM8bXRmrn1jyRJDs58QXSD3wEqM3Ask5+R\nlGSnxpz35liY5W18QM5Csjs4E+DUHx2tvY72PmeKQVWSY9KorvAuUZFvLskxNXXJmTcBAMDb\nx9qzUrUP3lAR6wBZhl0+BDTqZt+AjMFgMBhzhzktcERjMBg2b94cGklNTe3o6AgN3nHHHevX\nr//iF79os9lKSkp+//vf19fX79y5M8FDnQk4UypnCpnzKzQTP5e8x0FoHdvwgGQlsR01mwBj\nNHubEpi7mext4A6rQ8GstWAoHP06s4Y6PowsVEGeK9o2OrTzH0r7/wz2AQAAfQq39i5ccl1i\nioSDCMPU+xJIvmDEP0S9L2P2vaC0JHQk04CsTXT2+aDjSV89dR7iljwM2nj8F0xZ/Ef+DzXu\np4FmIMK0QizbOKlfSMa08PRK3bvBNZpEg6aFmLUFlDNlEMhgzBBZqdrq4tT6lqFAZKRcJT9D\nv6ggZZwTGZOAswCfDWJ3eBRROUVPq84B109erDvR0D+ymWZU/8Oti69bFmysTkQ9Q26Vkg90\nk0XE//Px5f/y1JGO/pg+Ta8dap24wPHO8Y6n37zQM+QGgOw03SM3LgwdAIPBYDCuZpJM4JgI\nHMe9+uqr3/jGN77//e/bbLbKysoXXnhhx44dsz2uWUXsDaobASQn+c6helk8b6TQcos/Rb1H\nwNZAfgdq0iFjBZoWBA/gFNzST1LTG9RzEoAAAHSZWHYrmIsAgI69Ju19Nniw0yq98zsc7uM2\n3HvlWws+38E3xcsXQBL5/DLluo+gJuba4PiQ/WSYujEaFWn4GFpumNo1E43kp/N/ifRz9QxJ\nF17klj0an1vwSqzYjLA5PldjTBzvgNTyXOivKA1fIHc3V/owcBPVK2mgkRrfJ0c3KrSQXsaV\nXw9KtmDOmAX+aUf19//3ZGOXfUTGRoCcNN0Tdy3l5lzRYhKDqhUgNJC/EUAAAODMqKwCbipi\nt9srfO1XB3utwQ+XQbv3+8+d0Kj4dYuzhP+fvfsMjOO87oX/P8/M9l30DpAEwQJ2SiRFUhKL\nJKpLVrNk2bIVK5Zr4sS5SZx6c8ubvI5fJzc3sZ3IiWPHiS3JVbIkq1OdIiWKoiiKvYEFANHr\nLrBlZs77AX13llgsFv38PnHOtIcgCOyceZ5zTH5yV83jr54K9sYAlOR5v3Db8q2rSwBUFPq+\n/0fbXn6/7ldvnjnfZLOUta070hM2vMk71A567JVTP3rx+OBmQ1vP3z72QUtn+L7t9qXWW7vC\n4ahZmudVSr6phBBi9pvxCY6dO3cmBnNych555JFHHnlk8sczPbER37q1n3kRyGiCAwBpVLwZ\nxZuTfo5w+Kj647T4dvS0wOmDa+A1XSxsvfPLxMP5/Wdx2U3w5YCZT7zNJ/dwdzP582nhelpx\nXV8PXau5tuf7/9Nq658YEvvwrchbT3kf+u/a/LHNd+0XbbKPR5JUSJ1+uP00onbFULrOo7cN\nHikFOoNxy54RCTgiKIVYJ7d9QAWbU7rCiZes4y/0/xlAZ61Z+7529Vfhm3stgcRUywu4vvXw\nxt1Hmk7WdRoWLyoNbF1Vomuy2C2zFPSlpC8F9wI6KP01HS+/Xzc8uzHo0Z0nr1xR/E9PfPTi\nexcGgw1tPX/94/f/8N41N2+cB0DX1C0b551r7LZNcGiKnCn0i+3qiT76ysnE+H+8cPxQTXso\nHJtX5PvYlQuqSrMA7DrU8G+/OdrQ1gPA49Tvv3bRJ66pku8uIYSY3WZ8gkOkJmIf5oR5CpNG\ncyEwoqQFN9YgZjdOy+T647ToCuulb/P5g/0Hd7fwxeN0eq+67Y+hO3sf+z+D2Y3+A7raeh/9\nlv9Pvgdtgr/J2QJbUNPsv1KkM9kejnRQ2gkOM4JoBxwB6GnOjhHjxwMrU6A0+LzkGHhWiZ6A\nuRqab5Tzg43WiZfig5Fu69CTalOGZvcIMRZEdPXK4qtXFk/1QOaAcfdMOVHbYRs/Vd9Vc7F7\neHZj0A+eP3bDhgptYPbE+iWFT7xVk3jY2kX5qRQZPXquI2bYdI40TGvPkQYAB8+0vrD3wudv\nXRbwOv/+5x8OHtAbNX704vELzcE//WSm3+sIIYSYTqbZU5mYIMk+09B0KppgJM+2GFE+8fZg\ndmMQN57iQy9z6Wrzgs37HKu1wag5rC9eO+aROIsQsZvz4hrRs5A7zvOR33D7WbAJb4Facj3N\n2zDZ5UKScSRNQJBjtAdgW7EurtvJncf6N30VqvwmeOSBZCqwCQBKUXZgRFVXFeW2pyn/nksv\nVOGGw2CbxwNuPgEjAj2TRXmEEDNaKGz84o3TR891RGLmorKse5OsAelz6GyrbbwzFD3X2N03\npQLAhurCK1cU7zky4p2Ex6l/8fblqQwpHDVHPca0+PvPHfO7bSaqvLK/7r7tVYODEUIIMftI\ngmNOIL2CY6dtdujzJn0sSVFe8h4l+RW8/ynbPVzzvuVO+pjNbUkWm1x6JIG1HDoaX4aDNAqs\nH7py01Fr7w+GHhRDzdaBx6mzVq26O4072uMg81kgCOhALqkFqf+HpZxFrDlhJuSMPPnwFdmd\ncUlmxDr1Y0SHzQoJ1VqnfqyWPAR3wZivJsaHXAVshOBx2/SsMUPcc4j86+3O68eR7iQ7LERD\nkuAQQvQ529D9Z99/t627f2blkXPtL753Ycc6+9/US8qzDdO+WwuASGwoqUqEv3pw/a/frnly\n19nmjl63U7tsccEXb1teUZhS8n1+cUqd4yyLu3rs35ocPN0mCQ4hhJjFJMExN6hccq7g6NH+\nup59tEJyVk/dmBJkFVDVOj6zPy5MZdVUVMlh+6cy7u0ib9JPKuRLq6+Enk2Fd3Lba4i1DERy\nKO8aOIce5q1DTya+Buezu1B5FfyZmNfAtWwdG/bv1c5mHWkbgNTWhji8tOg2PvnUiEEqh1p6\ndxpzTLj1/RHZjT5WlBvfpgV3jvVqE84yEO2Be/Z+fs2/AqFzpCdZRR9tsI8PIFcgSc9IHS7p\nwyKE6PcPvzw4mN3oEzWsNz+6WJTjbuoIxx386R2Lk1XQ0DWaNzJ5oWt077aqe7dVhaOmy6Fo\nLAVlF5YEVlflfXSmLfVT4sRMmylsQgghZg1JcMwZjqWkFbNxFlYQ5CatGHrFVI8pnrrpy9bz\n/8JnDwxGqHyZuvWrAMGbC9is2iVfnqpYRFl53BX/cYfcXm3R6jSH4iqh0vsRbYXRBT0ARz5o\n2Ee3njaEWmzOYubmE5SBBEeYreMjslEAEGHrGKl1KV6CStaTr4TPv8bBBiiNsubTguvgzh39\nzEShWtswh85PjwU5/bizjg8+wS1nwBYcHlq8XS29Hlr65fSmJ/JXofQm9O61381J5m9bFh95\n1Tq0E93NIMCpI8sNbei7mkpXz76vlRAiPY3tvcfO25TbCPUav3VD9btHG/efHGgTm+X68u0r\nNq8oNi1eWBKoaYh/G3HD+gq/xwGgqyf6wt4L5xqDPre+uip/y6oSt3P0qqKJ/vKBdX/z6P5D\nNaPkOJQiy7JJ51YWSyZXiJlB0pEiPZLgmEtUNjnHXpBiMrl86q6vc/1xXDwFtlBcRfNW9M04\noMWb+Gz85A4AtHgzlOa596s9//kNmMbQDqXcd36R3GnVmxi4BJyFcNr0lWAj/uXVECNJPdcx\n4WbA7mc6twExIOWn0EA5rfxMBnIQyZ6ZE0s5GE0cOw8rCOUlRwX0svHfPEXcdtZ667swY/3b\nsV4++oLVWqO2fGW6FEbJHMpdy1yPaJ3NPt0+h2W98gjXvN+/wUA4hoiBAj90DQAFStTKud1L\nWwgxTGtX0l9zlmV98wubaptD55uCuQFXVWnA5dAAaIr+90Mbvvn4gSPn2gcPvu7yst+5cyWA\n9443f/PxD7p7+n9E//rts2uq8v/3Qxt8KfSFjZOX5fo/X75y34nmU3WdAA6cavnglE35j6tX\nFL91KH5GW2VJYN1SWVkphBCzmSQ4RHJsoucoxxrBJvQ88q2EGm8B9lRQWTXK4tfOUNUGWr6d\nj76RELwGgL5io/+/fTv84k/M88dhmtq8xa4bHkizR2wqI/TmQWmw7B77/WNttMnc+SGCp9jo\nJkc2slaSv5qTdrdhIDqU4IjUc/gMzBC0AHmXwDFhPT7dRei2mz7jGVl1tXc/oqeGNqNn4ZhH\n3s2Tk1/gQ08PZTcGg03H+eJhKl01CQOYZORfw22JCQ5F3hWJB3Pt4aHsxlCU0Uu0dA3lL6IF\nV0Gl8ypVCDEr5QWSluPJy3IDqCj0JVbNKMnz/t/fueqDUy2n6jpdTm3lgtzF5dkAOkPRbzy6\nPxQ2hh988EzrI08f/uNPpPPehQhXVBdeUV0I4MYNFf/tX/b09YIddO+2qodurvY+cejFfUON\nXZbPz/mzBy4f7OcihBBiVpIEh0jC7Oa252F29W9GznPPYcq5Dq6pqktKautnueoKPrkbXc3w\n51PVBqocWq+hSuZ7P/sXkzQW3U1ll3Ptvvi4O4eKUqoD349jVt2v0Nv/mMrRNoRq4D9JJWvZ\nvkwCAX0fOpk73kTPscEdHDoE/2WUtXEMd08Z5a/j1vdhGfE7CofdLlY7PLvRNyjELiBaBOei\niRjVCJbJrWds93DzyVmZ4ICznLK3cte7Q82elZuyroYjP/FYrj1kf5GeoFr3oKQ2hBBxSvK8\ni8uz+6ZIDOd16xuWXiqZToR1SwrWLRkxS+LtQw1x2Y0+rx+o/727V7mSFO9IUX6W+1//cOvP\nXz+z73hzZyg6r8h319UL+3Iff/SJNXdvrTx6rqMnYiwuz7psUcFYyn0IIYSYkSTBMasYlhVj\nSwEOpanx/RrnzreGshv9oRh3vk6F94Oc4xrlOFD5Ciq3eUE9+dTqe6xIkJuHUgzw5qn1n4U2\nhi8Ot78/mN0YCgaPI1QJj7JZpUL5/f9ne44Nz270nYfgB3CVTkgGypVLlfdx7bOIDnxLaG4q\nu44CQ5kLjp1POI0AcOwcTUaCw0CSnFBmFg1NT55qcs1HpA5WEFoWnBVQSb79oklmm1sWjAic\nqVWuFULMJX9035o//bd3h/ci0TX6/btXB7xjLtYTN71iUNSwWrvCZfnjWUwKAB6n/tkbl372\nRptpm1WlWdIzRQgh5hRJcMwSFnN7NNxr9E/RJ1CW0xlwpNvx0epBtN4uHkHkAtwT/7w6/elu\ntfmL3HyMW8/AjFKglMrXjbVGIwdP2u/oPkO+9WyN7HoDN6ll/Sf2nLC/YM8JmpgpNhRYSNVf\n4uBZRNrhCJB/AfSRT8WW/efXpPHM0l3wZKM3odULQFklkzGAqaI88Cwe9SjKLrJP/3iy4JyM\ndWdCiBlnUVnWD76+/WevnT58ti1qWFWlWZ+8dtG8opS6tMbxeZL+cvQn3yWEEEKkQRIcs0RL\nuCc6rCQEgzujEQBp5jjMUDq75h4qXEaFy9I/P8kXk40QUTlpOWydA7oBnSgXtAAYmMdrBpNc\n0L6ZbmYoB2UtSbqXknynkTuNW0XN7qjVZXJMQdOV163nEtQoo6vaah3+TXzU4aF5G9IYwCxD\nizfj/adgxNd2oWXbZl8FViFEpmT7nF+8fSzrLgEAlsVEI3q/XlFd+IPnjiUeuXx+TpZ3yuaE\nCiGEmJUkwTEbREwjalfwsjsW9TtccY8vvUYkYkZMtjRSHt3tsl1SoZI/lF5ilxgrzQ/DJsdB\net8rMh+pJOtxlMs+x6HSnbMzbuQoZ+OibXysl+qJNUat/r+dCdM0ozEr6HdUKLrUOm1auoN6\nO7nm7aHeLp4cdcWDcKXzvnG28eWqHV+2Xv8BIkPfb7Roo1p/xxQOSggxy7x3vPnHL584U9+l\nKbV0XvbDtyxbNj8HQFVp1t1bFj65a0SxapdD62uwIoQQQmSQJDhmGsvkppPoboQ7iwoXwZ0F\nIGrZt4m2mA3Lcqj+V98MdEQ6I2b/W9wYEDYjHt2d7UzoCa8FoOfBSGgyTxpcFZn7y8xVlgGl\nA6BANUcabQ4IjDYrxL0AMZuueORakIHhpce5ELE6DOQ4uG9igJYP59h62cSsnsHsxuClLDbC\nZqtXL0p2FgCQUpfdywuvQvMJjgQpUELla8ZUEmV2owWXaff/LZ/cwx31cAeoYiWVxvcqEkKI\ntP3yzTP/9pujA1vWh6db/+Bfdv/lpy/furoUwFfuWLFiQc7PXj9zrrHb69LXLsr/3C3LyvKl\nAJAQQogMkwTHTMJt56x3f4yugb7uulOtup2qr0vx9J5Y72B2Y1CvEXZpTrcW/+afsrdw2/Pg\nEX03KbAJSj6OpMuMcO0b3PIRot1w+Ch/JVVsQ8957jk7/CjKXkP+5CtB+o7xr+XwWcRGZqBc\nZfCOY73MeBH5tiBaw7FzsIJEPnJUwLUYoy0tiWNY8VNa+qYgxVKr5UHZZcguk0UX9tx+Wn2D\nfHGEEBnXGYr+6IXjcUHL4u8+efjKFSW6RgC2ry3bvrZsKkYnhBBiDpEEx8wR7rbe+GdEhz3m\nGVHrwBPK5XPOW297hiLS1dDjZdi076QQNiKJCQ44iqjgXg6+j1gj2ICeT/7L4AE+Ay4AACAA\nSURBVLjkK3RxCWbUOvQD9DT1b8ZC3LCXO06q1V9E71kET7LRTY4cZK0kb+XoVyMnFdzNwQPo\nrYHZDT2bvEvhXQkaWzYh0wjOKnJWjecSFttPR2K2WYQlhBBiOvjwdGvUsPnp3R6MnK7vrJ6X\nM/lDEkIIMTdJgmPG4Jp3RmQ3BuPHX3VVbnJpWsSMfwIMjCzAkezR0RwZ58Gqg5qPsreNY8hi\nCDfsHcpuDAq3c/3bNP96BJaP+b066RTYgMBsq6CpyP6HUrK4EEKIKRcKG8l2BXtjyXYJIcQl\nxOzSpkKMSp4ZZgzutOvbCnDnRbCV7/J2RMM9A21iFVGWw+V3jChAoEiZdjkOjRQAi9HaG+2M\nGDGLdUUBp17odWgk89kzpPO0bZg7TtP86yd5LNOZU/NHzA6buEqoFCOEEGJ6uEQ1je/95khl\nceDWTfMvX1wwmUMSQggxN0mCY5pjmHVstQAxIElnUFIgUqA8lyfb6YpZFoEcSqmE3IRbc8Us\nm3csbs3FjHNdveGBRKlhcXs4FowaC3M8kuPICDYj9juSrBuaszRyefSCXqMV4MGgQ3ldeu4U\njkoIIeaISMx0OS7VssrWysq8ikJfbbNNX7BzDcFzDcE3Prx495aFX7kjSWswIYQQIkMkwTGd\nxTi6F9zZv5XnwDmbg6iwamhNCSlNS1qFwevwRMxo1BoxWdStudy6qy0cCydMA4tZ3NITK/bN\n4T4UVgShIxxrBTnIVQrvksEv9ViRO4+DNnNwyJ0/viHOQi4tW1eeqNltcZRIdyiPQ0mrVyGE\nmEBt3ZH/eP747iMN3T2xgmz3TVfM++S1i1LPdOga/dWD6//Xj/ZdbEtaEPrJXTWblhetWyLz\nOIQQQkwgSXBMX2wcHcpuADSvDKdruKt7xEFKo1W3p3hBAuW5c3qMcNiMWGxqpHl0d1950VDU\nvoJjKDaHKzuGL3Dby7D6Z1hw6DCCB6ngdih3OlcrWoeWQzbxYvsCsXOcRk6PLqkfIYSYDC2d\n4a9+Z1dbV2Rw89GdJ98/0fwPX7lST/7WJM7CksD3/3j7zvdrT9Z1vvZBfU/EZsbomwcvSoJD\nCCHEhJIEx7TFMC+OCChFV2/EoeN8oa5v9j5ll9H6T1DB2JpWeHW3V49/RDeZbQ9OFp/9rAi3\nvQRr5LqSaBO3v0n5N8YfzCbCRzh6HlYPND85q+BeGjfXg7KrsOBGvvAqBlcJkaLybZS3fOL+\nEpMhGjZrPuK2RsrOV5WryDu9KmVYbY2Rva9ajbUqJ9+xerNeNQtnR3f3xN44WH+hKZQbcK5f\nUrikInuqRySEmGF+svPkYHZj0LHzHS/tq7110/zUr+PUVd/xL++rtT2gtUtWZQohhJhYkuCY\nrjgKJMyecLlo/Rq6/CaE88mVBW/G+q45NdVrV6nYmfKrm5mCEbG4lTkCOBTlKEqy9iF8Nj67\n0af3DKwo1LBlOxzjrhcxWBfTiLDRilgtBa6Lz3GUXUV5y7j1CMJtcOdQ7jJ4Z3bbXfPUgdjT\n/8KdLX2b5PE7bnpIu/y6Sbs/uJk5ROQE5QG+uN2Rt57t+cX3ONb/79j7wk9dW271PfA1zKKy\nMnuONP7DLw52hqJ9mz968cStG+d/9a6VSs2ev6MQYqK9f6LZNr7vePOYEhyDcgOuxvbexHh+\nVlpTIIUQQoiUSYJjuiIHQMPrLA5x+smTzgeOS8hx6Z12s0lzXLPqO8TkJsO6MPhVNblBo0Jd\nLUg8ko2uJNewYAah8oaODB9GYteP2EVETsO1OD7uzqPyLekNfrrhlrro438LY6ikC/cGo7/+\nrisrTy26bOJv38bWYSAMgBkAgeaTWjq43zh/KvT4tzFyClJk13NaWaX72rsmfHiTorG99//9\nyf7osNSkZfFv3jlXkuf5xDWLpnBgQoiZxXY5SV+8tSv845dPflTT1hsxFpYE7r920Zqq0dcP\nbltT+os3ziTGt68tHe9YhRBCiEuabe/nZxEFVWgXJ1LFGb+Z16EV+5xxL7bzPY7sjCc42s/y\nh49ab/9ffu/7XPMG7Lq6TBCLQ4Z1Pi5nZHKzyTZvrki5kl4oblfUfiIuJ4nPGsbe54dnN4bi\nu5+ekPt1NfH5D/jiUURCQIStD/uyGwMYfA58fnA7+s5LsFtgFdn9woQMbyq8tK82ajfx6tl3\nzicGhRAimbL8+Blwffwex+f//o3n3j1/oSnY0hl+73jzH3/vnZ+9bt/1fLhP71hSPS9+kuld\nWyqlU6wQQoiJNqvez88ypK/gWCd45EIJbSEoayJul+d2+B1aV9SMmpZDUcCpu/Uk+S8riuBh\njjUDilwl8C0HpVRonU++yKdf6f8zgNaTXLdPbfwynPYfrTLL4hbbuMnNGiXkktzz7WfQOAqh\njRwtJ+n/miw+W1hN9k/RVmOmn657O609j/G5/f2bDpe6/g4U2izBYOs8af2Tm8xmm541AKwk\n8ZmorsWmIyOAhvYew7RSLw0ohJjjbt00//iF+KmIStH5pmAoHP8e4j9fPL59TWlJnvcSF/S6\n9X/83auefef8O0caW7vCFYX+2zbPl/KiQgghJoEkOKYx8pJjK5unYLUAMVCAtIVJpnVkhlNT\nBZ7RHoqiDdz8HMz+JysOHUX3ASq8A/poaZfOC4PZjSHBRj7xPK26N70BjwnDPuPAtpkIPQdZ\n69D1/ogg6ZS7Lf5I5YNls9IYajKyNlOItCQ/PZLF08OW9fJ3uPUceKCkSSzCXeeosNLu6F7A\n6puYRh77rz95Zk/HWY/LPrHo1DVNSXZDCJGqm6+Yd74p+OSuGsvqT+u7ndpDN1V/75kjiQcb\nJu891nzHVTarO4fTFN1x1YJRDxNCCCEySxIc0xs5SZ9OfR/Y5JYXBrMb/WLt3PoyFX98lFMb\nP+K4qpt98YaDk5PgAOyfBinJ/wLK2gRHMXfvR6wVygFXGWVtgh7fooJcVWzYzA0h19i628w4\nav5y89QBm/iCTPaF4dpD3HoOGPmtYyTrXkyDxzlWbIi+91riEY6VV2RweFNr3ZIC29Uoly8p\nmEV1VIUQE44IX7p9+fXryt892tTc2VuW77v2srJQ2GYRYp/O0HSZotjdE3tiV82JCx0Aqufl\n3L1lYcDrmOpBCSEyw7BbhyvEqCTBIcYiUgej2y5eD6Mz8eF/5DHd9s9cRhhmFJrTdmcirvuI\n6z9Cbwf8hbRwM+XOS/FERdkWt9vGk57jqSRP5SjXdS2B0YLI8GpqBM9KOMpSHNgMpW28xXj/\n5cEWKv1cXsf2+zJ5m77sRpzGJlTbFdGk3MEEh2vjjuiel2InPhy+X2Xne25/MJPDm1JbVpVc\nvrjgg1Mj/gm8bv3hW6qnakhCiJlrUVnWorKhyZhup6YUDc7pGK4o15PB+9Y2h57cVXO2sTvg\ncaypyv/YlQscyVbIjnTkXPv/+I99XT39baTeO978zJ5zf/O5KxLLfwghhJg7JMExVzAsiw1F\nDrKZRZGypL1FAKNrlASHK2Afd3hSzW5YprXnh1w79MjKp95SK2+mlbemcrZG+Ra1WBwcHiQ4\nNTXOou5EvqvgXMjR87B6oPnJWQV9ZJF5y+TTb3LTMUSC8BfRoq2UP+Pnd5DH73r4G7Hnf2ge\nfacvouYvd9z+Rcqf8MwOn6tFcwsVxi3nVkRLhm2pwO/9bfjVX4V3PW+1XFSBXMeazZ47HlKB\n2fPBl4j++nNX/OL100/vPtcejLgc2vqlBV+4bXl5wSxfHiWEmAR+j2PTsqI9Rxrj4j63fuWK\njBU7f2HvhW8/ecgw+9/T7j7c+Oy757/1xU2jNpQ1Lf7/Hj8wmN3o0xmK/u1jB3749e3SKlsI\nIeYsSXDMWEYLjHaQA47CS5d7sDjWa7TErJ6+TYfye/R8RWn906vkHzgusQsAQCVr+MzrtvEU\nb84nXhue3QAAtqxDz6miJVS4JMlJI27lUNUmN5hWCyNCcCrK0VRZsiUqY+MoJUeSREk0ZL35\nHe662L/ZUcu1+2n5zWr5LRm475Si7ALnJ/+Ewz3c3kjZ+eSdgPK3hQttgsy860O652HwBcAE\nCJRDVA0amUTTdfeN97tvvB+WCZVSHdwZx6mrT1+/5NPXLwn2xrwuXT7TCyEy6PfvWVXfGjrX\nOPRiwOPUv37/ZVneVCddXlpTR+93fj2U3ehzoSn4L08d+asH11363GPnOy629STG61tDx2s7\nl8+fPblsIYQQYyIJjhnIDHJwN2INA9sKnhXku9yuwAUsjnVHaxlDnx5iVtCMhQPOeZRGk2B3\nOUgHJ/R21fxwjlYdPauCFt/Ap14eEfSX0NJUn/P53Hv28bN7U0twACCNSjWtFLCtBzIhrEPP\nDGU3BvCxF7lkJeXOn5wxZBhHQI7BJtPk9lKpXRoiE6hsJRVWcfOZuLhadTOpJcBiIAI4khVY\nGTh6dmY3hvN7ZNm5ECLD8rPcj/zBtuf3nj94pi0cNRaWBO64qrIge5T3Gal7+1BDzG6N/Z4j\nDZGY6XJc6kd3S6ddeW8AQHNHryQ4hBBizpIEx4xjcdcrMDuHR9B7iEkj79rEo8Nmx/DsBgAG\nLDYiZqdbyx3zzZWHcrdw2+sjgqQo77pU8gW0+AYqWMrn3uZQMzl9KKim+Vel/vDJoTb7Hcni\nl2Y2sNUBEKkcaBmbbZuI6z60izLXHZhpCQ6TI8cROw2OAgp6IbnWQI1l1oYZ48OvccMpmAYV\nzKfVN8A92mIKInX971l7f8qn3+2POL1q3V207Jq+3UDGPmrPbNwDbmIOE3lAxSD5sgghMkDX\n6GNXLvjYlRPSDKWlM2wbN0xu646UXrITbY7flXxXZiaYCCGEmIkkwTHTROtGZjcG9B6FdzUS\nJmUYCR1MaTCeRoIDgH81OfK5cy+iTSAFZynlbIZjZMkJs4WjZ2B2Q7lJL4Vz4VD6I2cB5SxI\nc+6Ey4eY3RubZNU9kuEeju6D1f9lZAAqj5wbQEk/LaXPjCFmM4cWAMJ2/47TF3PP2zCbBzYt\nGI1svkqea6Cl9qKss9F85u/Q2dR/uTP7cPAldcvvU9myUU50+9W2z2P9PdxeD6ebcivgmCtP\n71Znq3nhFFuWXrFI5RUlP+4Mm6cAC33fzzhB2jKoVOvvCiHElEiWpFCKcnyjJClWLMjN9bva\ng/H9XPKyXCsWpPXxRgghxKwgCY4Zho0ksxU4CjMILfF1uk3980vGU+Aqo6K7ku3k8EeIHO3f\nMMGxOkTPkm8bxlD1g7n1OIJ1ACFQTnlL+/IjVL6Gj7+aeDSVp1rFo//q0fcHsxv9rDaO7ifX\nlWO6Tko0Bxwe+7yM+5I1Wacbo35YdmMAmxw9RJ4tqVzAeuX7g9mNfuGg9dIj2mf+DnoKb9t8\neeTLS3Gws4ER63ny38OvPw3LBAAi15ZbvR//ErkSkjtWI5snRoZMNg8T+UHyKV8IMX1duaL4\nB88fS2zUsn5Jgcc1ymcGh67+8L41/8+P98UMHhbEH92zSNfGvgJXCCHEbCEJjhmGoJJnJmx+\no2vktBJLZgCKMjGBM9rFtW9wdy0sg/ylVL4VTm0ouzHIbOHIEXKnloaIdFlHH0fXhcEAZ81X\nKz4FZ0CtuNm8eARdDcMPp/I1NP/yMYzZ6oDVYRdvAYdAmW8/QeWX8dk9tvGM32visJGQ3ehj\nNKdU0KSrmS+esImH2rn2MFWO5V9wbgj99LuRt58f2maOvPUs9wb9D/9l3JFsXYAdti5QerO0\nhBBiUlQU+h66qfqHzx8bHszLcv3uXStTOX3TEvrXzwcf2+U8flEjoLrMfGBLpCxvP6wSqEw2\nshVCCDGDSIJjpnEU2seVF5o/MezSsgf7pwxDLm3c0we6z1tHfwKzv0Mbh1u59QjmrSWbUQCx\n80gtwWEd/enw7AYAdJ23jv5crX0YTq92459YR19G/Ufc00H+Qqq6ihZuHlu5UA4lv3c3tMwn\nONSqj1ltZ+PqjNKym4YKcLDBkaOI1YJ7oPzkqIRriW26akrZpMkAABZgjVLjE+Bga9J93S3p\nD2qWsrraIrtfSIxH971hfuwhrah8ZDjJt/QlvtWFEGJ6+OS1i1YvzPvFG2fONXb73PraRfmf\num5xilWTOXK0PM/8+h0j50hylKMnU32nIoQQYtaRBMdM4yiBoxSx+K4c5LN/B64rr1cv7DVa\nB0uNEmlevVAb7wwOtk4/DTM64u09W6j9CEsWQUt43E0oBWIv1ICu8zbxzhr0NMFbBM2pVt2G\nVbeNY+TJH8XTa507KqdPXfd1Pv0mNx5FpBuBYqraSgWL+vdyjIOvwOrq3zQ72fwQRh35rplW\nOQ5SAfupQ8o7anYDANwJeS9FIAUiPvIKh7vVZbfOncoaozIvnAbbf73N8ycTEhzJvv7y410I\nMQOsrMxdWbk+nTPNJIt2jeQpdSGEELOdfAKeeSjrGu45gN5j/XU0lJd86+FK2qfTqWU5lC9m\n9VgwNHLoyptOg9g4Pc3o7XvxPnL2hGVwKERZCaVAVErPrtyT9GU+97SQN3mRxdSpPEABiX3p\ndKgJm8+vNFpyLS25NnEPR44NZTcGGS2I1sC5KPH4KeOYj+ixxA7B5FjEbYfRfpSjXeTKRt5q\nylmaeDbllSO7aKgGh1IYWCPNnU344Dfmyd3aHX8Of37iuWKkhPlKVAAO2hyn5IsphJibJqkN\nvBBiQsViNm2khRjVNHpFLFJFDvJdQfmfpJxbKfdOyvv4JbIb/WeQ5tQCbi3XofwZyG4AbPQt\ne7F7yWzYrWXQK1K6rpZ8XomW0oTV0ZGTHNU2YceKlGYiZJwRPxmnD8fqJ3kgoyAPuTePbD5K\n0Kv4/Ad85gluP4pQHbcd4VM/45qnbM9X1z7c/49IQGIFuGCbtfvxCRr7jKMvWGrfPplIXxjf\ndIa0KpumsOSHmpC2jkIIMV1oSdK4uqR3hRBi7pIEx4xFDugF0LKn5E0FOfvmaNjd2jt/8I/9\n+Q8tm9wpFQyjrPlQdokMzUlZ823i6dEXk3M9+ouFEFQWOTdCn6KnQY6OLT6F9GLy3UTu9XAu\nJddq8u1Ad4w74kuHcutBbjuUeDaVL9c+9Q1aciU89m19+fxBmLHMD3sGIn+2e/vHEuOuzTeq\n/OKEsJP0K6HKBzJ0OtR80jdNTcJOCCEmC7lX2HyOJRe5lkzFcIQQQkwLskRl1rE62DgDDgJO\n0gqgV01IGsudB385gnXxcYdfFV4Pq5Ujp2F1k3KTXgrX0lTHoLupcgefiS+vSJU3QHNlYNiD\ntDLSygCjbzpBJq88VsoHmyqwgPLB7OWuvQjXgyNw5FPWOrjKbY6cTKTDUTmY1uK2I/aHtR1G\n3iqbeHaxuvEr1u7H+NBOm72WgUgI3pzMDHUminZy8CyMEFx53nseJrc3vPOXHIsCgKa7r7vb\n87HPJjnTRdpqaKuBKJCJBklCCDHxDNOKmZbHme5nUS2f/Nu5Zx+s7v6IXkCeK2wmtQkhhJgz\nJMExuxinOHZscOYEW80wzpPralBGswMAALX4LuvIjxEdVj9Cc9GSu6GcUKWkl6Z3WarYAncu\nn92JnmYA8BZS5Q1UsGL4MRb3mtzNMAhOXeVQ+t/GU//9T45K2w6spHL54k9ghfu3jW7uPUs5\nVyJrw6SO79Ji3bZhjnZfYloReXPYtrOs5rCpRTqbsMmNh9FVD92FnPmUN6LGCje+xY27wWb/\npjPbfd3t7uvvNWpPw7L0iiryp9L5SLIbQogZ4KMzbf/+3NETtZ2mxWX5vs9cX7VjqY5IO7my\n4SuBSvm3s15CWbfC7ITVA+XH+DvECSGEmOGm/gFPZIzVPTy70Y9DHDtMznWZv52nUF32VW54\nF90XYJnwl1LpZjgy8IBKBSupYCXMCIDEiRtRqz5mtQ3+NWNWo1Mr12kSXvtbQDcQBjxAIGMr\ng5yVMNsQPT0sRHCt4M7DQ9mNAdzxLnkXQ582cxx0HyIdiWG65LcBVW3AvidhmfHxhevH8KF2\nxgk2WAceRbBxKFK0gtZ8CroLALd+wA1vjTg+2sk1v1DLvuRYunZyByqEEBPrrY8u/vWP9w9u\nBszGyou7OdSDvl/t7jxacgflLk75egpaLrQJKxMuhBBiRpm9jxNzkHXRvuqn2QDbF+bjpzmp\nfGvmL9t/cZtZJzGrJWaNaP/GsCJmrdI8agJmqQzTynwSiAxseomWAhl5U0TkWQ/HfDbqYIWg\n/ORYAHJz5FW7gy30nkXgskzcNwModzmHEpYpAchbfqnTsorUVZ+ydj8Ga1hx7NwydeUnMzy+\n6cMyrP0/Qs+IjobcdARHf02r7wfALfvszopy20EqvnpyxiiEEJOAmR95emh5Y7En/M3NRzz6\nsJR3uI0P/Zgu/xL8ZVMwPiGEEDOcJDhmD+b4F/4DTHAMNBvmrhvcbhdmg9uclOaimBR0MR8e\nmTzqYf6IaAOQoYW+eiHphUObhs20iD5s9k6f9ndUvBGdp7j77Ihg7jLKXzPKiSuuUyXVfHgn\n2uvh8lHZclp57SyevsEtJ+KyG/3x+gO07A443IgkaZActlm+JIQQM9f5pmBL59DHlbsWNozI\nbvRhky+8Sctnb9ZbCCHEhJm1TxRzEJHbbv4GAA2UoR6rGcfRMWVeLI4kiU9gzxHmWrupMSZz\nLVHqc2jHQnlBCmzT/Zt0+xYkU4M0qv4Mmj9A+1GOdpErG/lryLa8aOKpeeW0NVnJzFknlCRP\nwSZ6WpFdAdLBdh1kZm/SRwgxN/VGRqQzFmeHbA/j7rrpk80XQggxg8in51lElQInbB7FtZIp\naSV7KRzj2FEYF8AxQINeRs6VqZQ9J2gMwy4+oQ2P7UtpAsGJuqFywrMQPafj4+SAp2qibpom\nosJ1KFw3zb7DphkteRZPdwIg3zzuPmOz1zdF3YuFEGJilOZ7lSLLGqiGnvRA+a0ihBAiHRP6\nWCgmlwqQY1l8kHzkWDkVo0mODQ6/idiZgVfWJowL3Ps6ki6xGaKRffVKTWV4XgOHe6zaE1bd\nKUTDth+z2PZDmRVGpAFGl92+saHc7XDkjQzplL8Dmnf8FxeTjPIXgew+rLtz4C0EQKXX2EzW\n8JZS7jT7zyuEEOOT7XNuXV0yuHmiw/7XOgUqJmtEQgghZhWZwTG76ItJFbBxBtwNOEkrhL4Q\n0KZ6WCMZNUMt6wdxmGMnyDlK7QanKjbNoAVj+MOiRj6dsgEgFrH2v8ANp2GaKK7U1t0Mz9gT\nH6YRe/3nxu6nYMQAwOnWt23Xr66Oe0AlApA17Kwgt7+FnlP9m448yrsGrvIx332Q5qOSTyF0\nmMN1sCJwFpB/DabV+hSROl8RzbuSz++OC9PyO/q/rzwlavFvWXUvI3QBAEij/MuoZDtIctBC\niNnma/es7u6J7T/ZAuCps6U3z2/0O0aW4VA6zd82NYMTQkwbMSOhQI8QKZAEx6yjciakKWzm\nsJmkHkFcnC0+vZsvHkM0hKxiVX0NskqInB59cdRsMLmbYRIcDpXnUIUAcUut+ctvcPdAKcfT\n71v7X9Tv/iOquGRHjwTRZ75nfjCsg0k0bOx8EZE2fcdVIw90EA28X+IYNz4Bo3NoZ6yNm56i\nonvgKkHaSMG/mvyr07/CZGOr9gS31JLbRxXV5JemfUNo+Z0IlPLpVxDuABGyyqn6NspbNHSE\np0QtfhBmBEYIzhxJbQghZiu/x/HNL2zad6L58Nn2aMw8ll21Xu2m4EBPLk8BLbkDvnH89hRC\nCDGHSYJDTDq2KaIRH4+GrJ3/xG3n+zcvHjVPvqU23E9LtxEcLm0eAMAavsbKfPa7Q9mNPuGg\n+Zvv6J//J+gOAAjWcf1u7mmC5qSsBVS+FbonfgjtjeaB1xKHZuz+QLtqC3kGq35mES0FBgor\nBI+OyG70X8vkznep6E77v+ysw631xjP/bNWd6N/WnfrVd2tbPi7rqPsR0bxNNG8TYj1QDmhJ\n6v5qLtsGyUIIMctsWFq4Yelg+7A1CDVxuJ1c2fAVS4ZXCCFE2iTBISad8sOyaZmJYXU0rP1P\nDGU3+kOmte9nWkk1sooHTxjcya213HQ28ZLc3cYXjtDCtXzxHT730mBfEg7WcfOHauVvw1Mw\n4ia1J+yra5gGGrKoagkQBjzAiMwIRy/a/00jSeKzTywSe/xvuHPYHBwjarzxMzg92sbbpm5Y\n05JDqqgIIUQcgq+YfMWjHyiEEEJckuTIxWQjR6V9XB+Is8Vn99kcYZl8fr/9Rbtak92Ou1sQ\n6eBzL8d3XY2FuObZhKOTFnRniwEvkBeX3ejbl+SkZPHZxjy6Z0R2YzD+ztOXqpEvhBBCCCGE\nEJkjCQ4x6VQeuS4HDZ89pMi5AnpZ/1a0F0bE9lQO2U39AODNso8D8GRx+wmwTZki7qyB0Ts8\nQiUL7S9CpEoqk92BnAX2OxxJ4rMON52zj3e3cSgDPWWEEEIIIYQQYlSS4BBTQV9AnuvJuQaO\nReRcRZ4dcCwd2utwQ0uyeMpt30aEihZQll02weVV81ciFkw6kpG7VNE8rfoKm/Gu20H+nKQX\n8a2EsqmbQFnrk54yyyRfL01qmjXxEUIIIYQQQsxSkuAQU4TccFSRczUci6F8I3YpjcrtW4fQ\nvMuSXE1pN3+pv5jo8OD1n4PLC2ff/I6EtRKk4IjPmDju+Zq2dvuwwSj9ipsct37hUn8XzUtF\nd8KRN+wsJ+VdC++i5OfMKqqi2jZOBeXw+Cd5MEIIIYQQQoi5SYqMiulIrbvXbKlBT8fwIK28\nifLmJzuFFqzWf/vvrd2/5PqTsCwqXqiuvIeKFgCg3GpWL8KKxZ+Ssxi6Oz7o9jrv+Rpvu9e6\nWAOlVNkiyk2h7JmzmEo+hUg9jHZoPrhKoRJKdcxeaukGVb50qIXKAP2aB6ZkPEIIIYQQQog5\nSBIcYlry52u3/w/r8Au4eIwjQcoupWU7qGzFpU+inGLt1t+12eEMUNXtfOYZWMM60brzaGHS\nBh9UUK4VlI9tzKTgrgAqxnbW7EDKcf+fG6/+xPzw1b5CrZRdqN/wkKrew4699QAAIABJREFU\nONUjE0IIIYQQQswVkuAQ05XTqy6/B5dn5mJUuJYCFVz/Dvc2keZEViWVbIRyjH6mSJHHr9/2\nZX3Hg1ZrHXkClFt8icIcQgghhBBCXIIRmyvtCEVmSYJDzBnufKq6jaZ6FLOc26fKl45+mBBC\nCCGEEEJkmrxiFUIIIYQQQgghxIwnMzgEEG7g3gYA5C6Gp3SqRyOEEEIIIYQQQoyZJDjmNjPC\nF5/nrv7mFwxQYAmV3QItvreIEEIIIYQQQggxnUmCY07j+me5+9SISPdJ1Jk0/96pGpKIw511\naDjCvR3kK6CKy+HJmeoRTTUzAisCR9ZUj0MIIYQQQggxvUiCYw6LtsdlN/pw8AxFWuHKn/wR\niZHY+uhpPvU62ALAAI4+p9Z8nCo3T/XApkjoglW/Ez0XAUA5qXAjFV0FJT/EhBBCCCGEEIAU\nGZ3LONKSfFfzZI5E2OKz7/LJV/uyG/2MqPXBz7ijduoGNWW4u8Y6/Wh/dgOAFeXGXXzuV1M6\nKCGEEEIIIcQ0IgmOOYy0dHZNuWALH33Jeu9xPvICd9ZP9WgmEJ/dYxe17OOzHV8cmevpC3ad\n5u6aKRmPEEIIIYQQYrqR2d1zF3lKmTSwmbBDkad8KkY0Oj7xmnXwaZgxAAzGoedo2fVqzR1T\nPa4JwcEk82iSxcd1MwM0jX8amBH0NtrvCp5DYOHkjkYIIYQQQggxHU3jRxox0TQP5W/klvjp\nAJS/Ebp3SkYUj8PgLoBAWSAXN5+0Phi+JIHAFh99iXPKaP6GKRvkxNFdiIbs45lidHDXHkTq\nwQa0APlXw7dyOk7sYiPprt7T3O0lz1Loc778qhBCCCGEEHObJDjmNCraCt3HzbtghgFAc1Ph\n1ZS3bqrHBcBk4yjMC321NQEFrZLP7LU9lE/vHnOCI9zMrR8g0grNg0AV5a0GaLxDzjQqXsY1\nu23jmblBrJlbnh7KHZjd3Lkb0QbKvSEz188UI8rtTVAuWJHEnUy9FDrIoUMU2AjfqtSvyr1B\n84NXrabz5PKoBSu05ZtB4/gesCzz4lmrtUHlFGhlC6E70r+UEEIIIcScFzPi1yYLkQpJcMx1\nlLeO8i5HtANgOHOnyXM+xz6E1TAsYME8gyKFs3YHB5vGdvGW97j+laGCDh1HuO2AWng/tMzN\njMgEtewm8+IhhLuGByl3AS3ITBcV7txjMzOi9wy89XCVZeQW48dHXrfe+xUiIarIQ3le/G6l\nyNX3r2Zx97vkLIGjIJXLWmcORn/5Dxwa+Nq+86xasML5wJ+T25fGIM3zJ3t++k9m7en+QRWW\nee/7XX3ZdEgUCiGEEEIIMYdMv7noYgoQnLlw5k2T7AY4ODK70Y9Kc+F02sQdnjFcPNzM9Tvj\ny1WGarnhjbENchJ4crTrvk7zr0DfX9CdRdU3qK1fhcpECVg2ELX5IgPgyIUMXD8T+NBOa9d/\nIRICwHVtaOwcmNEDANA0ysmGGvymZe61aXtsc9neYPTnfz+U3QAAWOeOxJ779zQGabU3Bb/7\nZ4PZDQBWc33wX/+neeFkGlcTQgghhBBCpE1mcIjpx+qyjxNRdoCbW+PjJStSvzZ3HAGzTbz9\nMJXfmPp1MsAMctc+RJsAC45CylpvU0XCnaU2fAYAjEgmS2+gr6qFzdcBADiWyRuljS1r/zPD\nNsFnm6mpkwMeqipFlo9czviUnNmdyoWtY3u5N5gYNw/vxu1fgtM9pmFG3niawz0J1zLCO3/h\n++2/GNOlhBBCCCGEEOMhMzjETEL+woE/Djyc+/LV8rHUjIglyZ6YvbCi6Y9srCJ13PAYQkcQ\na0GsDT3HufGn6E3e8TSz2Q0AygVl/yRPenaG75WermaE4xMW3BNFYyc6DXK5bCYcqZS+SlZ7\nkoYsRoy7EtJnozHPn7CPn7OPCyGEEEIIISaIzOAQ049K9oBNtO4hcr/CZ/eitxMuP827XK2+\nncNh89Ae7mpVOUWqegN5sy51cS1JgxjlgLJZ/zIxmNtejat/wWyh/TVyz5usdq0E7zIEDySE\nnfAsnpQBjMZuok2/JCkYci1I5cKXKrSRVg2OJLeZHgu+hBBCCCGEmDMkwSGmH/JBlcGqj49r\nldD9as2dWHMnzBg0BwDjnWdjrzyKaLj/VI/fcevntTXbkl47u5qb37WNZ2z8o4o2Jy6mIABW\nGJF6uOdPzigosIHNIIbXrVAeyr0Oaiw1TSZOVgGcXkQTVn8AVH4ltJPxX0P3QrhTSnCoxZfh\nJUpMoKiyReQfc69Zbf5S49RHtvGxXkoIIYQQQggxHrJERUxH5FgNrXLYGgQFbTHpw9qjag4A\n5on3Y8//YDC7gb76kU9+26o/jWR8FVSQ0FPWmUWl12Zm6KmwbB7aB3b1juVCDCS0QUkdaZS7\ngwruQGA9fKsoeysVfRKuivQvmFlKV2tuSgxT4UKquJwK7oZvNfQckBOOQsreSjnXpXrhovn6\n5tvjow6n49bPpzFM1/Y7yZMw70N3uG/4RBpXE0IIIYQQQqRNZnCI6UkjfQW0ReBugKCyAEfi\nQebe521OtSxj7/POu76a7NJUfiMCC9Gyj8Mt0H0UWEhFV01qj1jNn3xXakskuJv5BLgDsAA3\nqQWgeWk2wXGWkrM0nRMnHl1+u2LL+vB5GP3lUWjBZWrrZ0EEOCmwCYFN6V3ZcfNvq4qlxq4n\nrKYL5HSrypWOGx6k/HSa46rcQv9Xv9nz028Ptk1RheXe+7+qVSxKb2xCCCGEEEKI9EiCQ0xj\n5AJdKu9gNdfaxjlJfOjCWUuQtWTKaiQ4CqDnwmiPj2t+OMsAwOqB2QaYUNnQEhZNcAdb7wOD\nnW7DbB0HdZJaPc5xces5dF6Ew0MFC+G5ZCmTyUFE6+/UVlzLzTWIhpFfQbnlmbq2tupqbdXV\nYB5/sQxt3uLAH/+TefGs1dKgcgu0soXQ5EerEEIIIYQQk00+hYsJZ7U2mOdOcDSslS/U5i3J\n5KUdScqCJotPG5R3Pbf8ZsSCFOWkvOtBxOEPETk5lL/QS8l7BWio4wnziWHZjcFoA7gClJvm\ngIIt1p4fc+Px/k3NoVbfSqtuTnNWSGZ5smj+2om6eKZKgRJpZQu1soWZuZoQQgghhBBi7CTB\nISaSafQ+9YPIm8/AMvsC7muudl65hlQEWoB8S+GpGs/ltYWrDLvJGtrC8c5lmHDOQip5gIMf\nItoIZjgLyb8WmpfDBxE5PuJI4yKHdpF/x0CuwQB32l6SuY3SS3CYhvnqd9A1rHmqGbMOPKU0\nJy3fkc4FZ47YiYORPS+bTfUqr8i1fqvzsqumekRCCCGEEAJGzJzqIYgZSRIcYgL1PvOjyOu/\n7t9Q5L3tMkd1NmLnAAAN3HMS3qVUcGPa0wT0rR83D+/h0IgHfsor0TbdNp5hTxLloqyNIyJs\nInrS5kizDUYT9OK+jeRXTLPgKF84MCK7McA68qK27LpZ3O409Nh3e195cnAzsvsl14ZtgS/9\ndyhtCkclhBBCCCGESI90UREThaPh6FvPDG46V1U4qhOKWfacQOhY2regrHzXF76pLdvU/0Sq\n6dpl17oe/ga5vWlfc2yMWOy9neGnvh959kfGoXcSO4+OjdUNTpK/MDsG/uRMlpckSq1AaQJu\nT1KypLcLvfazRWaB6AdvD89u9Inse7P3taenZDxCCCGEEEKIcZIZHGKiWM31HIsObjqW2beo\n4NAJ8i1P+y6UW+z81J/CNLi7jQL50Cbv3btZX9P7X9+wWi4ORrTKFd6H/pL82ele8hJzJWjo\nD1QOPpdwgANUnO5tk9+XZm0ONPLuq/bxd1717Lh7kgcjhBBCCCGEGL9Z+/Qipp4a8d1Ffrf9\nYWbwEtfgYGd019PhJ78X2fnTwTacNjSdcoomM7sB0+j9zxHZDQDm2SO9v/h2+tfUAqAk5VH1\ngsE/kloMKhm5201qrW0n3VRQYV8llITpJ/6CadFLZWJY7c2Jf2MAVpvNah0hhBBCCCHE9Ccz\nOOYWZt65v+6jmrbunlhlSeD2zfPzs5LkHcZNK6ogb4B7uvtvHQojz24NhZZ0YUXsw13hX36H\ne0N9m5GXHnNuusl991fiUidTwjj5odV60SZ+ZC93tVFWXlpXVeReyb0fxIcd5dDyRhymVoPn\nAW3MMSI/qARIP7lDZauocBE3n44fzWV3pn3N6Y8CObYzZlRWup1ohBBCCCGEEFNq6h8UxaTp\n6on+/nd3/93PPnxh74W3DzU8uvPk5771xlsfNUzU/TTdfetnBreME/Y3Iu9i27jVUt/72N8P\nZjcAwLKie56PvjUtSiRYLfX2O5htEx+pci4hzwaQa2BbwbWUPJtsjqQcUBWpalD5eLIbAECk\nrv0dWnz10FoVT7ba8jBVXjGuy05vzsuvThLfMskjEUIIIYQQQmSEzOCYQ7739JHjFzqGR3qj\nxt/97MDKBdfmZbmSnTUerm13kNPd+/QPOdgZ+fC8vrhEX5A/4gjPAvhX2J4be28nTJu2INF3\nXnBuv2siRjsml6pj6hpfiVNnFTkrYXYBJlQWKM2FJ2O8qU9tfhCX38OdF+H0UFYx1Cz/4eC+\n8obovjejB98BD1U40RdWe266b0rHJYQQQgghhEjTLH+GEYMiMfPNgzYzC8JR861DF++8qnKC\n7uvcfKPziuvMpjpEw6pkHsXOcs8JGN3QA+SrRvLyonHlLYbirfVgnvLepdriNVAarPimJ5Sd\nr5UsGPflFbSccV9k7Fw+KrKfUDMLKZX1+38T3vVCZPdLZnO9yi1ybdjm2XE3dPmpKIQQQggh\nxIwkH+Xnio5gNGpYtrsa23on9t6arpUOPPO7VlCSKRvxXPbFQcjpnvLsBgCVU+jacV/k5Z+O\njCr3XV+aDiVCxsyKwuyG5oeakLk80xSRe+st7q23TPU4hBBCCCGEEBkgCY65wu9xEBGzTd+I\nLF+Szh1TSl+6Lrb3ZZt49brJH4wt102fUSULIi8/bjXVgpQ2v9p920NaZfotb6eG0c2du9Bb\n07/pKqecbXCkXGjTMrjxDLqb4c+jokXQp+P3khBCCCGEEGIukATHnGCY/PK+Wr9H7+6Jxe0i\nos3Li6ZkVJfmWHN1bPEa49TB4UHy+F23/NZUDSmRY+1Wx9qtMGJQCmoSm9RmitXLzU/AHFbJ\nNVLHzU9Q0X3QR28Qy/XHrLf+E50DfVV9uWrLg7TgsokZqxBCCCGEmCtiSeaeC3FpkuCY/TpD\n0T/9t3fPXOyy3fuJ7VWVJYFJHlJKlPJ+/n9FXn8yuuc57mwll0dftsF120Mqr3iqR5ZAH28d\n0FN1Xa8dqKtv7SnK8WxZVbK6Kr0us2PGwYMjsht9rAh376fca0Y5ubPBeuEfYUSHIqF26+V/\nVnf8BRUtzOw4hRBCCCGEEGJUkuCY/X74/DHb7MaSiuwHr1+yecX0yxcM0p2u6+93XX8/R8LU\nV5LDMtHZAKXgLwDNwFIXdn704vGfvnbasvpXDz25q+bWTfO/ds8qmoRSI5EkHW2TxYexDu0c\nkd3oj5p88AW6/ivjHpkQQgghhBBCjI0kOGY5Zrxh1zwFwBXVhdM6uzEMudxgi4/stA49h1gY\nAFw+tfYOWrptqMPnzPTesebHXjkVF3zu3fPL5uXcvHHehN+ek839i+8OY6PlvP0lW86lPx4h\nhBBCCCGESJckOGa5cMzoCRu2u1o6w5M8mPGw9v2cj78+sMWIhKy9j6tIkFbfNoWjGr+X99cm\ni09GgsORh2iDXTx/9HNtZ9DoGpxsffRL8uRR6Wr4Csc7wtQYJj+9++yL712oawkVZHuuWln8\n6euX+Nzy800IIYQQQog5ZJZM8hfJuB26x2n/mJcXmDkNQUNtfOKNYdv9szasQy/0T+iYsRrb\nemzjDRPduxcAQP7VtnkK8q8d/eSiqviIz41CPxwxPrvbOvob8/Vv8alXMzHMUZgW/8UP3v3e\nM0dqGrqjhlXfGvrlm2d+5x/f6gwlrKARQgghhBBCzF6S4JjliLBldYntrq2rSyd5MGnjljOw\na3ALM8atZyd7NBkV8Nr3VQ14xlu4NCWOfMq7GZp3KKJclLcDrrJRT1Wrb4DbP7TtdiDLPWLF\nkGVaR3/DTUczOF5br+yvO3CqNS54sa3n0VdOTvSthRBCCCGEENOHJDhmv4dvXVZR6IsLPrBj\n8ZKK7CkZTzqs5CUhLPsFODPFpiQ9ejevmKzevZ5KKvk05d9M2VdT/k1U8hl4q1M60Zerbv9T\nKlnSv5kkU8Pn3snQQJPae6zJPn50IG5FET6H0CFELoBn9jeMEEIIIYQQIhlZoz775QVcj/zB\n1l+9WbP/ZEt7MLKgyH/n1ZVrF6VQZGH6yClPsoMop2JSR5Jpt2yc99qB+kM1bcODlSWBT2xf\nNHmDIAc8CetNUjkvr5zu+DN0t3JXk3X8SYTip1EAQKh5vMMbTbA3Zhvv6okBQO8p7nwb1sBS\nJs1POdvgmvj6JkIIIYQQQojJJQmOOcHl0B7YsfiBHYtD4VhNQ5CZg70x/+QsgsgEyq2gkmXc\ncCw+XrkB3pypGFHG6Jr61hc3P7mr5pX9dXUtoeJcz9Y1pfdfs8jt1KZ6aCkiBAooUEA1L7Nt\ngsPhjo/EwtzTSu5suPw2x49dSZ7XNl6a70X0Ire/BgxrFmMGue1FKvw49NyM3F0IIYQQQggx\nTUiCY64wTH78tVO/evNM1LAA6Jq6e0vlZ3YscegzY5mS2vp5a89/ce3BwQhVblSbPz1Fw+kr\nCJKZDrW6Rvdtr7pvezpzKKaRwmq02zSIpcJlQxvhDuvwU1z/Yf+u3AW0+uOUPd45ODesr3ju\nXZuetTdtmMfBgyOyG33Y5NAhyt46zvsKIYQQQgghphVJcMwV33/u6DN7hp4/DdP6xRtnunpi\nX7t71RSOagxcfnXN73DrWbRdABEKFlLSdSsTyWzh2FGYHQBDZZNzObTJKpYxvamq7WbdB/EL\nUvxFVLWt/89G2Hz7u+gZXIzD3H6O3/6u2vo1Coyr3u3Kytwv3Lb8P144ZphDlWhv2jDv9s3z\n0fy2/Tkxu8kmQgghhBBieojFktfgEyI5SXDMCR3B6LN2r7hf2nfhgWsXFeZ4Jn9I6aH8SuRX\nTtntjfMc2T+0abVzeDc518Ax7skXVgNzKxAFfKTmgWbMv8gQh0fb+gfW8Re4bj+iITj9VLFO\nLb0Jev8SFT67e1h2A/3zX8woH3+JNnx2nDe/b3vV5uVFL79fV98aKsh2X7miuK/KDNs1wQUg\n9ZWFEEIIIYSYfSTBMSecqu+0LJs2q8w4Xts5gxIcU8rk6EeJUY4eJn0eKO2CJiYb+8FDEwrY\nOkfaCqipmJ8yTg6PWnU3Vt0NMwotvqkKt9XYnsRtZzJy83lF/s/dktD/xVkCo9PmaNeM6ZEs\nhBBCCCGESJG8xpwT7JIb/ZiT7xPDme1g224dJqz01zuweXJ4dqPvgmweBvekfc2pl5DdAABO\nMs/wEj2Ax438l4MSBqN5ybd64m4qhBBCCCGEmBKS4JgTFpVmEdlXxFxcnj3Jg5mxjKR77BMf\nqbHqbKPgi+lfc3pKUmiDssrGfCmzCz0HuXsXhz6A0XKpI/VsKrgDzpKhiGse5d8BldDbRQgh\nhBD/f3v3HiRXXSB6/He6ex6ZycwkGZIQw3PCxJAXCSQCAUFd0RCWNYGsxa5w3b0Cq6xF7ZW9\nu1JirVVAWXDL19bWWpYuRmBllTCFgKziuiJcTBBRTMKbcJE8eIQkM3kwr+4+94+Jk0nSk0yS\nmen5ZT6ff8z5ndOnfwOHtvOd8wCInEtURoXG+qoPnzn1Z09v3G/8A2dMmdLPIzbZX1Lb76rM\nET/uNB9C6TiSpu8OzjNaRozMKYsKrz0RCl37jSfTPnB4O2p/Ln33d73ng6Tt60L19GTs2f1u\nX9GYHPexUHw35HeGXIO0AQAAxypncIwWf/uxWZeee3I2s+dvzUmSXPy+E69f5kT9AcvUhcyE\nUuMNITPuSHea7f+/wVJXeUStpjHzvk+Fmj7/DCuqM3P/PJk88zB20v12uvs3+17tkoaOF0PH\ny4d4YaYmVE5WNwAA4BjmDI7RojKX+cylM5df0LR+U1shDc1T6ye5t+hhSqoXpB2rQ3HH3qFM\nXVK1cM8DQY5olyEzMRTfKrEicww+fTY5rjn7gX9M33k57H4nVDckx00LlYd38kva+Urp8Y6X\nk+rmwZgjAAAQK4FjdJnYUD2xwS+xj1RSk4z5YMhvTIutIaRJZlzInXCUp0Elmfemxe0h7Hvh\nRuaEkIw/qqkOh2LofD0ttCZJLlRMDrmJA3pRtuLwTtnYT2FXWrInFXce+T4BAIBjgsABhyUJ\nuROTcOLg7a8mqTg/LbwS0ndC2hWSsUnm5JAZ8Q8xzW9Ldz4WCjtCCHsew1N1SjL2vJBkh/Z9\nk4rSZ8sc+KgUAABglBE4oOwqk+xRnNQw/NJ8uuMXobh7n8HO19Kk6mA3+xwMSeXUtGtDiRWV\nJwx0F4UtadeLodAWklzIHpdUzQyJa7UAAOBYIHDAkcq/m276v+muTaGYT2qPT95zfqge+deV\nDIaujfvXjR6d60PtgqE9iaP6tNC5PnRv2WcwOzYZM7Db5Xa9mHau2/PnNITirjS/MRlzQciO\njn9xAACRyOeL5Z4CURI44Ijs3lx87q6Qb+9ZSndtSrf8Pmm+PJlwennnNQzSQls/K/KhuCtk\nG4byzTNJ/UdC+3Np5yuhsCtkxoTKk5LaM0JSdeiXFt9NO5/bfzDNp52/S2o+NBRzBQAAhpPA\nAUei+MqPeuvGH4fy6foHkoamkB3AX7ZjliS5tN91Q/+RkmRDzZykZk4Ipe832q/CmyGU+lVA\nYXtI212oAgAAsTuqB0DAKNX+Tni3xLNdQ749bXt12Gcz7CqOLzWahmx9yNQO4zwO8+m8aWf/\nq7r6XQUAAETCGRxw2NLuXT3/W+Lv2F2j4HmlucZQNS10rt93NJPULhjEN0nffi19Z0PIVSZT\nTkvqGgdhj0lNfyucvgEAAMcAgQMOW1IxNg2h9BkElXXDPJmySOoWhdz4tH1dKHaEEEKuMald\nEComD87ed7fmf/Kt9NXf7VnMZDMLlmTff0XIHN3tS3NTQlIR0u4Dxo/3lFkAADgGCBxw+MYc\nF2oml7hKJVeTNDSVY0LDLwljZiZjZoZiR0hyg3nrjTTNt9yevtnnSp9iofjrB0OSyV7wF0e1\n56QyqV6Ydjy1T+PI1CfVZx7VbgEAgJHBPTjgSGROWxpy+17ykMkl0/6sbHcYTfOhe0vIbw/p\n8D5SK1N9+HUjLa5/pvCr+wtPPlTc9NL+615ft0/d+KPib38S8kd9p4zclKT2I6FyRshNCbkT\nk+r5Se2fhKT6aHcLAACMAM7ggCNSOyUz/7PppifSXRtDMZ/UTkmmnheqxpdhJmkh3fl02L0m\npIUQQshUJ/XvCzVlflpt+sarhed/lbZtSRomZk8/N5kybc9469v5+79e3PRy75aZGWdXXPq3\noXLPXTDSt/9Qeo/dnem2N5JJJx/tzJLqpGrW0e4EAAAYeQQOOFK5muTkiw7zSR6DL239RWjv\nc7/PYkfa+lhS7A5j55ZrSvn//vfC6gd6zyUprH4we/af5v7kqpAWu1f+n/St1/puXHzhye5c\nZcXHrt+znOn/tLKjvAcHAABwTBM4IGbdW/epG3+U7no6qZ0VkqMoAvnO0N4Waht7skL62tPp\nhjWhvS3UTUymn5809nsmRfGF1YVV9+87m2Jh9QOZ9zSHmrr96saelzz7RHrRXyU19SGE5D3T\nS+93TF0yYcqR/zgAAMCxTuCAmHUdcKPTHsWukN8WKiYewS7T1s3p0/emb7wQQhoy2eTU94Ud\nW9PNz+/d4MXHMvP+NJl3acmXF9b8sp/xX2ROO6uftyymWzftCRxTTstMO6u4/un9Nsme9+fO\n4AAAGCW6u4f3vnIcK9xkFKLW/0d/mh7J/treLP709nTz8yGkIaShWEhf+lXfuhFCCGmx+LsH\n0rdeLrmDtPXtfsbfCrmK/t42ye19UGv20uszZy3ZmzPG1GU/em1m/kcO8ycBAABGF2dwQMwq\nGkuPJ9lQMe4I9ldc81Do7gh77iyShBD6eyxLuv7JZHJziXeuri1ZVpLqsZmTZoYkObC8JGPq\nksmn7N1z++7Mwkuz5y1Pt24KucqkcWrI+qQCAAAOwV8bIGaVU0Ll5BIXqtScHpLKUi84hPTt\n/Z/bGvo7EWT31pLDmdPOLG54vsR481nJ+MnZBYsLT/3nfquyH7oyZLIhTbuf+q+On9yV7tgW\nQsg0HFd18f+oWPChw/sBAACA0colKhC3ZMJHQvVJf1xKQ0hC7ayk/twj3F0hf8Ab9POgmOr6\nksPZhRcnx5+6/z4mn5JduCSEkLvor3If+Z89t9sIISTjj69Y/r+z8z4UQuh85PvtP/xGT90I\nIRTb3mn/j692/ve9R/ZzAAAAo40zOCBymZpkwsWh+53Q/U5IcqFiUsiVTg8DkTRMSbfs+1iW\npPRJHMnJ80rvoqKq8pO3FFY9UHj28bR1SzJuYmbmeblzPxYqqkIIIclkF16cXXhxumNryFX0\nlo50V1vJltH5s3sqFy1JqmuP+CcCAABGCYEDjgkVx4WK445+N8mMD+0fOLJJCCEU94kcyakL\nk5P6CRwhhFxl9v3Ls+9ffrA3qt/n7iGFP7xQ4uSREEJ3V+EPL+bee+ahJg4AAIx2AgewV3Ly\nWZl3txV//2DId+0ZGjMus/AvwrYN6eu/T99tTRomJ9Pfn0w7O4R+Ll05Iml3V7/rDrIKAADg\njwQOYB/J6RdlT16QvvF8eHd7qJ+cvGd2qKgOJ81L5l06dG+anXxif6syx5/U3yoAAIBeAgeM\nWB0hZEI4koehHK2a8cm0RcP5hpkpp+SmzcmvX7vfeG7Ggsxx7xnOmQAAAJHyFBUYadIQNqXp\nr9L0yTRdlaZPhrCl3FMaDmOu/MfcaWf0HclNnz/mLz5XrvkAAACjzlWFAAAYCklEQVRxcQYH\njCxp+koIm/ssdoTwXJJMD2FKGWc1DJK6cTWfvjX/yprihpdDErInvTfbNLvckwIAoAzy+UK5\np0CUBA4YAh1b0+0vhc4doXp8MuH0UFk34Fe2960bIYQkCSGENH01SY4f3Pt6jky50+aG0+aW\nexYAABza448/fvfddz/22GOvv/76+PHjFy5c+E//9E/z5vX/rD0YYgIHDLJ042Ppxl+GdE91\nTl//r+SUxcmkAT7otK2f8XwIu0IYeCgBAIChddttt73++usf//jHp0+fvmnTpn/+538+++yz\nf/7zn59//vnlnhqjlMABgynd+my64b/7DoRCV/rqQ0nNpDD2hAHsoHhEqwAAYLh9/etfP+20\n03oXly9fPmPGjNtvv13goFwEDhhUb/1m3+WeK0yK6VtPJwMKHLX9jCch1BzdzMogbXsxbFuT\ndm1PKupCw3uTCfP3XHIDAED8+taNEEJTU9Mpp5yyefPm/raHoeYpKjCY0vat/Yy/M7AdNIQw\nttT4pBAqjnhWZZG+/kD62n3pjpdDxzvpzv+XbvxJcf1dodhd7nkBADAkNm3a9Nprr51xxhmH\n3hSGhjM4YFBlK/f+OQkhl00ymTRfSPqOH1SSzE7T50LY0WdsYpI0D+Ykh17a9kK6fd3+o7s3\npltWJ5PffwQ73N2Rf2lj647dXSdOGts0pX4QpggAwOApFovXXHNNZWXljTfeeMiNr7vuup/+\n9Kf9re3s7BzUqTGKCBwwmJKGpp6TNZIxlaG+OmQyoecylUw+5NtCrmEA+6hKkvkhbA9hVwiZ\nEOqjvLdo6wulRtO09YUjCBwPP/n6dx5+YVf7nrM/5jY1/q/lc6Ye19/lPAAADLJCobBz587e\nxfr6+kxm79UAaZped911jzzyyA9+8IP9rlsp6ZprrvngBz/Y39rrr7/+KGfLqCVwwGBKpr4/\n3fZ8kmkPDTX7PNS12Ja+eV/ynqtCZoBXmowPYfyQTHFYpPldpYaT0F1y/GB+/ttNX79vbd+R\nNa9u/fy3n/z2DRdWV2aPdIIAAByGtWvXzp8/v+/i7Nmze/6cpulnPvOZ73znO3feeefll18+\nkL3Nnz+/7972c/PNNx/lbBm1BA4YVJV1mTnXpJv+PSQHnFmX3xF2PxfqRsVFiUmuLi25omK/\ns1HSUNgZirtDdmzIlD5R5T9+8cqBg29tb//5bzddcs5JRztRAAAGoLm5+fHHH+9dbGpq6vlD\nmqbXXnvtHXfc8b3vfe8v//IvyzQ72EPggMFWWRcy3aHU3+/TzjeT0RE4wrjTQ+uzBw4n42fu\nXchvSXf/OhS271nMTUxqzw7ZcX2378oX//BW6ZM+Xt7UNliTBQDg4Gpraw98+GuapldfffWK\nFSu++93vXnnllWWZGPQlcMAQKH32wiiSNEwPjfPSrc/sMzj2lGTi+/YsFFrTnf8V0sLe1fkt\n6Y5HkoZLQ2bM3peEkCQhLfXP0wNnAQDK63Of+9wdd9xx2WWX1dTUrFy5smewpqZmyZIlR7nn\n7u7iUc+O0UjggEGXhKqJofOtEisqJw//bMolOWFJaJgRtv0+7dyeVNaH+unJhDnhjzcmSduf\n3adu7BntSjueT2rO7B2oyGWaptSv37xj/y1DmHHSuAMHAQAYNqtWrQohtLS0tLS09A5OnTp1\n48aN5ZsUo5rAAYMvaXhf+taD+9xkNKQhVxfGzuzvJcekpK4p1DWVPtOi8M6BY2kISX7/8asu\nav7S957eb/DESWM/OG/qoEwSAIAjs3r16nJPAfaROfQmwOGqmZZMWhyyNXuvVak+IZl8echU\nlnNWI0qpy06SEA68vGfRrOO/8IkzG+ur+4xMvu2asytzPr4AAIC9Yj2D44knnrj11ltXrVrV\n2dl5yimnfOpTn7rhhht617a1td14440tLS2tra0zZ8686aabLrvssjLOltGodkYyZlroejsU\n3g0V40PlceWe0AiTmxC6St09NNd44NiFZ0xZNGvya2/ubN3dddKksZPHjzlwGwAAYJSLMnD8\n4Ac/+MQnPnHuuefeeuutdXV1r7766ttvv927tlgsXnLJJWvWrLn11lunTZv2b//2b8uXL29p\naVm6dGkZ58xolKkI1S6jKC2pnpl2bQxh39tHJbmkekbJ7StymeYTGoZjZgAAQJziCxxvvvnm\n1VdfvXTp0h/+8IeZTIlz1FtaWp544okVK1Z88pOfDCF89KMfPfPMM//+7/9e4IARJHdcUndh\nuvvXobh7z0i2Pqk9J2TGlnVaAABArOK7iH3FihW7du368pe/nMlkisUSTw+6//77q6urr7ji\nip7FbDZ71VVXrV+/fs2aNcM7U+CgKqYmDX+W1F2U1C5K6j+aNPxpyE0q95wAAIBYxRc4Hnvs\nsRNOOOGZZ56ZMWNGLpdrbGy89tprW1tbezd49tlnm5ubq6qqekfmzJkTQli3bl0ZpgscRJIN\nFZNDVVPITYzx4wgAABg54rtEZfPmza2trX/913/9xS9+8ayzzlq9evUtt9yydu3aJ554oueK\nla1btzY1NfV9yYQJE3rGD77nhx9+uL8nNnd0dIQQCoXC4PwMQAgvbWx7cUNrvlBsntow+9QJ\n5Z4OAAAQtxEdOAqFws6dO3sX6+vrey5L2bVr19e+9rW/+7u/CyF8+MMfTpLkpptueuSRRxYv\nXnyQvSVJcvC3u/322zds2FByVZqmIYRsNnvYPwNwgHc78l+59/ePr32zd2T+acf94xXzJtRX\nHeRVAAAABzGiA8fatWvnz5/fd3H27NmNjY0hhL4tY8mSJTfddNNvf/vbnsHGxsZt27b13U/P\nYs95HAfx6KOP9reqq6urqqqq51IX4Ch9deWavnUjhPC7V965+e6nv/qZRYfqkAAAHPu68yVu\ntgiHNKIDR3Nz8+OPP9672HPhydy5cx999NG+txft+XPvE1VmzZq1cuXKjo6O6urqnpGe24vO\nnj172GYO9Oedto7H175x4Pizr21/aWPre08cN/xTAgAAjgEj+q5+tbW15/dRU1MTQrj88stD\nCA899FDvZg888EAI4ZxzzulZXLZsWWdn5z333NOzWCgU7rrrrmnTps2dO3e4fwDgAK+/vStN\nS6967a2dpVcAAAAcyog+g6OkCy64YPny5V/84hd37ty5YMGCVatWfeUrX1m8ePEHPvCBng2W\nLVu2aNGi66+/vq2tramp6Y477li3bl1LS0tZZw3sUZHtt6tWuM0NAABwpOILHCGEu+++++ab\nb77zzjtvu+22KVOm3HDDDV/60pd612YymR//+Mc33njjl7/85ba2ttNPP33lypVLly4t33yB\nvZpPaKiuzHZ07f9MokwmmXPq+LJMCQAAOAZEGTiqqqpuueWWW265pb8Nxo0b981vfvOb3/zm\ncM4KGIjqyuxVF03/9o+f32982fmnThw3pixTAgAAjgFRBg4gan9+YdO4sZV3/OeLW3d0hBDq\naiqu+vD0P1t0crnnBQAAREzgAMrgorNOuOisE97a3l5M0+PHj0k8HhYAADg6AgdQNpPHuyYF\nAAAYHCP6MbEAAAAAAyFwAAAAANFziQoAAAAjSL67UO4pECVncAAAAADREzgAAACA6AkcAAAA\nQPQEDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0cuVewIAAACwV3e+\nWO4pECVncAAAAADREzgAAACA6AkcAAAAQPQEDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP\n4AAAAACiJ3AAAAAA0RM4AAAAgOjlyj0BAAAA2CufL5Z7CkTJGRwAAABA9AQOAAAAIHoCBwAA\nABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAAQPRy5Z5ATH70ox+t\nW7eu3LMYPg8++GBdXV02my33RBhFduzYkaZpQ0NDuSfCKFIsFt94442pU6eWeyKMLlu2bKmv\nr6+qqir3RBhFOjo6qqqqLrjggnJPhBJ+/etfl3sKcCwQOAYkl8udc845K1euLPdEhtWrr76a\nzWYzGaf5MHwKhUIIQVZjOKVpms/nc7lckiTlngujSD6fz2Qy/k+W4VQsFkMIq1atKvdEKO3i\niy8u9xRGioULFz563/XlngVHpVzHc5KmaVnemBGuWCxms9lf/vKXMj/D6eqrr+7q6rrzzjvL\nPRFGkVWrVi1atKjnF5vlngujyJw5c/7mb/7ms5/9bLknwijyjW9847vf/e4zzzxT7okADBW/\nNwAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0BA4AAAAgegIHAAAAED2BAwAAAIiewAEAAABE\nL1fuCTBCZTKZJUuWnHTSSeWeCKPLggULuru7yz0LRpcTTzxx8eLFFRUV5Z4Io8uFF144a9as\ncs+C0WX27NkXXHBBuWcBMISSNE3LPQcAAACAo+ISFQAAACB6AgcAAAAQPYEDAAAAiJ7AAQAA\nAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0BA4AAAAgegIHAAAAED2BAwAAAIiewAEA\nAABET+AYpbZu3XrDDTdceOGF9fX1SZLcfffdB27T1tZ23XXXHX/88dXV1WeeeWZLS8vhbgAH\n95vf/CY5wOrVq/tu4zBj6Di6GGo+5RgGvtQB9BI4Rqk33nhjxYoVlZWVF110UckNisXiJZdc\ncvfdd3/hC19oaWk59dRTly9ffv/99w98Axigz3/+8/f2MX369N5VDjOGjqOLYeNTjiHlSx3A\nXimjUqFQ6PnDz372sxDCXXfdtd8G9957bwhhxYoVPYv5fH7u3LnTpk0b+AZwSE899VQI4cEH\nH+xvA4cZQ8fRxTDwKccw8KUOoJczOEapTOYQ/+rvv//+6urqK664omcxm81eddVV69evX7Nm\nzQA3gIFrb28vFosHjjvMGDqOLoaTTzmGji91AL0EDkp79tlnm5ubq6qqekfmzJkTQli3bt0A\nN4ABuvLKK2tqaqqqqs4777xHHnmk7yqHGUPH0cWw8SlHeflSB4weAgelbd26dcKECX1Heha3\nbt06wA3gkGpqaq6++up//dd/ffjhh7/61a9u3rx58eLFfS/6dZgxdBxdDAOfcowEvtQBo0eu\n3BNgyBUKhZ07d/Yu1tfXH/JUxoNIkuQoN2B0Knkczpw589vf/nbv4BVXXDFnzpx/+Id/WLp0\n6cH35jBj6Di6GEQ+5RjJfKkDjj3O4Dj2rV27dnwfzz333EBe1djYuG3btr4jPYu9gf+QG0Bf\nAzkOJ06cuGTJkpdffnn79u09Iw4zho6ji+HnU46y8KUOGD0EjmNfc3Pz4300NTUN5FWzZs16\n6aWXOjo6ekd6bjQ1e/bsAW4AfQ3wOMzn86HP/dIcZgwdRxdl4VOO4edLHTB6CBzHvtra2vP7\nqKmpGcirli1b1tnZec899/QsFgqFu+66a9q0aXPnzh3gBtBXyeOwu7u77zYbNmx46KGHZs6c\n2dDQ0DPiMGPoOLoYBj7lGAl8qQNGD/fgGL0eeOCBrq6utWvXhhCeeuqp6urqEMJll13W82ul\nZcuWLVq06Prrr29ra2tqarrjjjvWrVvX0tLS+/JDbgCH9PGPf3zMmDELFiyYMGHCK6+88q1v\nfWvnzp3f//73ezdwmDF0HF0MA59yDA9f6gD2SBmten991Fd7e3vvBtu3b//0pz89adKkqqqq\nefPm3Xffffvt4ZAbwMH9y7/8y9lnn93Y2JjL5SZOnLh06dInn3xyv20cZgwdRxdDzaccw8OX\nOoAeSZqmQ19RAAAAAIaQe3AAAAAA0RM4AAAAgOgJHAAAAED0BA4AAAAgegIHAAAAED2BAwAA\nAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwAAABA9AQOAAAAIHoCBwAAABA9gQMA\nAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAAQPQEDgAAACB6AgcAAAAQPYED\nAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0BA4AAAAgegIHAAAAED2B\nAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwAAABA9AQOAAAAIHoCBwAAABA9\ngQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAAQPQEDgAAACB6AgcAAAAQ\nPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0BA4AAAAgegIHAAAA\nED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwAAABA9AQOAAAAIHoCBwAA\nABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAAQPQEDgAAACB6AgcA\nAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0BA4AAAAgegIH\nAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwAAABA9AQOAAAAIHoC\nBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAAQPQEDgAAACB6\nAgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0BA4AAAAg\negIHAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwAAABA9AQOAAAA\nIHoCBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAAQPQEDgAA\nACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0BA4A\nAAAgegIHAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwAAABA9AQO\nAAAAIHoCBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAAQPQE\nDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAAAED0\nBA4AAAAgegIHAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwAAABA\n9AQOAAAAIHoCBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6AkcAAAA\nQPQEDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJHAAA\nAED0BA4AAAAgegIHAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDoCRwA\nAABA9AQOAAAAIHoCBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA6Akc\nAAAAQPQEDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAAgOgJ\nHAAAAED0BA4AAAAgegIHAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAAAIDo\nCRwAAABA9AQOAAAAIHoCBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgAAACA\n6AkcAAAAQPQEDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4AAAA\ngOgJHAAAAED0BA4AAAAgegIHAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANETOAAA\nAIDoCRwAAABA9AQOAAAAIHoCBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADREzgA\nAACA6AkcAAAAQPQEDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAERP4AAAAACiJ3AAAAAA0RM4\nAAAAgOgJHAAAAED0BA4AAAAgegIHAAAAED2BAwAAAIiewAEAAABET+AAAAAAoidwAAAAANET\nOAAAAIDoCRwAAABA9AQOAAAAIHoCBwAAABA9gQMAAACInsABAAAARE/gAAAAAKIncAAAAADR\nEzgAAACA6AkcAAAAQPQEDgAAACB6AgcAAAAQPYEDAAAAiJ7AAQAAAETv/wMXnRAaLJWWuAAA\nAABJRU5ErkJggg=="},"metadata":{"image/png":{"width":720,"height":720}}}]},{"metadata":{"trusted":false},"cell_type":"code","source":"g='ENSG00000111057' #### Plotting the log expression of KRT18 \nplotExptSNE(FG,g)","execution_count":10,"outputs":[{"output_type":"display_data","data":{"text/plain":"Plot with title “ENSG00000111057”","image/png":"iVBORw0KGgoAAAANSUhEUgAABaAAAAWgCAIAAAAnwnOfAAAACXBIWXMAABJ0AAASdAHeZh94\nAAAgAElEQVR4nOzdd5xdZbno8edZu07vM0kmvZAe0oAkQEggdFQQOKKi2PB4UGx4LKj3cAU+\nKDbs16soR1APXpoUqRKkJYEAKRPSSJ9MZpLpfbf13j/2ZM/MnrWnt535ff9xred917uemcxH\nZj/zFjXGCAAAAAAAQDKzRjoBAAAAAACAgaLAAQAAAAAAkh4FDgAAAAAAkPQocAAAAAAAgKRH\ngQMAAAAAACQ9ChwAAAAAACDpUeAAAAAAAABJjwIHAAAAAABIehQ4AAAAAABA0qPAAQAAAAAA\nkh4FDgAAAAAAkPQocAAAAAAAgKRHgQMAAAAAACQ9ChwAAAAAACDpUeAAAAAAAABJjwIHAAAA\nAABIehQ4AAAAAABA0qPAAQBwVlJSoollZ2cn6vn00093HOeKK66IxufMmRP3iscff/z9739/\ncXGx1+vNyMiYNm3aOeec84UvfOH+++/vmk9dXd3Pf/7zK664YtKkSampqV6vt6CgYMWKFV/+\n8pdffPFFY0zHzuXl5bfccsvcuXNTU1MzMjKWLFnyve99r6GhoeuwSdHziSeeuPnmm1etWpWa\nmhr7Pm/ZsqXrgEPRs/d5Tpw4MdEPzC9/+ctYt2uuuaabHy1VfeaZZxzTAAAA6I4BAMDJ9u3b\nu/nPR1ZWVqKeCxYsCIfDsdbLL788Gp89e3bH8W+66abeDB51//33Z2VldZPPgw8+GOu8adOm\n3Nzcrn1mzJhx8ODBjsMmS8/zzjuva7d33nmn67/aUPTsfZ7FxcVdu0X94he/iHW7+uqrE3WL\nevrpp7umAQAA0D13979hAAAgIkVFRVOnTu0YycjISNS5pKTkvvvu+/SnP93NgK+++uqvf/3r\n6HVKSsqZZ56ZmZlZVlZ28ODBqqqquM6/+tWvvvCFL8Ru09PTly5dmpOT09jYuHfv3sOHD4uI\nbdvR1rq6ug9+8IPV1dUikpmZ+dGPfrS1tfWBBx4IhUL79u370Ic+9Prrr1uWlUQ9RURVJ0+e\nvGzZMtu2//73v3fzjR30nn3KM2bdunU+n69jZNq0abHr5cuXt7a2xj2ycePG6D+91+tduHBh\nN5kDAAA4G+kKCwBglOo4L+Pzn/98L3tGTZgwoampKdrqOIPjv/7rv6LBtLS00tLSjqNt3rz5\n+9//fux29+7dHo8n2llVb7/99tjIUQcOHPjBD34QXaVijPnRj34US+PZZ5+NBn/1q1/Fgk88\n8URy9TTGxL7kjot3HGdbDHrPPuUZm8Fx7Nixrm/sRkVFRUpKSvTZG2+8sU/PAgAARLEHBwBg\nMI0fP15EysrKfvKTn3TTLfY3f5/Pl5qa2rFp2bJl3/jGN2K3P/vZz0KhUPT6m9/85ne+8524\n/lOnTv3617++du3a6G1sMkJhYeFFF10Uvf7IRz6iqtHrRx99NLl6ikjcl9yNQe/ZpzxjVq1a\n5ff709PTFy5c+I1vfKO8vLz7t9xzzz0tLS0i4nK5Ov7rAwAA9B4FDgBAzx566KEVnd1zzz2O\nPW+55ZboZhl333338ePHEw24atWq6EV1dfX06dOvu+66H/7wh88//3x9fX1czxdeeCF6oapf\n/epXe0w1tk3mzJkzY8Hs7Oz8/Pzo9datW5Or58jqX54HDhwIBAJNTU0lJSV33333woULE21f\nKiJ1dXWx9UrXXnvtjBkzBi17AAAwllDgAAD0rKKiYlNnBw8edOyZl5f3rW99S0QaGhpuu+22\nRAOuW7fuk5/8ZPS6trb2wQcf/PrXv37RRRfl5eVdccUVb731VqxnaWlp9GL8+PGxD9Uikp6e\n3vHcjYkTJ4pIOByOHe0Rtylp7LaysjKJeo6sfuRZVFR01VVXffnLX/7Upz41bty4WJ9rr702\nHA47vuXXv/51XV1d9Dr6wwMAANAPFDgAAIPsS1/60qRJk0Tkd7/73e7duxN1+8Mf/vDQQw9d\nfPHFsc0XRCQcDj/11FMrV6589dVX4/rH1kR0w3Q4LNZ0Pjg2dhsdJ1l6jqy+5vnYY4+VlZU9\n8sgjP/3pT++99969e/cuXbo02vTee+/961//6vqKlpaW2GygK664YtGiRYP7JQAAgLGDAgcA\noGddNxlNtERFRPx+/x133CEi4XD4m9/8ZjfDXn311c8880xdXd1bb731y1/+cvXq1dF4KBSK\njiAiHfet7HjAyve+97277rprwYIFHQf0eDyZmZnR67jVLrHbvLy8JOo5svqa5/LlyzueqJKe\nnn7zzTfHbt99992ur7j33ntjS5luvfXWQUocAACMRRQ4AACD7/rrr1+8eLGIPPbYY2+//Xb3\nnT0ez9KlSz//+c+/9NJLsYLF3r17oxfr1q2LXti2/fOf/zz21Fe/+tVvfvObs2fPjhvt9NNP\njxtBRGpra2MrKWIdkqXnyBpgnh3nfXQ9TTYcDsdOaVmzZs3KlSsHnjAAABizKHAAAAafZVk/\n/OEPo9fHjh3r2uG3v/3tpz71qfXr10cikVjwxIkTsY/N2dnZ0YsvfvGLLpcren3HHXf86Ec/\nCgaD3bz6yiuvjI323HPPRa//8pe/xD5pX3XVVcnVc2T1Ps8HHnjg97//fWtra+zZxsbGX/7y\nl7HbhQsXxg3+5z//+dChQ9Frdt8AAAADpHFLagEAiCopKYl9Ii0oKJg8eXJch4cffnjKlClx\nPf/4xz9+4hOfiF5fcsklzz77bKz/7Nmzd+3aFb2+5557vvKVr4hIWlraokWLCgoKGhsbN27c\n2NzcHO1w66233nnnndHrH//4x1/72tdi42RnZy9dujQzM/PEiRObN28OBAIiUlxcHN2OtK6u\nbv78+UePHhWRzMzM66+/vqWl5YEHHoieNXvWWWe9/vrr0akEydJTRP70pz9t27ZNRHbu3PmP\nf/wjGvz4xz9eUFAgIpdeeukFF1wwRD17n+f3v//9b33rW1lZWWvXrp05c2ZNTc1TTz0VOyB2\n8eLFb731VsdJHMaY+fPn79y5U0SWL1/+5ptvCgAAwEAYAACcbN++vfv/guzcubNrzz/+8Y+x\nEbZu3drxA+3s2bNjTd1s4SEiS5Ysqaur65jMb3/729TU1G4eWbNmTazzpk2bcnNzu/aZPn36\nwYMHOw6bLD2vvvrqbr7222+/fUh79jLPu+66K9FoM2bM2LdvX9wP2MMPPxzr8PDDDxsAAICB\nYYkKAGCoLFq06IYbbnBs+sIXvvDKK698+9vfXr169cyZMzMyMjweT0FBwZo1a372s59t2LAh\ntrdl1Gc/+9lDhw59//vfv+CCC4qKirxer8/nmzBhwpo1a2699daNGzeuX78+1vnMM8/csWPH\nV77yldmzZ6ekpEQnidx2221btmyJTjlJup4jq5d53nTTTQ8++ODHPvaxhQsXFhQUuN3u3Nzc\nc8899yc/+cnWrVunT58eN2ysIDJ37txRsh4HAAAkNZaoAAAAAACApMcMDgAAAAAAkPQocAAA\nAAAAgKRHgQMAAAAAACQ9ChwAAAAAACDpUeAAAAAAAABJjwIHAAAAAABIehQ4AAAAAABA0qPA\nAQAAAAAAkh4FDgAAAAAAkPQocAAAAAAAgKRHgQMAAAAAACQ9ChwAAAAAACDpUeAAAAAAAABJ\njwIHAAAAAABIehQ4AAAAAABA0qPAAQAAAAAAkp57pBNIDsaYiy++uLq6eqQTAQAAAHAKWrt2\n7Q9/+MORzmJUuPnmmzds2DDSWWBARurnWY0xw//WpBMMBn0+39e//vUZM2aMdC4AAAAATinP\nPvvs0aNHN27cONKJjAqLFi36mys40llgQD7hyx6Rn2dmcPTBBz7wgVWrVo10FgAAAABOKTU1\nNY8++uhIZwEkPfbgAAAAAAAASY8CBwAAAAAASHoUOAAAAAAAQNKjwAEAAAAAAJIeBQ4AAAAA\nAJD0KHAAAAAAAICkR4EDAAAAAAAkPQocAAAAAAAg6VHgAAAAAAAASc890gkAAAAAANDO8rhG\nOgUkJWZwAAAAAACApEeBAwAAAAAAJD0KHAAAAAAAIOlR4AAAAAAAAEmPAgcAAAAAAEh6FDgA\nAAAAAEDSo8ABAAAAAACSHgUOAAAAAACQ9ChwAAAAAACApEeBAwAAAAAAJD33SCcAAAAAAEA7\ny+sa6RQwMOGReS0zOAAAAAAAQNKjwAEAAAAAAJIeBQ4AAAAAAJD0KHAAAAAAAICkR4EDAAAA\nAAAkPQocAAAAAAAg6VHgAAAAAAAASY8CBwAAAAAASHoUOAAAAAAAQNKjwAEAAAAAAJKee6QT\nAAAAAACgneVxjXQKGJjwyLyWGRwAAAAAACDpUeAAAAAARESkuVZaG0c6CQBAP7FEBQAAAGOb\nMWb3y/aWx6WlXkQkI99afrVOXT7SaQEA+oYZHAAAABjT7M0P2RseaKtuiEhDpb3+t2bn+hFN\nCgDQZxQ4AAAAMIY1VZsdz3cN2289KpHQ8KcDAOg3ChwAAAAYq4yxN/1VjHFoCrWYqkPDnhAA\noP8ocAAAAGCMMtufNoe2JGwOM4MDAJIJBQ4AAACMSca2SxwWp5ykmj1++JIBAAwYBQ4AAACM\nSc21Ekh4KKxOXSap2cOYDQBgoDgmFgAAAGOSJv5TX2aRdfbHhzEVAJ1YXtdIp4CBaRmZ11Lg\nAAAAwJiUmi0Z+dJQ2bXFWvFh8aYM6ctNw35pPCx2UPz5mrNALO+Qvg4AxgIKHAAAABijrKVX\n2v/6fVxQx8/R4nlD+FY7aA4+ahr2xQKm4jVrypWSNmkIXwoAYwB7cAAAAGCM0ulnWWv/XdJy\nTt5bOnetdf5NIjp0LzVlL3SsboiIhBrsgw9LJDB0LwWAsYAZHAAAABi7dOpy1+Sl0nDcBJs1\ne7x4hnZlithhU1PiEA83m7rdmrtoaN8OAKc0ChwAAAAY2yxLssYN4ZyNjkINYoedmwLVw5MC\nAJyqWKICAAAADBdX4s1EXb5hzAMATkEUOAAAAIDh4k6TlCLHFs2YPsy5AMAphgIHAAAA0Het\n9RJs6sdz1oQLRV1xQc1bmqjwAQDoJfbgAAAAAHrNGHPwdXvnM9JaLyKSlmcteL8WL+7DCOmT\nrdM+acpeNE1HxA6LL0cLV7K9KAAMHAUOAAAAoLfsksfNnhfbj5FtqrI3/dFafI1OP7cPo/gL\ndfp1KkbsiFj8Qg4Ag4P/PwUAAAB6p6XWvPeSdDlwxS550jVlhbg8fRxOqW4Ajlze+GVcQG+w\nBwcAAADQK6ZynxjboSHcamqPDHs6AIBOKHAAAAAAvRMJ9acJADAsmBQHAAAA9E7muAQNqhmJ\nmjoztjRXmeYaTcuT1FzputwFANBfFDgAAACAXtGcKZoz2dQcjo8XL5KUrB4fN9UHzPaHTX1Z\n21O503XRNb2tjEQ1nDA1R0RUcydLel4fHgSAMYACBwAAANA7qtaKT9tv3GeqDrTHxs23ln64\nx0dNfZm94Tdih9sj1fvN679ynfef4s/s+dWhVnvz38z+DWKMiIhaOusca9m1fd/ZFABOWRQ4\nAAAAgF5LybbO+5Kp2C21R0QtyZumedN785x5758dqxttgk32gVesuZf3+Li94b/N4Xc6DGeb\nPS/b4ZC16oY+JA8Ap7Rk3WT0tddeu+yyy3JyclJTU+fNm/fjH/+4Y2tdXd1NN900btw4v9+/\ndOnSRx55ZKTyBAAAwClHtWiOzr5QT7ugl9UNEem6sKVNzSEJNZn6A9J4WCIB5z71FZ2qG7Ex\n92+U5tpeJgAAp7yknMHx4IMPfvSjH125cuWdd96ZkZGxf//+48ePx1pt27788su3bdt25513\nzpgx4957773mmmseeeSRK6+8cgRzBgAAwJjmeL6siASq7G0/FWOMiFgeHX+ujl8Vt/moqSlN\nNKipOaKp2YObKQAkqeQrcJSXl3/mM5+58sor//a3v1mWwwyURx555LXXXrvvvvtuuOEGEbn4\n4ouXLl36ta99jQIHAAAARopmTTQtNV3jxrSoSWm7sUPm6Itih7R4TeeHEx+20k0TAIwxybdE\n5b777mtsbLzrrrssy7Jth0L4Y4895vf7r7vuuuity+X62Mc+tm/fvm3btg1vpgAAAEAbnblW\nLFd81FJN88XFTMUGsUOdns2b6lzIUEvzpg5ikgCQ1JJvBsfLL788ceLELVu2vO9979uzZ09O\nTs7VV1999913Z2e3zc3bsWPHrFmzfL72/1QsXLhQREpKShYtWtTNyNXV1bW1zosYQ6GQYxwA\nAADoDc2Zai3/hL3tYWk9+QtnSpak2uLq8hdHOywtxyWtuD2SlqszzzF7X4kfc84F4ksfupyB\nkWJ5ulQDkWQSLMobYslX4CgrK6utrf3kJz/53e9+d9myZRs3brzjjju2b9/+2muvRVesVFVV\nTZ/eaben3NzcaLz7kRctWnT06NFuOmzYsGHVqlUD/goAAAAwFmnRfNf5p5maQ9JaK6n5Eqo0\nR5527ho9C7YD64wP2b50s/MFiYRERNxea/7FuuDSIU4ZAJJJ8hU4bNtubGz86U9/+uUvf1lE\n1q1bp6rf+c53nnvuuUsuuaSbB7WnBYrvvPNOQ0ODY1Nzc/PChQvT0ymQAwAAYABcHs2f2Xbd\n5IsvY0SpS1IKo5em4ZiU7zAtNZqaZ512jsy/yNSWiahmTxCPf3hSBoBkkXwFjry8PBHpWMu4\n7LLLvvOd77z99tvRYF5eXnV1dcdHorfReRzdKCgoKCgocGxqbGwcYNoAAABAJ2njNWuGqdsX\nF9ais8TlFRF71z/Mey9Gj18xIrL7GWvuFTp99fBnCgBJIfk2GY3uo9Fxe9HodexElfnz5+/Z\ns6e1tTXWIbq96IIFC4Y1UQAAAKBbOv2Dmnd6h3uXjj9Hi88XEXP0HbP3hU6Hy9phe8djpiq+\nIAIAiEq+AsfVV18tIk8++WQs8vjjj4vIihUrordXXXVVIBD461//Gr2NRCL333//jBkzut9h\nFAAAABhuLr9Oe7+18Gadca3O+rC16EtavDZ6YIo5vNHxCXN40/CmCABJI/mWqKxevfqaa675\n7ne/29DQsHz58g0bNvz4xz++5JJL1qxZE+1w1VVXrVq16otf/GJdXd306dP/8Ic/lJSUPPLI\nIyOaNQAAAJCAL1t92XEx01Tp3LnpxJDnAwDJKfkKHCLywAMP3H777X/6059+8IMfjB8//pZb\nbrnttttirZZlPfXUU9/61rfuuuuuurq6uXPnPvTQQ1deeeXI5QsAAAD0jboTbEHqZm9RAHCW\nlAUOn893xx133HHHHYk6ZGdn/+Y3v/nNb34znFkBAAAAg6ZwjjQcE4k/B1AL54xIOgAw+iXf\nHhwAAADAKc+aeb6k5sUFNXOCTlk1IvkAwOhHgQMAAAAYfbzprnO/olNXiTddRMSfqTPPt86+\nWVyekc4MAEappFyiAgAAACSfpj2mbrOEqkQ94i/WnLPFk9tdf2+atfAaWXiNRELUNTCmWF7X\nSKeAAbJ77jIEmMEBAAAADDlT/bI58Q8JHhcTEbtVmveZsr9Ia1mvHqa6gVHvjjvuUNVx48aN\ndCIY0yhwAAAAAEMsVCX178QHTdhUrx+JbIBBtnPnzjvvvLOoqGikE8FYxxIVAAAAILFI0JS+\namr3S6hJUwu1eKVkTe3zIC2HRJxOfQ2ekEiTuNIGnCUwYmzb/vSnP/2JT3xi7969JSUlI50O\nxjQKHAAAAEACgXp76++ktSZ6Z5pPmModOuV8nXJ+n4YxdiBhmx2gwIGk9vOf//zQoUNPP/30\n1VdfPdK5YKxjiQoAAADgzOx/OlbdaA8eWi9N5X0aR91ZCRpc4sroX279ZkZo8z+ckvbv3//t\nb3/7F7/4RVZWgh9yYBgxgwMAAABwYmxTtdOxwVS+q2l92UwxdYZYfrFb4+Npp4k1bBuImkCk\nNmjX2Cakoi4rNcVVaKl3uN6OU9ONN9540UUXffCDH+zTU9/4xjf++c9/JmrduXOnzFsx4NQw\nFlHgAAAAwChmjF1xyNSUa0auVTRVPMP4gTzcKnbYuSnY0LehLJ8WXm5OPC2R5vagv1hz1/Q7\nu75qDh8L2W1pGzFhu6nRPpjmmexS/7DlgFPM7373u82bN7/77rt9ffDSSy/NzU14RvLWrVsH\nlhfGLgocAAAAGKXsikOhv//aPro3eqtZ+Z7Lb3TNPmOYXu/2i+URO+TQ5Mvs82j+SVp8gzTu\nMMFKsbzqnyipM0VEjG32bjIV70kkrPlTdM454h78Ik7Ybo5VN2KMmJbw8XTP5EF/HcaCysrK\n//zP//zmN7+ZlpZWW1srIuFw2BhTW1vr9XpTU1O7eXbNmjVr1qxJ1HrbbbcNdrIYKyhwAAAA\nYDQyjbXB+/6XaW7/WG7qKoP/c7fvhtusqfOHIwO1tGCBqehyvKtamr+gPwNaPslcqh0jTTX2\nUz8xJw5F74yIvP2kdemXtGBKf8ZPLGyaHeMR02LEVjbmQ9+VlpbW1dXdeuutt956a8d4Tk7O\nDTfccN99941QXhjTKHAAAABgNAq/+WzH6kYbOxJ+5WHv8BQ4RHT6JabhqDQfbw95PVI03wQr\n1O0Wb84Ax7f/+X9j1Y02DZX2s79wffj74hrMX9S721jU2KIUONBnM2fOXL9+fcfILbfccuDA\ngUceeWTcuL7sUAMMHgocAAAAGI1M2T7HuH30veFLwpNmLfu8KdskNftMpEG8ITEBadlvWvYb\ntTR3mRatEdEeh3HWUGWO7HCI1x03Zbt0Ur8miSTgEuetTFVcqnwiQH+kp6fHLTPJyck5evRo\nN2tPes/yugY+CEaU0+K+oUexFgAAAElF+1tQ6OfrXFq8Sud/WFJdYgLtcWObqjfNidfbAxIJ\n2rUtkeOBSHXEdDkwpQtTfzxhW13FgHLuwiN+V0uTGBMX97o42hPAqYN6LQAAAEYjq3hmZM9m\np/is4U/G1O+VUJ1DvPotzV8paoXsxpZwuZFIW0NEvFZ2iruou0G9iXdh7Kapj0zdUbP1EVO1\nL8UYcbmDU+aHZp1hPF4R8VgZflf+YL0IeOGFF0Y6BYx1zOAAAADAaOQ64xJN63JYicvlXn3N\nCGQTOOEcj7RKuNE2oeZwWXt1Q0REgnZtIFLdzZCaP0nSnHbxcLl14rz+p9qBqT1iv/RTU/le\n29yNSNi7f2vqG0/7rdw0z+RU94T+r68BgNGHAgcAAABGWmu9vf0h++Wf2C/dbb/9Z2moEBFN\ny/R+4nZr8pxYL80p8n74Wx0jw0cT7wigVtCuix6BEidoO0z66PigtfrjYsWPbJ11jaQOzsoR\ns+NJicSvhNeao95jh92aMiivAIDRgyUqAAAAGEmm5qC98bcSPrm9RUN55NgWa/GHtXipVTjJ\n96k7TeVRu/qYZuRaRVMG92yRPkid6Bz35og73Q7XOzbaJihiupklodOXWVd/12z8f6Z8n9hh\nzZ+iZ1ypU04flJRFjKl03qjVnNirk5YN0lsAYLSgwAEAAIARZOwtD7ZXN6LsiL39IVfhXPGk\niKoWTHQVJKgvDBdNmyLp00zjgfh40RoR0QTTolWsHteAaM54mX2GjCsWT4oWnqbFCwcjXxER\nMUbssHNTJDhobwGAUYMCBwAAAEZOw3FpdDoxJNRqKvfo+MGayzAIdOKVcuI1U/2WmIiIiDdb\niy7QjBki4rbSHFejuK0e9go15bvsjfdJoLHtds96LZptnX2jePyDkbEl6QXS4HBWi2aOH4Tx\nAWCUocABAACAEWOCDQnbAombRoTl0aI1WniuBGvE8oqnfQNUj5XhtlLDdnPH7iqW31XQ3YDB\nJvv1eyXU0jFmKnbbWx62zvjo4KQ8fbW95aH4SSRun045c1DGB4BRhU1GAQAAMBKCTWbvv8y+\n1xN28GcPYza9pi7x5XesbkSluif6XfmqLhFRUY+Vnu6ZYqm3m5FM6da46kZb/NDmrjuD9jPZ\nGefonHWd9jFNybZWfkb8g7OJKQCMKszgAAAAwHAzpVvtN/8swWYREb9brC5/dfOla/6s4U+s\n31TU58rzufKM2Im25IhjGiudGyIhaamV9G5nf/Q6L2v++2TKWebEXtNarxlFOm6BuLsruwBA\n8qLAAQAAgOHVVGVvvK99kkIwIj4V7bCOwu2zFn9E3L4heLeRhh2meY+EG8SdoWmzJX1ej/uA\n9kkvqxsioh6/w9GyUZ4OZ7gG6k1zpXpSJLWw65myvZJeqOmFg/lFAkPM5enXjzrGPAocAAAA\nGFbm4KZOSzBsI60hcbvE49P8qZJZbM1Y47CGwkRM+ZtSt8+EmjUlX8edKenFfXxxxBz/u7Qc\nbrsN1ZiWw9K0Rws/IDoSC7fHz5Ntj3cNa+4U8aWLiAQbzJ4nTcU2ETEi4svU067QwsE7ZgUA\nTi0UOAAAADCsTMOJLiGRUERCzdZZnxXL6RfUcLO947+lue28FdN41FRu10lrtfjcPry4YXt7\ndSOm5ZA0bpeMETiuRbMn6qzVZu/LnaIujy69RkTEjtjv/EEay9ubAvVm+19k4fVaOH9YEwWA\nJMEmowAAABheidaeuDyJlmCYQy/EqhsnQ7Y5/KI0lTv2dx6k+T3neJNzfBhYS6+1VtygWRPE\ncoknRYsXuS7+luZNExFzfHun6sZJ5sDzw54mACQHZnAAAABgWOm4uWbfq07xOQm2wzCmakeC\n+LuaNq63L440J4g39XaEwac65QydcoYYO36ZTP0R5ycaKyQSlHDA3vNPqS0VEcmeZJ12vvgy\nhiI/U33AlJdIS42k5mnxUs0cPxRvAYBBQYEDAAAAw0qLF+m4uaZ8Z6eoJ8Va9AHnByIhiQSc\nm4L1fXixO11C1U7xISkN9E3XTUBMwh1ITeV79ht/aj9i9sTeyMEN1qrPat70wUzJGHv7w+ZQ\n+zm+Zt96a/bFOuvCwXwLAAweChwAAAAYXqrWuf9udr9o731ZWmrF7dNxc63Tr5T0fOf+Lo+4\nfM41Dm+0NmEkfNREakSMWtnimShiSaRGwuXGDqgrUzyTRD2aNtt03YNDRNNmD5r9YQkAACAA\nSURBVN7X1kW4xRx40VTukkCDpOZr8Vk6YXmnI2MSyZjgHPfn2u/8rb26ERVqsd+833Xxdwdx\nt1RzZFPH6oaIiLHtXU9b2VO04LTBegsADCIKHAAAABh2llvnXuSae5GEA+L29nRQq2ruXHNi\ni0M8b66YFtOyQSI10ZARkeAu0QwJHoreGRGxtmnqCkmfJy2HpWl3pzHS5kj63MH5oroK1Nub\nfy2tdW23DUfNrkekarcu+miPZ9Nq0enm4IvSUhPfkD9fKp51eKC52lQfHMRJHObIm4ni8QUO\n0yzhMmOaVVPENV6s9MHKAQD6hAIHAAAARk6iDUc70ynrTGOptFR2Ck5cLWkTTPPLsepGG7tB\nTH20snEy0mKaXtWMy7XgUkmbbZp2SbhB3JmaNltSB3VZR2dm//Pt1Y1Y8MQOObFTC+b18LDL\nYy35tP3uw1J7oC3i9un0i0RTEq5dae3Lgp2emGan5Twi0lzV6Ta0zwTfFYlI23d8p3rniIcp\nHgBGAAUOAAAAjHqedGvRv5tjG6Vuvwk2amqBjjtLMqeI3SiRLofOioiqqCXGbo+YkIQOim+u\npE7XoSxqdGQqdzk3VO6UHgscIpKSZy27URqOmaYK8aRp5kTxpJrK90REjNMUkEHdZ1Q9KaZL\ndUZExJPSfh2pNMHtnZttE3xXrQxxsR0pgOFGgQMAAADJwPJo8blSfG6nz/V2Y+8HMJH6Xmx9\nMahCCc5tSRR3oJIxQTvsx6G508Sf1XViiKRka+7UPmfYjaJ50uBwTq0WzY9dm/BBx0dN6KBS\n4AAw7ChwAAAAYLjYrWL5B3NA7ctvs33qPCj8OdJS5RzvN8tlLfuIvfFeiQTbg26vtfyjYrn6\nP2zX98y8IFJeIo3HOwY1d5pOPqv9PlGByfSh8AR0ZXkH84cZYwcFDgAAAAwxO2iqN5j6Eom0\niuXRtJmav1rcg7EVpStX1CsmGB830ml9ioiIqLtoEN7YFzp+qdn/fJeo6vilAxq2aI7rwlvt\n3c9LzWERkZzJ1pyLJCV7IGM68KS4zv2Kvfd5c2yrtNRKar416Qydfl6nMkrCmtHJeCRslx80\n9ZWaXWgVTRVr0A55AYCuKHAAAABgKJmwXfo/Eji5U4YdMg07TfMha/L14h74nhGW+haZ1s1d\nNqWIr26Ie5x4Jg74dX2jU86ThjJzYkfbvRFxuXTmZQmPgO291Bxryb8NdJAeuX3W3Ctk7hWJ\n2tVVYCKVDg2uQhGx928LPvFbU30sGrOKpnje/x/WRPYfBTBUKHAAAABgCJm6be3VjZhIs6ne\nqIUXDsILPFPUSjWBEonUihixstQ3TzTVtLwt4eMitqhPfKepb8jOgu2G5dJF10vVbqncJYF6\nSS3QCcslNX8EMhki7hkSLhW7oVNQU9Uzyz62P/DAHRIJx8J2xaHAn/63/z9+ojnDPZUGwBhB\ngQMAAABDqfmwY9g0Hxq0LT9dBZq69uSsjbZFEJq+VsQWExTt5a4fjgeTDALNmy15s4di5BFm\nghI8LJIpliWmWUxI1COuYvXOFfWGX3usY3WjTaAlvPFJz6WfHol0AZz6KHAAAABgKJmQc9zu\nsnHGQHXd38HqubrR0hh++UF7z5umoUazC12nr3WteJ+4PIOd2ykndMw0bxTT2h7xTtHUFbF/\nBfvoPsfnEsUBYODY5gcAAABDyZPgxBBv7vDm4cA01QXv/Xpk8zOmvkqMbWrKwy/9NfTA98SO\njHRqo5vdYppf7VTdEJHgIdNa0n6baDbMcB/VC2AMYQYHAAAAhpBmLTR120RMl/jpI5JPR5FX\nHzJ18fuD2KW7IlvXu5ask5Yas+9ZU7NPwi2SVqiTz9Vxi/mALiISOiSmy/ITEQnsE/+i6KU1\nYWak6ljXLlbxrCFNDcBYxgwOAAAADCVfkRZdLJa3PaKW5q7QjJHY9bMze9+WBPF3pKHM3nSP\nqdgqwUaxI9JwzOz4m9n12DBlFm41VQdN1QEJtQzTG/vCxO0q2t7QGjuy133OVeKOX+mj/lT3\nyvcNaW4AxjJmcAAAAGBoaeZ8TZ1qGvdIqFbcGZo2zQTrTNUmsXyaOlF8w3GqiGnYK40HJNwk\nvlzNXiTeHBGRQLNz79Yme8+TEonfJcQcfUMnLJfMSUOZqG3efdbe+Xzb210enX2BNf9SsVz9\nHO/4ASktMS11mlWkM86SlMzByDLRhwgVbWuyxk31fey7wSd+ayqPtkUmzPC873OadQodIgNg\nlKHAAQAAgKHnTtPsJSIigSq79AlpPR4NG1HNWaTjLhQdspnFJmyOPGYa97fdNoip2qzjztec\nJZpTZJrruz6h2YVSd9DxWBVTuVuHssBhb3nU7Fnffh8JmXefsQON1vLr+jyWse1X7zc7/9V2\nJyKb/26tvkGnnzHAJNUz3gR2OTS4CzvOELemLvB//h674rCpr9ScIqtgkigLfAAMIQocAAAA\nGC4mbB9+SEJ1HUOmZqtYPi1aM1TvPPF6e3WjLRQxx17Q1InW6efbR/d2fcRasFL2/c15t41w\nq1N0kAQazd5/dQ2b/a/JvIslNcF2rQmY7c/Fqhttgs32i79z5U+RzMKBpCnuceKZKKHSTkF1\na8qS+J6Wyxo/TcZPG9DrMPZYnn5OWcIYxx4cAAAAGCam4b3O1Y2T8ZotYobq4BJTt8MxbOre\ndS2+wLX8kk5hl8d90SetKYvE5XV6SiQl7vAXI+EyCe6U4B6JVA401erDYmzHZE3Vwb6OZu98\n2Skatne/2ufMutC0szVliVipIiLqEs8ETb9YXH0rwQDA4GIGBwAAAIZLoMpp2YeIHZRQ3dAc\nHGsklGBHzGCdqLov/rS1YLW9501pqNKc8daCczRnnIjouCXm6Kb4R1w+LVrYIe0G07JJ7A4l\nG/cE9Z8R24ei78k6VTdiX0jfhjJSf9y5qa6ib0M5s8Q3R31zxIRFXRwuA2A0oMABAACA4aKu\nhJ+De1UUsKW1VMI1oj7xjRd3Vm9eKZZP7IBDiysl+r9W8ayuZ5fqrEulpdpUd1jA4k7R+f8m\n3oxYMqZlg8SdJxIuM4Et6l/ei8Sccs2eKKKOtQzN6ePGH6ri8UnQ6RAWr79f2SV6ER8oAIwW\n/P8RAAAAhommTnKeh+DNFk9Pp3uEKk31PyVUdXIsS9JP16wVPc4d0IyZzqtUMmZ295jLp0s+\nJZU7pXqfhFskrUgnLBNPWnuH8PH46kZbnofFt0jUaYWLHZbGChNs0rQCSXFazZGarVPPNAfj\nZ47opCWSXtBdtk504nyzf7NTfEFfhwKApECBAwAAAMMltVgzTzP1e+LCWrS2hwdNyFQ+KZEO\np7oaWxreEcsvGV02towbvHC1aTok4cZOwax5mt7zzpeaP1fy5zq32Q2Ox6yIGLEbxRW/3MaU\nbzW7npTWOhExIlo4T+ddKf7suG7W8utsl8fsf02MERFR1alnWUuv7THVrqwzPhg5ulMCTZ2+\nouJ5Oq2fE0wAYJSjwAEAAIDho8VXiG+TqXpT7KCIiC9Pi87vudbQvLdTdeMk07hVeypwiCfD\nmvEpc+I107hfwk3iy9OcJZo94FkM6ko4d6TLqg1zfIfZ8ufOkXdN0wlr1ZfF5enU1eWxll8n\ncy801YdFjOZMlvT8fmaYNc71wf+yN/0/c2S7hFolLceat1YXXcJZrQBOVRQ4AAAAMIzUrQVn\na8EqCdaKyyeu1N48ZELVzg2RZrFbxErp4XmXX8ddoHJBH3PtfswEa0bUL1ZGXMy894JDz6YT\n5tg7OvFMh6a0PE3LG2CCIiIZ+da6/xARCQXE4xuEAQFgFOOYWAAAAAw/FW9OL6sbIiLqStzW\nTdNQsjLEM71rWH2L4heu2BGpL3MepO7IEGTmhOoGgDGAAgcAAABGO/WNd27w5IvltJ3nsFD/\nYvUvFj15KImVpSnniKfrcSeJT3g1fTz8FQCQGEtUAAAAMOr5p4hvggTi5kFYmrViZPJpo+KZ\noZ4ZYgIilqjHuZfllvQiaSx3aMqa2LcXhgKmtkxENWe8uAdjUoYJiDK5A8ApggIHAAAARp9I\ntQnuF7tRNFU9xeIp1vzLTd0b0rRdjC0i4s7RnHPF18cCwRDpqUagM843W/8SH03J0QlLe/sK\nO2LeedLe+rSEgyIibp+19Ao9/VLRfs3INq2mdZuESsWERd3imaz+hVQ6MHpY3hFaeoYkR4ED\nAAAAo4tp3Sat78ZWdpjgPvFM1rSVmn22ZK2UcK1YPnGljWySfaLjF0skaHb/Q0Inz4LJnW4t\nuEZcvV1fY7/+F/Pu+vb7cMB+42FtbbJW/FufszEB0/iC2NFMjJiwBPeb8HFNvzDhJBQASAYU\nOAAAADCahMuldUd8MHRYAvnimy1qiSd3JNIaKJ14po5bZOpKJdio6UWSkWBXEUdNNWbnS13D\npuQ5WXyZ+NP7lIkJ7DpZ3ZD2/VDtRhPco775fRoKAEYVChwAAACnsur6wNb9VZV1rRPyUped\nVuAf9RO/TfBggvgB9c0e3lwGm9uveTP78Zw5vt95O1LbNicO6KSFfRsufDxBvEIocABIZhQ4\nAAAATll/f/3gH57e3RIIR28LslO+es3CZacVjGxWPWifXBAXbxrePEaT6LYjjuxI30cL9y0O\nAEmCY2IBAABOTa+WlP/qsR2x6oaInKhtue2/3yqrSlBBGCUSbXUZO4117NG8rkfPtrVo3uTo\nlak+ZG/7u73xPnv7E6auLEF/ERFxZTjHrcwB5AgAI48ZHAAAAKemR1890DUYCEWe2njoxsvn\nDn8+vaSeYhM67NDgKR72XPrPVO+Q+gMSbhZ/vhYsFV/2gIbLGqdTl5qDb8eFddYKSc8VMfY7\nD5u9L8cmepidz1vzL9X5lzoOpt4ZJuRQAVHvjAElCQAjjRkcAAAAp6ZD5Y2O8QPHGkREjC3B\nUmneIa0HEq4KGRHeKQ61DFeW+ueNRDZ9Fwma3feb/Y+YyndM7W5T/pq94zemcssAR7XWfkZn\nrWq/V9U551rn3iAi5uCbZs9LnZaxGNsuecqU73Qeyz1e/YtFOuzGom5NWSbu0b12CQB6wgwO\nAACAU5PH7fynLK/HktBxU/+KROpFjIiKujVtqXhm2Dufk4rdJtikmeP0tPO16LRhzllERFTT\nzpXgfhPcL5EGsVLFM1F9c0VH+/aoUeboetNwsFPIDptD/9CMyeIbwPkvHr+19jOy5ApTeUhU\nNX+KZBW1vfHARudMDmzQcQmm6vhOU0+xhMqMaVIrTdzFYqV2HUCCVSZUq+4M8eYny/cfwFhG\ngQMAAODUtGBa7r+2OqxEWDg13dQ+LyYoIm2nhJqwqXzF3nK/tLRN+jCNlaasROddYi28YtgS\nbmcHxHjVPVV8WeIpHIEEBsBUb3eKRkz1Dh1/bq+GiDRKa6lEmsSdLSlTRDv8xp49TrPHxY/d\nVOU8TmOCeJSVJr5Zmqg1cMI+/py0louIERFPthZeoKlTe5U/AIwQChwAAACnpuvXzXpj1/GO\nm4yKSHF+2mWnt0gwGNfZvHc0Vt1oD+581kxaotnDu/lF8w7T9I6YUNutp0AzzhH3wPawGDaR\ngIRbnJsCtb0aof4tU7Oh/UATd6bmXyj+RJuMioioJ8XpCFkRb9dJGb0TbrRL/yZ2a3skVGvK\nHtWJHxZ/fHkFAEYPChwAAACnpilF6T+9aeWv/r5j+/5qEVHVtYsnfPbyuX7ZEN/VGFPp9PHb\n2HJ0qwxngaNlj2l8o1MkdMLUPqd5V4l6hi+NfrO8oi4xTke3untRbmjaZapf6RQJ15vjj+uE\nj4s7wdEnIjJhvtSWdg3r+Pk9v9GJqdvSqbrRFrVNzRs6/v39GxPoE5eXJVHoDwocAAAAp6zp\n4zN//LmVjS2hyrrW8XmpPo9LREx9l705whExjpMA1LQ2JFzFMARMs9P6DrtJWvdJypxhTKS/\nVDV7lqnZ5dCS3fOGJqb+HYeoHTKNJZq9MtFT1px1kdItUl/R6XW5U3TmOQnfFGyytz5pykqk\npV4zi/S083TGKtGT/9St5c7ptZYP5w8DAPQVBQ4AAIBTXHqKJz2lffqDeopM675OPdxusSyx\n7fgnRTQ1Z6jTa2fCEqlP0FKdLB+tdeI603hEQk2dggVLJb27ZSZtggl2zQhWdveUJ8V14X/a\nO54xpVukqVrS86wpZ+qcdWIl+FW/uSbyzA+kuW3Ojqk+bDber2U7rNU3tu3JkpDzUpg+CzeJ\nHRJvVk+vA4C+ocABAAAwxvhnSsu7Eu6wJkVFC3NNeZdP0ZZLJy0Zxsy6+bg72J+ETdjUvGka\n90qoXjzZmjlXs5aIOp870ze+HGv+50zZy6Z+n4SaJSVfC8/S3N6dcauu9t034uLdc/ut06+U\n06/szUvsLY/Fqhsx5vDbpnS7TlwkIuIrkuZDDlkMeAMO07DXVKyXYK2IiOXV/BWadwbnswAY\nLBQ4AAAAxhh1afalpvFNaX2vLeLOtZattV/7q6k92t7NcllLrpH0guFMTNx5EnaYxaC9P0vF\nhCTSIFa6WN6EfeygXfpXCZws6AQqzIkK0/ieVXzN4HzYtryauUgLVklKZt8e9BdL8/6uYfVP\n7Ns4wdbwhifsw7tMOGSNm+Je+X7Nbv93NKUljg+Zo20FDs1eYuq2xW/DoZbmnNW3NOLGrysx\nR//Rfm8HzfGXJVClxZcPZFgAiKHAAQAAMPZYfs08VzJWSKRerBSxUkXEuvDrZv9rpmKPBBok\na4I16zzJHO4jMzR9ial9IT7qzhX/tJ4fjjSY+o0SODn1wFusmSsdj18xNW+2VzdiWkpNfYlm\nnd7npDsKtdrvPG52rhc7LCKSPcFacZ2On9vLpzV7hWk5HD+Jw5Mr6T1NAGltFF9adBMNU3k0\n8N+3mfq2OpF9sCT81gveq7/smhstTxgJNTsPEjh5jI473Zp4rV3xnARO7uvhydLCdQM7QsWY\nipcdonU7NO8M8SfZYcAARicKHAAAAGOEkfAREzkqpkU0RV0TxD1Z3Hnt7ZZLZ67WmatHLkMR\n7yTNWmcaN0rk5Idt/3RNP1Okp8UjdoupelzsDke0Bo+a6ic07ypxpcf1NU0OsyRERJr2yUAK\nHMbYL/zClO9pj9SW2c/dY627WYsX9GoEb6GOu9ZUr5dAdJtPlfS5mnOOaIJf2oOt9qZHzY6X\npLVR3F6dvtQ69yPBv/8qVt1oEwqEHvulNW2B+tNEVNJypdFps4+Os3V8hdbkj0rghAnVqTtD\nfPkJc+ilQLWE488hjjLNR5QCB4DBQIEDAABgLLBN4A2xT7TdmSZjV0rkmPp6UTsYZr5J6iuW\ncL2YVnFli+XvzUOmqaRTdSPKDpimrZp5dqyXaToogeMSSrCVaaRlIFt9mNLtnaobbTnYZvMj\nvS1wiIivSMdfJ3arhBvFkx0tK5j6ysiWf5qqY5qaYU1fbM1aJiISCdkP3W6OH2x7MBw0ezZG\nDpeYCocdSU1rk733HdfCc0REp680256I39ZELZ0WtwJFxVeovkEqPTgendvW5LTtCAD0HQUO\nAACAMSB8qL26EWOfkPAhcbet/ojU1lgZmeoaDTs+Wo5LS7oTcj7ZVIIn46E6u/zJRAegRqk7\nq28v7cxU7HWOVx+RYIt4U/owluUXb1tlJ7LtpfAzv5dQoO128zPWrGWeD37VvPtye3UjprXR\n5XeHm0MOaZyc1mHNv8SuPmJKt7a3udzWsms1p7i36UVaRWxxpfb66xHxZou6nMocxjRWSP1j\nYnklY2pvd2MFACcUOAAAAE59JuL8wd5Eyk14fPUf/m/N3/4SqatVjydtxdmFX/2Gd2ov9rwY\nVYzDGbciJycOGNsue6yHw1ZFJPPkp+tAs31sv7Q0av4ELZjc2xwiDmWFNnY/JymY6mPhp/6P\n2J3qAvbet8KvPGQ1dalYiYiIelwiDplo+smakcttrfkPc3S7OVoirfWSWWTNOFsyerWbrKnf\nY47/S4I1IiLudC08R7MX9eorsbyavcjUvNMpGLElGJHmrW3Hz554yxx/w5p1nbh6NW0HAOJQ\n4AAAABgLAs7hSHPpzZ9pfntz9M6EQo2vvNT89ptT7vsf38xZw5fdwLlzJOT0gd+TKyKmpbSn\n6oZq7pmaNl1EIm89G37pr9LaFG2wpi1yX/45zer587/mFBvHhpQs8cXvA9JLkZKX46obUfa2\nl6zJM5zTcJyD4/VbMzud+KvFC7V4YZ+SMdVvm/IOW8CGG03ZMxKs1cJe7dui49aKCZna2Bku\nRoJhMZ2/Z41HzJHndOr7+5QYTj2WZzRMJUPyGWVLLgEAADAknP8kXv/i9lh1I8Zuajrxy58O\nfUqDSVPnOf1ma2nqAhGRoNOemtEHU4o1e6k16aOad46IRLa+GH7m97HqhojYB7aF/nK7hBPP\nzogNNe0MSXVY5GLNXxc936Q/ap2naZjGGsly3h1DcydoSud6iuXyXvYZTevjmbXxrwyb407H\noFS9kWj30C6ZuXXCZdb0T+j4C7XoPMlcEl/diA5YvaPfE14AjHEUOAAAAE596nbeXqHlrcOO\n8eZNrw9lOkPAk6/Z54vVYZ8Ly6dZq8U7TkS6OQFEC87XgrXiL4reRl57pGsfU30ssnNDL3Lw\nWxd+qdNOFpZLF16iCy7u3dfgJCUtwbt8uuh8sRz+yq3LLvfd/Av3WZdZE2ZowSTXwnN9n/uR\na8n5/c8hqqVC7KBD3Nim+UgfxvEXas4SzTtLJcGf6O2whBr6kyGAMY8lKgAAAGOAa6K4Tkjk\naOdgsd3qPLPADgZNJDI6NhztNf9U9U2QwFGJNIorXbwTxPJFWzR1ovPiEVeKePPbb1ubTE2F\nY0dTvk8WnlyIYSKizt8ZzZ2k7/9fpuxdqS0TX6qOm93LvS0SsWYui7zxD6f4Us2fYl307/b6\n+yTQfDLq0mWX67zVIuK57DMDeW9Xxgz2DiPuxBttuPuyISsAnESBAwAAYCxQ9S6VSLGJHBXT\nLJqqrmJxFXmnOs/U8E6anGTVjSj1it9pe1RPjmYvNrVb4rvnnyfaYUZzdwtJVMRIyx7TtF3C\ndaJu8Y7XjDMdTnuxLJ24QCb2+lzYblnTFlnzz7F3vNoplbQs9/nXi4jOOds1eYHZ+4apOaYZ\nuTptieT2+iSUrsKt4vJ2+oZ0fKk3z7lIJKK+vH68TTNnGnnRxB9XK5I+iU1GAfQPBQ4AAIAx\nw1WkrqKOgaz3XVX1x9+bQGtcx5x/+8gwpjUctOB88eSa6g0SaRER8eZo/mpNm9mpky9V8yaY\nqrKuj1vFs0zdK9Kyp+3ehCRw2ATLNPcy8TjvhTFYPB/4YmTawsjmZ0xlqaZlW9NPd513naad\n3OwjNUtPv7C/O3yIiIgdNodfNUdel2CDWG7Nma6zLpO0ovhungzNmGUaOh2Fa0Q0ZbykTOjP\ne1OLtOgsqdjUaTR1aUrIlP5OvPmauVz8k/ozMoCxigIHAADA2OWZUFx89z3H/ve3I9Unt+G0\nXDkf+kjOddf35nFzYo/Z95JpKFePX/JmWrMvEm8/jwsZeqrZSzR7iYQbRN3icl4E4T732tBj\nP4t/snCyNWOmqXkivrcJm/qNmjfER36ouk4/33X6gDfRiAo2m/deM7Vl4vZq4SydstRse8BU\n7W5rtcOmao+pPWAt/axkToxPZMKlcjRsGg+0R1ImWBP7/+XrpIskfZIpf11aTojltdxhSfOJ\ntoot0lpqWks1e6VkLu/3+ADGGgocAAAAo58txiTa92GA0levmf7Y0w0vPBs8eMCdl5+28hzf\nrNN6ldPuZ8ye56LXplWkoTxS9o7r7C9Iepc//o8q7oxuGq3557htO/LPP5mmurbInLPcF31a\nQgedHwgdFzsQ2+xjlDPlu+1Xfy+tbVt4mj3/0j2TxNXlDJRIyLz3tC69MT7u8uvka6XpsLQc\nE7HFX6Tp0weYkubM1Zy5ImLK/0eC8UfGmNpNmnqauAd2/guAMYMCBwAAwCgWOmEaN0v4uBhb\nXJmatkj8M7vuWjBArozM7Kuu7dszDRVm7/PxwWCTXfKoteJzvXi+1dilIk0iHtVc0XF9e/tQ\nci1c7Zq7whw/bJrrtWCSZhWIiGnYk/ABExIZpAJHsNnsetFUHRRjS+4Ua+4F4hu8GTGhFvuV\n30mgczmjtVLSHDa8MLUH1Y44n9KSNlnSJg9aVlGRxq7VDRERsaX1kKQvHOTXAThFUeAAAAAY\nrYKlpvafInbbbaTe1L8qoSrNWDGiaYmImIodYhw2nTSVeyXc2t0BGSJijhl7p0ik7c4cFS1V\na/Eo+tXU7dUJMzuWkdSV5bzFpnrESh2Ud5rqw/aLv4hNr5CydyN7/mWtuUkLZ3b7XOx5I8Fa\nCTeKL1fcDofLmtLt8dUNSbyvqrHFDjkWOIZEJH4XmF41AUBno+a/IgAAAOjMNGxqr27EtOyS\nlDnizpbgCQkcExMRb34/92I0pttzQ7p9NNiUcMxgc7cFjhZjvxv/dZkaY7+n1pz+JdOzUJ2p\n3mgCx0VEfYWau0I8WT0+1Il/qjS+IXYwPp4yK9GxI31k7Nf+0F7diAo226/9wfWB74nVwy/t\npvGgKX1WAm0bqWjWbC2+SDydF+M0Vjo8GLadfwK8GT1UqQaXO73tnBqHJtanAOgtChwAAACj\nUqRBIvVODUaCpaZmozTtao/5J2j+Jb39KBgJ2jtfMIfekKYqScnWiYutBZeJx3nTzUQ0Jdt5\nRoPlFl93m1yIqXCo2oiIOSYye9BX34iIaT5oyv4uJtx2GzhuGnbphA9o6tQ+jGL5NfsCU/uS\n2C3tQd8kzThzcJKsLpW68uhBIp0amqrN8X06bnZ3DzcdMfv/R0z7d9XU7Tatx63TPiOWp72b\n12mmSUtQ0v1ixX/bdeJZff0SBsTyS8pUaTngHMfYY3mT8JhqjAKDUm8GAADAYDOhhC1NuztV\nN0Sktcwcf9L5D+BxIiH7n/eYHf+QxkoxRpprzJ71ked+IIlmZCSg4xeJYlMWcwAAIABJREFU\ny9P7eHvypiVBS1gk3KccesVETMWzserGyWDYVDwrJtK3obzFWnCtZq6U1DmSdrrmXKI5F4sO\n0t8Lm2tExLm+01zd/aOm/JWO1Y02gRpTva1jQCfMc5hsYtsS9ou3U01Kxy/TqWt7znlQae5a\n8eR3Clk+zb84WTZwBTAaMIMDAABgVHJliFjOkx0CFQ7B4HFpLe1xrYrZ96qpORwfbay0dz5v\nnX5lH9LzZ1mn/5u99W8SaS/EaOYEa0GPgyQqf6jIEPzNtvWYhLtsPCEi4UZpPSYp8Seh9kC9\nkjp/8CeZiIg/8bSXbppERMQ0H3VuaD4qsqz9NqNQ519sSp7u1Mfl0WXXa+4kU7FNmirEm665\nMySzXyueBsiVpuM/JI27TKBMTFA9+ZKxUKy+TSwCMMZR4AAAABiV1CP+6dL6Xpe4X8J1zo8E\nq3oucBx717nh2LvSpwKHiBYvc+VMsw++Kg3l4vZr/iydfFaPG1KoFhjTZSWCiGj+UEwuNpHm\nxE1NQ1Kq6BfNmyKpOSfncXTgS9fCnk7t7Tp9oy0eP0XFWvwBkzfFlDxjao+K26dFs6zFV0pm\nkYho8Rn9SnxwWZI+T9PnjXQaAJIVBQ4AAIBRSjNWGLtFgh3+Pu/K0NRFpukfCR7oxQyIkPMK\nERNKWAjoTmquNe/9fXtEs0QniTnSOepRq6eP8f2i7vRE63bU3cPMiGGllrXy4/ZLv+44I0Ys\nl7XienF7e3g2pVCajznFHQ7f1f/P3n0HxlVdCQM/573p0kga9S5ZzXLvuOAGoXcMyUISQhxI\nlhA22SQkG8huNl82pO1uCtldQiAhCQECcUw1EAMG44KNuy139d5G0kjTyz3fH5JHo5k30qjM\nSGOf31+e89qVLUvvnXfvOQWLsWDxZEbKGGMzFic4GGOMMcZmKlRjyjXgbiFPB5AXVSbQlgAg\nSDoQSr0zdXljnzMxA8z1CpdKzIh4WETth6njKDjNoE3B9LmYtzqi3Ir/WlIlkImoAcgGoAZM\nQ6kUYKzHeADw2qhjN1mbQDhRl4EZqyGxcIxDtNmgTgZPyJwXdTJoFZ7/pxHmzJFv/ndxYht1\n1wMJTCuSFtwASWMPEjNXU/3W4c+DhUpVBkxdFMXhMsbYzMMJDsYYY4yxmU2Th5oRmQs0XU7m\n94J3M84HdeqYJ8NZq6nhgEK8ZE1EgyFBJ/9MPeeGPrr6qb+Ruk5IC+8DOYIMxfD1shCzxrE/\nADi7RPWfwTc0A4Xc/dRfgzkbMXPUkaMkZd8gWl4ekRKSdFL2DVPU3nVKJaZLq+8d70GYXAn5\n11Pb++BzAgAggC5TKrwJVEptU4LY2snWDrIWjflBpUYZYyzucIKDMcYYYyzeGBegpKXeXeAd\nAACQNJh8GSQtjeRQzKqQFt0mTrwO4kKBBpRw9hVYtDySw6nj6HB2w2+ghZp3Y9GVEX8BEyFa\n/u7PbgyPp/1DTJ4DWtNoR+pypeIvUO9BcLUDAGiz0bQc5IuqeiWmLcHkSrK3gNeK2lQwFACO\nVWDE1U/nX/H/a5Kkwvy1WPyJaHTqZYyx2OAEB2OMMcbYzEAu8HSCcIBsBHXWGBU3EyowoQK8\nA0A+UCeP66EUK6+S8xZQ42GydoHBhPlL0BRxMxGzco1S6j4V3QSHzwHWkOYvAECC+s9jxmVj\nHC7rMX1dNMY1g6j0mFQW6c4kxMlnwRpQuUN4qfEDQEnh31H4yNwMdguaciApYCmTx0bWZvDY\nUZ8GxgKFb0Lho7aD0N8MJMCYi7krxjfNhzHGxokTHIwxxhhjM4DzPNkOAbmHPsrJaFwDqrHq\nYky4TKYxC+ddP4E39WFrkXpsExxJhLzKtVEBALwTKo96aaPe6hHZDX+8ZS8WbgisqEK1h8UH\nf4L+rsGPWLRQunIzJGdS235q2gE+FwAQACTmSqW3giFg2ZHDLI7+AezdQx/bDlPjLmnh58CY\nG62vijF2yZt5Kw8ZY4wxxi417iayfjSc3QAAn4Us74GYcY/uqE1W3hAuPlXUiWFLZmiSonvp\ni5JNqesKAHid4Ojxf6LGKvHGL/3ZDQCghuO+vz1GbQeo/q3B7MYQa6s4/eehIiAAACCqXhzO\nbgxyWkTVC2Gb2jLG2KTxDA7GGGOMsZggB7nPg88CKKOcBuoy/3tycpxU2t9NzvNoiFUjDPJR\nby04ukBjxOTisPUmMxdC14nQMGZGufOopMGkcrKcDYmrMXl2dC99cQr/mjOgeAft3zpcq8Wv\nv5sadigc6B6grmOYvRIAwNYJ/U0K+9i7oa8BTLPGP2B2aZE042jMxJgfJzgYY4wxxqLP20rO\nA0DewU/kbQNPLerXgZQIAODtDXNUj3J8yvU3irNbwTH0vp1kDRZdiflrQ3fEtDmQt5paPhoR\nzJiPuWNVwZg0zLuWXD3gHJ5NAJIKC24EVUK0Lx3HXHbRfI4sZjRlSYWVIA/d/GNSASnur0kE\n3XAvHuqoVdhHQqAwK4Zs7UMHOsN8SwOQsweBExyMsajgBAdjjDHGWJSRh5yH/NmNIcJOzsNo\nWA8AYUuExqaPqdsqqv4E3oAuqj431b4N6gTMWqIwqNIbMX0edRwlpxm1KZA+D9MqYzFOdaJU\ncR+ZD4OtCXxO0GVi+nLQRHlpzEzjdYGkAimil9u+k3s9bz5N1r7Bj5iWo7nlQal4HgBAcjGa\nyqi3OugQLPpEwHcdgWIWhAAozPfshWNRpVdOoACA6qLqX8MYm1E4wcEYY4wxFmW+9hH1NYbj\nXUAOQD2oM8HdHLodVZlRHxsAdRwakd3wx5v3KCY4AACSizG5eBq6iaKE6cshPaKOthcVImr6\nWFS/CzYzoIQpBTj3ZkwtGeUIUX/SveUXgQtMyNzmeu4x3YO/QFMWAODcu6FuO7UdGCqKoU7A\nWddg9rKAcyBmFlFbcBIEiEBOBGFVuKqxYOgPSXmgMYJ7IHgHWYum0YbNGGOTwUVGGWOMMcai\nTITvACIcAICGhYF9K4bIRtCVR3NYF9g6leP2DmrZRq1vUs/h4Oknk0ZAXuHxUUh9B6ZEnHpV\nHHsRbGYAABLU2yD2/h+1KxRD8fPueVWhfIbb6d2/bejPshbLbpZWPyIt+qK09CvSym+NzG4A\nAOCKWxVOnZCCJdcrTC8yZGLa/AtHylh5a+g+WHETqHSjDJsxxiaDZ3AwxhhjjEVRe4/98Blb\nd68qL5VWV/j0mpGbUQsAoErHpKvJtn+4GIemEBNXAMbkVi00tzKEyFIFgABVZD4gFd4B2vQx\nT0bgFDQA4EXQSZgSupKBgGyeAbt3qK2shLJRnaSV+aE3PFsX1e0KDpIQVa/IWfMDa4IGEh31\nyvG2kXGVHpKLwl0ZS5ZK131ZfPgc2PuHIjlleNUXMS0fVBqqfwucPYPrVTB9PhZfB9Lwdyxm\nzMMVD1HtdrI0AQlMysNZV0FK2GsxxtjkcYKDMcYYYyxannv3/PM7qj1eMXjTlWpUff0mz2Vl\nF9pkyikgXSiQqc7ElJvBZwVhBzkZJG3sRplcBB2HFeJq1XB6wmMRza9JpZvDlgsBACCvaPZR\nJ1yo3ICgU0uzEEcUAbW4et1iqL0oAQjyWdy9SZoUncylGZRR93kgpYoWjl6wdUHiyHVM1k7q\nribXAIgwk26k8c3gxsq1culyaq8BmwVSczGzaPB7AE3lmFIKrl5wW0GfDmqlUq/GHFx07zQs\nZWKMXao4wcEYY4wxFhXbDzb/cfu5wEjPAP7HX9VPPuDONRGgGrXBKwJATgQ5MXZDBAAAzFxE\nLXvB1hG8wTAyyeLqBkc76HPCncdH7T4acRICp0ec18gLAIYmibiFy5/dgIBkic0zcPEnOEhQ\n6wFq+gjs3aBJwNRyLL0WtGHa8QbyKhVwGTyl1xmQPiBx6g2q3TlYUwPVHsUyn1LB+LvqqnVY\nME8hjhLo0kCXNu4TMsZYdHANDsYYY4yxqHh1b31o0O3Fvx81gLoEE64BOSXmg1IiqaSFX8Cs\npcMJB5UMKQZQBS9dIY9llNP4hEItDwKvj4ab3XqER/lY8omLvR4HVT1PZ14BWweQD1z91HZI\n7P8VOMxjH5mYoRxHCROGN1HtLqp5f6hiKICckwxS8OQJTExRrbxxYuNnjLGZj2dwMMYYY4xF\nRWOHUpsJgPqedNSF6U4yXdQJOHsTlt0EDjN5+6j1deXdRquU4SVQTl4QOYanaigutRjcEuFQ\n4xOZz1LnyeCox0bVb+OCz4x+LGbMBkMq2HuC47lLQD087UXUj6jTgXqNena2t8FM9qEJIFLJ\nQs1NX8KEpIl9CYwxNvNxgoMxxhhjLCq0atnlUZiVoFWHK+o53WQNJOagSKcOHfhCGsfKOtTn\nhT94lHnBw/MIVJJa+WCU5LC1Ti8K3WcVw2Q+i0CjVjYBkFTSii+Ig38EW5c/hhmV0oI7hvcR\nvqEeKwEwQauemwupczBvDZqyMHnsGrGMMRbXOMHBGGOMMRYVi0pTd51oD40vLp3ZNQskNWZf\nRS3bRk6qQMy+CsKkJwYPQ0wgsiltGC4zoZG1Mqp8IU1n9SqlEpUXE29IwmiQzwPCF9h8RBEm\n5cobv01tR8nSCrIGU2dhRsXIPSSQZIW+sABoNEnFShU0GJvBpBmbCGYzGyc4GGOMMcai4nPX\nzD54rtvhGvEwX5qbdM3yfOptoJZjYDNDQhrmL8WU/PCnGev1fhRg8lzUmKhrNznaAAD1OZix\ndpTyooNUmO+hc0FrTSRMknC41AgCpmhN/W6LR7j9Eb0qIUEV69KqsaZPVY5rk8fMbgyRZMxb\nhnkhhWkHIWJqCXWfV9iSXhbhGBljLN5xgoMxxhhjbLJO1vfuOtHW3uPITTNsWJQzuyAFAIqy\nEh9/6PInXjt5pNpMRBqVdN1lBZ+/tkI+9aqo+dBfDJLOv48VV0rzbh55SgGO0+Q8Bz4rSFrQ\n5KNhCUgx7DOiz8HCT44rsyKhUS3N9lIjkX0wIGOmSsoN2k1GlUmb5hFur/AiolrSXOSLUwAA\nALMXU/0HEFJIFXOXT9kl5txIe/4nqDssps7CnIVTdQnGGJvhOMHBGGOMMTZxRPC/r1a9trfB\nH9m6u+6T60vuu6ESAIqyEn/yxZUuj693wJWZopckpKZDovqDkacQdPZdMhVj7oLhs1reA0/b\n0CfhBGc1uZsx5UaQZvRSDgkTNTgXwEfgQdCOMvdELWnUkiaWY5tmhnScewedeQV8wz1fMXM+\nFl8xVVfAlEJp7Vep6hXqqQMgkDVYfLlUcQ0gt01kjF0qOMHBGGOMMTZx7x1pCcxuAIAQ9OIH\nNXOKTGvmZQ1GtGo5O9Uw+Gdq/FjxPNSwfzjB4WoYzm4Mn9dJtiNoXDt1Y48eGeHin5QxXpi9\nBE0l1HoI7F2gSYS0Ckwtn+JLJOfj5Q+Bzw0uK+hNgLFe3DRBJDgLwxibEpzgYIwxxhibuHcP\nNSvG3znU7E9wBCJbcLPPIfbhFhjkblHexxMmzuKFNhlnXRn1q8gaMIQp+TEJ1NdMZ7dTXwvK\nKkidJc25DvQpYx82Co+NmnZQ7zlwW0FnwqxlmLMKLoH1Soyx6OEEB2OMMcbYxLX3OBTjHb12\nxThq9KS4QWMY/jO5FXcB4RnX2GaewS89TqYVsABU/5E48tJg4RgCgP52X/Nhae2DmFo8wTO6\nLKLqaXAPDH109lDDO9BXjXPu4dkcjLEJ4x8fjDHGGGMTZzQod0416pULTGC2csPOEXHZqLQL\nXYgTCGvYJMjMRH3k2U+u7eR6m9y7QSh0z2UxReRrPOc+8J73zGGyW8fY2WUVx7f6y+IO8brE\n4Rcmfv2m94ezG/6gpY7MVRM+J2OM8QwOxljc8/rEy7vrPzze1tXnyDLpr1yad9OqIlniN4SM\nsVhYNSfzbFOfQnxupuL+WLYRW46SpXVE0FSIJeuGP+rKyHE6qN8qAKK2lJwnwHUGyAsAIKeg\nfimoFBbCzCyinTxHhr8c6ifPYVSVgzzFFShYhHxtDfbnf+FrODv4EQ2J+lvu06y5Ltz+1HkW\nvEoJtf52GOgEo/K3+shTBFfZoL5q5T37qiGd274wxiaIExyMsfjm8vi+9eS+M41DTxc9A67T\njX27T7T/+P7LVDJPUmOMRd2m9bN2nWivbesPDM4tMt20qkj5AJVW2vh1ce49ajoM9h5ISJMK\nlmH5lSAHzASRUzBxDdn2DyUyBunKiHrAGVCGw9dH1vcxYT2og1uxziRE3lMhyRogbzVKBYC6\naRnTpYwcNtv/Pir6h2vBkN1q/8uvUG9QL1mvfIwr7BQPcg3gKAkO4aGW3dR1HFy9oEnCtLmY\nvxFUOgAAn0v5EK8zkq+CMcYUcYKDMRbfXt5d589u+B2rMb/1cdPNq8M8XTDG2NTRa1S/emjN\nSx/Ufni8tb3HkZtmuHJp3qa1s9Sq8DlWWSPNuR7mXD/aeXWlqMkBVy35LIAG1OQDElnfC9mP\nyHkUZ3KCg6xAio+sBMIMcl6sx3PJc+9/JzC74efc/mLYBIchbDFRNJjCXkl4RNUzYLswWcnd\nT237qPectOB+UBlAZwJ7p8JRuqkvj8oYu3RwgoMxFt8+OtmhGN9T1c4JDsZYbGjV8j1Xl99z\n9VQvuJAMoJ/vX25HzjC1CXwWIOfMnQoROAklWLzXTI0HwieOviPqjoFjAE050pJrfM01ijv6\nWutA+EBSaGKCmZWgSQC3LTieVjJKuxbqODSc3fBz9lDLHiy6GjOXUv3bgbsDIKCEmUsi+rrY\nRU/N/XTYRHCCgzEW3yw25ftjiy2uyu8xxtjYfGG3kG/mdiZBQ/hNCTEcx/Rx2UTVu9DdAJKM\nGcU47ypQa2N0aafV++IPqbN+8BO1VYvTe2RjjvLOKIVtcKPSSivuEfv/MGL9iMEkLfv0aFcP\nU2WD+s5j0dWYfRnYO6nzsP/yIKlx1vVgmPE1ZRhjMxgnOBhj8S0jRddqDn6nBABZJn3sB8MY\nY9GDUrJyf1nUgBQ+iTDtUAtSJoiQxQhoACltOgYUU9R+Xmz/NTiHalhQ3SE4uUO6/huYGou1\nOb5dL/qzGxcGJKSBFkkjC3dwvkxVNBuksOuqMGuOfM13RfVOsDSDpMa0WVi6DmTlVkFDl/KF\nqaYxmCVBCUtvwcwl1HsO3P2gT8f0haBNjuTrYoyxcDjBwRiLb59YknesxqwQX8rruhljFxd1\nPkgGEPbguLY87Iv3mQFVC8l7EERAvSQ0oHopwMVeCtrnFTue9Gc3hth66f2n8I7vx+D64sxH\nClECdVqqq61rRFCSddd/ZozT6ZKk+TdHfnXUptJAs9J5Ala1GAvQWBD5ORljbHQX++8VxtjF\n7toV+dcszw8K3rqmeO38MFNwGWMsTqEKEzaAHPiKG0FbjroF0zakCKEG1WtQvRTkWSAXomoB\natYDJk33sKKO2s+BVaGcJ5kbqaclND7FfJ7g3MoF6spF6oWr/R+l1KyE+/9NVbl0igcQppoG\nZk71hRhj7AKewcEYi2+I+PCnFl2xOHfXifb2Hnt2quETS/IWlHANdsbYRcdjBls1eFWI6aBN\nBrUJVJkj8x0zm5SNUvZ0DyK2lLIbFzaZIdqrVGQ16BLAqbCKE01ZCbd/VvR1i85mTEyRs/JB\nnvqHAkyeBUVXU+MOoAvLYVDCnFWoyxRHXoKBdlDpMKMcS9aO6JHMGGOTwAkOxtjFYFlFxrKK\njOkeBWOMRQv17YO+jwEEABAAWCVMXgkpU923hU0tvXEim6aOVLFSHN+hEJ+9CgCklHQpJX2K\nL+mxUf271FsDHisYMjB3lbToATKfAmcPaJIxbQ511fve+xmIoZQHtZ+Eur3yuodAHz+pOsbY\nDMYJDsYYY4yxmc1eA337RkRIUN9HqM0E/axpGtPkkJlEN4ALMQEwbxp63LrrydMIwgpSIqoL\nQVM89iHefvD0gmwAdRpgRKu8MWc2aAzgDimbkpiG6bFoZC6vu4tazpF5RCEMadVtmFMWles5\nesTRJ8FzYc7IQAud/RtkLsbKO4ci9l5xdIs/uzHE2imO/U1a9YWoDIkxdonhBAdjLO71Wl11\nbQM6jTwr26jX8o81xtjFhqxVyvGBKoy/BIcg3wkQbYMfCACgFuW5IEV5vYbwgKcHQIA6hRwH\nwHOhBIbPQp4W8DRiwrqwtVq9FjLvAEfD0EdVEqZdEVFqSa2T1t4jPngKhBgOyippw+YIUyST\nZUhSfe7H4tBbov4Y2PowLV9acg0WzovS1aju78PZDX+w8yhkL8GUUgCg1uMgvAoHtleB1w2q\n0XqyMMZYJPhJgDEWxxwu72+3nX7r4yYhCAD0WtVnryq/c30Jzuh+AowxNk4ei3Lc26ccn8lE\nnT+7cYGPfFWIyYCJ0bkkgeUQWT4G4QYA0CaAPuRCnhZw1YBWaV6DcFP7FvAODEe8/dTxOmZv\nAl1wietQWLZSSs2lw69TVz2ghFml0rJbISlz4l/NeKnU0spbpJW3RP9KRD3nlLeYz8JggsMZ\n5jtZ+MA1AKqLv20wYyzaOMHB2ExxorbnlT11DR3W5ETNkrL0T24o0arl6R7UTPfD5w4fODPc\n6M7h8j617bTXJ+6+MjqTbxljbFpIWuU4honPYCSU+oYCkWhFuSIqV+z9CCwfD39WK08TIE8j\nKiY4rCdHZDeGCOrbj9ljJzgAAFML8KoHIxprvBIAEggvCI/ydu/QIh3UGklxB5RAkxClwTHG\nLimc4GBsRvjj9nPPvXt+6EMnnKjtefdQ839/eXVaUsyXJcePM419gdkNv7+8X3PH+hKNittg\nM8YuFvoicLWFhtFQHPOhTBIBOcNsCilUMSWEG/oPjYhIYV4ekEM57GpX3l/pX+TSQgPkPQOi\nB0AAJqBcCuqE0CUqAAC6odZmmDMfql4DEkHbMXM2qPmGh42k5gdVNhH8AMDY9Ktu6X/+veqg\nYKvZ/tS209Mynnhxtll5brbD5W3sCH3bxhhj8QqTloI6pPu1Og2MS6ZjOJOBAOEmJ0anUai7\nc7hH6aCgCpd+qA9zCuU5B5c6YSb3HhBdAD4AIrKS9xhmhFROJQCUMXPh0MfEDGneTcH76JKk\nxXcGBxljbEI4McbY9Ntd1UakcP+0p6pDCJIkriehLOQN0DDBt6OMsYuJpMGcu8iyH2znwDsA\nKiMkVGDySpCikxSIKikjpAYHAABilFp9E9DI4qEep2IxS9QotzVBTSbZzgWfBAA0MayjMfOQ\n99Rg0+JBQ383ecnoLKGe2uH9ZDWW3wz64Wa0WPEJKb2Uzr9P/W2o1kFGuVRxFajDZZcYY2x8\nOMHB2PTrs7oV4y6Pz+b0Gg1xeP8aE+X5yYpxrVouyopSpTrGGJsmkgZN68C0DkjEqAFHdKBU\nQcIMMPIXn5QJUnTyBep0QBwxC8PlBJUmeEGEugA0JcpnMM6H/sPgG7mChgBTVkz1WOMHOYGU\nZkpKiHOugL7V0Hse3DYwZGD2ctClBO2FqcW4cnMsxskYu/RwgoOx6Reu0IZeo0rU839SBX1W\nt14rzy0yLSpNO1ZjDtp6x/pZXJ+VMXbRiufsBgAA6lF9OfnOA3UBuQETUCoEqSBal5P1YJwP\nAycCQgQ2CyQmgT4ThBWkRNQUgbow7BkkHWZtIvM74Oq4cE4DmtZH1Cb2oqXQ6nUQkRfT5kDa\nnFiOhjHG/PjZibHpt35h9nPvnRchyyrWL8rB+Ol3and6a9v7HS7frGxjenJUSoV5ffTKnrqX\nPqjps7oRsSI/+bNXl5mM2g+Otg7uoFFJd24o+dzVUanDzxhjU4kEnd1FjUfA3gdJmVi+FgsW\njn3URUKL8vyYXQxTNxJK0H9ieElFQiWmXhG2N00oTTrm3AWuTvD2gpwAmuy4XBw0GVYzmRsA\nANOKIDENUA8gBS5R8UM0xHxwjDE2jBMcjE2/oizj/TdUPv3mmcAcx6xs4/03VE7jqCJHBFt3\n1T777nm70wsAiHDV0vwv3zI3UT/F93//+eLR9y/kMojobFPf9/9w6AebV3z+2oqa1n6tWi7P\nTzYlxl/TRMbYJcfjEtt/QZ01Qx97W6jhCFasky7/3LQOK8bISxZBDgCSQK+SUkKqXEwRlDH1\nCkhaBq4OAAGaTFCbJnAW0GaBNmvqhzfDeV1i/4t0fjcMFgtDCWevky77B5BzwNcSvDNqQb70\n/ooYYzMJJzgYmxHuXF+yuDT91b31TZ1Wo0G9tDz95tXFKjk+pm/8dWfN02+e8X8kgncONXf2\nOX72pVVTOAHlTGOfP7vh5xP05Ounnn54Q25awpRdiTHGokyceGs4u3EBndtFhYuwYNG0DCnG\nCDxOX70IaBnroS6tVCRh1JLUqiRQJUXr5BcvsfuPVHdg+DMJOrNTeNzS+s8ROUEErBJFHaqW\n8MMFY2x68c8gxmaKsrykb34y/uYne330l/eDb9MB4FiN+USdeWFJ2lRd6GhNt2K8sdNq7neG\n1jHp6nPsOtHe3mPPSNGvnpuVn8EZEMbYTEH1h5TjdYcukQSHy9cUmN0AAEEul2jUy+XTNSSm\nYKBrRHbjAqrZB8tuw4SVIDpJ9AB4EY0g54fvAcwYYzHCCQ7G2KS0dNusDo/ipt0n2rNMhizT\n1PR+c7p9kW96Y1/Db9847Y8/8/aZu68sv+dqvm9mjM0Mjv4w8b7YjmN6CHL5yKYUd/rILnMR\nhxii/mroPkAuM8gGNJZg1hqQhnvokrkp3HFkbsSEVJAyMcL2Nx4rSCqQo1KiizHG/DjBwRib\npODaqH6v7Kl/ZU99UZbxodvmLSqd7FSOwkzlzq96jSozZUQO5WR97+NbqwIjXh89+865wszE\nDYtyJjkMxhibAoZkcNuV4sENNS9KBMrN0QGAwAXACY4YoZbt1H3wwicLOdqp76RU9jlQG4di\noyw0jXANKgnqPkAde8HnAADQZWDuVWi8lBvQMMaiixMcjLFJyUtPSNCpbM6wHeMaOgYeeXr/\nz/5x1fzi1MlcaPW8rPRkXbfFGRS/fmWBWjWiaeKb+xsVz7BtfwMwWqHEAAAgAElEQVQnOBhj\nMwEWL6ejryvEZ62I+Vimgq+FvLUgBgDVIKWjuhJw9Ll7YTvdIq9xiBlbc0B2A4YqvLot1LYD\nC28dCqUXA+JQedFAKGF6cSQXoeZt1BPQo9fZRbUvQNHtmMJ9ZNlY1PzTgE1EnLdSZ4xNN5Us\nfWpj6ej7eH30p+3nJnkhvUb1g88vDyqlceWS3C9cH9xrpqVbYeYzADR3KccZYyx6iGDHkZYv\n/3LXDY+89cn/984P/3y41WyXFlyHWcGL5rByI+YvmJZBTgZ5TpD7MIg+AB+QE3zN5PwARJg1\nOAAAIKMhTCJDQtT5yOqlPkGOKA2YDSLL2TDxgN/XCSasWB+6D865AvTJY1/D0TEiu+G/ROt7\nEQ5ymHcAKOyrFMYY8+MZHIzNIK1m+/nmPo+PSnKMJTlxU+z9rivKAOCFHdWjlMk4Wd9LFOmE\n1nDK8pJ/+431u0+013cMGLSqhSVplYUK07n1WuWfbIYwccYYi57/eaXq9Y8aBv9ssbk/PN72\n8ZnO/3pgdcX136Lzu6nxGNh7ISkTy9di3rzpHepEiD7w1odEveQ5idrV4Q9DjZzr8gXXd1Bh\nktNbTTD0q0RCg1bKk5CrNkSHL0wKSXhAuP2VOKRVdwldIlVtB58HAEBW44LrpEU3RXIFsilP\nqARPP7h6QBvBvE7hpp6PqO8YkAcAUZ+PGVeANiOSqzPGLk18u8/YjODxit+8fmrb/kYhhiaC\nXj4/+5/vWJCcoBn9wJkAEe6+suzGVYXVLf1/fvd8VV1P6D5CEAANzYCdBJUsbVycO/o+S8vT\nD53rCo0vq+BbIsbYRLi9QqOayKTX6haLP7vh53T7/u/Vk7/8yhqsWIcV66ZigNOGRKfyBmEG\n8I3SU0OFKSirPaLDR3YAkFAvY4JHjPjRLcju9NXpVRVxs25FeMHVA2ojqKamunZ0+QttBJH1\ngXVGQVJJS2+D+ddSXwsAoikX1BF/dUK5BjkAgIhgOgYJ0bIFnG3+z+RooqYXpIK7QBtZZVPG\n2KWHExyMzQj/99rJbftGvOjYU9VudXh+9qVVk5z1EDNJBs3S8vRzzRbFBMesHCPG6iu5eXXR\nO4ea69sHAoMZKfq7rhxjKQ1jjAUSgt7Y17h1V11bj92glReXpX/xxsrctHH0nD5wViHZCgCn\nG3utDk+iXj1FI50+FO4JloA8gKMlJmRMkOWSC5Wq0eGrVjgLeb2iVy2lT3qgUea1UdN7ZD4+\n9OUk5EmF10PCjK76hClzqXNvaH0NNCnNJNLoMbNs3NfQhvmHQxm0Y9fTJevZgOyGP+oh827M\n3TTuwTDGLg1cg4Ox6Tdg97z9sUIntmM15rNNcdYy8LoVBUaDwi37JzfELrmg08i/eHDNpnWz\nkgwaAEjQqa5dXvA/X73clKiN2RgYYxeBH79w5H9eqWo124jI5vTuqWr/8i9217SOVl0iSLgu\n2kSgUJvZ4xJ7XvQ983Xfr+7x/f6fxZ4XwRNcVnmmwbAtXVWAEc5AxMHJfYKUvliEOCjGIbzi\nzJ/IfGy4rZitRZz9A9jbp3VYY9FlYO5VgCOfBQx5mL1hqq6ASaWgMQEE91vD1EUjJomEY1du\nUkth4owxBvGe4PjhD3+IiNnZ2UFxi8Xy4IMPZmdn63S6pUuXbt26dVqGx1iEGjoGfEK52eq4\n7qRngpREzY/uu6woa3jiq16revCWeVeMta5kaiXoVA/cPHfL969++QfXvvyDa7/5qYWc3WCM\njcuxGvPOY8Fvjx1u75NvnIr8JDlpys//WrWcahz5Q8ntFC/+Ox14DSydQAL6u+jAa+Iv/w7u\nmZ3jkHMAZIVfYKq8CzeZPoBugCaAToDwCxbiGXUfBWd3cFR4qXXnBM5mcXjOdg6cbOvvGIj6\nPz2mr5Aq7sP0ZZhYjClzseAmqfxzII/169Lnobpd4sjz4sjzVLcLfENNf6mnSpx6Shz6kTj6\nX1SzBZw9gLI065Ogywxcn4op8zD3qojGF66qKPmARERnYIxdeuJ4icrp06cfe+yxrKysoLgQ\n4sYbbzx+/Phjjz1WWlr6u9/97s4779y6dettt902LeNkbEySFHbthhx+04w1uyDlN19fd7zW\n3NRpMxm1C2alpiROWyWRBF0c/5RjjE2jg2FWlxyv7Ym8JMe6BTlPv3nGHjJZ4xNL84L6W9PR\nt6k7+L00mZvp6Nt42Qy+gUEdapaC5+iItSpSOqrnAgBAD9EZAP+DugqxDEA53y2jfrAeRxAp\n7CSRGcOqXEqTBhrH9SvcR7S7uruqzeJfNZKTrL9uTlZCVCtk6zIw79rId6eBNrH/aXD0Dn1s\nPgg1H0gr74feY9R5YGgnr4N6T5OlWpp9DyTkSRX3Uf95cHaCpMaEQjBEvHJHE6YKqcYUPPGE\nMcYuiNefDkKI++677/Of//z8+fODNm3dunXPnj2//vWv/+mf/umGG2546aWXFixY8PDDD0/H\nMBmLSGlukk6jvFB5brEpxoOZErKES8rSb1lTtG5B9jRmNxhjbMLsLuW3x0KQI8ymUMkJmu9+\neungcjm/JWXp/3jTnKA9qf6Y4hmo7miE15o2cjZqr0B1Jch5oCpGzXLUrgZQAdiJjgdkNwDA\nS3QGwKx4GrWUFVqIWkKNWgr6PUgAM+ztPYXpIDbOtqZ7a8wnWi2BNTHaLI4/fdxwtmMg/EGx\nRUSHnvVnN4Y4eunAM9RxIHhn4RGNfwcAQMTkCsxaixkrx5HdAMCkeSApLHrF5CXjHDdj7BIS\nrwmOxx9/vKGh4Sc/+UnopldeeUWn0911112DH2VZvueee2pqao4fPx7bMTIWKa1avvtKhdpd\nVy/LL8xMjP14GGOM5YZZXZJk0CQpVRoKZ0VlxjPf3vjFG+dcvSx/07pZ3793+U+/tFKhm7VL\nYfICAFCY+MyCWlCVo2YpqheAPPQES9SimIkgUi6gIGOiTi5C0AREknRyScDNah/RYaKdRDuJ\nPgZQnmIzDXRhWnTpx9Hpw+MTVW2W0LhP0DtnOqpaFTbFHlmaaEChsAjZusmrlM2xtYB3EiVU\nVEbMuRVUATdCKKHpMkxZPPFzMsYudnE5ebu2tva73/3us88+m5ycHLr15MmT5eXlWu3wAsIF\nCxYAQFVV1cKFC2M3SsbG464rygw61R//fm6wIp1GJd25oeQznyif7nFdhIjodGNfc5fNlKit\nLExRLInKGGMbF+f+/q1zHl/wy/nrVhSMtyeU0aD+5IaSMXZKzgRzc2gYk+O3HaZ1nHGQ0WhQ\nVQhyEXgl1CIE/nzuIDoZeBKiE4glAMVTMdRJwYyl1LEfhDs4nrUq8pP0OTzhqnEBwL66njk5\nSfK0t1ULmrsRAH0Eir9Ofc7JNM1FQxEWfYFs1eDuATkBDYVh160wxhgAxGmC44tf/OI111yz\naZNygyiz2VxSMuI2IjU1dTA++mlvueWW1tZWxU1CCADo64uzfhYsjiDCrWuKb1xZ2Nhp9XhF\nYVaiXhOX/z0n7PD57qPV3VaHtzAz8apleVHqnljXPvDfLx071zz0KixRr/7C9bNvWlUUjWsx\nxuJa74DbKxSWHiRGJyuK8zZQ7WGF+PyN0bhcTISbJjz69GGUUBcSJKLzobsS1SPmAkz3QkhN\nEpbfRfWvgevCjaKkxryNmKrUbzUMadTkhdPr67G5M6JXLdttI2c/JqSDPOq3tzp8PRTF4Usq\nUBuVNoyHpEZj8JIudklQj9ZnmrFw4u8J6qmnnjp48OCpU+OoYT5ozPctmzdv7upSnu7odDqP\nHDmSmMiLBVh0qWSpJCdpukcRay6P78fPH9l7ssMfee698/9y9+LlFcGTfh0u75v7G6tb+9Uq\nqbIg5ZrlBSp5HK+zrA7Pd367v9fqCow8vrUqUafeGNsmL4yxme+dQ82k9EL9vcPNd10x9X2v\nsXQ5XnYbHXhtuD0ESrjiFixdPuXXmgiyk6gG6gUSgEaUSwDHeJGOmEyk+G4pZfyXtwIEz48A\nAAAB0AsQXG8+9tBYhPO+TP014OwBdSIai0Ezvgd7k0GtU8lOb5hyHgCjzO+YDOppoGNbqLcR\nAAAlLFopzb8ZNAmKO6OpGNQG8IQsm1LrQZ8IwhW8f+oCkOLvWYMxFtfi7IdOd3f3t771re98\n5zsJCQmD8ym8Xi8R9fX1aTQag8EAAGlpaT09PYFHDX4cnMcxittvvz3cJqvV+rWvfU2lirO/\nLnYRa+m2vf1xU1OXzWTUrJidsWZecLPkOPL7t84GZjcAwGJz//DPh//w7SsCC5Seaez7f386\nZO4fqlf39sdNf9tV96P7LssyRTr3dfvB5sDsht9f3q/hBAdjLEh7j3Lxi1ZztIpiSGs+SRUr\n6dw+6O+GpHSsWIXphVG61vhQL3kPAlx49iYXebtRrgSpeNTD8gFaRxYZBQAZcfSjFI1SrXN8\nhTyjSFJhyuyJH424clbqzvPKb9okRJNh6ieqUE+D+PBxEBf+DklQ/Ueit0G64pvKiQlZLS3Y\nJI48P6JLK0rSgjvBmEy1L4N3+H8HGoux4OopHzNjjI0uzp7Ym5ubLRbLo48++uijjwbGTSbT\nvffe+4c//AEA5s2bt2XLFqfTqdMNTXEcLC8a2m+FsTj1+kcNv3n9lMc7dHuxbV/jZZWZ/3bP\nUm0czuXzCdp+UKHgnN3p3Xm89dY1xYMfPV7x2HOH/dmNQU2d1p/+5ejPv7w6wmvVtPYrxuva\nB3yC4rEjL2MsesItlDNGZwHdIEwvnClJjQDkOzmc3RgOnkPMARxl0YQKcSnR+YBqoMmIswEm\n0PZ1lET2jG8iG7EFucky4gfnO0PnaszNTtJG1pl4XOj0m8PZDX/Q0kqNB7FYuYAI5i2VEjLo\n7NvU1wgAmFKIs6/DlAIAwAVfoe6j4OgEWQvG4smkexhjbMLiLMFRVlb2/vvvB0a++c1v1tXV\nbd26NTt76A327bff/txzz73wwgubN28GAJ/P9+yzz5aWlnKFUXZxqGsf+N9XT4qRtz8fn+l8\nYUf156+Nv5sJi81tcyq/f2vptvn/fKzW3NGrUIm9qq6nrceekxrRDa4UJoUhofLaYT+vT3h9\nFK6VL2PsonRZZcY7hxSqfq6ojJOqnySo/xQ4uwAQ9DmYVBGmTMKY57EDKZYFFUDdgHmjHqxD\nXADgBXAA6EC5CmUkdABpSv1lDRNa8DJzzc1Jyjfp3z7V3jkwPN9wTnbSurL0aFyOumvCxKvD\nJTgAAFMKcOUXFTbIunHVVWWMsWiIswRHYmLixo0bAyMmk6mlpSUwePvtt69Zs+arX/2qxWIp\nKSn5/e9/X1VVtXXr1hgPlbFRdFucL+yoPtvUJwjK85LuvrIsO7JHdADYcbhFKC3E3X6wOR4T\nHHqtjIiktNI9QTd8K9zRE7bPXJs50gTHnMKUvx9QmC1SWZgSLvdxvNb89JtnzjdbBFFuWsKn\nP1F21dL8aS9jzxiLgfULc/5+oPnguRFLBtKTdfdeUzFdQxoHl1k0vwKu4YwA6XOkgttHdNyM\nlGLxizE3BVIBTLbSJOIcouMAgRPxDIgLJpi1GS/hjVktiSSd+lNLC1otjm6rW5YwO0mXlhCl\nKqoESmV0ASB0WgdjjMWLOEtwREKSpG3btj3yyCM//vGPLRbLnDlztmzZctttt033uBgbcqzG\n/L1nDjrcQ3cP1S2W9w63/Ns9y1bOieitYEef8qN+t8Xp9dG4im7OBHqNal6xqaquJ3TTitnD\nRUZHaaqSFHFHg08szdvyYW1zly0wKEv4uTCPKzuPtT323HBTg5Zu23++eKyhw3r/DZURXpEx\nFr8Q8T++sOLVPfXb9je2dttSk3QrKzPvvbYiOVpPm1OIgrIbAACONmp5E4s+Ne6T4SjLQybe\n/jOEALIDIoAhTM5Cg7gcoIvIAiAQjQBZYzVkmTT3ANVsp65T4LGDNhlzl2PRhqE+I+QGVwP5\n+lEygCYP5CkuEJ6brM9NnsK/XkWISTlkaVHYkMR1qRhj8SruExzvvvtuaDAlJeWJJ5544okn\nYj8exkYnBP3XS8f82Y1Bbq/4+V+P/+mRKyIpohFu+XeCThV32Y1BD9w895tPfOTyjHiPdPWy\n/LlFJv/HxWVpWrUctA8AZKToI+87o1XLP/vSqideO7nrRPtgJCfV8JXb5i1RmvorBD3x+snQ\n+JYPa29aVRj5jJtA/XZ3VV1vz4AzNy1hYUmqSo7yrTljbHJkCTetm7Vp3azpHsg42ZuDsxsA\nAEC2evRYQJ08ztNpAdNAoR+KBqQpWTdBQA0k6i6UC1WjVApYEGbnDMTgBltRQQKcveLQk+Aa\nGIq4LFT3HvWcl5Z9CdzNNLAXhBMACABQAsNCTFgSi4FNKSzbQIeeD46q9aOsT4kMUfXHdPxt\n6m0FrQFzKqXL7gBjVFbZsGl36NChH/7wh0eOHOno6DAYDLNnz37ooYc+/elPT/e42KUr7hMc\njMWXc80WxVoSvVZXVV3PspDGqKFWzcl6/aMGhfjc6e+TNzEV+clP/PO6p988fbTG7HD58tIN\nd6wruWHliLvb5ATN/TdW/u8rIzIOsoRf2zQ/3OoSRenJun+7Z1lPv6u525qSqM1LTwhXW7S+\nY6CnX6HlihB0tNp83WXjTnC8ub/x6TfPWB2ewY/5GQkPf2pRYBKHMcamBLl7w25y9eK4ExyA\n8gLyHQAKnP6mRtWiKbmNJHEWKHDxoIfEGUA3SlPfizci1hbR8A5Ym8HlAk/ISg1LIzXvAV3d\niDYiJMB2FGQj6MpiOdLJw6KVkmtAnH4bfEO/myAhXVr+GdBNakKK2P0cndox9MFuoZr9vsaj\n0s2PzMACumzyGhoaEPErX/lKTk7OwMDA888//5nPfKaxsfE73/nOdA+NXaI4wcFYTCm2KR3U\nMxB2U6AVlRlXLM59/2hrYDA9WXff9XG8biI/I+H79y4HALdXaMIUir91TfGsbOOz75yvae2X\nJZxXbLr32tmzsieyrjs1SZuaNErlfwAApzvMymQAu2vci5P3nmz/5d9OBEaau2yP/u7jp7+5\nIT1ZN96zMcbYaKSwP99QntD6GtSh6nIQzUS9AALQiFIhwJQs1XECKVRyBaoHKIr9bSr1nqOz\nfxlKXnjD/KjvPg55CQrHOk5jvCU4AAArrpILllNXNbj6ITETsyonWW2EuuuHsxt+Hhd99Dze\nzE+8F6FNmzZt2rTJ//H++++fO3fub3/7W05wsOnCCQ7GYiotKezTbOQPut+5e8ny2Rmv7W1o\n6rKaErWXVWZ+9qpyY8SlKGaycNmNQQtL0v7zH9NiM5LctIRw1U8LMsddpW/LzrrQoN3pfWNf\nQzyWhmWMzWRoKCCUgUKytLIBdBOe6yeBVIgw1W/gyTK4yCOEALIAxugHvn80VPdWwNQMVB6b\n1wGgkOAAryV6I4sufQoWLp/44W4btRwjWzfqkjBrLjSeUNyL2s6D2wGaaBcWYdNMrVZnZ2d3\ndnZO90DYpYsTHIzFVHleUl56QmAD1EHpybr5xakRngQRrl6Wf/Wy/KkeHRuWkqhZOz/LX63D\nLy89YUnZuO+5a9v7leNtA4pxxhibOJUBMy6nzg+Dwph9JeBMa3etmN0Yc1N0OHvAFbC6BxGU\nctygU8puAABeijfV1HxEHHkJPHYY/AeTXgN1uIrpBG47JzguVi6Xy+Px9PT0vPDCC3v37n3y\nySen4KTqS/H/FJs8/r5hLKYQ8dt3Lf7u7z7212IAAL1W9e1/WKwedfICi72v37nQ6vAeqe72\nRwoyE793z9IJFAcNV+lDNZ4CIowxFiFMXwXaNOraDS4zAIIuS8raAIZwlTunUfgJcTiBjrZK\nhJc6qsDaCWo9ppbCKP1BfCMXiqpkcAsK6umCEuYsJnFW4XBNzpSMN45Qf7s48KcR5UiED/oa\nlfdWacEQVP+FqOMo9NWCxw6GDMxZAfoYz9lhU+a+++577rnnAECj0fz617++//77xzzkG9/4\nxquvvhpuq8sV0cJtxkJxgoOxWJtTmPLMtze+9EHNuWaLT1BFfvKnNpSOWRKCxV6iXv3TL608\ncLbrZH2PxytKc5PWL8ydWKuauUWm/acVpmvOK+Yio4yxqEBjORrLgXwACDhTE+iYqNyiBbMB\nRi7bJA/g+Fdi9jWIYy+AY6gTOaGE+Stw7u3KfyFaE6A0/LiuQhAyen3gT3LIaqy4GdKWQm8X\neEd2N0dNPHZRmSSq/2hEdmOQVgUOCURwHMtXjaju4XWKqj9B/4VsSM9Zat2HZTdh9iQWy7Dp\n873vfe+BBx7o6OjYsmXLV77yFbvd/vDDD49+yB133FFZGbZ+3EMPPTTVY2SXCk5wMDYNkhM0\nX7xxznSPgkVkxeyMFbMn25XwnqsrDp/v9nhH3PDlpBpuWMkl5Rlj0TTj1qQEQ2kBiVNAASlg\nzEHpwq9IcpDrBHjbgLyAWlAXo6Yy0sUgHrs49Mzg6okLZxPUtB+0Riy7RmF/lR5T55K5yj8O\n0KhAJQOq0VQJhkzMWgS6ZABA0/VkOwKOc0BeAAm0eZh4GciT6jwSlwaU6ixICMlGsHvBNbwa\nF3MrpVX/ELgX1W8fzm4MEl46/zomz+J5HPGooqKioqICAO644w6v1/voo49u3rw5LW20f8rL\nL7/88ssvD7f1a1/72tSPkl0aOMHBWFQQwYfH247VmG1OT1GW8YaVhSmJ4y44f/BcV1Vdj8sj\nZmUbNy7OHb0AZ/yyO727TrQ1d9mSEzXLKjIm1hhFUX37wJ+2nzvfYiGC8vzke64uL8mZnhvQ\nivzkn31p1a9frqptGyrGsXZ+9pdvmavX8g9hxtglTo3SIqABgMEfj8nDi1OEnew7gFwAgwUe\nXOA+S75ONGwEGPsXIrUdG5Hd8McbP8LSqwEVpuNhyY3gtZEloCy0PlWa/SlIGLn8BDWYuBIS\nV4Kwg6SLZDBxSjSc8u59XXQ1oS5BKlmgWrsJdQFd0lVhbmwSjPLN36Izu6i3BTR6zJ2Ds5aO\nXOtD1Hlc4UDyUdcJLNw4hV8Ci70VK1Zs2bKlvr5+9AQHY1HC99aMTT2rw/O9Zw5W1Q/PX93y\nYe23/2HRqrmRlq+3Ob0//PPhQ+e6/JHn3zv/r59dWpaXPMpRMdbcZdt3uqOrz5mTali7IHti\n7U4PnOn6778e87fIlaQzt6wp+vLNc1Hp1nNcdle1P/bnwz4xVCKus8+x71THI59esn7h9CyT\nnlds+s3X13X0Osz9zvyMhCTDlHRYZIyxiwIaAYKz2+Q+PZTdgICnY18veOpBXTL2OW1h+ji4\nbeCxgUapxodKj3Pvhd5zMNAIwgOGLExfOFrbVMkQdlP88+78q2fHC4N/JgDRct537APtFx5D\n09DNDGZWUMvR0AMxswJ0Rlx8Q9hf5F4neJ3Km5x9kx03iy2fzyfLcuDHN998U5blWbNmTeOo\n2KWMExyMTb3fvH4qMLsBAFaH58cvHP3jv1wR4TyOX798IjC7AQCtZvv3/3jod9/aoFXPiPnG\nf373/PPvVXt9Q2sunnn77AO3zL3+svFVsOvqc/zg2UMuz3AvQyHold31mSn6O9dHcPManscr\nfvW3E/7sxiCfoMe3Vq2ckzmNf4dZJn2WiWvIM8ZYBHyKGQoibwdGkuCQw9XsQJBGK+eBpgow\nVYx9/osadTV5PngxONjf43nr95pPPzL4EYtWYt1e6msesZMmQZpz/Rhnl7UgqUB4g88PgOow\nTWrYTLVp06bk5OTFixenp6e3t7e/9NJLhw4deuSRR1JTI20OyNjUumjn1DE2XdxesfNYW2jc\n4fLuOqEQD2V1eBTP0NnnOHi2KzQeezuPtf1p+zl/dgMAHG7vr7aeON04vhcv7xxqCcxu+L3x\nUcMkR3iqoddic4fG++3uk/W9oXHGGGMzDgU/AAMAAIaJh0gtU44n54OKC3uPwXf649BCoQDg\nO38YPBem1Ugqad0/YfkVoNYPfsS8RfIV34SEsRYmoISpCikkBMD0MBXKiMDSTG1HqKcGfAq/\n39l0ue2225qamn7605/ed999P/nJT4xG43PPPfejH/1ousfFLl08g4OxKdY74FJ8aAeA9h6F\nxcBKuzmCph74NXXZFOMxtm2/QgJCCHpzX+OcwpTIz9PUZVWMt/XYvT4xgW6sfv12T9hNSokP\nxhhjM46UCL4e5XgEML0cMiqp68zIY2Wp8qapGNxFjmwW5Q3CRw4rqi9kiNQ6acFtsOA2cA2A\nJiHyfj1Ych31N4J7xG0A5q0GY77C3v2tomoL9A9NFSGtEStvxpzFEV6LRdXmzZs3b9483aNg\nbBgnOBibYgk6FSKQUoLCaIioxZ1BF3YBhWFm1KRsDpNnCZewCEenUf5KVbIkS5OaXzbKMpBM\nXiHCGGPxANWzSCHBgaiJcG0/4pLPQe0H1LALPA5AhORCac4tkDy+1ZSXJkwKMwtDpUaDUrlu\n7TgLhOtSpeVfo4Yd1FsDXjsYMjBvDaYpTd9wW8WB346oF+saoGPPg9qA6Zf6SiLGWKgZ8bDE\n2MUkUa+eV5QaVIMDABDxssrMSM6Qk5qQnWoIne6BiEvL08c8vM/qNhrUsjTZIp2jCNf7Y7w9\nQZaUpW/b1xgaX1yWNskao+V5SUVZiQ0dwQmXgszEyoJxzDGZEk2d1pd21ta29es1qnnFpk9u\nKEnUR5TqYoyxmUAQOTxCr5akSZd/Hh91MQgruM8N9lABAEAVaheDFPGPcUmFZVdh2VXgtIDa\nEL4qBwsmz13lee858AWvBpLnrALVFP01qvRYeuOY31LUtE+5G07tDk5wMMZCcYKDsan34K3z\nvvmbjxyuEbcFm9bNirBHKSJ85dZ53//jwaCFKrevLc5O1R+rMXf0OtKTdXOLTIEzIBwu73Pv\nVb+5v9Hq8KhV0orZGV+6aU5u2tQU6yKCjl57Z58jM0WfZTIsLUtv6lSYrBFJ/iXQ2vnZC0vS\njteaA4M6jXzf9ZWTGi4AIj7y6SX/+vsD3ZbhOu2pSdpH7oKDyQIAACAASURBVF4iRTP1E+qd\nQ82/2HLc6xv6pzxea37746b/fGBVYWZEU6wZY2waOTy+s13WLquLABAgI1E7OyNRH8M6zaid\nD+pC8LYRORATQZ0POJGOXaCbQT3I4gKastQ33O958ynwDa+6lTIL1NfFfDFCf8v44uyiMTPK\n6rO4wwkOxqZeWV7Sb7+x/pm3zx6vNQ/YPbNyjJ/cULJuwTi6k66ck/nzB9c89cbpM019PiFy\n0xLuvrIsM0V//3992GoeWh6Snqx76Lb5a+ZlAYDXR9/+7f6zTUM1Pj1esfdkx/Hanscfujw/\nY7I5jtq2/se3Vp1qGKrNOacw5d5rZ++uajf3j+jxVpxtvGVN0bjOLEn42H0rXthR/dreBqvD\no5JxcWn6P948tyhrCh7+S3KSfvetDa/vbTjbZAGg8vyUW9cUjXeOySQ1dNge31rlz24M6rW6\nfrnlxM8fXB3LkTDG2Hi5vOLjpl6Xd6jSJAF0Wl0Wp2d1UapmEjWSxk1KAk1SbKeOMAAA1fJr\n5KK53v3bRGcT6hOlWfNVK64DOfbPDuH+8fmbgjGmgBMcjEVFlkn/nbsnVf5qTmHKzx9c7fWR\n1yd0Grm5y/blX+4KLF/abXH+x7OHfv7gmjmFKe8dbvZnN/ysDs8ft5/77meWTGYYHb2Obz7x\nkc05PBvldGPffzx76Ef3X/b63oY9VR0OtzdRr756Wf49V5dPoP2qVi1//trZn792ds+AK8mg\nUclTeb+i16g+tbF0Ck8IAEKQ2yvCVQ/xe/9o6+/fOtPR61DcWlXfY+53piVN6D0kY4zFRH2v\n3Z/d8HN5RX2PvSIjMA3tJXACqBC0/Mx5kcGMfPVN/zjNg0gphI6q0DCmFA79Sfio+TBZWkBS\nYWox5szj70PGLmWc4GAsdvqd3j6HW6+WTQaNKrKFEioZVbIMAK/sqQttzuITtGVnzb/ds+xI\ndbfi4UfDxCP31501gdmNQTan952DLd++azERDDjcSQbNJK8CAKnGmd60r7594LdvnD5ea3Z7\nRZZJf8e6kpvXFCnWOvnbh7VPvnF69LOZ+12c4GCMzSyeNnI1grCDnIS60l6HQpdQAOh1+NtU\neX3UKKhz8AOCVsJiCU0xGetMRU7yngOfGcALaER1GUjjW7zJgmD+SmrYA86RXV1QxtKrAAD6\n2337fw8DHYNhAsC0EmnVfaDldaCMXaI4wcFYLFhd3r115qYL7/P1avmyIlNZxjh++9a09ivG\nq1v6AcDpVm5MG1QHZAJONwZPDBk0uGIFEaYkuzEuTZ3WE3U9Aw5PUWbi8tmZUzvpI5xTDb3f\nfnKf+8LLzI5ex/+9dvJUY++jnw6eION0+/64/dyYJ0xLmukJHcbYpYRoYDe46oY/O07lqmf3\nO4tDd/UN9QkjrzhNMNxUi8Dlo7MAs2d0jsNjB58nWiU5RD+59wJdSACRk1xdqK4EVXlULncp\n8LSQpxXnL4eak9TbPhQ0pOHc28FUDMLn2/87GOgMPILMteLwC9LqL07DaBljMwAnOBiLOp+g\nt0519Dv9r7zA4fHtrO6WJZwVcRHQcKUxB+P5GYkAHaFbCyddzEIIpYa3AEKxEW6UCUFPvnH6\ntb31/vKrRVmJ/3LXkrK8iKq3TsaTr59yh0zV/uBo682rihaUpAYGzzX3hcs3+c2flcrTNxiL\ne45zZKsCXx+gFjS5aFwBcty+NHacDsxuAAAAFajPdKiSe73B2YpEjQoABPUGZjf8BDXNzAQH\ndZ2mM9vA1gkAoDZg6ZVYtBZwKouJkOfEcHZjOHgW5VzAqan5PXHCR1XvUvVH1N+FiWlYsgIX\nXguqWL+iGAfykfVD8LQAAEgA5ZnoSgcowsQKSMwa/Iej7uqg7MbQoW0nwWEBPVeWZexSFMMa\nUYxdqmq6bYHZDb8jzZbQYDhzCpXvF+cWmQDguhUFKqWSbzetGl/Vz1Clucq5g7Iw8ah6fkf1\ny7vrApvLNHRY//WZj+0hi2imlsPlPRNS4mTQofNdQRGXR3lSt1+qUfv1OxYERqpb+l/8oOY3\nr5/atq/R6lD4VmGMzTRk2UmWD8HbAyRAOMBZQ91bwds73eOaIHLVKsZztMGNKhCh0KQHAIIB\n5VOBHWCMJG/sUcshOvTMUHYDADx2OvMGndw6pddwgwjuED+4AXwKbyBiyucVb/xM7HuRuhvB\n7aCeZnHwZfHqY+BxTfPAwiPH8aHshp9WAm0LGBKG01K2cOtwiazBv50ZY5cITnAwFnXdVuUb\niF672yfI4xU1rf21bf2ekAkCgW5fW5ycEPymRaeR77qiFADyMxIe/cySwNUisoT/sLH0+ssK\nYXI2rZsVmjpRydKdG0omeebxIqJX99SHxnv6XTuPt0X10g6XL9yEFXvIIqAEXdiZceV5yXdf\nWfb0wxsKLvSIFYJ+/XLVQ7/e/bs3z2zdVferrSc2/+yDfaem+1aYMTY6dzs4zgcHyU0D+6Zj\nNFPBp9D5GwDSdR51wDJAtYzzs5JSdGoAABhlHt80TPEbDQk6u00h3PwxWKfw523Y9DSRe+qu\nMhF0Zie1B3/HkrmJjr8dk8sT2Mxg7QYa4wXACG7FpJsgd8BUIzn8Yk81rwNl7BLFS1QYmzZE\n9OIHNS/uqHG4vQBg0Kk+84nyO9fPQlRYjZKWpPuvB1Y//vKJE7VDL4jK8pK/tmm+/1F57fzs\nRSVpu060NXfZUpO0K2ZnTlWz1R9sXv6rv53wNwTJMum/evuCkpyoz+AYXB3jX5vTZ3NbbMr3\niA0dyi8Sp0pKoiZRr1acW1EQUEjl/aOtT2073W1xhu4GAPOKTT//8pqgf9u/7ap7/aOGwIjF\n5n7suSNPP7why6SfgqEzxqKAXM3KG1ytQD7AcfeTmn6SFnwKP2C1Gv3a4rRum9vh9elVcqpB\n7fGRT5AsIYJB8UwI2hl3e2nrArdyBod6ajAxa2qugloACUDhGR5xmn+eU8PRMPFjuOzWyZ7c\nYaWedjSaMCktdCPV7RUnt4HbBgCg1ktzrsfS9aB0nzPyOB8I5TZkIIYXRmFGOUgyiJAZQ7ok\nTM4bx9fAGLuIzLDfQIxdjNITtaD0BH76TOeJM8OzK+1O71PbTvfb3PfdUKl4nqKsxP9+YHVH\nr6O9x56Ros9J1QelQowG9Q0rJztlI9TyioynH95wuqGvo9eeadLPLTJNoB3suBw61/XH7edq\nWvsRoCI/ZfN1sxeUpKqV1uAM0qiiOxlNkvDaFQV/+zD4bVKiXr1+Yc7gn9/Y1/D4VoU+doMW\nlKQ++ukloXd02/Y1hO7s8vjeOdT82au4KB1jM1XYF/IE5ImTBIcAEMP3gZp8cCj0fkJNvlqW\ncpJ0Do/vaEvfzho7ESBCbpJ+WX6KVq0lCJ6iKGFutIc+br7wS/9G2TRuKpCzwBc6o1AFcvbU\nXWVCXArVUgCAnJN6PUAWs3vb096qvYMfpbxS7S0PSAUVwzuc/rs4/f/ZO+/AuKor/59z35ve\npBn1LlndsuVu425jg02HBEIJIZQkpBeym76bTf+l7W5YloQawoYAIRiIKaYZ494tF0mW1XuX\nZjR95r37+0Oj0ZQ36mUk3c9fmvPalTQz797vO+d73h4+wOMQz7+KTjMpuWmUUyMHyAGVqnXC\ngGxWlQELdtKK0DwUsvS2qXVXYTAYcwgmcDAY086iOE1ZS78l2CfC4fRcquoJ3/kfB2s/uSUn\nvBrFT2Ksauaf7Stk3LJcE0D4w5mp57XD9f/7+iX/y4v1vY/88ei/fKp058q0nGR9bZtEN5ml\ni6Z9YPfvKug2Ow6UDc9cTXrld+9aNvif8gr0z+9Id065bVP22qKEZYviwtUNQaRtvXbJo5q7\npB82MhiMaAA5nXQNBlEAifbEeAoWQWykYAOgCHKCKQQTUbWUultACP6ClaeCIhsA3IL43uVO\nm9t3F6MUWsyObptrd1GejKun4P++IhymEpxcQoQo0MZjtK8eRC/qUzFrA8gmfcvTxEVcLY83\nfcMzAJ4BkMcCLzEqlC2h1AZi4J+RQ/kywNl+V2hN0FUfHkZ9/IRPSV0OxxPfo33DNT5iS43j\nqR+oHv41Sc4GAPA4xMvvSRx4ZT/kbgXlaHmgslRwN0qMWRaUmkGKdlNdkljxNlg7AQnGpGPJ\njRiXO/5fiMFgzBOYwMFgTDscwd3FSYdqelrMg/mWVMnzerl0gxKvQKtbektzhaHZp4ojSQjj\n9jl3eYS3jzddaTEjQn5azK416dOd5jAlWB2ep9+qDI8//kb55qXJD11f+KNnTgrBf7eV+fEr\n8yY+RRsjcp784J4VN13Ve66me8DhyUrUbVuWolL4vkIbOwcsduknuqlxmuW5cZKbOIJynnN5\nJObcSjn7cmYwohhlDlhPAw2zN1blAcxE4+oJI9I+gVb5PTIouAVaT8HOkRyMuZ7aL4C7CUQb\ncHpU5oKyYPDXqeq0+tUNPy6veLHNszqjhMIApQ4AnqAOQDap8dl7hGNPgM1nD0lby6D2Y7L6\ns2icnOsTr8TUlbT5RGhcm4hxY86Vs7eKze+Aw9epFA35mLIT5MF9OlCBis3gbaRiD4AXUId8\nNuDs98zCvPW07rRkfMLn9J7cF6hu+PC43R+8qPz09wCA9jWCKGUBTkXaW4cppSOfH9XLqbcT\nRF/VJx18L8qzQJYcumfaci5tOQgeQAJkTuRPMcbGNOcLM+YrbA7NYMwEWgW/qzix3+Hpd3iU\nPDFp5EcvSRubZSbyhZltwvCDpgFB6JaRHIIxY79cbZvl35491dnvq19991TzPz6u/dkDq/2G\nHYM43N4X3q8+cL6ts9+RGKPauizlru25Svls3k4u1vVKLvitDs/lpv5V+fG/+tzax98oH8zj\nUMi4Wzdm3X113qjFvJGwOb2VjX3dFmeyUV2caeS5UU60JMcY0hR2kBEMYkf2jl2eFydpKboy\nX1oTYTAYUQGnQcNWav44qFZFkY7aVbM3pjEh0vpwB1CRdhJMQlSjZgVoVoQf1WmVthbqtLoA\nAEGHqJua4Z170a9u+HBbxdPPc9u/D9ykpBMsuhkEN20LsKIwpJHSe8ZaT+ToEGv+L3C5Ts1V\n1NFB8h8K87lE4DMRJtvCbGrBrOVYupue3xdo84lFWzB/w4TPKTRI1DQBgOiPS6obvoODN7la\nqLsVRDfysaDOA5QBABAd6m+gjnPgaQPqRM6AigJQRJa6JvcOYTAY8wYmcDAYM0eMShaj8t2A\nI5l0fv12A8+HrPBFj1iv4JYAjGkeJor0538961c3Bmnrtf/yb2cf+9omvxZgd3q//tgRvz1n\nW6/9bx9WH6/o/K8vr59FjSO8KYkfm9MLAKWLTH/85iaL3W2xeZJNao5M/GHp/nOtj79xqd/q\nW59kJGi/dfvSwba74yUtXstzxCtIaBkju7E+sKugrKbHEfxbL8s1bSyZ7YJtBoMxMsoslCeC\no4p6+wCVqEgBxdRbIE0tFFzhlhm+TdSMKG0aCgCCVL7hCPEJYu+lPTX+R/XDOM20uwoTF0/q\n5JwMS+/GrE2DxS+gS8a4gtGtLoegHYcklutuM+05gwlXTWpgMwVZezvNXkWrj8FAF2iNmLMG\nk/NHP2wEhAjNgIdsTVAfmmoxjN8BlHpp3wfgrPe9AgDraYy5GhQpAABEiZp1kxokg8FYeDCB\ng8GYHdLiNVcVJx4NfnqfbOLyUiU/lV6RWgiOae1d2dTf1Cnh4FDdYqltsyxK8a23Xz1UF958\npLbN8trh+sHWszNAV7/jjSMN9R0DagW/NMe0a016ikkTaefUuOFNerU8sCfuBDhzpfuXL5wN\njDR2Wr//9ImnHtkSZxh3OrFGye9anb43zDE0N9WwVCrjw09Wku6xr2184s2K01VdHq9o0Mhv\nWp91x9YcyU46DAYjuiAq0JTOqc9qhBUpgGTvDz8GpaxbqomVQTWV00jq6AMA6Rofe+/UXMOQ\njob0CRxHbRH65kSKRyWYkI0J2eM7RvB6WxrEATOXnM4Zg6pBSVKWUHky/AifAQcAqI2YspS2\nng8dRmIh6n0iPrUc86sbQ1e00753MeGu6LezYTAY0QkTOBiMWeNf71z2369e+Ohcqz+yY2Wi\n5ATU7YUzNV1tPf0mvXLpImOsdqS7vr+fazjtvXa/wHGyslNynxOVnTMjcHx8vu23L5c53b7f\nd/+51n8ebfjFQ6slnURLsowZCVPQ9dbPyx/VhAftTu8bRxoe2F0wgRM+fFOxRxD3nWzyR5bk\nGL9z5zIyWo5JWrzmJ59dJYrU5vTq1CzDlsGYObrNzoaOAa1KlpWkm+7mUNEAQsQ+pgAjGXnm\nxmtre200LF0jPz6wMkUUaZtIeyi4EBQE4wgmj8+RRB4xhQTkEbXvGYJGEIAkjUvnC67TBwee\nf1To9c0WFCs36e79ml/mkK251nvkn9QdWr4k23Sr/2ey8m4RkbaU+SOYtJisusf3gopgl/Ln\nFl3grAV10ZT9JgwGYyHBBA4GY9bQKPnv3738nqtzq5rNiJifZkhP4N1CaKvRC7XuP/zD0t7r\ny/VQKfgHdxeuX5y471RzU5dVq5ItzTFuKkn2P/LXqiIukgPXz1andDGIzRGxZ15nv6Otxx5n\nUCYb1aOu20emd8AVqG4MUttmefyN8h9+esW/P3cqMAklJ1n/3buXTeZy4dS0SnRjAYCaVvPE\nTijnySO3L/3Epuzyhj63V8xJ1i3JNo09D4MQZOoGgzFj9Fvdj79x6aOy1sFFu0Ejf3B34a41\nE3m2P6fgCMaJNFTdRlCMbPNkVMvXZ5lONfW5hkyFeILLUmOS9f58N8ErllPw9SKlYBdoo0h7\neVIMMFZ/a9QlgdoE9rD+YpwM4ydXTDFe3A3U0w7UBZwBFXlA1KhKpNZ6iT1Vk2sZE8W4zh3t\n/+8fBUVOHxRa640/explcgDAmHjFff/mfvVRscfXXwyVGvmu+7jC1cPHyFRk7QO0rxH6m4BS\niElDY9bwVtEOVHrKQb3mOZUbxWAwoggmcDAYs0xmoi4zcfghGKKK0uEUjK5+4SfP9Tvdww/O\nHC7v/7x28dl3L/ubsLx3uvntE00/umeFWskDQEl2rEbJ28L0C4NGXpQxXOSSbFRLVrIkSxWJ\ntHTbHt1z8cyVbv+Yv3ZriaTd5rnqnsqmflGki1L0awoTIq3wj1xqD1E3Bjl8sf1bty/90zc3\n7z/XUtVkRgKF6bFbS5MnqaeEE8m8Y5IXykrSZSVNjdkeg8GYJkSR/uCZE1eah9VMs839+1fO\nI4FrV41N4xAFsJtBbZhzLRs4zALwinS44gNBxZG8UWWIjFh1kk7ZanEMuLwaOZ+sV6oCcl5E\n2u5XN/xQsIq0g2BkI4ZQkCy9XTzxVIjbBSm+CeRTmcE3EtRDBz4C73D1KHVWoGYdxK+FcIGD\nyNG0coYGNuPY9vw5POhta3IeeU+15frBl1z2YtXXHxXqLopdLag3ctklqJGwncLYDIiVcqjB\niLI+4mhVqM5+8DpBbQIScBLBTRtOUXMb8HKMz8UklgPCYCxEmMDBYEQXMsxy0yp/oco7JxyB\n6oYfh9OrCLACLW/o+/N7VV+6sRgAVHL+Szcv/s1LZYH7E4Jfu61EFtAp9trV6SekqlR2hz3D\ntNjdj/zxaK9l2JquoWPgu08d/68vrc9LMwTu9ou/nvWLIABQmBHzw0+vSIiRyHzu6pf25PcK\ntNfiSo3T7FyZtnNlmuQ+U0JRZuzhi+3h8YmZjDIYjDnEsYrOQHXDz/PvXrlmZfooiVe2PvHI\nS7TmBAheIBwuWkXW3wXakax2ogzCYT7BAUotFAQEDUHjGAtJ5DzJMkqXioi0TzoOfQTGLnAA\nxudzW/9FrHyL9jWA4EFDOubvROM4bSMmAbWfDVQ3AACol9qOouEmSL+Btr4PwtDNSxFL0q8H\n+Ug20nMYweuplyoeAfDUlKuu2gYuG2hNgAR4GZe3nMtbPpGrEAXI4sHTJbFJEXECQLsradVe\ncPQAACDBtHWYcw3wCtpTJx55Buy+tyKFfZhcTNY/APzst+llMBgzCRM4GIzoAlEj50oEsU0E\nG4DY2G6X3E0M864/UNb6heuLBhMTdq5My0jQPrevqqrZjAiFGTH3XZOfm2oI3H/TkqQ7t+W+\nfKDGfyqO4N1X564tSgg5896jjYHqxiAer/jXD6p/fN/ww6vfvFQWqG4AQGVj/0+fP/OHr6wP\nt8zURyjHQIRJuoeOkbu3556s7HQH93CNMyhvvCq6evsxGIwp53JTv2S8s9/RO+A06SMvh5wD\nwiv/Adah9AdRoFeOC61V3Kd+Cqq5tNCdwsauQ0RogEW943LhAADQxJOV9016PBODgrtOKiyA\nuwGNS9GQR61N4LGAwoTajLG2mB0TAgCNnmk5DTdcAQAAmVGjUrYJe/4VAICTYd4Wsng38BN3\nA0XDetqzN9TKRF0A8tCpiG9gXeX0/PMBr0XadASs7bj0XvHQk+AMKj6lbeXimX+QNfeEnoXB\nYMxrouWblMFg+EGQ8cSXzCnjzwCE9joBAAybMzrdgsXu9vuPFqTH/OKhNSNf6IHdBVuXJX98\nvq2jz5EYq9pamiJZXlHRKP1orqJhON7Z7zheIZEPcrmpv7LJXJQRWt29tijxybcqw2WaJdmm\nmbGiyEsz/Orzax999WJdu+/Puyo//iu3lgQ6mBwoazt0sa2r35kYq9qxIm11YXyEkzEYjLlE\npMUbAETeAgAgnts3rG74sfWJZ98i6++ciqHNWVABVCIvD3FONcIQXREtIQQrAgCnQsOQG4ho\nA08fIAeccXL9PnoprQawAVAAFWIWjCfnZZpAXsanZnmbg+QeWZzWsDoTyFA/HcFDK98X+5rI\n1q+Mz002EHkSxt9GzcfA3QbUC7weNaWgiVhaQmv2SQT7aumV90PUDd+mhpOw4nbgZ+LBCYPB\niBKYwMFgRDUl2bEHL7SFxzHMKoIjqFGM+xOdk6zPSR7lwWO4DOGLBywFJO08BmnsHAgXONLi\nNffuyHvu3aAMWK1K9pVbFo8y4sj0WV11bQOIkJOsN2hGn82UZBkf/8am9j57V78zNU4T2B3W\nK9CfPn/a38S3vKFv/7nWa1enf+uTS1n/VgZjrhNYWxeISa806UdcqbaUS8ebI8QXDATiBZCo\n+kGYU7owygAQQOqWFyhhUDe1ngTXUCsu5EBVguqlE1rkt1Ma+OZxUFoBYEecoWbtI6C58R7z\n4z8LihQmQdjcg3Zcpq2XMKVk4lfijWi6DoACFQBHnMZ47GCTbgCH5qYIMxUv2HthqCstY26B\nMrZQZUwE9r5hMKKaXWvS3zjS0NIdZN6GAHJZ6CSjdJFJPj1tDhel6E9eliiRDax5CTQECUEZ\nYVT37Mgrzop9+aPa+vYBtZJfmmO8d2e+UTeR52Auj/DM25ffOFIviBQAZDz5xKacz1yTz3Oj\nTDcJwRSTJiXMV3Xv0Qa/uuFn38mmVflxW0pTJjBCBoMRPVxVnJSZqGvoCE2Ou2t7bng9XRBe\nt2SYekOL+BYaBOMoWEUaaGyEBJMIziF3EgDkQJYMnlaJLbJhSwhqOQCegAcPVAB7GQWK6vF2\n+6KUXpGKNwKkAsyyc4Tyqh3Uabe+/KRoGwAA5AlvUALQcB2HdlVPSuDwgaOoGwAgRm7KO8In\ndxIVNAwGYy7CBA4GI6pRyfnfPXzVn/aW+9sZFqTHpMVrjgQvv7Uq2YO7C6dpDDdelfnG0QZ7\ncFsWjuAdW3P8L/NSDWolbw9r3cJzuCTbFOnMy3PjlufGTX6E//nK+Q/PDk9JPV7xxf3VDpf3\nyxPNB/moTGKCCwD7z7UxgYPBmOvwHP7yoTX/9Y8LfqNllYL/zM68m9aPZsETmwrdjeFhjE2d\n8kHOOTjMImgSaQ+AC0BJ0IQw/tYn3nbq7QLqRc4AsswpNbkYE6heSS3dQIOVLGUB8EM3Mm9X\nkLrhx1EOqpLRl+hBWAEkK2IoQF80FKqott2kXLvdU1shmvu4xCQ4/YR0looQsbv8FCPXglwL\nbomMURqfD/UXJQ7RJ4GaeYczGAsLJnAwGNGOUa/43t3Lv37bktYem1GvHMxxOFLe8fqR+qZO\nq04lW5pjuvvqXL/7xpQTH6P65UNrf//3soYO36wiVqv40s3FgdqEQsY9uLvw0T2h04s7ti4y\njpzyHcz52p6/vHulutVMEAvSY+7fVZAfIZncT2uPLVDd8PPPYw337MiL0U6k8rbHIt3kpdvs\nkIwzGIy5RZxB+bMHVjd0DNS3D+jU8rxUw1jcf7BkO71yVCK+dOc0jHHugaDjJuxdSj3UfsSf\nPUEBgFxCzXrgIkrk0wJnQMON1FEGnnagLuAMqCwCeYDy5Q0zYRmEekEwD+sgYyJyPgKIkTfN\nKKjWyktWAwAAFS7pJX0u0DBTuj8ipm+QsOFQxpLMTWJfN63+GACG00wIR1bcPkNjYzAYUQMT\nOBiMuYFayQeWhKwvTlxfnDhjVy/KiHn8G5srG/taemzxBlVRRowqzO/jxqsyjTrF029XNnfZ\nACA+RvWZa/KuWRnadHYE9h5r+MOrwxLJ6aqus9XdP7hn+aYlvgdZZpt7wO5JNqm5gDLgqiaJ\nwm8AEEV6pcW8umAiFeAxWkVHn4SWETuhChoGgxGdZCbqMhPHsSDHlAKy/SHx0F/BPfT9IFOS\nDXdhakRPRMYYoY7TobUhopXaDqLuhnGmRUwaokLNuhm5kjqi5QeoZ2QA4wJJ3hbxwj9Dw0o9\nZqyYuUFkbgHBRRsPDper6NNI8e3AyciK26kxXax4F6zdQHiMy8HSWzB2HJMQBoMxP2ACB4PB\nALPNffBCW0u33aRXrClMyEiQyCvmOSzJNpZkj1RQvaEkaUNJktXhEUQ6FpvPQKwOzxN7K0KC\nokgf3XPxquLE87W9j79RPlgzL+fJbZuy79mRp5gezxEA2LgkSbKRpF9qYTAYCxMs2sxlltL6\ns3SgB3UmzCwFDUuAnzTUA+4GibjoAG8ryDJmfECR21CtXwAAIABJREFUiZSjgTxwoXbaoyEH\niAMIt7jSAIz3VDMBFl2Dbhut+gjoUIKJPpGsuw9kqhkcBOKiazFlDTXXg8cB2kSMyfEZcCBi\n9lVc9lUgeIBwgGTmRsVgMKIJJnAwGAudA2Wt//3qRavDV0P79FuVt2/JuX9X4RjbhVjs7vL6\nvj6rOzVOvTjLyBEM7LQ6iCDSN47Uf3i2taPPnhCj2lKacuvGLJ4Lmnycr+11uiXydfut7j0H\n6595p1IYaubi9oov7q+pbrH8/ME1iBEbIhCCuamjNIiJxC0bsk5Udl6oDUpFXr84accKVmnP\nYCx41AYs3sr6KU0loj1iUYZggZloHT5m+DiQp4K7JSSMqpIJOIYgFlEqAATea7SIJRPvujqt\nIJJlt8GiDbSzGtw20Cdh8mIgHACA4Ka2LpSpQTUNep/gAiRAAt4Hqlgc4UJcVL1jGAzGTMME\nDgZjQdPQYf3V384JAY1gBZG+uL8mxaTZtWb0xM69xxqefqvSNuQtmp2k+/YdpSGKg8crfufJ\n4xfrfBO4fqu7qtn88fm23z68LjAFw+aM6FL2+lB7lEBOVXWdre5ekReXGqfZUppyIMwW9Pq1\nGRP2JVHIuN98ft2bxxsPXWjv7HckGdU7VqRuX57KesQyGAzGGPACHQAQAXUAY8jmG6EIBaNu\nsYq6zdR2CpzVvuoS5FG1BNQTayPCIy4D6AWwUCoi6gDiolTd8KNLRF1AhazHLla8SRuPwaAR\nujaBlNyG8fnSx3pt1NEJQFEZD7LRq8No30XafhDcfQAAygRM2Ya6qWmgS/s7xLoLYOtHYwrJ\nWwkyVn/KYMwfmMDBYMwOXoG+e6rpUn2f0+3NSdbfcFXmeGs6poS3TzSGawcAsPdYw6gCx4Gy\ntkDLDACoax/43lMnnv6XLYG/y5vHGv3qhp/LTf17DtXfuW14ppIUG7HkuLNf2trzfG3Pirw4\nAPj2HUv1atmbxxtFkQIAz5FbN2bdv6tg5PGPDCF441WZN141WlcFBoPBYAxDQayjQs2QgyYC\nSUcuf5QJJ9EA0YEY2rgXAIBPmo5RTgR3BzWfAHcXIAF5Iup3AIiAHPBGwEnevo0AxjkpoFNR\nPPYn2t84LMpYO8XjT5C1nw/VOKhA2w/QrpNABQCgiGhagcnbg/IyQs7dfoB2HB5+7eyktS9B\n+vVoLJ3kqL0fvywc2QOC79kM6k38DV8i2UsneVoGgxElMIGDwZgFus3O7z99or7dN5k7eKH9\nHwfrfvjpFYPL9ZmkpdsmGW/qlI4H8vcDNeFBi9399onGO7fl+iNHgzvaDscvtQcKHMVZsWnx\nmkGD0kDy0wxVzdI2oh6vL6VZIeO+emvJXdtza1othOCiFL2RuYEyGAzGjEOFGhCrAwMgNlJw\nIjeKDyWqVlLbgVDHTUUecKM00pohrOW098Ph4XkHqKMO428A5ZTXLVIQWintBwBEA3Cp0ZzQ\nQdvO0/6m0BFSkVa+FSJw0Nb3affpgNeUdp8GrxMzb5Y+tcdKOyU6FtHWDzB2ItVAfoSz7wsH\n/x50TkuP5++/lj/836if2ZY9DAZjemAGPAzGLPBf/7jgVzcGsTo8v3zhrL/WY8ZQyqVnCSrF\nKLMHSqG2TeppG0BNa1AbOYvdLbmb2RYU5wj+8NMrEmODvMrSE7Tfu3t5pNyWrKSgBNc4g3Jt\nUcLqgnimbjAYDMZsIIBYJxEWO4FKtBcNQpaMup3AJ/impkSL6jWoWjn1Y5wAoov2fRwqvlAh\nSPKYEqidug9SbxkIDSA0UO956j4I1D6Vl5gUbqADAMMTFdpbK7kf7W8CMWA+47XRnjNSu10C\nVy8AgOClF94R3/mduOffxf2P07ZKamsctjINRHCCvX2s4xXctPp9euJP4uH/pGUvgLkJAIRT\n70js6XEJ5z4Y62kZDEZ0wzI4GIyZxmxzn7wc7poOZpv7RGXntmUz1U8eAACW58YdKGsLj4+a\nS4IIHEGvhCsocCRIOU2IUYVIHoMkhtWk5CTrn/r2lndONlW3WDiC+WmGa1al8xzetD7r+feq\nQnaOj1GxniYMBoMRRVDrUGVK+CYz4Giuz5wJtVcDiEDFmW4NOzLOZqBSLlHeAXB3g3wizcgl\noZ6zQK3BISv1nEX5hqm6xASh/VQoH1apSApyBQCK4V6tYQeAKADx/ROpo9Pn0BG+n6MNqUx4\n+7fQP2Sk1d9K689g6VUQ4TkLFd1jymlx9ovHHwdHn++ogTbafh4LdtPuZunTRogzZpNpa5bH\nmN9E0/2DwVgYdPY5aIQ7fUffTD+ouWZV2lvHG0NqQPRq+b3XRHAIC6AoM+ZcdU94fHFWkLf5\n9uWpklUqV0t1JFHIuJvXZ4UE77k61+b0vH542Go0J1n/nbuWRUo/YTAYDMZsMEI6w9gzHUjU\nNfgUnZE3SVtETQRqASpVj0nNQLsp7QEwA1AAPZIs8ADtugy2blDFYHwBKEY37JzMwKj3RFCb\nG7GVUjPy61GfLP1/VZuAH2sqpXh6z7C64afmDORLP+9BmY7Wv0d7r4B7ANXxkLIW4xZLjLri\nDb+6MRQS6eW3UaOmAxL5p8jPgg8ag8GYDpjAwWDMNPrIZqJ69UzfX3mO/OYL6/76QfVbxxut\nDo9Cxq0pTPjc9YVJxoiWn34+szP/Yt0xrxA0vUmL11yzKi0wsnlp8sW6rNeP1AcGd69J37ly\nrKXLhODDNxbfsC7zfG2PzenNTNSuzI/nSPSWJU8eSuHghbYzV7rNNndGgva6tRkhxTsMBoMR\ndaAOgEg3fMXosNKYGHxk+YCfYDNyCSKXolDhHKD/VmsRW87QSxXgGtqfV5LiGzHzqikbSejV\nr0j8T6kNxBZMXQlV74ErVC8gi7YGvkRVIkWUTOJAVYpY/4TERa1upApAV+j+2mzx/HPgHMrL\ncA9Afy0krcL8W4L2Ezy0s0LqlxFJRrpwqVxiJJkSKgmDwZiLMIGDwZhpEmNVWUm6EA8OAOA5\nsqpgyjJdx45KwT90XeFD1xWabW6dSkbGLByUZBt/8eDaR1+72NTpS6ndtCT5izcVK4JTChHh\ny7cs3lKa/OHZ1o4+e3yMaktp8vLccduppsVr0uI14z1qLuJwef/9uVP+7JjDAK8erPvqrSUh\nyhGDwWBEGRyQLBDDfBkwbm4LHIpU4HXgDXvsr0gCPmbqLjPCnFzwu+bRAQs9eyZILPA6xfN/\nJ6pYTCicusEEQPulw7QPZRlk3efp2b9Ry1AKBicjeTswK7imhlejaSXtPhVyBowtAZkO3JLK\nDoUBLSTGgmPYcQO1WeDh/erG8K7tpyBhCcYEdJD12AfbtYTDpecJVTXgCZJOMDmHW7JZcn8G\ngzHnYAIHgzELfP22Jd998rjLE3T3vX9XQULMbD6ln0Cf2mW5pqce2dzW6+i1ONMTtCOcoSTb\nWJJtnNwAFwrP7rscUvvj8gj/9Y8LJdmxKaYgicfq8DR2WvVqebJJPb9TWhgMxpwAuTwKAGL9\n8DN/koJc0SwOaQpADk3X0u43QQgoSOH1aNo5lVchsQB8oIVnwAACanbqGyVTIWjdwekSOCRT\ncobiqE/Fzd+iPTUw0A4KHRqzQSkhZmHKDiBy2nXcpzsgQdNKTN4GhAeFFlzW8ENAZSJ5n6CW\nKnC0A3KgTkFdjnjk59Jj6a6AQIFDrgHCSVuExKbJ7/uZ991nxcYKAAqcjFt+Nb/lTiCs6JXB\nmCcwgYPBmAUWZ8U++cjmP++rulTf63QLi1L0d23PLV00J/uTIWKKSZ1iGr2khTEWKIX3T7eE\nx72C+NG5truv9vXfHbB7nnyrYt/J5kE/l/gY1RdvKt5YkjSjY2UwGIxQELl84LJA7AF3CxWd\ngC7gmkCeNbc79ymSMflesF6g7k4Agopk0C6eaidUDvki6r0QGg5ZeFultAAAOjDm3iLjBXUR\nkjiGKneQYFwexOWNeBKCyVsxfg11dgKlqEoA3qfXY/YqWvlR2P6IWasAEQ0FYCjwBakI3gim\nJ57gHvOEx4Ri2h72x0QOk0pAHSe79z/A7aS2fjTEM2mDwZhnMIGDwZgdkozq7961bLZHwYg6\n7C6v1SFl1w/Q1uvL4xVF+oNnTlQ2Ds84u/odP/nL6R/du3LTEqZxMBiM2cZrpvYTQH1VABTq\nwFWFms1A5rIUThSgXzXlmXIitVNwIfAE1cClI6qpUAWiGQCAGJCkUagMHkYEnYhM15QeSRYV\nzoWFeSTjr5rk1ajNComRFbeIXbW0pzEouOxGjAvdE5CAXAduqRb1itBaISy8iVpawR6QDokE\nC68H9VCFrFyJcnbHZDDmIUzgYDAYjChCJedkPPF4JVKC/RVAxyo6AtUNP3/ed5kJHAwGY5ah\nArUf9asbPkQLdZxEzZZZGlM0IlKnR2wUqS/1AIGXcakcMSG5aqjpDAJQEBoBAlwqjEbokehf\nhnG50zVQkoRQRIWq4R7AqEZuCaByas6v0JAbvk8vH6DNF8E5AIZELNyGCYsk98WEZbT5YHgY\nE5aGxpQGsuFbtOEQ9FRTjwN1iZi5EfRjdTdnMBhzFyZwMBiMmcbu9MplhOfmcrpyMFaHp6nL\nxhNMT9BOsnktIbi6IOHIJYlk43XFCYM/lDdIW741dVoH7B6dWjaZATAYswKl8OHZluOVnX0D\nrtQ4zXVrM/LT5rIt5ULG2w5Uqq+qtwNEBxDWEAoAgILgFq7QAMcNCl630CDnOA4NAP5MEURS\nQsUzfm8OzMqiLS1gDzbmlGtI3o5pHC7JRJIEYi+AC1ADaJrigiPCYdF2LNo+6o6YuY1am6G/\nLiBEMPta0CZL7M3JMGcb5GxjDlUMxoKCCRwMxlzC5RFe3F9z9FJHZ78j2aS+ZlXaDesy54q7\nJKX07RNNf/uwuqPPwXNYkB7zueuLijNjZ3tcw/RZXX/7sLqiod/jFbOTdXdtz81I0I58iMsj\nPP/elVcP1nkFEQDUSv7eHXm3bcrBSfxPPn9DUXlDb7/VHRi8bm1GSZbPpVUQJRzmRt3EYEQt\nTrfwo2dPltX4nkuX1fS8c7Lp0zvyPr1jxKp+RnQiRmx3CtQGwAQOAABB7KFSfqJesZ3jgqU9\nNCC3gYr1ABYAEeR6bsMqsXwfbT0HVARATCgkJbeAarpvpgogUiLCDMPJydIHaOd56K0CjxVU\ncZi8BjSJsz0sBoMRRTCBg8GYM1gdnm/+75GGDp/B2JVm85Vm89FLHT9/cM2c0Dge3XNp77GG\nwZ+9Ar1U3/fI40f/47Or1hQmzO7ABrnc1P+9p074/S9q2ywHylq/fcey7ctTRjjqd38//9G5\nVv9Lu9P7p70VDrcwmYVZikn95CNb/vJu1emqbovdnZGgvXVj9pbS4ZnlohS95IFxBuUEWuEw\nGLPOCx9U+9WNQUSR/uXdqhV5cVGlgTLGBEZOIkPFDI4jOhEHcx9EKi0DiVTSRFOOJH/4lRLI\nik/DsjvB3gOqWOAW2tc+YkIpJJTO9jAYDEaUwgQOBmPO8OL+Gr+64efMle73Tzdfuzp9VoY0\nduraB/zqhh9BpP/7enmUCBy/+/v5EHdPr0D/sOfCmsJ4rUp6vt7QYQ1UN/y8tL/mk5tzJlOr\nYtDIv3prSaStm5YkPf+u2u856ueOLYsmkznCYMwW+89JdA4CgI/OtYYIHIJIHS5vpI/kOBAc\n1Hwe3N1A5KBMRX1RQFEAY3LIksBBJHqLEi0QndQBCwEKzhrquASCBVAGskRQR3LopCD0ArUB\naoCLGakShPCgZZkLjHmNjDW4YUwEJnAwGHOG4xUdkvGj5Z3RL3Ccq+6WjLf22Dr6HImxs5y0\n3Nxlq2+XMGa3O72nq7oDsycCqWzqk4y7PEJNq2Vx1kSePNud3o/KWpu6bAaNfHmuqSA91Bke\nABQy7hcPrfnty2WX6vv8kbu25968IWsCV2QwZp1us5RlA0Bn/3C8qdP6p70VZ6u7PV4xRiu/\neX3W7VsXyfmJGAFQez1tfxOEoZObz9P+MyT1NuDmco+P6AGVqFxKnSF9NwiqVs3OeKIAaj0K\nzuqhF25wNyE6QRXqCU1Eh8zTQ8Xaodc6VC4HLn4GR8pgMBhzHiZwMGaHHovzn0cb6tutGiW/\nNMe4c2UamQtFFrOLxSbdPdRic0vGowqHS4i0ye6SqEOeYfqtrkib+gYibhIlWp34N03EC+NU\nVddvXyrrDbjirjXpX79tSXgJUmqc5vdfXF/R2NfYYdWqZcUZsUY9y/1mzFUMWnmvReKDFqvz\n5d7XtFq++b9HnG7f10i/1f3cu1UX6np/+dDacWctia4gdWMQVwft/ACTbxz/2BlSKPKRi6Gu\nchD6ATngTKhcMoXpGyKlHpFyBPk5kbTm7R5WN4bg3T2CIp6S4QfUhLrlrvah/ikAACBaqP0w\naq5eeJkvokjbRdpFwYUgRzRymArAHuYzGIwxwQQOxixw+GL7r18qcwwta9873fz6kfpfPLg2\nRrvQ6kjHR0Ksqk9qHZ5knPr0B0qhusXc0GmN0crz0wx69WT/NWnxGsm4jCfTMf7xYjJEbHcX\nFxNxUyQvDJ7D7ORxz0d7LM6f/uWMwx0k97xzoikhRiXp6IEIxZmxzKGAMQ/YsDjpn0dDS9gA\nYGOJ7xH3029V+tUNP2eudB8tb1+/eHytkamtJlTd8MWrUXQBYULhFMEnID/15Ydekfa5XQ6v\n780gJyRWoVBEeU8ut0QlI1BRbq32GlYJ1Jc8yHkHgtQNAAAEEKj7CipXTPsgowjRK1ZQ8P1Z\nKDgpbaW0jyeL2bKFwWCMBfZNwZhp+q3uQHVjkOoWy2OvX/zBPQvqFj5udqxMvdwk0R90x8pI\npbwTpLnL9vu/n79Y3zv4Uq3k799VcPP6rMmcc21RQpxBGZ6IfvWKVJV89r+Iko3qgvSY8D+v\nXi1fmRcX6SijTpGfZqhqNofEb1iXOQGPgA/PtoaoG4PsPdbAekkw5jefuSa/rKansTPIY2jX\nmvSV+fEAQCk9F2xB6ufMle7xChzgsUjHqQieAVAwgSN6ESntcDgEOqwCuEWx0+FIVKvkJHo1\nDkqlsyxRdMsxmZIMEZwIMnQdlj5e8JUiUlsD7ToMzg4ABFUSid8E6tRpGvMsItJOv7rhh4JD\noG0cpgcHRae31yPaKRUIyhScXs5JP3VgMBgLitlfVzAWGkcutTukShIOX2x3uL3RsNaNNhwu\n78sHas/X9lhtnqRYVXvfsMU6IXj39twVkVfgE8DpFr775PHO/uGr2J3ex167pJLz16yauJKi\nkHE/+eyqn/3fmdaeYWvMtUUJX7ypeOwnaey0PvvO5Yt1vW6Pr43r2qIpe0L47TuWfufJ44F5\n8goZ9y+fKlUpJN6Tbb32/3nt4snKrpA4R/DG9Zmfv75oAgNo7gp1kB2k1+KyO71qJftoMOYt\nBo38sa9v/MfHdccqOvoGXKlxmhuuyvSnb3gEOtiGOZwRat8iMkKOBhcxXYsRDVg93kB1YxAK\nYHF74pTRq0whp5MuWUQZEBUC4UAOYckbAbsBANC+s7TtveGgrVG0/RVTb0DDOO6hcwIRJB7k\nAAClfRAgcIjUa/W0iNQ3nxSoy+7t8oh2jSxI8aRtlbTsTdrTCLwM4xeRlbeCYZySKIPBmGuw\nGTNjeqEUjpa3lzf0u73ComT9tuWpgSvnQLwC7TG70uLZezKI9l77I3881tUfJGoUpcdq1Xxq\nnGbHitTcVMPUXvHDsy2S/6MX91dvWpq072RzXZtFpeAXZ8VuLEkeV/lzbqrhyUe2HDjfVtdm\nUcr5JdnGZbmmsR9+rrrn+0+f8K9zyhv6fvTsyfuuyb9nirIbMhN1z3x76z8O1l6q7/MKNCdF\nd/vmnPgYifIZq8PzyONHQ7JROA7vv7ZgS2nKhA1TJZUUACAE5cxInDHfUci4u6/Ovfvq3PBN\ncp4kxqo6+iS+l9ITtOO9EGqyaRdKLCcVicCP+2yMmcQlSutZLmH8OtdMIs8EPAvheRyKRUFN\nUjgjiKH5gAAAxAiii3YcCN9C299HfT7g/Jo40Ui2XEFxp9Arhu3pFm1y0SYjvpJYeuk98cTL\nQ9uANpwRms6Ta76GyRN5CMFgMOYK8+s7kRFlmG3u/3jutL/SAQBe+KB6/WLprmaIoFNPuvPf\nvOOx1y91BcsNokgvN/c99e0tKSZpV4tJUtMqnb/d3GW7/9cf+RMcXj1YV5wZ+5P7V43LnkPG\nkx0rUgEmklX7hz0Xwp/i/t/7V3asTJuqJixqJX/vzvxRd9t7rDG81kYQ6KX6vju2Lprw1Zfn\nxr16sC48Xppj4rm5YKTHYEwb25anvvhhqE0jQdwaocPRSMhi0LiO9h4NCqKMJFw9iQEyJoGz\ni1qqwTMAilg0FIEssswUIclhIpbOMwlRom4ztR4CMeDGIU9FTVBZLsrzqbcpdHmPPMrzqa0Z\nRKk6F8EJjjZQR3sbtXGBqKRUMp8xKMHKI4Y2SgcABPCIdp/AYTeLp18N3UP0ioef5z75c9YW\nmsGYx0RvySJjHvCfr1wIVDcAoK3X/vH5dsmGKcWZsQYNMxkNwuH2nrocWgQBAF6BHrkk3TJ2\n3Jdwea80mwMfjY6QlBHS5qC8oe8Pr16ckmGMSmuPvbnLFh4XRHq6SuJPNK1UNkp3hy1vkI6P\nkTWFCasLQ9sBKmTc5yZU8MJgzCesdokeUiKlp6uk+0+PDJrWY8ptoEoFIgdei7pCknkfKMev\nlTAmDW3bL1Y9Tdv20+5TtOU9sfKPtPd8pJ1lEcxEo9mAw4c8BWNvQc1qUOaCajHqr0b91aGZ\nF0SLqk1AArIySQyqNgHRgBixmReVcsyd0xCQrrolGHR/pFQ6bccfp23lIEglgwx0QX/bpIbI\nYDCiG5bBwZgu+qyuo+USi/Aus2PbspT954JMxdVK/iu3lMzU0OYMFptHiNBtVLKl4vhObnc/\n9VblvpPNlFIASIvXfPnmxSvz44syYt84ItHOQJJDF9utDs8EDDXHi80h3SIXACxSKx9KqcMl\nTJNvRaR/SqT4GEGEH39m1Ssf1752qL7P6uI5sjzP9PnrizMTWdo8YwHRbXa+dri+rt2iVvCL\ns4w3rMvgOXLmirSOefJy1/XrMiZwFdRkoyZ7ciNlTBbae552BqfSiG7a/CaqEkAlYZSglckG\nPF4aZsOhl8+F9E+Ug6polLQBzoiaq0EcANEGRANEN5hogPKYSHcXlM+3RlqIMRykCbQFgALQ\nwb8AwcQQgYOgTKQSd3+CQ28Gl0SKxyDUZWf5GwzGPIYJHIzpoq3HHj4LGaQgPWbnqrSX9tfU\ntw9olPySHNN91+THRe7TuWAxaOQ8h15B4s844T+XSxBtHsHjFf/9qRO1AdUozV22Hz5z8ucP\nrtlSmvzSRzX17aEe5pKIIm3vtU+5D0g4CbEqRJR8R6WY1IEvW3tsT+ytPF3V5fIIsVrFjesz\nb9+SAwCNHVa3V8xI0E6+EmpRiv54RWd4PDd1sv7tMp7ctT33ru25A3aPWslzUrlODMY85tDF\n9l+/eM7fEfZAWds/jzb8v8+ttTmly/JtzojSJyP6ob1npaKU9pzDtF3hW3jEBKWix+X2ir5y\nRYIYK5cruTntUiSCuw0EMxAlyBKBaIDogQTfTVTJoIgDV1i+kiqJWitpRz0INpQbwbActRIu\nNnMOgmmIRpF2A7gA5ASNCKGd1+WczuntDT9Wzg3tqY3kv46om0prdgaDEW0wgYMxXWgiPzxX\nK/hV+fGr8kOz8ceL0y0cutje3GU1aOTLc+OykkLvf3MdpZxbU5h45FJ7SJznyIaScduAUwqt\nVmevw0MBzlZ21YZ5bQgiffady49+dcOvPrf28TcuHSjz5XDGx6hWFcS/fbxR8rSa6U/fAACD\nRr6uKCE8J8ioUwSWdTR0DHz9sSP2obVQn9X1l3er3j/TYrG5rQ4PDHU5eWBXoVI+8QnxDesy\nXz9cH77i+tQkDDhCYH40jAWI2eb+zUtlfnVjkKZO6x/2XEwxqc02CQ+C1LhpsSJizBCuCGV9\nLumuwACg4LhktcolCF5R5JAoOELGZXYdbXh76MAhEIa8RZEDVQmqS8P2Q5J2s9j0KrgD/mLy\nWKAO2nt88BX1WsHeCIZSTNgx/eOedhDUHI6UnKXkYgTR5REDa1dRxZs49BU7Y0oRqGPAHtqT\nxRdnzAlkbKHKmAjsfcOYLjIStPExqq6wfhwcweUBbU0FkXb1O2J1CsU4+0Scre7+zUtlfq9H\nQvDm9VkP31iEc3quE8aXby6ua7O09Q5nWnIEv3Rz8QRsNdttrp6hQo/6FimrdoCq5n6PVzTq\nFD+4Z8XDNzqbOm16jSwjQdvZ79h3skkMK8FIT9AmG9WSp5pyvvHJJd3POK80D488Vqv44adX\nBLYWfubty/Yw3aG1e3gCJIj0tUP1nX2OH9+3asIjiTMof/nQ2t/9vayhw2eEZtDIH76xeOWk\nNTsGYyFzrLxDson4icrOh28srmgMW6gg7lo9r+wVFxxEDiBVR8CN1PMVAZQcB3M7awMAAKiL\nWt4P8tegAtjLgChAWRi6s8JEFj1A+y+Csx0AQZUCjkZqrQg9pbkMdYWgmnhP97kDamRJHtHu\nFe0i9XIok3P64foUAODlZOsXxP2Pg2P4cQ7GpJCNn535sTIYjJmECRyM6QIRv3xz8U+fPxNi\nTPCpbYsGF+cDds+f911+52STxysi4rJFpi/dXJyZOKYsjF6L68fPnQ6cCosi3XOoLs6gHKxH\nmDfEx6ieeGTznkN156p7bE5vVqLuts3Z2QG5Kk2d1kv1fU6PkJ2kW5pjiiTviBR6AmwsBKmy\nFwCgFLyiKAMCACa90qT3FcKkmDSf2rrob8FdDHgOvzqDzimxWsWjX9nw4bnWS/W9LreQk6Lf\nvSYjJFHozJUxOQ4eudRR1WzOT5t4ZU1hRszj39hc3tDb1mM3GZRFGbEjpCwxGIyx0BXWnGgQ\nUaSlOaZbN2a/fqTeL7MqZNznbygqzGBPYud98cdvAAAgAElEQVQwqMumPVJVKtqFYY/irJV0\nD6WOCgwXOAAAOYwtBfDld4g9+yXPSq3VuCAEDgAAGVHLSMSnLJiYy932M3r5AO1pBF6OCYsw\ndz2QuS+NMRiMEWEzcsY0sn5x0u+/tP7ptyorGvsEkabHaz+9I29LaTIAuL3it/94tG7I6IFS\nera6+2v/c+QPX9kwFkvF9840Sz7oe+NI/TwTOABAIePu3JZ757bQwlqvIP7v6+VvnWj0z/iL\nM2O/c9cyyZQKtyCKAQYWyfHSed0pJnVgQkQg9+8qyE3V/+3Dmvr2AbmMlGQZH7yuMHtmy4II\nwR0rUneskO4yK4rU7ZW2VQ+nvKFvMgIHAPAcLs0xLc0xTeYkDAbDzwiNtGJ08i/eVLxjReqR\n8o6ufkdavGZraUrSTKWPMaYJTNxIzVXgDe6QpUpC07JZGtGMQoXQpCQfwgBQb2iPlbCDpRvH\nAoAQ0VxzISJX4ZJd8yqzl8FgjAYTOBjTS1FGzG8fXieKVBCpjB9u5Pbuqaa6MBtLh8v7l3cv\n/+jelaOetqlTskc6dPQ5XB5hvNUuc5Sn3qrceyyo3Ul5Q9+/PXvy8W9s5rnQu3lIZseKooQP\njjVawmrab98ykovEpiXJm5YkCyKNTvNLQjDZqGntkegmG04kB1wGgzFbrClMkLRVLs6MjdUq\nACAvzZA3OV0yCLed9jSD6EVjGqgm6xDMmAgyHcl/kLZ9SC1XQHABr0ZjKSZuAFwQN3GASN1t\nEXC0xrfIAaeW1jJk7M3MYDAWNEzgYMwEhCAJXhKX1Uh4X48QDyGShMER7B9wJcSq55cRhwQu\nj/DmMQnXz4YO6+mqrrVFCSFxBUdkBD1DuR4qJf/QJ0tefKuqtcsnFSlk3D07csfScDE61Y1B\nrl+X8eSboTXJkuQFd36hlF5uMjd1WY06RUF6zAw0vmXMH6hAO8/CQCMIHlAnYNJqkLHOvhMh\nMVZ1/67CkI+wViX72m1TXQpHRXp2r3juLfC6AQAIweLtZM0ngY+YQsKYLmRazLgJAUBwjWy9\nMf9AWSJ1XpbYwMdF1j4CDtcV0f7T4WHUSpW3MCShIu1vBVsPqGMxJpVVrzAY8wMmcDBmB0+E\nUgKXZ0wlBsty4/55tCE8Loj03l/tVyv52zfn3L51kZwffYowR2nrsUf6W9W1W8IFDgBI0iqa\nLMMl7inx2m/eu7y9w+q0uWM0ipLsWL/jxtzlE5uy23vtbx4fLtuR88TtFUN2K8k2Ls4y+l/W\ntQ/87uWyqiH7Up1a9tB1RbvXMPNCxhhwW8SK/wP7UNvg3gradhzzPoGxebM6rLnK7VtyijJi\nXtxfU9c+oFZwJdnGe3fkG/VTvO4VT7xCy94JeC3Si++LdjPZ8cWpvRBjHCwwdQMAQJEJjjjw\nhlhHEdQsH8vRaFpPXR3gaA4IEYzbDIqF3QPVbaaWWvDYQGHEmFwgEVVL2ttAT75A+1t8r3Xx\nZOWdmFgwQ+NkMBjTBhM4GLNDRoL2yKXQlp8AMMZWrxtLEpfmmM7XSneSszu9z71bdaXF8uP7\nRq92maPwXETtRhZhU6xSRhBbrS6PIAIAIiToFEsS9VGckDFuCMGv3lqya3X66StdvRZXeoJ2\n05Lkvx+o2XOozp/3vrEk6eufWOLP8Rmwe77zxLF+63C1zoDd85+vnNeq+E1Lkmf+V2DMLWjt\n3mF1YxDBSatfxeVfA37crY4YAFCSbfxZtnH0/SaM204vvB8eprUnof8WiFmgn3raVkY7K8Fp\nAU0cZqxFfcpsj2ghgGjYQW1nwVkFQAEA+FjUrAFZ4piOJnKSdge1VIC9DgQHyGPRUAryBa1u\n0LbDtO1jEH0ebVSuw6wbUS9Ve2vrEfc/Ct4AY+OBLvHjx8nOb2PMQrFoZTDmK0zgYMwOu9dm\nvHqwLvzR+s0bssZyOCL+7IHVf/uw+vUj9XanF9A3NwjkyKX287U989UDMjVObdQrei0SBuxL\nIv/KBgVvUPBuQRQpKDgyXwt5Qgr1P3d90a0bsyub+t0ecVGKLqRTzzsnmwLVDT8v7q9hAgdj\nFLx22ndFKu6gfVUYXzrjA2KMDu1p8q9/Qjd11uICFDhEr3jyWdo5XBlE6w+Twt2Ye/UsDmqh\ngHLUrgXNKhAsQJRAxquKIuqLQV88LWOba9Ce87Tlw6CQe4BWv4yLvwiK0HZLYtV+8DoAgqdB\nopdWvI9XfXZ6B8pgMKaZeZvAz4hyko3qf//MqsDEYxlPPnttQaQGGeEo5dz9uwpe+8m1f/zG\n5nB1Y5AxOnrMRRDxwd0SdbZbSpNHbQ4i54iSn7fqhiRxBuXGkqTty1PC+xDXtFokD6lttTAj\nUsYouCwS2qpvk3lmh8IYMyN8rhfkR1688kGgugEAQEWx4k3aJ1EHypgWkAM+dvzqBiMI2nlC\nIip6afcZiXhvY6i6MXiSXva2ZzDmPCyDgzFrrC6Mf/Zfth4t72zptpr0yhV5cRPr+adRRXwb\nj9HRY46yc2WaUs7/6Z/lnf0OAFDIuE9uyblre2g3WcbIRPJMRUTJ2Q+DMYws8oJExjqYRilo\nSgfCgSh1d4jPmunRRAG05ZR0vPEI9J+j1iagAqoSMHkTaFnqPiOKcXRHiHdJRSPc39ltn8GY\n+zCBgzGbqBT89uWTLfQ16ZUaJW9zSqQcZybO814Gm5YkbVqS1NHncLqF1DhNeHdYxqgUZsS8\nd7pZMr6gklwYE0FuAE0y2NoAaNC8GDmMYVJjtKLQYPE2ejHUhgOzVqBxQS7gHf2SYdpZBh6N\n72e3hZqrMWM3JqyawZExGOOB8CB6pOPhGDOguyY8jMasKR4VYzJE6JnIYIwMK1FhzHl4Dm9Y\nlxkejzMoNy5JGu/ZLHb3X9+/8tPnz/y/F8/982iDVwh1CYlCEmNVmYlapm5MjJ0r01JMoQ/b\nOYKf2Zk/K+NhzC1Izg3AyUOe+mHG1eEl34zogaz7FJbuHl72IGLBJrLtoVkd1Owh10jHw7Lb\naPN74LFO+3gYjAmBOompIACALis8RvK3SaTgcTIs2jnFw2IwGDMOy+BgzAfuu7bAYne/faLJ\nH0lP0H7/7uUq+fje4eeqe376f6cH7L4nAB+caXntcP0vH1qTEMMqY+ctSjn3my+se+z1S/62\nPslG9ZdvWbwsd37a0zKmGG0qKf0ybdpPBxpBcKMmEVI3oT5rtofFGBHCkbW3w5JraHcDiF6M\nywTtwv28Y2IJbTgSHKMAiEpZ6K6il1pq0bRU+kRUpD01YO0EuRaN2aDUT8doGYxIYMoWaq4F\nccg1fDCvTpWAJim/Z42RbPsqPfk32jc0ddQnkpV3ooH1D2Iw5jxM4GDMBFaHp8vsTIxRqZXT\n8pbjOfzmJ5fevCHrfE2v1enJStKtK0ocb0aDw+39xQtn/OrGIE2d1t///fyvPrd2SsfLiC7i\nY1Q/vm9Vt9nZ3GWL1cnT4rWRjDkYDAkUBsy9hb1jooFus3PQNnhRij7OoBxlb7UBMyKs1RcS\npGCX0F0FtkD/AgSVHBRhAgcAeO2SJ6GWFnr2b9TS6nvNyUneTsxjfVgYM4gqgRR9VmzcBwMN\nAACEYNwyTN0uXaICgLEZuPNfqbkVbD2gjsWYVECW2M5gzAeYwMGYXho7rY+9dulstW/mtH5x\n0hdvKk6MnZaEiJxkfU7yxB8ZnbrcLdku9Gx1d4/FadKPNldmzHHiDMrRV0QMBiMqcXmEJ/ZW\nvHm8URQpAHAEb1yf+dB1RXKerVhGQ6HlNj8iVn8AHRXUZUZNPKYup90fSu8sl+rS5XGIx54A\n18BwRHCLlW8SuRozr5qWMTMYkqgSScFnQHCDZwAUsaMLFogYkwoxY+3fx2Aw5gRM4GBMHFGk\n759puVTfa3cJOcm669dl6NXywB3aeu3feOyI1TGcE3HkUvvlpv4/fnOTQSMPO98s09XvkIxT\nCh19jnEJHC6PcLqqq6XbHqOVL1tkimcVLgzGrCKKtKXb1m1xJhvVE+vWxIhy/vOVCx+ebfG/\nFET62qF6h0t45HaWozEGeAUpvA4Kr/MHqKvW9xg8EJkGDYvCj6bNp4LUjSHEmv0cEzgYMw8n\nB27hFp0xGAwmcDAmSO+A6wdPnxhMBgaAA2Xwyse1P7hnxYq8OP8+L+2vCVQ3BumxOPccqvvs\ntQUzN9YhKKUYuTGGTi2VjgsAACHCzciU1fT89uWyjj6fXKKQcffsyLtzm8SkkMFgzAAXanv/\nsOdCQ4fPHLF0kelrt5akJ8zzFksLivZe+/5zLeHxd0813XdNPsvMmgAk6ybxyl/B2Tsc4pSY\nfSsQibshtbRJn8XWDYIbuKh7nsGYe1APHTgHzkYQncDHoHYJKDNme0wMBiNKYQIHY4L84dUL\nfnVjkAG755cvnP3zd7Zphow2Ltb1Sh0KFyLEpwmPV/zHwbp3TjS199mNOsW6osT7rs0PTyFZ\nmRcv44nHG9o2JSNBmxYfwWQ+jG6z89+ePeVwD/esdXmEZ96uNOkVO1cuyAaEDMaMY3V4mrts\nsTpFQoyqusX83aeOB36uy2p6vv3HY088snmceWQ2ADsAD6Bjt85o40qLhVKJOKVwpcXMBI6R\n8PRQeyUIFiAaVGaCcqgPhSKGLH6Ydp0GazOIXlAnYsJq4CNkP5FIrRxxYZsaUNpykfbUARUx\nJg3Tl0f+QzFGRLDTrj3gNftees3U2QDaUozZMKvDYjAYUQqbpTEmwoDdc6yiMzxutrmPV3Ru\nX+7zoI7UY9UbJiJMH4JIv/vU8Qu1Pkml2+zce6zhWEXHo1/dEFJ1YtQrHrqu8PE3ygODcp58\n4xNLxn65t080BaobfvYcqmMCB4Mx3fRaXH/cW/7ROZ/TYWaiTq+WhauWfVbXG0ca7t2ZN7az\nuiitAvDLshxiFgD7OEcRkTPzgJm/jsTAaTpwGsAnDlF7BShzMPZqnyqBHCasgYQ1o54Gjdm0\n/rBEPDYjkr/j/Mc5IB5+knbXDr6iAFD+Drfxc6BLnNVhzUmo+eiwuuHHWgbqRSBPmvrriQKT\nohiMOc1CvfEwJkdnv2PQyC2c9t5hi/WsJF1rj4TjevYkrEDHywdnWvzqhp9us/P5966EKxe3\nbszOTTG88GF1dYtZKeeKM2Pvu7YgxTSOiv2GDok6ZACobx+gdKRZOIPBmCQuj/DtPx1t7rL5\nIw0dA5E+dJWNfWM7K6X0PEDg95hAaQ06rdSmREMSyFh2wOxTkGZAhPAkDkIwPwlBdAJh/6Yw\n3G104FRo0FkLtnjgMoCTgVw3xjNhcikaPqLm5uAowQBTj4WGePwvfnXDh6VdPPwUufZ7M5jV\nQkEcAOoAVAHRzWG5z1ErGaaOWpxCgUPw0KoPxIbjYO8FhRaTl5DF14NirJ8CBoMRPTCBgzER\nRrKr0AxvunVj9pFLHSE78By5eUPWNA0snJOXJTJNAODk5S7J+JIc4y9zRn9gFQlZBMd+Gccx\ndYPBmFbeO90cqG4MIlm5AABCBIk2jO5gdWPotGK9+M9XgSAWbiMrbmEyx6zgdAuIoJBx8TGq\na1env3OiKWSH64vNsZYXqAVAnoDGraBMmZVxRifUXiUd7zlMG14BAFDGYPYujC8Z/VyEI+u+\nIFbspU0nfB85bQIpuQ3jxpgkNe+w9dL2ivAwNbfR7hqMn5E/i9hLPedBHHroQvQoWwokdiYu\nPbVQL9BQNzcfgnTT4okgCuLB/6G99b6XLiutPyp0lHPbHgGlVOcgBoMRxTCBgzEREmJU2Um6\nuvbQbAWew9UFCf6XpYtM/3rnssffuDRg992cjHrF129bkp00rIjXtlleOVBb1z6gUcpKsmPv\n2LJIrZzKt6XNIVEwAgDh7qdTwpJs4wdnJLzuli4yTsflGAyGn/L6MSZlAADkp41pzkqpdE4W\nqFWgVoLdQcs/EM0d5Jqvj/3SjLFzsa73uXerqlvMiFiYHnP/roK8NAMAHLzQ9ty+qqYuKyJm\nJeoe2F3w1VtKdCrZnkP1g6WRMg4+Udp375oe34ncnbTjFUy8DZSstmgIwSod54Zkemc/rXgR\nvDdj8urRzybXkNJPweKbqbUT5VpQx87hfIFJQ63ST1AAACydMJUCBwVPKwhmQDnwCcANpcfS\nAeo6BuAd/i+IFuo6hsrNgGP1FIsWkAeiANElsYmbMq9o2nhiWN3w4zCLFfvI8jum6iqMcSNj\ntUKMicAEDsb4sDo8GiWPiF+7bcl3nzzu8giBWz+9Iz8xNqgl6o4VqWsK489V93T2O5KM6pV5\ncSrF8Ltu77GGx1675H+Uer62Z9/J5t8+vC41bspuwMkRCkxS46alVeTOlWmvH64PkX4UMu6+\na2ahawyDsaDwjjUpAzRK/qb1WWPbN/IibSg5hLZcpO1VmJQ/xqszxsibxxr/+9UL/penqrrO\nVHf/4J7lLd22Z96+PBiklNa2WX74zMlvfGLJTeuzegZclQ19QL2FcT27i/t5EvCWoCLtO4zJ\nn5rh3yJ6iVS2E2yeRevfw6QVgGNbZvBKjGG9LQB4RcRNssibAhCozSN2itQBgATVcpJEMOxA\noY9aD4PQP/QaQVmA6pUASD01AELY15eXemtRNg5bsWhBnQfWi2FRRHXuVF2Bdkhk3IwQZzAY\n0QwTOBhjwukWXtxf/eaxRrPNrZBxqwviP3dD0Z++tfnZdy5fqu91uLw5Kfq7tueuyo8PP1av\nlm9emhwe7+p3PP5GeUiieI/F+eiei7/63NqpGvk1q9L2HmukYXnqu1ZPyyRMxpNff2HdM29X\n7jvVPGhTUpQR8+VbSnJTZ852hMFYmOQk6/32ooEkmTQej9BjcQ6+/P/snXdgHNXV9s+d7VWr\nVe+9WtW924DBGDAYbEoghJIQAjEEEkjCm5CQN5BCypcXSELvGEwxxQaMO8ZNLmqW1XtvK23v\nM/f7Y+XVandWVtnVrqT7+0t7ZnbmStqdmfvcc56TFCX7+c0FE+ysgZCMvchFpweTefRlfyMQ\ngcOn6E22l/e4Ty0YBj+367zRQnvu/9/d1YDBqbl3D8mONUl+d3XvkiSXqiVLD2B6onP1uQ4S\nJmFTo2ccG0xjXtuMYOgHKctNnOANFJoAAilYPHJkKM5E6lNsjMrKjF7KaKw10TohJ4mDXPwg\nsB3rDgPjWqOBwVyLER+JCgCrgRXGSzy4QfJl2NoPVteKY4RCVgAv3GfnsJm9xE3scQKBEMQQ\ngYMAAHC8qvdQWVfPkDFSIV5bEH1ZUZyrYQTN4F+/UlLdNpL+bbHRx6p6y5tUz21f9Zs7iqd8\n0lM1/Z7dDQCgrFGlNVrl4kl1cPRKVoJi+5YFL+2utrqc69rliZtX+GuVKUTCf3RbwfYteV2D\nhjC5cBy/EgKB4EOuXpLw0bdNzoI4Jz+8OnNZTlRtu3pAY4oLl2QlKDjUxJPnwwBkAO6FKri8\ncsxrhmXKTZgO51uGWDtSaQzs1YUWq/u/wEpT/zwS+fb3W3kc1zwOInBcRJQGpkYwt40JWm0w\npHXfk/FLRedchuJQRTcxJW+7hxdsAtEliuMw2K1Mr2fYQneJudmjAWv7WHXjIuY6EI2TozHR\nNLfgghKgyK1gqMHmdqBNwAtF0jxfqhsAIAkHqPMMIynLuh2BQAhyiMAx38EY//n9cueyZ2OX\n9sSF3kNl3U/dtZjLGZkDHCrrcqobTvQm21v76n5zx8Ipn3pIx1ZRCYAxHtZZfCVwAMDmFUmL\nsyIOlXZ1DRrCQ4TLc6Nyk/zus8XjUsnRxHybQJg5FFL+n3649O8fVjqbGYmF3HuuzlpXGAsA\nRelhUzoqQigf40aAi4uHZjM+V4qbxrr6K0lavo/xiU3SkIFb3SssjLu4BsuVA+WzO8vsByHl\nRjDWYmMt2LUAPKzqgmEdMGMXHhAHxGSON2lQ8lJKpMCVn+PhDgAMsigq71qUcOk1IRobAFjW\nfjDYGGym0EjqGaa95GJgCzAmQCGesiwAAKWY8G8QbCCQ5CJJrr+Onrwct57wdKVGKSv9dEYC\ngeA/iMAx3zlY1u2Z1H26tn/PybYtq5MdL8saBlnfe66ePT5BlDL2SlSEkFLm45YEMUrxHRvm\nq507gTBvyEpQvPjomvPNQx0D+lCZID9FGSKZ/oSWh1AOQCqAkak7jkv2AD0mWQCFxqH4CXSa\nIEyGGKVvbJLUptF8DSSfesrhHAWBOAeJcwAAMIO7/wuMxn2P6GLgjrHWAlsf0GpAPOBGAIfo\n+F5BUZnoyseBoQEzwJlwLif2mg6GwSWnaZxEJMRB3DRMd48VSjAAF3HTJjqMeQYKTaSKbmYq\nPwX6orSKKJS6BiUvD+i4CATCVCACx3znaGUPa/xIRbdT4DB5pP46MLPlD0+cFblRbpUjDhZl\nhJOyDgKBMDU4FCpKD5tqvsY4CAAEVOY1jM6MLxxw1qSgmGxq9d1AkaoHH5OTFBofIfHs+5uT\nqLAzuKHTfR7ujXAJDQCAKJAvAiJwjAOiqNw7mLqPQDNatIIii1DataP70FqsOw52Z4sQBKIc\nJFk0nxumXBqKAzCJ6wNCXp9/KNoAFAJKDIAQN5K92oSjACQAJECCZdhaCfjiNwjJEb9g9rVQ\nmUFQyipOVA7uLMX6ARBIQBFFhcQDYyNpXwTCrIMIHPOdIS27r5LKJR7vpadJfMS0GnSFhwi3\nb8l77tPzdnr0Nh2hED10E1kLJRAIQQmiqMXbIPsyPNAMVhMoE1BESqDHNDfhUOi331/4+zfP\n9g2PmvzFR0h+fXsxAvjfd841do1aReQmhqq05j61ux1gTCgvJ3sR4olAGA/cCTUGntcIFVTh\nj7C6BfTdwOEjWcIYb1FMY+0hoF0dOjCYqjHiInHRjI91zsJBUgQ8DO4lWhzGDLaTGACoECQo\nAl4M8GLB5paBi5D4YuEwFY6E64HRAjYCEgMlB6CAMD5iJUpbA+0HcN850DIMYOCKUPxlKGYp\nUfEIhFkEETjmO6Fe6kTC5KNFIhuXJHzyXYuddk+12Lwiyfmz3mSr79RoDdbEKGlqzEQ7hly9\nNCEnKfTjo80tPVqJkJeXoty2LkXEJx9LAoEQxEjDkNSXGSKdA4ZPj7W09unEAm5BatgNq5L5\nXDIVgdQY+auPrfv6dEdjl4ZDoawExVWL47kcCgBeeGj18Qu9DZ0ahFBOomJZTlRN+/CTb5xx\ntZiVini/un0JJ9TvjktzC4QUqaBIZdli7RyrblzEVAviAjJ59h1IyEk0022uBSkUtvFtfSMv\nGA02HUOi9Ui6FpurwFwzUtXCUSDxYuBFuxyKAkoBMHt9NyYPrQFbP2AbcEKBP5XWP7jhYzzc\ncPEVArsZt34N2I5iV/lwmAQCwa+QmeR8Z21BTElNP2vc+XN8hOSJ24v+9cl557MjRaEtq5Kv\nWz4icHxxou3Nb+qcnnCFaWGPbiuIDZtQBXVSlPQXNxdM63cgEAiEIMHQh5v2Y20nYAbJYlHq\nBpDHj/+OfWc7//XJeaeCXFLT/+Wp9r//ZPkEG9nObQQ8zpZVyZ5xikJr8mPW5I/ep3KTQl9/\nfP0nR1vqO9UYQ1ZCyNa1qRN3YNEZbR9921TTrrbY6Iy4kJvXpUb7yARkzoBpd6/xixusQBuI\nGYcPoZBYxM2yM0MMNgHWULZ+LqMb2wCFxrZ6JFiEREUgKgRaBxQf0Dy/YjBYVwKmOgAMgAEQ\n8CKQfC1wJrrkBgCg73RRN0bBnd+hmOWkAROBMFsgAsd854riuFPV/d+dH+PEsSgz4vqVSa6R\nNfkxBalhRyt7OgcMYXLBkuzIlIstQr452/HCZ1WuO1c0qf7n1ZKXfr5WwCM3g6ACM7ifATVg\nK0JCCqIQmsyNn0AgjAserMGV7wIekSqwqg4PNaCcrSjGa7epQY35uV3n3fLjulWGFz6reuqu\nxf4d7pwjRMK/d1PWFN7Y1K399SslGoPV8bK2Xf3NmY4nbi9elXdxMRxjsJmAP68lDwSU1xaj\niKRv+BgEFI8KBwBsbgOGLXGGGRrdd1Jz+DkK1p8DU+3FVwgAwDaA1QeQcsvEP59Y18W+gTaD\ncQAk0exbCQRCkEEEjvkORaEn71z4bUXPwbKuHpUxMlS4riD2ykVxCLlXG4ZI+K41KU7eP9jk\nGexWGY9UdG9cnOCXQROmAm1navHFvnEYGxhQURDDQSz/UwJh/mCzM6dr+zsGDCESflF62NSb\nd2Aa137qVDcuBhlcvxtFLACugGEwRblfV09c6PM0WgaAkpp+k8UuEpB79Ezwj48qnOqGA6ud\n+efHlcXp4WJaw5z/HPdVA20DvhilrKSyrgIue2nnHIcXyR7nSIEi1pX+w4us5NHQdF6DaRd1\nwwVaA9ZOEEy8jfc4f1XyBw8EPHITJEwF8rkhAACsK4xZVziVYkWTxd6tcne5d9DQqSECR/BA\n4y6nuuGEwT0UhJI8DsK85ULr8N92lnerjI6XXA51y/rUu67K8hB4J4C2CyzuXzEAsFmtn+wt\n/abK3DtsUsoEK3Kj7r46Uy4eKZ0Y1LDbPNMMHtJZ4ojA4X+6Bg2ufqVOdEZbdXV9cdc7YBv5\neIDViOsOMAON1NqH52PfHF408OPA6rHEbQsPxGjmDVQI0Cr2eFCBMa4/jhtOYG0/kihRyiKU\nt2Hmvia0DrCXvn72oYkLHEgSzS5jUHwQkc85gTBrIA9P8xo7zXQNGhVS/sQLld3wTPRwQk1l\nikDwDQNqU1XrsFpvSYyUFqWHcyiEMdsTEgADgxwYI3AwDGYwcDnk30eY46j11iffOOM0DwIA\nO83sONgYKhPcsDJ5skfDzmmwC3aG+vWZvAvqkWRylda851RbSW3/8w+tUsoEABAqY7/2IoSm\nfFkmTIohrcXbpqjew+Dxb8VDrbjjLEpa5udxBSNItg7rz4G5Fhz3d5sNuvqwpgaie9CCW0ib\nCX+AuKnY1gZAOyMYAAFC/IwAjsodzDDfPI/bK0Ze6YdwXyNqOk1t/uUMpTuN444xKeMMeRLI\nEkHX7n6MmOVAeW3fSyAQgg0icMxT9EydLOQAACAASURBVCbbW9/Uf1nS5mjRmhYr/+mWBXnJ\nyskeR8jnJEXJ2vpY1i2zE/3uXY8xHC7v2ne2s3fIGKEQrVwQdf3KZI5HEvgMYKcxAHbY+wcW\njGHHwYb3DzU6896To2W/vLUwKca959zI/i696C60Dr++t7auXc1gnBwt+/6GzJULomZi0ARC\nIDhQ2umqbjj59LvWKQgcSBjqufR3oDvygto9Q2pAbXrvQMNDN+YBwPLcqFe+rHFtle2gOD1M\nKiLP0zOBt1ZiABBh7WCN4/66+SlwAOJCrwp31IGADzQDNrsjbx/3lkNkPorIDfT45iKUFAlX\nYGs5MHpHACEhEhQCNekHNv+BG0461Y3R4EALrtiLFt0wEyPgyICSAMOWUMyblHEGorJuxS1f\nYlX1SIDiopgVKGH9tIdIIBBmDiJwzEdoBj/x6um6DrUz0tSt/eVLp/563/L81EnfMn9wVcYf\n3yl1C6ZEy9YW+NeNCWP8x3dKj1X1Ol52q4wVTaoj5d1//fFyIX/mkofLG1Wv761t7NJijJOi\nZD+4yseigN5kO1c/2DNkjAoVFaeHK6RjFnVNVrtbV93dJ9ve2lfvGmnt1f3Pa6ff/U0YQmOK\nzB0gGJlEfVvR/cx7Zc54Y5f2qbfO3rsp+7bL0nz2yxAIY+kcMLT360Mk/NRY2cz3h27r1bPG\nu1UGq52ZdKNWaRTIYkHX7Ro7O8jeoPFM3YDjhxil+EfX5Ly4u9p1q1ImcMgfhBkgPkKSGiNv\n7nGvUpGKeDzkLenda9LHnAcPVAPGYPb4C/RXARE4/AQnHIkuB3oIsBGQCDhKgOCqkMKtZV7i\npTMkcAAg6SKsPeoeFSQDL2JyB+JJUOYtyDSADX3A4SFJLPBJhyACYZZBBI75yLHzPa7qhgM7\njV/fW/v/Hlw52aOtyY/5n9uLX9xT7Uz0XZMf8+ANuf5OZzhc3u1UN5zUtKs/Ptr8/Q1TT92s\naFJdaB02W+2pMfLV+THjV2rsO9v59w9HVy2ae7RPvXX2R9dk37LeN6LAd+d7nttV5XS/k4p4\n91+Xs3FJwqDG/OpXtSU1fQazXSkTXL004XuXpzt61uz6rtnzOGq9taWHmxrLInBQEAYANIP/\n83m159Z39tdftThe6X2FcyYZ1Ji7Bg2hMkFcuCQgeToEH9I3bHpu13nnPD9Ewv/xdTlXLrpE\nR1XfwuexX6M4FOJO6QNGLbiNKX8DzKPdNA0M+3fHNXPkpjUpC5JDPzzS3NKrlQh5BanK712e\nTtI3ZpLHbin41Sslzj7oAMDjUo9uy0f95VjD1lVBNgEV26LHqlaw6EEWgcJSYc7UbNrYZUGw\neokTfAMFnCD2gDCzpPECADax9X/xE8I0hHhYXwK0HgAAcUCcj8QFUzyaKAKJJqmMEAiEoIEI\nHPORyuYh1nh1m9pO4ymYL6wvil2ZF93Wq9MYrUmR0giFaNpjvDTHPdQNB8fO905N4DCa7X9+\nv6ykpt8ZSYhs+O33Fzob4rphtTNu664O3t5Xv3FJwvTr5xu7NH96r4xmRnPX9SbbPz6q5HM5\n/919Qa0fUSuGdJYdBxtLGwb/+cBKmmGcdoluHCyFtFgJBkcCJ3YUS1MoCqEQAGjq1g7rWdYk\nbXamokl1WVHsNH+XadI3bHrhsyrnvyYuXLJ9y4JFmeThY7ZisdG/ernE1Z9YY7D+bWcFn8uZ\nmtvx1MhPUe4+2eYZz0tRerY7mRCSCGrFo7jrNGg6ADDI4mLV8rIBlhlyXPiYrhNZCYon7/Ta\nSpYwRYwa3HUBDEMgj0Dx+eM0eU2PC3n98fU7DzdVtw1b7Ux6nPzW9Wlx4RLcvBKXf+S+N8Wh\nkpaPf2bccJQ5vxtsJsdLFJqAln4fKeKm9/sEB4IQsPezxIVB5nlJmEkk7CXJSBo2o8MQJCJB\nItAGwDbgygECXzVMIBACAhE45iM2mqUrIQBgjO00w+VMJfWRz6Uy4mf0+WZYz5KPAADDuikm\nD7tOoR109OufevPsq4+t47Elq9d3qlkL+K12prJ5aE3+dCt0Pj/R5qpuOHnt61q1x+9e267e\nf67T0d8Xs3WPYxiKSy1gcB8Dw4CtCIkoiERoJH/eaPaSiT3uppnBZLE/9uLJvmGTM9I1aPjt\n62ee/fFUKqoCAs3gHpVRLOQGSS5MwDlS3s3afendAw0zKXCsKYjJOdZS0z4mnY3Loe65Omvq\nB6V4KGEVXOwftRGpvyxhETiuXkI6TE0CO818eqx1/7nOHpUxQiFcnRfzvSvSxq9pwrVHmLMf\ng+3i7UAopVbcgZIXe9vfkUPkFkSpq5BhEDd+O9r9ly+mim8FmZeGqY5Tt51hSj8cExnuwN++\nwNn05Dgiy2wBRRXi5v0sG6KLZnwshGABpS3DTadZ4umXkAInAbYDPQzYBhwFUON+jzikaTGB\nMN8hAsd8JDmKPSUhRimeSfeKaRIeImSNRyjY4+NjMNuPVHR7xnuGjGfrB1bksiQkm8y0Z9CB\n0cLu6DkpWK1bAUClZe8rea5+YNPShOyEELcJm4P8FCUAolA0BdGeVvex4V4fF9yWmmeevWc6\nXNUNBzSD3z3Q8NcfB7vPn8VGv3+o8ZOjLRYbDQDxEZIHNi9Ykj3fc0/qOzWs8bY+ncVGO4qt\nZgAOhf70o2Vv7avbc7LdTjMAkJWgePCGBTmJ7MYZUyA7UbF9S97Le6qdpr8AsHlF0jXLJtq2\nkGCnmV++XFLVMpJ42Dlg+OBw47Gqnue2r/JWyIM7zzMn3xsTMuuZb1+h5FFIOSlpCVH5W3DS\nMuitxmYtkkWiuCLgX+KSiGsPsETNOtxyCmVdPpmzByMoaS2oW/FQw9jgOhRK3JrmLyi5GOVt\nwFVjPvkoZRHK9dEH3lSDjRWAHUs7CIRpSLIYEOkzRSAQ2CECx3zkioVxOw42ao3uWQA3rU0J\nyHimxvrC2CPlLJLE1OopelRGz0YGDjr69awCR1yEV1EgPlw6hTG4weeyT/MwsI/TkWpxz9XZ\nT7xa4pb6kZWgWJ0/3sJ4pEK0KDPiXP2AWzw+QhLwLIlaNr0GAKrbhlnjQcUz75Wdqu5zvuwc\nMPzm9dO/+8Gi1Xn+teAlTBCJkPvg9Qt+fG1uj8oQKhP4w/ni+pVJS7IjDpd1dw7ow0NEq/Ki\nshJ8JqDMB7450+lUN5x0Dhg+ONz0o2uyMYbTtf31nRoAnBmvWJodiRDg6kMsB2IYXHsErbxz\nsgNA8hiQx0y0ZgkzWMNyYwIArO6cCz4cFBcV3wN9lTBQjS1aJA5HMYtBkRToYRECDLXydpyy\nCDecAE0fSMNQyiKU7KOyO1M1Npx1eY3B3IhpPQq5yjfHJxAIcw4icMxHQiT8Z3645K8flHcO\njGSJcznUbZelXb8iOaDjmhwrF0RdvzLpixNjSuiX5UTesGoqMs04qStCL4nQsWGSovSw8kaV\nWzw1Rp6b5IMJTH6qsrLZ/eAAIBfzPUtUACA+UgIARelhf/7Rsn9/XtXWpwcAhNCmpQk/3JR9\nSVfOx28p/P1bZ13dZ2PDJL+7c1HA7TwZtoobAGCtxAkqqlqGXNUNJ69+WTPPBY5ML+VsydGy\nGUvfcIXLQQmRPhAlvRGjFN9+Rbr/jj+3Kallc3wAKKnp37Iq+el3S121ztyk0CfvXKhQs0sM\nMOwl7ksQAAJWGRrNGUcAhKIKIapwLug1BN+BYrJQzDTq+9jB2HieJWzrBVvvJFvAEgiE+QIR\nOOYpWQmKl3++9mzdQMeAQS7hFaaGRSv9WBtsp7HGYFHKhL41kt++JW91Xsy+sx3dKmOEQrQ6\nL3rKBfxx4ZJIhahf7V4KgRAqTvfqkvWr24qeeuucqyiQFCV78s6FyBe/542rk7850zGoGVOQ\nEiLhb12T8trXdW47cyjkrOovSg975RfrBjVmjcEaFy6ZYNmRUi54bvvKo5W9tR1qm53OiA+5\nvCiO1XxkhkmLDfm2oscznhIjP1TW1TVoDA8RLs4Mnxlr20nhzc23W2Uc1Ji91VjNB9YXxX5w\nuKlr0N2GYzr9jwhzFb2RveJPZ7L+aUeZWyZXddvwn3aUPRvhJROH6//eNAihsGQ8yNLNCoWn\n+v3sBMIcg9YC9mKsZh8kAsfcJxBrHoQ5ABE45i9cDrU8N8p3BlDsdKuML++pPl07YKcZIZ+z\ncXHCXRszfZgHXpQeVuRdgJg4CMED1+f+4e1zbvEbViaNs7QbJhc+t33lsaq+ug41w+D0OPm6\nwlhfpTzIxfz/++nKF3fXHKvqxRgjBEuyIn+yOTcuXKI22D491sJcrEMRCbgP3ZiXGiN3fXt4\niHCyU2iE0LrCmJl0eZwIm5YmfHK02dkr10nngP4v75c7fhbwOHdvzNy6NrjmD97cfAHgxT3V\n8eGSpdmRuUns5vNzGwGP85f7lrm2iVVI+fddm7O2ILg+e4RgIFopqmpliSulAs/SFQA43zzU\nlpybpGVJnkLRPl9eZgHlXYu//feoL6mja5U8GiUtmYGzEwgEAoEwzyECB8GPdKuMDz1/THdx\n/c1spT8/0VrZrHruoVUBSUQfn1V50X//yfJXvqxt6NIwDI5UiG6/In3T0ks40iGE1uRHT79n\nCisRCtGTdy602OjeIVNkqNDZNeD+63KuXBR3urZ/UGOODZOsL4xVyt3bcwxqzB8fbW7q1nIo\nlJMUunVNij/8BWaAEAn/L/ct+8dHlY1dI86UEiHPbLUZXNq7WGz0S3tqopTioCr9SI7yKo0d\nregBgB0HGzcuSXh0a/4Um5LOZqJCRc/8cGnngKG9Xx8i4afFymeRwzFhJrlycfyBUpZONNlJ\noY3dWta3dIUtTho6Axb9mKgsAuVMwvKQZnBzt7ZnyBipEKXFyieezoaisqi1DzDnPgS9Q79D\nKGEhVbwVOLPyCkwgBBKODBD/or3oWLgz24OWQCDMHojAQfAj7+6v13lkF7f06r4+3bFlVXIg\nRnQJClLDnn9olcVGW22MTBwsD6MCHifJY6qcGiN3S9lw5UzdwNPvlJqsIxJAacPgl6fa/nLf\nsnHeMvPQDD5bN9DSq5MIuXkpypRo9uY+AJAWK//3w6tqOzSdA/owufC78z1fnmr33O2zY61B\nJXAsz42KVop7h4zj7PPNmY7ESOnN6wKTe2Ky2K12JkQSMC/6+AhJfARp6Td3sNmZQ2VdDV0a\nHpeTnRCytiBm+vV6xenhd2/Memd/vat38jXLEpfnRO452cb6FqEslHPdE8zpnbjzPGAMFIVS\nllCLbwb+RAvZatrV//q4sqV3pJVVjFL88E15izIn2v8IRedwrnkS9IPYrEWyKBB6vbgRCIRx\noZBoATaWuYd5EY76FNxRgWv2Y3UP8EUoKpMquh5ExMWZQJjvEIGD4EfKm1g8MgGgvFEVnAKH\nAwGPE4QJJhPHYqP/trPcqW44UOutz35Q8eKja3x4IobBvcNGiZA3hRlya6/uL++XN/eMLMBS\nFLp6ScL2LXlcDvt0CCGUk6hw9O/88EgT6z7eGusGCgGP8/S9S/7yfrkz94SVvWc6Zl7gOFc/\n8MqXtY6/v1IuuOOKjGuXJc7DRJJg41R1345DjS09OgGPsyA59N5NWUleunoHnEGN+YsTba19\nOrGAk58StnFJQseA/g9vnetWjVqrfHy05Q93LfbML5sst1+RvmJB1KHSrm6VMTJUtCovKi9Z\naTDbBTyOowGzKwIeJzc5FIRcasNDYLeCYQhk4UBN4mmnd8j465dLXC+hPUPGJ984+38/XZkW\nJ7LhAQabABAHiXlUBPL2HIUokEUiWeTkf10CgeCCOB8BYNN5wBe/koIkJFkGgJhzn+Ca/SNB\nqwHrB+mOcuqqx5BiKt30CATCnIEIHAQ/4vno6cA8du49QRgG9w6bpCKuXEyan49HeaOKtc1K\nc4+2rU/vmQwyBax2Zufhxo++bTZbaQBIjJT+5PrcxRNe3rTY6CffONM3PGrpyjD4q5J2qYj3\no2uyL/l2b7niQShLJUZK//3wqnMNg629ukOlXawZ9d2DBozBt/6743OorPsv748uiA1pLc9/\nWtXWp9u+JW/mBkHw4J39De/sr3f8bLHRJ6v7ztQNPPPDJcXp4YEdmCfHq3qf/aDCKQEcKuv+\n/HirxU73qMbkK9V1qP/2YcWff7R0+mdMiZb9cOzFQSLk3rUx8+U9NW573r0xUyK8+GzD5UPI\npLO6Pj3WavK4SdlpZue3tY/eKnF2SGGw0c4MCzkpFBovMQQPtePG46AfAFEIiitAicWTHQ9h\n/oD763HdN1jdDRweCkumcq8DopEBgDgfCTPBrgJsA24ocOQAgIc7R9UNJ1YjPrMTXfloAAZJ\nIBCCBiJwEPxIQoTUzeLeQeIk59gWG/3B4aZPjo5Mp1OiZQ9cv8An3qJzEpXWPM4mnwgcf3qv\n9MSFUQ+/9n79/7x6+qm7Fq1cMKG5xMkLfa7qhpM9p9ru3pjJ5Vyi1r0wLaykhqVzZGFaMH4k\nEEKLMyMWZ0a09epZBQ6xkDuT6gbD4Je/rPaM7z7ZtmVVCikYCRR9w6YdBxvcgnaaef7Tqtce\nWz+Tn5BLojFYn91Z4SYBtHrJnzpXP9A3bIoK9UuTo21rU6NCRW/srXO0PI+PkNy9MWv6VrUN\nnewpV/XtaoAx7cYw0BamU8Tx2v2HqdyDz3/lNBzFTSdRXD619n7gkKcvgju4/iBT9cXICxvg\nrgq65wK16n4UkRnQcQUOmxEPNoJZA5JwFJYO/LF5GV1s7WMBcH892MzAm799yggEArnFEvzI\n5hVJngIHl4OuXZY4qeP88Z3S07WjE9qWXt0Tr5Y8ddfiZTlkZYMFhdRrQrhC6oPkl6rWIVd1\nw8nLe2onKHC09etZ40azvV9tig27xBz72uWJX5W0O6Y0TqQi3h3B3WR0cVb4N2c7PONLsmb0\nY9wxoB/SsnTdwxgqmlRE4AgU5+oHXD0mnHQOGLpVhrjwIPq/nLjQZ7JMIguvW2Xwk8ABAGvy\nY9bkxzgsh0cTN6aJNzmJLc5gM4MtFGK56uL+Bly5xz3YdR7X7Ed5m6Y5RsJcw6Rhqr90DzJ2\npuxDzlW/8f6hnLPg9hKm+guwXVwLESupwltR+OhdHlu8+FthDFYjETgIhPkMETgIfuSKhXG9\nQ8b3DjbaL/bLlIl5j2zNn1RVeXmjylXdcEAz+NWvavwhcBjM9mGdJVop9mYGEfwUpYVJRTy9\nyd3eNT5CkhLtA5PR880srRkBoFtl+PBI06DWLBHyClKVrnn1NIP3nGo7XNbdO2yMCBGN49nB\n5166zETE5/6/B1e+9nXtgXNddpqhKLQoM/yBzQtiw8SXfG8AWVsQ8/XpjtKGQddgiIR/z9Uz\n0brSiSMNysumqdSOTROLjd57pqOxS4sQZMaHbFycMPF2FXMJ165Abnh+lwPLgJol/WocxAK/\nP2n4TNoAAICsBAVrA9qMBPYLFwY7AJvA0XLa0SLWDablNIcIHISx4P5aYNguzvoB0PWDLGrG\nRxRIcH8NU7FzTMg4xJx+jbP+cRCPpGoiqZJFEgYADo/Y+hII8xwicBD8yx0bMtYXxZ6tHxhU\nm2PDJavyoibroHG+hd2ptK1Pr9ZbfZKS4KCxS/Ofz6urWocAgMtBG5ck3Ht1dvD0Upk4YiH3\n0W35f95R7tSVAEAk4D52S6FPEt2tdsbbple/qnX88B7AygVRT9xeLOBx7DTzxKunKy46zrKm\nDziIC5eEh0xo1SVEwv/5toKHb8zvV5vC5AI39w2zlW7r05mtdHK0LIAtQtxACD1975Jd37V8\neaq9d9goE/GX5UTeuykrTD6jC01x4RIOhViTBRIjZ/qhsKVX9zsXN5a9pzs+Odry9L1Lgiph\nYWaIVrLnOCCEopXBpdxN6hqulAnS40L8Nxh/cOPq5L2n290kJz6Xumkdu0Ds1WfUMMy+7m5g\n14gJ8xqbV90QW43+WW/BeLABdN1A8ZAiCULi/XKSKYGbjrBEaSvTepzKvd7xCiUWQ9mnYHd3\nHEPJi0lLZgJhnkMEDoLfiQuXTGe6YrV5nU5b7V7XoidLY5fm0f+cdLqi2mn85an2mjb1Cw+v\nuqQlRBCyJj8m5efyHQcbmrq1HA6Vk6i4/Yp0X02kkyfm4nHiQt8rX9Zs35L3ZUl7hZd+Om78\n+LqcSY2Ey0GeWRufHW99e1+9Y9GbotCmpQn3XZMj9ukC75Thcqhb1qfdsj7NZmcClacgFfEu\nK4o9UNrlFk+IlBZnzKiZJcPgZ94tdXNj6Ro0/GlH2b8fXj2TIwkGlmRFKmWCIZ27/LciNzJ4\nRDoHS3MiXtrDopFFhor6Pbx1frplAWe2deeJVIj+dv/yf31yvv6iGUdipPShm3JTogcwuN90\nKCRirU8BABB6ufGR5WWCJ2Ila74PAEISP9hLmYaYih2gHmm4jgFQTCHKvhbsOuBKgK8AFMgn\nH6zrYd+g7R79WaSgVt7NnHgL7KOXTRSeQi262c+jIxAIwU5QPPQTCOOQFM3+LCgT83y49P3W\nvnrPni/NPdr957o2LU1o7dWdrR8Y1lniI6Rr8qOlogAvDpgs9tY+vc1Op0TLveWYxEdIfnlb\nkT/Ovjw3KipUxOoS6sa+s533X5d7ks2wAwAiFCK9yeYo5k+Olv1kc+7CaU+wPz7a7NpVgWHw\nl6fa+4ZMf/JFHwcfEtgqjIduzDNa7K5GKqkx8t/cUTzDZVm1Hep2NjeWhk5Nc482NcYH5VQB\nx1GA09St5VJUTpLi8uI4b7N9IZ/z2zsXPv1OqavGkRkf8sjWgpka7ESJDZP84KrMN/bWuQYl\nQu7/3r24b9j0zv6G1l4thVBWouKHm7Jzk0IDNc7pkB4X8vxDq9r7DT0qQ2SoKDlKRlGIxgIz\n3QEwqrkj4Aoor+veKL4IN5ewxBMK/TJowmwGRWaDSA5mdyNqFJUFQl9fDDHDlL4FriICRWFT\nI679z8hLfiiKuwrJ03x83omDvBSrjo2jxIWc8FSm4TtQd4FAgqIyUfLSGe1JRvAzKPi64xFm\nBUTgIFwamsFDWrNSLgzIQtzqvOjXQ4SDGvfOIDesTPbheCqb2HOGK5tVbX26z463MheXK9/Y\nW/vzbQXLcwNTEIsx3nmkacfBRoeTApeDtqxKuWtj5kx2SBXwOE/fu+TPO8qbe0YexRBCGLOU\nPJittEpr1hhYetYCAI9Dffa/V/UOmyRC37T+tdN4x8FGz/jZ+oHqtuFZOtHyByIB96m7Fle1\nDF1oG7ZY6dRY+YrcqJn/dvcOedXIeoeMc0DgaOjUPPX2OadjxZ5TbY4CHG91WHnJytcfX//1\n6faWXh2fy8lLCb2sKBYF5cP69y5Pz0kM3XmkqaVXKxZwC1LD7rwyI0wuTI2Rr8iNstOYQkDN\ntsQNNxBCSVFS17ZTHCQXczJseJDGJgSIQmIeFYHA67UXJRahxIW4vXRMNCSayr/Gf8MmzFa4\nfGrp3UzJG2AZbUiE5DHUwtt9fio81DxG3UAIwqTgKrtbh3HrR5ByK5Kl+PzsEwEpU3B3OUs8\nLNU9JFZQhZtnYkwEAmH2QAQOwngMaswvf1lz7HyvnWa4HGpdYcx91+Qo5V6bdPgDIZ/z9L1L\n/vp+eUvvyF2fotB1yxN92DIDY7DR7NUurT26pp4xKypqvfWZ98pefWyd//oCjMMbe+s/ODw6\nh7fT+OOjzUM686+/VzyTw0iKkv3nZ6vP1A209OqkIm57n/6z462se4oE3AiFqImtPWpUqAgh\nFOM7f4GuQb03O8YaInB4kJeizEtRBnAAUpHXG5BMFFxFGVPATjN/fLfUzY+zuUf77M7yZ3+8\n3Nu7xELu1rUeT/BBSVF6mLde3bPXofmSIMTno9hL73dxd2rtfbi5BDcew9p+JFag+AKUexVw\nZ/3Hm+APUHgaZ91DuKMU6/qAK0DKFJS01C+lIvqxaZVCHngmFWIG9x4NmMCReSXuqwZ67OqI\nKBQlrQrIeAgEwuyCCBwErwzrLQ+/cNyZOmGnmYOlXeebh/7zyGqfrLdPnNQY+X8eWXO2bqCl\nVycT8fJSlEkTs4GYIAhBYqSsuYdlEj6gdc8cAQCLjf7mTMcPrprp1vQGs/2T75o944fKuu+4\nIiMh0pd/k0tCUWhZTqSjkU1dh5pV4MiIDwmR8K8ojj1VzVKlcsXCON8Oic0389KbCIEiP0Up\nFnKNHt1DQiT87ERFQIbkQyqaVL1DLF0MyxtVfcOmgMijcwy9yfb16Y7WXp2Ax8lLUV5WFBOU\n2S4IpS5HqV4lLQIBAAAzuPMUbjkENgMAgDgcpWxCEbn+Oh019uGf7yULydQNmAmIGQeSxVCr\ntuPKj/FFlxAUnU/lbSHNXwkEwkQgAgfBKx8dafYsDOlXmz452jLDjS0BgOMynfYHN65O/sdH\nlW5BEZ/rLSOgY4DFO8DfNHdrbV46mNR2qGdY4HAlK0GxcXHCN2c7XIM8LvXA9bkAsLYgtrJ5\naPfJNtetGxcnXLnIxwJHXLhExOea2HqdZsbPsj4O8wGRgPvA5ly37x1Foe1b8uZAp9geNnXD\nQbfKMAGBgwGY9X8E/1HeqHrmvVJn+dueU22fHmt5+t4lrIasfcOmhi4NhSAjLiRCQaQlQtCB\n63fjzlOjr42DuPIdyN2GYhb543RImTpW8/eiDGIMELDFARQSj9Y8AmYtNquRJAJ45JtLIBAm\nChE4CF4p99L5YoIdMbwxpLXsPNJU36kGgMx4xS3rU2e4TSYrG5ckqLSWHQcbnD1Qo0JFj91S\n+Ls3zrJOmIW8AHx3GDafi0tumhke3ZaflRjy0ZHm3mEjj0Plp4bdd222w0YBIXjoxry1BTGH\ny7t7VMZopWhtQcyizAifj4HPpbauTXn3QINbfEFyaEGqH1zoCdNm45KE+AjpW/vqGjo1CKGs\nBMXdGzOzEmZ9+gYASIVerYhlHi7FdhrvP9dZ0z4cKjRsKlBFyvQIM8AJQaJ84Cf6eaSzD6PZ\n7qpuOKjrUD+36/yTd46ZEJqsZiLMEgAAIABJREFU9he/qN57ptNhEuQob/zxdbn82a+gEeYO\nxkHcyWJGixu+RtFFXu02p4MkAiUswx0XT+qtIZ0wwi9nnxRCOfK5xyqBQJjrEIFjnoIxPlrZ\nc6F12GyjU6PlG5fGi/juHwarR1cRBw57y6lR0aT63ZtnHY0zAOBC6/DeMx3/e/fiwrTAzz9v\nvyJ9w6K4sobBYb0lLly6NDtCwOMUZ4SdYGsCMsMNNR2kxMg4FEt3RgDIiAtwhgJFoeuWJ123\nPMlio3kcytNfsDAtbAb+y3demYEBPjrS5BSqVudFP3xTfjCmrvuH881DB0o7u1XGSIVodX70\nigC54U6cBcmh43hSzF4K08P4XMrqkXIVHiJ0808dUJv+57UzbX26pWm2h7fqOdTFRVN6GOuP\ngigfiUjTjTGcqulntS4+fqFPb7K5drn6+87K786PmikyDP7iRJvFyvzilqDrTUOYt2B1C3ui\nhM0A+j6QTdzzZRKg3BtBEoGbDoHNCCYryESeaRwocoU/Tk0gEAj+hggc8xG13vrkG2fqOtTO\nyM4jTU/eudDNhTEhUsraxDFxqv4Xdho/u7PCqW44MFnsf9tZ8eavLgsGX7pIhWjjkgTXyD1X\nZ5U3qdxsAvKSlesLY2Z2aAAAcjH/2uWJX5xoc4svy4n0ecsJjOFoZU9Z46DWYE2Mkl6zLDFy\nYqndM9nPxROE0F1XZW5ekdTQqTFb7akx8gBW7sw8//7swucnWp0v95/rXFsQ88TtxQHpfzQ+\nBrO9rU+HMSRHyyRCljuRyWpv7tZpDNb4CEniLPwnhkoF91yd9ZJL02IA4FDo4Rvz3OS/f3xU\n2danQwh+eqWR45lYYKoCQRpQs+8v4D9YzU0AgGFw37DJKXB0DRpc1Q0n+8513n11ZjBkDhII\nAAAMS5boxU3sRbI+AFEoeS1KXgMmNXB4wJiYjj1gvPh94QhQ9DoUmuevsxMIBII/IQLHfORf\nn1S6qhsAoNKa//hO6Zu/Wu86O928Iul4Va/n269fmTS189Z2DLv1FHDQrzbVdgznJQeyoYM3\nkqJkLzy0+qU91efqB+00IxXxNq9I+t7l6YHqgPiTzblcDvXFiVY7PbLgs2Fh3E+3+PgpxGSx\n/+7Ns6O1SFXwydGWR7bm+9wW1E8oZQL/2bUELScu9LqqGw6OVvbkpypvWJkcgAF5wU7j9w42\nOLNs+Fzq5vVpd1yR4Spx7j/X+fKeGucq/cKM8Ee25kf7ruHOzLB1bWpytOyd/Q1N3VouB+Uk\nht67KSt9bLLVoMZc1jgIAPFKJlrB6rCDwdYLgvQZGfLsQOpR48O6qbGLxTcaADDGjV1aInAQ\nggWxl7sVokDs+1pOt3OAyLGyJaUy7gZDF7aogCtG4jjgzrLrLYFAIDghAse8Q2Ownqzu94yr\ntOYzdQOr86KdkYUZ4Q/flPfKnlqnCYVYyP3JdblTUyLMVnpIa/G2VaXxuslXlDeqjlR0O/oX\nrCuMKU6faI1JfITkj/cssdNYZ7KGSt1b5GqN1ncPNFQ2DWmN1sRI6bZ1qYv94C7hhMuhfrI5\nd9va1PpOjZ1m0uNCYsN8/xTy+td1bk4rFhv9z48rFySHzrpJ5vzhUFk3a/xwWXdQCRz//eKC\nq+Os1c68d6BBZ7Ruv6jTfVvR87edFa5vKW0Y/PUrJS/9fG1g84OmwKLMiEWZERiDtyKpvmGT\nwz9HzPduo4P9too7O1mcFU5RiPEo1kuJlrm6t1LefTao+VO0Rgh6UGgKlkaD3n09CUUXAW8m\nb7gIJPFIEj+DZyQQCAS/QASOeUffsAl7MaTsUbnn/V63PGl5TtTp2v5+tSlSIVqeG6WUuc/w\nx2dAY37zm7rShkGjxT7OsluY/NKH7Rww1LQPW2x0aozcrZpmfDCG/9t1/quSdmfkq5L2TUsT\nHtlaMPGnXC4HeaobnQOGn//3hFo/ss48qDGXNgzedln6vZv822UmPEQYHuKv5UeM8YHSTs+4\nzc4cqei57bI0P52XME08ex456GdLmwoUKq15z6l2z/juk+3fuzzdsai+46C7TSwAdKuMh8q6\nNy1N8NwU/IxznZGJR66KvRrkVQch9SljiQ2T3HZZ2o6Dja5BPpd66KYxuWyZ8QqEwPN2x6FQ\nZgJpq0QIGhBFFdzJVL0P2tE7L4rMQ1k3BHBQBAKBMHshAse8g7XcfZxN4SHCa5ZN0ca/W2V8\n/OVTzk6rOpMNIeQpr0QqRNmJ4wkWNjvz788v7D3T4VyyK04Pf/zWwglO8g+Xd7mqGw6+Pt2R\nnxq2YXo1Fy/urnaqG052HmlaXxTjc1OMGUNvshvM7CXB3krfCcEAa4NMAFBI2eMBob5Twyqw\nYozrOjQrFwhtdqa1T8f63roO9SwVOMYhIUIaGybuVhk1RupkI29lhkeyBiUBnl9cBmc1d2/M\nykpQvH+osaVHJ+Rz8lOU927Kjo+QuO4TFSrauCRh7+kOt/fesCrZ25eFQAgMIiW1+EE8VA+6\nHqC4SJEE8rl2rSMQpgI1y9I2CUECETjmHbFhktgwSbfK4BbnUMjnnTvfO9jgVDcAAAHweZTV\nymAXw3ARn/v4rYXjO4y+uLvaTaEoaxx86q2zzz+0Ck0gB+NgWRdr/FBp13QEDouNPlc/4BnH\nGJ+80McqcGAMJTV91W1qO82kxsrXF8ZwWUwFA4xIwOVykNPjwxW5mMwKgpeVeVEnq1k6/qxa\nEO0ZDBTe0sfG3zSHQQgevin/t6+fsdPMv74WK8SG3Dg7AAZHSwNKgqRrA9+pMShZkRt1ySZB\nD9+Yp5DwP/muxWZnAEDA49yyPvX2KzJmZIDumCz2D79trmoZMphtKdHyrWtTZq8OTvA9CKGw\nLAjzb/ongUAgzAeIwDHvQAgevinvt6+fdpvB3n5Fhmv1sk8oH+vjAAAUhQQCTkyoSMDnAEB2\nguKW9WnjJ2IYzfavPZbgAKC+U1PRNFSUfunOowNq9tT9vuml7htMdtaOrQDA2sJQY7D+4e1z\nVS1DzsiOgw2//8GipCjZdIbhc7gctCgzoqSGxahlee7cce4cUJsQQv6r9Jl5rlwY/11l7+na\nMf+4rATF1rWpgRqSJ2mxIaxVAwiBw32Tx6VSouXNPSz2kNkJihkY4cyzMCP8pUfXvLG3rrpt\n+HefCLYsoTYWiyJCuIgbCvxUQOQ2PXW4HOreTdm3rE9r6dEhClKi5ePkMPqVbpXhsRdPOevI\nGru0h8q6tm/Ju3b5FBMkCQQCgUAgsEKenOYjCzPC//2zNa99VVvdNmJp8b3L01cuGFkKs9P4\ny5K2yiaV1mhLjpLdsCrZLe934rh1hHWAEKTEyn99W9EED9IxoLfTrM0FoLlHOxGBw1s28jSz\nlOUSnoDHsdhoz02RbFLRPz6qdFU3AKBzwPCHt8+9/PN1wdAi15X7r8utbVe7yTTXLU+alPVJ\ncIIxfFXS/ta+OkdtUXiI8J6rs65cNBds1SgK/fGexd+c6dxf2tmjMkYqRGvyo29YlRJUn66o\nUNFlRbGefqiXF8c5BdY7NqT/8Z1Stx3iwiWXFc/ZSo2ESOnvfrAo0KOYs0hFvPzUAHfpev7T\nC24uOTSD//PFhaXZERET68BNIBAIBAJhIhCBY56SEi17+t4lAMAw2LXj6bDe8quXS1p7R2rg\nK5pUX5a0P3xj3tVTKn2PCRN3DrjXwgDApBp/8LheizjG2eTKityoCo9cEgBwbRkzBbgcam1B\nzP5z7n6cfC61Jj/GLajSmktqWMoHOgcMFU2DPi8OmibxEZKXf7H27X31pfWDGqM1KVJ605qU\ndYVzYXr5xt66Dw6PehMOasx/21kxpLPcun4umKcihK5emjC1b+uM8ei2AiGf+/XpDkdNCkJo\n09KEB67Pde6wJj/mV7cVvbynZlg/0lxpSVbEwzflz7oWKgSCA73JVtow6Bm32ZmT1f1T7rxO\nIBAIBALBEyJwzHdc1Q0AePGLaqe64cBOM89/VlWcET6FApaNixJe21vrFuRyqEk5XyRGymRi\nns7I0igxL2VCi3KbVyQdq+p1y57IS1ZuXjHdx8r7N+e09ukaOjXOCJ9L/WxrvuffqnvQ6M1h\noHPQEGwCBwCESgU/uyk/0KPwMcN6y8dHmzzj7+5v2Lw8SRyg3PX5hoDHeWRr/vcuT6/v1ABA\nZnyI5/flioVxq/Kim3u0ar01MVI65SQyAsG30AzuURl5XGpSN0S13urNYmZIx15BSSAQCAQC\nYWqQB3rCKFY7c6zKvRM7ANjszHfne7ZNvpJ/84rELpVh75lRBw2xkPvT6xfEhk1iusLloLs3\nZj3/aZVb/IqFcSnRE3Kv4HGpv92/fPeJtsPlXX3DpqhQ0fqi2M0rkqefui8X85/fvuqbs51l\njYN6oy0xSnr9ymTW/BSRwOt3Tex9E8G3VLcOs5qnWmx0bYd6YUb4zA9p3hIVKhp/iijkc+ZA\nSRRhzmCn8cdHm98/1OgovQyTC++7Nvvy4gmJ9Qopn7WDmOM4Ph7ofIMxYXMl2HuBMQMnBAky\ngZ8y4tFLIBAIhHkJmVkRRtEarA6reU+8+XSOD0Logc25GxbGlTYMDustsWGStfkxU2hauXlF\nkkjAffWrmiGtBS464d92WfrEj8Ch0JbVyVtWJ0/21JeEotCmpQmX7F6ZEiNTygRDOotbnMtB\nRWmXthEh+ASrl483AFjZvFQIwYDGYP3yVHtLr1Yi5OWlKK8ojp1I7yQCwbc89+l5146zKq35\nL++X6032iRSYSEW8RRnhZz26bvG51CUbwRDGg9Fi3QHAIzdWTA+DsQTsfUi8IrDjmltgsA2D\nXQfcEODNTadnAoEwxyACB2EUmZjHoRBrZ5BQ2dT9ODPiQjLiQqYxLgCADQvjriiO7Rkymq10\nYqQ0CLurjg+HQg9cn/vMe2Vu8dsuSycOczNGUpTU26bkiWUDEWaYkpr+v35Q7uw2/VVJ++fH\nW5++d8k0HYIJhEnROWDYy9bM681v6jYtTZiIG9RDN+U99uKpAZfWXRwK/XRL3lxq5DTzYFO5\nU90AZ9qGtRX4qcAlypEvsHRj1SGwXnSQEcSgsCuAT7IdCQRCUEMEDsIoAh5nUWaEW5tJAKAo\ntCJ3PD/OvmFT16BBKRMkREo5lL8WVxFCk6pt8SGNA/rWIaPeYpcJuenh0iTlJExSnawrjFXK\nhK9+VdPQpaEZSIiQfH9DxvqiueDcOVtIjZEXpIZVNrs7zq7IjYqe0v+U4Fe0Rutf3i8zmMc0\nY6rrUL/w2YXf3FEcqFER5iFuFk5O9CZbS68uM/7SCn6MUvzqY2s//rblfItKb7Knxsi2rU0l\nuur0wGDrYd9g60ZE4Jg+1kHcuwuwyxXY0oN7P0ZxdwKH7WHM0I61zWA3gCAMKfOB67sHNmwF\nRERtAoEwUYjAQRjDgzfkNnZrHJUgTu7ckOFt6btnyPj8ripn5m1smGT7jQsWB59l5pShGXyg\nrr/z4rKbymBtVRnTI6Tr0qeygpGfqvy/7avsNKYZZm63hLDTzIFzXfWdGoQgO1FxeXGc/5Sv\nSfGjGxe88PH5+rZhZyQvPezhrXPNTnVucPJCn5u64eB4VY/Jkj+Or800MVnsOw42Hq3s6Veb\nokJFlxXH3XZZ2tz+whLGx+alVTkAeKvr9ETE5955ZQZAho8GNe/BNICXPz62sscJkwFrzgK2\nuRuaMGasLUOhq8fuSuOOPXj4wmig/ziK34QUuTAdaD3WloC5DbANKCGIc5BsISAyc5lPUOTO\nS5gK5DIxR8FmwDoAACQHJJj4+2LDJK/+Yt37hxrLm1Q6oy0pSnrzutSCVHaTiN5h48//c1Kl\nHbXn6FYZfvfGmWd/vHyC/U2Cn+peXadLUrGDxgF9vEKUFj7F1QkuB3E5c+2SbbHRHxxuOl7V\nO6gxK+VCjd6iMYw8Yu4+2fbhkeY/3rM44FkSJjuDudRPbyts7FC39+gohJJiZSlxIXoGEx+U\nIKR32P2r58BO4wGNOTHSa8HRdDCY7Y/8+3hbn97xsltlfO9AQ0lN3/97cCXROOYtyVHsqRYc\nCvnpczhpbGas7gbMIEUs8OdHPhriAhK4lqiMbqGC458y27H2etq1YgBkcU+cwf0nXdUNAADa\ngtt3I1EMCKbqFU3r8MAuYC4+YTJm0JdhaxcKuwHQLCtSJhAIMwwROOYeNLbXAt0O4LDSQMBJ\nRtwsgIneD6Qi3n3X5oy/z3fne1/7qqZbZfTcZKfxuwca/nLfssmNOlhpURm8xacscMw99Cbb\no/854ZwTOh0TnLT16f68o+z/tq+a8aGNwXDRSTQ9QZGeMGqWZrTTDMYUsa4MMmQinrdNJTX9\nYgHXH/4Fnxxtdn6SnTR2ab840Xbzukl3kiLMAKdr+9870NDco+NxqZxExb2bstNi5b49RV5K\naGZ8SL1LR3AHVy2Ol4m9fkpnCMzgqq+ZC9+A3QoAwOGi7A1UwXXAmQcPePxUsNR4RCngT7cH\n/GyFseGOo3igCsxqEIaiyEIUvwqoqX4SMEuCDAK4+HjpsuNQOdvbaTx8HkWvneLJdWdH1Q0n\n1n4w1YH4Es+oBAJhnkNE0LkGtp0Hus3l9oOBbsH2C+O9ZywWG93Uo23s1pqt7H0ldp9s++M7\n51jVDQc17epJjDi4MXlprmH08seZn+w83OQ5J3Sjpl3d1qebmfF4g9VAd2ST1y2EgLEkK4Ly\nUtn0ypc1P/zbt3tOtfn8pGfq3FtdOPA0JyIEAx8cbvrt62dq2tUWG6032c7UDWx/7lhJjY//\nWQihp+5aXDi249WGhXEP3rDAtyeaAkzpLqZi94i6AQC0HV/4mjnzfkAHNUMgYT7wxvpYIS4S\nL4f5mcFBW5iyF3HbYTAOAGMDYz9u3c9UvAoMS6HfhOBHeomP9TfBDFi17Htah9njE8HSyRrG\nXuIEAoHgZB4I/PMKbASmmyVOdwInA9AlVjsZjD893vrJsVaLjQYAAY9z48qkm1anuLonWO3M\nG3vrLnEc79PIWcHp2v7K5iG9yZYcJeN7aR8j5pNk9VFO1fRNZLdulTHJS6b3zMDnsM+WKYS4\nwWERQnAlIVJ622VpOw42sm41We3P7apKiJAW+rTRsmf+0fhxQgAZ1Jjf3lfvFqQZ/PynVUuz\nL/NtO+HwEOHf7l9+vnmoqVvL41I5SYrUGB/niUwFiwHXHvKIItx4AvKuAelcr71DHCRZB7Yu\nbO8FbEGUHPhpQM3TxmS48zgYPO7Fuk7ccxrFrZzCAVHIYmxqds/joHhIPtbjGVFA8YBhu0Jy\nppFk502XYT0RgUAguEAEjrkF9iKiAwasvaTA8faBht2n2p0vLTb6g2+bNQbbjzZlOYNN3dpL\nPuhnTMBSPjgxWexPv1vquoRbnBeVmcHiJ5rii34u7f36mrZhq51JiZbNatcSnXFCDxwSYYAv\nODI+t99oY7C7ABci4BJ5Izi5e2NWTmLoB4cbazvUNFuazRcn2nwocFjtjDc33Niw+eFrECD0\nJltjl9ZgtiVFyeIjJnp1LWsctLPZf/arTa19+hQ/9CjJT1XmpwbRtRoPtbHWEQBgrGpBc17g\ncMCLQ7y4QA8i8OAhL4tPqjqYksABgmgUsRkPHQL7xexLnhKFbwDuWGkPM0iWjDUNLEeQpU3l\nvA64IWBjy6fjztYnTAKBMGPMPoHju+++e/fdd48ePdre3h4aGrpkyZLf//73RUVFrvtoNJon\nnnhi165darU6Nzf3t7/97U033RSoAQcJnQMGO9bFR0i4HPa6JK3R+tXpDs/43nOdN61OVspG\nnEqtXko2XPneZenTGWoAeWlPjVuCekV1v1IpDh87t0kNl6ROz4DDZmde+Kzqm7OdzmyXwrSw\nX95aGKGYlUtPkaGiIR2L05srcjE/O3GqZmM+gkuhOJmgW2ehXTQOCY8TKSb954KXZTmRy3Ii\n7//n0ZZelhKn9v5L1EZNnK5Bw29eO+2t+G7jkgRfnYjgxmfHWt/aV+fsmLMsJ/JnN+VPxGCF\ntcmOA/3EVNdZj4dc63UTtoG5BdvViBICPxZ4U2kERghq7B6OFQAAgO3GqSv44hQkugssvWDX\nAU8B/KgxBp+WHqw5CdZ+QDZAlIvWhgEQkmcg+dSfBpEkB6s9BQ4KEQMOAoFwKWafwPHXv/61\nvb39lltuyczM7Orqeu6555YtW3bw4MHVq0d6VjEMc+2111ZWVj7zzDNpaWmvvfbatm3bdu3a\ntWXLlsCOfCZA7MI2xvDYS9VDelBI+T/clM36pN7YrWN1KMAY13dplmePlGImREgR8vpMpZDy\nf7I5d0n2rGwTa7MzB0u73IIMgw9+1/rjG/NkIQKD1S4T8NIjJNNP33hxd/XXY+WkiibV7986\n98JDq7yZDgQzGxbG117KeOXBG3L53MCb/kh5nLRQkcZsN9MMh0ISLkdKqo1mAwIv/yah7/59\nz35QzqpucCh0+xUZS7O9lKMTpsfuk23/+WKMS1RJTf9vXjv975+t4XqpKXMS46UxE0IQMz8y\nblBoPHi5JSNl4ugLaxdWfwuMEUYMuhCIs5B8JTFim0sgoQKbVGxx96UFbNTh9gtYr0bKGCpp\nwSX8aBEXhPEscVMzVn0z8oHicJBCBkYjtgPQdhAoUfgiFLZoqr8KAACIc8CuAX3lqKkc4iPF\nauAFUQoVgUAITmafwPGvf/0rPX1UEt62bVt2dvazzz7rFDh27dp1/PjxN99886677gKAjRs3\nLly48LHHHpsfAocIOPFAuzsw7TmDh/QAAGq99R8fVVrtzOYV7h7jDOOln/xYTw2lXLAmP+Zo\npXuTsCil6NGt+TmJoSLBTHyoTFb7iaq+zkGDUiYoTg+feErzOAzpLBbW/BSMh1WGbcsTWTZN\nCaPZ/jVbskxjl6aiWVWcPvsW1q5bnljXod5/bvSDhygULhdoDTaEIDNecc/VWQuSfZa+0Tts\n+uBIU22H2mSlEyMlN6xIXsxWRuQNDkJK7+05CMFJUVoYq4jmq+9L16DBmzvy9hvzrl3ms68/\nwRWM4f1DLB4rLb26k9W9a/Jjxn97cUZ4pELU79HJe3FmhD867AQjohCUugI3nXALo8SFIL/o\nBMmY8PABwK4pLRiMtUBJQFoMhDlDVDEMN7HFF7q+os/tsx9+Fywj3xqkjOFufpCKzwYAwAag\nuzE2IiQGTjSgcYq8MFYfG9NOhUIglSBAKOpWX2kQSL4cRJlgace0AXFDQDR/DVYIBMKkmH0C\nh6u6AQCpqanJycnd3aPOmp999plQKLztttscLzkczp133vn4449XVlYWFBTM6FgDAeLmYeAB\n3eq48dhp+OQEvH1wzD5v76u/ZlmiW6l5cpQMefb+AgAAt0rmR7cVWO3MqepRL6vcpND/uaM4\n0p/lFXUd6oNlXT0qY6RCFBUq+vRYq0o7ko3J5aBta9PuuTprmo5yEiHXW3KKROjL+XDnoIG1\nbhwAWnp0s1HgoCj0+K2FGxbFHTvfO6Axx4aJNyyMT4+TMwxGCHxr9VfXqXnm/TKrfeQP2NCl\n/fvHlVtWJt9GWnjOabauTT1U1u02lY0KFW1bl+KT4/cNu0+SR5ndpslBzbDOMqhhz6uv79Bc\nUuDgc6kn71z4h7fPuR4kPU7+i5sLfTnK4IZa+j2Gy8f1R0cKBBBCqSuoxbeM7mFqHKtujICN\nNYgIHHMIFFkIui7cdRLAUSMCgCiUsAaFZTv3YWpP2fe+4vouPNRj++BP/Pv/hUTD2FYLwDje\nDbY64GYgXhawYlMDzVoeiMHa48skC54SeMrZl9dKIBACyuwTONzo6upqbW298847nZELFy5k\nZGQIBAJnJD8/HwCqqqrmg8ABQCFuDnDSAGu/q+x97rM2jUfOtcZg7RwwJEWN6aMWHiJcnRf9\nXVWv287LcyLd0oAlQu7/3r34QutwddswzTBpsSGLMyN8Ood159Wvaj8+2uytOYudxh8cboxQ\nCD3TUiaFVMTLSlCwrhIv9WnRzTh517wgKOKYMsXp4W7qjD/Kbd7YV2e1MTD2wF+cbFuXH+0t\nX50wBwiR8J/bvuq1r2uPVHTb7AyPS60riPnRNTlyH/mnjHMcuYRYtPgN71eICd5TshIUrz++\nfu+ZjqZuLZ9LLUgOXV8YG+SFfhjDqfqB0pYhtcESKhUsSlUuTZ/GPZTDo5bcBrlXYVUbAEbK\nRJCOuQ5jWsP+RsYI2AaIpLPNGRBKuxZF5OPBajAPgTAMReaDdEwbXfrUbpb3WUx06RecpW6W\ntBjs9UDJgcOmM7JJZiMwVq+bCAQCYUaY3QIHwzD33Xcfn89/4oknnEGVSpWaOmYtV6lUOuLj\nH+3555/v6nK3YHBgtVoBwGK5hI1iEIH4gMJ71BpPdcMBzVaQ8sB1OXwedai8B19MY1hXEHPf\npmzPPQFgQXKoD4sOxuFUdd+HR9iyLsfy+fHWaQocAPDA9Qt++dIpt0KV65Ynpcf50rU7MVIm\nF/O1RpaHgFndS2UGGNZbWvv0npMiBuOK5iEicMxqhvWWYZ0lNkzizVZDKRc8fmvhL24uGNJZ\nlDKBbyexabEy1mIHEZ9bnD4/WlEEglCpgPXPDgBZCYoJHkTI52xZlezLYfmT5j7dKwcajJYR\ne9R+jbmuS1PWMnTfhkxvHXwmhESJJF5uH8jbkx4CRByIpgLWNoC6Blu1SKAAxQIk800emW+Q\nJyK515I6pr+NNY77GgBYLnSYbkesAgc3BIBypHu4wwuwmzhhTkGRaxRhKsxigQNj/OCDD+7b\nt2/nzp1udSusXDJPvry8vL29nXWT3W6H2SVwAABAWqycNS7kc+LYmoAIeJwHr8u9cWVyc48O\nY5waIw+GzogHPIw/WekYMDAMnuacJydR8d9H1rz6VU1Fk8pspeMjpDevS71yEZu91jTgctDd\nV2c+t6vKLb5hYZw/+hrOJcZrmnCp7sWEoKW6bfj5T6uaurUAgBBcuSj+vmtzQrzkTVAU8oe9\nAkLokW35v3vjrFv52E/XzG3nAAAgAElEQVSuz5USxxa/gRB8f0PGPz+udIunx4Usz41ifcus\npqxl6M3DLJ4j/5+98w5sozz/+PPenbZkWfLee2c6y9kkIQRICIGEDWWUUSjQ/ii0paV70k1b\nSKEhhF0ghJ0JhOwdx4njvfeSrb3v3t8fdmRbOsnbkuz380+i5053r23p7t7v+zzfp7RJd6K8\nc1nOhBjZImEsNrnfbgAAhDGBazLKOrC2FTgWhUaDIJBsFzCHGz7B2tK+V6YG6L4IYXNR/HX+\nHddwoRlw8D3KUl5c2DgTf5wSgTQNzB6tYZkQEJGGUwQCwc8Eq8CBMX700Ue3bdv2xhtvbN68\neeCmsLCw7u7ugZHel715HD549dVXvW0yGo0KhSIkhF8vCFjmpoenxymrmt3TUzctTREJvGqi\nMWppQK2E8y7ueSJkqHFZ0Y2PkP3y3vkA4GQ5by11x86GgiSpSPDfL0p7nUREAvrWq9JuXzWG\njvEjB2MAwONrkDHRhClENIV42/1EqQLpIZgwbErqe57+z0mXrIAx7D/bVNGke/HJZZNcsTU/\nM+Ll/1u+Y19FWaPW4WQz4pV3X52ZkzjcPALC6Lh2YQKH8fY95a6kthWzYr67KW9M6QwBCcvh\nD0/yr58DQFF99wQJHCBKBFE82Aa7jyMGKRZOyOnGCsYlX3OFn4LdDABAMSjvamruRqADQmfE\nmkKXujEwCPJkFBoE7Uup+Cyu6rxnHMV5Wcjxmv4DKHQl5mxgHbAuyISisHUkLYhAIPidoBQ4\nMMYPP/zw9u3bX3/99TvvvNNta15e3s6dO61Wq1jct8p38eJFAJgxY8ZkD9TfUBT69X3z//bB\nxbMVfb3EGRrdtCzlvnWZ/h3YiBhmjf2codLI69sNl+t6LDZncrQiPyN8yIn9xKkbvayeG7tq\nTmxbj9nuYOPC5UM2RBxHiuu6d+ytqGjSYgxpsSH3rsucOGdTJ4s/PlZ7qKi1U2eJVknX5Met\nX5Q4ailKImIWZkWcKO1wiyskgvwgNGclAMAb+ys8PXfr2gxfnm++buHoVwLr240fHa1taDfK\nJMzstLAblyQPRy5JiJT/7J78IXcjjC/XL0pcNTe2ttVgMDuSoxVTVaxs7jYbvCea6ceQg4Yb\nCnFLMZi1oIhAaUuR2v2Lg0LXgukiNl8GzgpAgTAGhSwCJhArInHRbu78x/2vOSe+tJcz9VAr\nH/TfoPrB2hL+DdrLEAwCB7PiVnvdJXAO+rAhdQwzZxUGvlpgyrvoRglR+AawNmJ7G2AnEoSB\nJB1QoOYEESaSI0eOvPXWW4cPH25oaFCpVAsWLPjFL34xZ84cf4+LMH0JPoEDY/zggw/u2LHj\ntddeu/vuuz13uOmmm95+++133333/vvvBwCWZd988820tLTp4TDqTrhS/PsHF1Y166pb9GIh\nk50YGhTPjhjj5i5za7c5Ri1dlBNxusx9QuuGWEjff60Xr28AJ8u9+MnlPacbXTalmfHKH98x\nd1yay44FhGDyk2W+PN/8p/9dcL0sqe/50SunnrhpxtgdTDyxOdhnXj7p8m3t1ttK6nuOXGr9\n/bcXjVrQeWBdVqfOWtWid0UUEsGTm/Jk4uC7mhEAoLi221t81ALH5yfrX/rkspPt+76fKu3Y\nfarhz48UhIVMj9ahQYhEyOQm9ZfuF1Vr3vqysrpFLxRQOYmq+6/NSoyU+3h7UGC187Uhv0Lo\n6OxyWSd35BXc1F/jgysOU7M2oJnXD9oN0SCfi+RzgbMCJQzcyhSnnbu4xzOMa07h2dej0FjP\nTZONw8AbxjYNMpUDLQVhNFABkWzCC4pJE9z1C+febbi9rjdC5Sxh1t4LkhCwdQA3+KdDMsQM\nlVgqTkBiUpMy3Xn++ecbGhpuvfXWzMzM5ubmf/7zn4sWLfrqq6+WLVvm76ERpinBNyV46qmn\ntm/ffvPNN0ul0p07d/YGpVLp9df33c5vuummJUuWPPnkkzqdLjU1dfv27cXFxbt27fLfkP1P\nepxyfG0yJ5SKJt0Luy5VNvVV1qTFKjPila6XvShlQqudtTlYikKzUtWPbszz4V6xfU/5FycH\nuatUNOl+9tqZV55a4ceuJc1dpv8drO51/s9JUt2+Ks2b6cA4YndyWz+97Bn/7xelq+bEjrvX\nwEdHaz270lyo0uw53TBqPUUhEfzmW/OOl3SUNmqtdjYxUrZ69viPnDA5YIx5C44AwNUJeKS0\ndZtf+qTEpW700tRp2vppyXN3k+yMIGDXkdr/fHZlndwCx4rbTpV2/PaBBfkZwZ2lFR4i8rF1\nXupovGxx6YGB6gYAAOa4ok+pqEwUyedNRgW0xod7msDpxeysoxoCQeBgpGDnabWG2B7cuQcA\ngJYi9UqQeV1x8TtUfJbwwT9jvQaMPUgdA+K+lR4kWoYdFcA2AbYBEgEdi5gstyY7GMwcbsbY\nBEAhJKdQHAJfn2rCNOEf//jHQDPELVu2ZGdn/+lPfyICB8FfBJ/AceLECQDYtWvXQM0iLi6u\nqamvvpSiqC+++OLZZ5/9wx/+oNPpcnJydu7cuWnTJv8MlzBCWjTmZ14+abH1e0lWt+gkIubO\nNelnyjtbNebIUMnymdFbVqYKGapTZ1XKhD78RADA7uTc1I1emrtMp8o6ls2IHv+fYRgcKmp9\n/n8XXJn5JfU9+840Pv/Qooz4idWhyhu0BjNPIrTVzp4t7zRZnQ0dRpVCODc9fPgtDHxw/HK7\nt/hYEkYQQkvzopbmTUEPwukGQigpSlHTqvfclBozSsPdo8VtnjUvAHD8crvNwfq+XBDGF53J\nfqy4rbnLFBYiXpgdOZykuW6DbfueMregk+Ve+PDSjh+tCirLIHfUclFWbEh5C8+nfV5q2IJR\nFdlxNad447j2FL/AEeDw9XcbetMkgpRZ2NziGccM0/fZZM24Yw+KFoEkeVJHNkJQSBiEuGlq\nDBLkgiAXgAXguU5yuIvF1QC92jHG2MzhLobKRhBk/nSEccet1UNqampycnJLC883hUCYHIJP\n4Dh58uSQ+4SGhm7dunXr1q2TMJ6pB8vh3acaTpd1dBtssWGyDQWJs9Mmr0vih4drBqobvVhs\nTp3J/uKT7kpwZOjQ5TYdPRaLnb/1Rl2bwS8Ch9HieGHXJbc5mNHi+Mv7RS8/tWJCT22yei3z\n/vuHl1y/eYqquHZBwpM3zRijb6vexNMKFwB0piBrSESYODavSPnze0VuQblEcO1o61O6dFbe\nuJPltEZ7UNToTQ2+udDyz4+KXe2Ntu0uvW1V+r3XDOEAdaGqizd5p7XbXN9uSA7yPlN3rUj9\n75eVjV39nSlEAvqGeQnLc0drL2ru4Y+b+Cu/AhykigWKBo6vlifMa+vTyQSFL8D6SjA2DexW\njhgKBAMepxFg3RkU2AKHT3hVYJbFtVfUDYC+n59juRqGIlYLhEE0NzfX1dXdc889/h4IYfoS\nfAIHYUIxW50/+u+p8sa+DMzKJt2hopbNK1If2eDLPUtvtn9+oqG2TS8RMXlJqqvnxY/aAL/U\no6KhF89Kh2HiowjFX/UphVVdvD1Na9sMTZ2mCXUGiQnzevCBuhLH4d2nGqLVkttXjWkNMCJU\n0qIxe8YjQwOoTQ/Bv6ydF6812t/YX2Fz9M1q4iNkT986WyUfZeazSsH/RppCk1AFRuilrs3w\n/P8uDKw/crL47S8rY8Okvhtv+7Dh5M0+Cy6UUuEPbsgrrNXUd5qcLJcQLpufFj4mh2mRjL+m\nQxycliVCKcpcjsu+cQujmGwUkeKPAXlAMVTa3VhzHmvLwKFFwGHaDkKPZ2nbEMZhQQeHdQA8\nwhMGKwYTAj87mhECB47jHnroIaFQ+Oyzzw6585NPPvnFF19422qzkcUwwighAgdhEG9/VelS\nN1x8eLimICfSWx7H+cqu37193vXoufd040dH637/4EK1l2mGbzDmL8jnvBTqD0lkqCRKJWnv\n4ek1O2tUNc9jp9vg9ZKt0VsnVOBIipLnJIZ6U5Hc+OJkwxgFjtVz44qqNZ7xNflxYzksYYpx\ny8rUVXNiL9V29xhs8RGy/IzwsfQwWpIb9drecs8rxoLsSLGQ1KdMErtPN/C6q3x+ot63wBGt\n8qp+RqunQvYNQpCfGpY/TncfFD8blx/kiwfrojq16DYOUbj8G1dNCkrKp+ZuALsJhIExi0YU\nCp+PwucDAO78Apkq+faZ7EFNPPyZsAAA2DkVf17CaMAYP/bYY/v373/vvffc6lZ4ufPOO330\nuHz88cfHdXSEaQQROAiDOHyxlTd+qKiVV+AwWZ2/f7vQbWGtplX/z12Xfnnv/FEMID1OWc1X\nopwZP0pLCITgweuzf/d2oVt8+cyYnMRxsJkYBT5aOUxCl4ef3JX/s9fO1LX1m6VLRYzZoywI\nANp7LHYnJxxDnsu1C+KLa7sPnGsaGLxxSbK/rE8IAUu4Urxqzvg4CCZEyu9bl+Xm4xCuFD+2\nMXdcjk8YDk0dJt54Yyd/3MWc9LBwpdizzmhuenjEMGoSpxvUrPVsawnoB7kdocR8lBisAgdQ\nDFVwB+StxV11wNqxpR3aC7lT/wIAkEWg7A0oIoC6sSJRNOYVOARhYK4AWgHCSEBTQVdF4D39\nDZHMOAIAAMb40Ucf3bZt2xtvvLF58+bhvKWgoKCgoMDb1u9973vjNzrC9IIIHIRBaPT8yQVN\nXabfvHn+cl23xc6mRCvuWJ2+KCcSAE6VdujNPD4LJ0o69GZ7yMj73m1ZkXKwsNmtBlvIUFtW\njj49deXsWLGQ2fppSYvGBAAiAX3LytTbV/vNgC0/PVwhFXimW6fHhUxC59oolWTr95d/Xdhc\n1qDFGDLjlZXN2s9O8PiwChhKMJbcaQCE0DO3zV41N/bIxda2bktMmHT13Fh/Jc4Qpg+3r0qb\nlar+8HBNXbtRLmbmpIffdlWalDQSnmAwhtNlHeWNWgzAq5kCwJBJNCIB/dO78n/9xrkeY//N\nKClK/vSt07HR+9CI5PT1P+Eu74fmi9isRYpIlLEMpRYEfQqBIhwpwnHRO9Da39QcTJ343Gsw\n+y4UM9t/IxuMfAboC8FpHBxF4OzC3V8CADAhKHQFiAPCQGQsIBQCWADg/tyCQIaAKI8EwBg/\n/PDD27dvf/311++8885xOy41FfRBwuRDHvgIg1ArRB1anmqOizUaV8p3SX3Pz147861rMu++\nOqNDy+OwAAAY444e6ygEjqQoxW8fWPiPDy/1ihEAEBsm+97mGUlRY/KWW5QTuSgnUqO3Wmxs\nTJh01BYh44JUzPzglll/eOeCy3QAAJQy4dO3TtJDG02htfPiXYniIZcFvALH3PRwNB5NC+Zn\nRszPjBj7cQiE4ZObpMq9Z56/RxGgNHeZiqo1OpM9IVK+KDtyXNyIunTW3751vqTei+flFYbT\n6jUvWbX9h1ftPtVQ2awTMXROUug18xPGZFQxtWFE1OwbYPYN/h7HeKNrwgPVjSvg8i9QzKxA\nUXAoEYq+BWu+AovrHoqApgFd+U459VizG0VuAUFwNzkGoBiU7sQVA504EAhpKmCb9dgxWwO4\nB4ADJEdUKqDgtigOZDDGDz744I4dO1577bW7777b38MhEIjAQRjM8lkxHx6u8Yx7FrS//WXl\nmvw4uUTguXMvITKvm3wzJz1s29Mryht1bd3maLU0M145Xm6gk1AAMkyW5EX/9wcrPjhUU9ms\nEwnonMTQW69K8/HLnFAW50bPy4w4V9E5MCgRMd++Pnvcz3X8ctvpsk6N3poQIV+3ICEpKjid\n8AiE4ARj/Oqe8l1Hapxs3yU9Nkz6w9vn5Capxnjk379TOKS6oZQJ71k7RBeVXmRi5paVqWMc\nEiGowT08jyIAAFYtmLtBGjCZgIwSRd0MTh04erCpFKy17jtgDhsuAD0ba5qRNARFJADtn3v9\nGEFIKUBzWNyGwQhAUSCnULSXliv+Buux80x/vgk2YK4N0TOAIv5fE8JTTz21ffv2m2++WSqV\n7ty5szcolUqvv/56/w6MMG0hAscUx2B2FNVounTWuDDZrDS1SDDErejuqzMu1mgqm3RDHpnl\n8NnyzoXZkTSFPM3kUmNChtPD1RsMTeUlq/KSx/rMHchEq6VP3OTVWmkyQQh+fd/8nYdrPjlW\n122wCRkqPzPi4fU541svY3dyv3nz3KnSPm/5U6UdHx+r/fZ12ZtXkGkMgTBJ7DpS+/431QMj\nLRrzz147s/2Zq8bSYqa+3VBcy9+XVCyirTZWwFAFOZEPrc8hbXoJw4Xz7mrJBV4/HUYJjBIM\nZz23YJOVPfgp1/By70ukUDNr76NyFk/u+MYLAY1G2cB7MsHsZY9qGozZEkRFgA8zEcJoOXHi\nBADs2rVr165drmBcXFxTU5P3NxEIEwgROKYy+842vvxZqasjaZRK8tQts+am+8qTlImZF767\n9PMT9SdL2zV6W0KEbG56+L8+Lubd2WB2RKkk37om87W95QPjIgH95M0BMXUnDBMBQ92xOv2O\n1elGi0MqYqgJKOF572CVS93oxcniV74om5kalhmvHPfTEQgETz4+VucZNJgdX51vvnn5KH2O\nuvW297/xstgO8IMts2amhillQv8WBhKCD1kUf5wWgCRg0jfc8fiQs5xzfxHW9xfzYkO3Y9ff\nBLf8iMocqRE7h7EDIQHAuHe4x8AZgTMBJQVKESjlP6MGWwHzrtKxwHWSJI6J4OTJk/4eAoEw\nCCJwTFnOlHX+9f2LAyPtPZafv3b25adWxIZ57cMHAAyNNi1L3rQsufelwex48dPLvF1aY8Kk\nAHDH6vSshNB3v66qbtFLRMyMZNV912bFqH2dghCwTFyZzJfnmj2DGOOvzjcTgYNAmASsdpa3\nYTYA1LcbeOND8nVh8wu7ii1eLEUBQCxkRtcynDDNQRFZWBoO5i73ePyiwC3xEEaBfVBHG7am\nfaC64cJ55APhsAUODHYH28xiHQAGABopBVQcQuP0tWJ7sK0Q2Cv1ZZQSiecCHbAS0nDgN8sH\nAAAeU3wCgTD1IALHlOXDIzxLajYH+9mJ+kc2jKDLmkIqKMiJOn65zS0eKhcuyo7s/X9+Rvhw\nrOMI0xle81oAaO/h96klEAjjC0NTCCGMedRqITOaQvqmTtNf3i9y2Xl4IhLQgVxp2K23vXGg\noqhaY7Y6E6Pkt65MW5BN7JADBoqh5t3PFb0D+n5xHMXNR1mBW9WPFLOxuRy4ARPsLp629wCA\n22qBdQI99EM4BqfNWYEHFFywWMexJhGTjWDMQg9nwpYjgAdUc3A6bDmCpGuACl5LTh/SDxFb\nCYRpARE4piy1rfwrcrWt/LdbH3xv84xOnWWgMYdSJvzpXfmk7aJvHE7uwLmmiiYdAGTGK6+Z\nH8/Q455ZGjSESIUD+z72x8dQ+U8gEIYPQ6O8JFVxHY9Zxqy00SzYHjjX5EPdAID71mX6yzt5\nSGrbDD/YesJVwtljtBVVa+5YnX7/tVn+HRihH1kEtfhJ3FUOhlZgREiVAooYf4/JJ7QCRdyI\new7153FQPqzNfX13XDi5DuzRnBWD08m1C6j40Q2z/ziOykHqRl+UxfZyJB5pBU3AgMSAVIA9\nPY8ZoIiCSSBMC8gEdcpCe2mqN4o5tkou+tfjS78qbL5c12O1O1NjQq5flBiwj60BQmOH8ec7\nzjZ39TW73X0KPjhU85v7F4yvc2cQUZAbued0o2d8Sa6XQmsCgTDePHB91g9fPuVkuYHBGSnq\nZTNG8zV0NfP2JDFSfu+6rOUzo0dx2MnhpU8uu9QNF/87WL16buwYu5ITxhOEUEQ2RIx/S6+J\nQhCOIm8Gpx6cOmBCUMopKONJp0WRicMstOGwcUTxkcF66XzE8nsGBxoYnBhbEQgQEg60DkH0\nDMyeAWwdsC+F6Jkw9pwXAoEQDBCBY8oyI0X9zYUWvvhoEoYpCq2dF7923liXCwIEJ8t9ea65\nN7ciK0G5Jj+e8aIHjQ6M4XdvF7rUjV6au0y/e/v8S99bjgLDwKtFY6po1NmdXHpcSGpMyESf\n7t51WYVVmrbuQQUpK2fHFhCBgzBVcdqACayM6BnJ6r8+uvilTy6XN2oBQMhQG5ck37M2A43q\nqiQT888WpGJm29MrxzTQCcZkdV6s4ZnCYYxPlnYQgSNocHRiayk4tUCJkSAaJLmAAuGxFvU1\nVQGgZ6xkj3+CdZ1uezDLbh720fgTPTBwvPFxgzVi/XmwdwAgEEUhxTygA6gFEgaHg21isbb3\nJUIiIZVAoSvfXCRDzDLg6jCnBWABKRCVAiiAxk8gECaUQLgTECaEu9aknyxpt9rZgcEoleSG\nxcmeO5uszpL6ni6dNUYtnZGimtqVFE2dpp/vONPU2ac+fH4S3j9U85v758eGjVtuRWWzroav\nFKi6RV/VrMvwt6emw8lt/bRk9+kGl3fs4tyo/9syK1Q+gdUiaoXoP99f/vZXlafLOrsN1vhw\n2YbFSVfnTxHJjEDox27iSnbjxvPgMINAihLyqdz1IAwU3+WcxNB/PbHUaHHoTPZotXQszU3m\nZUbsPtXgGV+QFTmGAU4GRouD14sEAPQmYkMYJJgvYvOF3v9iFsDRBrZqpFwHVKB81wAAhGLB\nnT9zfvEfrqGkN4AkCnrN3VTOkmEeAIEYgMepioLxmK5ToV6SNYS45a3+6hVbCzaWoIiNIAqQ\nnCzOxlZi3F/0irHNxlaL6HQKya/EGKDS0VR+mCUQCF4hAseUJSlK8bdHF//ro+LShj6Fe+mM\n6Ec35so8jDO+LmzZ+ull3ZWnuvgI2VNbZs1IUU/qcMeVTq1lx76KizUak9WZHKW45arUxVfS\nBDCG37513qVu9NLYYfztW4UvPrlsvHIr3PIUBtKiMftd4Hjp08tfnBw0LTlR0m5869xfHlk8\nodklUjHz0Pqch9aPwOOWQAgyHGb24N/A1OV6iWuOsh1l9KqnQRBA64dyiWDsZYbLZkQtyIo4\nUz5odVopEz5wXaDbWITKhQKGcjh51sCjVIE0PSZ4w9mDzUWuV303LtaATWeRYoWfxsQPUscI\n7vkV7qjHXc0gDaFi00A4gksBQ4WzbI9nHgczHnYSSJiOnfWA2cFhCgz17t4cnA1rDqDYuwOh\niayT6x6oblwBO7hWEZ3hhwERJg5qNAbYBAIROKYy6XHKFx5f2m2wdWotceEy3sfZ85Vdf3y3\ncGCkqdP001fPvPKDFVGqAHocHz5Vzbof/Oekq21hcV138Y7u21elPXBdNnjPrahq1lW36NPj\n3Cs1nCweRfWKj5mDQurnElCD2bGXzwvjUk13WaM2JzF08odEIEwZuMpv+tUNF8YuruobKue6\nSR6Myeps6DCGSAUxaik1hkwNbyCEfnXfgo+P1X52vL6tx6yQCBdmRzxwXXa40oexYkAgEtDL\nZ8Z8Xejeu1ospJfNCJA1aoIvsL2Bv3bD3gjAAQTcwj2KTEKRSaN4I4VkQjrJwTZi6JMhENAC\nKhZ1tmNtEQglKCodJKMtMqUUSLIMW88Dd8WWnpIhlIDtX/Ps7NSCvROE/s/P4jC/+4+3OIFA\nmG4QgWPqo1aI1AqvdeA7D/HYX1nszk+P1wXpSvuLn1x2qRsu3vumZk1+XFKUolXjI7fC5BI4\n6tsN23aXXarttjvY+Aj57avSVs2JG352Q26SSiZmTFb3YcglAr83TazvMLAcf252dYuOCBwE\nXkxW5ztfVR4rbtforXHhsnUL4jcuSR5LdcOUpaOMP95eBpMocBgtjm27y/acbuwtxIgMlXxn\nY+6yGdEmq/PT43VVzXqKgqyE0BsWJ4kEY1ofY2i0ZUXqlhWpDicnYAJuVumDRzfmNnUaKwZ0\nBxML6advna0OCSzbFEIvuP0yaCrBaQFZJIpfABx/33HALHB2n71Lgg8aqShGwWEDxjaEhJTB\nig++wrVX9W1mhFT+RjRntN1z6XAkuxpYLXBGoGRAq8BU4XVnNkAUBB8NaHAg5JgQCAT/QgSO\n6Q5vOgMAVLeMuJtsIGAwO0rqeVzBMcanyzqTohQyidfPvCvtoqha8+y2065GA3Vthj++e6G8\nUffoxtxhDkMspB/dmPeX94vc4o9uzB3jdGLs0JTXSYiPTYTpjNZo/96/j7VeKbyqadVv/bTk\nVGnH77+9cCLyAoIbJ08vZF/xCQBj/Nz2MwOvhB1ay6/fOPfAdVkfHalzdWs+VNT68dG63z+4\nMDFS7uVIIyC41A0AUMqE/3x86YHzTReru40WR3K04obFSYGfezIdYe34/OtYU+kK4NpDaNZy\n/iwNRAM1BSUqBAyNVIAAnHb2iz+CYUCamNPOnd5JCUQob81oD08BrQb6SmEy7b1Ky96C7e2I\nCQVJKlB+a/FOISnL0wUWKCQh6gaBQAAicBC8zU+CdG3WYHF4cY7rs47LS1ZJxYzZZ27FS59c\ndmujCAAfH6u7bmFCcvRw3fWvmR8fGy59fV9F7wphZrzyvnVZfk/fAIDUGIVYSLu5z/YSCMMj\nBCDvfFXZ6mErc76y66vC5inTWWnckEeAvo0vPnl53adKO3h13tf3Vbhlb3VoLX9898JL31s2\nWUMLLCgKrZufsG5+gr8HQvAFLt89UN0AAHBacfkJlJPCs5IvTJraU1xcc2aQunEFrmgPnbea\n/2dnHbjiCNY0AgIUlowylgLt8+FfFAu0FNjB13wKAdDYUAS9v3T9SaRaDeLE0f8kY4Cm1E6u\nHYP7gxxDkaZsBAIBIADLFAmTTG4S/5w2LzkoTUbDQkTeFhKj1VIAkAiZ72zgScRw5VZo9Nba\nNoPnDhjjcxXund58MyNZ/edHCj7+9bqPf73uz48UBIh8IBLQd67hceG6Oj9uXBZyCaNDZ7I3\ndhidrI/MW79xqrSDN366jD8+nUEp/M0RvMUnAl51AwB4a9OqmnV1fFc8AiEgwBxuOc8TNxnB\nphown8cAALQSyeZP2tD8Atbw9C0CADB2g5WnfgT3NLO7fsadfBdXHsUVR7kTb7Ef/wL0Pi/d\niEHqqwEx/foRQqUSa7kAACAASURBVADUIDmJNWPNXmD9c+lAwAjpdGpA21cEtIBKoBGpsSUQ\nCAAkg4Nw55r0U6UdNseg9fxwpfiGxaNxw/I73qzjpGJm2cw+67hrFybEhkt37KuobNIhBBnx\nyvvXZbm6xngmd7jw9NQYDhPal2R03HZVmkzM7NhXbjA7AEDIUFtWpt7Fp3oQJoHCqq6XPimp\nbzcAgIChNi1NvntthkQYQBdng8XBG9eb+OPTGRSVS828kbv8BXBXLhe0gMpdj6KyJ20M3kx2\nvNHeYxl+bhqBMKk4LOC08m/SOVDmemwpBVYHlAgJYkCcBWiq91zw0fjUs8gUc9w3L4NRMyio\n7+C+eYXa+Jyvs0iSUMzdWH8W7B0ACFgDeHYtwU5sKkMhC4Y/9nGEQhIRncVhE8Y2QAIKyRBM\n9T89gUAYNgH0DE3wC6kxIc8/vOhfHxW7TDcWZEV8d9MMvzf7GDWP3ZjbojGVXWmOCwBSMfOj\n2+coZf31orNSw/726OJe+z00WIGICJV4ax8YFy6bsFFPKgjBDYuTrluY0NBhdLI4MVIuFpIn\nA/9wuqzjFzvOumakDif3waGaqmb9Hx9aFDjSWLRaUtXMo2XEhJGGmjygjNV07CzcXITNPUim\nRrGzQRY2mQNIjRlZS4WB18YpjJPFl2o1TZ2mULloZoo6VD4tfuqghxEBogDz3JFBIAVGjRRL\nJ31M/gRFpWPYx7NBFQtC9wsy7qwFbavnvlhTj7ubkNpngSGjQOpVvbvjpv/w7+PkTxabLBCF\n5IBI5imBQHCHCBwEyE1SvfS95e095i6dNTZMFpge8idK2j8/Wd/caVYphAuzIzevSBV6KUUJ\nkQpf+O6SrwpbLtVojBZHcnTI+oJE3j4yiG8GKRbSV82OPXCuyS0eKhcuzp1S5Z0MTY10IkQY\nd17dXea53l5Y1XW2vHNBdoRfhuTJ2nnxVc0lnvFr5hMDDi/IwlHmGn8pVMtmRr+xX+ppmyIS\nUDaH+0QxXCnOiFdO1tD8Rnmj9i/vF9W3G3tfSkTMfesyb1qW4t9REYaGYlBYOu7i6euBIrIB\nADgjsK0YmxGSAh0D1BSf7qLkuSgiBXfWuNltUAtu5tnbyOPWcWVTJ/gWOAacExANmC+DFQXr\nShiBQJjaEIGDAACAEESrpb0uFQHIPz68tPtUX91pi8Z0ua7n68Lmvz66OETKvwSHELo6P+7q\n/LjRne6xG/O6dNbCqv4nA3WI6Lm78qVi8n0hjCdGi4PX8AUALtVqAkfguHFJclWzfqDqx9Do\n/muzvTn4EPyLSED//sGFf36vyGXGIWSo21alR6ulf/2giBsgqDE0+r/Ns4LUUnr4aI32Z7ed\nNg6otLLYnFs/LVHKhKvnjvI2QZg0UPYN+NRL4BjUFxYlLILQJHBUYnspAAd9/hClSJgDgild\nboko6rr/406+hyuO9/3Q8jBq8e0oOZ9nZ4+cjgGbRpKRKooHax3PWETBoHFjJ3B6wE6gQwCR\nNkkEwrSATNgIk4TF5mzuMofKhSPtw3e+ssulbriobze+vq/iiZtmDPMgTg6jYbeGkYmZ5x9e\ndLKk/VJtt8nqTI0JWTsvTiIiXxbCOOPwaNbjws5XJOUvKAo9c9vstfPiT5S0d+kssWGytfPj\niSVtIBMXLvv7Y0tKG3rq2gwKqSA3SRUWIgaAlGj5Gwcqq5p1NIWyEkLvvSYzIYj/jhgcGmB1\nQElBEAHI6yV675lGI5+PzAeHaojAEQTIo6ilT+HKvVhTBXYTyKNQ8nIUOxfYNmy/PHhXDtsv\nIyoE6CmVbumOWE5d9W0ouA1rW5FYDiFRPO4bAACAojJAIHHThnqPgCJSh39CpFyEbc2AB3+J\nhDEgTR/ZyCcfezW2X74ycgSCJCSaCYiUpxEIUxwyZyNMOD1G2yufl35d2NzbwDU1JuSJm2YM\nv6XI0Ut8PRcBjhW3DUfgqO4yHq/WaEw2AAiXi5amhaeEDWvhoiA3qmBq1aQQAo1QmTBULtQa\n7Z6buvU2rdEeUDYBc9LD5qRPqpcEYSwgBLlJKrcsm/Q45a/vmxJtJhwarD8Cjit5dpQUhSwG\nMX/JSW2rnjde12bAGPPWKhICC7ESzbzN7e+EHfW8+2JHHZraAkcvYjmKHipXRSCmFt3KHX19\nUBBRVMFdQ3SKdT9OGIrcgnXHwNYEmANKCLIZKGReoHfktVdhW9GA1xgcdZgzIekKvw2JMFIo\n4hBHGA1E4CAMlx6jrbi2u8dgj4+QzUoNY+i+G1tzl+locVtHjyVSJVmaFx0fMUg+sDnYZ/5z\nsqHD6IrUtOqfefnk3x5dnJ04rIZeOpOHd3ffeOwYD9Gj5FxDz+HK/t6unQbbxxeaV2dFzo4n\nvcRGzNHitp2HamrbDCFSwcwU9f3XZkWESoZ+G8E7CKFNS1N27Cv33PRNUcvZis7vb565YlbM\n5A+MQAhoOAvu2Q3cgFsDZ8bag0gtBCFPRgZD869vUxQK9BkawQfY6CXO0y112oIyllHKaHz+\nE6ypB0AoIgXlb0LhySM+kECFwjcA5oCzAB0UhusY20sBsPt3nO0EthPoQKkAJRAIEwEROAjD\n4qOjtTv2VljsfS5TiZHyZ26bnZUQ+v431Tv2VTivZNq/vq/iW9dk3r4qzfXGL881D1Q3enGy\n3Ov7K/7w4MLhnDrMS0lLhFLsW92wOtnj1Z4OW/hoVVduTIjAyyPv2BlSdglG/vNZya4jtb3/\nt9ic7T3Nx0va//qdxWmxxKZ0TNy+Ks1gcXx8tNbTatRocTz/vwvpcSGxw8s5IhCmCdhcOkjd\n6IPDxiKk5hE48pJVnr7RvfGpd62eTnhzuBz8ZMuasbUNOCsShoFoGmR2eIAi09G1PxinY1FB\nom4AcEbAdn4Fk+0mAgeBMLWZqDkeYSrxdWHz1k9LXOoGADR0GJ/ddvqbwtZtu8ucA3wEnCy3\nfU/ZqdIOV6S4rpv3mMW1/HFPrpodyxtfPZc/7qJVa3V6TBoBkJ3lWnXWYZ59+FQ26Z7ddvqm\nn+/b+Nze7/372JnyzqHfM0JMVmdpg7a+3ehkPX+uCaSmVe9SN1yYrc6XPr3Muz9h+FAUemRD\nzranV/JWozic3IFzzZM/KgIhoHFo+ONO/p4Ra/LjPC1jGJq695qs8R0XYVKhI/njTH8c95zh\n6rbhlo9w2x6u4S2u6T1waPnfRSAQCISpAsngIAzNzsM1nkGjxfH2QZ7ObQCw+1TDopy+Jwxv\nU3GW44aZ6ZCbpLpnbebbX1UONP+fkaK+c80Q1ad2Hw6O3jeNjuOX23/z5jnXInxpg/anr55+\naH3OLStH4OPlA7PV+dre8s9P1veeQq0QPbg+Z9RtYobE7uR2Hqo5WtzaqbVGq6WqEH4niOLa\nbpPVKSPNZQBOl3W8/WVlTatBwFA5iaEPXJc9otyWGLVUZ+IxQQSApk6Sbk0gDMbrjYN/g0hA\n/+mRglc+Lzl4oaXXCio5WvH4przhW0ERAhAkSMNsE3CDU0QpOWL6ckixthB3He7fhAEsTVzz\nh1TSvT4saacDuKcZOirBbgJlLIqfNTVtDigZIIG7MWrfJvLFJxCmONP6Ek8YDhhDnZdOll06\nfneMpq7+KVlKtOJQEc8+KTEhw88NvmdtxsLsiC9ONTR2GNUK8aKcyLXz4oZ0hlN5aSLre9Mo\n4Dj874+LPUsMduwrv3penEouGuPxMYZfv3nufGX/4mS3wfan/11gWW7dgoQxHtwTs9X51NYT\nNVds+XQmHgtM18CMFgcROP53sHr7nrLe/9sc7JnyzvOVXb+8d75L5hsSikJiAT0wScoF+fUS\nCG4gQSS28hlMCrx+49QK0Y/vmPv4phlNnSaVQhSlIv5BwQ8SIPEK7CgDZxNgOyAhMPFIkAOo\nr3QF95wZvD8AADi02FiBFLmTPdoAgWO5M//DVccAX1nmCYmmlj2A1Il+HdZEQCFhJrZ55JnS\namBIfQqBMMUhJSqEIUAIaC8dyBgvXVelAzqqXrswQS7hKZSNj1ScKu/keEpI+MlKCH1qy6y/\nP7bkZ/fkXzM/fji+95EKUXQIj39HXKgkTDaeAkdNq6GLr+bF4eQuVHlJpR4JF2s0A9UNFzv2\nVWA8/rUqHxyuqfHSdMANkYBWKcYq3wQ7XTrrG/vdU5lYDv/ro+IR/XXmZ4V7iZNHMQJhMJJs\noKQeUQrJ5/p+n1wiyE4MJerG1AEJkXAWkl6PpBuQ9HoknOVSN4C1gJN/bQasHfzxaQBX9Bmu\nPNKvbgCAvo07+CI4xr9u1/8Is5EoD9CA/BQmDkmWEGthAmHKQ9YGCUOTm6QqrOKZYOckqU6W\ntHvGB07J1ArRbx9Y8Jf3i1yZ9hRCCoWook1fsUf/+RnZExtyIkfSjANjfOBcc1G1Rm+2J0Up\nblic5ONpdf3MmM8utnQY+jNNokLE188Y57YUJit/cQEAGC1eNw2fkvoe3rhGb23rscSoPR/0\nx8SJyzx/U16umhMrZKajSMpyePephsLKrh6jjaEpJ1/FU4fWUtduTIlWDPOY91+bXVilcfvA\nzM+MWJoXPQ4jJhCmEpQIqddj/TGwt/RF6BAUstRHBgdhisNTcuJ9EjsVrWWxvgY6zmBrNwhk\nKCQVRRUA5fE7wRyuPMzzZosONxSitMWTMM7JRpiNBCnAagEcQCmBGu4dmUAgBDVE4CAMzb3r\nMi/VdrvN4pKi5P+3ZdYPth538wiIC5dtXp4yMJKbpHrlqRUXqjU7DlR26W1CEU1fSf1o6jK9\n+EXpL+/Mdz1vtGhMVc16mkKZ8UrPLqQGs+Mnr54ub+wzCTtV2vHJsbrvb565xosbRYhYcOeC\npKpOQ7veBgiiFeK0CPm4P9vEhHmVGMKUoo+O1ta0GoQMlZesump2LOUl7cUHnsUv/ZsmwG1U\nb+avSaERDDxbXrLqkQ054372wEdvtv/4v6eqmodOcjGaRyBvxUfItn5/+at7ys6UdZisziiV\nZMPipM3LU6bioziBMGYYJVJfD6wenHqgpcCEkozUKQtr5yq/hI4ybNUhWQRKXozi8odehKfF\nIFSBnW95QBiKTSeB1QISICYSxDnBbsmBG/fj9lN9L6xd2FCPNZeo7HuBGfxwYtGB3cx/BF3L\nlL3VIBEw07F7DoEwnQnuazphcshNUj3/0KJ/f1xc22YAAIRg1Zy4hzfkqOTCfz6+9I0DFQcL\nW3Qmu1ImXDUn9lvXZHrWpDA0lRgpNzlYicT9I9fQaapu06fHhJiszhc/Lv7yfF/PCJpCGwqS\nHt6QIxiQI/DK56UudaMXm4P9+86LM1PV3tJAEIKMSEVG5ATK9pGhkrnp4Z5JLuFK8b8/uuyq\nXvnsRP2HR2p/c/8C9QjLOrzZVcolgujxTt8AgMhQCW/FzaK86IzYkLp2g0wsmJMWtnJ27PSc\ne2/fUz4cdQMhX8oXL1EqyU/unAsANgcrEkxF1zcCYXyhQ4DuvzzWtOqPFbd3aC2xYdKVs2NI\nf+WpgM3IHvsXmPq6kmGbAXfXoPZSKv/uId+K1Etx2+fuUZEKnJfB2bdggx2tYKtEIWuDeG3f\n2NivbriwduHmgyhp/aAg7a2xLgA9nnW7AQHrxN0tYLeisFgQuzdRIhAIUxsicBCGxcxU9ctP\nrejQWnoMtvgIucv4UC4RPLYx77GNeVY7Kxb6mpK19Vh8bEqPCfnju4UD+8uyHP7keJ2D5b6/\neWZvxMnib4paPN9ud3KHL7ZuWTGmfiVtemuPxUFTKEwmDBu5Bekzt83+2Wtnqlv6570RSrGD\n5bTGQdkQlU26v++8+Jv7F4zo4AuyIpOjFZ5WrzcvT2Ho8dcY1uTH8RbFXL8wYWH2dM8Axxh4\nP4SezM+MCFfyWMAMB6JuEAgjAmN4dU/ZzsM1Ll+nt76sfODarM1juy8Q/A5XsdelbrjAzedx\n3FwUlef7vUiRBQC46xtw9nVaQfIsEOoBBhcVcmZsOo0Ua8Zt0JML7in1Fh8kcBi6sLYNKSKw\ngaeHPYqeUi2TueJD7KG3wawHAEAUNXsNveIOEI3/ghCBQAhMiMBBGAGRoRJviRK+1Q3fO4iF\ndF2bYaC64WLvmcb71mWFyoUAoDfZbQ6W9wgd3tWTITHZncdrNV0mO76S85qoki5KUnuzUOUl\nXCl+8cllBy+0lNT3OFkuLVYZqhD+9s3znnueLuvoNthGlMTB0Oi3Dyz46/sXXUkiDE3dsjL1\nrjXpwz/I8Fm/KLG0vseVSgMAFIVuWZFK1A0AsNicZitPrxM30uNCfnDL7EkYD4FAAICDF5rf\n/6Z6YMTh5F7+vDQjXjkrNcxfoyKMHdxW7C0+pMABAEiRheTpYO8GzgZCNThbsOkkz36OVsA2\nQAHvmW3rxPYuoIRIHA30lQQlh5dW4k4LYBYQDQYNd/h1XHcBAIChQeb2Y2IUPwdFZU7gsCcX\nrugrdv9/+19jjrtwAPe0Mrf+lNiLBh9euhwQCL4hAsc0RWeyS0WMYBIdIlOiFFIRY7a5Tw4F\nDJUdpzzJp24AAMfhmlZ9fkY4AMgkDE0hXkOKkNF2RcEYDld3aS0OGHDfa+gx0xQqSFKP6FAU\nhdbkx7ncQHYdqfV2xrZu80irVCJDJc8/vKi8UVvXZpCKmZxE1aizA4aEotAPb5+zJj/uaHFb\nh9YSo5aunReflRA6QacLLiQiWiSgeYW2/IzwiFDJWMxWCATC6NhzupE3vvd0IxE4ghu7l9m7\n3TjcIyAaRH3G59iLAwUAAGcBOoAFDqcBt+/H5rreVxgxSL0IqQsAAIReimsYGSAanHb2kz+A\n7sojlpMFkxXEIuhN/xSIqdy1KPeaiR7+5IExe+wDnnB9MW4oQYlDi2IEAmEKQASO6YWT5XYd\nqd15uEZrtNMUyklSfeeG3Mx45SScWsBQt69I3X7AvaHmpoIkuUTgYzLoMnoQCej8jPAz5e7Z\nlQihxbmjdJDqMNq0fI1O6rpNc+NCRWMQgHib4w65yTdZCaGTJjTMy4yYl0kalLrDYZifFX6s\nmKfRzAPXZU/OV4lAILjRquGfuDZ3eZkeE4IFSSiY+LqtS0a2AtELoiRefbnRRK0ZjAOY45p3\ngX2Azxd2Ys0xQBRSLUSqXNx2ArB7dgJS5wEALjvar2704uTAaEFRqdR1T4JMNcWSGrCuE0xa\n/k0tlUTgIBCmCSTzZ3rxh3cKt+0u6zWGYDlcXNv9/RePXazhe3qYAJbnRT1984ykSDlFIYQg\nLkz2+Ibc6+fHA0B2YiivYyVDUxlx/ZPGRzfmKT2SNW5ZmerNhnNI9F46vGLsddMwyc8I5zXI\niFFLEyKI31Xw0am1/P6dwo3P7T1W3I48Pqy3r0oj6gaB4C+kYv7VGpl4lGoyIUBAcfO9xPNH\nczhBHCC+allBFFCTInA4W7HpGDbsxaZDYK8CGFYfNGyqGaRuuOI9ZwEwyGJR7Ep3mUIWg+Ku\nAgDcUcN/zM56kIZOMXUDAADzdG3vg+OvcSYQCFMPksERNFyu6/n4WF1jh1EpE87NCL95eYpw\nhPkFF2s0Ry61uQWdLH7589IXn1w2fiP1RV6iKu9OlZPlOAwDxx8bJrs6P/7AuSa3/besSBmY\n7xAfIdv29Mo39ldcqNYYzY7EKPmWFamLckbvDeGjjmBEHhyehCvF96zNfG1v+aBj0ujJm2dM\nz+YjQY1Gb33i38e69bbelxhjABAyVJhSnBSluGlZ8tz0cL8OkECY1szPjPC0YQaABdl9aWjn\nKjov1XZb7WxKtOKqObHExzdYoDLWcNoG3DHARxNRVPZ1SJU0qsNJkXQ+Np0epCxQEiRdNNaB\nDgNsOQO2qr4XbA92tIC9DsmuGrpJrY2vhhcDsBZw6EGgRLErkDIdd5zBVg0SyECZjsLnAqIA\nvE/4MQcYB5G+gdvLccM5MHeDNAwl5qMofldUpIwAkQxsPKlbKCplgsdIIBACBSJwBAdvHqh4\n80Cl62VhVdf+s41/fXSxSj6CktELVfyZGpVNOqPFMeq6iVHA0DzSzPc2z1QpRLuO1DpZDgDE\nQvrONem3rkxz200pEz5x04zxGkmkl1+giKGUY/6F3LE6PTUm5O2vKmtbDQKGyktW3X9tVmrM\nKJNNCH7kvYPVLnXDhd3JXTMv/q6rM/wyJAKB4OK2VWlHi9vaugcVqqTHKdcXJJmtzt+9fX5g\nbeNbX1Y+d3c+8RUKDiiGWvQQbinCHSVg1YMsAiUVoJDY0R9QlIGYcGwpAVYLSACCSCTOBTTx\nTVIdLf3qhgtnJ7aVInFfqzgwabCxDYRyFBIzqG8r4lvNQoP+AVksSrnRU69AEcm47CjPu8OT\ngAoSmQ9j7sw7uPZEf6DmGEpbSs2/nScDhaLpBevZo++7hVFkMkohzt8EwnSBCBxBQFWz7q0v\n3e+LTZ2mbV+UPXPbCK7XVrvX9DyrnZ1MgYMXIUM9eH327avS6toMNE0lR8klogn/fCpETHq4\nvKrL3a4sWS3jMIzdJnJRTuSinEiMgWRtBDUXqvnFwQvVGiJwEAh+RykTvvjksjf2Vxy51Npt\nsEWEStbOi7t9VbqQoV748JKbc1N7j+VXb5zb/sxVQzb/IgQIKHY2ih2/2SmtQvKl43a04YEd\nDfwb7A0gngkWLXdpJ24v6QuK5FTuRhR/pTxHHM3/XkYOgiGWTFD2Mjj/BZjcW7+jeTcMc+R+\nB9efHqhu9AWrj+GIDJTEU8FEFdyEWSd35jNw9hUao5TZ9LpHgkbQIRAIY4YIHEHAkUttvSnx\nHvHWp2+d5WkH4I34CBlvXC4RqEbY1GPikEsEM1JGYx42auYnqELETHGb3u7sz+Qs7zA09Jjn\nJ6rilfxtcUcEUTeCHW/ioA/RkEAg+KBLZ33/m+ryRi1CKDNBedtVaWEhYzJBUEgF392U991N\neQ4n52oQZrE5D15o4T37qdL2lbPHkAhAIIwIzksze2wBzsme2AqmTnA5hdqMXOE7FMWg2DkA\ngKTJWBIPFvcaXhQ2DJlGKKVv/BH39au47UoWsFhOLbkdpS0Y9Y8yyeD6s17ip3kFDkCIXnYr\nNWctbq0CuxVFJKDI5AkdIYFACDSIwBEEdBvcc+N7sdpZk9U5/MyLFbNiXttbrjPZ3eLrFyXS\n07ilJUKQFamIUoj3lbVzA4Qki4M9VqNZmxWplk588iohsEmIlLllv/eSGEn8YgmEEVNY1fXL\nHecs9r6u4SX1PfvONP36vvmz08ahpevA9uftPZbemkdPmjpJgxXCJEJ5WSxBEtxcCKbeJKNB\nT2JcxT46dk7fu2NvxF2Hsa64zz2ElqLw5ShkeOW6qlhq83O4sx56WkCqRJGpIByHlZtJA5vd\n00/64t0N4Nk85gpIrkIZQSPiEAiE8YUIHEFAWAh/eoVEyMi8WMfzIpcIfnXf/D+8U9je07+S\ncHV+3L3rMsc6xKDCbHV+drK+qlkHgHISQ9cXJIoEdHmHgfNIk+EwLuswLEkeh2duQlCzcXHy\nmTL3/sQUhTYsHpXRHYEwjXGy3J/fK3KpG71YbM6/vF+040erxldt91Hn6K3xCoEwESBBArbX\n8kzIhQlY6+HN0YuhHVh7nxkHJUaR16Cw5WDXACUEoXpoa9LB50cRyRCRPLrB+xckkvE3m7EZ\ncdURlL5iksdDIBACH3KDDwKWz4x59+tqzyqVFbNihl+f0ktukmrb0yuPXGqrbzPIpYI5aWE+\njNa6DbZLNd0avTUuXDY3I3ykTVsCk/JG7S9eP+syjDxU1LLrSO3vvr2wx8LfFFZrHlOzWMLU\nYFFO5HduyH1tb7nN0VeTIhUzj23My0kkPoUEwsgordd26aye8fYeS1mDNi9ZNY7nilJJ4iNk\nnskaCKH8DNL5iDCJCOJAmA72wVoGE45EuRi8CBwA4PbgR0tAEj8hwwtgUNws3FnNu4mrPEwT\ngYNAIHhABI4gIC025N5rMnfsG9RwNClK8eD67FEcTSSgr86PG3K3j4/Vvba33GLrW2SLUUuf\numXWuOQP+xGWw394p9CtHUaH1vLHdws3XcufxkLsMwi93Lw8ZemM6DPlHR09lmi1dHFe1Ih6\nGBEIhF66DTzqxpCbRs1jN+b9bPsZlhs0UbxxSVJSlGLcz0Ug+ABJF4AgDttrgNMDkiJBPIjS\nABAKTeDPUJBHATMJdxkMZh2IFQHrwYnSV0DpAbC5m8EDABg6gLi4T20C9WNJCHCIwBEc3Lkm\nPT8j/ONjdQ0dRqVMOD8zfOOSZMGEpVQcvtj60ieXB0Zau80/f+3sf59eERkaTKWbbpTU9bRo\neJwUqlv0TquTt5QzgkxiCVeIUkk2FJCaFAIAgJPFAMDQ5MF6xKgVXs1EfWwaNfMzI/7x3SXb\ndpddruthOS5GLbtjddo186fdMjghIBDEIoG7tS2KnQOVB8DU5RanMtdO7GAcFq74C1xzHJx2\nQBSKzqbmbgZF1MSedBTQAhSbh2tP8W1iiLpBIBA8IQJH0JCdGPrjxDmjfrvNwV6u62nrNkeq\nJHlJKt8dWHcdqfUMWuzO3aca7luXNeox+J12rRcbcwAZRVkpGNhIBQCENJVDVvkCFZ3J/r+D\n1WUNPU4Wp8WG3LYqLUYt9fegCFOfU6Udr++vqGvTA6C02JD7r80aWOyAMXx5vulkSUdvcd91\nCxMmuS1U4JOTpApXij2rVKJUkuyJqfnKSgj98yMFLIedLCcSuK8HVjXr959tbNaYwkPEi3Ki\nluQF3gSPMLWhBXTBd7iLO3FnWV9EKKNyNqC4/Ak8KefkDr6Ae650ZsEcbi1hu2rpld/FVgtw\nLApLAKlyAgcwElBkFq/AgSKD+ImUQCBMHETgmBacreh84cNLLm/RsBDx45vyls7w0lkdoLbN\nwBuvaeWPBwshUq8dZyKV4rlR8rMNPR3GvgKWCLlofoJKJiTfkUCkvFH77LbTxivOKeWN2i/P\nNf3krnwyOSFMKB8drd36acmVV7i8Ufvj/556asusaxcmAIDNwT63/UxRtaZ3c0l9z4FzTbes\nTH1ofY6fblbbFgAAIABJREFUxhuIMDT64W1zfrHj7ECfUYmI+eFtcya0nxdNIdoj2/mtLyvf\n+rKSu1LAsud045K8qOfuzmfoqeA5RQgapGqq4GEwtmNDOwhlSBk/0cUpuPZUv7rhwmFhdz8P\nehMAAKLQjNXU4ttA4P88VpQ0H1Udxpq6QVFGhGbd4J8BEQiEwIZM3qYUVjtb3qjtMdjiwmXp\ncSG9FqR1bYafv3Z2YKs8jd7627fO//2xJd6WywQ0xZvqIAjyZ76ZqWq5RGD08BNVh4gy40MZ\nGq3JjLQ4WKPNKRMxUo+FPkKAgDH85f0it7+j3cn97YOLczNWSYgmRZgYjBbH9j3lnvGXPy9Z\nNTdWJKDfO1jtUjdcfHCoZn5WxNx0YmnZz5z0sO0/XPneN9UVjToAyEoIve2qNLWXfmETx6Wa\n7jf2V7gFj19u33m49vZVaZM8mAnBqscmDZKpQRwoS/EEX8ijkHySNHrc4f7J70Nw5TEPc/jS\nl5xZR137xOQMyReIoq56nLu8B1cd6SuoicxAc7cgZYy/R0YgEAIRMhOYOnxd2PLyZyU9VxIQ\nshJCn7plVkq04qOjtQPVjV5YDn94pOand/EnQM5IUR+/3OYZn5ka3LnWEiHz+Ka8P71XxA0w\nnKMp9P2bZ7pq6SUCWkKkjcCmvt1Q387jN6Y32y9UaRbnkiQOwoRQXNvtaqMzEJPVWdagnZ0W\n9k1RC+8bDxa2EIHDjbAQ8WMb8/w7hq8Km/nj55uCXuAwtHMXPsCdlb2vUFgqmnMLUsYCAFj0\nwAhBMP52J4Rggh1WhzhcfQZ3NyF1AHjWMGJq9k0wexOYtSBWAEXmLwQCwSvkAjFFOFXa8cd3\nCwdGyhu1P3rl5LanV3qrK6lu0Xs72j1rM85VdLo9ysdHyK5bmDAuo/Ujq+fGJUbK3zhQWdGk\npRDKTgi9d10msdMPLroNNq+b9F43EQhjxGzjUTd6MVmdAKDR8X/89p9tZGjq/muzFN6r5Agu\nOrSW7XvKL9ZoDGZHUpR8y4rUlbNjJ8JJsNOLK5OrnDNYsWjZQy+Avb85LtbU4MP/pBIXcxVH\nertRIHUCyt+CoomFwXQlJAp49T3Wo6NLew0EgsDRBwLpeHaSJhAIUxIicEwR3jvI0yRca7Tv\nPtXorarZR7VzWmzIXx9d/K+PissbtQCAEKyaE/fw+hxPe7ZgJD1O+ev75vt7FIRRYrE7PzrK\nY4LbS9ikZ7kTghvOApSYr4ESD3HhXl1s4yNkABAqF1q6nZ5bOQyfn6wvruv+1xNLp8ZVdOKo\nbtE/tfWEq0N5RZPu9+8UFtd1P75pxrifSyEV8saVMv54sMBVHhyobvRh0nOX9rhe4e5G/NUL\n1IqHUcLozcuDBYvdeeJye1OnSR0impseHhcu8/eI/A+VsoQtPwicx/XKQhYJCARC0EMEjilC\nZbOOP96ky01SldT3eG7KTfKlgmfGK//1xFKdya7RW2PCpMTXYCpR22YoqtboTfakKHlBblRw\nzbj+9sGlU6UdvJuUMuEcUghAGA6cDZvOg7UasAMQDaJEJFsA9BDTnsx4ZXpcSFWze+7bjBR1\nYqQcAJbNjP7gUI23t9e1GfaeabxxSfKYRz+V2fppiUvdcPHp8fpr5idkxo+zkcTi3Kiv+apU\nluR5deAODro9JGCOA6d7pSpgjju3k06YPUyBL0g5W9H51/cvavR9XXsYGt16Vdq912QFYndR\njCev6akiglrybe7sO2AdkORrtoHNo3QlMmWShkQgEAjjBJm1ThG83RMpCm5ennLgXJPOZB8Y\nl4qZ21alD3lYpUw4xrUsg9khEtJCZrjupFY7+/GxutL6HpuDTYsNuWlZSrhyepUKG+1OvdWJ\nMQ4RCxQ+u/mOAo7DL316+fOTDS4Xkmi19Ee3z8lLDo6czy6d9fBFfpsDIUM/fetssTCYxBqC\nf8AOrN0NTu2VlyxYa7G9Dak3AuWr0zBC6Lm75/3y9bN1A/pMpceFPHtH3xr4nWsyCqu6PBUQ\nF0VVGiJw+MBic16q7ebddLqsY3QCR2FV13sHq+vaDWIBMzNVfe81ma57yopZMd8URR8rHmQ4\nlRApv/vqjFGcKIDgPGqpOA91oxdjFxg1IJ+yunB7j+VXr58bWG/rZPE7X1VFhkquX5Tox4EN\ngmNxzSGu4SSYu0EkR1EzqOzrQDjyNBOnDRfvx03F2GpAIVEoZxVKmOVjdxQ3k478OW4qwsZO\nJFbilkpcfMh9n5R8FBb0tckEAmG6QQSOKUJ2YuiFKnf3fgDISVSFK8W99SYue//cJNUTN82I\nDfP1KD9GOA5/frLhfwerunRWikKZ8cqHN+TMSB7Co7Sp0/Sj/55y1UWfr+z6/ETDT+6auygn\ncuKGGjiwHC7pMLReWWgCgEi5KC9KMY7Na979uurT4/UDI23d5l/sOLv9hytDvGRrBxS1bQbs\nUSDcy7evz5omnxPCWLGUg0PrvmjNWbCpCCkW+35rbJh06/eXHypqqWjSUQhlJSiXz4yhrpT7\nycTMPx9f+vGxuld3l7EczyeV16OU4MJkdWIv33CDeVieiG6881XVjn2uxje2Fo3pyKXWvzxS\nkB6nBACE4Of35O8/27TndGNzlylcKS7IjbrtqrSg10mVcaAd3AHUy2UTALDTHoCpDOPFvjON\nvF+6T47VBYrAgTnu5H+w5kqVsVWP64+z7SX08u+DOGQEx7Ho2N1/An1feiM2dOLmYpSzmiq4\nw9e7BBKUUtD7AUBpSzlRCL6wB1gnAABCKGcFteyuEf9EBAKB4G+IwDFFuGtNxqWabrdH6shQ\nybULEgAgMVL+50cKunTWtm5zpEoSGSqZ6PH8Y9elvacbe//PcbisQfvMf07+4lvzCnx2uPjr\nB0Vurm8Wu/PP7xW98eNVUvHU/6xebje0GawDIx1GG8vhefH83XxHCsbw2Yl6z7jebD94oSUo\nFpZdzW48USumV6YPYdRgewt/Sr6dPznIDZpCq+fGrZ4bx7uVoaktK1KPXGwtbdB6biV+xr4J\nlQvFQtpq55mRxoxckW/RmN884N4L02x1/vOj4n8+vrT3JUJo3YKEdQum1Bo1Sl+JG88OyuOg\nKAA+cY0WoKmbvgEAjZ08/bYAoKHDOJnlID7ATef61Q0XVi1XsY+adcvwj8Od+8ilbvQfvPRr\nnDwPRWcO6xAUTRXcArOuwV0NwDpQeBIowoY/AAJhQkBBLjcT/MS4rQwT/MvstLBf3js/Wt3/\nCJifEf6nRwoG6gLhSvGMFPUkqBs1rXqXuuGC5fB/PivxtvwOAB1ay+U6Hq8Qvdl+rrJzfEcY\ngFidnJu60YvGbDd4VKSPDpPV4a3/SANf19UAJDM+VMBX7kRRKFiqbAj+B3v5QnmLj5wblyZ7\nBgUMFSiLxoEKQ1O8ypFEyKyYFTPSo50u6+DNoylr0ProxDQFQMo4quDbIBmgjMtUIOYR11D6\nUmCCIHdv1AgZ/tmRkKEDQd0AANxROqK41+PUF3qJnx/ZgKRKlDgTpeQTdYNAIAQvU39VfPqw\nKCdyXmZEfbuhW2+Lj5TFqCewAsU3vMUyANCiMbf3mKO9DKxLxzO976VT63XTlMHoXcUw2pzj\nYsYhYCiEgFdjChafUZmYuX1V2psHKt3iNy5Jnm5eLZOAzmRv7DBKxUxChJxXVwpWGCU42vnj\n48TquXHtPZa3v6y0X3F2VMlF398ys7fZyljQ6K0tXeYwpShaJaW8d8IKXh5en9PcZXIVVAKA\nTMw8c9sctWLEDZJ8VLUYzPZRHDCIQNF59NqfYk01mLpAFobC0rCxizuyDXSt/fskL6DyN/tv\njJPB7LSwA+eaPONz0gNm9u708njjGEmvYo4Fu5l/k8WrJRCBQCBMVYjAMaVgaJQWG5IW6+dh\n8CYY92IZvMlocVS36DGGlBhFqNzrOlKoXOhksUZvDQsRMeNnSBFQ+JiqjNdCk0hA5yTyt9QJ\noKe9obj76swQmfD1fRVGiwMAJELmzqvTt6xI9fe4phQGs+O/u0v3nWnqNURQK0QPrc9Zk89f\nlBF0IEkWtlR62hIgSc44nuWO1emr5sSeq+jq0lvjwmWLc6NkY6uza+s2//vjy6fL+rLQk6Lk\nj2+aMTstaL65Q9Labd53prG5y5QUqchNUhktDqPFkRSluG5Rgko+GjEiUsUvetIUCldOeCaj\n/2GEKKr/I41C4+j1z+Gmi6BtBkYEURkoLNl/g5skVs+N++xEfW/DexeS/2fvrQPbvM737/s8\nYrAsy8yMsRPHYWZu0kAh3dqVeV1p7bZ36/A3+G7tOiituLZrU26SNkmbNNiwE8dJzMxsi1l6\nzvuHElmWHhlkke3z+Ss6ouOIzrnOfV8Xl33X+uxATckZIXOLEBKNpXWIYgE/ZEgeih3xCN5n\nBAKBMPkgAgdhXCi1psv1/b0KfbRMOCszQsBjA4C7U0oOm4qRXVtWWqz4f9/VfHa8wXbCyWah\nrYtSU2JCHOMJbPA57DPl3c9/csVipVkUmp8X/dDmvOiwybY8lfA5FEK0S30FApAKON56lvs2\n5jz7+jmLdYij/uysyDnZE8aeEyG4cWHKDfOT2/u0VhonRoomn+Zls8uJlAqipAL/11FjjJ97\np9hRCBtQG//vo1Ia4zWzEvw9G1/AjkCSxVh9DvD1bCnEQqJC4CV793liZMJN873Tk6I1WJ5+\n7ayjRVFzt+YXb55/8ZEF2Yne8egJLPvPtbyyp9zkEGW6cFrML384czylQzZRSWtwLo4bv9g0\nUaFYKGkmJM0M9Dz8B5uF/nL/vPcP1ew722I0WxFCM9Jlj2yZlhITLG44KHEObj7lWlqJkuaN\n7XHS5uKKwy6jCKXOHc/0CAQCYSIyJX/jCV7i0MW2V/dW2A7SAUAm4T2+vWBBXvS83KhIqcDJ\nLhQA1sxKEHCvveX+81XFntNN9qssVvzZiYa5OVE9Cr3OYT1KUYjLQcevXKuqtdL4VFlXRZP8\n5ccXT7KWBDaFUmXC+n6t03iiVMB300XsAfmpshcfWfDq3orKFjnGIOCxty9J3bkiPUi6kUcP\ni0JJUeJAz8ITLtf3V7bI9UZrepxk4bQYJ9vUlh6NY+BRdqL08e35tsQHv1Fc3ctY5vPewZrV\nRQkT7q3CDD8DcePB2IytasQSATcRWMGy4WFk39lm129Ui5X+33e1f7h7TkCm5EWautT/+rKM\nHuqXcbq866OjdXesGZ0/IhMSIfdnOwv/8lGp429KWqzkx9vyPZ8rYaIh4rMf2pz3wKbcXqVB\nKuYGWz8mkiZS+Tvoij1gHeyoQskLUPKiMT0OVXQj3deEexz8ShFFzdqGwonvD4FAmHIQgYPg\nIZfq+v728WXHkQGV8Q/vX3z58SWpMSG/u3P2nz4saesd3K4vyo95aHOe7d8KjemrswxxHsXV\nPf94dOGhC+0VzXKDyZqZIBELuPtcbinXGD8+Wv/o1mne/psCTHq4iMOi6vs1ZisGABaF0mSi\nlDAve6lkJ0r/8ehCg8mq0ZvDJfxJsl+dCOgMlj/vunSuctDoPjla/Kvbi+zJGn1Kw1OvnFHp\nTPYbVLcqnn7t7CuPL46PGK93w+ipYor/AIBuub5fZZg8wiIlAEHORHn7M2ayAACjMfOE43BJ\nO83kBnrwQtt4BA4AmJ8X/fZPl+8909TUpeZz2dPTZOvmJLImo3cJYXgoCgVt4SdKWciKysHt\nJVjTA4JQFJ2PwsZeTcYRUBt/hmtP4fYy0CshNBblLCfqBoFAmJoQgYPgIV+ebHIdtFjxnlNN\nT+woyIiXvP7U0hNXuho7VUI+Oz9FVpA22Aha265kXM5iDD1yw0+2Dx6vPfOfs4zPXlrP7GPq\nLS7X91e1KqxWnB4nmZsT5TcVIEkqSAwVaM0WjEHEZVE+e2I+l8XnBtdB1qTnpd1ljuoGADR3\na37z34tvPL3UVof/5clGR3XDht5o+eho/dM3T/fbPF37pEZzFcGnMH5hwmR5RbrkzH6KPQo9\nTeNxeqnKJLy71gWN4QKBwIhQhjJXj/f3HiGUtRhlLXZzNQZ9LzbKEUcCwiiSvkkgECYxROAg\neIirWYbTOJtFrZwZBzOZLE/dr8nx0PW60czsV2pyMz5+VDrTnz8svVgzGEybmyT95e1FfojX\ntYEQiLnkgznZ0OjNR0s7XMc7+rUltX3zcqMAgLExBAAq3Yx7THuftrpVaTJbM+JDM+IlTtem\nxzmP2JCKuRGSyVK+MdFIj5OcqWBIfsmI82v7ko8IcWMzJOJzJmVSDIHgb3RddPM+0HaAbf3F\nC0PJG5AkPdDTIhAIBJ8w2cz5CH7DyTvAYXzkN1V6vAS5qU3IShyyXnfns5AU7Sv/hec/ueKo\nbgBAZYviD++XTIqDUkLA6OzXWd0cwrf0aGz/cHdK7+6OHmCy0P/84uo9fzv+l12X/v7ZlUf+\n+f1z7xTLNUbH2yzIi2b83N26PJ3sNgPFpvlJYiYV4Nblk2GLYhP4Rj9O8DJW09hCSScKVhPu\nLMe1x3HbZbcpqlMBs5quft+mblzDKMe1H4G2PXBzIhAIBB9CBA6ChxSkMmePObaiuEMWwtsw\nN9F1fEVhXFz4EK+BzQuSGfdUNy5KGdUsx0iPQn+W6Zi0ulXhlDNHIIwJnvuGIHuvUJqb0gl3\nJRUe8Ore8n1nWxzrpM5V9jjpd2wW9cd75xZlDoYU8jisu9Zlb19CsngDRriE/+f75trtWgAg\nVMR9dmfhnJzIAM7KW8zNiVpSEOs0KJPw7hl3lqfJQh+51PHON9WfHKsn3+Gu4N4q+tSL9KHn\n6MO/oU/8BbdfHK7AckKBuyqt+/9An3iVLvmUPvWGdd/vcHNxoCcVGHBPMVgNLqM07jwViOkQ\nCASCzyGV8AQP2bky4/urXfYIFRsRofxti1NGc/dHb5zG57L2nm6yWDEAUBTaMDfR7kJqJztR\n+uytM17aXW5/Ij6Xdf+m3NlZPlnWt14/S3eluUedkzQZ4hgJASExUhQRyu9TOq8yEUIzM65J\nCVsXpR660OaYlAkAbBa6eZl3lAWN3vzN+VbX8bLGgcoWeV5ymH0kOkzwl/vn1bUrm7o1Ij47\nO1EqC+F5ZQ4Ej8lOlL725JKKJnlHv1Ym4ecmSRlrOiYiCMGvbp954HzE12ebW7o1Mgl/Tnbk\nj9ZmhYq443nYqhbFnz+81DkweHS/qij+qZumjyd6djKB287jss8GL+sG8NWPQdeHMtcFblJe\nQtVNf/8foB0Sgk1a+tz7FD8URY/LtnZCou1kHMa6DlKSRwh2KGIWQ/AEInAQPCRWJnzxkYUv\n7ykrrbvm97lwWszDW/IkwlEtSTls6qHNeTcvS6trV9EYZ8RJIt2YXKycGT8rK/JsZXfXgD5K\nKpibExnuMyOA4Y7ZgyxbjjCxQAg9vCXvD++XOI3fuDA5IfJa1VJytPj3d8/5x+dXu65vycIl\n/Me2TctO9I6y1tytcdft0tCpchQ4bGTEh/onodZipbvl+kipgEt2nsPColBBmmw0VXITBYsV\n27odEUIb5yVtnOe10AetwfLr/xYrNENcew+XtIeJeQ/ckOutZ5nA0BZcvd91GDccRYnzgT+x\nvV3ouuND1A0bmMY1R6aiwOGOSVKsQyAQCM4QgYPgOcnR4r8+MF+jN/co9DFhQiF/zG+ncAl/\nNGpFqIi7bjZDS4vXyYwPFfLZOoPzwojNQgWp4X6YwFTAbKG/PNlYXN3brzLER4g2zUuanxcd\n6En5gyUFsX97cP7r+yrr2lUY44hQ/g9XZW6cN+SNXZQZ8cbTS8ub5F0DuugwQV5KmGAcjrMm\nC/3N+dbaNiUGnJkQmhbjttWFTQVGWehTGl7fV/n9lU4rjSkKzcuJemhLXqzMy9HIhGBjQG18\n55vqsxXdKp0pOky4cV7SjiWp3i2sOHm100ndsLH/fMs9G3LceUhNIVQdYGaypcA0ljeg2Jl+\nn5BXUTA4OgMAVkxJ1wlRLKgaXIeRiMkDnkAgECY+ROCYurT1ahu7VEIeOyM+dDyVwGIBx7VS\nGmN84kpXdavCZLFmxoeunBkfVFXBerN1QGey0lgq4Ej4g5PncVj3rM95aXeZ0+1vWZ4uk/ik\nRL+8SX78cke3XB8tEyyfEed6ij7J0OjNP33tbEOnynaxrVd7rrJn47ykJ3YUBHZi/mFGevjL\nP1lsNFuNZqu7Wiceh+Xof+Exzd2a594ptheDHLzQFh0m4HNYBqYEomkpAXjjqXSmx18+3au4\n5m5I0/hMRXdFs/zVJ5ZEhJK4lklLj0L/2L9O2a1tuwZ0bx+ouljT+3/3z/Oii21rr5ZxXGew\n9KsM0WF+SsUKWrCVQf25hsX9VaPEpAVEASdw/8nuytqDudzdrMMdxaDpBjYPpCkoajp4KSce\nRc3BPRedbTgQhWJdAmXVrVhZD2YdCMJRxHRgT/WPCYFAmKAQgWMqMqA2/vvLslNlXbaLfC7r\nB6sybl2e4aUfU+hXGX777kVHR7ddR+p/c+es1JiQYe7lHzCGq53Kym41jTEABkCJUuGcpDDe\ndf1ly8JkmYT31v6q9j4tAESE8u9cm7XWB/UjGMPLe8r2nm62j+w+2bR9SeqDN+R564XwG1Ya\nn63obupS87isglTZMC0V7x+qtasbdvafa1lSEDPLN74qQQiPw+L5uOMJY/jzh5e6Boac0HbL\n9bIQnqvAsWZWQqKbuCKf8uXJJru6YUepNe06UvfYtnz/z4fgH979tsYpuAcALtf3Hy3tWFUU\n761n4Y/C1ncqg0SRGBBjlwISexxeg3HrBbr6G9DLAQBEkVTeDShm7OI1NmHdVTB3AjYBKxQJ\n8oDj7EE7AhFp0F3tOowigjR1CPfX4PKPB2tq2s7i0NPUjDuB441yNk4IlX0H3fz1oBkHLwwl\nbQDHCg5sxfV7ce/lwYHW4yjjRhQ2XqNfAoFA8D9E4Jhy0DR+7p3i2jalfcRgsr59oBoh5K3E\nwb99fNnJr76jX/uH9y++/tSygBcGX+lUVnTZN9gIAFoVOoPFujprcEm3OD9mcX6MWmemMR6n\ny90wHC1td1Q3bHzxfWNectjS6WNczAWU5m71nz641Nilto8sL4x7+ubpjHv4768yu519f7Vr\n6ggc7sAYSuv76jtUfC4rLzksLdbz8JS6dqWrkAQAA2rjzhXp+8+1qnQmAOCyqZuXp/9gZYbn\nkx4HVxr63YwP+HkmBH/ilMNtp7i614sCR2FG+HsHGcbTYiW++1afSPBDUVQu7qlwHpfEgTTZ\ns4ekqw7g2u8GL2t76eJ3qIIdKGXRGB7FqsbKb4C+Ln1aNdjUDsLpSFg4+segMpdZG06DXjlk\nlMOn8oLSP9Wsw2W7wDK0wkLZgqv3ovyd3nkKYQyVey/oerBRjrgSEEYDGvIDjdu/d1Q3AAAs\nOlzzGSr8MfAmtiELgUCYghCBY8pxsabPUd2w88mx+h1L0sYvQHTL9SW1fa7jbb3assaBwgzf\nOlmcq+z54HBtY6eaw6byksPu2ZDtuEu00ri6R+16r16NsUdjjBIPaUIJEfo2oeC7i8zNwIcu\ntk0ggcNkoX/9zoXOoZUCx0o7xHzOT7YzHMIzdsUDgFztfKI71ejo1/3frkuVLYPK4NrZCT/Z\nXuCZ72aX3Lkywk56nOSj51a392lMZjopWuzrWpJhMJlpN+MMTTSESYPWxeTo+riZcdwz8lNk\ny2bEHr88RFFls5BrVteUBRXcAqX/w/11g0OSeKrwdkAe9ZMaVLj+qOswXbmPlTgHWKMVlbC2\neFDdsKO7CtxkYI+6k44nZq16ii75FHdcazhFEWlo1i0Q4nFxig/BvRXO6oZtvKcMWY3A8lZ7\nLAJhNBIyO17hbmcDbAAMtBn3XUHxS7w0AQKBQPATROCYctS2M6gbAKDWmbsGdPZAB4/p7Gfy\nLQMAgPY+rU8Fjg8P1/3322tVqUaz9XxVT0lt7+/umjMn+1ppgMpgdpciIdeZnAQOX9PtUpx/\nbdz91jQIKa7qcVI3bBy80PrADbmupeDhEh7jHzjFPRcsVvyb/xY3dw9JKT54oY3HYXnWrBHi\nPkA0RMhls1BydOD7xZKjxU6lXtfGfdPLZqXx/nMtxdW9crUxPkK4aV5yQZpMa7A0d6sBICU6\nxAObZIIHxIULHQu+7MRHjPfXx4mf7ZyZlSD95Fi9UmuiKJSTKH1ocx5J+x6EI0RzHoC+GlC2\nAG0FSTyKyvNQ3QDAAw1AM0mTFgNWtKLw0dWHYiuYGP1BMZhaxyBwAIAonFryEJh0WNOLhGHA\n97wgzucY5Mzj2AoGJYh8L8rQZjC5VvwhAAA9c50dgUAgBDNkPTflGMbfwSvWD8NsEkS+3D/0\nKvT/+67GadBixS99Wfbfn62w/WkomMwt3G1BR5mzGyS09GgYx00WunNA5+q6sqIw7qOj9a63\nX1EYADt3s4WubJF3y/XRYYKcpLAAZpRequ1zUjdsfFPcet/GHAFvzB+c3GRpiJCj1jkfiYsF\nnID4iTKyZWHK4ZJ2V81x66IUbz1Fn9Jwqa6vX2WMCOV9ebK5tu2anlLdqjhyqSMvJayuTWmy\n0ADA47BuWZ5228rMgLfRTXrWz018da9zZwRFoXVzvGx1xGahm5el3bwsTa4xCnnsABYrBTMo\nIgsivJGcanVfgDPMVU5gEwBzYRemDZ58MrlCJPOw48Z/DOPl6RUPjhGh2IBYgJn0Ka/VjxAI\nBIL/IALHlMOdAaRUzB0mnRHj0cof6XGSMDHP1UOOw6Zm+LJ841Jdv8XKUJ3ROaBr7dUkRYkB\nQMJnc1iU2cqwfooQ+ftXfH5edEUzw7nNgrxgrKF1xzB7BsarbluVUdYoL2saYrJw28qM/FSZ\n9yc3LCW1ff/8/Kq9/CRGJnx8e763fEBUOpPBZI2SjtaCvqmb4UAbAMwWur1PmxE/5hZoHof1\n6I35f/24lHaQDygK/XjrtODZ5mUlhP5sZ+FLu8tthiAAIOSzH9iU65UQGQD47ETDu9/WGN03\nvFSEHJxkAAAgAElEQVQ0DX4AjWbr+4dq1TrzIzdO88qzE9xx48KU5m7N/nMt9hE+l/Xo1mm+\nc6EO82913hTFvTUpChl1EDjFc7fTRqxRbfVpGnf067rkurhwYaxMFEyHGm5B4Vm4bj9glwWM\nJAG4/rF/Rig0DStqbc7rQ66RBqktK4FAIAwDETimHDMzIvJTZWWNzjZ+t6/Oco3o61Ma3j5Q\ndbGmT603xUeIblyUsnFu0vBJfiwKPbp12v/7n3M/54/WZnmwytSbLAfOtda1K9lsKidRunZ2\norvzVY3e7RmR/SoKoWkxktJ256r4+FBBuN9t57YtTjl5tbNmqB9KbpJ088IUP89kPMxIZxat\nosMEjHqZgMt+4eH53xa3na/q6VcZ4iNEm+Yn+7+moLFL/dw7xWbLoNTVNaD79X8v/PuxReOx\n9gSA0+Xdb+yrtEXwSITcH6zKuHFRCmuk8MthQpQ5bA/1iJUz4+IjhO8drKluVWLA2QnSO9dl\nDRNwExCWF8bNyoosru7p6NdFhwlmZ0d6ay96rLTj9a8rx3qvvWead67MkIWQ/bAPoSj0xI6C\ndXMSz1V29yuN8ZGiVTPjIketBhKCEyRNQtJErGh1Ho+eBoLRf8NTwE0Bo2uVHwXclBHvXNEs\n/9cXZXZ/5dwk6eM7Csb5lT5eDC1g6gVAwIsGnhsPXVEUSlqKm48PGWRxqOwtfpigDZS8Gqtb\nwDrkaAqFZaGwTL/NgUAgELwFETimHAjB7+6c/dpXFd+VtNkODMQCzl3rsrcsdC7jbOnRPPHy\nabs60Nyt+dcXZaV1/b+6vWj4p1g6PfYfjy5860BVdYuCxjglRnLHmswFeaM+w7lOVYvid+9d\n7FddM9/65nzrF983/vHeudFhDEvh2HDm4x2EIMZhp50bHUIhuNqpstVxIAQZEeIZcQHY9fE4\nrBcfWfjZiYbjlzs7B3Rx4cLlhXE7lqSyWQFrlPCA9DjJ2tkJBy+0OY0/uNlt2C1CaP3cxPVz\nvZ+8O3q+ONHoqG7YMFvoz080PnPrDI8f9tsLrS98csV+UaUzvfZVRWuv5vHtIwQlTk9jLmCR\nhfASx2GLk50o/eO9cz2+u38IEXJWzvRadoad3aeaPLgXTeOaVsX8sX9ZEcZKbpI0l9hhTCYQ\nonLn0Vf7sGbQZQmFRVCFt47tYUSzsVUOFsczGAqJ5wFrhFqG5m7Nz14/51ixVdmi+OlrZ//z\n5JIIKcdMK2lsohCbhcQs5Bc1zarBfd+C0cFShJ+MItYCxWA4hTLWQ2gSbjoG2m5g8VBYKkpf\nBwI/FjYKo6npD+Lmg1jZAFYT8EJRzDwUO8+5oINA8DNUsNScEiYWROCYioQIOc/cOuOeDdmN\nXWoRn5MSIxZwGd4Jb+6vci2LOHGl80JN7+yRKvnzksNeeGgBTWMag2c97WYL/ccPSuzqho2W\nHs1fPy594aEFrrcvyoyICOX3KZ2tyOfmRDmdx2ZHhaRHiJV6s4WmpQIuL3DOCxw2ddvKjNsC\nFNLpLZ68aXpqTMiuI/W2RoO0WMkDN3inywBjOHGlo6S2T6k1JUWJN85LinHfRTUm6jqYrXbr\n3FjwjgYrjd/cV+U6vv9cy/bFqYlRw63O02Ilq4vivytxDta5d2PO8AVTBHe46/oZETc2xAQC\nYVhM3dh0FWUnglIHOj0ABWI+ChGCuRG4Iyi8Q6B4SLoJDDXY3AW0EVhSJMgB1shVGJ8cq3ft\nR9PozZ+eqPrRBgEetPbo5VBhfJbPo8pw3wEwdg0ZMjTj/u9Q5A2Mt0eReSgyoBE/fBnK3okA\nA20GaiJ5gREIBIITROCYuoRL+OESt9EVGOOLNb2MVxVXjSxw2KAoTw3ZAS439DPGbVxtGOga\n0LludHkc1q/vmPX79y86ahyZCaFP3jTd9UHYFPJ/T8rwaPTm9j6tVMxjrE8JZlgU2rE0bcfS\ntH6Vgc9le8tKVm+0/Pq/Fy7XX7NwP13e/cX3jY9ty/eKE6E71WA8akJzt1qpZQjBxRguN/QP\nL3AAwFM3T0+MEn90tF5vtABAjEz44A25i/JjPJ7PFIfLptzmObkHIciID+K0BQIhWME6W4QZ\nQqEiCBU5jiPRWAQOAAAE/GzEzx7TfRgjmQCgsmUAwxA5w0zLKcTjUr6sjzB1O6sbNvSNYFED\nO9AhVhYDbjoJylYADJIElLLYwc0UEXWDQCBMdIjAQWDGZKFda/htDON24UW6B9ympXYyCRwA\nkJMkfeuZZd+eb6vvVHHZ1LQU2YrC2KBKTmFkQGX8z9cVxy532DqGkqNDHts2bXqaDw1Zx4/B\nZC2u7mnv04aF8IoyImz988PoZR7wzrfVdnXDhslC//OLsoI0WVz4eOMkcxKltW0MxRq5SZ67\ngZjMzJ8Xh6toALeKH5tF3bYy49bl6Z0DOj6X5d3/zKBFozf/77va0+XdfUp9jEy4bk6it7q0\nClLDv7/aOdZ7rZoZP3prWAKBMIjVTc2Uu3FvQ7uadLofN9Ny3wocZjfJrwBgHgiwwKFqpy+8\nBabruV29VbjlNFV0F4SlBHJWBAKB4D2IwEFghsdhyUJ4A2rnMBQAiI8Y7/ZyNIjdpKgCQIjA\n7fGCgMveujjFJxPyDUaz9af/OdPWq7WPNHerf/7Gub89uCB44jydKKnte+HTK72KawoUj8P6\n4erMnSu86bWOMRx26dcAAIuVPnqp44erx2t7dtOytCOX2rUGi+OgkM++eXmax4+ZECliUcg1\n9BQhmJlpwfgcgA6AApAilAHA/CGiKOSfz1cwoNSafvLvU/Ygm7Ze7Vv7q85V9vz1gfnjz2r9\n0drMC9W9etOQlzhMzEuKFsvVxsQo8drZCWcrur8pbsMYAwBCaMPcxIe3BLREnECYuFBM1rwY\nAPnJsjcjPtTxl9ROehzDBGjMUG3nTZD71TUV0IU3xvSVXYPqhg2zjr6yi1r6LCDid0AgECYD\nROAgMFBS21fTpkiIFLsKHBw2tXJmnB/mUJgRzuOwXFtqo6SC9LhAl3d6j0MX21zXZBYrfvdg\n9V8fmD+mh2roVJ2v6ulTGuLCRUunx0aE+qQEoFuu/81/Lzi+Lkaz9e0DVZGh/FVFXrOK1Bst\nah1zoVAXU+PSWImVCf/24IJ/fnHVXtWcnSh9fHv+MEnJIyIWcFYVxbv6rT57qzAluvV60LIV\noB9jOUKFAFPdYXHXkTq7umGnrHHg4IXWjfOSxvngydEhLz664JU9FVca+gGARaHVRQn3bsyR\nigfl0QV50betzKhtVwJAVoJ0wnWHEQjBA+InYb1L+gkC4Dv7l/uIm5emnbzaZRkaA8/jUDcs\nYmg6Q+6L6bwDLxYQBdilrI/iAjegHsbqDtD0MIzr5VjehGQkFJZAIEwGiMBBGIJSa/rzh5dK\navsYrxXy2U/umO4to8fhCRVx79uU8/LucsdBFoUe314Q/F0no6eiibmQtaJJfn1LPDIYw5v7\nKz//vpG+Xj7w7rc1j2zNWzfb+0klB863uKpOAPDlyUabwEFjXNmm7JTr+VxWWpQ4zqN3C5dN\nsdmUhalJKtRL5ikZ8ZJ//Xhh54Cua0AfIxPEyoTjf1/9eGu+0Ww9fnmwM2J5YejKmWYAp5eS\nxrgGoWDPN/E156uY1tkA56t6xy9wAEBarOT5h+ZrDZY+pSEuXMiYxRsjE/rnC41AmOQIMkFX\nA8ahlXesEBQyyz/Pn5kQ+rs7Z//ry6t2966ESNFj29Jjwxl6ZNjUCKZI44UlgpAiUF2wD2AA\nBIBC5w9X3OF7sMG9kfYwVxEIBMKEgggchCE8/8llV3VDFsIryoxMjhGvmZXglEjiU25cmJIa\nE/L+odr6DhWLQtNSwu5cl50aE6TlG1YaHzjfcqm2X6UzJUWJtyxMSY4eeQllcZPZYKXx9RXR\nyHxb3Prp8QbHEb3J8uJnV9NjJRnxoaN5hNHT3K1hHG/qUgNAl0L/3rH69uvH8hRCczMjblmY\nwhq1eeeA2vj2gapjpR2M6gYAeJA37A6EUFy4aPyOHnb4XNYvf1h001JFeZPcZLGmx4bOztYB\nuJxqAgBoAIwA/vs0BSHu3Hy86/Ij4rNFfB9vZggEAiAUvhG05VhbBVYlsETAT0HiWf50rJyT\nE/nWM8urWxTdCn2sTJidGMpmUTpLkxUPqRRDwOJSUb6eDJIuALYEK8+BVQsAiC1B0oUgHG9/\n5XhnxRW5zYniku9JAoEwSSACB2GQbrn+XCXDmeqA2rhlYXJOUgAq6qenhf/twaC227Sh1Jp+\n8ea5unaV7eLl+v4D51se3jJt84IRqnPTYiXHSjtcx1NjJaMvKNh/vsV1kKbxgfOtj23zssDB\ndZOqy+WwzFb69UM1/Q5tTTTGZ2t6BVzW1rmjOpBX68yPv3SKMT3Hxqb5SUFrTWInO1GanXjt\nw4Jxo/sbWqa4wBErEyo0DJ3w42kUIhAIgYMCUcHYM1McwEZsqgHrAAANlBRxs4Ea27cBl00V\npMkcZyBkJ5vofhMtx9iMgMWmxDwqCiG3Jl/eRDwNiaeBVQuIAio4OuBCE4EfylCswRWhsNRA\nTIhAIBC8j4+7EAkTitZe5sN5AGjpcXsVAQDe3F9lVzdsWKz41b3ljJ5njqyfkxgiZFhp3ToW\nt8v2PuZnGfHZPaAgjdl5fnpaeHmLop/JlfZ0da/F6vbQyJEvTzYyqhscNpWXHPbLHxY9vn0c\nS+dAgJC7RS0F4O+cFIxxU5f6XGVPY5eadlM65E/WummhWjcnwc8zIRAIgcc6gLUHwVQD1j6w\nDoC5AesOgoUpbHVsIC4VIWZnijk5Yk42nxXvJ3XDDksULOoGACAK5d/sbHSKWGjaDmD597+F\nQCAQfAap4CAMwue4NdDmub+KQNP4xGWGQEqLFZ+40vmDVRnD3Fcq5v7p3rnPf3Kluftan7CQ\nz757ffayGWNwchXy2IyWnEK+9z/ga2cn7j3dbGtIscPnsu5cl1XdqWK8i9FsHdAYo0Zhelpa\n1884nhQl/sejCz2Y7WjQGy0cNuWVaFImIgA4AK6vTjSAXz9T1a2Kf3x+tb7j2muUGhPy+I6C\nvORAlsNsnJdY36H6+myzfYTNQnevz8lP9WV8I4FACEqw4QI4hZtgKzZcQKL1XvGt8LmxaJBj\n0YNRDQIZishCi3+K6w9hZStgjCQJKGMViHzes0MgeAJFdh8ETyACB2GQzIRQIZ+tG5qdCQBs\nFnJ3bj8iFis+frnDZqKRnSRdNC1mEjmEXkNjMDulUdrpVY4c+ZGdKH3tySVXGwZaezVhIbyC\nVNlYfTSLMiMOnG91HZ+dFTmmxxkNXDb1/EPz3z5Q/W1xqy0StSBN9siWaakxIfXuy3w4o5MP\njBYG+1IAMJiYx8cDxvib4rZdR+q6BnQsCmUnSu/flOuD/hc2QgUYlwMYAWBAhcubaY1emBwd\nmZc8WhPZ8dPRr332P+cc36WNXeqfvX7ulccXJ0YFrO8aIfST7fmrZ8WfLu/uU+pjZMLVRQkJ\nkVMlJdczzBa6vU8r4rMjpUFzJkwgjB9aCTSDGyhgI1j7gB3j9wlNItTtdPVeULYAACAKxc5C\nGetQwa2Tbi1GIBAI1yACB2EQHod174acf39Z5jS+c0WGZ96irT2a37130bG9JTdJ+ps7Z/vT\nqdQPCHkcDpsyM5liholH9ZeyKFSYEV6Y4aHbyO2rM89V9jhl+mYnSn1U6i8Rcp/YUfDjrdM6\n+nXhEr7oep1Ihhv/1/AQXph4VJJNYqS4to3ByH00dq1j5ZU9FXtON9n+baVxRbP8p6+d+c2P\nZs33nonpdaQIzce488PDrR8dVRjNNIAJ4FxukvSZWwv9s5///ESjqwZnNFs/Od7w9M3T/TCB\nYchLDgtsIclEQW+0vHuwZu/pJlvDV1y46NEbp83J8b6ISSAEANrg9irshWjwqYu6g77wH6Cv\nVxFiGncUY1UrNedR50YVAoFAmCxM7YI9ggubFyT/5kez7JuuKKngp7fMuGNNlustLVa6rVfL\nmBhqg6bx798vcTLvqGxR/O3jy96dc8Bhs9D8XIbyToTQwmn+OHeKlApefnzx6qJ4m9YQJubd\ntjLjbw/O91nbBQAAm0UlRYlFDl0wKVHiQqbmglE6jALADQuYbzmiV+tYaenR7D3T7DRopfEr\neyuwT7wpWLtPmd89OGA0D6pglS2KX7x5bphPkBepblWMady7mC10Q6equVttsTIn44xIn9Kw\n93Tzf76u3H2qaRgP2kkMxvDr/1744vtGu51NR7/21/8tPlPRHdiJEQjegXLfw4j8bVc0mcAN\nhwbVDTuaLtxZEojpEAgEgj8g8i3BmUX5MYvyYzR6s5XGjL0S/SrDG/sqj1/utNIYITQ3J/Kh\nzXnxEc4H0eVNcruvhCMXa3q75frosGCvr7ZgWmPWm2gLAHAoVghbwHbfCvjADXlVrcpexZCt\n184V6RnxEp9PFAAAwiX8Z3cWAoDeaBHwAva5vmNpelyY8PDVTtu+PUYq2DYvKWfUUbX5KbKn\nbpr+6lcVeuO1cgM+l3XvxpxZ3u61uVTXh5mUjK4BXeeA1ovBsTYwBqccXxvdcv3xy51rZzMU\n2lhpbDJbvfVSuhNtfKPmDGKl8afH6z88XGdrMhILOHesydy6KGX0CUEA8NWZ5jf2VdrblN7a\nX3X3+uztS6aW4f+Fmt7L9c4ONVYav7W/yovByQRCwKBCgQph6FJBPGCRMiXPwQo3SV6KRoif\n69+5EAgEgp8gAgeBGbGA2U9bozc/8fJp+yEqxvhcZU9ls+Llxxc7aRbuoj0AoK1XE+QCh9Fq\nHjBqMGAAwAAW2mqwmMJ4Yj6LudUiOkzwxlNLPzpaV1Lbp9SakqNDti9JLcqM8O+sAQACqG4A\nAJuF1hXGrZ0R168xCjgs0dhdTtfPTZybE3W2srtrQBcVJpiXE+ULr4FhTD10Bu+XVKj1pj4l\ncwG23fXTTl276vV9FWWNcouVjpQKbl6atnlhMosaV7t0ZnwoY+9PVoKXU4SdeGVP+VcOlTIa\nvfnVvRUqnfnOtQxFYYxcru93apozmq2vfVWRGCmeUt0Z5U0DjOMtPRql1jRW1x4CIQhB/NlY\nfxJoMwx+27EQf5ZXHEYnOhjjgxfbDl1o7+jXRoTyF+fHbFucynGT2j4E2s0vGs1sHEYgEAiT\nAPKzQRgbjCXiKp1p15G6J3YMifAcZqct5Ad1GhkGUJi0+PqxN3IYjBZwEDBvNYV89j0bcu7Z\n4K9ZBjEIQcQ4bFZkEt7GeaPtavGMBJeCIxtsFooNF3r96ViU22Wok3JxpaH/52+ct7dy9Cr0\nr+wtr2qV//y2meOZwE1L0w6XtDu1w3DZ1C1jSSMeK70K/ddnW1zHPzlWv2NJqjsJ1Yl95xge\nAQC+Ots8pQQORosfGx43/hAIwYWFj+vYIFCBhIf4fBAkIG4WUMR1GGga//a9i2ev96P1KQ1V\nLYpjlzteeGjByEcaoihQdzCMi4lvK4FAmLQQDw7C2LjSwBzk6To+PV3GeLwQKuJm+rhxQ2u0\nGMZhbWCmLVbMsGegMTZZyaHHhKe8Sb7/fAtjTcSKwngPqk5GRMRnJ0czO7Dmpw7x13ztq0rX\n/eqRSx0VzfLxTCAhUvTn++YmOQSmJESK/njvXHez8goVzQrGPiCzha5hKidhpK2XOZqn1X1k\nz1jBGJfU9n15svHghbaOfrd1Z4ElxY2Dr1TMnWSezYSpCW4pN7/1pPXQe9a9X1v/97nlzQ+s\ne74GMwn6AAA4Utpx1sVtp65d9fGx+hHvixIXMYyyeChutlfmRiAQCEEIqeAgjA2Tmfm00HU8\nTMy7Y03m2weqncYf3pLnI/NLjPGFpoHjVT1aowUAIkJ4a6fFZrrZGAwDzaRuXL/Kx6YFBB/z\n2YmG17+uZLxqTk7kj7dO89Hz3r0+67fvXnQazE2Szs8dNFBQ68x17cw7/4s1feOMGslPlb32\n5NL6DmVnvy42XJgeJ/GpBy0A2FKEma+yjvZzxOcw/0gJeG4NcRwxW+iWHo3eaEmMEjP2cTR3\na/7vo1L7fzuLQlsXp96/MYcaX0+Q19HqLQghV8Fo+5LUMRmaTFAwxg2d6m65LiJUkBYb4uu3\nLsHfGLWWvf8A/RADDtxSbj3yLmvDw4GaVPBwuqyLcfxUWfdd67KHvy+KLQKTGjccHrQa5Yeh\nvJuAN1x/osWK2azJ/8VCIBAmK0TgIIyN5Ggx42Ey4wHjzhUZCZHi9w/WNPdoKASZ8aH3bMiZ\nke5hGOqIfHO187xDIUmf2vjh2aatsxJmJI5tZ8hCwzQUkIX1BKZbrndV3AAgIpT/zK0zZmb4\n0DNl4bSY3945+9W95bYOL4TQujkJ92/MddxIuya5Dl5l9ELpEJuFshOl2YnS8T/UaEiLZdYW\nEYJUN1e5MjMzvIzJfmI0L9ahi21v7quSa4wAgBBaOzvhgU25IcLB1hij2fqrt8879txZafz5\niQYhj33HmsxRztAP1LWrXv2qwlXdKEwPv3V5ekCm5E9q25Qvfn7VLkLFhYt+sj0/IA5HBB9B\n115wUjeujVeeZq2+BzhTvUZJqTMxjis0RsZxJ1DyMhRdiOX1YFKDMAKFZwHF3CGIMf6muO3z\nEw3tfVoBjz0jPfy+jTmuFvIEAoEQ5BCBgzA2Ni9IOXihzfVsduviFMbbL86PWZwfY7bQCCGf\nHggMaE3FjQztMwfLugoSpNRYDjk5FJtNsSwu1lwsRHFIbvyw6I2Wxi611mBJjQmJCA26bL/i\n6h5Gw4I+pSE5yofNGjYWTouenxvV0a9T6UxJUWJXEwpZCF/IZ+sMDFpGokN3yUQhJSZkTk5k\ncVWv0/jKmfGjf29sW5x6uKS9o1/nOBglFdwy0sb+4IW25z8ZTKTGGH9b3NrWq/n7wwvsJQ8n\nr3Yxhs7uOdX0g1UZ4zR29SKHLrYxNvvINaZJX77Ro9A/+/pZrcOHoqNf+6u3i//56MJMH1vk\nEvwGVrhJO7aasbofyeL8O52gI9LNF2bU6M3a+aEotmjEW73w6ZWDF9ps/9bozafKui5U977w\n8AJf21GPAUMP1rYBbQJ+NApJATeeaAQCYYpDdmuEsZERL/nFD2b++8sypfbakYKAx75/U87s\nYYM83Xl9N3SqPjxcV9eu4rBRTlLY7aszPU5Xae7TMraP6IyWHpUxZoyb7TCuqN+ocexVoRAK\n44nJb6kLZgA9AB+Au/tU03sHazR6MwAgBCtnxj+8JU8iDKJ8B6WG+RwMABRao0zi83NCikIJ\nkSIAtxan62YnfnnSOdUvVMRdUjAhDeF+cdvMFz+7+v3VTvvImlkJj23LH/0jiAWcf/540Tvf\nVB8t7dAbLXwua+n02Hs25AyfG4IxvHewxnW8vEl+obrP7k7a1MVwaAwAKp2pX2WI8kF8j2d0\nDuiYx4PVMcSL7D7ZpHWR/CxW+qOj9c/dMfKGjTAhQDy31s7DXDV1WFEYf+QSg1HoqpnxXnyW\nima5Xd2wYzRbX9tb8fdHFnjxiTwEW3DbN3jgyuCAMJZK2gq8cTVvEoIdalTtqASCE0TgmJxg\nDKfLu6paFBYap8WGrCiM82LT8tLpsUWZEeerejv6tVFSwezsSM9c7g5dbPv7p1fsxSDN3Zpj\npR1/unduQZrMg0czu48SMFlaAQQA4QCj3bFwKHYUP1RrMZhoCwBwKbaIzR9TGcgUwIBxDUCf\n7cKXJ9mvfTW4XcQYDpe0dw3oHA/MA467xFmEUGRoUOxm792Y068ynLgyqAhESgW/+EHhKDNH\n7NR3qL6/2tmrMMTIBEunxyVHB6YARCzgPHdHUUOnqrZNSVEoO1GaNPZSlFAR94kdBY9vL1Bo\njVIRbzTvpn6VoUfBUJoBAJUtcrvAMUzI4qjyF/2F2I3xrVgY1HFUXsGdH21Nm8LPMyH4DpQy\nnXk8KgVEvmqpU+nN5W3KfrUxTMTNiZeEi4O3EWZebtSOpWmfn2hwHFycH7NlYQpo+rC6C7hC\nFBoP7HH9CRdrnKvtbJQ3D+gMFqEP7LfHBO447KhuAADoOunGT6js+wCRPTCBQBgCETgmIXKN\n8XfvXnR0yth1pO43P5rtxU2OWMBZOXNcVaMavfml3eVOrS5Gs/WFT6+88+xyD3bE7nJJEYII\ncTvGNACFUApAyigfkEIohBMUm95hUOlM3QP6aJnA74USFowvAlxrAKZp2HWEIdWivEleUts3\na9jqHn8yPy+KsQdkbk5kSHDsFbls6le3F5U1Dlyu71frzakxIctmxPG5Y1u9vbW/6tMTDfT1\nD9euI3V3rMm6bWWGD+Y7KtJiJWmx4w1OQgjCRr0DGcYM2PGq/FRmLTUhUjT65/IDc3Kivitp\ndx2fmxPl/8kMj8ZkaVUatGYrYBByWQkSvmTEGEvClAdFJlFF6+mSb4aMsjms1Xf76BmL6/u/\nKmkzXU9fPlCK1hTELsuLHv5eAeTBG3IX58ccutjW3qeNkgoW5UcvTOPT59+0dpZduwVXROVv\nQSnzPX4K11IpGxiDzhhogYM24f7LDOPGfqxuQJIgskwiEAjBAFl5TEKe//iykw9oW6/2D+9f\n/M9TS4Onq/xSXT+jb2JHv7axS+XBdiglQhQZwu9VG5zGCxIMAq5tEUNj3ICQECDodgUe0Nqj\neXlPeUnttQKKosyIH2/NT4j0mxlYm13dAIAuOVZqmbeU1a3K4BE4JELus7fO+MuuUoNp0GAl\nIVL0+PaCAM7KlfxUmbu994gcK+1wyg60WPE731RnJoQO30c2mYiQ8KViroKpIykjfrCZfGZG\nxKysSNdzy/s25vh2fmNk2fTYb4tb7R92G+ES/o/WZAVqSoz06Uz1Dt00aqOlsleTGiaMGraf\naHiyE0MZs8n95pVL8A+sVXei2Az6/F7c3w4cHpWQSy37AQr3ZguGneY+7efnWxxHLDQ+cLkj\nQsKfdt1sYkBlbOnRiPjs5JgQbnDUc01LCZuWcr0dg7ZYD/8V1A7eJSYtXbKLotgoycP816Vz\nBIUAACAASURBVNhw5m4gIZ8tDbjga1IAduO0begDInAQCIShEIFjstGj0BdXM9QZtvRoyhoH\nfJdgMlaUWrfu34zbkhGhENo5P/mz4pZOh9L07FjTxulDKgswbkfIhwLH1YaBY5c7ehT6KKlg\nRWGcx9vU4elV6J985YzKwVm9pLbvyVdOv/rEEv9Ye2LsVDc+YdJzF06LefuZ5btPNTV0qAQ8\ndkGq7IYFSRMidVKjN/cqDTFhAsGwR+LfujRRXxsvbp06AgdFoZuXpb+xzzkPOCUmxDGXFyH4\n9Y+K/neo9suTTTb32fgI0cNb8oKtMoKi0P+7Z+7uU437z7V09OtkIbx5uVF3rcse3ohkPJgs\n9IHzLTWtSouVzkoI3Tg/ScAdYbVAY9zM1BbUotCHCzgea+tbF6XsP9fidLbMZlE7V0z++Jgp\nBqLyFlN5i4G2+rrr/nxdH+P4ubq+aQmhSq3pta8qjlxqtxV7ScXc+zbmrp2d4NMpjRXcXjpE\n3bgOXfUNy1OBY9n0uHe+qXatcFw7KyHwkbHDNKGQ/hQCgeACETgmGx19zHZ0ANDWqw0egWMY\nywOPvf1kIu79yzJqu1VdSgMLdSTI1EnhZpdb+cqWD2N4aXfZV2ea7SNfnWnesjD50Rvzve5B\n8enxBpVLbpxSa/rsRMNDm/O8/GTMDFE0osMoiRCpdAwyRxAmHUSE8oPtiH54Wno0L+8uv3R9\nUb5wWszDW/Lc2fG6M55sd//NMCm5aWma2ULvOlJnNF+r1inKjHjq5ulOK3UBl33/pty712e3\n92lFfE4QRv/YYLPQTUvTblqahjH2talNW6/2l2+dtzubHi3t+Pz7xt/dOXv4z7LGZLW4pGsB\ngBVjtcki5XvYAhYpFfz1gfn/+OJq7XUzjvgI0WPb8h0rcQhj4nR59yfH6pu61WI+Jz9Vdvf6\nbI+9vX2C7z0Fe9XM5yu9KgNN41+9XVzdOujwotCYnv/kMkKwZlYQaRxY3sp8haYXzHrwqLtW\nKub+8odFf/2o1G4hDwDzcqPuDYafS54MOGIwM3TCInGy/6dDIBCCHCJwTDYEPLeLg4B7RDlS\nmBEuC+ENuKwzshJCx9NngRBkxUiyYiQYdwK4qhsA4KvF09HSdkd1w8be0835KbLlhV5OuStr\nkjOPNw5494ncIwYYfC4WBbeuYL+xz/k/PCdJOjsrwl9Tmpx0DuieePm0LZvGxunyrpo2xatP\nLGE8wBe5sSMVC3zy8bfS+MD5lku1fQqtKSlKvHlBclqsRGuwHC5pb+lRiwWcwvSIwowA6KoI\nwQ9WZWyYl1jdotAYLCnRIRnxbhvf2CxKLODUtCnLGgdSY0OSo32eGewxfrDs/fOuS065LX1K\nwx8/uPTmT5cNc5Drmh1uh1H4GD2ZCaEvPbaosUvdNaCLDBWkxkooSm2mazHWA2JTEMKm4nz3\nxT7JeHN/1SfXW9h0BsuRS+1nyruff2h+ECrRvsNdywmXTZ2r6nFUN+y8d7BmdVFC0Phl+4o5\n2ZHvPLv84IW2pm61RMidkR4+JztI6v4Qil2JW/Y6j4blgyB4nVMIBEKgCKIdL8ErpMeFMjaf\ns1nUjLRgKd8AAB6H9bPbCv/wfonjzi0ilP/szkKvPD5CYRgzdG4D+CpR7DCTCyAAfFfS7nWB\nw+omMsZi9VOrCELxGLcBDE5jxxI2xvDBd7T+ur3FkoLYx7blB0+EygTl46P1jp8RG31Kw+5T\nTXeuZbBgmJMdWcsUPOGLtgu1zvzzN8/Zn+5qw8A351s3zE06VdYl11zTLj88XLcoP+bntxXy\nOAHYgoaJefNHMg6kafzuwZrPTjSYrzsOLpwW8/iO/KDyGfUbzd1qxvdPR7+2vGm4JkdM46Pn\nWupaFFq9JSKMP29GbGbStS9b/rgtDBBCdqtaC91upq/HDGGTFXS0dYDDykEwFV+vMdHcrfn0\neIPToN5keXlP+T8eXRiQKQWEjOiQui41AAYY8vOUERNS1cKcztMt18vV/ogSHyUoLJH5x14c\n6Vn5xuADCDjbl6SO5xEYwDTurQV1D/BDUHga8D3xnEZh+cDi4Y7vwCgHAGDxUNQCFDnPy1Ml\nEAiTAiJwTDbYLPTwlml//vCS0/jtqzPtv83N3erqViXGODMhdPzpBh4zMyPi7WeWf3mysbZN\nyWFTucnSGxeljNjpPWriATpdGlK4CKV46fGdcRdL2S33fmtAamxIY5fadTwt1m8nzwKEZmBc\nBXDtr0aIc8vy3A1zI+o6lDqDJTU2JC7cb46nk5mrbqpyrjYwj9+0NO3Elc623iHv/Ix4yeYF\n3q/jfetAldNm2Erjr8861zGdKut655tqfzVPjZl3D9bsOlLnOHK6vEuhMb74SBAlHPuNrgHm\n7zEA6JLrZgCzwNGvMjz96pmO/mvfdf0KfXWjfNHM+PVLUgQclthr3+qAsd6KO50HwWyhWziU\nX40GjWZrQDS78XC+qgczBQxVtshVOpPfo7gCxoLMiIuNA72qIZbkEgFnRV7MJ9117u41TDaT\n/0HxhVB10NWGg8pZH5D5DAOWt+CLH2BV17XLbC6VuwFlrvTgoZAkE0kywaIFqwl4Uid9ikAg\nEOwQgWMSsqIwLkLCf+tAVU2bksY4KUp8x5qsJQUxAKA3Wv75xdUjlzrsN15SEPvkTQViprL2\nktq+8qYBvdGaGhuyfEYcxwdG4lIx9+712V5/WAAAYCFUhHEDQBeAFYACiEAoA8BXPfYhblaH\noSLvn/lsW5x6/HKnU004m4W2Lfb2wctwhCE0D0AOoAfgAUgBOCFCmJkRyJ4Updb0+YnG2nYl\nRUFOonTb4lTG9/YEwmJhrtYxW6yM42IB56XHFr//Xe33Vzp7lfooqWBVUfzOFRle34xhDMcu\nd4x8OwAA+La49f5NucGT4mS20LtPNZXU9im0xqZOBq2woll+pWG4goUBlbG+Q0VjnB4nCVrn\nDg8YJi95mA3wW/ur7OqGnVOX2mdmR6wpiPXa5ABoYD5gp7EKgAbwuVuwSmd699ua45c7VTqT\nVMxdVZRw++pMUTC1fw6Day2YDYxBrTNPHYGDx2E9vCbr0JWOkia50WzlsKiCROn6wjgxn+3O\n20Um4YUHTfkGAADFZi1+hL70Ce4qvzbCFVH5mz2OUPEVJi196lUwOXw5WEz01T0UV4SSPS2+\nYIuATY5PCATCcEyMX2XCWClIk/3j0YUWK8YYOwoTL3x65cSVIcdf31/tNFmsf7h7juOgzmD5\n4wcljmksH3xX96vbi4ZpYvcnGAON8Sg2SxyEsgGyAYwAXF+L/QvyohktMBbkeb81IDtR+svb\ni176ssxuYiIL4T22Ld/vfdQUuDnRDQhXGvp/++5F+yK+uKr3qzPNf7p37oT2I0yJCXEyRLCR\n6r72SshnP3hD7oM35NI0pnymKeiNFle/fXdoDRaFxhguCQohQK4xPvPa2ZYeBr86R2rblYwC\nh9lCv3Wgau/pJltHGEWhTfOSHrghd8Kd5zOSlSBldEcS8tnT3TQ5YgynyhgCHQCgq1MtKPKm\nNSPGzFt0AIzBgsC3W3Sl1vTYv091Xf88KjSmz080XKju+ddji7xXe+hD3JmJctjUZBLpRoOQ\ny7pxduKNsxM1BouIx7aXai2cFp0cLW7udv5y2LkiI+jquQRSauEDoOnB6m7gCJE0AdjBJMEA\nAABuPjdE3bCP1x7xXOAgEAiEkZgAP8kEj2GzkOOuvluud1I3bJyr7Gnp0SRFie0j/95d5pQ1\n29Gv/e27F956ZllgF/HVrYq3D1RXtSjMVjotNuT21ZkjNtgDAPilN3vLwuSTVzsrh3bw5iWH\nbV6Y4ounW5wfU5QRcbmhv2tAFyMTFqaHD58eOumxWOn/++iy0xGlQmP6y67SN55eFmxL09Gz\nbXHqmQrn3SObRd24KGXE+/pO3QAAAY/F47DsGSXDgxAIg+b9+ea+qhHVDQCg3LxpXt5Tvv9c\ni/0iTeOvzjSr9eb/7wczvTbFwMFmoZ9sL/jD+xedCsQe3pLnrk7BZLHqTcxSl8JNXIXHIMRl\nyqTGACwEPq/V+vR4Q5eL2tjcrdl9sum2lRm+fvbxsyg/5vV9la665NLpsZNDnvMA8dB3NZtF\n/fHeuf/47OqFmmtLIAGXffuazK2j+L4NDOIoJA6uWGtHsJK5yg+ru/2QB0yYDFA+r8sjTEqC\nZcVJ8ANNTK4NNho7VXaBQ2uwHGeqPO9R6Iurexfnx/hqfiNxqqzr//2vxL7srmlT/vq/F+5e\nnx0kK0seh/XCwwu+PNl0tLSjW66LkQmXz4jbtjiFzfLVt7OQz14wKn1nSlDWKO9lskFp6dHU\ndygnbhFHYUb4M7fOeHVvhV27kUl4j28vSI0JcNIHQmjhtOijpaPqUslLDgsSAQ5j/P1VBp3X\nldxkBkPiAbXxwHmGgMZjpR13rcuaHL4zC6dFv/z44ne/ralqUVhpnJ0YevuarNwkqbvb8zgs\nsYDD2P4Q4T4O3DMoFAbQDs4iB2KhMD805JfU9jKOX6zpC5KfoeEJFXF/cdvM//uo1PHFykmS\nPrJlWgBn5QUM7VhZDOZ+QBzgxyPpfGB5/kmMkgr+dN/chk5VU5cmRMjJSghlzKsijAp3H0pE\nASIbVwKB4CuCYsVJ8A8s9wl/LIdNeNeAzl0YR1vvyMeePoKm8ct7yl2TCN8/VLt2dkKQlL6z\nWdTNy9JuXpYW6IlMRXqVbs0Re5WGiStwAMCaWQlzc6JK6/p7lfroMOHsrIggEQvu35Rb0Szv\nlg/5n0+OCmnuGaKl8jis4HEY1RosBtPIVSfzcqMYt/T17SpGm0YAqG1TTg6BAwDSYiW/u2sM\nzfwrCuNcQ7IpCi0v9KYBBwAg4LGpZAvd7KhxICRkUYnefSJGdAbmd47O6K5xJuiYlxv19jPL\n951tbuxSiwWc6WmyFYVxQdd8MSaUF7D85OBF8wDW1qCYm4A7rnxTe2oPYVwYVIzDSJoIE/pd\nRyAQgpugWCUT/EN2gpTNoiwuCaMUhRyX8sPsnQK4rWrsUvcpDa7jFitdWte/qije/1MiBBVS\n96Gew1w1UQgVcZfN8PJecfxEhPJff3rpJ8caLlT3qrSmpGjxtsWphenh+861fH6ioXNAx+Ow\nZqSHP7ApN9GhAy6wiPjs4TtrKArdMD/p3g05zFeTNTkTd6/Prm5V1DhE6lAUundDji+2iCwU\nQbHEVroHgwGATaEQForwzwsTHyns6NcyjEcEy9t7NEjF3B+u9mvijA+xqLDitPMgbcT9R1Ds\nrYGYEMEBTOOBFuarwryf6kUgEAh2iMAxhQgRcm5eluYUiAgAWxelOFZAxMqEsTKhq68hQqgo\ncAEZw9gZakftdOiISmdq7dEKeKykKLHvukgI46RzQPfBd7W1bUqEUGZ86A9WZ8TKhIy3nJ4m\nkwi5Kp3JaTxKKsj2t/fqFELAZd+5NuvOtVmOg5sXJG9ekGw0W7lsVrCd0iGElhTEfFfS7jr+\n89sKZSG81NiQYeIkMuNDKQrRLqVkCEF2otsmjkmPWMD5548XfXO+9WJN74DamBITcsP8ZN+Z\nUiPgs6kkHz34MGycm1RcxdClsmleACZDAADQNQJmypkydoJVByzmHwuCnzBpweyuspI5HYxA\nIBC8AhE4phZ3rcsKFXHfP1RjEwUEPPYPV2XctHRISwVC8PCWvN+952wyt3VRSgCPYWPDhQgB\nY214XMTYFjEavfnN/VUHzrfaSs3DxLz7b8hdTWpAgo/zVT2/f++i6XpOakOn6mhp+2/unD0n\nm6H2mMdhPXlTwR8/uORYo8TjsJ6+ZbpPvTYdaevVXmno1+jNydEhs7MjgycVNSAErW3hfZty\nq1oVbb1DjuLvXJu1ojBuxPtKxdzN85P3nG5yGl9dlBDjRnqbIrAotGl+0qb5k3mrvyg/5s61\nWR8crrV3cXLZ1D0bcgozgihMakqBabediUDrJ57AoVdgVQdQXCSNB46X/WtGhXaALt2De2rB\npEPSeJS3FiXO8PzRWDxwt24LyF9HIBCmDETgmFoghLYvSd2yMKWtV0Nj7K54YX5e9AsPL3hj\nX2V1q8JixXHhwp0rMtbN8Wba31iJCOXPzop0ynYBgLhwYSFTlKM7MIbfvHvhasNgnqtcY/zr\nR6U0jdfODuQfSHDCbKH//ukVu7phw2Sh//7plfd/sYLxfbsoP+b1p5Z+cLi2pk1JIZSTJL19\ndaa7ZETvQtP49X2Ve0412WXB5OiQn99WmB5HuriDDlkI77Unl37xfeOl2j6FxpgcE7J1UUoe\nk6UoAJTW9e8/39LZr5OF8OblRm2Ym/jg5jwhn/3ZiQazhQYANgttWZhyj7uWFsLk4oerM5dO\njz1V3t0j18fIBEumx7qrKSP4AcQOYXbEAQSsidQ3BGYdfWU3bj5/zVyGzaNy1qKsVf5sisMD\nLfShv4P5Wi8w7q3Hx19F09ZSM7d7+IhsLgpPx33OVcMAgKJzPZ4ngUAgjAgROKYibBZKuR7B\nYLHi81U9LT1qsYBTkBqeHH1tTZCXHPbiIwstVtpsoYPE0fDpW2Y8905xrUObd4xM+Nwds8bU\nYFJS2+uobth592DNmlkJwVZOP5WpaJYPMGVM9qsMlc2KgjQZ470SIkU/21no46kxsOtI3Rff\nNzqONHerf/V28dvPLAuSjw/BES6b2rkifeeK9OFv9ureii9PDr6sZyq6D11s+/N98+5en71j\naWpDh5rGOD1OQkIWphSJUeKdUWKMoUeh71XoOSwqIjQoXK5HDQaDHKxmEEYACtIyq9EiSAOK\nC7RzZyII04CaQNZLmD77Du6tGRywGOmyryiaRjlr/TeJ4o/t6sbgYMUhnDofSUeubmMETd+G\nT/wTLENeIJQ0B4UTL3YCgeBDyMp7SlPXrvrLrkstPdeyUSgKbZ6f/PCWPHtJP5tFBY8/RU2r\nQqkZ/JmMlPKf3TnmE/LKFgXjeK9C36fUR0pJ2WSwoNC4rFmvI9cwCB8BBGO8+1ST63i/ynDi\nSue6Of7IdyB4nZLaPkd1w0Z5k/yjo3V3rcuWCLmTsjFBb7R0yfVRUoGIT5YHbqlpU/7ri6t2\nU9XCjPDHtuYHj5PuMOCeUtzwLZjUAAAUByUuRonLgJqwrzVLgCLW4t5vATsE2XBkKHxl4OY0\nZnB/4xB14zp0zXesrJVuXx11J+4uA70cRBEoZjoIx2eRZtLi3gamyWHoKAOPBQ5pAmvlz+jy\nvbinBixGEEdSmStQ8vxxTZVAIBBGYsL+qhHGjd5o+dU75wdUg3tFmsZ7TjdJRNw71oxgsV7d\nqjhd3t2nNMRHiFbMjBtrja5Sazpc0t7ep5WKubOyIt0VhztS1jTgZAvSqzD8+p3i159aOqbT\nM1d3QDuuGbTBgxVjlclsomkEwGNRIRwO5eNqE63J2izXaU1WDgtFiLjxoQI/V7cM87IG23mp\nQmtSapnlmMYuNeM4Ifg5frmDcfxYaedd67L9PBk/0Dmge21vxZmKbtvFOdmRD2+ZlhA5SYJv\nvUhLj+anr51xDBsuret/6tUz/3lqqSwkqKsGcMc5XPfV4GXajJuPgn4A5dwcuEmNG2EGio/B\n6lIw9QHFRbx4CMmfGJUp2Ip7ykHTifvcRI1YjFjdjUIZDMJwzQHceNxusIrrDqHM9Sh1meeT\nMeoco5eHPJdRO65ff3EENe8eAADaCtREeF0IBMLEx5sCx4kTJ55//vmOjo4ZM2b84he/yMjI\nsF+1f//+Bx54oK2tzYtPRxgnJ650Oqobdvaebvrhqgx3vowY45d2l391ptk+8uHh2gduyNuy\ncLShX99f7Xrxsysa/bXzlvcP1a6dnfDkTdOHd2T8+Gi9qwCh0Zv3nm4aU/e7u4qPUBE3KljL\nN/QWa5/BgAHb2nENVqvGbIkS8DmUr4prWhT68i41fd0brF1paJbr5yaGcVj+UzlykqQxMmGX\nS5pPXLgoJ8jiKjjuq5y4bLKeCxZoGncO6HQGS1K0eDQGqIwdUgDQr2LIq57oyDXGJ18+7fgn\nF1f31rxy+pXHF5O6Nic+OlLnqG7YUGpNu082BqkVi74XGwcQS4CbvnO9EvdcRolLQBTj/3l5\nDbYYhS0O9CTGiLabvvohaHsAAIzmkW49BNxZihuODhmirbh6H0jiUXiGmzuNhCAUWGywMmTS\nIbGX4vOIukEgEPyF1wSOixcvrl69GgBSUlLee++9jz/++N13392xY4ftWp1O197uHMtHCCzN\n3RrGcaXWJNcYHYNjHfn6bIujugEAJgv98p7y7MRQ15TErgHdp8cb6tqVXA4rJ1F6y/J0jcH8\nl12XzEOdIw9eaIuPEN22crgf5rp2FeN4jYMlx2iYlxudHB3S3O18rn7zsjS/ZW2MCRrjfqMR\nAziajVkx7jcYY4Q+2XhoTdayLpWT8blCb67qURfE+s8yk0Whn99W+Nw7xWrd4OIvRMj52W2F\nwfZKiQWctFhJQyfDW3R6OrNXCMHPHC5pf2N/pU3SZbOobYtT7liTxecOt+CWiplP44P8lN4z\nPj/R6CroKLWmT441PLp1WkCmFLRUNDP3ObobDySGfrp5H6ibAQDT2MkKwQ5WNqEJLXBMOGgr\nffl90PfbLmE2C4BJ42DzUEg0w3jbeeaHbSsGjwUONhclzcKN55zHOXyUGABbKwLhGkQXI3iE\n106Af//738fExFRXV9fU1DQ2Ni5ZsuTWW2/98MMPvfX4BK/D5bh99Yc53vy2uNV1EGP8bbFz\nec6psq77Xzjx1ZnmyhbF5fr+j4/V3/O3Y58ea3BSN2zsP+emRPM67hoy0Bg7Ndgs9Md75xRl\nDp5I8DisO9Zk3bxsBLtBDzCarXtONz3/yeXnP7m893SziekPHxGDlaaZUtZMNG2hfZIk36ky\nMMa6dagMfu7hyUsOe+fZ5betzCjKjJieJstJkkZK+X/9qPS3716saJb7dy4jcO/GHNcSpFlZ\nkbP+f/bOO7Ct6nz/77nay5blvfe2k9ix4+w9IIEsQhiBEMpqgTJa+P5aShctqy3QQkMKhD3C\nCIEEshMgCdmOHTu247331N665/eHHEWW7lVsWbJl537+QudK95428tU9z3nf50mmSLRlGGP2\nnW1++fOLtoI1s4X86lj9i58Vu/7U/Oxw6vEp4QDg3p+zz1LWQGG9DACXaMavZyhvyABg8c4N\n2X0sBrL6E6u6AQB0DQgAAKRjQQqDV8EDtTZ1AwAQiwCqWj8iZSmlAQfW9jkPAgDW9o5mVkT+\nRhQ81PuTIyDm3gd8yWhOy8DAwDD2eKyCo7Cw8Mknn4yPjweAqKiovXv3Pvzww5s3b8YYb9q0\nyVNXYfAgUxICPztKEd+VEO4nFnDoPtXh1DJgpb1PY/9Sqze/urPUYBry2KTQGH+iaWvvluvM\nFtKFoWlqtLRX0ek8nhYz4m6FEKngpQcKatsUDZ0qEZ+dFh0g8/P8lmxTl+qP7xfaOiwOFbbu\nPF7/t3vzbTk1w8SCaR+azRh7w0RHb6Z+2DWT2GQhuWNrOusn5N57Q2pzt/q3207bfC7aejWn\nyjsfWJV+6wJfcWLPTw1+6YGCN3eXW003uGxi3dz4TUuTmVyecYck8YcHKQz8Tld0VTQNuDAA\nyk8LXjUzZu+ZIdprQrike0B7y18OqbSm0ADBqpkxt8xL4LB9xYnZbUwW6vuMkeZucD2TEO7n\n3DoH9P2P4wXuKwGjXVkZgQAhoFRnxNdd+UZhdc+hwtbOfm2QH392VtiSnMgxvVc7KRFIyMV6\nExjNg62obC6RuhylLqX+OJu6wBbRjA8XrohY/jRuPI+7qsGkQ9IIlDwP+OPxrTbpsaoLWBwk\nCWU28BkYGNzAY+uj/v7+oKCru+IEQWzbtg0ANm/eTJKkQMA08focOUlB+anB56t67AdZBHpg\nlasuYhGfY98yYD9u/7KotpfybTbrDQe4bBbLpaPE7YsSz17uNg99CvcXcdfMjnPxKRckRfon\nRfq799lrgjF+/tNih4fgzn7ti58Vb3ti3ogepFj073ZxaDRwWARc8fuwh0CIPU69If/9tszZ\nxfP9A5Vzs8NGanDrPaYmBr71m/kKjVGpMUYEiVx7yjCMGe19WrrYnfJGVwIHADy+Pntmeuj+\nc82tPZogf35GbMCeU031HYMNbl0Duvf2V12o7n35gQJfa5saKXGhkhqqdr+EMexKmyhsWBB/\n9nKXgycUj8NaOyd+vKZEjaZj6GsELAKcFStxBPL3sZl7E4zhP7su2ReN/lzWebSo7bkted5U\nKjFuLsQtF7C2HwkDQOR020EICbjAY6Pk1SAMRtIo4NL+tKGgFKzqoDgQPGrzY4RQ/AwUP2O0\n53Ebs5Gs2IvrTwxWFXFFRNbNKG7WuM2HgYFhYuKxu3l0dHRNTY39CEJo27Ztv/jFL7Zs2bJj\nxw5PXYjBUyAEf9o8/c4lSbaGlIRwv5ceKJie4qqoPj+V+uiMtCHjlPalLpieEux6tZ4aLX3+\nvnx7S//MuIB/PjRTKuaO6EJjQ3WropEqPqO+Q1nbNjLTED6LRRmYwiEIL5mMhop5zuoGAISI\nud6ObqFErTOV1FHUyZst+MyVxAffwV/EjQ4RM+qG7+CicWA4PQUF6SF/uSdv+1MLXnqgoL1P\no9Q6Cm0ldX0/lVAtNiYUa+bEOX9pWQRaN/c6WvoOk6w42TObcu3r/iIChX+7N38CJM5wWMBm\n2d/ekTSByLwL0IQvQRo+p8o7nVtiL1T3OGdCewzSQp56hyz8BHddBlUX7qrE9aexxkDRMySQ\notjZKCTFhboBAChhEQidMqr9IlH0hBcCyPMf4tqfrvZMGTVk0ee47vi4ToqBgWHi4bEKjnnz\n5u3du/f555+3H0QIvf322yRJvvfee566EIMH4XFYW1akbl6W0jmglQi4EiFtZ4qNTUuSz1zu\n7pHr7Aez4mVLp0fZjwTQ+/BlxwdcahhioCAWcO5feW3z+ZykoHd+u6CxU2WNp40MfqQAbQAA\nIABJREFUEvls/X/XgI7uUEe/NjlqBJUjBEIyHrdPb8BDBwP5Hm6r0erNLT1qAIgJEcfLhA1D\ny094bCI9dHwacRUaI6Zpeh9QUXvm+QhGM3mxtretVxPkz5+SEOgv8kUxbtITHijic1nOsRcw\n8vKEC9XULe4XqnsW50S4MzmfISXK/5lNOW98UyZXD/5NSYScR9ZkZsZdO8P7OmRedlheSlB5\n00D3gC48UJgZJ+P6YJuSKAL6SoeMIARcNkiCUfAMIE1IHAZ+w40/mzTQyZE/lbRvXOh5Ky4A\nwE1ncGe546jZAkYz8NgAVysmUcrNtH5j9nCExKzHcO0h3FkKBhXwpSgiFyUuBta1H+F8GTzQ\nhDvKnMfJy/tZ8XOYXhUGBobh4zGB45577unq6qqtrbVPhwUAhND27dv9/PxOnz7tqWsxeBaC\nQBGBw916kvnx3nx87ocHq09VdA6oDGEBwhtmRG+Yn+Cw+5ebHCQWcJwbUuLDJC8/OPObnxv3\nnGrsGtAJ+ey8lOAHVqWHBgyriYlFoMQIP4+3OhvNZGFVd2uPxl/MnZYYNMzJuMCFiclwVCQH\nhGw2R0gojSYjSSIAHovlx+V4sD/FbMGfHa356li91TOFz2Xdvihp8YyopgGdymDmsoggETc5\nSOQiDNWryCQ8FoGcQ4IBIFg6upZjb1JS1/fqV6U2zxoRn33fyrSbZvrEckKrN395rK6sYUCj\nN8WHSTYsSJjEnQhcNrFmdtwXP9U5jCeE+7muVnNGo6fIUAQAjX5kKY++ybzs8JykoOLa3s5+\nXYhUkJMc6CeceJIcxnj/uZajxW0dfdpgf/68KeFr58SzvZBvLeCx80b4/RljUOBU3HkajI41\ngyhmKfJ3N2tj4tOvoo557lV4K/4Zt5VQH+AFgIADun4gCJBEoKQbUcCwXaU4ApS+BqWvAdJM\naUQ6WrAB9HXYogDEQ9ww4IyJgNvXSD1u1GJ1N/Kjdn1mYGBgcMZjt8X58+fPnz+f8hBC6LXX\nXvPUhRjco1ehP1LU1tKtDpDw8lODpyY61TcOG38R97H1WY+tz7KQmK4UX8RnP7kh+6UdF+0z\nUyRCztO3TWWziFsXJNy6IMFgsriIaxkzyhr7//VFSXvf4EKUwybuWJx019Lk0ZwzMy6AUt+R\nCDmue/7p4BAEZcmGWmc6Wd7Z0acNkPCmJwe7VyO9dXeZvZOi3mj54GCVUmv85c0ZbpzN4wh4\n7IL0kFPljt0oPA5rTqaPeuN1Deiefe+8vcmuRm9+fVeZTMKfnUkV+zeGtPVqnvrfmT7l4NN8\nXbvyx4vtj6zN9BHxxRtsuSHVZCF3n2y0yWTTkgKf2jh1pJ1EkUFCynTtyCCf700YHmIBZx5N\ndsyEwELiP71/3mYs1avQX26WHyvp+OdDM11HAk9OWFwi9S7ctA8rrzRfcCQoZvl1om70KvTf\nnW5q6lKJ+JwpCbJl06MIAvUq9GI+9R6DTOI1uVxP0a8KAJjErNlPg1kPBHsEIgVpwRU/4vZK\n0KtBFkFkLoUAT6sPxlasOgl4sNEY68qAG40k8wF5+Y+I3lLd1SEGBgYGJ7wRwsDgcxwtant9\nV5nOOLj9+OVPdYumRTx927RR7mtRLg8qmgY+PVJT26bksImpiYECLqujX8dlExmxAbctSrSv\n0ndQN7oGdO/uryyt61PrTDGhko0LEhZMjfB2E0q/yvDH987bb8yazORHh6plEt7Kghi3T8vj\nsB5Zk/ny5xftBwkCPbo2y4OazumKrtd2ltpKytkstHFh4pYVI7MZ65Hr9p2liP7dfbLx9kVJ\nPmJx8ujarOZudWvP1aQeDpt44pZsj8ff9KsMRTW9PXJdRKBwekqwi0oc1+w90+QQIWTl6xP1\n4y5wvPFNmU3dsGIh8bY9FQVpIcHSyekGzSLQL2/OWDMn7nKTXGcwJ0T4pY88fQkAbpwR87/v\nKpxPvjwv2hPTZBgthwtbHWyzAaCqRf718fpNVzTrbrmuo08b6MePCBSOxhpWoze3dKvZLCI6\nROQLSj01PBlKuQvp+7C+F3EkIAjxym6/73G8tONfX5bYGtMOX2j9+EiNwWhWaGiLrazxz15B\n4AdKivw4xJcC0EaiUKNTknv/ifvbBl92VlsqTxCz70AZi0c/zUFILVYdBzy0Ws3YgrXFSJTn\nsatQ4h9JPc7iInEwAADWAbYAIaK0CWNgYGCwcV381F3ntPZoXvmqxGwZUuH/48X2mBDxptHV\nKTiz90zzf3Zdsr3sluvYLOK5e/OuWcpb26b47bYzNgmmtk3xwmfF5Y0Dj6zN9OwMHThU2EpZ\ndr7rRMNoBA4AWJIbGRUs+uBgdXWrHABSo6T3rEhJjXZnWUVJe5/m758U2RfImC34s6O11qah\n4Z+nskVB6XBhIXFVi7wgPcQDc6VBrTcrNMZgf/4129eD/PlvPTn/u9NNpfV9ap05IVyydm58\nRKCH81O+P9P0zt5KnWHw+yAVcx9bnz03y50ikQYqi1kAaOigHvcSA2pDQ4eKx2HFh0mEfDYA\nqLSm4to+53eazOTpiu7VsydtEQcAhMuEo8zcWTsnrrlbbe9QKOCxH1uXNdLsZwYvcbKcIkoc\nAE6Wd21amtzep33jm7IL1YMKSEyI+NfrstwoZjSYLB8frtl1osGa6iUWcDYvS1kzJ85nPaGA\nH4j47tdsTjj6lQZ7dcNKN70xFgBkxgXcMt9bfrooMgd3VjovyVF07khPRZ754qq6MThkIU/t\nYEVlgZ+HfqwNjY7qhhV9HYime1VZQMHJKCAGDzhawKKk+YB7sLYMsA4AALERJxk4yR7MSWBg\nYJhkMALH5OdIUauDumHlwPkWzwocSq3xre8dtzfNFvLfOy99/PtFyOXT35u7K2zqho3dpxpX\n5Ed5L8wVAJq6qBecLT0aFw04wyQ1Wvri/d6KW9t/tsVe3bCx53TTiAQOOv9OACDpD42Sph71\nFz83NHarAQAB5CcHrZ8Z5+fSnYTDJtbPi18/z1vPoOcqu1/fNcTeTK42vvBp0X8fm+uGPwVd\n5rE3vAAo0RnM7+yt3HeumSQxAAi47E1Lk25dkChXG+j+xela08eS2jbFNz83NnerxQLOtKTA\ntXPifGpvnCDQE7dk35Affbaye0BliAwSLc6JCPTzXSOY6w2lU5i0FbnaoNaZfrvttH3tUnO3\n+vfbz7328KyR6s7/+rL0WMnVDXm1zvTmnnK9yXz7ouui9cP3OVneSWkq7ACbRYTJhCFS/pys\nsFUFMd5LekaxM1B3JW4tHjIYOQ3FFozsRKQFN1ygGjfj+kI0beUo5ngVbKFR4bEBSAMQ3rzd\nIUTMeoAs2oE7rzxJEiyUOJ9IycaG83YzMWPjZSDViDfdi5NhYGCYyDACx+Sno09LOd4t15kt\n2IMrrpK6Psqnim65rq5dlRRJu0rU6M3lTRQ5oABwrrLHqwIHXe49i0DjEok6fKyJJ84000g2\ndNA5tiIESZ42c7XS2qd5dU+5TZ3BAOdqehu71c9smDqOSQS7TzY6D5ot+LvTTY+vzx7p2bLi\nA05RbSZnx8vcmJsbvPBZ8dnL3baXOqN5+75Kk5lcMycOIUSpcYz7Qv3r4/Xv7Kskr9hkXKju\nOXCu5V+/nDnuE3MgLUaa5laHC4O3CfIXAMidx0Okgu/PNDl0ZgGA2UJ+cqTmb/fmD/8SjZ0q\ne3XDxmdHa9fNjfcpPe66xXWxhg2zhXz6tqnudauNDEQQM7bgqFzccgG0/SCUoagcFDltxOcx\naMBMKeEhrOn32PMKoutLRYC8n9LC9yNmP4TlraBoAzYPBcSAMABrD1C809wCnGQgJq09NsMg\nTHoOg1sw9V2THxGNpRafw3atbuiM5tL6vuOlHXXtyuHs5au0tN2tSq2rOE+t3kR3fpWTT6dn\noVtwTkmQ+ba+4ehgYoPPHZlqGRkkmpdN0YWxJCfSS44M3xe2OteedMv1JysdbUTHkkYq80gA\naOqkHnfNqoIY524IHod117IUN842UqpbFfbqho0vfqrjsInpyUHOh7hsYlbGeJqDtHSr7dUN\nK229mv99d3m8psSg0BgbOlVGqkox32RJLrXb4pLcyMpmCuEDAOjG6bhM83690TLGDWgMdPgN\nO5DbSOWU5CVQxBSi4F5i0W+JgnvdUTcAgCuks1BBAo/tAyEujWUpJ8zrJqO2OUijUGwBipwG\nQhmQKpvdqSMW6txuBgYGBqaCY/KTnxb8/Zkm5/EZ6a58MY4Wtb31fYXNwDIzLuDJDVNiQly1\nmofQp6uGuWx9l4p5PA6L0pTRIz4L/SrDBwerimt6+1WGmBDxmtlxK/KjrC0zi6ZFfnuysaZ1\nSJAeh03ce8PIrDrHnmlJQT9epNhIzEkeca/1UxunCnjsQ4Wt1pcIoRtnRP9qtbciVOo6lRSj\nCGo7VIuyxi3Bga54hMtxRwUW8Niv/GrWW99XHCvpsI6kRksfXZspk/A+OVLT0KHkcViZcbIV\n+dHeaFqpaqFdgzV1qX+9Puup/53pkV/d5GQR6NG1WUH+41ko8XNZJ0mVBHyqvNNkJukqrRi8\nREXTwNbd5dYbI0GgFXlR961M85HU2H6lobJFrtGb4kIlyVFD1nWzM8PWzo379udG+8GF0yJW\nFcScq6SQ/ACAMn/aBZTfUvdOxeAlZqSFbN9X6aL70gpCKNrlI43PwWKjmCm4schxHBEQN2I7\nD1o4ocBLAEP90EtwvO4wSosLgXXCaK8MDAxjjFcEjp9++mnHjh1NTU16vd5h3BuXY3DNzPTQ\nmRmhZyqGbI/7i7i/uDGN7iOnyrscEkDKGwf+7+0z23+7wEWuxNSEwBCpoFvuWB2aGRfgWqfg\nsInFORH7zzlmeQj57NFnFrb3aR7/7ynFld7sunblqztLi2p7n7kzBwDYLPTyAwXvH6jaf67Z\n6lSSGi19ZE2mB91AvcSy6ZHfn2lykGZEfPY9y0cszQh47Kc2Tr1zSVJNqxIhSI7yH6UXo2ss\nVI4wAGB17BsvpiTI2no1VONumvMF+fP/sCn3iVvM7b2aQH++TMIrrO75/T9+soUHHylq+/Zk\n44v3z/C4suDiwR5jHC4Tbn9q/s5jDWUN/Rq9KT7cb8P8+NhQiWfnMFL6ldR7dCYzqdQafa1L\nZXJT0TTw9FtnbGVWJIn3n2upalH897E5bNZ4Kk0Yw6dHa774sc6mhuckBf3m1imhdtr6w6sz\n52WFHy1ua+vVhAYI5mWHW52SkyL9KcuakkfYAplA07jHZhFxYeP8R8RgJTZUfMfixM+O1rp+\n24Kp4TKJh3O4vA0x+w5LbyOoh7T0EtNXIxlN/ohbIMkc4IRg3WWwKAFxgBuOhLnAGqevNxID\nENRaBtOfwsDAQIPnBY6tW7c++uijMpksJSWFx5tgPx6TEoTgz5unf3e6ac+pxvY+rZ+Qk58a\n8osbU12sGXb8QPFk0K80HDzfcsv8BLpPcdjEM5ty/vrhhQH11bVKRKDo/26/djXmQzdltPdp\nS+qu5juIBZz/d/u00ceUbt9bqXBynvvpYvuKvKjpKcHWC/16XdavVme09WplEp7EpdWl78Bm\nEf94cObHh6v3nW3WGy0sAuWmBP3q5syoYJF7J4wIFEUEuvnZkV1IJqynMgqJ9HQqyoi4Y3HS\nz2WdDm1WYTLhmjlxozmtiM+2bjJr9OaXPruoHtpy1dSl+s+uSyOyABgODtvaNngcllXIEHDZ\ndy/zcILSKAmgWWlw2ISPFA5cP3x0qNq5iay+Q3mkqGFpnhljFQCJQECgSAKNkaeMlS+P1X10\nqNp+pLi29w/vnvvfk/PslZfsBFl2guPEbpoZs/tko9qp5/G2RYkjmkNatHRqYqD9T9Xg+WfF\niPhMSayvsGVFamas7MtjdY2dKhGfkxrj39ajqbbbD5iVEfrEyM2Vxh9xIGvD38iS/dBWjg1a\nFBCBslegcI93PiLgpyB+CgA5/p3siA2cWDA1OI4T/sC6RjwfAwPDdYvnf49feeWVDRs2fPLJ\nJ4y64TuwCLR2TtzaOXEkia9pFY4x1LUrKA/VtFGP28iIDXjv/xZaKwvYLCIjNuDGGdHDKS8X\n8tn/eHDmyfLOi7W9Gr05NlRy44xo/2F30tKBMZyv6qE8dK6yZ7pdeC2bRUy4rEcRn/3LmzMe\nuimjT6mXirnju7k6fBZPCa8/7Chw8Disuenj6QERJhP+55E5274rL6zqwRgIAi2cGv7AqnRP\nLVrOVXZTOtGcq+xRaIyj/6rbkx4jzUkKKq517E9ePy+ez/VRv6652WEfHa52rv+flRHK9KeM\nMWUN1JbPpfUNS6YP3iQxaCy4GkMkC40gs2k0kCTeeazeeby5W326ouuatX6BfvwX75/x2s5L\n9R2DLXIBYt6vVmfkUlnSuAAhePau3Nd3XTpxadBFmEWgm2fHPrgqfUTnYfA2+WnB+WlXf+Ix\nhgs1PbVtSjYLZcQGZMQGDOsspAXXHcPNhVjTC8IAFDmNSFkCrKu3a6xsAG0HYAyiMOQ3MrHM\nTbgCIn895K8fi2uNu7oBAACIm4WxBcx28bGsQMTzbmYtAwPDhMbzAkdXV9emTZsYdcM3GXYQ\nGgKgLHO/9sdFfPZtC935mUcI5maFzc2iMLx0G6PZQmntAQDOW3kTFIRgfN0TRkpuQuDGOfG7\nzzXb/mkCJby7FybJxB64aZgt2EKS7mUZRAWLnv/FDL3R0i3XhcuEnl1XO7duWcEYdw/oPCtw\nAMAf787977flPxS32Q+W1vXVtimGGUuk0ZvbezX+Ym6Id7xmHYgJEf/ihtTt+yrtB8Nlwl/e\n7C0vmLFBrTM1dqm4bFZMiNhn1SV7MMZmGi8Jk9lxnMTtBApGMBb3n16l3rkWz0pdu3I4zYyp\n0dKtj8+tapG39WoC/fjpMVIBz51HIH8R9493T2/sVNV3KNksIjVaGkrvP8XgIyAEeSnBeSkj\n2fMnLeSJ/+K+K7KashMrD5BtJcSCJ4DDB7MW1+/CyquVBVgcQySuB84E6VTCJAAA8gkJgxpV\nGx6oBaMKBEEoZAoIkoDsB2wBwh9YI9MlGRgYrjc8L3Dk5eXV1dV5/LQMYwZCkBrtX9444HzI\nIRyxX234oayzo1/LZhEJoZKFmaG+FpLH47BkfjzK9n7XvqcMXmVhVlhOgqymXSnXGkP8BOlR\n/qNXE8obB7bvu1zVorCQODJIuGlJ8uKcSDeicPhclmszXfeQ0JvXDN/zf/iIBZynNk6p61DY\np8CUNw08+ebp1x6e5VrjGFAb3v7+8g/FbVYvj7gwyaNrM932Ihk+GxcmTk0M/PpEQ1Onyk/E\nzUkKumX+BM7dNJgs7x+o2nOq0WruI+SzNy9LWTc33sfjmRBCsSHihk6KJrK4cOcHBoyxAqGx\nEDhY9Or88CO9WcRIdu9dEhcmYUw3Jje48dRVdcM2qOwga34gMlbi+m/t1Q0AAHUzWfc1kbZl\nzGboHrinElcewMo2AED+UShtJQryrY5FwCSu/Q53XhhUYQBw01GUshYFZY7vvBgYGCYKnhc4\nXn311Q0bNkybNm3JkiUePznD2LBpafIz2885DIYGCFbkR9leXmzs/+RYvemKMWRlm+JUVfcj\nN6SF+Fg1wfLp0Z//6GgpwmYRdIGCDGODv5Cbl+SxTZifyzr//kmRrcGhtUfz8ucXGzpV96+k\nddIdY/JSg9kswtlINSHc75rbv10DuuKa3gG1ISpYNCMtZJhr/h+K250zbg0my/sHq57/xQy6\nTxnN5P+9dabJLjS3sVP1u3fO/uOhmVlxXjdcSI2WWt1/JwH/+PyirYsBALR68/++q9AbLXcu\nSRrHWQ2H9fPjX/my1GFQLEDL8ihrrMwevHRJXd/+c83tfdogP35BRsjy6YNxVwAgk/BDAwRd\nAxSVUBlxHhAsGBgcwB0V1Ac6yiBxJlZS7eSpW0DbCUJPFqJ6FtzwM1m26+rLgSZ8ehsx9TYU\nUzCOs3IAt53CHeeHDJl1uPIrND0cBGPq+8PAwDBB8bzAkZubu2HDhqVLl4rF4sDAIZt+jY2N\nHr8cgzfISwn+yz3Tt+6usGVJ5qUE/3p9loA7+IXRGsw7TjSYhq7W5Brj5ycbHlt5jVZkpdYo\n5nOG3SwzWu5altzUpTptFyLD57IeX5/twlBzQGssah7oVRs5LBQpFebGSDkTxN7i+gRj/Obu\ncmf7hp3H61fNjPFqIszwCZEK7r0h9Z29l+0HeRzWY+uzXH/wkyM1O36otZk+hgYInto4dWri\ntespSusdfRAHx+uoHRasHC1qs1c3rJgt+MOD1f98aOY1L8pgpb5Daa9u2Pj8x1rfL0tZkRct\nVxs/OVxjayKLDBI+eSs3QEx5G/SYov3mnnL7hNefyzqPXGj7+y/yrf93IQT3rEj9x9B4LwCY\nkhA4PZnxGmTwAiaKXC0AwEYN1jk6HF09qu9FPitwmPTk5e+dh8mK3azIHHtvkfHFUd2wQppx\nVzGKY7ZOGRgYro3nBY5nn332X//6V1ZWVlZWFuPEMXGZnRmWnxrS2KnqVxmiQ8QOOa8VrQo9\nlbdFXadKoTX6U6Ue6I2WHT/Ufn+mSaU1cdhEfmrIgzeljUFsB5dN/HVL3rnK7sLqngGVUak1\nqnWmjw5VHylqW1kQMy/b8UHkcqfy8OUuy5XVclO/9lK7fENutJS+xYBhfGnu1vQq9M7jJImL\na3rDC2LGfkqU3LogISXKf8cPtXXtSj6XlRkn27IixXWr1L6zzQ6xEV0Duj99UPjuUwuuabxi\ndArCsGKykBhjRFPVX95ILX9UNA1gDD7eXuE7VDbLKcf1RktDh8qh12+YnK7oKqzqkasNkUHi\nG2ZEu87eHiW3LUxcPC3iUkP/gMoQGSSanhJMsOpI7CyZcQjkmUTt81U99uqGlZK6vi9/qrcl\n/izNjSQQentvhbXrECF044zo+1emMV9LBq8glEF/k/MwEgUCQf884OLQeIMHGsBCZWRj0uOB\nJp9pVMGg68eUlm/0uhIDAwODPZ4XON5+++2HH35469atHj8zwxjDYRN0eZMKqjyIK4dMzgKH\nhcS/e+dsRdOgr4fJTJ4q7yyp633j13PdjjUdETPSQtJjAn6z7XTTlYDSjn7theqe5XlRT22c\nanubxmg+UnlV3bCi0pt/qOxanxMFDB7FaCH1ZpLHInijM+DQGWiL5F0cGhemJgbaF18YzeR3\np5tq2hQYQ3Kk/w0zorlD/6/YfbLR+SQ6g/ng+ZZNS6/xMBpL4yQSGyKmUzcAwGoY4YyFxC5k\nEQYHSEz9f6PrQ3QYTJa/f1J09nK3bWTXifqH12Su9KZ4FywVLM6JtBuIx9iIwd6bg8MmUgA8\nU41yvKSDcvxYSbt9pPHinIj5U8JaejRavTkmRDxRUr0Z3KZXof/m54a6dqWAx86IDVg9O3bM\nCqBQzAzcWkw5jkSRmOAA6WRVjlhIPEa5Qu5gotgJGMRMf2isQcDiIguFdRqwmE3T6xCfLnhk\n8Fk8L3DodLrly5d7/LQMPoWET/tYSXnox4vtNnXDhkZvfv9A1R/vzvXw5Gj4+Ei1Td2wcaiw\ndV52eEF6iPVlQ6+GcoHX1K/VmSwC3y4sn0DoTJa6fq1cP/h0KOGxE2VCMdfN21F4oJAgkHOL\nCgBEBftu9G9zt/pP759v79NaXx483/L18frn7s23xRVjDM3djt0iVhqdukicWZEf/eWxemeJ\nZ93ceBefSgiX/EDxSA9xoZIxayubBCRFUEvDbBaKCx2xM+VnR2vt1Q0AMJrJN74py4yTjWG4\nNZtNZJK4D4MKgEQgJFCwBx89+5TU66sep+IsNouIZ9w9rw9OlXe9tKNYbxwsFz1Z1vn96aaX\nHywYG49wFJaB0pbjqiM2q0sAQPFzUGwBIIQiF+KWw44fiZgHbJ9oiqRGRO975eLQmIMCEnEv\nlQFKwKCBUY9C//Xx+vpOFZtAqdHSW+bFi5kaWwYGBjs87ywwb968ixcdu2QZxh4LiXefanz4\nPz+v+9PBB1459v6BKp3RY7vZ6VH+XKpd99hgUYCYoj+luIa6sLC4duwKDk+WdVGPl19tlVfT\nb/hrfKwWYOJitJClXUqbugEAKoP5UpdKRxPoe038RVzKdOGIQGFu8ogf2oxmct/Z5je+Kfvf\ndxXHStrxyPfbhwPG8MKnxTZ1w0pHv/aFT4tsV0QI2DTmL5R/fQ4E+fOf25Jn72DKYRObl6fc\nMMPVBuOK/GjKXfGNCxOueUUGG2kx0qlUuTMkCfWdypGe7fCFVudBC4mPFrU5j3sVAgWyUBwL\nJRAozLMbawES6r3ZQD9mz/Y6Ra0z/evLEpu6YaWjX/vazktjNgciYxWx6LcoeTGKyEZJC4j5\njxE5G62teih0JkpYD7wrLVpcPxS/GoXPG7O5uQGSRiF/ilpUJItDkmsHLY8ZKG4ZsB17MJE0\nEQVnAsC5qp6HXju+53TTpYa+4rreL36qe+DV4/UdI76vMjAwTGI8X8GxdevWNWvWREVFrV27\n1sFklGHMIEn8h3fPFV2RFTR6dVNX7YlLHf95ZI5HanolAs4tM2O/ONVov20u5LFvn0O9Oayn\n0Vb0RvOYNfYr1FQVjwADqqvjIvoiAqG79QUMDrQr9SanMhkLiVuV+mR3PVmeuCVbpTXZ62WR\nQaI/3p070vTZunblXz+60Nl/VXfYebzhr1vyZDSrL7epbVNQPpA1dKqqWxWp0YMPzVMSZecr\ne5zfNhyTUevbtj+14Fxld0u3JkDCy00OumZii7+I+8J9M/71ZYnNalTAZd+zImVotwLDtVk1\nK6bEyeeVxPid7y//59E5wz+PhcSUFjMA0DWgpRyfiMzNDqPUceZle2LdZdEAwQHkKx6KDMPh\nXGW3WufUAwJwsa63X2Xw+D2ZDiSNQlLqBlUky0SyTDDrALBPF25cBRHT7yHPv4tVV/d1kF8E\nkXv3OM6JAmEwkfsIrj+AB2rAYgSuBEUUoKi5AEhvtLy2s9Qqe6ErNh0KjfHYp5ebAAAgAElE\nQVSVr0q3PjZ3XCfNwMDgQ3h+zZaYmAgA999///333+9wyEt7oQzOHC1uK3Iqmmjt0ez4ofbB\nm66RcjJMZqYExwSJjpR2tA1ouSwiPlSyfGqEmE/9jaLrFJD58T45UiPis6ckyJIiqYu6PUWQ\nP99hw9xKiPTqki8+SMQmkNmp2SEqQCDkMv0pnkFFo3YpR1EjIxZwXn6w4HxlT3lTv8FEJkb4\nLZwaTlf+QIfZQj43VN0AgKoW+StflbjIVXWPTqq0y8FD/VqbwLFleWppXb9haG1LUqT/8OUG\nHoc10iViarR02xPzS+v7mrvVMgkvK142ZmuJyURNq4JyvLJFbjBZhu8jwCKQkM/W6in+OiaT\nA8WsjNDleVGHCodoHKnR0tsXJbp/UmwBdQnWlABpBADgBCP/WcD1oZ1qBhc4dydZwRh6FXof\nuimxr6Ea+xaiQGLBU7itGCtaAABJY1DENEC+lxPHD0AZdyDAYDHaW2+U1PcpNBQecA2dqpZu\ndTSN8xQDA8P1hucFjj//+c8eP+dIUSgUv//973ft2iWXyzMyMp599tn169eP96TGlDMV3dTj\nl7s8JXAAQIRMuHnhsJ4+V+RH7Txeb3KKdege0H98uBoACAItmx71+PqskS5Kh8/CaRGfHa11\nHl80LcL232Iee0FKyA9VXfZanJDLWpIa6qVZXYdQeWUAeEIAzU8Lzk9zPzDyYl1fRz+FBFZY\n1dOr0F8ztWREiAW0916JnUdvcpT/aw/P2rq7vLxxAADYLOLGGdH33pDKZnm36onNQrnJQW40\n+DDYcCitt4ExjEjgAICZ6SE/FLdTjGdMqvvSUxunzs4M3X+upa1XE+TPn5keunp23Gi+6njg\nCOgbrqYxGHtw7/co8Abg+bANJMMVpFTtrtc8xHBtEIGipqOo6eM9j+GAHIxF+5TUpbgA0Ks0\nMAIHAwODFc8LHH/5y188fs4RQZLkqlWrSktLn3/++cTExHfffXfDhg27du1au3bt+E5sLFHp\nqFNOlBqKgs8xICJQ9IdNOa9+dUlpi19BAHbrWZLEB8+3+Iu4969Ms46odaY+pSEiUDjSLgM6\n7licVNE0cLF2SNH4pqXJWfEy+5Epkf7hfvzC5v5etZHDQpFSYX5cAJ/NlG94DDGXpaIq1nDb\nZNRTdFAV+AAAxtDep/GswJERGyAWcJwLsMUCTmZcgP1IUqT/aw/P1ujNAypDmEw4mvWe0Uzu\nOtFwobpnQGWIDhGvnh2bk8RIGN4ihsb+UybhSQQjW57de0NaSV2/gw3nomkReSnuy3m+yezM\nsNmZFH467mDsAH3jkKxJBAAkVpxGIYzAMQHITwnhsgnnuOvkKH/7ukuG6woZvbYlkzCyFwMD\nwyCT0FZg165dJ0+e/OCDD+655x4AWLFiRW5u7lNPPXVdCRyhUiGAY/s3AITK3HwsqGtXHi/t\n6JbrQgMEC6dGxI3cxH52Zlh2fODx0o6WHnW/0vBTCcWG5N4zTVtWpNa1K7buLq9slgMAi0Ar\n8qN/cWOqn1P07EjhcVgvPzDzx4ttpyu6+5T66GDRjTNi0mKkzu8MlvBuzGTKmL1FuITfpTY6\nhGUigEg/TyoIbiCiabACABF9bJB78DisR9dm/uOLEofwl0fWZFLu7Yv4bBfTGw4KjfG3207b\nYlmau9UnyzpvXZDwwCqPlXQx2LNoWsRHh6pVWkcN6+ZZsSN1HQoNELz1m3kfH6o5X9U9oDJG\nh4hWz45bNp0JrnYFNlD8xAAAmAeA1AHBrJB9HZkf76GbM7buLre/SYr47CdvyR7HWfkuZiNZ\n+B3UFmJVH/IPQRnz0JSlQEyqvRmMcYhMKOSxNQYTgiG30egQcUwIE67EwMAwiMcEDrVazWKx\nBAKBWk2bXygWj0Xx2Lfffsvn82+//XbrSxaLdffddz/99NOlpaVTpkwZgwn4AkunRx4sbHEe\nd/1MTJIYA7CcwiDf3Vf51fF620PGFz/W3bkk+e5lySOdlUTIWTUzBgDe219J+QaN3ny+qvv5\nT4psmzYWEu8721zZLP/vY3NG372CECzOiWTsEscXIYeVESKu7dPor/wrc1lEgkwo4Q3ejjDG\n+8627DvX3N6rCfTjz8wIvXNxknB0y/vhMDUxkM0izBbHDcMgf741lhJjaO5WtfVqrSOjrC1a\nnBMZGST64GB1VYscAFKi/O9ZkZo+VHFTao1qnSksQDj6iNb3D1Q5h85+dax+TlZYRmwA5UcY\nRoOfkPvclvyXdhR3XfFbIQi0qiDmjsVJ7p3tkbWZAJku3qPRmzlsYjgJO9cFmN7Tx8UhBl/i\n5lmxKVH+n/9YV9euFHBZmXGyu5Ymy5hgHWcMWvLLv+K+QQsb3N2AuxtQXSGx7neTRuM4VtLx\n1vcVvQo9gYAY+jQo5LF/c0v22NjVMzAwTAg8tmaQSCSZmZllZWUSCa2GOjYmo+Xl5cnJyTze\n1Z/A7OxsACgrK7t+BI6piYFbVqR+fLjaYrf1sSQ3cvWsWMr3X6zt++BgVU2bAgASI/y2rEi1\ntd8fK+n44qc6+zdbSPzx4eqUKP+C9BD3psel7z//5kSDc0lqfYdy/7kWiYDT2qsJ9OMPJwyC\nwZeR8jm5Ef5Kg1lvInlswo/HtslqGOM/fVB49vKgiYxGr27uVp8o7Xj913P8Rd4tQA3049+9\nLPn9A1X2gwSBHl2bSRCosVP1768vVTQNWMdDAwSPrs1y+0/ASmq09MX7qe1Li2p6t+2paOpS\nAQCPw9qwIOH2RYkjMm5w4MSlDsrx46UdjMDhJTLjArY/teBUeVdzt1oi4ExLCkwI9/P4VTCG\nQxdaPjtS29GvJQiUFOH3wKr0YebsTGIQO4D6gYPgAuFmWhPD2JMaLf3zZvfdInBfK758AuTd\nIA5AidNRtCuJcOJCXvjepm7YwC0VuPwYyl48LlPyLIcvtP7zixLrf5MYsIUkEBIJOJFBovQY\n6W0LE+lyphnGhr6+vhdeeKGwsLC4uFilUn388cd33XXXeE+K4brGYwLHa6+9FhQUZP0PT53T\nPfr6+hISEuxHZDKZddz1B8vKyjo7OykP6fV6mGgpMHcuSZqZEXK0qK21RxMs5c/ODKOzDNx3\ntvnfX18Nlq9slv/unbOPrc+6aWYsAByiqgQBgIPnW9xe3U1LDPyIajw+THK5WU75kbe/v2zL\nkuCyiQ3zEwgCVbXIMYaUaP/18+JH38PCMJYQCEn5HHDqSjle2mFTN2x09Gs/Plz96Nosb8/q\njsVJcWGST47UNHSoWARKi5HevzItNVoqVxuffuuMvXl714DuLx8W/vOhmQ4eLh7hdEXXcx9d\nsKmTBpPl0yM1de3K57bkuXdCswU790pYsY9JHkvUOlNhVU97nzYkQJCbHORDmQgehcdh2dsY\ne4O3917++ni99b9JEle3Kp5+68wfNuUsmOrd63oKhcZ4qaF/QGWIChZlxwd6zEBXEA+qs91y\nQ4eCHSS2hPuZCeumrzDDFzMjGLwAPr+HPL0TyMEnB3zxIEqfRyx/cBJ+AeqLKIdxfdHEEDhI\nPWirsVkOBB/xox2ijjDG7+2vGjoCFoyVGiObQAIu63KzfHbmpHJcnnB0dHR88MEHubm5y5Yt\n27Vr13hPh4HBcwLHE0884fAfvga6Vvna6tWrGxoaXLzhmhKJr5EQ7pew6hobhnqj5e29l53H\n3/m+cvG0SCGfTZkrAQCUkavDJCteNjcr7OeyIXISi0APrEr/4/vnKT9in5RpNJOf/XA1D6Ww\nuue7001/uzef2YieBJxxUjcGxyu6Hx0TF51ZGaGzMkLNFswirt40vjvd5BxNZyHxjh9qn7/P\nwwmyALB9X6XFKWzmTEVXSV2fezvzbBbyF3Ep0/U8a546TI6XdrzxTZltPkI++8FV6SsLYsZ+\nJhOdjn7trhMUP1vbvquYlx0++s4mb7P7VOP7B6psIbgxIeKnNk6ltEYaKW19xjd2xxfVDob1\nxgWZfr1InpUcjyT5oz85g++D2yrJk184Dl4+gUPj0bQV4zIl74H1NL3hdOM+hb4By38azHIG\nwOpiECQh6SKbDtU5oHOwWLbRrzL0qwxFNb03zYx9bL3Xt0AY6MjIyLAukY4cOeJZgYPEk9As\nkmEMmHQyNkBgYGB/f7/9iPWltY7DBfX19ZgGlUoFANYSlUlGRdOA7eHSHp3RbE2mpLNXFAtG\nZbv4+ztz7r0h1XaSlCj/fz40My81ODbUHZsoldb00o6LzmtChgkHXaHB1fydMYHNQvaSaHUr\ndWFRVYvC45ceUBtanMwyrJTW91OOD4cFUyl8cxFCY7/PX9umfPGzYnu1Ras3//vrSxeqe8Z4\nJpOAkto+ytLCfqWhsUs19vMZET9dbN/6bbn9D1Bzt/qZd88NqEdbVWS11LWpGwDQ2Mv53Tch\ndZppk3D3noEKXHFiROMTGiShCVTy81DQEjaBvgk0ZaBvAuzRMD6LCg8ctakbg+hqsfqC7ZXZ\nqW3Zme/PNJXUTbA9yMkEQTD3VQbfwsPCmFqt/uijj44fP97e3o4QioiIWLBgwd133y0SjV3L\na2Zm5s6dO/V6PZ8/uDNZWloKAFlZjLhLgZYqsNOKRm8CgBlpwVYTRAdmpI3qh5PDJu5YnHTH\n4qRehV7EZwuuGEyumxv3ylelbpyws197uXkgK87z/QIMYwmdu0pogHCMZzIcMHheUzPRP8yZ\nzBa6Q9dky4rUy83ymtYhiszdy5JTovzdPqd77DndSKlFfnuycfqkyz31Nnoj7VfCxSEf4asr\nnTX2qHWm/Wdb7lzijhWrje9ON/U79V6ZzPizo7V/2jy9tk3xQ3F7e582JIA/NytsSsL17lcy\nOVFSC6aYZnxCg9Ln4o5qynEPnF3fgOU/A3mlaJclRP7zgB/ngTMDgK4WMNWdSlsJV4qtwmRC\nAY+to39YtXKyvJPxHmJgYLDiSYHj3Llzq1ev7urqAgCRSIQx1mq1n3/++XPPPbdnz568PDe7\nx0fKunXrPv300x07dtx7770AYLFYPv7448TExOvHYXRERATSrhsjg0QAcMu8hOOlHU1dQ7aU\nEyP81syJ88gEHCrkV+RH96sMnx6psVmN8rlsvXFYpvc9cuoiRoYJxJLcyO9ONzmPL88bz1DM\nlCipszMIAKRFe6CW3oFAP75YwFHrKHbJ3IhntiEWcF5/dM7eM82F1T1ytSEqSLR6dpxHegFG\nSnMXdX1KY6evVxz4IFEh1JsHBIGsN3CfBWNo6KD+F6/vUI7y5JU0Xk4VzQMfHKz6/Mc6WyjY\ntz833jgj+olbsq/ZxMowXmj05oZOpcWCE8L9JMJhl47yaWL7+D79d+EeKHsR6qzFFcfthgiU\nvxrFTR3tqY3duP8IwFXZHVu0MHAYBa0DjgeKmrGZpgrSogVsAsQBAA6bWDM77vMfa6nfeQW5\nekzLPBk8wiOPPHLgwAG6owbD+HiEMUwCPCZw9Pf3r169miTJ7du3r1mzxtrN0dPT88033zzz\nzDNr1qypqKjw9x+LrcJ169bNnj37scceUygUCQkJ7733XllZGeN5Q0dCuF9qtNS5RiM5yj8p\n0h8AhHz264/O+fRo7fHSjm65LlQqWJQTcfuipNEEOrjmjsVJS3IjL9b2DagNUUGiXqVh67dl\nw/lggHhy+hSOEoXG+P3ppvoOFZ/LyowLWJ4X7TEbPy+QERvwwKr09/YPMaGYPyV83dz4cZzV\nzbNi95xqdPCwYBHIvchP17AIdPOs2B0/OD7MhUgFc7LCRnnm1bNjV8+mjlIaM+jidV2EKzHQ\nMS0xKCpY1NqjcRifPyXc26lDowQhYBGIsiZp9DcokqZd0WCyfHbU8S9r/7mWtJiAG2dEj/Ki\nDJ4AY2U5qC5jkxzYfqQwZUeh6Mtj9VYTLjaLWDc37p4VqcPJQkbxObjmLMV4Qq7nZz3uIIJY\n/hBOn4trzoGqD6ShKG0OCk249gedwQYAwqosAADWXLJXNwAAAQAmseYSki4a9bwBEM1tChGA\nrq5Q7lmRYjRbdp+krv6zEiJlwvUmHlu2bJk6lVaGe/TRR8dyMgyTCY8JHO+++65Cobhw4UJG\nRoZtMDg4+MEHH5w9e3ZeXt5777335JNPeupyLiAIYu/evb///e9ffPFFhUKRnp6+c+fOtWvH\nxJ9wYvKHTTl//qCwwW77NDZU/IdNubYNLQGPff/KtPtXpmEMY7PLFSIV2HbsjWZy/9nma+7p\nBYh5mXG0JqMd/dqD51vaejVSEW9GWkj+6PprJhAXqnue/7TYVg5w+ELrtycbX7hvxrhYS16T\nAbWBRaBbFyTMSAs+VNja0qMJ8uPPygzNTx3nfy+pmPuPh2b+e2epLeXHGhPrjQgVANi8PEWt\nM+0922xbp8WHSX53Z473VMWxZEpCIGWz9FSmU2DksFnoL/fk/e3joiY7x428lODH12eP46yG\nSWZcQFFNL9X4aP+skiL9CqksXXhslgYo6gEPF7YyAsf4g0nc/g3WNg6+NCnf+dH4zaWrXwaz\nhfzqWL1cbXz6tmsXJqD0OajyZ9xcBgCAretyAGkYMWPSPg2i6MxR5OBiMNRhXelgKwpLioS5\nwIkAE43xk8kzhheIH401lygO8KKv/JsBALAI9MubM1bPjrvcNHDoQmux030DIbRwguRGMdiT\nn5+fn09r/Pz444+P5WQYJhMeEzj279+/ceNGe3XDRlZW1q233rpv376xETgAQCqVbtu2bdu2\nbWNzuYlOmEz45hPzfrrYXtUqBwwp0dKFUyMo99DGpYaXyyb++dDM9w9UHTjfYraQCEF8uF9L\nl9pkubqrwGYRv7l1Ct3O8N4zzdv2lNt6XnafapyTFfbMnTl07580aPTmFz+76NDs0Nip+vfX\nl/7+Cx+KEsAYDpxv+ehQtdUpPUwmvO/GtAdWpY/3vIYQHyb59yNzGjqVHX3aIH9+fLjfcHYR\n3YNFoF+vy1o9O66soV+lM8WHSfJSg1k+n4gxHDDGIj6bwyYcrEb8RdzbFyeO16wmNDEh4v89\nOe/nS5117Qouh5URG0CXCO5rbF6eUlrfb7YM+SZEBYtG349286zYPaebHPyzWQQSCzjO3hwA\n0DngfigYg6fAyrKr6gaA0sDaU0axaXGkqHXT0mQX3bWDIIJY+3+45DCuOIYHOkESiBLziBlr\ngeuL4v64g7VFoLcL1LPIseoHJJpNb8rrod8jXjTw40E/NAqK4CK/Auf3RgQKIwKFszND//Du\n+bLGq8oLQaD7bkxLirxGaCADA8P1g8cEjoqKijvuuIPu6Ny5c//617966loMHodFoCW5kUty\nI8d7ItRIhJzH1mc9vCazW64NkPAEXHZHv/bTIzVVLXKMITVaeueSJLqG84ZO1RvfljlULJ8s\n6/z8x9q7l6WMyfTHjXOV3ZThI+erehQao8lMfv5jrTUHJC1aetuixGuWdRjN5M5j9cdLO7oG\ntOGBwkXTItfNjWOzRrvOf/9AlX17bWe/9vlPiwZUmWvnxo3yzJ4FIUgI90sIH6OnqNhQcWwo\nTRs5PSV1fV8dq2/sVEmEnOwE2aYlyb7TqoAx/OXDC6cruoaMIshNCvr1uiymwNhtWARaMDWc\nMijHl8mIDXj5gYI3vi2z2a8snBbxy5syRl+pFCwVvHh/watfldjco2QS3q9WZx6+0NpMFVHk\nJ/SVv5HrGnWN/au6Xr4FU6yiMYaqFvm1BQ4AIFgo5waUcwMAgNmIL3xPfvFnrOxBfsEodTaa\nvhJYowqD8zrYCIZGbFEigg+cCGB7zUOdVIO+kuL62iJgScBEUWblQZBsKWgqsKYMzEogOMCL\nRn4FwKI1nBLw2K/8auaRorZzlT1ytSE6WHzTrJgx+11mYGCYEHhM4BgYGAgOpi0jDw0NdYhu\nZWAYKWwWiggcVDHCZcKnNg7LPetoURtlP/ahwtZJL3B0y3WU4xjjY6Ud2/detuUsVLXIDxW2\nPndvngsTcp3R/Js3T9e1D/YK1bYpa9uUp8o7//HgTMpaGKOZHE6NQ59S/9WxOufx9w9U3TAj\nms+dDE0ZY8YnR2o+OjTopd8t19W1K38sbn/1V7OiQ0YslHiDE5c6HNUNAMAQESjycUdMBi+R\nnSB7+zfzuwZ0/SpDdLBolOnj9qTHSLc9Mb+yZaC9Vxvkz0+PkQp4bKXWeK6Swi14ZkaIp67L\n4DbYMuQHy0Xsu4UkW3s1LAKFBQiG5Q5r0pNfPod7Bu2rcV8rPvUlqr9AbHgW2L6qbRlbsPoU\nkAYAa1hXMfBTkHiGx0on7DF1AWUiGNYD0Px2WDxY9ESAKAuJsgBbAA3rFx8htGx61LLp42k9\nzuDAnj17jEbjpUuXAOD8+fPWIMv169czCbIM44LHBA6j0chm056NzWYzXrgM40IXTe1xt1xH\nkpiYgGX/TV2q2jYlQiglyj8q2NWyUEK/Wvj0SI1DiqTOaP7nFyUf/m4RXSvEruMNNnXDRnnj\nwN4zzfalFiYzufN4/d4zzd1ynVjAKUgPue/GNBe1IWUNA5S2YTqjubpVziQ4Dp/mbvUnR2oc\nBhUa49bd5S89QFHuO/ZQJtEAwJnLXY8BE+N9/RIaIKDLhx4NbBbKipPZZ4ffOCP6x4vtZQ1D\ntlviwiQbFzDtUeMPYoux4aoAmhCoRwgw1bp76+4Kq+2ov4i7ZUXqimv1NOHiAzZ14+pgZx0u\nOYKmrxztvL2BRY1Vx4fmp2LQVwFLAgKKTvDRgl0E1dEcIr0QWjc8dYPBN9m8ebNCMZiJ8/rr\nr7/++usAoNPprEoHA8MY48mY2JKSErrvcUlJiQcvxOCzKDTGhg4VBpwQ7ucjhfF0W4IiPmfC\nqRsavfk/uy79dLHd+hIhWDY96tG1WXRlDnmpwWwWMlscnxDDZcKOfgrdp1uuq2yW03m1nqFf\nndoEDpLEz7x7zmYhqdaZjha1FVb1vPHrOWEy6opiI2WOAgAAGIwk3SEGZ85UdFEWKxXX9mn1\nZiHfk3d796BsmAIApYaJ92MYC9gs4h8Pzvz2ZMORC23tfZrQAMG87PDbFiVODvveCY8kDTRX\nq/kCBJYbUuX7Kx2jrBFCBqPFWseg0Bj/s+uSSmfaMM9VzBZuKKYZL/ItgcOiB30/cMTY3DBU\n3RgE66uQNwQOFl3EIaJtFWExNXcMQ5DLqcO5GRjGBU8+8j777LMePBvDxMJsIT86VP31iQar\ndyCbhdbMib/3hmHFuXmVgrSQvWeaKcbTJ15N8ks7iu33wDGGQ4WtZgv5uztyKN8fIhVsWZG6\nfd+Q3loeh7U8P+rDg9WUH+lXDW7LmMxkW6/GX8y1he+qaFan9uGpP5W0OwdkKDTGDw5W/+6O\naZQfj6HvnogZuQOFx8EAWrPeaDFjAA7BErF5BK3p2jijoJEJMMZKrdEXBA66XfpQGvGLgcHj\nsFlow/yEDfPdStBk8CZIkga6Zqy4mqnx8OwuvkC6pwRZq/wQQgiBc8H7Z0drbiqIcdHPiPWO\nOcqD6CkMWcYHoxK3HMYDFYMv+RIkFQGf5/g2i8ouEsZzcEKBJQWL0wKVG4u4iVhDYc8BAqbo\niWEsIIFRnxncwWOPvO+//76nTsUwEdm6u9xeRzBb8NfH65WaYcW5eZWC9NB52WEnLnXaDwb6\n8X9xQ+p4Tck9mrvVlBX+P15sv+/GtGAag8aNCxNToqQ7fqita1cKeKzMuIAtK1J75LTFpYF+\nfI3e/OHBqu/PNFlLP+LCJI+syZyaGBgSIGjvo6j7sC/NuFBN7UZ2gSqy0UpKlDQtRlrZ7Pho\nNSsj1BtV6yPCgsk+g8pMDu6k6S2gMesDuGKeT1rTBftT/9/FZhEBEqcH5fFgaW7UnlOOheIA\nwLRSu4FWb/7m54aqVoXFQiZH+a+bG+8jRXPDpLZN8eGh6spmOcaQHOW/eXlKeozjXj3D9QYK\nWQ7iFFBdxiYFYvtxJam/Sku8daW+tk1hIXG3Qv/O3svOn9IbLbXtyiz6nHjkF4wHOigO+PvG\nPofFSFZ9BIaBqyN6Fe5So/Aw4A79o0Zsr3hwAELiBVh9Aix23VucSCQqAMQB8TRQXxzydm4o\nkuR6YRoMDAwMnsFjAseWLVs8dSqGCUe/0rDvbIvz+JGi1s3LU8Z3mYoQPHtX7v5zLd+faWru\nUsv8+DPSgu9eluKlxYBSa2zuUvM4rJhQsWfLnuud/C+sYAz1HSo6gQMApiUFTksa4mQR5C8I\nlgp6nCxIQwMEyZH+T791pqLp6pNWY6fqd++cffH+gqW5URdrHaszAGDZ9KvhOxo9db8u3TgA\nIAR/unv6858WlTdevWh+avAwTWS9ityosakbVkiMB4zqUL50WM52Y8vc7LDt+y4bzY59PXOz\nw3ykAj8tRvrgTenv7a+0b5ualx3GbKePlLp25R/ePWdLPD1f1fPd6aa/3pOXFe+1nAWPcry0\n44XPim0dVReqe4pre3976xRG6mJAwjgQxtnfXoP8+VYXp0OFrXSfcggbdjxn+jzcVEo1Pt/t\neXoQ3Fs8RN0YHMUgV0DIUPN+jteSklgS5H8jGFuxZQAhFrCCgBNqPYL8CoAfgzWXwaIAQoj4\nMSBM847OwsDAwOAZxr9omWGi0K80XKjp6ZHrIwKFeanB9t4WNW0KTGUFZo1zcy1waPXmunal\n1mCODRXT2TSMEoTQyoKYlQUx3ji5DZ3BvH1f5d6zzdandomQs2VF6s2zYkdzToPJ8vmPdT8W\nt3UN6CQi2qqBkZqJsFno/26b+qcPCnWGq7qDkM/+f7dPO3O5217dsGIh8bv7K19/dM7l5gH7\nOh2CQLfMi5+dGWYbiQyi/hd0nZER5M9/9VezL9T01LYpCITSYqS+4C1KYtJgMVGNYz1pErB8\nbrc8yJ//xIYp/95Zaq9xxIdJHl6dOY6zcmDD/IT81JBDhS2tPZpAP/6sjND8NNr4LQZKMIaX\nP79oUzesqLSml3Zc/OD/LRx9bLO3MZnJN75xjO4mSfzm7vK5WWECHlAvYZoAACAASURBVPNY\nwkBNXBi1HwRCKM5lPyNKm4U6a/HFg3ZDBMpdiZLyPDtDN1FT7A8BANbrh/y0Iw4SebVuAgE3\nGkE0xRFuOOJOsBRqBgaG6xnmSYJhWHx3umn73kqdcXA9LBVzH1ufPTcrzPWnrsm3Pzd+dLha\nrRtcRs6fEv7I2kyb6cPE4m8fFxXaNWKotKY3vikzW8h1c12Zn7nAYLI8sfWULbhErqJ2WGCz\nUGo0nUMYLVMTA99/euGXx+qqWuQIUEq0/8aFiTIJb9ueCsr3V7fKTRby8fXZi6dFHitt75br\nw2SCJTmRqdFDqsqX50XvOtHgnIqysoDqmckOhCAvJTgvxYfWuhZMuyVoIX20LXRpbmRmXMDu\nk42NnSqxgDM1MfDGGTFslm9ttcWGih9YlT7es5jANHQqGztVzuPdct2lhv6cpCD3TqvWmfqU\n+vBAkbeNk6pa5JR+MRq9+VJD/4w03+gaYPA9kiP9s+ICyhodJfhF0yKk13hsQMTCzTh1Fq45\nC4oe8A9BqbNQqM8UjtH+1hBA8IHUA2IBJwKJpgPLb0wnxsDAwDAxYQQOhmtz9nL3G9+U2Y/I\n1cYXPi3672NzE8L9ehV6td6EAGGnHHWEICWKdu397cnGN/eU248cL+3o7Ne+/uicUeabmC34\ndEVnY6dayGNlJwS6mAMAdMh17QM6AIgMEITRN3q4pqJpoJDKZuKTIzU3z4pzb4W5+2Sjcyyr\nMxvmJ/oJ3akmkPnxfnmzox+7yanBwQrGYDaTXDaRnSDLTqAtg48NFT+1cerr35TZ14asmhmz\ndk6cGzMcX1yYiRK+159iI1wmdP5nZZhMuPDQcXHIBTWtiq27y62lWwSBluVG3bcyTSr2Vo2S\nSkdRGDV4SEt7iOH6RK0z9Sj0oVKBkM9GCJ65M+eVnZfsTZ0WTo14dM2witRQeDIKT/baTEeB\nIATkVRTjwnAk2wjYCIjDtIQwMDAwDB9G4GC4NrtPNjoPmi1414kGADh8oZUyqR4AluRE0nWd\nYIw//6HWeby6VXGusntmRqjbs61rV77wWXFL91V39KW5kU9umMJx2pbUGS27L7SUtypsI1nR\n0jXTo/gjNyy47NTWYUWlNbX1qmNDaYLWXHK+itqYEyFkbQgS8Nh3LU3eMN/NChFK6GqAQwME\nw4zhWJIbmZMUdLS4ra1XE+jHm5EW4lDlMVFgIYJNsBw8OAAAAfimySjDdYIL6cENVaK2Tfmb\nbacNpsHvOUnig4UtlS0D/31srpesW1w0LYYHMnk6DIO09mi27i63aRkF6SG/Wp0ZESj825a8\ny83y+g4li0Cp0dJ4mt+sCQQKno67z4HF4DgeNgsAAPlcOyQDAwODj8MIHAxQ06q4UNMrVxui\ng8ULpobbm2tYaeyiKIcGgJNlXRo99YYbi0A3zYy9f1Ua3UV7FHqHHnIbVa0KtwUOvdHyx/fP\n9yqGbGMeKWrzE3Gdt7W/Ptdc1TGkRKKsRU6S+I7ZcSO9rlNPxlWc+zWGiZpmnzM8UPiHO3MQ\nQjGhYo8Xky/OifjkSLVc7VhAvn7eCGQUmR/v1gU+U/1Lg0Jj/PBg9bnK7n6VPiJQtGpmzOrZ\ncayhpUNSrqhPr3IoTfLjClm+mhSr1Bp75PrQAIHzX7Ez/UpDXYfSYiETI/xcmNQy+BrJkf4h\nUkG3k0mwn5DrhnnNx4erbeqGjaYu9aHC1lFaCNERH+aXFOlX2+ZYnhYTIk6bmGLo9YnBZPGe\ne3G3XPfE1lNKu2zys5e7a1oVbz4xTybhpcdIJ1XmDldCJN9BNn4H+is23iw+ilqCpBMs642B\ngYHBR2AEjusaksRvfFu2/1yLze/tg4NVT22cWpA+pAuay6Z+iKFUN3hc1u/vyEmJ8rfano8x\np8q7HNQNK/vPttx3Y5p9EUevyuCgblipaFMMaIwBI8xYSYqk7owVcNlRwa7Mz1wQLhNStqhE\nBgqTXTbdjAaxgPPCfTNe/vxiU9dgCQybRWxcmLB2jifrRMadHrnusf+e6lMOflWau9Xb9lSc\nr+x5/r58+3gULsEOEfirTDqjxYQB2ARLwhFwCV+8bTZ1qf77bXlJ3eDzcUF6yMNrMsNp6qfM\nFvL9A1Xf/NxgTTNBCK0siH7wpnQB1xf/pzE4QBDoN7dO+dP75+3dZNks9PgtWXzuiBeclxr6\nKcdL6/u9JHAgBM/cmfvse+fsY6dDpIJnNuWMsjmRYQxo79O8s7eyuLZXZzCHBgjXzolbM8dR\nGh49X/1Ub69uWOlXGb4+Xj85HXzE0UTmQ1jVBPp+4IiRJBbYjOjMwMDA4CbM4+x1zZfH6u1D\nMQBAoTH+/ZOid59eEGK3ozslUdbepxnmOQ1GS5hMcE11I9ifL5PwKIs4RrOJ19xNXWyiM5p7\nFLqIwKtZHl1UOoiVTrlupALHtMTAtBhpZbPcYXzN3Di3iyxW5Ef/XNZJOT6cj6t1pqYuNZuF\nYkLFI1q4JkX6b3tiflFNT0u32k/EzY6XeSndZhz58FC1Td2wUVjdc7y0Y8HUCPtBFiKkXFcR\nML5AR7/2yTdP25f8nL3cXdOmeOvJ+ZRxyNv2VHx3usn2EmO890yzUmP6491etehn8Bi5yUHv\nPLXgk8M1lS1yiwWnRvvfuSQ51mWQBB10tjsms2NZhweJCha9/dsFB861VLXISYxTovxXFsT4\nSJgxgwvqO5RPbj1tsxvv7Nf+77uKSw39f9483bMXotPd6MbHDkyCqRfMSmBLgBMEyHNfWsRC\nfgng57XiR2wG3WVs6gXAiC0DYQbT/MLAwDBZYQSO65q9Z5qcBw0my5ELbXcuSbKN3Lk46WRZ\np4P9m5+QoxyFIRxC6I4lSVu/LXcYT/3/7J13YFvV2f+fc6+2ZNmW99524tiJkzh7b1YIBMpe\nbYFSaGlLoZSut/N9eWl/bWlfKBDKHmUTdgiE7OXYjhPvPeQpW8va0r3n94cdWZbutWVbktf5\n/BU9V/feE2ud8z3P831SIlbkTb6VxhgbmF6zZ4pfdpjEZhRC6Hd3Ff/t3Yunq3uHL0KjpYvi\nYpKVnQZrUvhktmJWLYy9fUfOG183uotcKAp9a2PmxsXjdGtzuNiXD9R9cLzVxbDgYdWB/LbG\nFNBo5YLYKbYzKKnTVLZobQ4mPT5s69LEia5eMADDYkFwdnRLarn9Tc7WarwEjlnBW980+RY0\naY32D4633LXLO8nZYHZ8eqYdfDh2sbujz5QSO8mEI0KISVDJHrlxydSvkx4fVtfhrcwCvx1P\noBAJqKvXpgEEJUmEECSe+6TGrW64OVHZc65eE9geWEM/Xr7w6XEhwt6Ftd+A85LIIoxEkVtA\nkjSdQ/ITlx7rvwR2eKcK29vAWoPCt4GQNC0iEAhzECJwzF9cDNur867iHqLdw6ETAOJVsicf\nWPevj6rO1WswBopCm5ckbiiI/92rpb7nyiSCFP+KMvasTccYXj5QZ7YNT5iG2sROJUt5SVY0\nAIcbeUqsIko5KqkkRSWnEGJ9/FFpCiVPyuguUiG+bmdORraqX2cTCKj4KJlMJmQByjsNKplI\nOqnNydt35K4vTDh8vqtXZ4mNkG1cnMBXC+PJX9+pOFTe5X5otbv2fVpjsbvu3Jk7iTFMAovN\n9cfXRjXNff2rhl/etszPqmmzg6nTDGotThZjsYBKjZClRUoD27jExOMdw+d7MsOpnMhuZ1OX\nkeXxhWnoNBCBY75x7fqMx98s9wpKRPQVq1KnZTyEGYuLwe4iOC/O1QVY4EiPD/OahwyRmTB9\nfVKdOty3H7BrVESzH8XfCMIJe9+EGGw84lY3hmFt2HgEqa6DmWopRSAAAIvJQpUwGcj7Zv5C\nU5RQQHHuh8jE3m+M5Bj5n7670upwafS2BJVMKKAwhsWZUReavac7N2zK8u1Xwsc169J3Lk9u\n7DJYbK70+LCpl0IsTI3YXJR4+HyXZ5Cm0H1XeTuMKiSCtbnRx306lazPi5VNyokAY1DrraoI\nqWq0XyPD4k6DLTt6kmUOGfFhGZdNwGmso8/kqW64eedw0w2bMqU+r2wweGp/lVfT3D699Xev\nnHvxZ5vHLZYx2l0lHTp30ordxTb0m/RWx9KkQFrKJahknLPnWdrEgW+30zW9u52E2cDWpYna\nQdvLB0asRuMipQ/fsCSW+M4SRuNwMnye2QfPqStbtAtTI2/elq0KE/NegrWDox+wC0TRQI/1\nm3jN+vQTlT1etxPQaBo7juPB86PUjeEo0950LC5r98wqsMIuAAzoktW0SwcuLhGcMYGzF0Tj\n5IQSCATCrIMIHPMXhGBpdvTZ2j7fQ8tyozlPkYoEqZc2eBGC/7pj+dMfVX1d1jkUkYjoW7Zl\n37g5m/NcPmQSwSSc/8fgZzcuyU5UvnW4aaimJic5/L6r8gszVb7P3FGYoJAIv6nuHZrZS4T0\n1kXxq7O5/+/j4mBYF8/kz+KT0xs8armyzQHA4WIbO41efwe1xlzVprXamYz4sMWZUQFJkrA6\nXN+c51BYtEb76eq+LUXjFIDUa0y+c2iN2dFvdkRP0BhlDHatSNn3aY1XkKLQzuLkQN0ilKTH\nKz39Gt1kcO12ZicpaQpxLlRmaUNfwhS5fmPm5iWJ55sGtEZbUrS8OC9mGldrA0bbS1/Ulzf2\nG8yO1DjFteszNi1O/KpMXd2mszmY7CTllatS/WkSRAg4MokgUiHWmTicswatzkG1oV5t+KpM\n/fg9q7i+STAYzmH9WWCdgAAAQdgiFLkRKO5v9YJ01c9uKnrqwyq31WiEQvTDawuDZ7A9Pg6O\nyRIAsLaeu/9y5Ld3FmclTl92ySWwuRkPHAd7PwCASIWi1iFFjnfuhicsh9BPIBAIsx0icMxr\nvnvFgostWqt91PJ7WU70ukV+dWkNkwkfvanorl15Ld1GsYjOTgwPk3nPOxkWN3UZuwcssZHS\n7ESl/8kdk0ZAUzdszrphc9aA0SYTC8bIWaAQWpcbszo7esBkB4DoMPFU6iAEFEIIfEpeAAAC\n3sx1DHyLbjgPuRj8zMdVn5xud1cr5KdF/uymosSJpDBgYBnWjoGhkIi+ZFfWp7PyJRSoNeNY\n1WIMOqu3c/4QA5ZAChx7N2S09gweLFW7I2Ih/f2r86cz/3kK7N2QfrLK249WQFOcu51KmWjP\nuvT3j7V4xbctS0qabJ4RYbYTHS7Zvmz6rQTa+0w/efqk2++pQW144j/nn/6wyl1TdqSi690j\nzb+5fTmnZk0INpevSnnj68YxnmC2uf76zoVnH9roFcf6M6A/DQAw/BuLYbASu0wo7hq+S20p\nSizOjSlv7O/TW+MipctyYuSSmThlxRj16qx/eLXs+Yc3CejQtgFinXigDCzdABikCSASYM2R\nkaOOAdz9EURvRIp03iugaeh2RyAQCMFmJv5aEEJGRnzYUw+uf+7TmrJ6jcPFRirEe9alX78p\n0383SgCIi5TGRXInM1e36f7+3sXWnuHOJgkq2Q/3FgS2UtcXu5MZ2n70Mt3gg6ZQrH/PHPc6\nMXJxH9fuVnxY6OYQ2YncG1w0hTISRlwDX/yi9qOToyxmq9t0v3mx5JmfbBDQfskxdsZgc2kx\nDGsZAkomE8RQaCxFadzpKYMxnz7Dlx0zOWgKPXLjkp3FyWdr+waMtqRoxc7iZL638cxncWbU\nz28uenp/tXu3M0op+dHeAj6fyHuuXCgVC9453DTUZ5Sm0NVr079z+QQqoQiEYLDv05pBH+9q\nL8ccg9nxpzfKXvrZlkn0xCVMkdu253QPWDhz9Ny09AyqNebkGA+1FLvAwOHYBdZWsPeAOJ7v\nUmEy4bim2qFDFMeZxFHXJwaArgFzZYu2KDuEZhw2DdvyFjgu9ZLX1wCFQCSE0S5meOAkUi4C\nSs6Rx4FEIOL94xMIBMLshQgc853kGPnv7ypmWWyxuwKb99uttTy276yn43q31vKbF889+cDa\nCWWZYow/P9vx2Zn2zn5zlFKyamHsrdtyZD5LZYvN9frXDV+VdupMdqVMtL4w/tuX5XH2yAwe\nBQnK480DjtH5C5lR8ogQJlRnJSpX5MWU+HiLXLk6VSkb/ms4XKxnl1A37X2mkjrNmvzx83cc\njNHi6vecRrlYi8nZpRSlxEZIU2IVHT4OFwihZTnjlP8IKCQWUHYu5wh5EBYzS7KilmTNdHM4\nP9m6NGnlgtjyxgGN3hqvki7NiR7D7oSm0J07c/euz2jqMrIYZyYoIxSh+KQw2GVnrCx2UYgW\n01IakUIDwgguBpfWc7c38kJrtJfWa9YVkLVZqBHQ1GO3LL1qdVppg6aiUVvVxm1vrBu0jxI4\nnFrAPP7N9t4xBI4ZBVIuxeZar/+I3YXeKRuux+kaMIdS4GDb9o+oG8MhDA4nSEZ/mWMXtnUj\n5Tps+Aqw528rQmFrgHwJEwiEuQgROAgAABSFAl7V/MHxFt9+ci6GfftI0y9vXebnRTDGv3np\n3Jma4W0Ts83U3mc6frHnyR+s81yS2RzMT54+2XIpVcRocXx2pr20XvN/D64PpcYRJhZsyYmp\n7R3UmO0OF6uUCLOi5YmBSA+ZEI/dsvT/Pqx0W43SFNq9Nu3eKxe6n9CjtdgcDOe5Ld2D/ggc\nNkbvm+TDYqeDMYnosAf2LPrVC2ddzKiciz1r09Ljw3p11sMVXd0DliilZE1+bHaSt9SVHC5t\nGvDeaBJQKCGEWTCzFIVUuKFwAkuFMJkwlNNxq2vQ7DICDL8rLC6jTKCUCWZlTRAhGNgcLq8v\njTHo0Y4ynXExuF6t7x6wxEZIc1PCZ5bj45yjMFNVmKk6lthT9Sq3wBEVPhe/rgXhKO4aW+9X\nYqwbCrQOiP5xOLpDNzx3Cqk1jLUXbD7pJBiAxcCyQI1Ow2QdIMpCqmuxuQyc/QAYBCokXwoC\nUudFIBDmJkTgIASLRrWRM17fYfD/Ikcv9LjVDTfdWsurB+t/eG2BO/LxqTa3uuGmV2f9zzdN\n37tqIYQQiYAq8lm0hxiFVPjzm5fetj23qctAUVReSrhXQ4QxilD8MUnBwLI823EMtgOELcuJ\nfvKBdfs+q6lu1TkZnBQtu2lL9o7lyR+fanvukxp3s4bXv264Zl36967K96yIylTJbU6m02hz\nR0Q0VRivFIfQx4QQcByszezy/uBbXEYBEoro2VoZRAgscolQIRX62apZ6aFcV7Xq/vbuBXdf\npNgI6Q+vLVi1MDYooyRcYnlOdJhM6FtSlJcS4e3lJFQBEnC0IAEA8ax6mUTxwqRbHv3XZ1LK\n2msUtOpE7KWUCLGQDmU+IHZwzaOGfklZDKN/LZFIBQBAK5Fyc7AHRiAQCDMBInAQggaPj8eE\nfDzP1PRyxk/X9P3w2pGHfInNpfUagJAKHDOH5Bj5qCRhDxJU0uhwSb/B5nuoIGOKWzrDr25O\ncvgT965mWexk2KHd1Np2/T8/qPR8Ksvi94+1pMeFXbYyZeR8BIvilckRUq3FYWewQkTHh0kE\nVGid2wiBxs5w+8vaGDMROAhDIAQ7i5N97W99EQood71b14DlsefPeKakDTWl/tv9a0lXoKAi\nkwgeuWHJn14vd2vWAKBSih++YYn3U5EAwpeB/qx3XJoG4hljseEfNEVdvan4j6+VsaNtob57\n+YJQposigZQ32clrjiWJB7FftvEEAoEwZyACByFY5KVEVLZw5K8uSJ3ApHOQZzfPaB7Va8Or\nEYwbsy10/VlnEQih71y+4In/nPeKry+IX+jHq4OAopCQM4lDQIk9H1IUElPDueJflHRwXu3z\ns+2eAscQ4RJhuGTG1QabrE6GxSE2dpkbMJybt/xxwjTi1aj1mnXp25clB6SH9LjctSuvvdd0\nzkOwFgtpF8syo0tXvnNZnttD+sMTLb4Fdy4Gv324+de3+1sLSZgcq/PjXnhk0ztHm5u7BgU0\nWpQeed2GTF+HLABAEWsAaGwoGcnjUCxEqs2hHG2gWF8Q/48frHvh89raDj3D4KxE5R07c8d1\nmAokLivurQGjA7ALBDRIhSM1KYgCzwxNcRwVvzt0AyMQCISZARE4CMFi7/r0AyUdXvnGIgF1\n45Zs/y/iVVvhJl41Kp4YLa9p1/s+LZm0veRh+7IkiYh+9uPqXp0VAEQC6vpNmTdv9felkQhU\nFqd3cg2NREKK9w/ePWDhjHf2c8dnFMcre/79WW1nvxkAopSS23fkXL4yNdhLvsoW7cen2tT9\n5giFaEVe7FWr00LdgzBwIOCuMEKIVB7NLHwbtf75rYqKpgGObfkgIBHR/333ypNVvaX1mkGr\nMyVGcfnKFJuDeelAXXWbzuZgspOUt2zN8fSO4auFbOycQC0kYdLEREjvv3qRH09EELEKhS0G\nhwawC0TRMJv9d3KTwx+/ZxUAYIwn1HUuAAx2sedfAMelnDgnAzYnKCUgFABCKHU3CCVg6waM\nQRKPFNm8ybQEAoEwdyECx3ykXm04frG7T2+NjZBtLkrITAjKPCMmQvrEvauffP9iXcew9JAa\nq/jR3sIMntaVnGxfnszZ72Nn8ag9/8tXpnxd1un7tCtWpU5kyPOL9QXx6wvie3VWm4NJipZP\naPEsohQgwFZmAOPhvVMhJZcKYsaYS/G1jx23d+y08/GpNs/imgGj7e/vXewesHz3igXBu+kL\nn9f+55sm98OSWs2X59RP3LsqZD52DItdl8qLpo6IkjhZjg7KImoumhHOZp77hKNR65fn1LuK\nUwozQ2RJuHZR3NpFo5Lqf3Ubby4Gn0QW4lUnwS9oKUjn1I9yqNUNwGz1OyPqxnAMw6ADpeWh\nhE0gjQcAkGeFdlSjwSyYKrC5Dpx6EISBLAspl5N2LYTJwWLiGE2YDDN9aUEILBjDvk9r3j/e\n4i4ffftI0y1bs+/Ymev/RZq6jC98UVvbrne5cEZC2O07cpbnxnA+MztJ+Y8frG3vM3cPmGMj\npelxYdQEzRQWpkbce9XCFz6v9XTX37Qk4dr1GZ5PW5wZdf+eRc9/WuO41GGUptDNW7M3LZll\n9b2hJy5ykg4IIjpMSCtY7GAxQyMRhcb5MlmRF3Oyqsc3XpzH/eaZIdidzAuf1/rG3z3avGdd\nenRwmgVUt+k81Y0hGjsNr3xZf/8efzZLp0Rdh37fp7XVbTqGZRNUspu2ZO9akTzFebxEoLAx\nFmZUWROmkVBKK6Y4WkIAcTG4rIHbz+hMbV/IBI4JsSAl4mIzZy1kZOgHQyAEF1MfmDh+RoFl\nILxoWN2YXjCD+z4E+3ATN3BqwaDFliYU9y2gSHUngUAIEUTgmF8cruh692izZ4Rl8WtfNeSl\nRPjpOX+6uvd3r5Qyl/SR6jbdY8+fvfeqhddvzOR8PkIoLU6RFjf5Zcz1GzNX5MV+ea6js98c\npZSszo9bwbUkvmZd+uqFsUcvdHdrLXGR0jX5cWlxE0gVIQDAqerew+e7+vTWuEjplqKkcd8S\nCBCNxH5mfuxakXzgXEft6Eqi6HDJrdtzJj3gIRgWd/SZtIP2pGj5pPUaPpq6jJxOLgyLLzRr\nty5NDOzthjhS0c0XD7bAUVKn+c2LJe4PeNeA5a/vXmjsMvzgmoKxTxwbBChCHGNxDdoZC4sZ\nCtFiSioTKINaonL0Qvenp9s7NCZVmHjlgtgbNmdJRGQvaCzGaNTqZ3OT0HPt+owvSjq8sk7E\nQvrmrdO6iU0gBAHs4C7IAgCw8R8KJeaaEXXDjVOLjaUoYs10DIhAIMxHiMAxvzjAY/R4oKTD\nH4GDZfE/P6xiWO8Z8Itf1G1dmqQKE3OeNXXS4hT3XDl+M5R4leyGzWRSOxlYFv/Pm+ePVAzP\nS6padYfKu7YtS/rZjUsClYIroKk/f2/1W980HTin1uit4XLRuoL4u3blRSimtKtTUqd56sOq\nroHhlN3i3Jgf7i1IUMnGPst/fP0L3Xj2Dggs2kGOBjcAoDPZGRbTwewp86+POD7gH59q370m\nbYqKIQLKaha/c6SzrW8QAcpPi9i7QSGXBEvg+PNbFQdL1UP/7jfY6tWGb853/e3+tVN8v80Z\nqtt0JXUavcmeFC13f3vLJULOxp8AEB+4z1RgiQ6X/OW+NU++d7G6TTcUyYgPe3BvIRG4CXMP\nJFLwNk8Rz4hsOGzl6YJkbQEicBAIhFBBBI75RY/Wyhnv1vpl9NjaO6jRc1zB6WLPN/ZvXZo0\npcERpo8D59RudcPN12Wdy3KidyxPDtRdxEL6jp25d+zMdbhYkSAAK9uLzVrPdAMAOFevefiZ\n0/se2sjp5D8JUmMVCAHmmlROJS9pbCJ5tMIIhSio6kavzqrWcPRzxRiX1vdPccV4pqbvT6+X\nuQWj0nrNp6fbH79nVfpETHn8pKRO41Y33HT2m1/+su5HewsBgGHxJ6fbztb26QYdSdHy3WtS\nF2dGcV1pDsKw+O/vXjxwbkTsfv2rhh/tLdxclIgQ7FjO0ahVJKC2FAUlWSkgZMSH/e3+td1a\nc9eAJS5CmhQtn2gtJIEwO5DHgywaLP3ecaEMRc6M3R0ur6Wx4gQCgRAEiIP9/EIh5V71+elc\nOEaWMl8/V8Ks4PB5n5zSMeNTJCDqBgC89lWDb7qBRm/97Gx7QK4PANHhkjX5HIXNucnh/rTU\nnRwbF3N7x/DFAwVfu+WxD/l55b+8XeGVDqMdtP/57YqpXJYPTrcXADhR2QMAJqvzh/888dSH\nVSW1msZOw5GKroefOb3v05pgjMQTjHHXgKVBbbA6prM57jtHmj3VDQAw21xPvFUx1CTo25fl\nedUASkWCR25cMlT8xan0zQQQgsQoeXFuTEqsgqgbhDkLQtTC64AerYBTNFqwF+iZkZsm4BGs\nZ3PTHAKBMOsgGRzzi5ULYuvVHM3z/DTgGCNLOVFFGrLOYvqN3DURGgN3fIZQ28HRGxgAatq4\n45Pj4RsW/88bTEndiPniwtSIX962LHj++QXpqm9tynznyCi7nMwE5V278oJ0xyHiIqUCmnIx\nrO+h5JgppauUN/YbzA7feIPa0NlvTgp0O2cj170AwGB2YgyvZTMlcQAAIABJREFUHmzw7SH6\nzpHmNflxBRnB8tE8UdnzzKWuzDSFdq9Ju2tXXqDyjCbEF1zyn4thD5aq79qVJxbSf/ruypNV\nveUN/UaLIyV2uFHrH14trWjSWuyutDjFtzZlbSlKJG1KCIRpICKdWvMQbjmEjR2AWRSWiNK3\ngCw6MBd32XDTSazvBEqAotJRxqpRbYpMvbjxS2xQA8ZImYSyd4DSO7ELyRdic73vhZE8iE3H\nCAQCwQsicMwvrtuYeexid1uvyTOYkxy+e02aP6fHRkiXZkeXN3qnR8ZFSouy50uC95wkQiHq\n6OOIB89XJSCwPukbQ+CAbjQrpMI/fXdlRdNAbYeeYXB2knJFXmywV3f3XLlwRV7sx6fa2vtM\nUUrxiryYPevSBXRQcu6crNPJ2lmMBQLBzuKkz854O/VEh0v81ED56Dfw5if3G2wBFziiw0e8\nZsNkVHaSUCGFtl6XzS5CCI5e4PVwDZLAcaSi60+vl7sfMiz+8ERrS8/gE/euDrFMgDHu5ilU\nHMrgGMKzUWttu/6RZ0+7HWeauoyPv1le06Z74JqgN/QhEAgciJVowTUB/+bAAy3siefBOiz+\n4uYTqP4QtfF+kEUCAO6rxuWvwqXG8Nimx5oatPgmlFA06iqSVAhfCYYSAI9fYcUiUOQHerwE\nAoHACxE45hdyieAfP1z35teNRyq6+/TWuEjZtmVJN2zOFAv9bS7wyI1LfvVCSXP3iF93dLjk\n17cvFwao6IAwLawviOdstbi+cAa0neMnJym8spVj2DnJ4QG/15KsqCVZIVXxirKjgq0bYsCD\nDoONGVnx3nZFvFCE9x8bMbCIV8l+eevSKfYfiQzjTZ/mMxyZCluKEj843oIQXLtBdtM2uVQ0\n/O3U2S9gsUM7yK22DPDkMU2dF7+o8w1WNA2UNmiKeXpsBwmEkFRMW7gaA8kl3IWKT39U5eun\nu/9k62UrU7ISSdo5wR8wmGqwpRFcJhCGI3k+yDLGP4kQShgne/IFt7oxBDZ0s2dfpTY/CCyD\nq95zqxuXDrO4+gMUm+9VHYPCV4E0E1vqwakHQRiSZYGYGLQRCISQQgSOeYdUJPjO5Qu+c/kk\n0wWjwyVP/Wj9ofLO6jady4WzEpWXrUwhzRdnO7vXpJ2u7vPKzSnOjbl8Zep0DcmT2nb9G4ca\nW7qNQgG1KF11x46cmAgpANy4JavyRW+BI1wuumLVjBj2zMfsNHmqGwDAAnPjjvh1+YkVTTqr\n3ZWZoNy0JMF/AZSPpdnRconAt+FuWlxYamzgvVoXpEZ85/I8nUn97ctHXTwp2uXCDZFhIq2R\nQ+NQKYOSr6QdtHcNcLs4V7XqQixwAMDynJhjFzlyWJZzjcRocXi1dnZTUqchAgdhfLAL9+4H\n26W8MEcfNjeAIh9F75zWYc0SWBdQoZio4756sOg44r31YNGB3QD2QY7TnFasbUYxPvNJUQwS\nhfqbjUAgENwQgYMwYWgK7VieHMDmGoRQ4mTYAZODYXGUQiS5tHAV0NTj96z8/GzH4YquXq01\nXiXbUpS4a0Vy8Gwm/OfjU21P7a9yV6OoNeajFd2P37tqYWrEqoWxP7+56F8fVbv9HXKSwx+6\nfnG4fGbYrc14bAzHwpvFbF5aWFFWIKenCqnwwb2Ff37rvIsZyVuWSQQP37A4gHfx5KYt2Van\nCcBbUsHYumGxav9xjhX+psVBaRTCV0gFAAyX3Umw+c7leeeb+r16wRbnxqwv4EjX4sz1GMJM\njKUJ/mAsH1E33JiqQZoB8pzpGNBsgHHi5kO4sxRsehDJUVwhytkFomA6nZkH+I5gUz8At6sR\nAIDTrx58BAKBEEqIwEEgzCNKWrTH6jU2JwMAFEIrMlQb82KENAUACKErVqUGI/fBYHbUdej1\nJkdyjHxhasSERBPtoP3Zj6u9lohWh+vv71549qGNALB1adKa/LjaDn2/wZYaq8hNDp8Josys\ngMUsi7kX2AwOfJuPLUWJWYnKNw81NnUZBTSVnxZxy9acICVNAAAGB0Vx/y9u3RZd2Wxu6jJ6\nBq/fmFmYGRQDDlWYOFwu4vRYzUyYhgyIpGj5Mz/e8PxntSV1GpPVGRcp3b0mbe+GDM7PjUop\nEQtp3xIVAEiI4vWcJhDcYHMDd9xSj4jAwQnrYs/+CwyX6gQdZtxxGmtqqbUPgihYvclBxP9x\nFskQJeeTaZEkWN3ECAQAYMlClTApyPuGQPCXxk7DxRat2eZKiwtbkx8noGfZQvpkY//h2hEr\nURbjM80DBqtzbzCTcfafbH3xizr3PnBeSsTDNyxOi+PpJOdDab3G4eJYhLf0DHYNWBKjZAAg\nFQuWZgfIQ366qWzVVrUO14asXRQf1PfYGEoQgqDcNzVW8ehNReM/L8gopPQ/f7j+o5OtZ2v7\ntIP2lBjF7jVpwbM7oSh03caMFz73tuFIipav5UqaCAExEdLHblkKAHYnM3b9kUhAbS5KPFDi\nvQOvkApnuEEPYabAmLnjLp74vAerz46oG25setz0NVq4J0g3RbF5QAuB8UnLkqtQeCIggLB4\nGPRpvy2Lgsj0IA2JQCAQJg0ROAiE8XG62Cffv/jluZE5R0qs4he3LJ1FJeguFp/0aX8DALXd\nxl6jLU4pCcZND5aqn/qwyjNS16H/+b6zzz+8Se5fg0zOfW/3ocQ5tIdsczBP/Of88cqRGWRq\nrOKXty3LiPdXDJooCJCQEjpZjkIDIT2ju+f4AwIRAhHmyqymkIKi0N4NGXs3hMjp8MbNWTYH\n8/bhZncL3vy0yEduXCKabm9mf9xVvr87v1dnOd84ksEeLhf9/OYipYwUghH8gJZxaxwC0lqe\nBw2HJzEAYE0dWhi0m0rCqMVXs+XvjQpSNLX8JkAIAKglt7Dn/g02DxdScRi15NZRfWQJBAJh\nZkAEDgJhfF74os5T3QCAjj7Tr18seeGRzbPFYFVjtHGmQgBAp84aJIHj7cPNvsEBo+1gqfqa\nden+XCE2QsoZRwhiI4Iy5uni6Y+qPNUNAGjvM/32pXP7Ht4UvGWwQqjU27UYRmUfi2mJiJoL\na1eaSnSxrV5BCikoFPgmO2ODELprV94Vq1Jr2nRGizMjIWxRmmq2lFLJJIIn7l19sqqnoklr\nsjozEsJ2FaeEybhbrhAIXiB5DnZoOOKy3NAPZlaAGZ6m2q5gtXkaAuVuocKTcNVnWK8GSoCi\nM1DhbhR+yZlIEU9teAS3nwJDBwAGZTJKXQOCOfUrTCAQ5gxE4CAQxsHFsJ+dafeN9xtsJyp7\nti2bHf3PeH0OATAe4+DkcTFsex+X7zqAl/3BGBTnxnD6FyzJiooKjigzLVjtrq9KO33j3VrL\nubq+tYuCVQsgpESR4qhBp9HJOgCAQpRMIJfOlZ1VGkUDBQyrxpesRmkURVMp0zWe2Agpn2A3\n81m7KD5I78NjF3u+PNfRo7VEh0vXFcRdsTKVomaJ9kPwB+UysLaDbXTNhWIhcRjlA8misJZj\nbwDkQa/ERHG5KI5feKJFKGNTsMdAIBAIU4cIHIRQ43Cx7x9rPlfXrzPZk6PlV69N42xPOHPQ\nGGxWO7dbYVuvKcSDmTTRCjFNIYaroUNceFCUAoQQAOKUVmi/FzAyieBnNxX96fUyz24OKqX4\njh1zavevW2tx8TTUaO8zrV0UxFsLKGGkOAoDxhhTcy7ZmEbRNK3C2IqBQUiKgOQdzCAwhsff\nLP/mfNfQw7ZeU2m95lBZ1//cs3LqnYkJMwUkQPHXwWAVtjSCaxCEEUiRD7Ks6R7WzAUlr8Tq\nEq74qtAPhkAgEGYjROAghBSD2fHTf51q7xvWBTr6TKeqe/duyLhvd/70DmwMxALeqfZsqU8B\nAJGAWp6uOtvs3QouNUqWHBkUJwuaQnkp4bXtet9D+WmR/l9nRV7MC49s3n+itaRO09FnsjsZ\nrdH+0L9Orc6P+8E1i2bvlrgnY7yRJMJQfEsjQHO3+wyFkHyu/t9mAmdq+i40DwxanelxYTuL\nkxVSf1Wkoxe63OqGm8pW7btHmm/dPuHtfYwxw8Ks836eHyAIK0BhBdM9jFlCRBrKvwbXfTpi\n+YkolL4BJS2b1mEFDscgOC0gjQKKrEEIBEJQIF8uhJDy4hd1bnXDzfvHWtYXxBdkBKVH4xRh\nWHyxZSBMJhq0cLgVBq/zQjDYujAWAZS0aNlLNSkLEpSXFSYE74537Mz99QslXmkjaXFhW4oS\n+U7hRBUmXpAa8eahRs/g6epetcb0rx9vmAObvQkqeVyktFdn9T3k53vMaHGU1vd3D1jiVdLl\nuTHh8rlgojHHMJgdRyq61RqTSikpzo3JTpo1FsV8WO2uP7xadq5+xGHhzUONP7+5yM+kvGMX\nfZoyXIpPSOCobdf/+/Pa2nY9w7KpsWG37chZP03taQiEgIBS16KYBbi7Aiz9IJBgqxEPtOJT\nz0BECpW1BUSztYoQ65tw4ydg0QAAIAolrETp24mRB4FACDhE4CCElBOVvDPaGShwqDXm379a\n2trDbSSxcXHChDIRph0KoW35cSszVV16q4vBcUpJdFhwm2UU58b89s7ip/dXdWstQ5FNSxK/\nvztfOHHXzFcPNvgG1RrzN+Vdl62cNleFQIEQ3Lc7/3evlHrFr1ydmu5HF5VD5V1P7a8ctAxv\n9ymkwvt25+8sDmL3X8JEOXqh++/vXTRZh1+jlw7UXb0m7ftX58/qxJlnP6nxVDcAwGB2/PG1\nspce3eKPxKYd5DZT7DdMwEzxRGXPH14rYy+pqM3dxt+/UnrHztzbJp4DQiDMIKQqlLkFa+rY\ncy+B69Inpb+BaT9DrboHRaRO6+AmA9Y14MpXAV8qxsQs7jqNzd3UkrshOI3JCQTCvIUIHITQ\nwbKYr+sn30x3GmFZ/LtXStt6OdQNhVR4w+bM6zdmhn5UUydMIsyLD50TwaqFscV5MV39Zr3J\nkRKriFBMJrPAxeCmLgPnodoO/RwQOABgXUH8n7+3+rlPaxo7DRiDSim+eWv27tVp455Y067/\n81vnPdNkTFbnX9+9kKCSFWbOONFwfqLWmB9/87ynzQrL4g9PtCZGya9Znz5945oSThd7qJzD\nGddscx2/2HPl6vEXYHw+wYNWxzW/OVCYobr7igVpcWMJfCyLn9pfxfpYC73xdcNlK1Kig+Mu\nRCCECMbJlr8xom4M4TCz5W/QWx6ddaIAbjkILOs9akMb7q9B0TO3SJlAIMxGiMBBCB0UhSIV\nYp2JQ8uI8ZiJYsyw4KKQEMF0Wh5ebNFyqhsIwXMPbSRTZ/+hKZQSq0iJnco1ePu8sMFpATMt\nLMmKeurB9TYHY3cy/teYfHSy1dc7dmj9TASOGcKX5zo4TWQ/PdM+ewUOncluczCch7oGzP5c\nYfOShCMV3h4cAIAxWGyuMzV9ZQ39j9+9aoy3cWvvIGe6h4vB5Y39O5aTJCbCLAZrm8DOlUBq\n6sPGLqScHR3chmEcYOri1mQMrUAEDgIPLJ71NciEaWGueeYTZjibirgdHzYtSQAABjtMzk6D\no3XQoTbYW8zOHhZzty8JAZ393HN0jKFDM2uap0yFAaOttF5T16G3OZiOPtPf3r3w4P+d+Pm+\nM68ebLA6Qvq6CGgqnWcjNzc5PJQjCQESET0hBw1OGQ4AWnnihNDD92Wi1phmr0AnEwv4ymvk\nEr9yxNYuir9i1ViJHk4X+9T+qjGeYOHpbwUAZtu0/XYQCIHBxt9P3cqd0jhzwdxi6DiHCAQC\nYVKQDA5CSLlzZ15Nm76uY1RnjTt25ualRDDYaXJ0YhjZ53SyZsZpDxOlTEsqh1TM++mQ8R+a\nGxgtjmc/rvmqTD20+pKIaKeLce9AlzX0f362/S/3rU6MCp3V2c1bs//7jXKvYFykdNuyWbWL\nFQREPF1+RBM3OiEECYmI+xtDyq8RzHwUUuHC1MjqNp3voRV5fpmMIgQ/vq5wXUH8l+fU1a1a\nDVcuRnO3UWu0q5TcbkGJKjlCwCkSJUXPViNGAmEYMX99lmS2WRQLJCBWgp1LspETS2ACgRBg\nyAyYEFLkEsHfH1j74N6CtYviFqZG7Fie/I8frBtyg7MzOk91AwAwAItddmaSOxUuBn90su2P\nr5X9fN+ZZz6udvtc+sniTBVnx8FwuSgrMWBZAwyLW3oGS+o0XQPmGbKXizH+zYvnDpaq3eOx\nORiv/Pp+g+3J9yqDcXeGxXoTh1HL5qLEh65fHCYb2RkuzFQ9fs8qKc/Scf5QyOPOuyRrNrX4\nmdssz43mifslBMxYvn91vm8Po6tWp+VMJK9qRV7ML29dumsFr5OOyebkO6RSilcu4Ch+S4yS\nLZ1VLa4IEwMbMHMRu05jVymwrQAc9V9zABSVxd0wRR41Q+pTcH8LrjuMaw9hTdN4z0UoaS1H\nWBSGYhcHYWgEAmFeM9/XBoTQQ1PoqtVpV/m4J7pY7x6ZaDhug4mX4GkH7Y8+d8advV/W0P/J\nqbafXL/Y/w3/KKXk5q3Zvs077tudzyl8TILyxv5/flCp1gynrxdmqn60tzA1VhGQi0+a0oZ+\nzl1ZL8439RvMjgC2I+0aMD/3SU1JncbpYhVS4RWrUm/dlu2ZR3PZypSNixPq1Qa9yZ4ap8iI\nV87e3W9fMIZvznd+cqpd3W9ShUlW5MXcMvq/z8d1GzO/KuscMI7a/Y5UiG/YlBWEQeKqVp1a\nY1bKhflpqslZxs5DNi9J/OR0e2WL1jOokAq/fVnudA0pIOSlRDzzkw3//qz2fNOAzeFKjlF8\na1Pm9mWTcb5IiJJxxoUCKjZCOsaJP/3Wkt++fM7zKytBJfvNHcsFNNm/mTFYjbjyMB7oBLEU\npeSj7BVTMshkmzHT4DZmwowG2E4kWAEw576OaBG15Ca29GVgPeqtBGKq6BaY9h8/h4U9+RJW\nX3AHUGI+tfbbIOHNOkHJ68Blwx3HRmpS5HFU3vVAB7ebG4FAmIcQgYMw85lMYsPT+6u8vAkc\nLvbv711ckhXlvz/obdtzk2MUrx6s7+w3I4SyEpXfvXzBshzuzdiJUtuu/+W/SzytBy82ax9+\n5tS+n24KoGowCera9eM/CQBjGDDaAjVUtcb84P+dcDfRNFmdbx9uqmga+Nv9az3lJJlEUDRH\nN2b/9z/lh8qHDRf1Jkdzt/Hohe6//2BtpGKcyV+EQvT3B9Y+90n1iapelsUIoTX5sd/bnc+X\n1T9pWnoG//J2RYN6OKNKKhLcuSt374aMwN5lTkJR6H/uXvnWN02fnG7TmxwSEb0iL+aeKxfG\nq7hX9bOIpGj5b+5YDgAMi2lq8ouutYviw+U1vm22thQlSkRjKdwRCtHf7l97oqqnpk1ndzI5\nSeFbliaR+qyZA24uYw/8C+zDGZS44iBKXkhd/VMQjaVb8V9u0FPd8AjWI7pgyoOdcaD4RfTm\nR9nGr0CvBoQgMp3K2T4T6lPYEy/izoueEdxVzR7/N7X9x/wnIZS+HcUvx4ZWcFlBGoMiswCR\njyqBQAg8ROAgzBQoJOK0FKXRhJfQdidzsqqXM368sueadel+Xgch2FKUuKUo0epw0RQV2Enz\nm4cafRsr6E2Oj0623b4jJ4A3mig+HTm4QYi3y+MkePnLOre64aauQ/91uXpX8VzoAjs2Z2r6\n3OqGm26t5eUD9T++rnDc0+Mipb++fbndyfTqrHGRUt+qgaljtrke23fGs52z1eF65uNquVQw\nH16gqSMW0nfszL1jZ67Z5pKJaTTtG7CBZirqBgDIJYJf377sv98o1xpH3mNF2VH3X71o3HMR\ngvUF8esLSCX/zMNiYD9/Cpyj8suwuoY9+jq1/e5JXA+zPdx7HmwPzEWBAwBAHkUtuXG6BzGa\nQY2XujEE7qnF+i4Ukch7om0Q2ywougDo0PWqJxAI8xAicBBmChI63MT62mQgMT1hwwvdoJ2z\nKSMAaPTehTD+EAyjBy+nVTe1HRzlIQazw+Zg4iInteU1QfzsS7I4MyqAmSblDQM88f75sH4+\nVc2hxwHAyaoefwSOIcRCOnj1TV+VqT3VDTdvH26eDy9QAJFLyM8uN4szo154ePOXpeqWbqNC\nKizIUK3Jj5vuQRGmBG4466VuDMdrT8CWOye1yuX4FgIAABeAi8xpQwM2dPMe03cBl8CBNU24\n5D9Y2wEAgBDKWkctvRbExAmYQCAEBfJjQJgpCCiZTBBrdfW7rUYpJJAJYig04TmQUiZCCGEu\n086I8RL+QwafpSgercycrOrd92nNUJtJhVR4y7bsa9dnTHGzdGxW5MVkJykbO/kb1AGolOIf\n7fV34e0PNp6+szbHnG0gZ3Eyzf1mo90poqleHt3NYHZiPP3V1gDQ3MX9fujoM9mdTDByRgjz\nEJlE4H+GHWHmg4193AdcDjAbQDmJek8+VZ2GSZh1zV2wpgk3HIfBPpCGo6QClLkmkD8kNP/a\ngesQ1jSxB/824iSCMW48zg60UZc/ChRZhhAIhMBDvlkIk8TF4Jo2XZfWHK2ULEiNDMi2pIgO\nE1IyF7ay2EUhoYCSjtsg1upwOZysVyqBTCJYmh1V1tDv9WSKQkNbgi4GY4yF01qnnZ2sLKnV\n+MZzU0YSKL48p/7L2xXuhyar87lPatQa87i7+hYHY7A5hTQKl4iEE7REpSj0x2+vfPL9i+60\nAomIvnxVqsnibO42SsWCwgzVDZuzArsRnRyjaO7mWEKnxEyz5WqQaNdZytR616VyIAfPSxQX\nKZ0J6gYA0BT3hwUhFFS5jTCfwRh/frbj87MdXQNmVZhkTX7szVv9ct4lzBCQWM6j5CMQT8aA\nBlHxmG3mOEDFT8m4NDhguw2JA1bI6T9s+Qe46sDIMNrLUPNpassDIAjMBg+KzgBaCIxPeyOK\nRjEc5ta44qNRPqlDQV0HbilBWWsCMiQCgUDwhEwUCJOhpl3/13cq2npNQw/D5aLvX71o61L+\nwku/QYgWIr/WtGUN/fs+rWnuNmIMqjDxzduyd69Ooy6ttR7Ys+ihf53ysqy7c2euxmD93/+c\nb+oysBhSYxV37MydrsrtGzdnldX3M6MdLxRS4e41w/1lWBY//1mN74mfnWm/dn1GWhz3X8nu\nYsvUutZLPXFFNFWYGJ47QZlApRT/7q7ijj5Tc/egQirITgoPtu/p7jVpT77vXdMrFFCXrZyD\n5Q8WJ1PaoWc8cniyM1QXudSu7X43/Qk2+emRn5xu843npYSTdhWEYIAx/q+XS09fklkHLc62\n3sGjF7qf/MG66bVhJkyA9CI4+Y5vGCXlTk7gAKQEKgvY4aakeEjVQHJE501hlAEGmwfNH75o\nP/sNNhkpRbh49TbZnjuRLERiPe6t91Q33EG28guqaE9g7iGUUoVXsOf3e4VR/k6uLioY93E3\nkTV1Nv2nNuJkVW+/wZYcI9+1ImX3mjSimBMIhKlDBI55AQbsYMwMdiBAAkoipKZk5dBvsD32\n/BmLbUSPN5gdj79ZHqEQBarDyLgcqej+0+tl7ofaQftTH1a1dA+6UxtSYhXPP7zp9a8bLjRp\nTVZnRkLYtzZl9mitjz53xn1Wa8/g718p/c7lC27aEviGmuOyODPqV7cte2p/Vb9huEQ5M0H5\n0LcWu5072/tMepN3T4EhLjQPpMUp9FanycHQFIRLhDIhDQAY4GiTpt9D1nEwbGmHDgAmqnEA\nQEqsIsU/Twet0V7e2N9vtCVGyYrzYiZhWXLFqtRureW9o81uxUcpE/3k+sKk6DlYo6vWW5nR\nFUoxKumqpYkl57tZj/iKvJibtmaHfHTcbF6S8O6RZq8sG5pCd+2aQesKwlzi6IWe0z7eNF0D\nllcP1v/gmjlqJznnQLHpaMlOXPHlqKhIgjbfOflr0jlARWG2HbAJgQhR0UClzZz6FGwZ1P/x\nAaavc+ghazJYv3rfUVkS8aunkTQUXZNwawlvPFACBwAquIyShrPnPwSrEQBAEkYt3o1yNnDd\nGI/0hfVA7xI/dEjZbRnOx2nqMj69v+pMTd+fvrOCIhoH4RIsnikfbcLsgggccx8XdpicfSMN\nShiDkJIohLHjVn/w8fGpNk91w83bh5tCI3BgjJ/9pNo3/tmZ9mvXp6fFDW8ghMtFng78Thf7\n25dLfc969WD9ZStSIhTTsCW4riC+OC+mrkPfb7AlRctzksI9f9ftTl77CReLyzoNBttwgigC\nSAyX5EQpegZt/T59FgGgqtuQE60IUrHDhydaX/y8znrJRCNKKfnJ9YUrF8RO6CIIwd1XLNix\nPLmkrk9rtCfHyDcUJoTJ5qbRupXLWGRJfmxKotKms/ZqLSqFZMWCmI2LE0I/Nj4ENPX4Paue\n/aT6UHnnkAiTGCW7f8+ikGmahPnG6Rpu593T1X0/uCbEYyFMHmrLnTgxF5d9hgfUIJahlHxq\n7Q2gjJnSRZEK0aoADTDAWA+841Y33DA9HdaD78muvj0kIzBwxy1+NYD3G4Sy1tJZa8GsBYxB\noeItEUIUCk/Aeu82YW/25nZbvNeupfWab853bZsxqYsEAmGWQgSOOQ4GPErdAAAAJ2szOwcU\nwknOMJp47AbHtqUMIJ39FnfWgxfljQNugcOLerXBtxEpADhd7MWWgQ2F07OYFAvpxZlRnIeS\nouUCGrkY7xJmmkKqeIVb3QAADNBpsNEI2Xg0EZuLNTtciiDUrp+o7Hl6f5VnZMBo+90rpc/+\nZGNyzISTL9LiFHylN3MJPvOXqEjJng2Zgpm6eRWhED16U9H3r85v7zNFKsQJKhnZZyMEj0EL\nx9c1ABgt3HlthBkLyluD8uaL1YKjimMfBQCcVecgNAIHR5EIAABIlUG5nXx8pQkt2IZPv+oV\nPDPI3SPpdE0fETgIBMIUIbXTcxwna/VSN4ZwsGZ3s5KJwreq4XEhDDxjdNYYI+vBYufu0wEA\nnAkp045CKty+PNk3vqowgfOV6zTaWJbH0A2A98DU+OB4q2/Q6WI5/RoIQyQquW3nYhWSGatu\nuFHKRAXpqqRoOVE3CEGFryt2XGQo8vwJXmj01uOVPd9jgAvIAAAgAElEQVSc73J7b00nWAu4\nGbMNgHvA/5kMZoL2S+hxE5tvt/uhuHnkgbMPzBfBVA62tgmM3z9QylLueCp3PASg7HVU0TWj\nugIr40yIewtkkCiYBAJhypAMjjkOi7k3wQCAwS4BmkxdxsLUCN/SaADITwtRymhilExAUy6G\nY1qQxu8ZMYabQ9LEcw1CwwN7FtkczOHzI7mdeSkR123O7LdxvKwMi+Vi7poOsYBSTNwXwx/a\negc546093HECAERIhbkxinrNqHWCiKaKksL5TiEQ5hvbliV9fIpDJ91ZzCH7EoKHi8H//rx2\n/4kWdzrhhsKEB/cWTJPVqwuzFwEPt0jDGADkiFoMY3mTY9x3AbcfAWs/UAKkTEOZu0AeLHNx\nOjaJ6W7niMelAABgFzYcBZtHIxiBCkVsBUFEoAaAkgpQ9jrceGJUMDKZKrxyYhfCDNaU4MEm\ncJqQOBKiliJlzuRHVXAZnbkK99aDbRCUcShhYVzTKc7WafEqomASCISpQgSOOc4YRhtosj3V\ndq9J+/hUm1eRiFBA3bY9RIaIMolg27KkAyUdXvGkaHlxHm/dTWKUbGl2dHmjd+/YzARlfmrk\n2Hc0mB217XqjxZEaq8hNjpiimYVaY37lYH19h55hcXZS+O07cjITuHNHxUL6F7csvX5jZlWr\n1u5kshLCi/Oi1QYbp8ABAPFhYpVMNGDx7jqaH68MkgEHX7XF9LbgnfksTgyPlovrNSajzSkS\nULEKcX68UkL+aATCJfLTIu++YsGLX9R5tpraUJhw7fqMaRzVPOT5z2reP9biGTl2sdtocTxx\n7+rQN7HGbK1b3biEGbPnEb2WLyUZN32OO08OP2AcWNeAy5pR4Z0oIjMYI5RsuspRcYojvvkq\nAMCDp0epGwDg0mLdlyj6OkABM1OkVt+OkxbjhmN4sBdJI1BSIVqwFeiJTPgZG9v0Glj7hh5h\n+wAYG0FVhFKumPywZJEoY5X70c7i5Gc+5jBTIwomgUCYOkTgmOMIKO5keAoJaOSvg6PF5nr9\n64bS+n7doD0pRr5nbdpf7lvz1IeVJXXDjS3T48MevLYgO4T7zw/sWWSxOY9d7HFHMuLDfnnb\nsrE7Vj56U9FvXzlX2z5itZUWF/br25eNnWz/zpHm1w42uE00C9JVP71h8aS7e5yt7fvty6Xu\n9JNenfV0de/PbiraUsTbZDc3OTw3eeRvGy7hTdOQiuhNWdHnOnQdeutQUEChgoTwBbE8RblT\nZnGm6lC5t3kYAPAZixDcJIZLEsO5P54EAgEAbtictXJB7Jfn1Op+c5RSvDY/fsWCqZlTErgw\nWZ2vfdVwqqpXY7AlRsl2rUi5dn2GgEYAYLG5OPNoKpoGatp1+Wnj7A0EGifgHq64FfAAIK73\nhkWDO33kBszgxk9Q8YMBHx8AiJaslu/9rvmjl8E1PGdAAqFs73eE+csBO8HawHEOYwS7GiRp\nARwGSlmCUpZM+nTce9ytbowEtechYgEKC4wwtGddekOn4euyEUNWAY2+fdmCkL+pCATCHIQI\nHHMcGgkltNLGeOcBygX+rj/7DbafPH2yVze8YNaZ7JUt2h3Lk//03ZX9BltnvzkmQhIfGWq7\nQYmI/vXty6tadUOpDRkJyjX5ceO2T1cpxU8+sPZEVW9d+3D2xKYlCWOf9fGptn2f1nhGKlu1\njz1/dt9PN4qFE95vYVj89/cuehXXMCz+5weVq/Nj/eyuqpQIYuQijU+3lCyVHAFIhPT6zOhB\nu0tvdQpppJKKRMHMC7h1W87pmj4vE5OkaPlVa1KDd1MCgTBPSI8Pu/eqhdM9imnG4WIFFArS\nj6ze5Hjw/070aIedI9r7TPs+rTlb2/f4PatoCrX1mZwubpOIpi5jqNei2MpvomEG4BA4sK6R\n+xRLH9j1IA5YYYgn0itvERVvdJQdZ/p76Oh40fINdGwSAAAzyNkwFQCA0QEEUuCYIthQy31A\nXwsBEjhoCj16U9HO4uRTVb1DjeR2FCen+teZnkAgEMaGCBxzH5lAJUAiK2NgsBMACSixTBAp\nQGI/T3/xizq3uuHmYKl627KkZTnR0dO6Bb0oPXJR+sQmWAih9QXx6wv8rb99+3CTb7BHazlS\n0T2JRMp6tYGz/4vJ6rzYrPW/tWp+XFibztqut7IYA4BEQGVHy2PkI69pmFgQFoSeKb6kxCr+\nfv/apz+qOt84AAAUhbYWJd595UI/xRoCgUCYcVg0uOUbbOoGRCFlMsrYCuJp8MfBGL4u73zz\nUKNaYxbQaKhmJy8lwGvyNw41uNUNNxVNA1+VqXcVp4whqlChL1AZq4iD5xeH4W64BgDgsoO/\n86AJQ8clSy+/yTvKP37ccgbkZpS+BtDMKFR0clulgivAFrNLs6OXZpNe4wQCIcCQRci8QEQr\nRLQCA0YAvL3KeTjF5ScKAKeqepflzPGfJZPV6SvuDNHYZdgJExY4jD5pF24M/Id8oRDKUMnS\nI2UWJ0NTaHq9G9Ljw564d7XF5tIYbAlRsqAmjBAIBEJQwX2VuOo/wA7vtGNjF+45TxV9GyLS\nQzySZz+pdptfOF24omngx0+d/P1dKwJbp3O2RsMT79tVnJIeHyYVCdwVmp4sTAtK+sOYyAEk\nAL6aBQLE43Eu4UlWRTRIQj5+OhxoBWZMvpMw3NmCzdWo/Ry1/vtAT4t762iECnDoueLB6TVL\nIBAIAYUsReYRCNBE1Q2GxWYeP0v9RBbks5Qxdqgmt3kVE8Gb8BIbwd0TcQwQArmIniHOlDKJ\nIC1OQdQNAiEEGMyO09W9B0o6Klu0YzSHJkwYxoFrP3CrGwAACIBxsNXvDnXsCBlqjdm3CTfD\n4qf2VwZ2ICYr90/8oNUJAGIhffO2LN+jGxcn8HljBxVE5XFFUwC4W2+gqDwQcVQ9oNhCoIOW\nv8EPClvFoW70arHZCgB4oBk3fBP6UXkNB/dVIA5FCwAAReRP/sKsFSxNYKoEmzrgzXEJBALB\nE5LBQRgLmkJRSglnVUV85IQX5LOOoUV7Wy9HTubkao8z4pWZCUrf1mjxKtmi9BA12SUQCLOa\nD0+0vnSgzm18k52kfPiGJdOy2pxjdA1YBprLF3Em51sHwNQNYbxW0AGnrKEfcykZXQOWbq0l\nMSpgrTTjVVKjhWO7IuFSt84bN2dLhIJXDtYPSSECGu1Zl3HXrlysu4g1Z8A2ALQYKdJRwhYQ\nBb+QB8UiqhjjBsBGAAwgRVQGIP7XhRZT+TezNW+BfeRnF0Vkoqyrgj5UTiQZSHU5Np4BlxYA\nwMXgjj7cM9IXBneeRwt2Tc/YAIB14arXsK4REAKZGEbvWKDYNaCYrLvWYAU2nAb20jtNGIWi\ntoPI37JcwrwFY7JQJUwG8r4hjMO2pUlv+fhQUBTasjR0U71p5M5deb9/pdQrmJMcvs5vFw9P\nEIKf31z0i3+f9dSMwuWiX9yydMiynkAgEMbgq7LOp/dXeUYaO42P7Tv770c2KaT+Nsaaz2CM\nLzRr2/tMSpkwPy0yJkIKAE4X+8zH1Z+ead+R2LNoEc+JDo7KguDBWRUyhJep8xTZtSKlXm3g\njA/9AyG4Zn365atS2ntNDhebFqdQSIW44zOsPT/8VJcF66uxsZHKvh2kcRMegcMINi2IwkAc\n6ZcDBYpEaCUAC8D6NYlVplHFP8a95WDuAVoC4WkoasGEBxlAREkoei974h9Y1w4O7/QZbPPe\n/wgIuLcet5WCWQtyFcpYiWI4snIAAKuPY10jAADGYLaBSAACGhBC4RkoYSPIUyZ5e3Md1h0d\nFXEO4L6PUOKtQM39rTICgRB6iMBBGIfbduQ0dBrKGkZ2GAQ0uvfK/HmyYbi+IP7Xty/710fV\nQ5IEQrB1adJ9u/PH7djiCcZQ067r6DPJpcL8tMh/P7Lpo5Nt9R16hoWcJOWedelkZUIgEPzh\nHS7bY53J/uU59d4NGaEfz+yirXfwz29VuNfzIgF145bs27bnPPNx9VAz1AEbr/0BCq3PaGIU\nbyfyX714dmNhwh07cwPyw3HV6tTGTsPnZzvcEQFNfffyPK8sRbGQznF3K7d0jqgbblgH2/UV\nlXXrBO5t0+GWT7G+cfihNIbKvBKU6f6dTE2gyJoWocRVvEddDqAFoXb3FCp91Q0AQJJAv80w\nZs++gRuOjQTqj6AFW6nib/nWLOO+C6MeO1wwJLQppJNXNwDwYBlHlLWCqQaUyyZ9WQKBQOCD\nCByEcRAL6f+5e9XRC93n6jRaky0lRnHFqtSptPJyMWxpfX9nvzlCISrMUMVM3HsixGwoTFiT\nH6/WmAYtzpRYRYRiYgZgao35L29XVLfphh6KhfSt27Nv3Jwdegd6AoEwq2FY3MpVMQcATV1B\n2fidS1gdrseeH5U953Cxrx6sF9Do87PtQ5GLunCtXaQS+5RsKOJBEdJ0+pULYqLDuetDtUb7\nhydaT9f0/fOH68LlUzWkRAj95PrFO5Ynn6ru7dPbEqNkO5YnJ8fwyisAgI3N3AdM7cA6gfJP\ndnFZ2aoXweHxvrVq2OpXqYLvgCLJ//FPHoxx3VH2/Odg1AAtQPE5aPUNKCpEDc5R8jKsLueI\npwR4wY9bTnuqG8PB2kM4Nhul+tzLwfM14uDI8fF/COAY4D7g7CfzIAKBEAyIwEEYH4Rg05KE\nTUsSpn6p6jbdX96uUGvMQw/FQvrW7Tk3beHOlpw5CGiUHh82iRPtTuax5894tmKxO5kXPq+T\nigV71qYHbHyTpbnbWNOmtzuZrETl4syoWae5VLfpKlu0FrsrIz5sXUG8gKaauoyNXQaRgM5N\nDk+KHmuOPldhWXzsYk9jpwEhyEuJWLsoDs2615XAA4WAQsBweUxSE8kpm58cOd/NqRe8f6zF\ndelvameoP1/M/XVRjUzg4TMqUlD5N0zUonuKiIX0b+8s/sOrpXydvHq0ljcPNd63e/KmjwyL\nyxv723tNEQpxYUbkPVcu9PtMvt6rGBi7nwIH7i3lWE5jBquPogU3+zuSKcAeexnXXqqbYJy4\nsxp/8EfqiodQYigKWFBiIUpfg1tPjQrG5qHszYG9EW45yx1vPs0hcAgV4LRxvNMpIdZUAi1C\nikRO09YxQYAowAzXIeJKTiAQggIROAgAANpBe1OX0elisxKVcUFzD9WZ7L96ocTTs93uZF74\nvDZSIXKX+/rCsHjAZAeAKIV43MIQBuNBu8vuYsUCKkwsoKd7aXe4ootzevr24ear16RP4+js\nTubJ9y5+VdbpjixKj/z5zUuD9+oHFruT+cvbF45UdLkjiVGy6HDJhWbt0EOE0BWrUr5/9aJ5\n1dilW2v53culni62eSkR/3XH8uhw3vY9hFkEQmhBakRVq873UEH6ZGyP5xUtPYOcca8W3eUD\nEXcfX743rTNHaU6ODYtOzUWp60EwDZ+g3OTw5x/e9E151+uHGnq1HL8jJXWa+3ZP8uINasMT\nb513W2iLBNQt23Ju2Zbt18lingartBgEfrufmtScYTzYEYIfRtzfNqJuuGFd+OQb6PrfB//+\nAICoZTfhpCW47QyY+kEWiRIXo9TigOto2KzlPmDmyKpA0fm4w+fPAoAHamGgFgAwJUDJG1D6\n1omNUxQP9k7fMBIHYNuMQCAQfCECxyxmwGhTa8xKuSglRjFpi0oXg186UPf+sRYXwwIAQmjX\niuT7rsqXSQL/3vjynJqzI917x1o4BQ6M8cnG/qN1fQ4XCwBiAbVpQdzqrGg+XUBnc7bqrA5m\nuP2YiKbSI6SR02pv0dzFPaXW6K2DVodSNm3t7p/eX+WpbgBAVavuv1469/SP1s+KreB9n9Z4\nqhsA0DVg6RoYaX+AMf70dDsC9ODegpCPbnrAGP7waplXj566Dv3jb57/y32rp2tUhMByx87c\nXzx/lhndGjYtTrF1aUiy+mczY/xKSkUCT1NPrV30fH0GAOz76SYUN/l6zKkjFtKXrUz58pya\nU+Aw83R4HZdBi/MX/z7rqew4XOxLB+qUcuFVq9PGPR2FL8TdR0Y6YrjjkYUTcLLAfI1CQ9KO\nV13FGcZaNVgMIAuR3wqKW4ji/E6cmdwtxHLuP6iYIykVpW7CukYwdfkeGoZ14fZvAAFK2zaB\nMUSswn0fer/iQhXIp9XtlUAgzF3m0d7mXEJrtP/xtbKb//j1I8+e/t5fj975v98cr+yZ3KWe\n+7T67cNNrkuiAMb4i7MdT7zl4x8WCFp5NtDaek1e8/Uhvqzs+aqqZ0jdAAC7i/2ysvvrau7/\nqdnJNAyYHQzrnh45GLZBazY7OBMjQ8QYKSc0NW2fPpPVebCUY/esudt4vom7VnZGYXcyB0q4\nd/+8+Pxs+6BlkmuAWUe9Wt/YyVEpfaF5oKOP27iBMOsozIhalhPtGZEI6W/vWiCcT5lKk2MR\nT5JLZoLyZq7MhW3LktKmVd1wk8DTFHYMI9Kx+apM7ZW3MsS7R1r8Ol+oQGl7gB6V1YLCMlDC\nlgkMQs7diQzxxP3F3o21R3Hfx1h7FOy88yLssvNewcldEzRLQcmLJxCnxdTS76HMyyA8HSSR\nKIxbNsXqE8BOpJWPOAnF7AGhaiQiy0Wx1wKiJ3ARAoFA8BuSwTH7cLrYR/edbvOwmtPorb9/\npfT3dxWvzp9Yh7ZBi/OTU22+8ZNVvS09gxmTcp0YAwHNPQUX0Ijyycow2VxnmjkW26ea+tdm\nR8vE3m/dnkE7HlY2Ri6FMfSY7Fkqv5NmA01+eiRw5HtCenyYPAg5Mn7S2W92cdbxA7T2DHot\nn2YgGr3N7vRLt2JY3NY7WJChGv+ps5/OfjPfoQ6NOWUKxsCEmcPrXzeU1Gk8IzYn8//erSjI\n3DyNGWGzgtUL4/LTIt1+z0PQFPr2ZXkrF8TKJYKXDtQN6aFCAXX9xsxbt+dM00i9WVcQzylJ\nX7Zyko0t+DYbugbMdicjFo4sO7VG+6HznV39lsgw8coFMXkpw8UpSJmDFtyHtRfApgGBFBRp\nSDmxPxeKK8bdZznsPBLXT+g6nmDtYTCeB8BD0wBsLIfwZShyA8fdlbHcP4ECEcjn1O8FytuK\n2s/jgdZRwdhslLOR5wQaJa9HyethSMgY5CgtAcYB1n4+iYobSTJKuBVcRmAsIIwESjyBcwkE\nAmGCEIFj9nG4oquNy0j/5S/rJypwNHcb+Ra69Wp9wAWOwgzVgZIO33hBusq36qRTZ8EYY59C\nT5bFnTprjs/YLDwrXrN/K+EgsTY/Li8loq5D7xW/a1fetIxniDE2e2fFPrBENIFtn//P3nkH\ntlWd7/8992pvy3vvGY/ESZy9QwYzhLApe5cyWuBLFy2/0kFbRmmhtDSEXSCsMjIISciOne29\n95KHZO19z+8POYosXclLliX7fv6KjtZRdK17z3Pe93lCouPGL/A9VD8nnoIgQyiCMaaVpLUG\n68Hz3cHgWxzMEAR6/u6Fb+2u21XWTlEYAOLCBXdsyNYYLB/sb4gO4//riZUag8Vqo5KiRXzO\n9P/J2OzUp4dbPj3UrDFYEAAAwhfrEwkCbVmWctn8hIm9srfNBoSQa2nh/rNdr35RaTQP79V/\nsL/h8pKkn1w7Z9i3mCVAUZPofeNIiLzbqKavwNA3PMIWopSNSDZR03F9LWgcxafO33wM6jPA\njQWBe4UOSikG3idgchd6UOZSYM0soZDFITY+iWv247YzWDeIROEotQRlrwFiDKdRXz5hEzqx\nsiTAkkzkiQwMDAzjYfpP4QzjpbbdfbXsoKlbY7FR47JU9HHyQlPgGL92XtznR1rcPAJYJHHn\nJprVvh1j8HIKtWMaUcbbZwnQ6pYygV0PLCmgEX9TBIF+f8/CN7+t/e50J8YYAKLD+A9dnbd0\nzvikKP+SFCWWCDgaA02JcmFa+KhP71Ea3t/XUNsxRFE4M0F667rMABdyR0h5CZFCZxaPD/gc\nVnrcbLmcyk+Rc9mkZ22LgMfKTfZiCsgQUmiNVtrOAgBgupDGgojPfnRr/n1X5nT06cUCdkWz\n8u9fVjqdoYQ81oNX5flwvA4wz79/7njVcJMFBgDABIHmpYdnJcqW58dkJkzcJyI/Vf41nVKW\nlyxzOpW0KXR//eSCa/coReFvTrYlRgmvXZ464bcegSiBKHwQtB3YpASOBInj3dpexgXW1XgZ\nr0YeAgdw+MSGR6j9b4D+UkUPSioiltw44QkELwQLzdmI5mwc7/OQOMFLnQsPBMFe6cnAwDCb\nYQSO0IOiW94P30XnZOGD9DgpiyScBhyu5CT5f0XEIok/P7Bo+67aPaeGV/sZ8ZJHtuTn0r1X\ntMTrhU4MXSSEmMOitdsQT/XetbUfq4+C5eIeFD8TSRcDcakpRiLg/Oz6wgeuzG1T6KRCTmy4\nYNQsmKmGRaK7N2e/8lmF2/jGhYnJ0SKM8b4zXafq+lRaS0Kk8MrFSRnxl66kz9T3/+bt005j\nlK4B/dGKnmdunreyMKB26A9cmffbd07TWre4cuv6DNdy65mNWMC+fUPWm9+6X+Xff0Xu7PlP\nmNlw2SRCQHsG4AVBxUGowOewshKkVa2qlz4tdz1p6k22F3eWx4YLxqLzTjUXmgad6oYTisJq\ng/Uuui2BcbGyMPazw831nSMse1gkcn3l70530P7A7irt8JvAAQCIAEkykozubDo6No/QWZ/j\nKCaTvPEPuPEkVnYBm4ficlD8xDN3ZyaSJBSWiVUNbsMoaTVjn8EQGChgjjSGicBcEoUeGXH0\n+zaJUaJxle4DgJDH2roi9ZMfmtzGVxXFJfm1Y19ltLYO6rVmm4BD3rwx6+Fr5nQN6MPE3DCR\n1z7McBE3O0ZS1+t+aZIXJ5XRtZrHirkDBott5AUZi0Bx4qls9bQO4IGvALu4bRkbsLUfRV7n\nVsoh4rO9WdxNC5cvShLx2f/+pqZvyAgAfC7r5rUZ21amGc22X24/Vdk6HCxX3jy451THnRuz\nblqTAQB2Cr/0aYVT3XBgs+O/fV6xMCcykEXdi3Kj/vLg4n99XVPfqcYYh4m421al6Uy2zw41\nOaYnFrDv3Jg9lkSAmcT1q9LiIwRv761vU+gQgtQY8d2bcxZmR073vBj8A5dN5ia5u0g4mJcx\n/Wvy0OLrE620WwL/O9Y21QKH1UZ9dqTl4LmuXqUxWs5fVRi7bVWamwrpze+5sUutM1pFk0sH\nIwn0x3sXbd9du7usw7HZkBwtfmTLHNcP7hpK5YoPr59phuB59rQ6xmkejDFQdmBxUc6q2dLE\nOCFQ3k3QvAf3nB6uImLxUPI6FM/EcjEwMAQ1jMAReqwtjvvwQGP/kLvR901rJtK5evembB6H\n/Phgk6OynSTQVUuS77ncn9ldlT2ayl61c9exoV+XIhcsTg731d0JAADXzk/4+nxXZZfa8UAM\nUJggu3Iuva03hyTyIkWtQ0bNxYZhMZeVKuNzvDQb+wWsPTNC3XBgGwJDLQiDPZ10ZWHsysLY\n/iGjxUbFygUOr4q3djc41Q0HFIV37KkvzozMSpA2dKo9DzwA0BqsFc3KkpyoAE0dAADyU+R/\n/8kys9VuNNtlomHN65a16e0KHYtFJEYKvfWZz2yWzolZOifGbLUjhMbVsMYQEtx3Re7T/z5p\nHSkyLs6Lnp/FyFjjg9bKCgDaFPQGnP7CbLU/+cZJpzFTa6+2tVd7rKr3pYeWum5RmL3nf5ks\n9kkKHAAgFrAfv67goavzugb0EgEnwqMu0ptxzzTaY/sGCVLB3EM3nuZ6Eyvb8dnPcX8TUHaQ\nRBH5m1FqScCaWUMPkosyr0Ep67GuF5EcEEYDObM8ShgYGGYiQXqiYvABn8N64b5FL+68UNU6\nvI8n5LHu2pQzMb8xgkC3rc/csiylqVtjp3BarMS5VvQLCq2posc9urJVaQgXcrMiRykS4bLJ\nbQuTVmabuoeMABAfxo8U+2rQ5bPJ3EiR1U6Z7BSXJKZU2hjGQnM5BRiwpQcFvcDhIFLGd715\n4DyNZTrG+Ifz3VkJUm/9/wAwpPN615TCZZOuO59cNjmZ1vQZA5dNDqhNH+5vrOscoiicESe9\neV36hEMlGYKHOSlhf/vx0n99U1PZorRTWCrkbF2Rum1l2ujPZBiJN/mPM8X9XF+faPO0nW7s\n0nx+pOUWl7Rab7FHIj47zH9liVw2mRZL71I0PyuSNroleKU0yTzQN15qF3XAjQHxpTxU3F1F\nHXwN8EV9UN1LHduBlB3E/G0BnGgIwhaisImavzIwMDAEHEbgCEkSIoUvPbS0oUvdptDKRNyc\nRJlYMKn9HBGfXZQ+JUW5LUr6MteWQf2oAoeDKAkvyrsfhydskmAHbOse09iXAPIyHvRQFFZp\nzbR39auNABAp8/pF+LiLIfCUNw/++q3TRstweVFTt+bA+a5f3Va8ZJxBSwxBSEa89C8PLLbZ\nKa3R6qPLj8E3RenhNXSO3VN0KnRSWtNHO36yRuEqcKwoiNmxp9ZTOL5ycXJgXJxWF8XuLmu/\nMLJTRirkTN4BZKpAbBR7A6jPYH092NTAliJhDkiKXd0iqFMfe56dce1+yFwOkvGEnjIwMDAw\nBDGMwBGqIARZCdIsn5vVjV2a0hpFv9oUGy5YXRQXHcb38eApQm/x6ODwOR5isOVgUdCPTxOd\n/frPjjS39ep4HLIgTb51RerYDSYJAon59OkqjnVUaowkOVrsWcIdJeMXpE7bR57xUBTuV5tk\nIs4Yv0qKwn/9pNw48k/MaqNe2ln+/i/WMoajMwMWSYScumG1Uf1qY5SMHwztY9etTPv+bNeA\n2uQ6KJdwr181teUwWoN1LOMiPvu5Oxf+6cNzPS6bBBsWJNy+IWtKp+eEINAf7inZeaj529L2\n/iGjiM9elBt1z+Ycz2aWIAKxQLYIyRbR36sbAC2duoQx7qlBs1rgoACm/0+SgYGBwV8wAsfM\nBGP497c1XxxtcZqovb+v4YGrcgPvucj1ciHLZQXFKststXNY5KhuIN5AwgLsKXAgNhLkTnJi\nE2Pfmc6XP61wxuKcru/fVdr+lwcWx8gFvp/oZMWrrkUAACAASURBVFl+9O6yDs/xJXnRu0rb\nT1QrKExxWISrz6hYwH7mlrnBsGKZeWgN1rf31u051WG1UQiheRnhD109Z9RQ3sZuTS9d5ZRa\nbylvVjKeowyBp2/I+K+vq49XKewUJgm0LD/mgStz3frjAoxUyHn1kWVvfF19rLLXTmGCQEvz\noh+4Kk8+pb7UADFyvltW+sVx91/p3CTZm0+uOlrR26bQivjsuenhAe6/Y7OIW9Zl3LIuw2y1\nzwBhFFtNXu/zcdcMBluxqRKs7UAZgOADOxnx8gFN1t6FgYGBYdphBI6Zyd5THZ8dbnYdMVvt\nf/+iKjNemp3o//xXH8RJ+R10tpTx07oLRFH4m5Ptnx1u7lUZuGyyMC38vityR1030sBPR5QR\na8oAX9x8I8VIthpIf2bQjIrJYj9V19/Upd55qNkt9FehMr76eeUf7i0Z40vdtSmnvFnp5pN/\n+aKkd/fVOz1fHETL+EnRoqxE2bXLUyR0uTZjx2bHfUPGcAnX9Rq6a0DfNaAPl/CSo8UscjY6\nwFls1JNvnGjpHa6XwRifbRh49B/HXn1kme9j1Vufke+7GBimCKXW/Og/jik1w8eencKHy3uq\n21SvP7bCv5ZP4yVCyvvVbcVWG6VQGaPC+IEx5V0/P+F4FU3dH62LFodFrJ0XN/WTGonVgHsu\ngL4POCIUkQXSxBmgbgAAEoUDQQJF594qDqhDdlCArVi3D+wXLdIoI5hrsa0biTYwGgcDA0Oo\nwwgcM5PdZe2egxjj3WUdARY4UuXCNpWhVzNie0TCY+fF0BubBYYXPjp/8Hy3498mi72stu9C\n0+CfH1icmzT+/xxhPuKlgaUb7AZgSYGbEOB8+FN1/S9/Wu5WaO3KmYYBrcE6RpcWmYjzxhMr\ndh5qLqvtU2nNiZGiq5Ym13eqd5W6H1GKIeMj1+Yvyp3UdaFKZ96+q/bAuS6bHSOEFmRHPHTV\nHAD42+cVzt7vWLngkWvzZ2Hdwd5THU51w4nRbHv3u/pf/6jYxxPDvXvWBHV5eShz6ELP7rL2\nrgG9XMxdnBd93cq0kI6wqW5TNfdo+BxWbrJs8t60nx5qdqobTgbUpk8PN9/r18SuicFmEQmR\ngfPfXZ4fc/2qtM+OtLiG1G5ZlrJmbsCFDDpwXzWu3AmWYY0bN+xFCSVozlZAIXw8D8Pmo6T5\nuLXMfVwgQ/GhYQruJzCADcx1l9QNJ3YNNtciXsF0zIqBgYHBbzACx8zEXwn2BpNtSG+ODhNM\n2NUMIVidHtkwoGse1OvMNgGbTJDx82IkrIDYpNFS2aJ0qhtOzFb7v76ufuXHSyfyiqQA+Bmj\nP2wK6BrQP/fOaYvNl6cpxlipNY3dhpbLJm9bn3nb+kznyH921dI+8nB5z2QEDqPZ9sRrJ7oH\nL15JY3yqtr+69RiLhdQu1no9SsNv3j71lweWzEkJm/B7BRiMQa23SIWcCXc/AUB58+C4xp2k\nx0kSo0Qdfe4pmBFSXuCtUk5WKz462NTSq+VzyPxU+d2bsx0L5rqOoff2NTR0qhEB2QmyOzZm\neUtzCHIwhhc+Onfg3PBPikJlrGkfOnCu++WHl0w+yzPwKFTGv35ywSkvEgS6ekny/VfmTaaK\nytsR62pgaaWwwWq3Y8wlCQF7hpds3XdF7srC2IPnu3uVhhi5YGVhbF5ycPy4GVX4wgdgH+EG\ngjvLQBCO0tZM16R8gDUDwOIgwVh/OoiSmyiTGvfWXRoSyomV9wMrxOxsJoqJwq0YqwAoYGMA\nGWFWA8YjHmLtAUbgYGBgCHEYgWNmIuSxaBM9hbyxXnA3dKpf/6rK0ZXAZhFXLEq6fUPWxK7X\nEYKsSJFnZsqA3qIyWACBnM8JF/qhULmxS33oQo9iyBgt468sjPXWrny6vp92vKZdZTDZBLxQ\n+qP49mS7b3UDABAC2eTMCJUa+vKQQS/jY+TrE21OdcOJ3kTjwGez4w/3N/z+nrE22kwjar1l\nx566g+e6jRYbj0OuKIi95/KciXX1W6z036zZSldi7QJC8PSNRb/cfsrVL5bPZT1941x2YMsK\n3vmu/oPvGxz/Nppth8t7Smv6/nTfou5B/Ys7y5072CeqFWW1fb+6rXhZfuj5/J2oVjjVDSdt\nCu373zc8eFXetExpwtgp/OyOU651QxSFvzzWymYR910xcV8hs5cj2XLxSO43WAaNVgwAGAAB\nlyQECAk5pDgEFaIxkp0oC3A15VjA3Wfd1I3h8Y6TwSVwYMp+ao/96KfYqAUAJItmrbuNyFk8\n+hM5AmL9E7irAvc1gs2MwhJQSgmwprNPKoAY7VQ5wEXzaYSAI8EkF+n7AFw1DnoTXAYGBoYQ\nIpTWcgxjZ35WZPeJNs9xH3X+x6t695/t6lEaomSCjDjJRwcbnStnq4368lhrTfvQyw8v9Ysb\ngtlGnWpX9WovLY9jxLyFSWHcSay+3tpd+8mhZueSaefh5m0r02jrn00W+vUhxmC0hJjA4Rlo\n4klhWrh0cvpRmJirN9Gk3kzSje/CaJUIrtAGOgYbOqP1sX8cc9ZPmSz2fWc6LzQNvvbY8gl8\nBUlRohPVNL36ydHiUZ+bnSjb8fTqnYea6jrUFMbpcZIbVqXLJQHdpewe1P/3QKPboNlqf/Xz\nir4ho2t9PgDYKfzq55WLcqNCzq32eFUv7fixyt6QEzjO1g94dkUBwFfH227fkDVhI4bkaBHt\nL1VyjBgABo3WAePFNRUCADDbqUGj9Z1va6RCzvVLU+amTW1ua/BQVtt3pLxXMWSIlQvWzouf\n6sBaGgwD9ONGFVA2IILl5Gj7bof99B7nTTyksH72IuvyB8h568fydBRfgOJnXZEChTsuqRsX\nwSQX2EJkdSn3I0Y/vzAwBAwKB8vPDkNowRw3M5Nb12WeqFa4+TLkJMk2LqSxMcMY/+m/lzwp\nGrs0tJfsdR1DP1zoXl8cP/nplbUrFSPNDnu1plPtquUTvZA9Wtn70cEm1xGKwp/80JSdKF1R\nEOv2YG8t5SI+e5KVDoFn1A35MBH3sa2TvZJbWRj74X73lSoArCqaVNO4twqF0OWLoy2e3WF9\nQ8bfvnNm64qUJXkx49IHNy9K+uJoi2eFzpZlKWN5uljAvnvzdBocnKrrd1MxHNAuoQFApTPX\ndgzlp4RY3jBtrRyEpp9rSy9NugcAmK32rgH9hHuIrl6afLi8x22QINA1S1MAQElXtCXis7OT\nwiqaB1/fXXvH2sxlk/P6CX4oCv/54/POUqDzMLi7rOOqJcmPbMmfTJvbuCG96LAEC4hg8RnF\nql77mb2e47aDH5CFq4F0vazFuKkMNx7D2gEkDENJ81DumuD5IAEGYy+bBCwuuAgciJMeoAkx\nMDAwTBkhtlfGMEbkEu7rj624fFGSY984Usa/bX3mXx5YTLs7euBct6cnBS2uLdMTRmOyKegu\n/Xu1Jq2ZpkwAACqalc+8Wbrtt/tu+f3+3713xtNc4LvTnbRPpB1fVRQrpCvT2LgwccJWI9NF\noRdJSMhj5yTJblidvv2pVZP3z7txTbpni/jli5Im6TA6rtiaLJ/5iDY77hrQG+jKTALJuQb6\nP5CqVuXv3jv74MuHaeMhvREXLnj29vmuZRccFnHnxux1/hAZA4DeOO6vQ6MPvepob76tUWHT\nmYE6MXyUz7AnUVlTmBb+sxsKXX91RXz2z64vnJMSZsfYRqeCAUDExf/Az0+00iplM4ndZR2e\njU5fn2g7UuEuDE0t4Vm0wyg8c7i6Jgig2mvcbSMcGHW4z6VwFWPqwBvU4f/g7hrQ9uPeeqrs\nY2rXC2ALPeXRT3jZUbhkH4sQrwDYoXF+YWBgYPABU8ExY5GJOI9fV/D4dQUWG+Xbz/+Qx96a\nN6y2UZr/x4LWbHU0WruDQWOyirnux+SXx1pf/1+V8+aRit4T1X3P372wODPCOdirpDdV7aEz\nW5UKOc/ePv+F/55XuugsKwpi79qUPb5PEgRsXpT4zcm2zv4RThZ8LuvVR5YmRvktqpbPYb30\n0JJdZe3HKxX9alNipHBTSWJJzmT3VK9cnLyrtN1md79UFXBZhpFSF0mgm9bQ27hqDJYde+r2\nnupwvE5Bmvzhq+ekx02PXaXJ6mtJ396ne3bH6e1PrRp7qX9JTtSOp1afqO7rGtCFS3jzsyKj\nQ2fZ7G2qBAGUlyvtGHnIfDona+bGfXuSJrVqzdzQWyd48/ENE3HjIyalk25ckLgoJ7qstk+h\nMsTIBSU5UQ7x3cei2X7xKNEard0qQ8Kkw1yCmQPnu+jHz3WvLHQvQpw6UHQeRGThgfoRoywe\nyr48YHMYHZtXGRTbLM4jCreU4baz7g/ob6Eq9hLzrp6yyQUvCPgY3DeHAACQDDhiRAiBnQik\nr40EBgYGhlCBEThmPhSFa9uHdCZrSrSYdrNx7KXUY2n+HxUCIfqrWgSkRzHukM6y3SPCw2an\nXvms4p3/W40uPt6b+6m38XkZEW89tfrA+a62Xp1IwJ6bHj4N3c7+gM9h/fXBJf/5tubA+W7H\nJmdRevjD18zxo7rhgCDQlYuTr1yc7MfXTIkR/+LW4r99VuEs8ueyybs3Zxelh7/8aUVdx3A9\nbbiE9/A1c+Zm0HxBFhv11BsnXVseKpqVj792/G+PLJ2WSI6ESFFjl68ajb4h48lqxbhae/hc\n1tp5QZEf6Ub/kLGjXy8RsJOjxbStUovzoiUCjqvRqYNlc2JbejVuqhwAZMRLUqc1PXpiFKaF\n37Iuw62Hqzgz4sY1oVfpnZ0oW54fc7TSvUXxrk3ZxKSr22QizoYF7j2SBEI8FmEa2YflEMA7\n+y4dIdbRrJRDnYEhesPmviFjYCeCUPGd0HoEtx0DswZINgrPQjlXgCBi9KcGChRJ02kLAIAI\nFH7pLk91Y5i2szA7BQ4Ug7Fnqyki2HOAHXrKMgMDA4MPGIFjhvPtyfa39tRqDVYAQAjWzI1/\n6Oo8N7/DsLFZRQp4rI0LEic/pXABh0CI8igxJRCSexgxnmscoM2M6FUaWhW61JhhwaUkJ6qy\nRen5sMV5XqsMBDyWf5fr04VczH36prmPbyvsGdRHSPm03TdOBjWmD/c31rQP2exUWqzk5rUZ\n4+oT8TvL82PmpoeX1fZ1DegjpfwF2ZEODe7VR5Y2dWu6BvThEl5mgtRbycP3Zzo9DR3MVvs7\ne+ufu3PBlM/egysWJf0wWrdXm4JuDy2kGNSYXv9f1ZGK4WVwuIT34FW5nqqNkMf65W3z/vDB\nOVeXiuxE2aNb8/uHTM++fcrVJCg6jP/zm+cF1G7Af9y5MXtxbvTusvbOAX24hLckL3p1UVyI\nfpb/u3luwvcNnx1pcWgK4RLefVfkjldiM5pt5c3KHqUhJoxfmBbu27k5UsDp1JhczwcIoKtf\n39KtdtwkCRQtm+ELMKmI00NXhxgmCnjAB8FCaWtQ2hqwmYHkQPAdx0RiDopOwYpWt3EyfzkS\nuOzBmOh/abFxHH2CMwmEohGYMO5yyUxhESgDYIb/cTEwMMxCGIFjJvPNybZXP6903sQYDpzr\n6hnUv/LjpcjlqmVFQcxJurAGkkD2i53P0WH8p24s8ksKA4dF5ESJqj1M9XOjxRyPNm+d0Wsx\nqsZl1bRlWcrh8m63zfO0WMmWZamTnm9owGERo9bXVLepfv6fMuPF7o/WXu3h8u6nbpy7Zu50\nFgiI+Oy189yL+RFCGfHSjPhRymXLveSwjCufxY8UpYf/5Nr8N7+t8ZbUAwBcTmhb3Flt1DNv\nlrrKNIMa0+8/OEeSxHKPkFdHqdSusvaWHg2PwypIla+dF4cQkgo5259a9e3J9vpONYEgJzFs\n86LECYd0BAM5SbKcpKBL/ZwAXDZ59+ac2y7LalfoBDwyJkww3tqN41WKv39R6cyQlou5j1yb\n73lsOBGxyUQJr1dvsdgpAMAUvtA4eLyi26mBL8mJEni0Ls4wls6JqaULiprO4GRWsFpuI4J9\n/dO2L16hui610hA5i1mb7hvxMAH93yMShpiNsR8hUDKgKIyVGCwI+AhFMKsABgaGGQnz0zZj\nwRh/8D1N8kVN+1BZbb+rPeT64viy2r5DF0Y4cczPivzxNXnlzUqV1pwULSrJifLj8iMvRsJn\nk1W9GkdlMo9FzokVp8ppWqyjvG/cxcgFzn/zOOTLDy/95IemA+e6FSpjdBh/zdy4G9ekh/SS\nyb9gDC/tLDeO9Law2fGrn1cszI701sszdRyr7P3udGf3oD5Cyl+eH7O5JHECNfDeclimMZ/l\nqiXJS/KiT1Qrduypo5Xn5oZmM5STQ+U9tEUo731XT7uIFQvYN66madbgc1jbVqb5f36zAK3B\naqewbCr39jksIiN+Iu1CDZ3q598/42qso9Saf//+2Vd+vDQ70asAJGST6TK+jcKDWtO7+xsb\nXLx4SzIjb1ox84+Ta5ennKxWVLepXAfnZ0VuXOiHqsmZB5JGsu98nmoux4oWINkoPouIz3R/\nTFoJbi6jeW76ooDMMWjhIxQfdGU5DAwMDH6FEThmLP1qk3MPzY26jiFXgQMh9Mtbi1cU9Ow/\n29U9aIgO468qil1fHI8QSoicqv6F1HBharjQYLEDgMD7nva8zIgIKc8t7xYAitLD3SwMuWzy\nR5dl/egyehN4hvY+XbtH+gwA6E22c40DnmG6UwfG8MJH5w+cGzbVa1PoztT37z/b9cf7SsYr\nSCVFi6CSZjwlxg9mMaNiMNk6+nViASdWznctiYqQ8q5akhwp5T337hn7yPSHdcXxPpZ5IUEd\n3T4zALT0as1WOyMpjguVzny2fkChMsaFCxaMQWc8UtGzfVetI4o4XMK7Y0PWxoWJQdVD8OWx\nVk/bYDuFvzja8szN83w/l0WgaCn/ya0FNR1D7f06DotMjxUnT9k5KKjgssm/Prjkq+Oth8t7\n+oaMMXLB+uL4zSWJKKi+3WkBU2BTAqUHUgos1x9PRKQVQVqRt+ehxCKUuxbXHBg5WIhy10zZ\nXBkYGBgYggJG4Jix0MaoOfD0vwCAlYWxgXRrd+BD2nDAYRG/uq34uXfOqHSXnFATo0RP3uD1\nsmaGgTE+eL67qlVlttpTY8SbS5J8N7R7Y0jn1UpWpXW3gZxSjlT0ONUNJ5Wtyk8PNd+63n0X\nzjebS5I+O9zi6dJy7fKUycxwVLQG6/bdtbvLOjDGABAjFzx0dd6SvGjXxyzOi/7TfYv+9U1N\nY5caACQCzo1r0q9dHvI9U7S/HqPexeDJrtL2f39b48w2lgo5P7k238eP8JdHW1//6lKe1KDG\n9NKn5d2Dhrs3B1H8k7cg5KZu955EbyCAvERZXojrgBOARaKtK1K3rgj5nwh/Yu7AmuNgv3jw\nsKORdDmw6LN+PCEW34yT5+HGE6DpA2EYSilGKdPgzRQI7DpALCDoI6sZGBgYZhuMwDFjiZTy\nZCLOkI5m7ZqVEErXjnnJYW89vfrbk20NXWoOi8xNlm1amMjycOuYkSi15l/vONXQqXaO7DzU\n/KsfFeenjLuLmDZAZ9S7poIjFfSxxEcqescrcESH8Z+7c8GLO8v7L2YNcFjEreszL5vvxWbf\nH1AU/uVbZa4N871Kw2/ePv3bOxYsnTNC4yhKD3/9seVGs81gtoVLZsilZ6YXY5SESCGfM/ET\nCm1ydMhhs+Ovjrd+f7aza8AQJeMty4+5aU0Gj07GPdsw8MpnFa4jar3ljx+eiwsX0vaGmK32\nHXvrPMd3Hmq6Zlly8Bxd3n6ZWeQM+Hr9CUXhHqXBaqMSIoWz5HQ2biwKrNoH4NJvaFVg5bco\nYtvYV/IoNgfF5kzJ9IICDMYabCgHbAEAIEVIMB+4M8E9nYGBgWEyMALHjIUg0E1rMt74utpt\nPCNe4tqfEhIIeawb6Nr4ZzyvfFbhqm4AgFJrfv79s28/vYZ21eSD+AhhZoLU7dUAQCbizMsM\nqCuE0ksssbeOKt8UZ0Zsf3JVWW1fZ79eLuYWZ0X48G3xC8erFbR2gDv21LkJHA74XBZ/Blkk\nrpkX9+GBxl6PxIdb1o1PnHKAAeusJoPNbMcUgQg+yRGz+URoluXb7PiZN0udxrdtCl2bovFY\nZe/LDy/17D358lir5yvYKfzVidafbiv0vKu+Q+1moON8SmWLclzBw2PHbLV3DeiFPLZbP6AP\n5qSEOQOeXSlInb3OjjY7rmpVdg3o5WJuXkqYRMDZVdq+Y0+dI12IxyFvXptx/ar0adeAbHbc\nPahnkYRbz910gfXnR6gbDigTNlQjUfF0zCjowLpSMF1yWgW7DmsPIbwYeEyvLsMMgcJM3yvD\nRJg5l90Mnly7PNVO4ff3NRgtw1fGS+dE/+TafHL8bo4MgUett5TW9HmOKzXmU3X9KwrG7a7/\n1A1Fz/ynVKm5pC/wOaynbpw7mY33CeBtt3nChSQ8DhnI7qqakUaATtoUWp3RGni71gDDZZN/\nuq/k5U8rLjQNr+RFfPZdm7LXF7tH4YwKBjxg0lqp4V8nClN6m8lkt0TypKGocXx3usMz1qdN\nofvkh6a7N7vvIbd5xBs7aPUybrZ5zeUx0QVpTxKDyfb2d3VfH29zmMjEhQsfviavJGd0ZXzb\nyrT9Z7tcg4EBQCLgXL9q5huF0lLbPvTizgtOX14Rnz03I/zoxYhlADBZ7Dv21ClUxsevK5im\nOYLNjj/5oemjg42O+KcwEfeey3M2LJjCOrgxYR3wMt4f2HmMG6xsA003sHhIngKCsTbUjBu7\nZoS64Xx3/TnEywBgyoIYGBhmL4zAMZNBCK5flbZpYWJDl1pvsqbGSBIiaZJKJsbp+v7zjYNa\ngyUpSrRhQaJYMMPXdYFHoTJiL6YGPYP6CbxgSoz4rSdXf3akpaZNZbVT6XGSbSvTAtyfAgCr\nCmN/ON9NMz41u9B+x9ND0YmbpehMJS5c+JcHFjd1a9oUOpmIk5UgnZisY7CZneqGEzumdFaj\nhCOgfUowc5JOjnSMewocHDb98oPDot+tSory6rWZMlo+9HjBGP/67VMVzUrnSPeg/jdvn372\n9vluRjOeREh5Lz205B9fVp1rHF6dFqWHP7IlP3KK66qCE6XG/IvtZa5pSjqj1VXdcLK7rP2G\n1Wlx4X47QY+Lv31esfdUh/OmSmf+6ycXdEbrdBuChODPqUFJnfkQ9zcM3yRIlLGGmHMFoCmQ\nG6z0PziAzWBTASu047oYGBgYJgMjcMx8xAJ2cWaEH1/QbLU///5Z1+KC/x5oevqmorHs7zGM\nHRHf65+nWDDBhEgBj/WjyybSSuBHls6J2VySuLusw3WwODPiuhBx10uLo19PRkh5kol+L6FI\nepwkPW4iSaJOzHaaGF0AMFHWSb3uNKE10Jv1aujGC1LDadN2i9LpWzmiZHxH/LDbeG6SzO+e\nSmW1/a7qhgM7hd/aXTeqwAEAiVGiF+5fpNZbugcNsXLBlMbZBjm7ytpps6I9wRiqWlXTInB0\n9utd1Q0n7+6rv3JJMoc1fYUA7Egw00wM2JEBn8rYoOz24/8CTa/rCK7/HpNslLtpCt7Pe+kW\n9n9VFwMDA0MIwQgcDONm++5at9YJjcHy+w/O7Xh6tVzMna5ZhS4Yw4FzXUcrexUqY6xcsH5+\nvGMVERcuTIgUdva7F2uwSDQ/y5+KVYBBCJ7YVrg8P/a7Mx1OL8b1xQlB1ZTQ2qs9VtnbqzLG\nhQtWFMS6lj6tKox777uGvou2pk5uWJUeVB/Bgc2Op7233xveUle8FS4FOdFh/KpWmvalmDCa\napSb1qYfqehxa+WIkvG3LPMq8z15Q9GfPjp3qvZSff6clLBf3lrs96OuqtVd3XDQptBqDdYx\nFutJhRypcPZKGw6au+kzZWix2jz8JgKCt6/bYLI1d2tykqbNkhwJi7C5y92Gg+AhQe40zWgU\nsKJ6hLpxEapuLxo4CTwpishDSauB9NNlEknv9wyAgOXtLgYGBoZZASNwMIwPisL7Tnd6jhvN\ntsMXerZMcTznzMNqo377zulTdcOLlsYu9ZGKnvXF8U/dOBch+Mm1+b/cfspmH3GFd9v6rKn2\n0QwAC3MiF+YE6Ubc23vrPj7Y5Ow3eW9fw50bs5w2tzwO+cd7S/788QWnmSKXTd68NuOaZSlT\nNyWKwu19OoXKGBXGT44SEaPZ6Ngp/M3Jti+PtvYoDUIeqzgz8t7Lc5xWkRbKbLQZbJSNQASH\n5ApYQjQdGSYsgrR4tKgAADkV5dxTz/r5CQfO0fRe0Wb6RMn4r/x46RtfV5fV9mOMCQKtKIh9\n4MpcH/KBWMD+/d0lFc3KmnYVhXFGvHR+ZuRUaGo+mrCs9ulZhIco5Hi0xUnWQ00YH9+pbXq/\nbk4MCluPNcfBfrHWiR2JpCuACNbT35B7/PkwFAU2CxgHcccRPFBDzHsAWP74COxoYMnB5qFP\ncdMAedFQDCqs6QGShaTxwJmefigGBgaGAMAIHAzjQ22w6E00axIA6J6QMcQs56vjrU51w8n3\nZ7tKcqJWz42blxHxz8dXvLW7tqpVZbba02LFt6zLHG8IjsVGtfVqtUZrUpQo8I4bIcfh8p4P\n9ze6jtjs1H921WbES52tXolRolcfWVrVqmpT6CRCdl5y2JTmdNa2D73yWUVzz/BucFqs5NGt\n+XnJvrzrfv/+2aOVw3uJWoP10IXu03V9r/x4WXK0SGfVGGzDf6p2DBbKYrIZw7jhRMBlBQGL\na7DRROoIWSF5lC7IirxtfeaHBxopFyuWjQsTL1+USPv4+Ajh7+5aaLLY+4aMMXLBGHsBCtLk\nBWlTm0iSGkvfhCUXc8Nmcb/JBMhLDjt0gT4V242CtLDUmOkROLy9L0mgpGivzi8BgpuEIuLA\npgS7HkgJsOVjiJPGYDUCezpMfIgxxD0YB3DHYZS60R/vh5B4NdYdGeG6yk1BokU0j7UaqfIv\ncHsZOOrjSA6RvR5lb4AgLDtkYGBgmDSMwMEwPvgcFkKItoZcwGMOp3FzqJz+8vdQec/quXEA\nkBwteu7OBRN+/f1nu978tsaZzLqqKO7hXNpbogAAIABJREFUa/LCRDOnk0ilM9e1Dznkm6wE\n2eSv1vaU0XV9A+wu63D1skEI5afK80cLv+xRGt7eU1fdpjKYbWmxkpvXZozXEKdrQP9//y51\nBiEBQHOP5pk3S19/bIU3z+DT9f1OdcOJ3mT7z66aZ+8ocqobwx8EwI5teqtWzAl0VTOHYEk5\nAo3F4PprImLz+KxQXUXfviFrWX7MgXNdXQP6KBl/eUFMYdooVn88DunDQHRaWFEQ+87ees8m\nrOtWpgVDemgIsakk0VFF5TrIIon5WRGubZ5iAbuuQ3P1r/ckRgpvWZe5uigukP/NeclhuUmy\nGo/o6w0LEoLCVAixgB0FY+mLsmhxy3d4oArsFmDxUFQRSlnvn1qJMSL30l9GEEBeki/xYJ2f\nBA4AUoSkm8HaAzYlIBJYkd68Ramyt7Gi9tJtu4Wq3oUoO5F3uX9mwsDAwBBMMCtShvHB45AF\nqXLPNEQAYExGJ4BraKsrA2rTGF9BpTMP6SzxEULPHeAD57pe+Oi868ihC92d/bp/PLp8ZkQF\nf/xD0wffNziiDQEgP1X+5A2FkzTq6x400I53DYy7QKmqVfXMm6XmiymeF5oGLzQN3r05+6Y1\nGWN/kc8Ot7iqGw5MFvvOQ01PbCukfcppj5ogB2fqB0w2+uPKRJnEMA1t20IWj0uyTTaLDVMk\nIngkhz2WXdAgQKEyfnywqaFLTRIoJ0l245p0h244ee/VaYfHIX9/T8mfPz7f0Kl2jLBI4vpV\nadtWhoYNcPDA57BefGjJ6/+rcgqOKTHin2zJL0iTtyl0tR2qhk7NNyfbtIZhI9I2he6PH55r\n7tbcc7l77M7UgRD85vYFf/74/NmGS7Gsl81PePiaOQGbgx+w6Khz/wTzRdMTmwl3l+KhZmLe\ng37zvBgNFJmBonNG6AiOceHICdjcpcPJwo4Ftq+gdKxs85wVAOCGg5C9HsggkLEYGBgY/Aoj\ncMxq7BQ+Wa1o7dXyuaz8VHlWwphWOA9elfvTf55wriodbFiQ4LtmnoGWMDHXc6cUAMIlo1+T\nVTQrX/tflaNzgSDQ5SVJd2/Odg3sfG9fg+ezmro1xyp7Vxb6uh4KCb481rp914iLtsoW5S/+\nU/avn67ksie+SPZWiKQ1WH76zxOOXORrV6Tmp4zeJvDq5xVOdcPJu9/Vr5kb77TDGJW6Tved\n1eHxDrW3p3hrIrPZKZud3l0fYwoDnh4nDkSK2MHaVO+F0pq+598/6/xyq9tUe091/OHeRbnT\n58joX5KjRf/4ybILTcpWhVYiYM9JkY/9iA1+KAqP6mLjLyKkvGdvn6/WWzr6deESXkyYwFGd\nkRwtSooSffD9QcojW3rn4ebNixIDmagil3D/dN+i6jZVU7eGRRJ5ybJkf2cPTzW44/AldcOJ\noR93n0SJqwI2DWLR3VTVt7j5CGAKAIAgkJAL3JHnFN4EW8ywsgb3nARDP7D4SJqKElcDe2zF\nX0Pt9ON2C9b0orCkic2HgYGBIWhhBI7ZS0uv9g8fnGtTaJ0j64rjn9hWOGoreEa89I0nVmzf\nVXuucdBgtiVECLeuSN1cQt9nzuCblYWxTq9Kt3HfTyxvHnzmzVKnHSBF4W9OttV3Dv3tkWWO\n6gytweqt6KC2fcjvAofObNNb7AIOKeKyArB0wBh2Hmr2HO8eNBwu76G1dRwjC7IiG7totAOF\nyqhQGQGgTaE7UtF7x4asW9f7CtxVqIwtvVrPcZsdn67rv2LxWK8pvSWK+IgaiY+g7z+Xi7ls\nFstKp34QiJgWdSMUMVvtf/3kgpt0pTfZ/vLx+e1Prp4xPRwIobkZ4XMzPCreTVqq9gAoO4Bk\noYgUlL0GWKHR8mY0297/vuFweU/fkDFKxl9dFHfL+gw+JxBXQVIhRyp0X9Z2Deh7lTT1YhSF\nzzYMBD4yNi85LKR2KSgAl76PoSb6R6maIIACB7C4RNFWmHMF1vSCqQ83/s/zISimeAIvjFt3\n457S4Rs2AzYN4sFqIv9u4IdwpBoDAwPDFMEIHLMUs9X+7I5TjgWbk/1nu8R89lgKU+PChb/+\n0XwAsNkpFhmSkQdBwjXLUk7V9Z1vHNHys6ooLjqM/9dPLnQN6OVi3qLcqMvmx7t1v7+9p94z\n7KC+U33oQs/aeXEAgMHrAthbPOfEUBmtpa3KPt1wr02EkLM4RS6f4uZtjcHST1f5AgBN3ZrL\n5k/8la9flXa4vGdUx9z3vm9Ylh+TEuN1n9NZdu6JxmDxdpcnWQlSWsElK8FrscCaufHv72uw\neKRObipJ5JE8g03n+RQuGZK+ntPChaZBt3hXB539+uYeTaj3p/gG99ZSh/8NluFlOe44D3WH\nyHWPgTRmeic2KnqT7bF/HGvvGz74FSrjxz80ldX2vfLI0sBoHJ4YzPSVVgBgGFmEZbNjjd4i\nH0NZ38yH0mHjebApAFuBECFuNnAzABDY6X9Usd0yHWVpXCRPBkgGmwG37R+u5gAAABRbMhGB\nQ9d1Sd1wYjPg1r0o99bRny7zoqeTHCQJ9r9chlkOhZmFKsNEYJams5TSmj43dcPB7rIOz6J6\nHzDqxiThsIgX7lv0022FC7Iik6NFi3KjfnVbcayc/8TrJ7473VnVqjpS0fPXTy489a9S154g\nO4Wr21W0L1jZMhwaJxFwvFWV+1gbjxe9xba3VuFUNwBgQGfZW9un8dIl4S98eIhM0l5ELGD/\n49FlW5anyMVcAIiQ8mir2SkKexp5uhIVxvPmyBgjH4fD/3UrUj07brhsctuqNG9PiQ7jP3PL\nPNdmJQBYVRR72/pMFsEWst1FGRbBFrFCrCJ9GlFp6X1zwLulzgzBZqGO7XCqG8MYVNTxt6dn\nPuPh8yPNTnXDSUuv9n/HWqdjOgAAMXK+t04Zp39wm0L3i/+UXf2r3Tc9//21z+59a3etpyPP\nLMKuwtrdYO0AbAHAQGmx8TTWHwcAJKAvZPA2HhhQ0iqi+McocSWKzEcJy4mi+1Dm1WMIgnEH\nq+rpx9VNQJe07T4NeTKKpnF1QVlrGQMOBgaGGQkjjM1SPC/1HJitdoXKGGyu/jMbhNCmksRN\nF3t8ypsHPzroXm1b3jz40cHGOzdmO25SFPbs3HZgddm3v3ltxiufVbg9IDFKtKLAb5s21b1a\n90oBBFY7VdWrWTIGl4oJI+KzE6NEHXSH8eSrrEV89sNXz3n46jlWG2Uw265/bh/tw3wsdAFA\nIuAsyo06Wa1wG5cKOYtyogAAY3yypq+pW8MiUVaCzFu6SmKU6I/3lrz8WYXzwyZECh/bWpDs\nM8FxeX5Mfop835nO9j5dmIgzPyuyKH2410DIEnEIrtGmt2MbgUgOweWzpiNSMWSRi71Wu4RL\nZ/IeO1bUgZGmmAgPtoJGAZLowE9p7HimcTsoq+0fl+mvH5EIOMvzYw57BGlFyfjzsyIBoLlH\n88RrJ5yKht5k++hgU0WL8sUHlwTMQyRosAN0YeMFwB7reWs72DIgtgSUNJ5TKLYkELPzgTAa\npW6Y7It48yXFFNhNQIx+wUaU3ElVfInbSt1jYhkYGBhmIozAMUvx4cI4GYPGieEo7eayycx4\nKVOF+8N5+uDYg+e6nQIHm0XERwhpLTZSYy9txV++KMlipd7+rs5Z8FycGfHEtkL2aDYrY6df\nR7/I9zbuR+7YkPX8+2fdBrMTZUvy/LbQYrMIEcHmsknasqZwyShtHY9vLfiFyuhwgXUgFrB/\nfss8AY/VqzQ8//7Z+s5Ly8XizIhf3DqPNpcxP1X+rydWNnWrewYNseGC9DjJWCqnZCLO9V6q\nPNgEm82ZIXaYgacwXS4TcYZ07iXxiVGi1JiZ3J8CevqqMQDABhUKboHDm/Ou3ui1lSwAPLa1\nYEhncU0liw7jP/uj+Y6z8I49dZ71GlWtqh8u9iHOGgwYnwcwAV17HQBgaw8KnwupG3DbgUsV\nDSQHpW0GyYxw0OR6+bkmOTBGeZrNJ4pvhtzNWN0NBAvJEoDD6NoMDAwzFkbgmKU4t3PdiAsX\nBNItX623/OPLykMXhpf0bBaxbWXaHRuyZt/21CUGNfRBnv0jg2O3rkj9+xeVbo+RCjlu/ppb\nlqesnx9f36nW6C3JMeJU754RE8ObmYdfXT7oWVkY+6vbit/4utoRqYsQrC9OuP/KXP8ePCSB\nlhfE7D/b5Tk+qlGrXMJ97bHl+053VrYqjWZ7epzkisVJUiEHY/zcu2eaukd4/p9tGHhpZ/lv\n71hA+1IsEmUnyrITGVVi+uGyySdvKPrde2ddZS8Rn/30jUUzxmGUHp73Xw9usLc4xYULaAu+\n4iIC7eXpiljA/ssDi0/WKKpaVSaLLT1OunZenHOPwc2bycm5xoFZJXBgXAtgcnWy8HiEFQBQ\n4koUkY+VtWBSAS8cReQBd4YIjig8D3c4tBvs2uGCIgoAjWe7gi9DfOYkwsDAMPNhBI5ZSlaC\ndO28+APnRizbSALdf2VewOaAMTz37hmnZwQAWG3Ufw80Ygx3b84O2DQmhp3CRyt6m7rVHDaZ\nlxzmrb9gAshE9D2xDksIJ1ctSdboLf890OjsEEmKEj1901yxgO32RBGf7cfpuc9KwBmkM1yU\ne0xjKlhZGLt0Tkxnv05ntCZGiaTCKWknvv/K3MYudZvi0uqIIND9V+Y6++R9QBIj+o8cVLWp\n3NQNByeqFQNqU4TUD36fWoO1rnNIrbMkR4vS46S+F94dfbqGLjWFITNeErTxkBgoAEDB4RtV\nkhO1/alVn/zQVN+pZhFETpLsxjXpU3T4BQ8oJhvYPLB6KLDiKBQW7OvtjQsTS2v6PMc3LZzm\n/C+EYEletGfdGUVhT59gB24Z7TMdE8AQAAAiABG0MgciL/5q8eUofmkA5xYouDKUfjVu+hoo\nl4IjcRJKvmz65sTAwMAQvDACx+zlyRuK0mLFHx1s0hmtAJAWK3ngqtx5GcMrYaXGPKQ3x0cI\np65jpapV6apuOPniaMut6zMC3ykzdtr7dL9776xrwu78rMhf3upu6zgxlufH7i7roBn3MM64\ndX3mZQsSKpqVar0lMVI0LzOCRQZ6Bzk3Wtw8qLePNAQhEJoTG6CtMxaJfESZ+IUwEfefj6/4\n37HWc42DWoMlKUp87YqUtEl8wM4++ogWjKG9TzeqwGGzU2fqBzr7dTIRtyBNHiVzL7n68ljr\nO3vrnDX5BWnyn24rjKfbqTZabH//vPJ7l/qUNXPjHt1aIOQF0anBYjfqbWo7tgEAiVgCloRL\nTn9xdZSM/8iW/OmeRWDhCIiFN1En3h2xyGRxiCU/moBvYoBZnh9zy7qMjw82OX+sSALduj5z\nUW7U9E7MGwSB4iMEnf00vxWzzCTLpduRwwOzR7AuYgE7OZATmhZQRCESJ2PFaTD2A0sA0lQU\nkT/Nf3d2C+NRysDAEJwE0VUsQ4BhkeiG1ek3rE4fUJv4XJZzSVPePPjal1UtvVoAQAhtLkm8\nZ3OOZ13A5GnsotnEBgCz1d6u0GUmSP3+jn7BZse/fee023Xnmfr+v31e8ctbJ5Jv78bCnMjN\nJYluGkdarOS29ZmeD46S8dcVx0/+TSeMjM9emxl5slWpvRh5KOSwFiWHTXVMbIBhkcR1K9Ou\nW+k1uGRccDlexTue97scVLep/vrJBefhx2ERt6zLvGXdJZfEvac6Xv9fletTKpqVz7xZ+p8n\nV3mKhi/tLHc2iDk4eL7bbLV765QJPEa7Tm8dct60Y5vWqqSwnc/EvkwHKG0xERaPK3ZhZQcQ\nJIpMJwquABF9w2OwcefG7FVFcYfLexRKQ4xcsKooNmjrlRxcvSTl9a+q3Aa5bHLjwgTax89Q\nXE4lXBFQdrC6SB6IiwSLgAhcX+10wpWipHXTPQkAs46q3YV7ysFqAK4IJSwksjYAa7a7pzEw\nMAQVjMDBAK47xuXNg8+8WWqzD+9xYYx3lbY3dKn/9uNlfq8O8NE66i1fMxg43zRAu6t2uLz3\nkS0Wv5SpP7GtcFFu9LelbV39hnApd1FO1LXLU/3oDOpfYiW8qwti+7VmncUu4pCRIu4kg1pn\nPAWpcpJAdo8cHBGfnRnvS9cb0ll+9dYpnYstosVGvb23TibiXL5o2EvvvwfcI3gAQKEyHjzX\n7dYpo1AZPRMcAOB4laKzXz+WBpypBgM2WGliOww2DY8UonE1nzP4CRSWiFY+MN2zmCCpU+BD\nNHVcsyylb8j4+dEWZ2aWXMz92fVFnkVbMxo+gAhABwCAEAhkYLOA3QIUIDIDOBmAplVMt5vx\nQIWjqgJJU0E8IzxNfWDW2o+8DMaLorNZh5sOUv11xPJHmWoOBgaG4IEROGY+QzrLhabBAbUx\nNlxQnBnpe4v47T31TnXDSUOn+nB599p5fq4UyPHimCjgsXxHYE4vtOoGAGCMO/v1/urDXzon\neumcIEol0JtsGGNvPTgkQjGjRYqMC53R2qrQAoaUGLG3NzVZ7B8dbDxc3tM/ZIqRC9bPj79u\nRepY4kWmnQgp78Y16R/ub3Qbv++KXN8y1nenO3R0oQ+fH2lxCBxGi617kP74bOhSb4IRAkdL\nj8abF2xTtyYYBA4bZcF0PrYYsBWbOWhWLfMYZggY4xPVioYuDYFQTqJsQXakm56v1lu+P9PZ\n0a93RE1vXJhwpn5ApTUnRAlX5McKgql9LDAglIPxOYCLziMsDrC4CGUDTLPzCx5qwo1fgHXY\nngl3HEQRhSjjGkDB22A7SajG/ZfUjYtgTTduPY7SV0/HjBgYGBhomHVnytnGrtL2N7+tcXbj\nR0h5T2wrXJgdSftgisLV7fRBgBUtSr8LHNmJskW5UZ7Gb1cvTa5qVYWJOQmRoiCsBeB7V4j4\n3Bl4WXO0svet3bUOWSdGLrhjQ9baefHjrbDBAAabyWy3UZhiE6SQxWMR9P9XNjv+YH/Dzh+a\nHAZ7jmCdH12W6aZc6IzWJ14/7jT+bFNot++qPVGl+OuDi0NC47hjQ3ZcuPC9ffUKlREAkqPF\n92zOXjxawG1rr5Z2vKNfZ7NjFokI71+M510k4fU/igy4mQstDmNR+rsCkNPDwOBvFCrj7947\n45oPnZ8qf/ZH853e0scqe1/cWe7UMT862LiqKPbpG+cGbQVfQJAgtBjjFgA1AAUgQigFYLor\ncaw6XP8J2EcEouOB8ilvJLHqwawCjhg409DGi/vrvI0zAgcDA0PwwAgcM5nSmr5XPqtwHRlQ\nm5575/QbT6yk3Z61U9jbssHqxc59kvzilnnbd9d+c7LdUYLL55KRUv5HB5o+OtAEAPERwke2\nzJmfRS/HTBdF6eEEgSiP/gK5hJvi145ujPGeU51Hynv61cYYueCy+Qmj5pL6nS+Ptro2gfcq\nDS98dL5HaaA1BPEGhalBs9ZKDe+/WSibwWaWcIRCuq7df35V9fWJNudNR7COxmB5bGuB68N2\nHmp2jTVxUN2m2lXacfXSEDCcQwg2LEjYsCBBa7CSBBrjrqy3RQ5JIIeqw2WTqTHiFjodJDc5\nzG0kK1HKIpFnxRZBoNykoIgSJJFX6x8fdzEwBCcYg5u6AQCVLcoXPjr/x3tLAKBvyPin/553\njR8GgEMXeuIjhHdudE8WU2rNfA7J586SqzguQjnTPYcR4IFKN3VjeFxxBiWtnRL7T7MKt+/B\n6oulf4JYIvlyEAa2jMVG85EBAGz08fYMDJOEghm4ccgQAGbznsDM54ujLZ6DFhv1zck2z3EA\nYLMI2qgFAJhMZoQP+FzWI1vyP/71+j/fv/gvDywW8jjtfZdWrV0D+l/vOFXZSpO0Mo3EyAXX\nrUj1HH/oqjzCf/UmFhv19L9LX/60/HR9f5tCV1rT9/z7Z3//wdlAblybLPYde2i2az7c3zik\no4mG9YbaanCqGw4wgMait2H3sEOl1vxtabvnK+wq7RhQj7h+ok18BIDSWsXYJxYMiAXssdec\n56fKacfnpMidtjW3b8jyfEBarMRTHZMKOVtX0NimXrM0JdyvDUcThkQsNkGjgrEJDotgBA6G\nEKO+c8hN3XBwpr6/e9AAAAfOdbupGw52l17ynLbZ8aeHm69/bt9Nv/v+ml/vffhvRy40DU7d\nnBm8YvLy324zgM3o/7ezGanaty+pGwBg6KHq3gVjv//fyztI6GXDyds4AwMDw3TACBwzGW8F\n7c099PElAHAd3YJHKuSU5Ea+tbvumTdLn3mz9K3dtWr9ONa3oyIVcuZmhDd2qQfU7pcFNjt+\nf1+DH98LAMxWu9Fim8wr3Ht57pM3FMXKBQBAECgzQfrC/YtWFflzI+WLoy2el62HLvQcONft\nx3fxTV3HEO1/lM1OjV11wgAmG41tBO14Q6faszQGADDGdR0j+n5prSgAQGugH58ZrJkbnxHv\nLjWySOKuTZd2d5flx/zqtmJX5+BVRbF/uLeE1iT4ns3Z912R69wE5nHIuzZl339l7hTMfYKI\n2XI3jYNFcMTs0IjtYGBwpcOLfxMAdPTpAMCbgY5KZzZc7DN9ceeFf39T4zwFN3Zpnnmz9HhV\niAm7MwHCm98WgimQX3HfKafZxyUoK+454vf38gFKWkw/nrwkkNNgYGBg8M0sKW6cpXgzI/DR\nzXvF4iS13vLfA43OfaSkKNH1q9Ie+8dx59LxbMPANyfbn797YZ5H0ftkqO1wd65yUNNOPz4B\nSmv6duypa+nVAuD4COEdG7JXFU2k6cPZX6AzWtkswjN9c/IcoYu3AIAjFT0By4U1WWj2Eke9\nyw2MKVqfSACwY/e+J8p7fYqb7hEt5/cN0eySxcgFY5xYKMIi0Qv3Ld6+u3bPqQ6HEpQRL/nx\nNfluf4krC2OXzoluU+g0BktSlMhHOQZC6PpVadcsS2lX6ABwYpRoKg7myUAgUsqJtFAmG2UB\nABbB4RBBUV3CwDBefDh8O+4S8ugXxiwSObKl6zvV+892ud1rp/C/v6kOKlPqWYEsHbqP0YxL\nkqdC4ABdJ+0w1nUG0jAJxc9D2l7cuB+GT98YSA6RdxWS05S1MjAwMEwXjMAxk8lPlR84534x\nBAAFqb72P29Zl3HZ/PiKFtWQzpwYJSpMC7//xUNuG+M6o/VP/z2/4+nVniagGMBGUQDAGmfD\nBu3WvY/x8fLNybZXP6903uzs1//+g7M9yuyb1mRM+DW9BXxMHpWXHpBBjZcO2CkgMcprlk2S\n97vcQAghoFc4PG0vM+IkCIGnyoEQZCWMMFTbMD+hopmmimTDgoQxTiwI6VEaPvi+oaFTjRDK\njJfesj4j1kOvEQvYj19X8NDVed2DhjAR1+lN6AaLJNLjxtpWxmERnoUhQQWH4E1G1yit6Ttd\n1682WBIihJtKEiecsqnUmKvbVXqTNTlKnJ0oC+Iw66mCojBCwZziHdTkp8hZJGGzuwu7Ah4r\nJ0kGAAuyIj473Oz5xOLMSMep1ls3SvegQaEyRocxuUKBA0nTQJ6LlTUjRgk2kbxhSt7Po6Nz\nlPEpg8jZjOPnQW8VNqqQMBzFzQW+P/e6GBgYGCYPI3DMZG5dl3GiWmE0j+gyiJELrl4yigtj\npIy/dt7wpVJ1m6pHafB8TK/SUNs+NCfl0okNA2gtVrXV4ligEghJOWwxe6wSQHqc9EhFr+d4\nZoIfrMLNVvt/dtV6jr+3r2FzSZK/sl39iFzM7aerUHBtPZhq4sIFC7IiT9e7t/jmJsky48f6\npSBAXJJjstPoNTzS/b89UsZfX5yw74z7VtWaufFu1+4bFiTWd6pd7UgJAt26LmNBkFnSjp2y\n2r7/9+4Zy0U33+YezcHzXb+5fcHCHJpP5DATDewEQxKz1f78+2ddHVt2Hmp+dGv+ZfPHJ4Rh\nDB/ub/joYJOztC0/Vf7kDUVx4TO5YsiV0/X9731X39StIQkiK1F69+acIHGiDSFkIs5t6zPf\n3utubHTv5TmOyqn5WZErCmLczoMCHuu+K4b9NWkdOka9i2GKQFnboLcMd58EixoINpKmoOQN\nwJ+ac5AgGrStNHMQxEzJ2/kEiWNAHMPonAwMDEELI3DMZBKjRC8/vOQfX1ZVtigBACFYURD7\n4FV5Yzc1BAA3c8eRdxkBLgkcKrNZZ70kplAYq8wWCoOUMyaN44rFSZ8fadEY3FfCN61JH/ts\nvVHXMeTsYXbFaqMqW5TL8v12iaAzWr8tbW/q1nDZZF5y2IYFCRNLul1VFFtH17OzekI9NRPm\n/26e+/z7Z123DXOTZL+6rXhcW7hSjsBqsrk1pIjZfDZdUuyjW/N5HPLb0uFgHYTQ5pLEh67O\nc3sYQvCTa/PXzIs7WtHbpzLGRQjXzoubIivcAGC1US/tLLeMzCqy2KgXP73w/s/XevaaaQ3W\nXqUhKowfhNpcUPH+vgY3P1qz1f7yp+V5yWHeDJVp+exI8zvf1buOVLYof7m97F8/XcmZBfmd\nnx9peePr6ou3qAtNg0+8fvznN8+bWIvfbOaWdRmx4YJ3v6vvGtAjBMnR4rs2ZS9xyYf+xa3F\nX59o+/Joa4/SIOSx5mdF3LM5x9l55610jssmQ7R8w2ix9SqNkVLe1JVDjsBqxpUHcV8LECSK\nTkNzVgE5ifdFJIpdgmKXAGUBgj0lySnOt4paiPvPAjXSZAoREMOYXzAwMDC4wwgcM5y0WMlL\nDy3RGCwDalOsXDCBSLkwEU2KgQOZy102CruqG040FouYzfJsRvBEKuS8cP+iF3eWN3apnSMP\nXpVXkhM13jl74sMzwmCelOGoK+XNg79776zT/m3vqY4vjrb84Z6SCZRdbFmWcq5h4FTdiOqJ\ny+YnrCwMaCacVMj5ywOLT9f313eo7RTOSpCW5ESNt0CdREQkT6qzGc12G8YUiyCFLB7Xy2Ul\nl03+5Nr8G9ekN3SqMUBWgtRHQ0F+ijw/hT5YJLSoblMptTTNR0qNubpNVZh2qaese9Dw+v+q\nymqHF+2FaeGPbJmTEjTVHA7z1wCtVcaAZzUQANjs+MC5rh9dRhM3QwvG+NNDNI0DXQP6oxW9\na+cFNqYx4Kj1lrd2u5e/URR+7X/sIxaSAAAgAElEQVSVy/JjaM1rGXywZm7cmrlxepONQOB5\nRiYJtGVZypZlKVYb5emWtSg3KjqMr1C5F/dtLkkMNvecUekbMr7xVfWxql5Hyee8jIgfb5mT\nFCUCixq3H8DqFrAZgR+J4hajiAK/aAe4r5X66kXQDfc24qpDcHY3ueVpkE3avoTggKYLN+7D\nmi5ABJImoIyNIPLDpcsluGEo82bc+jWYVcMjLAFK2ojEKf58FwYGBoYZASNwzAokAo5EMMGd\n3txkmVzCVXpYP8glXNf+FDNFryBgAAtF8cgxXXulx0lee3RZXYe6o18XLuHlJMrGVWzig4RI\nr7u1Y7eT8I3JYv/DB+fc8mVae7Uv7Sz/w70ljpsY4z2nOk9UKwbUprhwwaaSRG8tFSySeP7u\nkgPnuo5W9ipUhrhw4fri+MV5E7wOa+nVfnywsaVHy2GTeclhN6/N8ObdQMuCrMhJtn4QCEnY\nAhjzsjdKxp+wUUIo4iN2V6W9dJdKZ37iteMq3aU/xvLmwZ/+88Rrjy33dOsIJBjDwfNdb++t\n71UaACA+QnjXpmzPYNoAY7baaWUjAOhVjiPHUaW1eHudpm71jBc4ypuVbrVFDoZ0loYuNdOo\nMjGEo53aaL3AuWzy/9218A8fnG1TXArUWDsv/t4rgij5yBOM8Q8Xei40D2oN1oRI4aaFiUIe\n6/HXjrvWh55rHHj8teOv358T1fUh2C+O67txw+egaUNpV012Dno19fVLTnVjmKFeas/rxE3P\nTfbFu8/iik8u+m4CNqqwohoV344i/fm9IHEyyn8Ia9vArAKOBImSgPS6/8TAwMAwm2EEDoZR\nYJHEz64veu6d067XuBwW8bPri7yltEwGhFBOkizH3xfNceHC4syIsw0DbuPZibKsBP+81+n6\nftpV0JmGfqXGLJdwTRb7L7aXOdqFAKCxS324vOeqJck/uTaf9gURgnXF8ZPPTNl7uuOVTyvs\nF71a6zqG9p3p/PP9i4PcV3JW4aPGJ1J26a4vjrS4qhsOdEbrxwebHr+uYKomNwY+3N/g2sHR\nNaB//v2zD16Vt3XFdFrrc1gkl03SehOMq8aE9F6kQBJB3Z+i1JotVnt0mGAytqAGk9foZb33\nuximiNQY8T8fX3myRtHSo+VzyYJUefb/Z+8846O4zrZ/n5nZXrWr3iuSEKACCEwHFzA2Nrjb\ncY/tuKQ9rnESpz55kzh5Eidx3I1bbGwHY4yNO8X0LiEJBKj3upJW2r4zc94PK5bV7uwiaVfS\nSjr/D/6hM6OdsTQzmnOd+76upLDTmFyK597y9o5ea7RG1tlvbekeTMA9dq5r++HGWSkRvt2v\nJqtz0xdHf5JnA8CeJRu44ziKLgTlKA2k+eZzzs9fxx01IpWAHIDbq6GnBXRB/J1lbfj0VvAK\nBcMcrvgIrXgaUEgraxCN1Omh/EACgUCYihCBg+BNY6eprNbQb3amxCgX5EYzNDU/O+rVx5e/\n9231ueY+AJiRqPXNdxD5f9EPsGk8eerWgt+9ffxUfa97JDNBM1I7iQB0+pQNu8AY2nstOrXk\nw901bnXDzacHG4pzohfkhrSW1YM+k+PfW09xQ5NoTFbn3zaXvfCTJWN0UMJIyUnWxurk7T5u\nvvF6eY7H7KWirheE8L2uXGAAJ8/zGEQUoscs+sJodry3s9p3/M2vzl5ZnDSKtrhQgRAU50S5\nLRtd5WAuL54R3XEahTher2g1mH03eVaxhRV7y9te3X7GdUUpZaJbV2VetzRtdH5Acf6NVOP1\nI/AxIYQKhkZLZsUuCZ11VGhhOf43bx13t9E1dA54rYU4Wf7wmU6hb4XyNsB5gHwaUnBvFRqV\nwME3nbW/+StgnZTY74MIGztREAIH7q0DVsiqzN4PxhbQJo/ks3hsaIb+LlBGIH0y0OQVnUAg\nEEYDeXoSLsDz+OXPKrcdqHfPhxOjFE/dUpCdpI3TyR+7aU6A7xVTlJSmbZz3YqmCYcZuZjUi\nIpSSvz206MiZzjNNfRyHs5M0i/JiQhh4GGBN2NUftKesTXDrnrK2sRM4jp3rEvQfqW4xtvdY\nYie0r4HghqbQz24teOaNo555zCq56KlbCz3Tll0BzL74Bk8CgJnleu0sdz50V8HQEZIxuRlP\n1fc6hVoYbA6usrGvKCsy5EccPt9fm1te15OVot2wKi06QgYAXX3WqlrjSM/qnjUz/vBuidfg\nrFRdsVDGzYTz+eHG5z4qd39psjpf3V7Z0m0eXZlPXqouKVrZ1GnyGs/P0E+fEJlxBmOobOxt\n7jKr5KLc5IgRdRROOJ8ebDjioV/QNOVVkQEgkAXuwsn5eUKxI+gpG/KBO94F1hnokAAgDa5N\n1SEQM+cCO83Df+Dizjq8eyPuOh8Npommlt+NkoQLPAmEaQKPyUSVMBrIdUO4wPu7aj7eV+c5\n0txlfuaNoxufWDGciu5IqcRgt1vZC9NpBcPopGHUI4oQLMgdq3KJoqxIhqZ8p5qJUQpXXoO/\nNn5Dv9+cmuDp9XNQAOg1OYjAET7MTInY+MSKj/bUueqkspO01y9L87LOSYtVnWkUyNZJ84mP\nMbNc99D2ATPLOXkcN1ovngAEyKcMYO47PsTr5f96dIkdLtyVUVpZVJGs3+FUDy/dycXy/HiE\n0EufnnbV1bvCfe5bmxNChTRUsBze+IV3ECkAfHGk8fqlaUkjtxyiKfTM7UW/eeu4Zw1LRrz6\nqVsKgjpRwnkOne749FBDS5clQiUuzokuzo3+x0fl7jtdJmbuuCLrhmWTpjHhu5NDpHzk/o/n\nIEIAAopDqtb3DxYGQCAdVakUxnxj5eA/Wd5HZgEAAIUWxQT3s5X5bRFCMp/TdvTivgpw9gGj\nBGU6UqQMjg8Y+E/+PEQrMXby2/9GXfcMip7IRr/zCP7sCAQCIUwhAgdhEIzhk/31vuN9Jsd3\nJ9uuWnjxMksKoSip1MHzDo5HAGKaCpPmlPEhUiO998rsVz6r9BwUMdRPrpvtmgRFqCSujAkv\ndEKNwSE8K7+b1KM8Lsfj0dW6EwKjUYjvvTI7wA7XLk795niLl4hGU+j6Zd5vwEaHQDaQg+ct\nLCdnQpy2kBTld84cKgffUcNj7ACB6hKjw6EcXrqTm2Vz4hbPim3pNg9YnCkxyvBJivGivn3A\nN2wbADCG8rqekQocpdWG4+e6+syONcVJIoZq6TLTFMpN0a7Ij6fIQyAU/GNL+fZDja5/txrM\np+p73/76nGdTodXBvvJZpVzCrF0wkmaHicOflO8Jw1AY874VadfN83WGQkCJkD5vNKfC83D+\nGBhjzuqgvRReiqZW3QNCgeXDB0WkYrkOLD59guoEUA5pI8I9J3DHLsDnZV/DUdDMRPFrAVF8\n+TcClSAci098htb8KJjTCwq2G9vKgDMA5oHWIuksEAVrCkYgEAjjABE4CIP0Wxy+/oUuGjoG\nhv85YooSTyddw5MblqVnxKvf21Fd09ovFlF5Kbp71mS7A1yWzo59b4eAVcHy/DFMYZifHaWQ\nMmab93R3VqouaoQxJRyPKzsGagwmi4MTM1SCWpafoJGNWTYhxrjLaFPJRBNo4hBupMepf33n\n3H9sKXf782kU4oevzfPKyuUxdvLC9dh2DstD/ePMiFfPTIk43eDtD1KUFRkgvWh8cPDCPwgM\nYOd42Qi1HppCEy7ZXBSnUL+SiwC1NgKfw/J/fr/Us7FOIWX+54Y5Ex6OM5U4UdXtVjfccELX\n7Ie7ayaLwKFTSTzthDgO+9r0Ugjuv2rmf7+rcQe0KWWiH6zLLSyMwWcN2OiRykxLUOZ6EI/K\nEpumkS4OG1oHz8TO8ZydkYmAQkgsRUkz0aIbUVRK4M+4OIim8r/Hn3gT7B5vSrIIKv/WIbvZ\n2nH7DgB+iH+q8TRIo5G+GDqHFM9e2MHP+HjgbMTmAxcKbbgebN6DZAUgCevIHgKBQAAicExn\neB6391q6+myxOnlMhEwsohASblMVzKsjCFKYGVmYKdDejzHWqyVKmciriOPK4qSxM+AAAKVM\n9NhN+X/eVOo5t4nUSB+9MZCjii8Yw67qrq7zEpiD5et6zG391ityYhT+zdtGh93Jvbej+uN9\nda4Gh9xk7YPX5JE0ShcLcqNff2J5abWhvccSrZXlZ+gnvJQAIfjl7UX/+58TnhpHfob+Z7cW\nTuBZufCj8wAAYKEK+SlAYqSCppDgJDktdgSzxHd3VHnZBplt7LPvl85I1JDWtlCxv6J9mHu2\nGixWOzsp1N6ls2M9HwVOlqcob/OfdZekXL80bW1x0vGq7vYeS5RGVpCp1yjEAIBm3gk9lWCs\nBdYGsigUUwSi0auKzMKrndtfcX+JWd45YEcypeQHf0cKzag/1htNErX0Cdx4EPqbAVGgSUZJ\nC4Ee8mTGfacEGz1wXznSF4fsTEIGxtbjvm1E2FqGxGmA/FaGEggEQjgwCf5YEsaCivqef22p\nqGsfXHAozIz80YZZWQmac81G353lEtFv3z7e3GXSq6XFOdHXLEpl/OcmEnxhOf6XG496hdRG\nqCQ/vX72JTNjfHbHDt7g5I0YOxESiSi1mIoMpv11yazYrMeXb/6utra9X8zQs1IjrluaNtIX\n5fpec5dPgY+N5ctb+xcOLR8IEozhmTeOllYb3COVjX2PvXjwz/cvmJ0eygNNXmRiRuiyuQCF\nEEMhVmiKKx6bOzdSI/37w4uOnu2sajYihLKTNHNnhIX7psh/G8Vkb6BjeWy0OkU0pZIwnrNH\nlVx0WVHiV8eavPbPiFfPGckd9OUR708AAAfL7yxpve3SzFGd8sRTVmvYWdLa0WuN1kqXzYmb\n8Ku0zyTQTOSPydITdM2i1KNnu9x/7zDGdgebHKOSiOgBqyMxSnnNJSkLcqIBQCZhBLNgkC4X\ndKEpE2Dmr8bmPnbvFuDY8x8eK97w41CqG4NHkqL0lYF2cAq8XAEAOIwAANFp0FLpuzFYf5BR\nw/UCL+gOxoOzA8RBl70QCATCWEIEjulIdUv/z1457PAIPiip7n78pYM/2jD7D+8eZ7kh86Io\njfStrwct6xo6TCequr890fKXHyxUSMnFM1w+O9jopW4AQO+AXcgBFFvYeg4POsZjbLdzXU5+\nQM6kIhj9fCwmQvbI+lH1MJ+nzY8TattAiB1SD1d2eKobLliOf+3zyn/8cHFojzWF0YgYg93b\n8EVEoZAbcLhBCIpzootzxrAcaRSIhNOdsIRmJkrgsDm4pk4TTaPkaKVXfOYwYXlc2mI80zHA\nYwwAchE9NykizSPQ5Icb8uwst7u01T0yMyXi6dsKhz9Dtjs5f04KLd0CcbnhD8bw/NaKTw82\nuEe+ONJ0+dzEx2+aM4FOsQE8krzITFBLxqwfMLSIGOqP9xV/fbx5z8m2zj5rrE5+WVHCmHZi\nBgIh0cpbmIKVfF05NhtRZCKdVQTMRFS9UX5Mr2gpAFBzruBO7wb7UBsOmkFFV4/9mQmBBfzC\nzhNgE4FAIIQFZI46Hdm0s9rhE+vYM2Cvbet/9gcLX9x2uqrZCAASEX1JXoznW7KL6hbjf76t\n+sHVpA9zuOyt8BsQ69VW7eB73OqGGx7bnLxBTE3kYqOX7OUmQMP/6DhZ62PVBgAAZ5r67E5u\nsrziC9LYadq6r76hc0AlExdk6K++JHl089vhoBTRGKDPccGLQ0pTeolociwBhxS9VNJts9s9\nNA4JzURORLqTk+Xf3VG1+bta1+NXKRPdtXpGtEb2xdGm1m6zXi1dkDus+rjvqrtbjBeeEhYn\nt7e2m8P6zMhBxxOJiP75bYU3Lks/3dhrd3CZCZrCzMgRzeLFDCViKMH0X4VsUr427C1v81Q3\nXHxzvHl2mm5NcdKEnBIALM+P80ou88ddVwSyHw43EEKr5yWtnjdhP1gvUEQMHRGo6m08UGWA\n8ZTvMFJlAAAoddS1P8O737hguqGNpZbfhaJSh+zNOXDtXtxTA6wdVLFU+nJQjo2mTPlvC6JU\nY3JEAoFACB2T8k2FECSVjd52gIPjDb13XJ717x8vMVmd/RZnbITsnx9XCO65t6yNCBzDx19W\nq+84y5sE92R508QKHEo/LS2qUPeEO/z4IGIMDicfhgIHh60ctmDM0UjKUCp/zUSfHWp44ZNT\nbp3owKn27Ycbn/3BggjlWM20VSJawVB2HvMYiyhKPEnq20MOjVCMTGrjODvHA4CYpmT0xFxF\nz31U/s3xZveXJqvz31svTHgaO00l1d07Slr+8sBCuf/6uE6T3VPdcIEBSpv7MiIVnr/jrERN\nVuIo6/ARQkVZkYcrO303haRIp6Ku53RDL8vhtDjVwtzocaih2FnSIji+o6RlLAQOnsdfH2su\nqzWYbWxKjOqaRSmCxRozUyLuuHzGuzuqPM1wc5IiYvWy7062uiyxYiJkD1+bN6ZWTYRxAKmz\nQZmOTbVDRkUqFDVYmYiiUtENv8E9zWDsBJUe6RKBHvocsPRwB1+4ENfSU8c1HaHm3ISS5of+\ndCkFMLHA+njEUCpgwqL9kEAgEAJABI7pCC9oJeoxrpSJXM6F/mbmPaFuTJja6NXS5i6Buu5I\nrfcrLwY/03vgAABjKKs11LT2ixgqN1mbmRDqLmL/pOsV57oGfC+cjMgQh0okxwh/oE4tmXA3\nTR+wjWt18hc6qyleLKUTaOQdT9NqsHiqGy4aOgZe/vT0mDpxUgjJiF0OAABIaVo6QbqGi+Yu\ns6e64Y+qZuN7O6vvW5vjb4dOoWcyArA4OZOdDaHgeP9Vuafqe71MkZfOjp03ct+KVoO5tNrQ\nZ3IkRStmper+/lH5odMd7q2ZCZpf3l4Yrw994E630bZpZ/XZpj4e485e4b9Znb3ealHwGM2O\np187Ut0y+GQ4eLpj6/66J27OXzpbIIDmjsuzinOith9ubOo06VTS4tyoK+YmIoQeuTavqdOk\nUYjjIxUklntKgFDSddBbintPgqMHGCVSZaCoJa4WFfcuSJ8EemHFjS//yDuMluf48s10VDZI\nR5UyE/h05QuxeQ9wHkeklEixBILoliUQCITxgQgc05HMeM2RfoGlOd8Js14t3CHsb5wgyIr8\n+JM13r4SrnGvEQqJeCzwIk6BqKvP+qf3S8s9OjhWFMQ/esMcqTjQtA1j2FXasqOkpbPXGq2V\nLc+Pv3xu4iiWS7Uy0YJk3bGmXrd1JUKQGanMigqxwLGyIP6dr6v6Ld7ee+sXp05cp7wwdq7T\nU90AAB47rGyTQpTpZZiyr7xNsMdnX3k7exM/do0qEwXP49IaQ0PHgFImykuNGIu566TDN0bX\nH/vK2wIIHPUGi79NfiJxR0lytPKl/1m68Yszx852m2zOeL18/eK0qxeOLKkUY3jjy7Ob99S4\nr3+pmHalI7mpbjH+5q3jL/50aWin8RV1Pb/YeNRq907I9kKrFIfwoC5e+OSUW91wYXNwf/2g\nLC9Vp1MJVGxlJ2mzk7xTojQKsSaN2CpPLRCFdEVIVzSa73VacdcZgXHOiTsqUMqiIE9NAEqG\nVFeAoxFzBgAO0ToQpQIKuyJKAoFA8IUIHNORG5enHz/X5ZUjKJMw1yzydsZenh/32SHvvmUQ\nmpkTArCmOOlEVdfe8iHVnqsK4y8rSvDaU4S0LAz4fgKNNL95+3jV0Iyb3aWtIpp64uZ8f8fl\nePzbt4+7F0sbOkxHz3btLm393T3zR5GDk6ZXxKqlDT2WATsrFVEJGplOHvq5gVou/t098559\nv7T1/ESOotA1i1JuWp4R8mMFB3byAlNWDCzL94uoIdOVbj8WrQ6W7zM5hm80OClo6Bj48/ul\n1S39ri8ZGm1Yknbf2pwJtHIMBwRzWwUx9AvXzQFAQ6+lxyqcuyGiKWWojZ+jtTJXhRHL4dEl\nZ32yv/79XdWeI17qhov69oGyWoNgwPbowBj/9cOTF1U3AGCRUIpHMNidnNej3oXVwe6vaF93\nCYmfIIwK+wD4qb3FVuOYPVsRiFMQkIuWMGHwmGhqhNFABI7pSH6G/unbCl/45JTbJz8pWvnY\njXOitd6l9fkZ+huWpW/eM6RrdGZKxG2XTdaYwAmBptAzd8zdW9723cm2jl5rrE62siBhUZ6A\n5xlDqcQ40sEPiVwRU7qzDXyVUILvjpKWB67O1SiEhYavjjZ5loK7OHau67NDDesXp47if0Qm\nonNixtxgbGZKxCuPLT90uqOhY0CjkORn6FLG/qAjhccsBmGDVR57T1D9GW0wNFL7+d1NUmwO\n7uevH+3qu1D2z3L4v9/VKqSiyZstGhLS44Z7DQcQvBp6/JZv5Mao6DGTkEadC751f/0w92zo\nMIVQ4Khp7W/1X+riJjdZO7onYQB6BuysH+vljjFohyFMFyRKAAQgoHEg6bD/PmLM156Ajjqg\naBSbgVLnhPIMCQQCIWwgAsc0ZdmcuPnZUZWNfV1Ga7xOkZsS4e8V9oGrcxfkRm8/3NjSbdap\nJAtzY64sThp+3CDBzdLZcV492BhDj8UxYGflYlovF7sqtCV0NEOpWd7Ig4MCMUOpaSSr7xCo\nowEAnseNHabZ6cKVzIILiQCwt7wt5K/1oUXMUMvmxAEItKyHCQj57Svx3bQoL+atr8/5dhAU\n58SImeH2p7Acv3V//Z6ytq4+W5xOftnchCuLk8KtLGJveZunuuHm4311t67KCLezHU+yk7T5\nGXrBVjUvVhT4rY+zOYVnzjSF8uPHz5FnmLAc39Yz3ExZaUj9g3sHhOtcACBer7A52CitbEV+\n3LWL00at3fhDJRMhJLzW7k+JJhAujkiOIjNxd5X3OMWgGKEMeIxx63FoL8PWXiTXQ1whKBK4\nT/6O22vcu6DkPGbdT0Aeev8OAoFAmFiIwDF9kUmYoqxhrZjlZ+jzM/QhOSjL8U6Wl4U6emMy\n0m12HGnoMdoGPfzkYnpeUkSCRgYANJLS9JBV3ADTYLHI7ya/HrH+a+AD0zNg//Z4c1OXWasQ\nz8uOCtVVMRlBQFNIKmiYQiNvy4mUGNWdl89486uznoORGulD18wc5uGsDvbJlw+fbepzfWno\nt1XU9+wrb//9vfND6FyAAQAw8hMEMxzq2wUarADAaHb0DNinuXfPL28veu6j8v0Vg7IjQig1\nVlnXNuQnNjtdd8tKv91YMiHDHQygljBhqB3RFKIp5C9h2hOE0JyQPkz0Gr/hRI+sz5ufPYYx\nEEqZaHaavqzWW8miKESSUAjBQM2+njvwb7B7PDEQReVdA7II7115Fh9/AxsG1RBs6oDO02BH\nuH3ISgluPMV+/gJzw8/G9rwJBAJh3CHzTMI4caq+99XtlWeb+jgex+rkt67KXDM/UXBFFwM4\nOJ7HIKbR2BVdTyxmB7urqpP1WNK3OLh9tYZLZ0RHCq3yzUrTCa4KKmWi9Di/yy96taS2TWB8\ndKYPu0tbn9tSbrENtrV/sLtmRUH8kzfnTz2PzGEioWOsbKNXzTBDqR2c1Gh3ODmeoZFKzChF\nNADcdmnmnHTd5j11LuvNwszIW1ZmBEgD9eKjPXVudcPNsXNdXx9rvjIUIZcOnu13WJw8iwFo\nRKlEMjkzmvxakX8lLsCmENJttO0rb2/tMUdpZAtzo5OiQ2yCGwwahfjXd86tbeuvbukXMVR2\nkiZerzhZY/j8cGOrwaJXSxbkxvh7KrpI1cnrDN41EQggVReONq4IoVlputLqixetbFiSGq+X\nh/DQabHq5GhlY6d36rZOLZnjp94thDyyPu+xFw96BdDctiozOZyuxqkKBt7B9XO8HRDFIKmY\nVvqL7g4DMFi6sK0XSbQgjwL/VYGDKKPplT/jq3eCoQZYG6jjUMZKpEkU+NzGA2514wISjCKU\nuHfITYHrSrGxE2kCSm+sDWjxkNPDPDZWgLUNMAZZDNLOBkRmE9Mdo9H49NNPb9mypa+vb+bM\nmb/85S+vu+66iT4pwvRl8j2S9u7d+5///GfPnj2NjY0RERHz58//9a9/XVBQ4LkPuc3CjQOn\nOn7/znG3zV57j+Xvm8tqW/sfWe9dWmm0s50Wh3vmHyEVRctF1JSTOc51mlifhgUe48qO/qXp\nAmU1CZGKqxak+Bq+3rMmO8C8cXl+/NGzXb7jo/CIbTVYnv3gpFdv+e7S1qQoxR2Xzxjpp00N\nGKSQM6l2rp3DVgBAQItpvdGm6rae79FwQp+NVUuYBKUEAGal6WaNNhbhwClvL5Xz4+3BCxw2\nztnjsSrIYb7PYXbyrEY84mlzXqrw/2BKjFI9Bpa0Xnx5pOnFbaetjkEN7o0vz9y6KjPcrs/0\nOLWnKDmi+rgkrWxGlPJc15ApSqxaOjM27ExqXNyzJufxlw462SHPjeRoJWBo7DIBgE4l+d5l\nWSMNZ7koCMGTtxT8/LUjnnlMMjHz5M0FVS3G4+e6+82OhEjFpUUJY9E2khareu2x5W9/c+5k\njcFsc6bGqm5cnhG4bARj2H2ytazWYLaxqTGqtQuSxyLeZcrD8lYz24HxoJGtA/rtXJ9CFEeF\n4fR7oJmv+gRMbeDSyBWxVNa1oL7Yw1wko3Kvuuhn4/ZywXFKI+d6vVU/MLTAUIEDm/v51hps\nG6CQEbrLwGYEikYRaShnHajiwNHHN30E9vPCZR/g7iNU8vUgCZmHDmHSwfP8VVddVVZW9oc/\n/CEjI+P111+/4YYbtmzZsn79+ok+NcI0Jfwe+hfjz3/+c2Nj40033TRjxoyWlpZ//vOfCxYs\n2LFjx5IlS1w7kNss3MAYXtx2yjdEYNvBhnWLUjwXtfpsbJt5SPdEr83p4PjkKVfZ3usnCqHH\nJx7VzSPr8+L08vd2VJltLABEaWX3r80J0K4PAJfPTTh6tuu7k62eg4tnxa4Z+ZR4x4kWQee8\nL440hdsEcjyhkUzOpGHgAXgEjMnBdVu9m1b67aycoSKkomAO1G8WvjD6TH4vmBF8uEPAjtHM\n2hWMlKFG5owwb0ZUYWZkSfUQl1yKQvetzQ3qFIfBmca+57aUexqdsBx+55uqpChl4NtkcrEw\nVZcUIa/pNvXbWIWYTo6QpzwYAmUAACAASURBVEcqwlYAzk3W/u2hS17cdtqVkitmqKsvSbnz\n8hlyKWOyOp0c789/N3hmJGreeHLFf7+rOdPUx/E4K0GzYUnqm1+d23Gixb3Pf76tevymOYvy\nQhykAgA6teSn188e5s4DFuczbxz1DBLevKf2yZvzF84UMKIm+AMD76luuOCww8p2KURhZudk\nNfBlG4HzeHqb2/nyN6iih0EWCpnALtwqiAWXQ8Qe71cYO/d+7Nj5AThs4tx4iDqvnPIcNlTj\ng/+i5j/A9+29oG64cBr55k+ojHvDuFiGMLZs2bJl//79b7755l133QUAq1evLioqevzxx8nM\nizBRTD6B47nnnsvMvODGf8MNN+Tk5Dz77LNugYPcZuFGW49Z0D0eY1xS3e0pcHQJTfvNTs7i\n5OQhtaALA4TfAwLYH9AUunF5+g3L0tp6rCIaRflE3gh8GkK/+F7hivy4nSWtbT2WOJ1s2Zz4\n5fmjedVr8ymMd9FttDlYfvhOmVMSBBQABQBGP7GURjsbpMARpZV2Cpl3xkRc/DIIDId5Fgsk\ndwKAnXeOVOBACJblx5bWGLBHPxWNkDxEtjutBstXR5uau8xapbg4J9rT1OCLI42+Nq4AsP1w\n41QSOAAgQSNNmDzRwtlJ2uceWWS2sUazPSZC7raMUcqCuiOGg0ouuvfKHPeX73wzRN0AAJPV\n+cf3Sl97fHnw91EwvPTpaU91AwBMVuef3i9966mVxJd0+LC8BQs9ypy8BWMOoTB6hcDN+4ao\nGy44B+7cj5IWABIBUgMEcYNIVGDtERhnfX4+YhmKvWD649y/zfHlWwBAa+R0lE9dGM/yVdtA\nKZR6bjeApRXkCaM/Z8JkZuvWrVKp9JZbbnF9SdP0HXfc8cQTT5SVlc2ZQ8J6CBPA5BM4PNUN\nAEhPT09NTW1tvbBGTW6zcMNqF54+AYDVY0Lo5LBv18bgbiw/xQQOvULcMSDwlqC/2OssQmik\nzeqLZ8UunhXsEqXCz/xcIqJF09WDwxcnLxxy4fRzYQ+fVYUJp+p7BceD/GQsmPcAAAC8/03+\nMFmdr3x2xusznRz/t81lG59YEWSr2WeHGl7adtpxvt/h04MNC2fG/PL2Ipe+1twtrME1dfmU\nZE917E6uqdPE0FRilCJMLHIUUkYxbMeZMeKLI02+g3Ynt+NEywRmGNudnFeRnQuLjd1X3n5V\nqJt3pjA8FtaXAYAHloYweoXAAy3eQxIpZGSCRoK5UgAAECE6C6hR/vZRXD7uE0hew33exXr0\n8tuAOf/WwfPOPR+5/klp/bxmWLpAKdwQh529CIjAMU05depUVlaWRHKhIm/27NkAUFFRQWZe\nhAlh8gkcXrS0tNTX199xxx3uEXKbhRtxOjlDC3vpJ0VdKN/AQgHv5zf5GcfQ0WvpMzkSoxTj\nsB4YQmZEKWu6Tfahrek0hfJiwzSwbV521CcH6gXHp5xByujxF2gSvFfuVQuSK+p6dpUOmQht\nWJK2KC/YInaaopCfW2yk5RsAUFLdbRUqY2npNjd0DKQGYRVR3z7w/NZTXjUah053bNpZfdcV\nMwBAJhb+cxaq4pFJAcvxm3ZWf7i71u7kAEApE929OnvdJSnkJrU7uW6j0MozQIsfaWx86Bmw\nO1hhYbStR6B3jOAPBP6ju/1vmiCGPnEpCs3MA6lnGZETc6ehpw0fP4yN7UiuRYmz0OwrgB7W\n0wwlXwLd53DXmSGD8YVUUgJ/YDPu6wQApE+klt1CZc67cE793dhkHPzCX1Wmn3I/AABq0pSV\nEUKOwWBIT0/3HNHpdK7xwN/40EMPff311/622u2jjPwjECb3mx/P8/fff79YLH766afdg6O+\nzebMmdPc3Bxgh4t+whTD6mC/Pd5S3z4gkzCzUiNG3Q8slzIrCxK+Oe79s42JkM3PuVBhLqIp\nCiHBRWOp0CLkiaru57dWNHeZAQAhWFWY8MDVuWPX0R1aZCL60qzoo029XabBx7dGKpqXHKEN\nV5mmOCd6UV6Ml9WlSi76vkcFOEElZkwOgfc/lVC654igKPT0bYWrChP2VbR39Fri9YrLihJG\nbVnqCQIkYyQW1vs1gkaUlB7x1RjAEyRIu5AdJ1oEO1C+OdbsEjiKsiKPnOn03WGYYdhTg//7\n78kdJy6oYCar8/mtFWab89ZVE1ahECaIGUrEUE4hKUEhm8gXIZVMJJiQBQAqeZj+OQhPGEoO\nIKDW0khMofD6SSJlAjZdSDhD+sih6sZ5qCbcUAoAuK8dt55BVQepdU+BdBhZPIhGc++B1lLo\nKMPWXiTTQ3wRislDAFTeMrCZgaJA7HNEj5wU3o9NGGAx0DLgfPolKTFShCDPizDFCBAK5uLB\nBx9ctWqVv63f+973cDjVXhEmEWEtcHAcNzBwwSpJrVZT1IXnL8b44Ycf/vrrrz/44AOvvhVB\nLnqbvfnmmz09Ql2LADabbd26dS6hZJpQUd/z/94tcS95fQhQmBn5qzvnjq7M+Ifr8/otjsOV\nF6YfCZGKZ+4o8vRuQAB6majLx2VTylAKn/lhabXhF68fcRuXYgw7TrTUtPa/8JMlYVKSfVE0\nMtFlM6IH7KzJzsrFtFoiCudVVoTgmTvmfnqwYduB+laDRSkVzc+JundN9nCsQKYPGgnTb2fN\nziEah5Sh9CHSrRbkDnGdCBUakZzHvI27kGpJI0onUQZwhPFHtP/rIToiqPW9DiELEgDoMlo5\nHtMUumph8tfHmmvb+j236tXS21ZlBXPcSQHP488ONW7aWW3oFyhSeG9H9YYladKgVbZJDUKo\nKCvS82+Qm3kzAuWbjDVKmWhmSoRvAxpCaEFO6G/2KQyFGCmjs7FeC1FIxoSdxIkSF+POk8AP\nPnKxQin8qFUpQSwCx/ndelv441upxbcP8yAovhDiCwU+WSocj4XUeqTW4f4eAOC7BnBqFPKp\n40AJxRCXjps/9RKSUMxKoCbH8hJhLNDr9V4TKNeXF5035efn5+fn+9t65513huT0CNOQsBY4\nysvLCwsLPb+cNWuW698Y44ceeui11157++23r7/+es/vGvVtVlRU5G+TyWSCYUgkE0hdl6mu\n22x1cHqlZE6SVh7cu6zFxv7u7eNeK64l1d0vfHLqiZv9PokCIJMwv79nfmm1oaK+x2pnM+LV\ny+bE+SoRkTIRxthgc7qXsxQiOk4p8f25v/X1Wd9Ylvr2gZ0lrVfME4iFD1tUEkY1SUroaQqt\nX5y6fnEqy2GGDt97IeR09Vnf/OpceV2PxcamxqpuWpFe7GfigQCS1dJeG2u0O508ZiikFjM6\naVhLVwCAENJJVHbO6eBZjDFD0TJGPAp1AwAKMvU6laRnwLseJCdZG6+/eOisyerctLO6rLbH\namdTYlQ3rUjPTtK6Nqn8iERyCePqDJKI6P976JJ3vjn3zfHmAYtTJmEW58V8f22OTj31X7v/\n779lviVybuxOrrrVOMtPgu/04b61ORV1Pa4UKjeXzIyZnz0WOoITc9WAuwE7ACkQlQyUX6fb\nR67Ne+ylQ169XTcsSwumpWt6IqW1DJLYuF6OtyOEaCSTMbpwK98AAJBHUbPv5M99Atbui+/s\nAa49BsMVOEYOQuJVt9i3vgAA2Mk5TzWLcuKRx/sJiitAWVcAxaA0De7cg63tABikMVT0EpCT\n8o1pTV5e3ubNm202m1Q6uJJRVlYGAO5ZG4EwzoT1zCorK2vv3r3uL92NJxjjBx54YOPGjW+9\n9dZtt93m9V3T7TZjOby1pPlc+4VSlwPV3Vfnx88I4t3oUGWHYD35rtLWH27I89frflEKMvUF\nmfrA+0TJxVqpyMZyPAYJTUmFGkExxmca+wS//VR9z+QSOCYj00rdqGo2Pv7yhblHWa2hrNZw\n26WZd6/O9vctEVImYqL9FEeBhBZJRt6T4v0hIvqpWwt+/84Jk/VCPUiUVvbETRcXRpu7zI+9\neLD3fNNWY6dp/6n2B67KvW5pGgAsyI3+9KCAbZ5n65xCyjy4buaD62YOWJxKWbjrSqHibFNf\nAHXDBSdkgRTOsBxfVtvT0m3WqSSz03VqeQjCRFJiVC/8dOlr288cr+qy2NhYnXz94tRrFqW6\nrpNuo+1kjcHQb4uPVMzPjpIEY2uNbZg7CPi8zIf7MNcH2IBo4eDYzATNK48ue+PLs2W1BpPV\nmRarunFFxpKgnaGnJwwlU1KToa5Qk0bN+xGY2rC1BylpDAIOuGAccJdvDGIdAIxhzB5tTPFq\nQMjx1dvYMsAZrdzxenHRPCYjG+RapM8EbcrgfrI4lHLz9Hi+EobFhg0b3n333U2bNt1zzz0A\nwHHcO++8k5GRQawPCRNFWL+FKxQKd/irG4zxfffd9+abb77xxhu33y6gZE+32+y7s52e6gYA\n2JzcJyUtD67MUI02mbKlW9jbjOX4zl5rSszYLiuJKCQKqKFwvN98B0Er0/DHaHa8+21VRX2P\nxcalxaluXpGRk6wd0yOabeyBU+0t3WadSlqUFZkYdfHV9fDEaHacazYOWBzJ0arMhNBbtD6/\ntcLXNfP9XTWrChM8E44JbgozIzc+seLjfXVVzUaxiMpJjli/OHU4/RHPb61wqxsueB6/9nnl\noryYWJ28OCd6eX68V96EXi29d42A0jStzAtOVF1kEZii0OSqBTjd0PvXD0+6/JUAQCFl7lub\nG5I8kTid/Jk7igDA7uQ8JYyP9tS++dU5+/nmsmit7NEb54zavQXzZy+oG274FqDiAQlL/DER\nsp/dWjC6wxEmK4gGVSJSJQLwwPYBHvDajk9UeH+LQjt26oYLZv4VTMFyvqMRW01UTApST/fK\nL8Jw2LBhw6JFi3784x8bjcb09PSNGzdWVFRs2bJlos+LMH0Ja4FDkEcffXTjxo3XXXedXC7f\nvHmza1Aul69du9b17+l2m5U1C9QyODn+VEv/woyLlEv4I4DRhr+40PGEoVFytKqhw/ttAAAy\n4sM0hSQAtW39T7x8aMAyuFDTajAfPN3xg6tzNyxJC/yNJqvzTFOfod+WoFfkpkT4i/Dw5XBl\n59/+W+aeTDI0umlFxl1XZE+6Fe/Ne2rf+brK6hgUIAoy9Y/eMCdWN7IY3QAYzY5KoVohnsdH\nznQSgcMfWqX4HiHdIQAmq7OkWsDFmeXwwdMdrnvh57cVzMuO/PRAQ1OnOUIlLs6Jvv3yrJCs\n7U9qAuRwu1gzP0lzsfzpUR2X3VfR3txl1ijEhVmRaSHSULqNtp+/fsTi0UhitrH/2FKuVYqD\nj7t246lu7C5tffmzSs+tnX3WX7957LXHl8dEjKoWgO8SHMZ8J6JH+UeZMKWhEDMPc5XAtw8O\n8Azeuw/Xe5d1oIzi8TgdkYRKzAIAYA1gOwuYB0YHomATuwhTGIqitm/f/vTTT//xj380Go25\nubmbN29ev379RJ8XYfoy+QSOgwcPAsCWLVs8NYuEhAR3AMq0us1sTs4qlNoAAL3m0WcWzJ0R\niRDCPlUSqbGqSE1YJIHduDz9rx+e9BrUKsWTsT/ln1sq3OqGC57Hr24/s3hWbADLxm+ON7/8\naWX/eU/WtFjVozfOcRsWBKDVYPn9O8c9gwlZDr+3ozomQn5l8WRqo/30YMMrQ6clpdWGn79+\n5KX/WSb2F3E3Qrx+L570m/1uIowCo9nh+8Bx0TsweJEjhFbPS1o9bzJdpeNAgPIrhNCVxUkP\nX5sX8oOeqOr+ywcn3Z6mFIWuWZTy0LqZwTtVfX640WITSBr+73e1IRQ4PPl4X53voN3JbT/U\neO+VI9PpAACABxA4fwAACCpIiDClkSC6ACgbgAlABEiFtQMAjZ57oOgMau6143Q62IlNB8Du\n0RUoikWqpTAp2n8IE4FWq33xxRdffPHFiT4RAgFgMgochw4duug+0+c2CxCtKhGNfo6XEqO6\ndnHK1n31noMMTT0yBi/Ko+OKeYkmq/Otr865V+/T49SP35SvDNeYVX/0muynG7z98wGA5fjD\nlZ3rLknx3QQAB093/OWDIfpOXfvA068dee3x5TrVRSwVvzra5BDKStx2oH4SCRwYw/u7anzH\nm7vM+8rbVhUmhOQoeo2EoSmWE/hxxeqCfc/bW962dX99c6dZqxQXzYj63qWZk+7qDSERSglF\nIcEg2JLq7r3lmqWzx2RyazQ7th2or20bEDNUXqpu7YKkyRLD5GbxrNhXt1cafRTtuVlRj6zP\nG4vus26j7bdvHXc/ewGA5/HWffVRGtmNy9MDfONwqG0TKM0DgJrWfsHx4KnvMPkZFz6Ti0EB\nSAB8WlQAAMjkkBAQJAUYXEOi5m3ASXNw5W7oawe5BiXNRjlLPZNcxxRsOjhE3QAAZzse2I00\nV47PCRAIBEIwTD6Bg+AJTaGUSEVdl8D7WUZw9fMPrcvLTtS+v6u6qcssYehZaRHfX5uTHhdG\nDSDXLU1bWRhfUdfbZ7InRytnp+moYfdohA9GITNXF4I+ry4+3C0wtzdZndsPNd5x+UVCMZs6\nhd/mG/2Mhyf9FkeXn9zQqpb+UAkcMjGzZHbs7tJWr3GFlAlyMfnvm8u+ODJYftxrste1D3x3\nsvUfjywKt9hdB2+1sAMc70QIMZREwWhoNCZ/NeRSpjgn+tDpDt9NZ5v6fv/O8UtmxjxzR1Fo\n1Yfj57r+8G6J2w91V2nr1v11f7xvQYDGhBNV3f/5pqqmrV9EU7kp2nvWZE/4U1EhZX5797z/\n925Jp8cdsWRW7FO3FgTllOmfb0+0eKobbj492BC8wOHPw3jsvI3FDGUVkiNGXwhGxQPvWxWC\nkP8gFQJhCJjHnZXQ34ZSZkDhaqQZ39JU3gr2eoFxZxew3RB+sbsEAoHgBRE4Jj2Xzox5Z7/F\nPnRNflaCJmUYoYwBQAguLUq4tCjByfIMTYWnO0OEUjJG67rDhOXwJ/vrDlV2GvptcTr5muLk\nkZ6PXi0V7AYCgGit326g2tbRL3KK/cx5xmguNEYEuCJDK3M9cm1ea7f5XLPRPaKQMk/dUhCM\nqcHJGoNb3XDTbbS9/sWZn91aKPgtE4KF7bewg1cUxtjBWZ2cTS2OFFFjErz6o/V5jR2mVoNZ\ncOvB0x0f76sPfv7sxmpn/7Sp1DPtBQCau8x/31z2p/sXCH7L5j217q4oK8Dhys5jZ7t+e/c8\nf7HB48bMlIjXn1i+u7S1rn1AIRXlZ+jmpI+h14M/kbS9x+Jg+SAbxGamROwpa/Mdn5U2VmaH\ns9J0+yvafcdH/TNEdCbGA4A9zV8pROcCIq49hGEw0MGfeAf3XxDWUeI8Kv8moMbrjZ0TTqkD\nAGD7iMBBIBDCHyJwTHqiVZL7lmXsOtNR1222O3mdQjw/XVeQFBGqzxeFyM5g6mGxsY+/fKi6\nZXDq29xlPnq267KihCdvGYEZvkoumpcdefSMtymdTMJ45l96Qfn5nQxnebswU7+zpEVw/OLf\nHDao5KKESEVLt8BkODclZBc/AGgU4n/9aPG3J1rKanvMVmdanPrqS5IjlEHN8A8K1SkAwMFT\nnWOZADgyOMy61Q03GLDJ2RchGRO3uSit7JXHln2yv37L3jq3uYMnu0pbQihwHD3b5dvWAQAl\n1d2Gfpte7S0v9gzY3/jyrNcgx+N/bql45+mVwXtPBIlERK+eP04tZv4ycRiaEgVdZ3HlgqRt\nB+pbDUOSvEQMdcflM4L8ZH/cecWM4+e6bEPdrJKjlavnj3rZnEbMPODbMe4GbAekRFQSoJCZ\nHxOmMjzLHXkNLEMcl3HzMV4ko2ZtGK+T8L/aMV49MgQCgRAMROCYCmjkovVFiQDAYxymtRYT\nTVefdVdpa6vBoldLFuTGzEjUBP+Zm3ZWu9UNN9+eaFkyO3ZR3gjqOH5y3ewnXz7suXAtEdGP\n3TgnQI3AzJSIo2cFjPpnDmNuf2lRwraDDVXNQ85cLmXuWj0KO72J5M4rZvzxvRKvwRmJmkX+\nhaHRgRC6fG7i5XNDViTsz7vU6mAdLDcOpTS1bf3fnWzr7LNGa2XL8+MEmyycvIDEAAAcdvKY\no9CYnKSYoW5cnn64slNQ4OjqEz6l0dHpp8UJY+jsExA4Squ7nULmNZ191rr2gQlvVBlPCjIj\nPz3YIDCeoQ9e6JGJmb8+eMmL207vLR+s48iIV/9w/ayQPLQFSYtV/f3hRf/eeqqivgcAKApd\nVpRw39rcYO9EKhbBRBYYEiYjuOO0l7oxON54CHKvAnpcQqMYHSAGsG8bGiJZKoRxxskKu48T\nCIEhAseUgqgbgnxxpOnFbafcC3Tv7qhetzDl4WuDNfzfJ1TVDAD7yttHJHBEa2WvPLbs0wP1\n5XW9FrszPU69YUla4HjC712WVVJt8DK/jImQrV2QfNHDMTT17AML3/nm3OeHG20OjqZQYWbk\ng9fMnHShpysL4jGGV7af7um3AwBCsLIg4cF1M8PfiiXKT/ORVikeB3Xjza/Ovr+rxm3n+cHu\nmltWZtztI28JWhcDBkDAY36MBA4X/qQ9bXCFM14EyJfVKATcXk1Wf9EYgdJ2piRLZsXkZ+hP\n1gyZhklE9L1X5oTk8yM10mfuKDJZnc1dZp1aEiBMaqQ4WL6l26xTSbyusYx49d8evsRkdXYb\nbfGRilDFMBEII8bUKTzOObGlB6nGRTJDDJIXYPMx73FZDlChNy0mEAiEkEMEDsIUp7rF+NxH\n5Z4mFzyPPzlQnxyj9JdRMkz6TII++dAzIDweADFDXb8s/fplw91/ZkrEH+8r/tfHFW5n0IUz\nYx65Nk8mGdYdrZAyD66b+YOrc7uNNq1SMnm7kFYVxi+dHdvQYRqwOlKiVTr1mHhDhJyVBfGe\nEoOby4rG3Eluf0X7ezuqPUd4Hr+3o3pGosZLlaMFJQzkf1PoWDwrxr167zUewqPMnREpYijf\nooyUGFW8kIFRgNycOP306j5ACP3vvfM37azedqDBZHXSFMrP0P9g3cy0WFUIj6KUiXKSL557\nPUz6LY6NX5z98miT677LTND8cH2eV8mbUiaazklGhLDAv9EGosfx4pTNRJQEm0uAtwAAIDGS\n54MsNAomgUAgjDVE4CBMcb462ixo4fnFkaYgBY5IjdRsEzDbi9KMRxBGfob+lUeXtfdauo22\nxEjlKOb2CKFwy+wYBSKGykyYZN0BKTGqh6/Je+nT0541OPkZ+juvGCuXATdfHWsWHP/yaLOX\nwCGmpRRL8dh7/i+hZWiM27BXFsTvLW/38n3MStTcvDIjhEfRq6X3rc15cdtpz0GJiP6fG2YL\n7l+QGRmtlfk2thRlRYawxGCyIBHRd6/Ovnt1tqHfplGIxy5bt9toO3i6o9VgidHKFsyMjtON\nRktiOf7Jlw/Xtl3wlKluMT7+0qG/PrhwOG19BML4EZkpPC7XgXysfHaFkWQgSQbwZsA80MpB\neZtAIBAmA0TgIExxWnuEQxkE/SlHxPL8+Le/Puc7vrJwnLIAKQrF6xWCq82EMOeaRSlFWZGf\nHWpo6jJpFZK5M6JWFsSPQ4dZ21DvRo9x79sBAaUS6QecBk+Ng6HECmbMJ4QIoV/dMffbE81f\nH2tu6TZHa2WLZ8VuWJI6/Fk0jwEDpi/2A92wJC0rUbNpR3VNW7+EofNSI+5ane2vO0zMUL+8\nvei3bx/39AdJi1U9flP+MM9qSuJrVhJCPj/c+PKnle5I2tc+r7xrdfYojGZ3lrR6qhsuWI5/\n86uzzz6wMAQnSiCECKRJRAlzcctxr3Fq5jUTIzGQnhQCgTAJIQIHYYoj99O1oZAGe/HfuDy9\nvLanpNozCxCuX5ZelEVC1MIIjsd2BycP+tcdchKjFA+umznOB1XKhH8OgpX5IkoSIY61cWYW\nOxEgESWR0OPUi4EQjM7Y1cry3VanneMBgEYoQspoJEyAacGsVN0fvl88zA/PSdZufHL5F4eb\nalr7RQyVm6K9rCiRDnvPl0nKqfre5z4q9xxxsPyr2yuTohSuhCmOxw6Wk4kvfmuX1/UIjlfU\n9Q7JLeLsuG4X7joFtj6Q6VH8XJS0CMa4IYtA8IIquAWr4/jqHeC0AgBSxaK8a1AUaQ8hEAiE\n4RJ2L/2EyQvP48+PNO6v6Og2WmN18ivmJS2dPfEe8vOyo747KdDPPz87KshPlojoP92/YEdJ\ny8FTHYZ+W7xevqY4aU76ZApbndrUtQ+8+lnlyVqDk+X1aul1S9NGVAUwJZmfHX2qvtd3vDgn\nWnB/hCgZE0pjhTHF7OTazQ53QxqHsUvsiPHvJzpSZGLmuqVpofo0QgC2HxIIagGATw81REfI\nXvmssryux8nyUVrZDcvSrlmUGkBp8vJjdsPxmMfnK32cFv7Yi2A5L1ibO3DV57irkir6vqDG\n0dNvb++1xOnlQYZGEwjeUDTKXEVnrgRrHzASEE0vix8CgUAIHiJwEEKD3cn97NXD7rlTQ4fp\ncGXnqsL4p24pDGHhPcb42xMt5XU9JoszLU511cIUneoiL5eXFiZ+eaTJa1KnU0vuuDwEfgcI\nwWVFCZcVJVx0T7uTO3S6o7HTpFaICzL0KTGTZtI4Sals7HvipYOO8xaShn7bq9srTzf0/vrO\nuRN7YhPLhqWp351srWsf8BxMj1OvX5I6QWcUSrqtTl+7nQEHp5XwEj/ClsXGVrUYzTZncrQq\nMYoUY4cRTV3CXYQ1rf0/+td+tztsV5/1xW2nzzT2PX1bob+P8ve8TY5WumURXL/7grrhpq8O\nt55ACfM9x+raB/61pcKVKQsAhZmRP1yflzTyFCpDv62l26xRiBMilQxN6oAIXiCQEYMYAoFA\nGA1E4CCEhv9+V+u7MryzpPWSmbHL8+NCcgij2fHLjUfPNvW5vtxX0f7R3ronby5YlBcoW4Gh\n0Z8fWPjf3TVfHGnq7LOq5eJL8mLuWZN9UWUkhFTU9zz7/sn2nkH7A4pC1yxKefDqSRBoOnl5\n9bNKh09Axv6K9hNV3YI9RByPGzoGuvpsCZGKhEgFQuBg+amXFikTM//44eJNO6t3l7Z29Flj\ntLLl+fG3XZopFU/6WGRTCAAAIABJREFUOnyWx06fYBoXVlZY4Pj0YMMbX541WQdDXhfkRv94\nw6wp4Lw7NfB3TZqtrG/2za7S1nWLUmalCrswrp6f+OHuGvcv2s31HsU42HBW+Dy6K8FD4Gjr\nsTz6wgGz7UJmcEl196MvHnzpf5YO346k22h74ZNT7qDxWJ38kWvzFuQKV1ERCAQCgUAYEUTg\nmBoMYNwKYAEQIxQBEDf+ZlT7ytuFxyvaQiVwvPDJKbe64cJiY5/9oPTNJ1dqlQIl6CyHP9lf\nd/B0p6HfFqeTP3B17sKZMaGasnIY8xgzw5AojGbHr9445vluzfN46756nUpyy0o/fumE4LA7\nuVMNAo0YACAocJysMfzz44qm85m7OpXEyfEDFqdeLb20KOF7l2YOM393UiAV0/esyb5nTfYQ\n94HJjx9xw++mL440/evjCs+Rw5WdP+858uJPl07zPqYwoTAz8mSNwXfcwXKC+5841+1P4IhQ\nSv733vl/+eCk21taIqJvuzRzTXHShZ2c3vk4LjBr9bxLPtxd46luuDCaHR/tqXvg6lw//ytD\nsDu5J1851OxRn9LeY/nNW8f+9975c2cE2zhJIBAIBAJh6ry1T0mcLE9T6GKT6EaM6wEGX+Ex\n7gZoQyh/nH+5vQN2wXFDv/D4SLE5uL1CGorFxh441b52QbLv+OMvH6puMbq+bOk2HzvXtaow\n4albCoKc1Nk5rtfucPCDS4hKkUgrFlH+P3RnSYvvyiEAbDvQQASOMcLm4ASzgQHAaveenFS3\nGH/++hHPNeGe8xezod/24e6ao2c6//mjxRLRpK9x8GIqqRsAIKIQQiD4axcL1f+/t6PKd7Ch\nw7SvvH1FwTgFIRECsH5x6rcnmpuHNqroVJIeP39rLD63ticzUyJeeXTZ8XNdzd1mrVKSn67z\nLtWRRYBjwPcbkWyIaHJayMIGAE7VC/uY+rLjREuzT/cNx+N3vqkiAgeBQCAQCMFDBI5wBGPY\nVdq6aWdVU5eZoVBuSsR9a3Oyk7RC+5owrhMcRChrzE/UA71G0msSeO+M1IQmRLBnwObPKM7d\n+uHJ+7uq3eqGm50lLUtnxy6eNXrrUzvHdVptnnMok9Pp4LlYmd+y9qZO4U7ybqPNbGODD3Mh\n+KKWizQKsdHs8N2UFOXdKv/+rhrfindP6toHth1oGEU4JWE8QQjUYsboM8tlKKRgvMUpo9nR\n0Su8Yn+u2UgEjnBALmWee2TRG1+e3VnSarWzEhG9dHbs3Wuyf/C3Pb41FACQfDEXDBFDueJX\nBEFxRdjYKLAhbohrD+unUsjfuC9nhtYheo7zPCZ9iwQCgeDG6efNn0AIDCnEDUde/+LMnzaV\nNHSYeB47WP5kjeGn/z5wuLLTd0+Mu/x8hsDOY8qyOcJTghX5oZkqqGRifwvOKqGIBHd7sxeC\nZSDDp88h4GLo4HgL63fxUCwSvssQghPnuvaWt7UaBAQaQjAghK4s9i7qAQCFlPGdu571M9/w\n5NhZfzfadKTXZG/uMnPDntGNG5FSkXKocYOYRvEKgUfHFKtemaqo5eKfXDd76+9Wv//MZdv+\nd/WTtxREa2VrhG5ttVy8dHZQvZAovtjLTBQQhTKuQBFDlM30OLXgt2fEC4/7wvu5cTAO1GZF\nIBAIBAJhmJDV47Cj1WDZvKfWa5Dj8fNbK4pzVvm8l/trAGEBOIDxK6q/bmnayRrD8XND5oFX\nLUwO7AA6fFRy0cyUCF8fU4pCgt5sfSaB1XsA6BMqMxk+dk64/dvG8XI/N1NBhn7LXt8qGwAM\nv//PCRiMYkl8+No8Us0RQu68IqvLaN1xosU9oldLn7qlwNeuxU8vyxDMNoEmo2nI0bNdL247\n5Sqwl4jo65el3boqM3yadxCCWLnYJuatHI8xFtOUQiScTqGWi+N08jah4q+cZMFaOYIwGOwY\nmzFgCuQIjYk/K0Lg6Ql9z5rsbqPtu5Ot7pFIjfTpWwtVclGQh0E516HYItxVAagP5BKQxSBF\nqtde1y9N21fe5qXuMTS1Yclww4Mz4zVfQ7PveFqsimSpEAgEAoEQPGRCFXaUVncLrvB09Fpb\nus0+QYYCxQsAAEC71Q0OY5bDkjHOgxAz1P/7fvGOkpYDFe2dfdY4vWL1/MR5QzuKMcZfHW3e\ndrC+qdMcoZLMmxF11+oZGoW//wVvHrl21uMvH7QMLU6+ZWWGYGWyXi0VdL4IpmUm4ETY78YF\nuTGFmZEl1d4BhO5vwBi+Od48YHX+7u55oz43ghcMTT11S8G6S1JKqw1GsyMlRrmyIF7QKzQz\nQdPZJ9yt4CYhckwCRG0s32tzOjheRFNqMaMM7yiT/RXtv337uPtLu5N7b0d1TWv/7++ZH+C7\nxh8pQ0mH8bi7/fKsv3xw0mswPU69KG9YLWw1rf07S1paDZYorXTJrNg56frRnOvkhmf5Jg53\nux9mFIpgqBQ0xu8VYob6xfcKr12UUlpjMFmdKTGqFQVxMnGIDqpUA3YCxwNYgavH/fUgikaq\nlUANKiw5ydpf3F70/McVbiuQSI30p9fPTosdbvL3ZXMT3t9d3ePjTnXrqozQ/C8QCAQCgTC9\nIQJH2CHYXXx+k/eMHaFIjJuE9o0CgC6T/VhTX7fZjjHIRPSsOHVOtGrsarMRgsuKEi4rSvC3\nwx/eLdlT1ub6d3uP5bNDDfsr2v/5o8UxEcNa98tMUL/22PI3vjxbXtdjsjrT4lQ3Lc/wF623\nsiD+za8EYv9WBtFdjwAYimJ5gYZAEeV3QoUQ/O6eeZt2Vn+8r95qZxECDAJ6yKHTHXXtA8N/\nSyYMh5kpETNTIgLvc/PKjCNnOlgukH7l62LrD7uTO3S6s6nLFKGUFGTqAygjnWZHh9nuPmq3\nxREhFSWqpYFvUAwYjXtGkovXPj/jO3i4srOs1jAZp/eXz03kOPz6F2fcXi1LZsU+sj5vOKvo\nb3997r2d1W4leuu++iuLk356/Ww0nVpfWL6JG9ojyeNelmdFVPY4HH1Wmm5WmnBmyujBHB7Y\nDZxpyKCzE5sOIPVK98CSWbFzsyLLanvaey3xOsXsdN2IUpaVMtGf71/w1w/L3M1xSpno+1fm\nLA9ROyeBQCAQCNMcInCEHf5mRBSF4vRyn2E1QBKAl8YhQyit1WjbUdXpLr+3Ormjjb0Gs2PJ\nBE1FjpzpdKsbbnpN9te/OPPz2wqH+SGRGukTN+cPZ88blqeX1RpOVA2pm7h+WbqATT22Ya4W\ncD8ABUiLmDQAv6XOKhHTa/dufqEQUjCBbiWJiL57dfZdV2R3G6217QPPbDwquFt1i5EIHONP\nbrL213fOe35rhdt1EgHC5yUoEUPduyY7P2NYd83JGsNfPjjprgdhaHTj8oy7V2f7TnvNTq7d\n7L2E22tzykW0XiZw+Tl5dsBpdnIODEAjSiGSy5lAsiDGUFHXU98xoJKJclMihqkhBqCn3+6O\n2PSirLZndAJHt9G2o6SlucusU0mKc6LzUi8iRYWcNcVJKwria9v6ByzOlBhlrM73ASvA8XNd\n//nWO4HliyNN2Una4Qthkx0MLIe9q9IAgMcDGJsRGpOKpzHH2eatbrhwNANvA+pC9Z9MwvjT\n1odDSozqnz9cdLbJ2NhpilBJcpK0wfbXEAgEAoFAOA8ROMKOuTMio7Uy35r5pbNj1QJumhxC\n6QARGLcCWABECEUAJAFQR5vafM0Fag3mnBhV5LC7QvzR0Wt9f1d1VbMRIZSbrL15ZYZefZHW\njyNnhH1PBc1Tg0fMUH+8b8HOkpaDpzu6jbaESMWa+Umz031W/PhO7CwBcDtrdGO+EYkWABIW\nGlQiEY+h3+Fw/2hFFKWXSgLExLpBCKK0sgCWotNq+TesWJAbXZi1oqrZ2NlnjdfLGZo6XNnZ\n3muJ18uXzYmL1w9rtmbot/3qzWOeMbQshzftrI7USNddkuK1c49Q/xQA9NqcvgKHg3f22vrc\nlxyH+X6HycmzGrHwVdrcZX72g9IzjYPrwwxN3bQi/a4rZgRzgQVwMg+cQeOPb443P//xKatj\n8Me1aWf16nlJP71hNk0hALA5uBGtio8aqZi+aI2PF98cb/Ez3jyNBA5s9deXx4OFhskpcHAC\nMbEAAICBG/AUOIIHIZSTrCWGLwQCgUAghBwicIQdEhH9m7vm/u6dE57pp/kZ+p9cN9tjL57H\nzRh3ADgAGIQiKJTl6cdhdXJGPzOotn5bkALHkTOdv3/nhN05KAqcber76ljzH+6dH7hgWNAR\nAwCsdpbleIYOvUUIQnBpUcKl/ltmADjMlnmoGwAAgB3YWYbEi/19j0YsUogYO8dxGIsQJR2h\nL1xGvJqhKd/IW4Qgl7zsThxihspLjciDwbnu8DMR3Hx9rNnqk1EKAFv31/sKHE6hRicAcAjp\nCAMOk+9U0sra5IxURHmrIXYn9/PXj3g+PViOf29HtUzC3Lxi9E3+erVUKRMJ3sUByo4whmNn\nu6pbjQhBVoLGXT/V0DHwf/8t8zIb+upYU0KUAmP86cGGbqNNJmHmzYi8/6rcYRZWjBsdvcIa\nZXvPRZxcphYBHnuTVqhF/ssofG40AoFAIBAI4QkROMKRzATNq48t21XSWtvWLxHReakRC3Jj\nPBZfMc9XYHCvNbEYd3G4j6byAQaN0Fj/cXOCFhLDx8Hy//dhmVvdcGG1s3/54OQbT66gKL+v\ntv5mKVFa2VioG8OC7wYsFLaCjYDN4L/KmkGICdiTEgClTHT90rQPdtd4jV9WlDhGTpaE8aGx\nU6i4HaC5y8Ry2EsHo/0UU/iO8xg7eWFfHjvn7DGy2w81NnYOqBXigozIlQXx+8rb24XCQT7a\nU3vT8vRRF3EwNFp3ScqmndVe47E6+SV+kpK6jbY/vHvCM/koP0P/i+8VaZXir481C1opv/tt\nlfvZYrWze8vbS6oN/3hkUZKQkbAXGON9Fe1nGvscTj4zQb2qMEE0Ns7KgrnUADCtugwoJAeg\nAAT+mlDo4r+sMEUcB4AEKlMoBdCaiTghAoFAIBAII4YIHGGKRESvKU4S3IRxp4e64cbJ40YK\nZbm+kItphkKCModGGtRbeEVdT69Q0mpbj6WqxZid5LcGYVVhwge7ajifU1o9LzGY8wkKbAu0\naczayO9Zky2XMpt2VtscHAxGDKbetXo8nPkIY4fYz3SaoSmKQgNOp4PnEYCYohQikUrCGO0s\nYO/VbrVPzgv2H9Czq6TtlU8uKAJfHmn67GBDVqLwTKzP5DD024NJEbrzihkWG/vpoQa3NpER\nr/7ZrYWCMbEYg5e6AQAnawx/2lTyp/sXtPlp1PJSTgHAZHVu/PLsr++cG/jcegbsv3nrmLsr\nBwA27az+1Z1z0+NGXIlzURbmRh863SE0HppI7EkCxVBxLO/drUOjSAShbOUYVygFyGeDpWzo\nKELK4klclkIgEAgEwjSDCByTAw73Y2wFQBRSAPQJ7oNxn/sdjEYoM1J5ptNbB5GL6OSIoOq9\n3dl4I9oEAMnRyp9eP/v5rac85zCL8mJvXZUZzPkEBZKMZlPQUBS6dVXmtYtS6zsGWI5Pj1Mr\nhXwlCZOL/Az9F0cEIo1WFsa3Wy2eamO/k42USpRi2uQYMp+X0FS0T3UAjSiEEPYx1Onqtb+0\n9ZyX/0VFfY/N6TeGKciKBppCj6zP+//s3XdgG+X9MPDnuTvtZclD3nuPOHYSZy9CBoGQBCik\nYZdVoBRogbZ00F/XSzdtKWWVTZkJBAKEEALZ21keife2ZWtYe914/1CiyNKdLNuSbDnP56/4\nkax7vJS7733HtQuyzrbrrQ4yO1k2qyCBK2mrrd/kF93wqG3W9gxZJWMJs55oGhr1OX/94LRv\ndAMA0Kez/eaN2pcfWxL2BLFVszN2n+w706bzXcxSS29aHmoFEEnR+84OtPebhHyiLFsZYgvb\nqQaHKQDDSbr3YpUfhkM1gcX2KBAonglwJWM/A8hhADFAJEJJNSASJntfCIIgCIKECgU4pjqG\ncbnoDpq5NL+Ah1Ec95JGXCzNyohzkFSHT7K6TEAszksguKtIQqGScV75j9pndPWcjJn5CTuO\ndfcMWZVS/uyixJriMTSip2mmT2cbHLYnq8QpKnEYOnJiCQDwAAhoKwClIPJZ1mIhMdbuhshU\ntrQy9eMDHX6X2UI+8Z1VeX65VCRN6x3OnDixzubSO9wuiuZhmFxAqCV81m61IlxoI/37Oxxt\nMLB29+zWsM86SU+UKCbcXRgAkJEkDaVgpJujYAcA0DVomV2U+OVx1vnWLJxuiqIZnPuNS2t0\nHD/PEgTp01lPt+pYBieNkcXuPtE01K+3JytFVQUJCgn/6XvmbjvQ/tWJ3l6tNUkpWlyRvGl5\nfohdUdv6Tb97q7Zn6NKPaX6p+qebZ4r4sfffMQ6TOnWiDq3RSVJJcmlFuhJM7P+XKUGQBQVZ\ngKEAxFDiBoIgyCQK0uAcQYKIvTOqy42LbqOZEdc2NEOztrWEYMQkSByDS/ISitVOjdnpomil\niJelFAe5SAhRRY4qXi7UmfyLO9ISJAVpLNngLpL2Td1XK0W3ryocx3EbOg3/3FrX1m/yfFia\npfzhdeUTzj8nIK+ccZ8eWUlOQB7nJFqSJm2kg2RIDGACnC8iYjYZGwk3HINP3z33ja+ath/q\ndJE0hKAsW/WDjWWQ7c/VRdNumk4Q8xM4Gjr4kvElNEM7qEsZUjjELGb20hUnSZVmqRo69X7r\nd11VPJavZqIE3Ff7Qj4+vzRpx7FEv9QMIR93uPxLVAAAKSpJ8DeuPp01cGKUR6/WNms87zeX\n7Dnd9+zH9UbrhWY9UhHv/mtLV85Kv35J7vVLcsf6ai6Sfuq1496BxB6HGjTPfdzw4xtnTGij\nUeci6Q+PdzUPePMETXvODV0/JyMrtKlDUx2MxhCfKchodX20v721z8TDscKMuA0Ls6MzzwhB\nEARBwgUFOKY0mrH4RTcAADTA8cCkAwAgZKkAT5IKkqThrLbgEdjjN1X+3+snvPMdAQBSEe+J\nTTN9+xe6SXrLvvbPDndqDHapiFdTnHj32pJx1/93asw/efGIb21LQ6fh8RcOv/Dokon0FAAA\nACwF8uUM1QJoEwAYwJQQz+OqT7G6bWb3pfuuDsppIx0qgQKNd0U8xELi++tK7726RDNsj5Pw\nRQLCRpJaB3vpFknTfCyk6gkIYGOrvbZNo5AQShlPKhTOL05VSLSsTyZw+OvbZ729q3n74U5P\nyxu1UnT/tWULOFqBRkhplpJHYIE5JkI+XpIZByH87Z1zPtrf/tnhzn69XS7mzStRr52X+djz\nhwI/5ep5o8xeFQs4C17Ewgn9H3e+e/jpd075dg6y2N1//eBMslIcOHP62Lmh0206s82VpZat\nmp3OWnd27NygX3TD4+uTvfdfWzrB3UbZzrp+n+gGAACYHe73j3Q9tLJQyNaWBZn6TrXofvPm\nCe+wpP11A58c7Ph/d8/NUsds41gEQRDk8hNLp1OXIZqtCybDYCTDJzDSJ+8AQpgCYXIUtqQx\n2Hef7I2T8qEN4BAmKUVV+Qk3LsuLk166Ec0wzM9fOXqq5UKNusXu3n2y7/h57T8fWpgaP54O\nIO990xrYfdBsc2/d137vNSXj/lougBJIcKZseLlp0je6cXHRbXZb5Xx08odcgmEw5eLMINaS\nk1Ef8vP3D8/4dffITm5/4NrS/33tP9MEADCrMDFOyn9wQ9n31hZ1D1qlIl6KShT9GJxCwr95\nRcFrX573W79jdZFIQAAACBx+Z2nud5bm+o6Y+dnmqr9/eMZsuxTAXTs38/rFOcGPlZsiU8kF\nepN/IInAsar8CbW32HagI7AvMk0zHx9o9w1wOFzU796qPXpu0Lvyzu6Wn3x35uyA6pjuIfYC\nIpKi+/W2cQwnniwkxZzuZukGZXORjX2mKlR8F4Ocburpd0/6jYLWGh1/fPfUcw8vmqxdIQiC\nIMhYoQDHlAYh6w1ehgYEhAUQ2BjggIAPoBKCaGQF17Xrf/byUd9Yg9VJXrsg2ze6AQDYc7rf\nG93wMtlcr315/snNVeM47jm2M2kAQGMXSxfDCPEtEPBblwMU4EDYCTAcg5AOqKDAIBTgId3l\nPt40FNi7tGPAfLBBs2Fh9scHOnzXFRL+99eVev4t4hOFHBNVouO7V+SrlaLXvjzvyVlIjRff\nuaZ4aWWK39N8B+guKk+uyFF9c6qvZ8iikglnFSYEGczkhWHwwfXlv33zhN/6LVcWjNoYKLgO\nDXsnkfaRmQsvfdboG90AABitrt+/VfvaT5b79T0Jku0vEsRS1oPZ4SY5SqP11mDdppEp63Sr\nLjBKCABo6TU2dRsLM9CgXARBECQ2oADHlIZBKQAQ+M+JhBDgGFQAEBfN27IMA/724Rm/TAqa\nZv7zSf2CMrXvefyJJvbkedZGgKGgOXoMcRXeRwLNsG+CZmgGMBD1okPYQAiUAr4uoEpFJeCH\n+Buz/+wA1/r/fr5iRl781r3tXYMWmZhXXZBw68pCv2jjJIIQrKhOW1GdZrK5IIAycUiTUxQS\n/oaF2WM91uKK5GceXPDKF+fPdRkomslOlt1yZcHC8okmtXGN/uUTl4IRJMXsqvWflgoAsDrI\nvWf6183P8l3kyihJVolTVLHUuoLrOwMAEBCxFKlBvIaGOeemv7+n9Re3VEdzMwiCIAgybijA\nMaVBwCewRJIe9FsnsNTod3fvHrL0sOVXO1xUbbN2+cxL0wFtTpYWIQAAm5NkGDCOZPmCdHmf\njuXQBWnRu6eEcXzDIYSe6EafzrbzeHev1qaU8WuKkwKz071omhkw2DAI1cpwzIJBpjYJQRAi\nzOhyuWgaAMDHsDgBP8TuGwCAYYuLdd1gdgIAFpUnL5rwZXykyUNopDpxpVnKv3x/Hk0zNMOE\nazTsjFxVQydLmtiMvEv1KcMWp93JPpq3V+v/rpWllq2bn/XpoU7fRRyD919bGltvBRIBkawQ\nDRhZ+onkhTBkB5mCFNyx0foO/6bFCIIgCDJloQDHVMfD0iAQkHQ/A0gAAIQCHpaKw+Bp2zTF\nOBjgxgAfg6KgzxwDk5X9QgsAYBx5DZaWwH4rMi1BMr6T+BuW5B6oGyCpEQkbQj6+cXH2eF5u\nXASEwBowqhMAIMQFAIBPD3U+/2mDtz/ix/s7lsxI+el3Z/pdaNE08/GBjrd2NXvqnOOk/O+t\nKV49JyO2rm2QsRLgWJJonLUS8XL2lreJcaP/aTd0Gj7a396psSgk/Jl58TcszRVM9+6PGAa5\nYpHjcN3i3K9O9PoNjYqT8m9cmuf9UCwgIGTPJpMIWZJWHlxflpMie2d369CwHcNgQZrivmtK\nynP8W5ZOfWtmpLx5oN2vR8msbFVKCL+ZyBQ0My8exyFFsfwqG63sNy0QBEEQZApCAY4YQGAJ\nBJbAADcAEI72IyMZk4vqYy6OWcGgSIClYzAM00yDXFAlKUe8/spZ6R/ubSMDzpPW1mSM79BF\nGXG/vn32P7fWDQ5fCDGkJ0oevX5GahTnEfIxnpgQ2UbGOHCIy3iS1j7Tv7fV0yNP9Pee6c9L\nlX/3inzfxRc/a9y6r9374bDF9bcPzwxbXZuW5wEEYbN8ZqrfDX/vevBP/N/XLb49Pk+36nYe\n7/nr/fMnOniIg5ukMQxOfBD1lBIn5T/z4ILnP204WD/gyT6rKU66/9pS3++hWEiUZinrO1gS\nPWqKWdK4MAxeMy/rmnlZZptbwMeD1HpMcVnxknuX5e+sG+jSWUmaVkkECwsSZ2ai9qKxSiri\nleeoTgf0zwIAROhNA0EQBEEiAQU4YgYEo1ewU4zFSXX59uygGbuDahMRhaNGRkalVorKslnO\n45VSQXXBiPP4jCTpYzfO/OdHZ22OS5nba+dmblw0yjSEIGqKk/77+NJzXcODw/bUeElRhiJc\nWeihk/OlApxvI+0kTWIQE+B8CU8MAdxV20sHjFoAAOw83uMb4NAaHX4tIT3e3tW8fkGWZ7QE\ngvgpz1FtXpHvNzClIlflFzvz0zFgfuOrJr/Ffr3the0NP785zLX0e073v7WruXvIgkFYnBF3\n19risuwpdJXrJOlDrdrWIYvVSSnFvMqMuIq0uNBzptRK0VO3zXK4KI3BlqQUifgsf6ffX1f6\n2POH/foTXVWTEbxDaohNSbhY7G4XSatk4ZwCPlZJcuEtC7IZBlA0Hf03ZCTsbr6igDXAsSyg\nNzCCIAiCTFnommpacdNDAR1JAQMoN63jY+qJv/7jN1X+5MUjnpkIHlIR76ebZwaOBriiKnVm\nfvzuk709Q1aVTFBTnFScOfo0hOAEPLwyb0JDHydOgPMFuH+hskZvY33ygN7m23OkvsPAGgdx\nuqmmHuOkf2nIlHXH6qKa4qTPj3T1DFnj5YKakqRVs9KDT349UDfA+st2sF7jO5Z14l7f2fT2\nrmbPv2nA1HXof/z8oZ/fXL24Ykp0BrE6ybePdBovTr4cMFED9QOtQ5aNVeljeh0hH89Sy7ge\nLcqIe/7RxS9/fu50q87uJNMTJTcsyV012/8QJpvrf1+3nGnTW+3urGTZjUtzx1eZcvTc4Euf\nNXZqLMAzjvfKgmvnZ2GTlzsDIUDRjelhZn789Utyt+xt810szVJ+d0WwcCqCIAiCTCkowDGt\n0AxLk4gg62OVGi95+bGlnx7qbOw0uEg6L1W+YWEO19QGlUxww5LcsBx3ipNy3ImVini+F6Fu\njqmKAAAXyfkQggAASrOUpVljSIvQm9lHdbpJ2mx3KaXhue0/NGx/d3eL3yJNM89tq19Ypp7E\nS26vAy1ab3TDq2XQcl5jLuIOWIxDWoLkqdtmAQBIij2XoXvQ8uPnD3lbxvbrbYcbNN+7qnis\n5Wm7anv/9O4p74dGq+u5bfXdg5aHNpZPYPuT7Ejj4McHOroGLQoJf1ZBwnevyBcL0cnJ5Ljv\nmpJ5JUmfHenq1VpVMsG8EvVVNRlT4W8ZQZDLUJAzZwQJAp1DXCbCdnYi4OFBwhYDetvZdr3Z\n5k5PlFQXJIaekwDJAAAgAElEQVTxRvGYmGyutj4zACAnReY7vzZC5hYn7TjaHbg+r2RE1kw2\nxwUVhCAnOZzXWggSL2evmRfwcJmI/S+ird/U3m/mEVhhuiJZJQ7lKKdadRRbnojO5OjQmHNT\n5KFvOEJahyys622DlvAGOLy4chn+va0+cCDO61+eX1yRzNWVORBNMy9tbwxc3364c8PC7IzY\nHF/y3Cf1H+/v8Px7aNje0mvcfarvmQfmh9JGF4mEyrx4lFGIIAiCxC4U4JhWMCimGDPbuoBi\nbBDgGORHaL4sw4DXvjz/wZ428mK0NUstfeKmmQXpisFhe127YdjizFRLq/ITItqGkKTo179s\n2rKv3bMNAsc2Lsq+Y3URb1yd/Jxuasve9kMNGq3RkZYgWVOTsaIqLbAyYEFZ8vxS9aEGje9i\ngkJ4++pC35X8NHllXvzpVv8K5yUzUlELNyS8Flckv/FVU2CVyqKK5MCw47DF9cyWMwfrL/wC\n4xhctyDr3qtLRq07sHGMRwUA+PbfmUSOkX0xvGwc6xFidZAn2VobUDRzqEETeqZb56DFYGHJ\nzWEYcLpNF4sBjoZOgze64TU0bH/583M/21w1GTtCEARBECS2oQDHtMLDEinKEtCGAzopPQB6\nAAAGeQIsmcDCf+ty6762d0Ymq3dqLL/477HVNelb97V7SzCy1LKfbKrMT1OEfQMe/95W/9nh\nLu+HJEV/sKfNYnc/esOMsb6U2eb+0X8OdWouBIx0JseZNt2hes0vbqn2i3FACJ66bdZnR7o+\nO9zVPWSJlwtrihNvW1UoF/vfKv/5zdV/fu/UsfND3pUlM1Ievb5irHtDYhfDAL3dbScpDEKZ\nAJexNa2cuIwk6b1Xl7z4WaNvjCMjSXrvNSWB+/nNGyfqOvTeFYpmPt7fAQG8/9rS4EfhSj2A\nEERzwlEQchFPzzbfOk40oQafY2WyuRjWQbIABKZ1BOHijss4XVEN2YSLN6wWuM4wTPBGMwiC\nIAiCIIFQgGNawaFEiGc56T6G8Z404743cWnGbae6RSAj7DGOjwLuwgEADFbnu9+0+q50asxP\n/vfoK48vk0bgAkNncnx+hKVUZMex7luuLBhrwvPbXzd7oxte+8727zvbv2SGf0t5DIPr5met\nm58V/DXjpPzf31XT0Glo7jXiGCxKjytIH0Osx+mmOgbMRqsrM0kaYhEBMqVY3VSLzurwabmi\nEvHyVGIsAhdy1y3OmZEb//GB9k6NRS7mVRUkrF+QHZjK1NBp8I1ueG0/3Hn7qsLgrRBm5sWn\nxkv6dFa/9fmlapV8Mqd7eJWlKvY1D41YYgDEQFlqpGKsrJRSAYHDwMnZAIDEuDFkb6UlSHAM\nspYFZUam4ibSjGzhJwCA0005XBSaLYUgCIIgyFihs4fpBocyMV5IMw4GuCna5aQHA5/jpIfC\nG+CwOcjB4VD7mA5bXLtqezcszA7jBjyae4yst0kZBpzvMY41wHGI+9ZiYIBjTMbaMNJj98m+\nF7c3eJtHzi9VP7SxHNW2xBCaYZq0VtfIjll6u5tndGRHpt1Afpr8sRsrgz+ntc/Euu4m6c5B\nS0nQ4UcEjj11+6zfvHGiV3spxlGerRpHwlSEzMlWaUyOJp9IJYbBpYWJydH9wxHy8fml6n1n\nB/zWBTx8YdkYxs1IRbwrqtK+OtHjt56lllblJ4x7eyTF1DYP9QxZpSJeRa4qJYrB00SOH4RU\nxBNGJrkJQRAEQZDpDZ1AxACDxdmlsUiEvCy1NLReEhCDIgBEJPA/n/agGQcDaAjCNtiPIDAI\nAUcKNov2fvZrqgkKcvzQ9+ZltLHfWhxmq4GPtH1n+59+56TvyqEGTY/W+vwji8fXXgSJPoOd\ndLH1Ax+yujIVwkgkcYQiSE+cUNrl5CTLXvzRkm9O9bX1mwgcK81Szi9VT53CAhyD62emtQ1Z\nWocsFiepkvDL0+LiI994ONCD68u7Bi2e2a4ePAJ7+PqKscYof7CxzO4k99ddem/PT5P//Obq\ncXd0bu4x/um9U96NETj2naW5d6wuis4PcWll6v92twQ2i2HtdoSMQ7/e1tRtpGgmL1UWZNQx\ngiAIgkwbKMAxpRmtrhc+bfj6ZK/n+lwpFdxzTcmV1WkhfjoT7JI/nPgEVpged757OMTnR+ia\nvCBNwRpngRAUjqUSxCNRIep0sHRsTVJOQm//N79qDlzsHrR8e7pv5az06O8HGQcHyd4lgWYY\nJ0mLeHiU9+NRksWeoyEWEtmhDffhEdiq2VP6lzA3UZqbOMkNOFVywX8eWbz9UNeZNp3Z7s5J\nkW1YmJMaP+ZcCRGf+NVtsxo6DfUdBjdJ56TI5pUkjbtXhcXu/vkrR337gJAU/c7uFoWEf93i\nnPG95phkqaXfX1f64vYG3/qdksy4O9cUReHo05uLpF/4tOGzI13e+NGSGSkPX1ch45hrjiAI\ngiDTAwpwTF00zfzilWO+UQODxfmnd08BAEKMceBQ6GZbxyA/jOkbHt+7qujJl4/6FYeLBbjN\nyXJRV5GjCu/RPRIUwlWzMr487t+G48rqdPXYoxIrqlNf+eI8y3pVqAGmcPG03mB96FzXMApw\nxIogORoRHS0UXG6KfGllyp7T/X7rN68o4KPkoLAicGzDouwNi7In/Ep0caagODMFgwIwsXfy\nb071sXY53bqvPToBDgDAhoXZ1QUJnx7s7B6yyCX82YWJK2elofaiE/fctvrPj3T5ruw902+x\nu5++Z+5kbQlBEARBogAFOKauw42DrDkRr395PsQAB4EpMFpLM/5RDj42/mptLlX5CU/fM/e5\nbfXtA2YAAIHDa+ZnVecn/t8bx/2iHkUZcYsqJtTDIoiHrisXCfFPD3Z6Dopj8Jp5WXdfXTyO\nl7p+cW59h+FI46UmJhgGN1+RX5kXH7btIpcTuZAARpZ1IYHxRxvIGlGP3VipVoq3XhyuLBPz\n7lhdNGrHXGQyMG56gKQHAfDUOuE8LJnAksb9clyR08Fhu9VBSoK2mA2jzCTpgxvKonOsy4TR\n6tpxjKXldm2ztqXXGLlBZgiCIAgy6VCAY+o618Ve8aEx2PVmp0o2+pACCDARnuWg+ijG5l3h\n40k8LFjjwHGrzIt/4UdLDBan0eJKT5QQOAYAePqeuc9+XOcp8MYweFVNxl1XFUfufjWfwB64\ntuzGpXme1ol5qfJxt+HkEdhv75yz7+zA4QaN1uhIS5CsnpNelBGRb11wAh6emSTtGrQEPjSO\n0htkskh4eKKEPzRybAQEICsyHUZDJ+Dhd68tvnlFfteghUdgmUlSYlIDLggXF91N0TqfBcpN\n9zKA4mHjDBlzVQtCCMbd1AOZdDqT8y/vnwrsbOLR2mdCAQ4EQRBkGkMBjqmLdSDIqA/5wSBf\nTGRTjJ1mnBDiOBRDENlSf6VUoJReCr5U5sW/9OOlWqPDZHOlJUgEUWk0kKAQhmu8yOKK5MUV\nYxhzECGbV+Q//c4pv8XUePGymamTsh9kfHKUYjEP7zM73RQNAJDy8UyFSDY1ZmGKBMSkxO8i\np6XXdL57mKTpvBR5eWTK4gCgJ1gkEjqGcY2MblxA0hoCSxrfG3t5jmrrvvbA9cL0uOi8VyNh\nZ3W4H3vhkEZv43oCNnkFcQiCIGPCOl4dQUY1JU6sEVb5aXLWdZVcEEr6hi8cinA4mXeJwxhx\nuDxdUZXmdNMvf95otl0oOKrKT3jkhgp0ERJbIADJUkGyVOCmGRwG68qBTITdRT7z4dlvTvV5\nV6ryE57YVBkvD9O7EONg6POA1gLgBlAEsQyAZUc60kEDK8cjDMPYIBzPgIwFpeqybGV9h8F3\nEcfgXVeNp6wPmQo+OdSpMdgBhKyDxSAEJZljHlKOIAiCIDEEBTimroXlyRlJ0u6AwoRNy/NR\nA7bL0FU1GUtnpDT1GE02V0aSNCe0CRfI1MSL+k1UN8W0663DdhcPxxIlgvTJrouJqH9sqfON\nbgAATrZof/tm7d8fWBCG907GxpCHALjY24ixM1QToHWQmA1ARH+s4b+RhWHwd9+reX3n+e2H\nOj03ynKSZQ+sL0OdhmJXXfuFcBWGwcAqlSur09MTJVHfFIIgCIJEDwpwTF0Ejv3hrpq/f3im\ntlnrWRHy8VuuLNiwMHtS94VMGrGQmJmPLjzGj6KZniHLkNGRGi9JUYkvnzjhoMV5sF1nd18Y\naXQOmJOkgsV5CZPb3DTsHC7KTdIkTftFNzwaOg3nuodLMidahsPQTZeiG5dWdYAeAOPthREK\nCDhjUnACCXoSIfHAtWX3Xl3SM2RVSPm+BYaR02uw9RvsOAbTVeLEcKXVIAAAANwXx1FjGIQA\nUMyFefEQgk3L82+5smAyN4cgCIIgkYcCHFOaWil6+p65rX2mTo1ZKuIVZcQpJPzJ3hSCxKTT\nrbp/flTnTYmakRv/w+vKM5Okk7urKHBT9P42rZOkfRcHLc4T3Yb52dMkXlbbrH3588bWPhPD\ngDipgKtLUXu/aeIBDsDWCAMAwDBaCCIY4MCgCIdyijGNPCrA8Xg44f/KCRzLjkpSmNVJfnSs\n+3z/ha8CQjAzS7WuOo03vWJtkyhTLau7WHMEMYgDCBmGAbC6IOHONUWTuzcEQRAEiQJ0ShED\n8lLlV1Sl1RQnoegGgozP+e7hn7181Lfg60yb7rHnDxlHzjSZlnqNDr/ohkeXwe7pdRrrvjnV\n99OXjrT0mjxhjWGLk+uZXENDvBiGaes3HWrQtPaZKI4hFACQY1wPGx6ejcMRDRQIPJ6PpYfr\n9Sma6R60nGrRaQz2cL2mLwaAdw52eKMbAACGASc79J/W9kbicJenq+dm+k7AgQAACCEE1y3K\nnrQ9IQiCIEgUoQwOBEGmv3d2t5IBF/PDFtcnBztvXTnNc7YtTvYLb5phrC4qThTbYW6aZl7Y\n3hDKMyGEFUFnqZzvHn5my1nPhGkAQGaS9JHrK1jGr0AxYFj7fUa8tQEEOB/PpplkBtgAgBgQ\nQxi2ihK/FKfqgoSHNpanJYTzi+rV2zq1LN+6052G1TNSJFNjnFCsy0mW/XRT1bPb6oYtF6K3\nIgFx15qiWYWJk7sxBEEQBIkOdD6BIMj0d67bwL7exb4+nfBwzl4j06AHR+egRW/iTNnwtW5+\nZrJKzPVov972xItH7D7BoK5By89ePvqvhxb61W5ALIOhzgW8AAaxtNC3PREYFAIQ5r4VjV3D\nP3v5qG8QsLZZ++P/HL5+cU6P1kLgWGmWcvnM1AlOGNUYHazrNMMMGh05l0G9WHQsKFNX5qmO\nN2n7dNakOFF1foJyjJPXEARBECR2oQAHgiCXAY5qA84qhGkkRS5knRipEPHE/JgfM+x0UVwP\niQWEzUkCACRCYvOKgusW5wR5na172+0BqS5ON/X+t61PbJo5YhXLAowV0N0+SwTEywCM4eEU\n//u6OTDFSW92vPR5o+ffnx7q/HBv22/vnDORgd84d3xkgqETxI9EyFs6I4IdYRAEQRBkykIB\nDgRBpr+8NLn+3FDgekGaIvqbiTK5kFeklp3TmH0XMQhnZyi5PsWjtc90sF4zZLSnxouXVqam\ncKc/TKLUBDHrOEwAwJM3V2eppSRFp6jEo14/N/UMs66f7zEGrEGIlwEsjaG1ADghlAAsBYDY\nvkN+rov9y/fV2mf6y/unn75n7jhe3+Wm2gfMOoONNdbGJ7CUaT23GEEQBEGQqEEBDgRBpr+b\nluXVNmn92kZKRbxrF2RN1pYmyOmmtuxtP1A/oDM6UuLFq2dnrJ6TDjkm31alxanE/IYBk8lB\n4hhMlApmpikUQh7XizMMePnzxi372r2Bg7e+ar57bcmGqdenUC7mL52REjgXNiNJWl2QQHCU\n5+jNzrd3NZ9t19udZHay7KbleVypPFwDWQCMg/hEB7LoTI6dx3u6Bi1KqaCqIGFO0aR1SaC5\nvsyRapu1g8P2pDEGIw7Ua17Y3qA1OgAAWRmKhIC+HstK1fzR+r8iCIIgCIKEAgU4EASZ/mbk\nxv/ilup/b6vXXuwCkJsif/SGionk208ii939o/8c6hi4kJShNzvrOwwHGzT/d/ssrhhHllKc\npRTTDIP5PMFF0QQGsYBP2VXb88GeNt8VF0k/90l9frq8PDtYn87xYRiGa9uhePi6CpuTPNI4\n6F3JTpb98pZqruhGa5/p8RcOW+xuz4cag/1I42BpNns+S1HGhMfKcvjmVN8zW85662I+3Nu2\noCz5yZurJuVSPz9VcbJFG8oz+3W2MQU4apu1T79z0htb7OoxutxUiloGMQgAEAuIleXJs3On\nybhiBEEQJIymx6w3JPpQgANBkMvCwvLk2UWJ57qGtUZHeqKkIE0Ru2X/737T6o1ueB1u0Hxz\nqu+KqmCtLj2xDJph+szOAYvTc9kp4xNZcSKJTz+OHce6WT99x9HuMAY4Boftr3xx/kTTkMXu\nTkuQbFyUs3ZuxjgiHWIh8ds755xu1TV0GhwuKj9NsaBMHaTdw78+qvNGN7yaug1CHuFwj2jD\nwSewG5fljXU/oejT2f783mm/thcH6wfe+qr5e1cVReKIwd20PC/EAMfhRs3pVl2WWrqgLHnU\nsbsAgHe/bfXNnGIY0D9gGdBYN63IXzMnI17KD4yvIQiCIAiCjBsKcCAIcrkQ8PDKvOlwr/hQ\ng4Z1/WC9JniAw6NJZzU6Ll3Jm11k/ZC5JEEquzins19nY/3EXrYZn+PTqbE8+txBb6Cha9Dy\nj61nT7fpntxcNb4XrMyLD+WHa7A4GzpZRueQFFi/MPNE05A3cpSeKHn4uoqckSNUwuWbk72B\nTT0BAF8e656UAEd1QcLPb676zycNevOFkTQQQtbynC172z3/SI2X/GzzzFEzXFp6A5uYAIZh\nejTmRDTaA0EQBEGQcEMBDiQirKTDSjpJmsIhJsB5cp4Igxfu9Vns7re/bjnZrDVaXZlJ0o2L\nsueVqid3twgSW0xWF+u6kWPd17DDbXS4ARhx25xhQJfRUXZxTqdYwP5fg4S7bcdYvfRZY2Aa\nxben+q6qyajKTwjXUQJxfesAAGIB/p9HFrf1mfr1NrVSlJcqJ0IYo0vRTJ/Wqjc7U+PFiSHX\nbvTr2UNIBovT7iJF/En4r3lpZWpNSZInxSktQVLXrn/588BpuJf06axPvXb8lceXiYXBdhta\ncw8EQRAEQZDwQAEOJPz0TrODunDpQjG0jXQ6KHeiUI5DbEBve/S5QzrThT4IOpPjZIv2usU5\n319XOnn7RaYnu5PsGrTweXhGoiSUK9UYkhgnZI1lhNIcweQk/aIbHhYX6e3QMbsosWvQEvic\n2WHqgknTTG0zy1AbAMCxc0MRDXDEy4VcU1eS4kQ4BgvSFQXpoc7WqW3WPvtxXc/QhcSWmuKk\nH2woS744bsZidzf1GE1WV6Zampsi9/1EqYg9VMQjMCFv0mb3iviE95tfmqXMSZG/9VVTW78Z\nx6HTRVEB3zS92bmvrn/17Iwgr1mYrjjbrmdbj1RzEwRBEARBLmcowIGEmYNyeaMbXjRDm1w2\npUD60meN3uiG19Z97csqU4sz0fkuEh5ON/XGzqaP9nd4qgAUEv6da4rWzs2c7H2FzRVVaS29\npsD1FdWj16ewXdpfesjTuWLT8vwDdQMag9330cJ0xdVh+h463RRJse/D4vB/9wgvqYhXU5x0\nOKDGRywkxppKVt9h+MUrR32/kKPnBh973vzij5aIhcQnBztf+/K8N0ulMi/+0RtmpMZfiH3M\nKUrcuq898DXnFCVOpN9qeM0pSpxTlMgwzJDRccsfdrM+p0vDEgjztWl5XkOnwS84Ei8XrJmT\nHraNIgiCIAiCXIQCHEiYBUY3PJy0m6aZwz6TDnwdrNegAAcSLk+/c+pA3YD3Q6PV9cyWs043\ntXFRziTuKow2LMxp7DTsO3vpa8QweNOyvOqC0XMfhByNIXk4RlxszBkn5f/74UWvf9m072z/\nsMWVGCdaOStt0/L8UJpKcjl6bvBI46De7ExLkKyZk6GQ8FmTUFLj/WeIjtuwxfXZ4c72AbNE\nSJTnqK6sTvPEDn64sbxXa+32SVER8vHHb6xUSPi+n05SzMkWbfegRSkTVOSoAgfuvL2rOTBM\nMzhs//xol0zMe/bjOt/10626J18+8sKPlgh4OABgVmHi0sqUPaf7fZ8jE/PuXlsy4a87zCCE\nAu6kEuFo1TRV+Qk/+27V8xfHxAIAynNUD20oD2O5EzLNuc3AbQYCFcBjcugVgiAIEmUowIGM\nWb/edqpFZzA7M5IkNcVJfue+rH3pAAA0w9icpJtkH/hktDrDv1FkjHQmR/uAWcjHc5Plwevq\np7LmHqNvdMPrza+a183Pmh61KgQOf3nrrMMNmoMNGu2wIyVevGp2eogDTRPE/F6TgwxI5FBf\nvLxnGGbvmYGmnmEegd1zdcmi8hSRYEJFE26S/sP/Tvr+ULbua6/IUQWO7RDw8CuqUidyLK8j\njYN/fPeUN4Hii6Pd2w50/P6uGoWEn6AQvvDo4u2Hu+ra9TYHmZMi27goxy9+cb57+M/vnfbW\n6Qh4+OYV+d+9It/3OY1dLM1KAQCNncOtfSz5NX0627en+7wFHU9urqrKT/hof0ev1iIX82cV\nJt65pmhqzi1WSPg5ybL2gME9AICq/NEbuy4oU88uTGgfMOvNztR4SWaSdMokqSBTm62X7vkS\n2C+8dUBFMUxbCXgR6fuLIAiCTBuxeg2DTAqGAW981fTeN63e/v9qpejxmypn5F46x8UxDFAs\nn0tATCIkJELC6iADH1UrxZHZMhISi939wvbGncd7PPEpkYC4dWXB9YtzY/E6pIHjstNid3cN\nWvxaIcS0eaXqcTToJTBYGC9pNdicF6ONEIAkqSBVLgQAaI2Op14/3txzafLFu9+0/t/ts9MT\nx59Y8f63rX4hJ5KiT7XqKvPiT7fqvIsSIfGj71SG0kZkVEar6+l3Tvq91TT1GJ/9uP7nN1cB\nAAgc27Awe8PCbK5Pf/K/R822S8loTjf16o7zCgnft9CJZo/WAjdF9+nYx8009xi9AQ4I4dq5\nmWvnZjIMmPp/aPetK/WrxwEALJmRUp4T0thgPg8PMQCHIBfYB+iWtwFz6a+YMZ5j7ANY0d0A\n4wf5PARBEOQyNx1uZiJRs/1w59u7mn2nG2oM9l+9dlxvupR/IcbZJ/+JCQGEcPlMlh4BBA6X\nVqaEfbcxSmt0fF3b+8GetoP1A043W6wo3BgG/ObNE18e6/Zm39id5IvbG9/f0xqFo4cdxdHc\nIfhDlxWZgJihluWrxGlyYVacqFwty44TeS6x//juKd/oBgCge9DymzdPsHblDNFXJ3oDFxmG\nyU2RP33P3O8szV05K/3utcWvPLFscUXyuI/i61C9hjWQeqCu3+5kWffz1Yke3+iG14d723w/\nzEtjD5blp3IG0TC2SMbUj24AAKoLEv7y/fnFmXGeMh+5mH/X2uKffnfmZO8LmbYYzX7f6MYF\nrmFGd3IytoMgCILEDJTBgYzBR/tZuuLZHOTOE92bll9I3iYwPI4vMbpsDLh0RSQi+BKeCABw\n19ri1j5jY9ew9yECx36woSwtIWyF9zHtgz1tb+xs8sY11ErRYzdWVuaNngQ+EfWd+lMtusD1\nd3e3XL84J+ZqOvI4Li8FPDzj4hjU6c1F0XaSBgCICIzP8ePDIIwX+98F7dVafVMqvDoGzA1d\nhvLskO7VBxocZh+JOqC3VRckhNI3ZKw0w3bWdZJitEbHqL8GHWy1GACAXq3VTdLeRiQ3Lcur\nC5gPIhfzr12Qve/sQKeG5UWKM5WjbH0KK81S/vMHCx0uyuYgVXL2QDaCjBXFMAMmp8VJEjiM\nF/PjLg4YYqw97J9g7QGJc6O3PwRBECTWoAAHEiqKZnq17HnXHQMjGumLCYEA59lIJ8lQOMQE\nGE+AXzhlkQiJZx5c8NWJ3trmIZPVnZEkXTc/ayLZ79PJrtrelz5r9F3RGOy/evX4y48tSQxH\n3j6Xpm4j67rVQXYPWXOSY6zgeUauqjgz7pxPEM3j2gVZQv6kDeCMDpoB/RaH3u72BhdVIl6q\nVMCaOBCoT8seiQAA9Gqt4w5wyER8g4WlyY48IMISLjKOIawAAFkIB+UK6kEIcezSd3JuSdIT\nm2a+8GmDt1tqbor8sRtnxEn5t60q+O2btX6fnpMsWzIjPCkqAACL3d3aZ6JoJidFppRGL9wg\n5OPT/u8IiRqD3X2y12j3yVVMUwhnpMgxCAHDkcDItY4gyLTD1bkPQYJDAQ5kFBTNfHqw83Cj\nRmt0AAgBWw9RPs//egCHmIzHfk0OIVw1O33VbDQj0B9rgozdRX5xtPu2VYWROy5XX9jgD01Z\nEMJf3z777x+eOXJxZA+OwfULs+9cUzy5G4uCXrPDMHLMqt7upmkmUxFSgCxIZ1mxYPwzL+aW\nJu042h24Pr8sadyvGVxNcdKLnzUGltWUZMbFSUcPcJTnqD4/0hW4XpatxLARoaIrq9Pml6rP\ndRl0Jmd6oqQ4I87zhMUVKU9urnph+6Wp2IsrUh5YXxqWfCiaZt7+uvn9b9s8qV44BtfNz7pr\nbXGQWScIMgW5KeZ497CLGnEB02t0CAi8JEkKhEnAyvK+AURjbjyEIAiCXFZQgAMJxu4kn3jx\nyPlu/5vhfipzI1tDcZno5EiMZx1eEEYF6QrWdbGQyEiMyZoOlUzw2zvntPSaWvuNQh5RnBmn\nVkYwBWaKcNOMX3TDY9hJJlM0V62Kr8J0hUzMC2w/wSewGbnjTN8AANyxqqi2STs4smxkUXny\n/NKwpTP4SU+UbFqe97+vW3wXRXzioY3lAc+lgbOdIXUAAEiogCAXAGxZZerWfW0tvSMmoRA4\nvHN1UeCxJEJiVmFi4PqymakLy5M7NWaTzZ2ZJA3jeJT/fnHugz2XuoFQNPPxgQ6DxeXpn4og\nsWLA7PCLbnh0D9uKE6UwcS4TGODA+DC+OhqbQxAEQWIWCnAgwbyzu3XU6EZRRtyymeGZ7HiZ\n4xGYi+9/imYAACAASURBVC0Zj09EtgtGRU58eY4qsJvAjUvzeBE+dETlp8nzOdpATkt27pa0\ndjKkAAePwL6/rvTP7532W79jTZFCMv5yEpVc8Pyji9/+uuVIo0ZvdqYnSK6el7VmTnpEm2ve\nsbqoNEv57jetHQNmiZCoyFHdsabIf0QLZWYs3wDqQiCDcQLgaIDSZQQuf/qeuf/9/NyOYxfm\nCuUky36woTzEiSFePALLT2OPHo6bxe7+aH9H4Pqe0323rizIvDy6zCDTg8XF3vHXTTEOkhIp\nCkH6WqZ/N6AupEEBgQrLuBqNiUUQBEGCQwEOJJj9df1BHuUR2NXzMm9bWYhjsTAGYMorz1F5\nqyp8VUzg5nkoIAQPri/76ctHjBaXd7EwPe6GJTkRPS4SXmGJF6yclZ4UJ3p1x/mmHiMATHay\n/LaVBeMYRutHKuLdd03JfdeUhGGLIaspTqopDlYFw1j3eaMbF1AmxrIPKq6Wi/mP3jDj/mvL\n+nRWhYQfLw9b/sUEtfaZSLab3gCA893DEw9wON0UgWPoLR2JApz7PYvAIAAAxs+EiiLG2gXc\nZiCIh9JMAFEdFoIgCDIKFOBAgjGYWVoDAgAq8+Ifvq4iWSUm8MieBzMM0NldFhdFMowAxxLE\nPBExbc9vbl9VeKpF5zcaNjtZtmp2RkSPS9PMn9877RvdAAA09Qy//Pm5B9aXRfTQSBiJCBwC\nwNo0RTyW7gyVefHPPLiAohmGYWJuhs4YUHpA+mctAQAAZQCkDhDxAAAhH89NiZkkoIk0zGEY\nZufxnnd2t/brrTiGlWTF3Xt1SVFGXPh2hyD+EiT8ZrbO5Qohj+d95yFEUMFSGoYgCIIgXKbv\nySsSDlz3LdVKUXqiJNLRDZJmmg3WPovT5CJtbsrgcLfobUM21+ifGZvy0xR/u39+SeaFiwoM\ng6vnZPz5vnmRLlE52aJt6zcFrn92pMvuZE8hRqYgAmOZ/AoAiBfxeGO/IY9jcDpHNwAAlIXz\nITqyXW8mIjdFzpVeUZQx/nKY57Y1/PWDM306K8MAkqLPtukf+ffBY+eHxv2CCDIqlZifEnCO\ngUFYokaVVgiCIMj4TevzV2TCllamsK5Hp+lGv8XpGNmTggFgIGBxOilIVzzz4MJHrp+RnijF\nIDhQN/DXD850DXJfiYUDVxNTN0n3DLEPBkamphSpQC0ReK9/IQRJEn6qdKqUV0wtkHsuDIzU\n/NqJk4l56xZkBa4vLE/OUo+zN0GnxvLJoU6/RYpmnttWP74XRJAQVaUqSpJkAgIDAEAIVGLe\ngmzlpUAtZQa2s4z5ILCeZs+3QhAEQZAAqEQFCebGZXln2vSnW3W+ixsWZc9mmxoQXgwARrb0\nAc+6kJi6VyAT9J9P6z++2ETQYncfbtCcaBr6471zy7Mj1YkjyI16IpabjF6GIABqCT9BzHOS\nNAOAkMCCVLlf7ohEAHmACZg7A3mAiNT82rC49+pSAYFv2dfuacYBIVwzJ/3760rH/YInm7Ws\nA6F7tdYBvS1ZJR7/XhEkKAhBbrw4N17somgCg5jv+5W9kbEcAwwJAGQAANZTQFwOpbMmba8I\ngiBIjEABDiQYAQ//071zdx7vOdQwqDM5UuLFV9VkVOUnROHQNM3QHDXlbnraZnB0DJi3HfC/\nleom6ee21T/38OIIHbSCYzaEQsJHQxliEQ7hmJpuXCbcNGNzUwzDCAlcSGAAElA8m7Ee8nsa\nFM8GcEr/z0jg8K61xdctyWntM7lJOj9Vnhg3oSnIdo5hFgAAGypSQ6LCf8yTW8OYDwMAAPCG\nPGhgOwOIOCDMi+7WEARBkBgzpU/jkKkAQrh6TsbqOZHtcxkIwyAGAc0W4hhHQ4FYcbxpiPVW\nakuvSW92qmSCSBw0L1W+bGbqt6f6/Na/d1UxGqaATAOe0jat3eX925ILiHSZkBDkQVzG2E4B\nSg8AALgKiisBMdGRMX5cJL39UGdjl8HlpnNSZOsXZiulYfhDVkoF4cqkS0uQsK4TOJaC0jeQ\nycDYm9jXHU0QBTgQBEGQoFCAA5miIAByATHsYLl/qBBwF8/HOCvb13vhIbvbG+BwkTQPhzB8\n1QeP31iZniD5YE+bZ4ZLgkJ4z9Uly6PSaQWZNtwk3dhl0BjsSXGikizlBJvjuimaCFOAbcDi\n9GtObHKSnbQ9TykGRBKUr7o4fCac4TyKZowWl8NNPfny0T7dhV42hxo02w50PHlz9ZyiRO/T\nth/u3HGsp09njZcJ5pYk3byiQCqK6lvc3JKkBIVQa3T4rV9RlSoSoJMEZDJQHI1+SZZ+2AiC\nTFdujrHoCBIcOndBpq4UqdDmtrlGvrupJQLh9G0MkRrPfr+UR2CJcSKGYb481vPet619Ohuf\nwMpzVPdcXRyWMZY8ArttVeHmFfm9Wiufhycrxah1AzImtc3af2w526+3eT5UK0U/vK7Cexkf\nOopmOgy2PpODpBkMwngxLy9eIppAxQ3NMDp7wOglBljdlNVNSS68cjh/3QeH7S991niwXuMm\naQyD9Mg8NKuD/NO7p17/yXKxkKBp5qnXj9c2az0PDRjs2w52HmwYfOb++cpwpGs53dSAwS4R\nEgkc87A8BDz817fP/t1btQMXf3wAgDlFiQ+iKdHIZOHqAYxN2/ZbCIIgSLigAMe0NThs33m8\np2fIGi8XzCpMrC6IRuOM8OJhsFAlHrK5LS6SZBghjiWI+ZKglzo9Q9a2fpNIQBSkKeKksXcm\nNL9ULRfzTQGjcJfPTBXy8We2nP38SJdnxemmTjQNnW7VPX1PzYzceO8zGYZp6jH2am3xckFR\nRpyQP4YrQwLHxj2IAbmcdQyYf/nqMbfPeCONwf7Ua8f/9dDCvNQxBOBoBpzsM5ov9n2gGWbI\n6jLY3bPT48Yd43BSNEulGwQAAPulAEfYDA3bf/DP/cOWC3/CbMcGRqvr2PmhpZUpe8/2e6Mb\nvq/w9tfNP9hQPpFtWJ3kh/s79tYNeDoZqeNENy/Lq8hWcj2/MF3x0o+XfHu6r73fLOTjFTmq\nWRHoJN09ZG3tM0EI8lLl6Rx1MQgCAID8NMbVE7DMAH7aJOwGQRAEiSkowDE9fXms+9mP6z3l\nBgCAD/a0La1M/cmmmQQeY/flMQjVEr5aMnqoYtji+tdHdfvO9ns+FPDwG5fl3XJlfhjrOKJA\nKuL98tbqP7x90mBxehcr8+Lvv7aspdfkjW54kRT93LaG5x+90H+0pdf4tw/PtvQaPR+q5IL7\n15VxzfpFkHDZur/dHTC8maToLXvbntg0M/TX0Zgd5oCuliTNtOttpeMNvcEg2RkReG94Z3er\nN7oRxOCwHQBw7PwQ66NHA9brOvR7T/drDPZklXjZzNSSzLggL04zzN8+qmvzGf+sGbY/80n9\nw+tKZ3B0FAYACHj46tmR6rVkd5L//qRh98lez4cQghVVafdfWyYaSwT2MmS0ug7Wawb0tgSF\ncE5R4mU00UZUBBwtgBwxwQ3gMiieMUkbQhAEQWIGCnBMQ50ayzNbzlIj7xzuOd2XkyzbvCJ/\nsnYVUQzD/Pr14w2dBu+K0029+VUTBsHNVxZM4sbGoTIv/pUnlu083t0+YBYLiBm5qvmlyRCC\n2mb2a6G2fpPB4lRKBXqz84kXj1jsl8Ze6k3Op985KRPzYjF/B4khLb3shfHNHOtc9PaAoa0A\nAAAMHOuhEOAYDiHF1rs37OkbAIBTrf4ZGaw8+WUWjq/Ld51hwLMf13166NJwpW0HOzYszA4y\nF7a2Vecb3fCgaWbLwY4gAY6I+tuHZw7Ua7wfMgzYVdvrIumfjiX+dbn55lTfvz6q8/4y8Ans\n1lWFNy27PFpsQhwqr2KsZ4CjBdA2gAmAIBtKqgEWrNgKQRAEQQAKcExLu070UGx50V8e657E\nAIfTTXUMmIctriy1NOy3oU616nyjG14f7G27cVkeL9Z6dkiExMZFOX6LQeY12hykUir47HBn\n4PUSRTPvftOCAhxIRHE1Ax1rk1CuydCsb2ghghAkSfj9PilRHnIBISLCH+BwuKhRnyPg4Z4C\nELWS/Z3Q9x3y65O9vtENAABNM1v3tZdlqxZXJLN+enMfe1ypa8jqdFOCqI8Q7tPZfKMbXnvP\n9N+5ukitnNCM2+mqpdf0x3dP+ZY4uUj6v5+fS40XL664PJLyIA9KZwHpLMCQU3xyM4IgCDKl\noP8zpqEBg41j3U7TDDYZgz931fa+tL3RW3Yxv1T90MbyBEXYbsVw3UC2OcherTU7eTr0leAa\n5Sjg4Z7vJNfdcq5vDoKES3FG3Pnu4cD1kqxglRSBxBzX3uKJFTIkivkYhAMWpyePAwIQL+In\ns/Xo0ZkcX53o7R60KGWC2YWJM/PjA58TXEaiNHAciS8Mg/deU+KZiHRlddr2w52Bz1k5K937\n710nAjsRXFjnCnBQFGc8iKSY6A+hau/nfAtq6zehAAerz450sjZw+eRg5+US4PBC0Q0EQRBk\nLNB/G9OQRMh+AisREpMS3fj2VN+f3j3lu3KoQdOrtf7nkcXhyq0I0mcjplpwBLOgLFkhaTRa\n/cv7V1SneW7Jcv1sp813AJmyblia+/XJXr8EIrGQ+M7SsaXTp8iF3UZ7YBpHWtAhIKGIF/GU\nQp6TomgGCHGMNbVk98nef2yts19MlXr/29allak/2VRJ4GN4m1q3IOtki3+VCgQwUSkEDMhN\nlW++Ir/4YgeNwnTFfdeUvrLjnG8Hkyuq0tbPz/J+6OnWEUhjYF8HAHD171TJBGLhJPynHySR\nJ0yDgKeh7kEr63qXxhLlnSAIgiBIbEEBjmmopjgpsBulZz36mwEAvLWrOXCxa9Cy50z/ldUT\n7YjuGZsCOG5YysS8tATpBA8xRUiExFO3z/rD2yd97w/XFCfdt67E8+/izLiDbHngJZmcoxP8\nMAyz80TPwTrNkNGRGi9eNTt9sn5nkNiiVor+8v15/9hytrHrQh5HUUbcDzeWc409DtSvt7X2\nmYR8PFsl7jE73BfvXUMIMhSilAkHOAAAGARBalJ6tda/vH+apPxbF2UmSW5dWRj6URaVJ9+9\ntviNnU2uizELiZB4cEM513vd+gVZc4oSdtX29uls8TJBTXFSZd6ItBGZiD1gLRNzZmLMK0rc\ndrhzOCAYumZW+qSEEwoz4gLH5QIAcAwWpo8tx+fywTUAa0yDsRAEQRDkMoQCHNPQ/FL1/FL1\noYYR17pKqeDONUXR34zdRXYNst9xOt81PJEAh9Hq+ufWS2NTMAgDC/hvXlEQc4NjgijPVv33\n8aW7a/vaB0xiAa8yb8Qox6vnZm070KkzjUiPJ3AYYuMVp5v6xSvHTrde6Frf0mvce6Z/TU3G\nj25AXeuR0eWmyJ95cGG/3jagtyWrxCkqkXeAEcMw35zqq2s3ON1UdrLsqpoMqc9Fu8nmem5b\n/e6TfZ4P+QR20xUFV8xJs5O0AMfixfwJ1qeE6OvaXpKtsmPHsZ4xBTgAADcuy1s8I+XYuSGN\nwZaWIFlQlhx8ZHVqvOQ27kPMK1V7w0a+5pequT5FyMd/vLH85Z1NnRffe3kEdvXsjCtnpob8\nRYSTSiZYNy9z20H/Ypz1C7Kn2jBvN0l3D1lsDjIjSaoIYXpX5FTmxR89Nxi4XoUaKiEIgiBI\nUCjAMQ1BCJ66bdb2w12fHurs1VripIKa4qTbVxcqpYJJ2A13c0AmyGOjvioDnnptxNiUi9EN\n6DmkVMS7bWXhhkXZ4z7E1CTiE1fPy2R9SCbm/fm+ef/6qM6bIZ+eKPnBhvLSrJAyOLbsbfdG\nN7x2HO2eW5y0sJy91D+iSIqZTsGpywGEIDVe7Je1MWxx/fLVY74dOj7Y0/rzm6s9eQoMA377\nZq3vL56LpN/ceZ6iqDtWRzUg26djb100NGx3k/RYi+lSVOJrF2SN/rwQbFyUs79uoLnH6LtY\nmqVcNz/Y66cnSH713Znne4y9OptMxCtIlXu6fkyWu9eWKCSC9/e0epqwCvn4pmV5NyzJncQt\nBdp9svflL84PW5wAAAjhyuq0u9YWc2XQRNo18zN3HOvuHnl7QCHhT9dRaAiCIAgSLijAMT1h\nGLx2QVa4zrAnQiQg0hMlPUMs5cQTSU4+08Y+NkXIw39xa7VMzMtOlor4LL/eTjf1xZHu5l4j\nhsHCdMWaORkxN2MliPREyR/vnduvt/VqrfFyYWaSNPQS971n+lnX95zpj2aAg2HA7pO9733b\n2jNk4fPwihzV3WtLstTTpM7oMvSPrWf9+o8OW1y/e6v29Z8sFwuJhk5DYFgNALBlb/t3r8iP\n5rwPCUdzCgEPH1MPjrAT8vG/P7Dgwz1te8/09+ttqfHi5TNTr1ucM+quMAhLMuJKMoK9zfZq\nrd2DFpVcmJ0s40fynRDH4KbleRsWZncPWQAAmUnS6A9zCe6r2t6/f3jG+6GnXq97yPqX++bC\nyehjJOITf39g/qs7zu860et0UwQO55Wo772mJCkO9WRFEORy4ebumY0gQaAAR6wa6zyUI42D\n//u6uX3AzCfw0mzlXVcVZamjNFtk84oCvyajAIC0BMmyCeRLN/caWdcdbjJBIchNkbM+2tpn\n+tVrx4cuNu378lj3lr3tv79rDteAkhiVohKnjH0Qr97MPvrBr+Yl0p7bVr/tYIfn3yRFHmkc\nrG3W/vHeueXZqmhuAwkLs83tVyvnYbS6DjcOXlGV6peY4OV0U50aS2G6IsIbvKSmOMlvGuvF\n9cRJb9PLJ7DNK/LDe+u+T2f9x5ZL2V4JCuGD68siHcoU8vGCtOj9TEPHMOy9ohq7DMebtHOK\nEgMfigK5mP/wdRU/3FiuMznjpAKU0YYgCIIgoUABjhjjJukt+9p2HO3u19tVMsH8UvXtqwtH\nLRV+a1fzGzubPP92uKjDDZrj54d+97051VGp5r2yOs3lpv77xTmz7cKQheqChEdvmDGRG4YY\n9zUH10MUzfz+7dqhkSMJ+nTW//e/k8/+cNG4dzJubpI+3jTUq7UqJPwZufGTPitRJRMOW/y7\nEnrWo7aHtn6TN7rh5Sbp57bVP/fw4qhtAwmXIaOdddQlAGBAbwMAgCDzjyKzJS41xUkLytR+\nbXrlYv5da4uju5FosDnIx54/7NuuWGt0/O6t2t99b45vW5/Lh97sHOKYVnOue3iyAhweEMIw\njlSfsk636r4+2duvsyXFiRZVJAdpMYMgCIIgwaEARywhKeYnLx6p69B7PtSZHNsPdx5u1Dz7\n0CKVnLO+WmOwvx1wb4qk6H99VPfK48uic3Ny7dzMZZWpzb1Gk82VmSSdePJIEUfetVTES09k\nL2do7DSwVso09Rg7BszZyVHKZ/Fo6DT8+b3TvdoL++ET2KYr8m+5siCae/CzuCK5rd8UuL60\nMmWCr9yrtX5zqq9Pa01QiBaWq7l+dgCAE03+8zU9WnpNBotzcprIIBPANbIaXBwCUszxyyAS\nEFnR/ZOEEPzy1lnbD3duO9DRp7PJxbw5RUl3rimalteWO451+0Y3PCiaeXtXy+UZ4GACRxNf\nxBWhm66cbqpjwKwzOTMSJRlJ0agNZBjw7Md1vvlTX53oWTIj5Webq9AUYQRBEGQcUIAjlnxd\n2+ONbnhpjY43dzU9fF0F12fVNmsptlO0Xq21T2eNWnWGWEj4jT+ciLJs5azCxBNNQ37rN6/I\n58rjHTCw36ADAPTrbdEMcAxbXL945ZjF7vauuEj6jZ1NSqmAq4FoFNywNPdEs7aufcQv2MpZ\n6QvLJpS1vnVf+3+/OOe+ODLz/T2t18zLfHB9uW9k7WC95vMjXX06q8tNc72O1U6iAEfMUStF\nWWpZp8bst45jcHZRIgCgODNuTnHisXP+f8ibludFtCUEKxyD6xdkr1+QPdYCwJjTxFEZdL5n\nmGHApJfkRF+8XBAnFXjai/rJT2UveJyW9p3t/88nDd7g18z8+Eeur0iNj+xJwv66gcDqsL1n\n+mfkxk+FPmIIgiBIzJk+7RUvB8fO+18GeBznWPfwvZAO/aGp75e3VK+dm+lt/yYV8R64tux6\n7rb8QZrhe24mR83O492s3/mt+9ujuQ0/Ah7+l/vmPXJ9xezCxCy1bEGZ+te3z378psqJXO3U\ndeif/7TBG90AANA088nBzh3Hur0rf/3gzK9fP3703GDPkHWQI0ucT2CJcdPwRvrl4IcbywND\nFZtX5HvbxPx884g/ZJGAuOfqkk3LJ3NUxPSObgAAuKZbcecxTHMQwu8syQlcz1LL5pZMt1oJ\nN22zugfMrm6Lu99JGb2/DEcaB3/7Zq1vas+pFt3jLxyxOciI7ufbU72s67tPsq8jCIIgSHAo\ngyOWWDnOM4LHKVLi2ftNQgiTx96KcuoQC4lHrq+4Y3VRW79JJCC4xqZ4leeoRALC7vT/HsZJ\n+Vx58hHSqbGwrvcMWUiKnsSRDRgG187NXDs3bFkkXx7rYV3fcbTrqpoMAMDRc4Nf+gQ7uCyv\nSptqMxcigWHAwfqBhk6D3UnlpsqurE4X8mP+q67IVT3/6JJXd5w72653uKjcFPmm5XnzfArs\nPX/It60q7Bgw83lYbrJczDHQBAmXgjTF7pN9geuF6YrLMH3DY8PCHDdJv/NNq9NNeVaq8hMe\nua6cwKGbpAeH7UlxomkwcstGDrmoi6WIjIukbS7aIuWlQIC9s7sl8PlDw/Yvj3dvXMQS/QmX\noYBqKQ+ueDeCIAiCBIfOI2NJarz4BOt60DKT2YWJKrlAb/JPvp1fmqSQ8PUm5/GmoaFhe0q8\npKY4Ucqd5hAihgH76wbOtuusdjI7WXZVTcbEXzOIOCk/xFapEiFx/7rSv/kMAgQAYBh8aGNF\nlGMKXGfJOAanWcnxhUaSAfp0F9b31w2M+iJV+Qn3rysN57amJJPN9evXTvjWoL2zu/WXt1QX\nZ0Y1+hYJ6YmSX946K/hzVDKBSoZKkKJk9ZyMD/e2Bw5ICu+gltgCIbhxWd6q2RlNPcMWB5mt\nluamyDUG+2/eOHGwQeOpWlpUnnzfNSWJMTuo1U1bL0U3LqJoh5McFuAqzsKlbvb1cOHqkh6H\nahIRBEGQcUEBjliyanb69sNdge3QPDfDuQj5+C9vmfXbt074xjgK0xWPXD/jk4Od//38nN11\nIalBIeH/8LryxRXjbyppsbufev342bZLF2nvf9v6s81V4xjXwjDM3jMD9Z16h4vKSZatmZMh\nEkz013VNTUZGkvTNr5qaeowYhMWZcXesLsyP+tjCihzV50e6AtfLc1RwCtw/1RjsTT3DbpLO\nSZHnTKw1CVePSW/My2BmKXoHAOAYXDU7XSriVeSo5l0e7fT/seWsX4edoWH7b9488eoTy6Zy\n9orF7m7tM9mcZJZalsqRLIZEjtNN1XcYBvS2JKWoLEsZ4pukVMT7071z//7hpV85lUzw/XWl\nNcVJoR+aYRiDxaWQ8KdTWDZOyvd+E/Qm58PPHtBffI/yxO5PtuqevntuQVpMNuZw0yxttgEA\nLtoiwCdtDvfCsuQjjYOB6wQff/KtE2IBUZAiXzsrPUiRKYIgCIL4QgGOWFKUEfeDDWUvbm/0\n5tACANbNz7p6tJqCsmzlq48v++Jod3u/mc/DyrJVy2emHD039OzHdb5PM1pdf3j75L8fluSm\njPPs7blt9b7RDc9r/v7t2teeWO7X54KkmF6tZdjiSkuQBM4pMFpdv3r1WGPXsHflvW9bf3FL\ndXn2RE/CyrKVT98zd4IvMkFLK1M/2t/ud7uMR2B3rpnkgZQkRb/02blPDnZ4u9IuLE9+5PqK\nUecQc5lTlHiwniVHw3sJwTWiQq0UPXrDjPEdNBaZbK4DIweUemiNjqPnhhZXTKjPa4QwDNi6\nr+2Nr5q9ZV+LK1J+sLEM9YKNmtpm7d8/PKO52D5ZJRc8uL48xN+WjCTpX++f3z5g6tValTJB\nfqoi9Hooi8P97jdtu0/3Od0UjsHq/IQ7VhUmT/ag67B7f0+rJ7oBIcTwC7Fnu5P80X8O3rqy\n8MalnP2epiyaoVjXGYaCEBSmKxo6DYGPFmVE9h7Aytnp++r6/ToNCwW4GzLDVtew1dWntx1v\n1f7o2vKUafc7hiAIgkQCCnDEmHXzs2qKk3af7O0ZsiYohPNL1SFmsIsExHWLR5TRbjvQEfg0\nimY+PdQZZCZLEA4XtedMf+C62eY+2DCwevalNJMjjYP/3lbvrV9YUKZ+cH2Zb97vP7ac9Y1u\nAAD0Jufv3qx99SfLgjfaiAkEDp++Z+6rO85/cbSLpBgAQGG64oH1ZSWTXYzw4meNH+/v8F05\nUDdgsrn+ct/88WWWrP7/7N13YFPl2gDw95ysZjejTbrbdO8BlFL2RkUZoiK4t7jXdV3vvV63\nXq/rU3DjRcTBEEGG7FloKaV775WOpNk753x/BEJITkqbpm1S3t9f5M16gTY55znPmBJ+oLjD\n6aBZxKPbM+HnZIb+eYYgmWVOVqgn7+e3euR6d9Mou2TEF13H3Y6TzV/urnZcOVHe3avQf/JY\n/jXQpHP8tfVq/vF9kcmhg69cZXx78/kPH52WEsUbyisgCJCEcIYbzjZZsNd+KG7rvdhIyIrh\nRXV91e2K9+/PFU2s88+yi8F6hERCcIADcPGn2orhG/fXinn0WRkjHaE9xlCEOIaFICQAwO3z\n4l77vsjprqBAuuN392ggocib907ZX9Rx8Hxnl0zLY9GURgubQ3P80tEaLL+ean5q6cSvVYQg\nCIJGzu/PFa9BIh799nleqJRu7SVuddkqJV6/KpnK4Dgvw1G37HIvhuK6vn/9cM5xcu3pyp7W\nHs2GZ2baUvHVOvPpKoKr2XK1sbC6b3amnx1TEmLRKU+sSHv0phTb5VMOw8MUCS/SGSy7Cwhi\nDeVN8uq2gSGeMjkhk9D3H87bdrxpf1FHt1wn5AZMTxXduTDBns6TGStYPTf25yONjs/KkAi8\n8hPuRwZpq8n0yY6bGIYTtiSsbVecq+sbVqUD5JnfT7aYXD5vrRi+7XhTytU6nozEwZLONpfv\nt+00MgAAIABJREFUDo3e/OvxpieWpY7e+449s8UKALA1aEKAc8xu15lWvwtwUFCWyeo8sxkA\nQEVZAICpycGvrs1Zv6vSXs2aGSt4amX6GDT9RRBkSW7EktwIAMCB0q4dZ5ynxgIA6rqUepOV\n7v99lyEIGjqLlfi0AoIG54uHztDYcJ3deHGd4mHHTXcNF5zu2nSg3upysbqzX3uwuPOGvEgA\nQM+A26vZ3XIfvZrtGTIJjRKNqMmFF7X2atx9kTR2qTwLcAAAqGT09nlxt8+Lw3GcsMPIfdcl\n5SYH7z3b3tmvFXBoucnBiyaF+0IvkrEUKmCGChhdMueerCiK5MQHjcuWBter0Cu1JsK76jqU\nMMAxBpq6nbtF2jR2Ea97S3mznHi9hXjdf0UGs1t7NC6RjYva3Vwh8GUUlEEjcY3WK6ojyWgA\njXzx4312ZkheSnBjl0qhMYYJmePy9eRuKhyOA63BDAMcEARB0FXBAMe1KzNW0NlPEC/IjBV4\n9oKBLGpCONe1EzuCILlJF0/ScByv61C4PBUAAKrbBmwBDiZ9kKvZsM3YaBmkUyDqjXDDIDGL\ntGj+yLur+DUEAY8tT/vnxiJbyZLdbXNiYedOiBCJRPwLNdr1QUYzcRjUaJpo19mWTY86Ud6N\nA+JPLqoPt/4dBJ0spKBMo1WF4WYUIVFQJpV0RY0SjULyOJztFe6a+JBRxBdSHSEIgiDf5/dD\n3SGP3T4vzvVwIYTPuCk/2uPXXLcs1XXiw80zY+wXgnAcuEnOAPasjRA+IzyIYPAtiiKTEnzx\navbEEC1muxvBkBI9nse714gpiUGfPj4jJ14YQCWRUEQSwnl1bc69SxLHe1/EggPpgSzik43E\nCL+fa+sXUiKJfytTo0Y3VhjuZio54Ye2X8uQCJ67NYNKJg4YZUo8vBIw7sgonUkRsanhTEqI\nU3TDF2TF8AknqWdJBO7STiEIgiDIEfy2uHaJePRPHs+fmhxsu7ROQpH5OWEfrcsfSc1/ShRv\nwzMzZ6aLOQwqhYzGhXFfXZvz0NJk+wNQFJGEEGe9Oo5rfWJFGpnk/MO5eu4EvJptsmDuUv3H\nGJWM2nt/OpqdGTrCYbHQEMWFcd59cOrON5bsemvJhmdm+ma7GRzHu+W6ypaBZUSR0JQo3uSE\nYc+EhjywYkaM63gjOo28el7sCF/ZZLa6m98MAFiYE0aY7XXd5PARvq8PWjw54ptnZws4zjkF\nbDplLdGnJTRygUzqHbNjnWIcYQLGLSO49AJBEARdU2CJyjUtTMh8494pRrO1T2EQ8+muMQXP\nXvO1OycBAHA3qb23zYl7a/N5p0U+h7bI4fg4O074xVMzvt1bU9kyYDRbY8Ts2+fFTU/zxWGZ\nHqtqHfhqd3VNuwLDcB6LduscybLpMWQ3aedj49bZsXQqeeP+WlsVNJmErpgRfdeihBG+rBXD\nj17oauxSoSiSGMGdkSa+1lpsDAuCAK/8Jo6GypaBz3ZU2Ls/RAaz+pR6vfHi7Mm5WaHrlqUi\nCGLFrVYcIyMkFPHRv8gEwOfQPlqX/9mOipKGfttKYkTgUyvTwtxkWAxFbbviu321tR1KHMdZ\ndMry6dHL86OdTjUjgpjPrEz/8s9q9aVeCRQSumpmzIyJ9flsF8yjf/7kzP/9VXvwfKfJgpFQ\nZFK88KGlyWL+RIu2+44pcUKJiH2sQto1oKNTSQmh3OlJwXA2EwRBEDRECI67KRiAHGg0Gjab\nvWHDhocffni89zIR7C1s//rPansvscSIwOdvzXDXzwzD8Il3ZFNU2/eP74ucmq3OzQp9eU32\neG3JzmTB2no0ZisWFcwaef/8jj7t6/8719pzuSFfYkTgv+6eJOAEjPCVoTHW0Kl8+vPTTpM7\n+OyAZ1el4wBEi9kiHt1kNavMagt2MeQRQKKxqSwSDHOMJpnK0C3TBQXSRziltbRJ9vqmYuuV\nXWDykoNfvp3gQ0mjNxfW9kkH9Hw2LStWIObRW/o0R6t7uwd0ZBIaIWDMTwsJYtMAAE1SdUWL\nXK03h/KZ05KDA/y2SSSG4f0qA49FIyygGG0Gs1VptOA44AaQ6V5t/4Fh+N7C9n1F7VK5zjZ7\n/ra5sa6lpmMDx3HpgF6rt0QEM8drDxA0jt57770dO3acOXNmvDfiE+h0+s9FTeO9C2hE3nlg\nxbj8PMMMDmgcXJcbMTNdXNuukKuNkcGshHDuIJf0J150AwDw1e4q11EyRy50LZ8Rkxw5zi0M\nqGQ0Lsw7hdk4jr+xqdgxugEAqG1XvPfzhfcfyvPKW0wwZU2yE2XSXoU+RMCYmxXqU/0sfjrU\n4DqXVK42NHSpbMVNJswsN17RQthgNZoNFiGd5zplE/IWASfAK+HCjfvrnKIbAIAz1b3lzfL0\nGOe+Hiw6ZV5WqP1mQX3/ngudl25ZKzuUNV2qtfnRR0q6Dpd22R/26/Gmx25MSfPPnj4oigQH\njiiE5BkMx6t7Na0DOtvVKASAiEB6iog9SFvoYbw4hr+2saiops92U6k1NXapTpR3f7Qun0Uf\n65beJ8qlX+6q6lXoAQBkEnJTfvTdixLcdYaCIAiCIHfgNwc0Plh0ilPH0C6ZduP+uqqWAb3J\nEhvKWTMvPivOX7u4DU6uNjqd89uV1PePe4DDi6rbFM1Stev6hQZZl0wbKphoXQlHAsfxT7ZX\n7DnbZl/5/VTLLbMk91+fNI67clTVNkC83npxXWN2HnMLALDiVr3FwCCPw5khNHRag8Xd3Nmy\nJoIAhyONwfJXeZfTohXDN+yr6ZRe8UE3oDF+uL38o4fzAq/sHqI1mFEEgaeyhCqk6naF3n4T\nB6BNobdgeLZD1yqPHSrptEc37Fp7NFsONzx4QzLhU0bJ4ZKud7eU2G9arPj2E82tPZq378+F\nFY0QBEHQsMDjCcgnlDfJX/7mrP368IUG2YUG2YM3JN8yW+LuKfUdyooWud5ojQlh5yYFe+Vy\n1tgwmq3u7jKY3N7ljwjnENt09MEAxxX2F3U4RjcAABiG/3K0MS2GPzU5eLx25QhzMwDJvm62\nmgkfYLKaYYDDxw32oWS2DP7cxl61xSX1AwAgJfr11xstpyqlN+RGAgBwHBy50PnjoQapXAcA\niBKx7lmcODXJJ37afYTBgnU4RDfsulSGhCAWc8T1PmeqegnXC6p6xjLAgePg+301ruvFdX2l\njbKJeqkDgiAIGiUwwAH5hE+2l7tmv3+/r3ZOZkiQS1aw0Wz9eFv5ofP2jGggCeG8vCY7SsQa\n9Y16g5ATQKeS9SaC04ZIP/krDNEgl2RH3t1jgjno8PPs6EBxh48EOGJDucV1zhd7AQDeKmiC\nxlEgk8qiU+x9kRxFBF3lQ4kwLIthuNVCHBHrkl3M9Nl0sO7nI4329dYezev/K35iedp1uRFD\n3fdEpzKY3bVJUxrMIw9wqHTEM7zGeLaXTGXoGSCI4wAAKlrkMMABQdcss9X51ACChgL2foPG\nX5dM19ZLULJhsWLFdf2u61/trj505dlgU7fqnxuLXEMkvolCRpcQHcELOAHT00Rjv5/Rkx7D\npxI15GMzKInhE6cSxyt6BgjqOwAA7o77xx5hOhWdRr5xWpTtz2SUOGhFcbMO+Q4URRYTzXnl\nMqkzUq8yHoXLcJ5WCwDxCC0bW/NIudq49ThB97jv9tX6yyf5GBikeY1XUhbddRUR8cZ0RozF\n/TnMIHdBEARBECEY4IDGn9rNRSRAdH3JaLb+da7D9ZFdMl1hDXG2rQ+6//qkedmhjiuhAubr\n90ymUyfUqSCXSb17caLr+iM3pozLJAJfxiY6SwQAcBhj3erPnZx44ctrsgNZl/cZEcx654Fc\ne44Vk0JwsoQiKJ0MJ+b4gbXz4udnhzmuiHn0v6/NuWqyVayIxXFpSIkgCN9N61NbR4+KZjlh\nYYvWYG7oVA5j3xMaN4BMFMjA5UrDzuPN//5f8f/9XkGYVzVE83PCCNcXTiJeHyVBgXR3PU1j\nQ2GCGARBEDQ8E+psCvJTIh4DQQDhwGIx3/k6klSuc1cu3tajAWle392ooJLRl27PXj49pqxJ\nptFbYkLYM9LEwzrn1+jNjV0qkwWLEbOFXN89gbxltiRMyPzfX3WtPWoEAbGh3PuvS4Ipx66m\npQQTntflpfhQUs/crNDcpOCq1oF+pSFUwEiN5pNJl8+/Akg0LpWtNmuwS7/MZJTMpbJROCbW\nH5BIyJMr0m7IiyxrkmsN5shgVl6yiDADywmFhN6aF7XldIvWeLnsLohNC7DicpXB6cFZEkFW\nrAAM2vVD70utiPpVhoPnOztlOjadkiHh541tixAqGY3iM1rkV6R3FVf0FJzvtI+8+eN06+zM\n0BdXZzn+MhIymKwIAhzHr+bEC9cuiN9yuMGxw86crNCb8qO99ncYAhKKrJgRs+lAndN6eBAz\nL9mHPgAhCIIgvwADHND4C2RRJycGufZy57FoU5KCnBapFLdVxzSKn51HJUUGJg1/ZgqO4z8f\nadxyuMFW+o4gyOIp4Q8vTWESXWjV6M1bDjeUNPQrNKbIYNby6dFjf8KcnyrKTxWZLRiCIFc9\nBL9mrZolOVXR4zTJIl3C97V+BMwA8pRE599KOzo5gEaimTEzhmMklERFfSX9BBqi2BBObMiw\nr5lHCZlPX5dU1CjrUujJKBIpZOq0pvUlzqNVAADpMXzbR0C4m9YeCAIig3yl/fCxsu4v99TY\nYzH7izsyYvgv3ZZJc/815HUpwWwKijTKdLa4obRPe7LIOYfxWGlXQjh3kJ7cJyukP+yvbevV\nAIBEiVj3LkmcdumL4O5FCfkpogPFHZ0ybRCXPj1V7Pq1OwbWzo8zmq3bTzTZ83pSonh/W50J\nc/0gCIKg4YIBDsgnPHNzxqvfFjqOFOUwqK+szXYt2RDzGCIenbAxQWascHR36Rt+PFi/6UC9\n/SaO4/sK22VKw1v35zo9smdA/8wXp/uVFy+i9isN5+v7V8yIefSmlLHb7iVmC9aj0AdxA9yl\nIl/j6DTyJ4/n/3as6diFru4BPYdByYoV3H99MpnkZ8f3KILQSMTlNtAEFkAhzXTIbvhgaxnh\nwwpre2/IjQAAJEVw40I5DV3Os2lzE4NdG0uPi54B/Re7q516QJQ1y3860njvooQx2waCgIQg\nVgyf2aXQnSiTnikhqNAEAPx1rsNdgGPr8aavdldfuoW3SNX/3Hhu3bLU5dOjbUvx4dz4cC8M\nnXUyoDFu+qv+QmO/Vm+JCGatmhUzSHgdRZEHrk+6cVpUZcuA1mCOEbNTo/lwQCwEQRDkARjg\ngHyCkBvwxdMz/zrXUdkiN5issaGcpXlRbKLuAwgCHlqa8samYqf1hZPCr4VpDkaz9bdjBJ35\nimr7atoUTvkgX/9ZbY9u2O042TwnKzR5+JkjHpPKdev/qCqo6rHdzIkXPr48LdxnLtL6DhqF\nJAnh7D7TajJb+5XWg+c7z1T3Pnxj8uLJvpXEAUFXNaAxEq7L1RfXEQR5dW3O2z+V1DtUZmXG\nCp5ZlTEW+xuCk5VSwg6XR8u671mUMMan3ufr+/77W5lab7YSNS4B7rsUa/TmH/Y7l34AAL7f\nW7t4cvggg65GqK1X8+wXBfYuWgMaY1mT7JbZksGnz4p4dBHPJ8JbEARBkP+CAQ7IV5BQ5Lrc\niKEk5M9MF7/74NQNu6papGoAAItOuX1e3IoZMaO/Ry/QGy0dfVoOk+rZYVxbj4ZwKCMAoLbj\nigAHjuNnLsUUnJyukI5ZgGNAY3z689P2sxoAwPn6/qc/P/3F0zPcNfC/ZjV1q978sdix86JG\nb/7w17IgLj0n/prIToImDC6TOIuHx6LZ/yzi0T9eN+1sdW9Dl4pEQpIiArPjvP9zbsXwfYXt\npU0yncESJWLdlB89xM/ePpfosI1Gb9YZLIQlgaNEpjK89/MFvcmC2MaqEIU4OG66FFe2DBC2\nO9GbLNVtitH7YFn/R5Vrj/Ctx5vnZYfBvqEQBEHQqIIBDsgv5cQLv3p2lkZv1hstPpLPfFVK\nrembP2v+Ku7AcRwAEBHMenx56mgc0NvojVZ30xYVWrdja7xux4lmx+iGjUpn+uVI4xMr/KQl\n7Fj543Qr4VyJHSebYYAD8i95icElDTKC9SubdCIIkpciGmFjoM5+7U+HGuo7lSiCJERw186P\nt4cwBjTGl78utLe2Kazp/eN067O3ZMzNCnX/ehe5C2GQSWgAdex6cAAAjpV1G0xW28hYFAGE\nEe68FOLup+4C4gAAnUNTWO8ymq0lDQQj3m1hdxjggCAIgkaVn1V3Q5AjFp3iL9ENixV/6euz\n+8+145cGTLT3al75pvAC0TnAICJFLHf97SRiTnFd366C1rPVvVqDhU4ju+t2MdqpEzgOmqXq\n05XS2nZFaZOc8DHlzcTr17LWHjXheouUeB2CfNbMdHGuSzPa9Gj+Im/PHz1dKX3ww+MHijta\npOqmbtW+wvYH/nPM/rn6xc4qp8a9RrP1v7+VyVzGu7iaHE/cazMnTkBCx7RCpUt2ufwEQRDE\n5d1D+Iw7FxK3BRmkGDDCTZ/XkdPozY5jWRwp3U+FhyAIgiCvgBkcEDQWjpV2Nbq007Ni+Mb9\ntR/H5Q/9dWgU0qrZks0H653WE8K57/9ywd57lcukrluWOjcrdFdBq9MjSSgylKuXHmvqVn28\nrbymTWG7SXHTI9PsJrvkWuaun6jf9RnFcXzP2fadp1s6+rSBLOqk+KB7lyTyObSrPxOaKFAE\neWZlekFVz4lKae+AQcil5SWJ5mSIEa/2jTSarR9tLXfqlGE0W//za+n/XpprtmKnKqSEzzpZ\nIV12tUmoyZGB87NDD105C4bDoNy9IH7EGx8ep1wSEopgCMAwgOAgVMjITxWtnR/PcJNvIgnh\nJEcGVl/6QLbLkAiiRFcJcOA4kA7oegf0QYH0ED596P93XCaVRiERlsaIec6j3yEIgiDIu2CA\nA4LGQmXLAOF6dZvCYsWHNTz1roXxZBT5+Uij/fBxeqq4sKbX7HCUr9Sa3v/5wuv3TG7sUlW1\nXn5rMgl59KbU0WvwKVcb//blWcfSazNRlz4AgCSEPUp78F/pMfzSRoKMngwJf+w3MxJv/lhy\norzb9ud+pWH/ufYz1T2fPJ4fKoCdZa8hCAD5KaL80ZxLXd4sVxIV3PUq9HUdykAWlbBLKACg\nl2gOl6tHl6akRfH3FLa192s5DGqmhL96Tmygm/Yio2dSvNCptzSKICgJZMcJ37pvyuDPRRDw\n9zty/r3pfG375RhHUmTgy2uyBn9iY5fqk+2XQ9WJEYFPrUwfYidvMgmdkxm6/1y70zqNQpqV\nETKUV4AgCALujyEhaHAwwAGNIgzD0bFN5fVZFoz4MxrHcQzHARjGvxKCIGsXxC+dFtXYpTKZ\nrTEhnK3Hmly/A6wYvvtM20frph0o7iyu61NqTZEi1o15URHBo5WWDADYXdDq2ljOFQlFVsz0\nj6awY2n5jOh9Re1Og29sPXTHa0seKKrps0c37GwNaP5x16Rx2RI0USk0bj9tBjTGiGAWgiD2\nqkBHgawh5RMhAMxKF89KF3u+RW/IkAhmZYQcL7vi14pOIz9wfdJQnh4USP/08fyCqt7adgWC\ngKSIwKnJosGzMbrluuc2FOgMl5t01LYrnv+yYP3TM0P4Q0rBePjG5NZedY1D5giNQnr2lgwh\nN2AoT4cgCIIgj8EAB+R9OoPlx4P1x8u6+5T64ED63KywNfPjxrgrm6+RiIkvfIUHMalkTwoQ\nuEyqvfFkY7dz8cvF9S4VgiCLJocvmhzuwVt4oK5DSbiOIMB+lsFhUNctS0mL9rOshDHAYVA/\nfix/wx9VJy/l1WfHCdctSxUP7YzCRxTV9rpZ78Nx4NUCBWjcdPZrtx1vbpGqaVRSWjRv5awY\nOnUcDieEHLdny0HcAGYAOStW4NrtEkWRaaOZVzIa/nZrZlo0//dTLd1yHYNGzooT3LckcYix\nBgAAgiD5qaL81KH+rbcea3KMbtjoDJbfjjY9uXJIzaFZdMonj+UfPN95oUGm1pujRayl06Lg\n5CwIgiBoDMAAB+RlGr35yf871dGntd3sGdD/fKShsKb348fyr+UYx4JJYT8drne93njr7NiR\nv7i7Cheyz6TPcBjUp25Ol8p1Ih49K1bIZhB3P4WCA+n/uGuS3mjpluuCA+nu2sT6Mo3eTLhu\nNFtNFqu7FrmQHzlc0vnhb2X2NjrFdX17Cts/eHjq2JcgpcXwggLpfQrnepPIYFZsKBcAsG5Z\n6vMbCpzKWO5cED96ZXqjBEWRpXmRS/MizRaM4lFMfFhq2p17dthUtxHXWhJCEGThpPCFk8Yo\nvA5BEARBNjDAAXnZthPN9uiGXVO3aufpltvmeOFkfsycr+/fdqKpVaphMyiZsYK18+NHclrO\nolPevn/qf34ttbf0p1FIdyyMX5IbMfKtpkTxCKexpMWMdZZEYgS3sIbgAn5SZOCMtHFO8/Yj\ndBpZEuKvkxRDBMRXlQWcABjdmAAGNMaPt5U7NQnuU+g/3lb+jzsnVbcq+lWGUAEjNZo/rNZC\nniGT0BdXZ/7rh2LHsBqHQX1xdZYtVyhKxPrm+dk/HqgvbZJp9OYYMTs3Kbi0UXbfB0cZNHK6\nRLBmXpwvx1tbpOptx5tbetQMGjkthr9q9hhlyribgeJuHYIgCIJ8BwxwQF5WRHR+CwAoqunz\nowDHxv21Px1qsP25V6Fv7FIdKen66LFpI7lEGRfG+fypGWVNsrZeTSCTlhbDE7jkV+PACgCG\ngOEdcK+YEbOvsF2uNjou0mnkse/dsHRa1B+nW52ul5JJiH91kYBGYl522JbDDRar84nQ4inw\nQu5EUFjdazARTMe40Ci7572j9hY8EcGs527JSInijfZ+MiSCN+6b8s7mkn6VAQCAogiJjFS3\nK+LDubYHcJnUx5an2v7827Gmz3dW2p9b16E8dL7jw0emjWpnIo/tOdv22Y4K66WYQklD/76i\n9v88khfCZxTV9u081dLRp2EzqJMTg26dI/Fu4CMulOs69gsAEBfG9eK7AACA1YIV78Vqz+Dq\nfoQThKbMRLMWABRGQiEIgiDP+dn0Qcj3aV0Kd23cJa77oGapesvhRqfFAY3xi51VI3xlEopk\nxwmX5UfPzgxxim5YcZXBWm2wlBksFQZLmQXrBWCo18q4TOp/102bkhhkX0mKDPzwkbyxT8Pm\nsWjvP5zneFYj5jNev3uK4wqO43vOtj368YkbX91317tHPttRQTgHAfJTYULmc7dkOtWjTU8T\nr50/1sM1odEgUxmJ78CBY4Ph9l7NK98WOnXMBQDgOCBq+uk5vdHy4W9laoOZRiXRqCQKGTWY\nrN/urTlU0un0yG657vt9NU6LCo3ps98rvLkhL+lT6D/fWWm9MmOiT6H/bHvFhl1Vr35bWFjT\n2yXT1bYrNh+sf/i/J2Qq53/qkVg5K8a1EIZCRlfNlnjxXYDZaPnpn9Zjm3FpI9Aq8e4G66Hv\nLb+9DazERxEQBEEQNBQwgwPyslABo7PfuUQFABAm9JtGiQWVPYSN94vr+ozmUWkiYMUHTNYW\n+00cWM1YJwYMVDRyiK8QKmC+dX+uQmPqkmmDA+nj2Kk+Rsz+aF1+a4+6S6YVcukxYrbjgTKO\ng39vOn/qUhNNqVy3q6D1VKX008enD73/nMWKd8m0DBoZNuT3TfNzwjJjBfuL2tv7tIEs6qSE\nIMfoG+TXeOwhzR8BAOgMll0FrfcuSbTdrG5T/HiovqFTCQCQhHDWzItL90YN3dHSbsJz+20n\nmudnhzmunKnqcU0sAgCUNso1erOv9bspqOp1qgOyKa7vP1fX57Qoleu+3VPzt9VXmfw6dDFi\n9hv3TvlkW3m3XGdbEfMZT61MixG7He+tMZgVGlMQdxiVaFjxXlzqfC0Bb6vEyg6j2Ys82zkE\nQRAEwQAH5GWLp0QU1ToffgEAlkwZ6rn6uHOXUGDFcLXOTON6P8BhtjpfbAQAWDEZhgShyDDa\nzgeyqIEsqvf25SEEAdFidjTRofDZ6h57dMNOrjJ+v6/2xSEcnRvN1s0H67efaDZZMABAqID5\n6E0pU5ODvbJtyIuE3IC1C2DKxgSUmxREJaMmonNvV/YyhyOlXR9vK7evV7UO/P37onU3pS4e\n8YCn5h41AASTtjv7tU7xaHczZXEcV2hMvhbgkKuJMzIIg+8AgFMVPd6dUpQTL/zm+dlVrQO9\nCn1wID05iudu4FdLr+bHIw2N3WoAAAJAXlLwbTNjuMyrfxNhDefcrcMABwRBEOQxWKICedms\njJDb58WhDvM7yCTknsWJU5L85hJuUCBxXgCNQhqN8AGOG3FAXL+D4Rqvv934OuumRcvZauJ1\nJ29sOv/zkUb7yVWXTPva90Unyp0jJhAEjRIBJ2DdslTSlROaUDcn1rYvApPZ+s0e59oQAMD3\n+2t1xpEWI5AQxDW6AQBAEOdduUv4IqEIf8hpKWOGz3aXnkb8T603WQxmL1d2UMhoZqxg4aTw\nzFiBu+hGW5/2nV9LbdENAAAOQEFN7zu/lRnNBI1anOnVxOs64sHnEARBEDQUMMABed+9SxK/\neGrG2gXxC3LC7lwYv/7pWWvm+1OPyZnpIYRz+GZlhJBJ3v+VwcEg10KHdJnUj7hrxaI1mK/a\nn7+sSUY4ouXbPdVe2BkEQUNz/dTIz5+aMS87NEbMTo4MvGW2ZLKbEqTUaB4AoK5DSfiLrzda\nqluHMXaUUGIEcdtLSQjH6WM8P1VEWD0xNVnECPC5bNa8lGDCr6EQPnFOH5tBGZsBK052nml1\nTefpUeiPuWTqEWALCJcRrt9cDoEgCIJ8kM99qUMTgySE479zLkU8+lMr0z/ZfsUoxLgwzkNL\nk0fj7VCECgBC2FIUQSZajwkRj7gVi4jHQNGrZFdXNBOfC3XJdP1KA+zHAUFjRhLCeen2bPvN\n+g7lhYZ+pxPd4ED60rwoAIDWfZqGu6bUQzcjTbz9ZEtrzxW5ACQUcW1qK+AEPHVz+kcVF6uH\nAAAgAElEQVRbyxw/2MODmI9fmrHiU4ID6Y/elPL571f0GRVyAx5bnvaPjUWu4eC5WaFju8GL\n6oiGrQAA6jpVi67sgeIKTZ1pbSdo3Y2kzPTCziAI8n9mosZJEHRVMMABQQQWTQ5Pi+H9cbq1\nRapmMyjZccLFUyJIVzsD9xSJjAosWL/TKoLQSIi/BoncWZATtvV4k+vR+aIhlOKbLG5zns3W\niZbqAkF+JD6c+95DeZ/tqGjqvni6OzU5+LFlqcwAMgBAzHPbSEjMH2nzaTIJ/ffdk7/dV3Oi\nvNvWniI4kP7QDcmT4oWuD16QE5YSFbjjREuzVM0MIGdI+DfmR7srvhh3S/OiUqJ42443N3Wr\nWHRKegz/ltkSOo38wPVJ3+ypcfwUTYwIvHdJ0rhs0urms9dyad2K4fuL2sub5XqjJUrEvik/\nyj5BDE2bg3fWYuVHHZ+I5ixBE3JHc8sQBEHQBAcDHBBELFTAfOTGlLF5LwoahuNWK345QwFF\n6FQ0xl25tf+KFrOfXJH2xc5Kx4u9M9LEq+fGXvW57hr4s+iUIO4wWrFCV2WyYNuON52u7OlX\nGsKEzMVTwhfkhHuxfyE08aRG89Y/PbNnQNevNIQFMXmsy10tokTs+DBufafS6SnRInZ8mBdi\nuIEs6nOrMh64LqmjT8tlUkMEjEGC0aEC5mM+mbJBSBLCeeG2TKfFVbMkOfHCP063tvVqeCza\npAThrPSQnw7Vn6ro6VfqQwSMRZMjVsyIHo2CSldhAmZDN0ESR7iQCQCQq42vfFNoD3udruz5\n/VTLi6sz81PFAACAIKQlj6DJ07HaM7iqH+EGo8nTkfDxidRAEARBEwYMcEB+w2LFLFY8gOr9\nISY+AKWSojE8GMM1AGAIQichnIkX3bC5fmpkdrxwf1F7R5+Wz6ZNSQweYgPaaaniED7DPrbQ\n7qb8KDJpYv5bjQuN3vzc+oJm6cWcf5nKUNYkO13Z8487cxAY5IDcQxAg5jMIkzKevzXzjR+L\nO/psE8RxAJBQAeOF2zK9+BPFZVKHMrljYpCEcJ6+Od32Z5XO9Phnp7pkF6ezt/Zovv6z+kx1\nz3sP5o3BB+PinLCGP50DHDQKaW56CADg898rm64Mf+iNlvd/Kf3hRb79PwuJSidFpY/2PiEI\ngqBrBwxwQH7gQoPs2701DZ1KDMfDhMw7FiTMzQqdeKdaKMJAkZEmbPuFED7jnsWJw30WlYy+\ned+U936+UNdx8VIwiiI3TYu6c2GCtzd4Tfv5SKM9umF3qkJ6rLR7zjjV+UP+Tsyjf7pu+tGy\nrroOJY7j8WHcOZmhhE00oeHacrjRHt2wK2+SHyjuuC43wvXxFivWIdMNqI3BgQGhAuYIv0gn\nxwvXzondeqrFaLbYgvJCTsB9C+MFHJreaCmoImg1qjNYTlVIr5/qN5PjIQiCIP8CAxyQW6WN\nsroOJYqAxMjAtGj+eG3jcEnnu1su2G929Gnf3VLS1qv24AwZ8ncRwazPnphe0iBrkaoZNHJa\nDD88iDnem5poTlcSjz84VdkDAxyQx0gkZH522PyrNZ6EhotwthQA4Gx1j2uAo6xl4McjDX1K\ng+1mbAj7nvnxtnISjy3ICp0SL6zpUA5ojCIePTWSZ2trIlMZLW4aBPYq9CN5RwiCIAgaBAxw\nQATkauO7W0ouNMjsK7lJwS+uzmIzKGO8EyuGb9hF0GX9lyONN0yNDAqEnRcuKmnoP1PVK1MZ\nQgXMhZPCIoJZ472j0YIgSE68MIeogyDkFUqtiXBdoTGO8U4gCLoqjY549rbaZTRvfZfq0z+u\nGMvS2K1+b2v5W3flcBgjqu7hMqlTXUYFsxkUBAE4UYjDw7fDdLixFlgVAKEgZCGgxgNkQpas\nQhAEQSMCAxwQgbc2ny9vkjuuFNb0vv/LhTfunTLGO2nqUik0BKdbVgwvbZIvyIEXA4EVwz/4\npfRwSad9ZevxpgeuT1o5M8br7yVXG8saZf0qQwifMSkhaIL2Q7nWBXHpaqJTpmAYT4Qg3yPm\n0weIgo8hLs1Q/ixqt7pMsNIYzIdKu1dMi/L6xrhManIkr6rVebw3CUWmJgcP++XM7biuAOAX\nZ2nh5nZgbEBY8wB6TdR1QhAEQUMHAxyQs8YulVN0w+ZsdW+3XOd6zDQEVhxrBpgU4HqAMBA0\nBKDRAAyp+tpgcjsZVG+0OL8Nhjd2qaRynYhHl4RwJliBN44DwrYjO042O0Y3AAAWK/bl7uqU\nKF5SZKAXN7CroPWbPTX2f/agQPqzq9InJQypPyjkR+ZlhzYRjUWYD+OJEOR7Fk4Or25TuK67\nzt5u6dUQvkJzj3PPHW95YkXa8xsKtIYrvqzvXJgQNtyiGNyI687aoxsXYWpcX4gw54x0lxAE\nQdDEAgMckLM2N8dAAIBWqXr4AQ4LbjkL8EvHT7gat6oB1ouQc4cS4wgVMtzluEYEXVGFUdU6\n8NHW8tZLB2qhAuZTN6dlx3leyCBXGTEcF3IDPH4Fr+jo036zp6aiWa4zWqLFrFtnxzr1QThw\nrsP1WTiOHyju8GKA43Sl9LMdFY4rfQr9v34o/vLZWaECeAFtQlk5M6aqdeB0ZY/j4uq5cbAs\nCPKifqWBSkFHWBkBAQBumBrZ0Knac7bNvkJCkbsXJ2ZIBE6PxAm/SkdTbCjn6+dm/+9AXVmj\nXGc0S0I4t86J9eSTxNwJcKJKHLMU4EaA0AjugiAIgq5VMMABOaO6T3ygUoZfkoC1XI5u2OEK\ngLUBNBoAYDRbC2v6Ovu1fDYtO07g1FZDwAnISxYVVPU4vUCUiJUuudz3tEumfenrs47pHl0y\n7d+/K/rsiemSEM5wt3yguOP7fbX9SgMAgMei3bUo4fqpkeMytKWyZeDFr86YLJjtZkOn6u2f\nSmraFY/cmGJ/jHSAuFtbt8x5nOpI7DjZ4rpoNFt3F7Q+tDTZ9a4BjdFosop4jIk37GbCI5PQ\nf909+WSFtKCyp0+pDxUwF0+JSPZqNhB0zbJi+M5TLT8dalDpTACA8CDmQzck56WIxntffgxB\nkKdvTl+QE3aqsqdPoQ8VMObnhEWJ2K6PjBGxS5sJ0jNjiB5MyNZ6HMfx+HDu4NcPMAxHUQQA\nIOQGPLsqY4iv76hXbSzvVCj0ZiaVHMM1JBD3lcIBpgckGOCAIAiCLoMBDshZSjSPTEJcm5/T\nKCQPTnJwrM/dOoJGlzT0f/hrmb2hOo1CWrsgfvXcWMdHPndrxhubzpc2Xu54GiVi/ePOSST0\n8qnz9hPNrsUsZgv269Gml27PGtaGfzna+O2eGvvNAY3xk+3lvQr9vUvGYWjL5zsr7dENux0n\nW5ZMiYgWXzwkZdMprtU6AAAO05sdYVtc5oa6Wz9ZIf16d3W3XAcAYNEpa+bHrZgR4/ifBfkU\nixWTq40CToDT/9GMNPGMNPF47QqaeGrbFbsL2gpreh0bRnT0af+x8dwLt2UunORcTwENS1oM\nPy3mKsPObpgSUdE64NSGgxVAmZ8ZAgDQmiy9aqPZivMZFCHLOWSg1Jre+ankfH2/fSUzVvDy\nmmw++4pHYhi+p7Btx8mWrn4ti06ZlCC877okD9r3nG2WFTTL7Bkn1VJyBSf+prhGMuL8hQjT\nNyBo3MlksrfffvvcuXMlJSVqtXrTpk133HGHV17Z7HIMDEFDAQMckDMei3brnNifDjU4rd+1\nKIFO8+AHhrjBOwDmXoX+nxvPOQYmjGbrd3trBBya48Euh0H94OG8wpreqtYBswWLC+POTA8h\nk644GavvUBK+R10HQWXyIHQGy48H6l3XfzvWuGx6tNOR3Ggb0BgbOgn+XjiOF9X22QMcU5OD\ndxW0uj5sarI3L4qSScR5PeQr8332FrZ/tLXMflOjN3+1u7qjT/v0zele3AzkFV0y3YZdVUU1\nvVYMp5DR+Tlh91+XxGXCkgHI+zYdqN98qB5z6XBp8/Wf1fOyw2AYdLTFh3KevDFl05HGftXF\nMbESMfueBfFsBrWwVV7WqbTHPiJ49NlxQUyHb/x3t1xwjG4AAEobZW9vLvnPI3mOi2//VHK8\nrNv2Z6XWdLikq7Cm76N1+VGiYQz26lYaTjfJnBbbVJyibvG00K4rVslCgMLmxxA0zrq7uzdu\n3JiTk7Nw4cLt27eP93YgaGiNHqFrzd2LEp++Od1+Pi/kBrxwW+YtsyUevZibgw+EvrewnbCH\nKGE1RG5ScGJEYGe/bsvhhn9uLNpzts3xWBlxUwjhbt2d2g6F0UywJYsVr2y5ohW80Wx1TXLx\nLq2eIC/j4l2Gy2GjOxbGi3jO/8hTEoPmZIZ4cTPurg2mO6xbrPh3e2tcH7O3sK3dfWMXaFxI\n5bonPzt1pqrHdkpjtmD7Ctuf+eK03uT2pw6CPFPTpth0oM5ddAMAoNCYWketzyXkKCOG//Zd\nk15bnfX40uR/r835+21ZEUJmcdtASbvCMbOjfUC/t0qKXcqg6OjTFtcRJGOWNckcGxKfr++3\nRzfsNHrzN3uqh7XJGilBk2MAQI38yq8hhIrQx3qyGwRBrlJSUmQy2YEDBx599NHx3gsEAQAz\nOCBCCAKunxp5/dTIPoUeRREBx/NGmwgahludL8UAABAkrFXaTvgU1yNdHAf/+bX0QPHFbpot\nUnVRbd/B4s53HsylUUgAgIQIrussOgBAUsTwampMZre5cCaz1baTwyWdmw/Vd/ZrSSgSH8Z9\n8IbkqyYGe0bIDaCQUcL0vBCHvp48Fm390zM3H2ooqJT2Kw3hQawluRE3TosabnBncHcsiC+s\n7nU6+w0VMJdOi7TfbO1RK7UEM31xHJQ1ySOCh3EFDxptmy81QXDU0afddbr11jmxhE+BIM8c\nLe266mMG+eyFvItCRiViNgAXcwAxHC/vIkgVlGlNHQP6SD4DDNp6vK1HY290VVRLXJFaXNdn\nb8kxFGqioksAgNoUACjhwDoAEAogByEBaQAZ5y7gEAQBAFAUXi+HfAv8iYQGExRIH0l0AwAA\n0FCARl65hABUAtBgKoX4x891vOvJCqk9umFX0SL/9WiT7c83z5QwApyjdTQKabinaoOk0dpK\nQr7dW/Pezxc6+rQ4DixWvLpN8ez6ghPlztesvCKASpqdQZCFwWFQp6de0RyBRac8vDR544tz\nd7993YZnZi6fHu31ZO8oEeujx6bZQzkIgszNCv3w0Tw69fI/+yClkoR5MdA4KmskCDsCAErd\nrEOQx2Qq4+APIJOQ8KBhzg2FvERlsJgsxNk1/ZcC1jQ3X9YAABr1cutxx9RCRxYrPsjEd1d0\nN+3MGVQSwpyJcG5C2Nch9MkwugFBEAQR8u8Ax5tvvokgiFjs3AlPqVSuW7dOLBYHBATk5ORM\njHowHMcrWwb+OtdRWNOr0pmaezWHy7v3X+iqaBsY89Fvw4OQUhByHkCjASoCaDRCzkNICQCA\n9BjnIXY2mS7D7dxFEOzZsCIe/f2H8uLCuPa7okSsdx7IHVbdLwBAzGdMTQ52Xc+QCGJDOVK5\nbuvxJtd7v9hZNUj29UisW5bqNOqPy6S+ekc2i+7NBqJDJAnh/PfRadv+tejLZ2ftfGPxy2uy\nnYJf4UFMd4EVe8cQyEcYLcTnGwYYioK8jce6SmOX+Tnh4/KZBgEAbJ/ZhB/c9o/zpEgejSjo\nQCGjKVE8+013I8N5LJrrFYhBxAURf3HHulmHIMhP3XfffXz3jMarBMchyB0/LlGprq5+6623\nRCLnTooYht1www1lZWVvvfVWbGzst99+u2rVqu3bty9fvnxc9ukVTd2qD38rs7fSpFJIfAGd\nd6kzebiAec/c2KARplqMKiQQITlXiyycHL7zdIvTGA4ahXT34gSnRw6oiT/j5GqD/c8J4dzP\nn5ze2qORynXBPHq0iD30hFhHf7st672fLxTW9NpXsuIEL92eDQAobZQRBjJkKkNLj9qDebRX\nxaJTPng471SltKxJpjNYYsTsxVMixvdMgM2gsBnEG2DRKQtywvefc648koRwsmKJg1nQeIkM\nZsmJrqtHD3lgJAQN0fQ08e+nWtzdOysj5LFlqWO4HegK7AAKg0rSEWVYiNgXDyqYAeQ7F8Z/\ns8e5xdKaeXGObYnnZoX9eKDedfLXktyIYW0pRshMFnOqr+zEwWNQ810ufkAQNMasVqtaffm4\nncPhjKQ+5dlnn73uuuvc3bt27VqPXxm6xvlrgAPDsPvvv/+ee+6pr6+vqKhwvGv79u2nTp3a\nuHHj3XffDQBYvHhxTk7O888/778BDrXO/NLXZxWaywXzJrNVKtWQUJTDoQEAOmTabw7W/215\nmn91oaeS0Q8ezvt+X+3+onZbe7O0aP66ZamukQIBh3h8ifDKmA6CINFi9giTBdgMypv3Tals\nGahpG8BwkBDOzbx0cq43ur24PchdI4QgPjqws0Wq/uGvutp2BYbh8eHcOxcmJIRzH1+RarRY\nj164XHKfHBn48ppsz4JN0Oi5KT/6QoNzNQoJRW7IiyR8PAR5LDNWsCw/eufpFsfFQCZ12YyY\nyQnCxGF2SoK8CwFgciTveEO/03oEjx7CvfwNe+ucWCE34Pt9tT0DegCAkBtwz+JEp+G+Ih79\nxduzP9paptFfrlWZmS6+Y0G848NwHFy1Q9SSVLFEyCzrVA7oTEwaWSJkTo7kO01PgyBo7JWX\nl2dnZzveTEtL8/jV0tLSBnn6XXfd5fErQ9c4fw1wfPrpp62trXv37r355pud7vr9998DAgJW\nr15tu0kike68884XXnihrKwsIyNjzHfqBX+da3eMbtjJ5DrOpTN/qUJf361KcqjRGD+4GZNb\ncTUOLCigkVE+CXEursYB0JmtJitGoqCPr0h7fHlqZ79OwKG5y02YlRF6uISgTd3szFDvbx8A\nAEBqNC81mue0GCYkrhJHEMTdXRPV6cqeNzYV27vun63uLarte+6WjIWTwl9Zk33LLEll64DR\nbI0P42bHCb3a7RTyjhlp4oeWJv+wv87eHoXDoD65Mm00EpEg6LHlqVOSgnYVtHb0aXks2uTE\noJtnxThVPeA4juHAv8L0fsRixQqqelt71Cw6JUPCd/xNTxZzUAQ52yLXm60AABRBksXs3Gjn\n5tnzssPmZYcpNCYMx90NTZ+ZLk6P4R8o7mjtUQeyaDnxwpx4of3e8ib5xr9qGzpUAAHxYdx7\nliSkubyLXYKInQATyiDIx8THx584ccJ+UyLxbMAiBI0uvwxwNDU1vfrqq5s2beJyCc7nKysr\n4+PjabTL377p6ekAgIqKCj8NcDR2EY9MMxgtuEPprFShH/cABw6sBmszhuttNzGgt1gVFDSY\nil6uJNJbsG6N0Wi9mMWKIiCITh28WUZ+quj6qZF7zrY5LmbGClZ5OLnWQ9nxglABs0umdVqf\nkSYOvFqR+URiseKfbi+3Xlmtg2H4FzsrZ6SJ6TRyfDg3PtwXYm3QYFbNkszKCDlX29er0IcK\nmNNSRO6KjyBo5HKTgnOTCDocAQAqWuTf7a2t71BaMVwSwr5zYQJhLyTIYw2dyrd/Kunou/jl\nhaLI4snhT6xIt+dEJIrY8cEshc5sxjAenUp1afVtd9Uvu0AWlXCo/N7C9o+2ltlvljXJnv2i\n4IXbMp3SQCAI8mVMJnPGjBnjvQsIugq/DHA8+OCDixYtWrlyJeG9MpnMKaDI5/Nt64O/7DPP\nPNPZ2Ul4l8ViAQBotc5ntmPDXXo/AgACLoc4BjkiGTNmrNce3XBcJCFsEsIAAFgxvF1lsDq0\nRcVw0KMzkVCESxvsp/Hpm9Onp4r3n2vvkmmDuPT8NNGiSeHenYR6VWQS+q+7J/17U7H9MBEA\nkBUneGZV+lhuY9zVdSjkRF1RtAZLWZMcnpn4keBA+vVTYU0KNJ6OXOh656cS+826DuVr3xc9\neEMy4Uky5AG90fL374sce+5gGL63sJ3LpN13XaJ9EUUQPnO0IvV6o+XL3VWu6+v/qJqVEULY\nwRSCID/yxx9/mEym8vJyAEBRUVFAQAAAYOXKlXCCLDQufDrAQdjJ5uuvvz537lxVFcE35eCu\nejIsFAp1Oh3hXbYAx3j9liZH8v465zwkFQBAD6DYoxsIAuJ9ILfcgikI16240hbgUBgtVqKh\nLzK9efAABwBgSlLQlKSgkW9yJKLF7K+enXWstLupW0Uho6lR/HHf0thT64hnAQIAVDqCWioI\ngiBCFiv2xc5K1/Uf9tcumhzOHbXz7WvKyQopYUfh3Wda716cMDY1QRUtAzqDxXVdozdXtQ5k\nxwld74IgyI/cddddSuXFSQiffvrpp59+CgDQ6/W2SIfHLFbnpsUQNBQ+HeBw7WQjFotfeOGF\nl156iclkKhQKAIDFYsFxXKFQUKlUBoMBABAIBHK53PF1bDdteRyDePXVV93dpdFovvvuOzqd\nPpK/jscWTArbdqLJMWsAAIAAEOTQ92FmssgXpqjggOAIBgCA4xdPiY1uPqqMVgx3M6nO15BJ\n6PycsPkgbLw3Mm6CeW5/EUTu74IgCHJS36lSagmioiYLVtYkm5keMvZbmnjaejSE6xq9WaYy\nBAeOxYe2zuA2LL73bBuNQnIcNwtBkN+xnZRBkI/w6QCHayeburo6pVL5yiuvvPLKK46P5PF4\nd99998aNGwEAqampW7duNRgM9qhhWVkZAGAkbX7HF41Cev+hvPV/VJ4ol9pWhFx6aCjbjOMA\nADadsiQrLN838ggQQCaMcSAI+dID3D3R56IbOI639Gikcl0Qlx4Twoat7+xixOy4ME5Dp3Nr\nmFABM9V9xzgIgiAng5z3avXE4XJouKjuC0DGrDZEzGe4u+toaffR0u6Fk8KfWZUB56RAEARB\nI+fTAQ7XTjZxcXFHjhxxXHnuueeam5u3b98uFl+co7lixYrNmzdv2bLl3nvvBQBYrdZNmzbF\nxsb6aYdRGyE34LU7J8nVxo4+DY9FCxUySSiiN1ktVoztZvKIq+Nl3bvPtHb0aXlsWm5S8G1z\nYgOoXj64IaNcM0bQ64SEXGw5SaeQFEaCw1b6lYdZCo3JYsWE3HHLSanrUH68rbyh82K6XUQw\n66mVaRkSwXjtxzNKren3Uy3N3SoyCU2KDLxxWpS3Dmdfvj37lW8LbfMCbfgc2qtrs2EYaCyp\ndKZ+pSGEz6BfrbwLgnxTqPv5U9faaKrRkxnL33SAYD1GzB5JEVBbr6ajTxvIokpCOFc9lkgI\n50pCOE3dxB3TAQAHijtEPPpdixKGtQeVzoQABHZHhiAIghz52WExi8WaM2eO4wqPx+vs7HRc\nXLFiRX5+/pNPPqlUKiUSyXfffVdRUbF9+/Yx3upo4LNpjrPZ6FQSAEM9X/3gl9IDxRcbefQr\nDfUdyiMlXR8/lu/d8R8UVGTFtRhuuHJRaJ8Uy6WS5SSzU6EKAkDQpQOUwyVd3+2t6VXoAQB8\nNu2uRQnX5UaO8ZzRngH9374641gw3N6reeWbws+enBEj9pupdRcaZP/eVKzRX7xAeryse+ep\nlncfnOqV04aIYNY3z8/ec7atuk2B43h8GPfGaVHwNHvMNHWr/m9HZUXLxVq8WRkhj9yYMo4B\nQQjyTAifkRkrKG10DotHiViug7ohz2RIBPmp4tOVUsdFEoo8tDTZsxfskuk+2VZe0tBvu8ln\n0x6+MWVu1uWp7TiO9wzoqRSS/YgFQZC/35Hz2vdFnf1um7XvOds29ADHofOdG/fX2oLsYj7j\n3iWJjhuAIAiCrmUT8IQERdE///zz5Zdffuedd5RKZXJy8tatW5cvXz7e+xpP5+r67NENuy6Z\nduP+2qdv9uYEEASQ6KQ4Myaz4mocmFEQQEb5JOTyCFgEAZGcgF6dSXkpj4NGQsVMKoNMAgBs\nPd701e5q+4PlauPH28qlcr1jp3d3TBZsz9m2mrYBK4bHh3GX5kUxAjz88d5+otm1HZrJgv1y\npPGl27M8e80xZjRb391SYo9u2PQM6D/4pfTjx/K98hY0CmnFjJgVXnktaDjaejXPfFGgd8iE\nOl7WXduu2PDMLOaQf+bLm+S/HW9q61Ez6ZRMiWDN/DjWkHPBIMiLXlyd9Y+N5+zpcgCAMCHz\n73dMcjc+DPLAK2uzfzva+NvxJttXW3w495GlKekSTyoK9UbL3748Y7sIYSNXG9/5qSSASpqW\nIrJY8R0nm7ccbrB9+4j5jAdvSJ6ZLgYAhAcxv35u1oHizm3Hm9p6CdqCyNVGncEylC/unw41\nbNxfa78pleve+amkX2lwN3lHpjIYTNYQPgP+UEEQBF0L/D7AcfDgQdfFwMDA9evXr1+/fuz3\n45tOV/QQr1dKvRvgAAAAgFBQIQW4bYpORpFQFk3MpJqsOAlFKJcOOPQmy//+qnN9/G/HGpdP\nj+ZzaDgOqqSq6m6VUm/m0MnxQeyMcC6KIACAjj7tK98WSuUXh+AcK+3efrL59bsnJ0YEevAX\nqOsgbpXkbt0HldT3E05yrWod6JLpQgVuy6Eh3/fToXq9S51Xz4B+V0HL6rlxQ3mFzQfrf3D4\nXavvUB4u6fzw0WmwKAAae0JuwOdPTj9e1l3dprBa8fhw7rzsUDIJThb0JioZXbsgfs38+D6l\nnhlAGXok1NWB4k7H6Ibd5oP101JEn2wv31/Ubl+UynVvbCp+dlXGktwIAACZhF6XG9HaoyYM\ncJBQZJB2IXYqnWnzoXrX9e/31VY0D2gN5ohg5o3ToiQhHADAyQrpV7urbccGdCr5trmxt86R\nwJ8uCIKgiQ1+yl8TCNvUAwCUWjPRzNaxgCJIABmlOFxOqWtXGkxW10daMbyiRY7h+M6yzr+q\npO0DOpXB3DGgP1LX+1txhwXDAQDvbimxRzds5Crj25tLLFZP/nru/k0I1zEMN1l8bopVr8Lg\n7q4+omPTIdIZLI1dxFMPoDFT0TxAuP77qZZ+pdv/d7u2Xs2mg86nB3K1kXBaJwSNAQRBZmeG\nPnJjymPLUxdNDofnn6MEQUBwIH0k0Q3gPtDf0KVq7lY7Rjfsvt1bY8Uuf31Oiu2UUbYAACAA\nSURBVCfuiZ4ZKxhKk9HqVoWZ6DvXYsUKqqRlTbI/z7Q99snJbceb/jrX8e//FduPDfQmy8b9\ntR/+VnbVt4AgCIL8mt9ncEBDERRIXJwfxA0Y4/YWgzCZ3YYJjGZrVbeq2aV2t0upP982IKJT\n6zqUrs/qlusqmuVZccPuDJoQwa1qJTiHTIzgOt6sbVd8s6emqnXAYsXDhIzb58UtyAn3kX9P\nLtNtuYFnXeX6FPoNu6pPlHfbbqZE8R5fnhYXxvFwf9AIuBsLL1cZn/r89IanZw7ecq+gqgfD\nCGJ1xfX9epOFToVfChAEXaQ1WH471ljdqjCarbGhnFVuakBsKloIWowDAJRaU2uP2pZSAQCY\nnBg0LUVUUHVFYimdSh5iTxDCCyFOrBj+9Z4aVgDBJ+Gh8523zJbYNwNBEARNPPA6yTXBXfOt\n+TlhY7yTQUSKWO7uihaxG/oIMloBAA19mt4BtykJPQM6d3cNYsWMGNd+mRQyeuucWPvNopq+\npz8/XdooM1swHMc7+rQf/FK6fpc3r4E3dave//nCox+feH7DmY37a13bggwiO15I2NY+TMiM\nEg27T6rWYHl2fYE9ugEAqGodeG59AWGaMTTaot13uu1T6HecbB786QoNcQIOhuEqrduZnRAE\nXWtapOr7Pzj606GGkob+qtaBXQWtD/7nmLtRWfFh3EFSJo0OFzAQBLx256SHliYHBdIBAAFU\nUl6K6POnZgwx6DDIoYIjDMNVOuLPurJG+VBeAYIgCPJTMMBxTUiMCLzvuiSn9lrZccI184dU\nsT82RDz6tBSR63paND8ujKt3c9FGZ7Jw3GcrsBmeZCuE8BnvPjjV8WArTMh8874pjiuf76y0\nulwG33mq1Vvn/LsKWh/75OTB852NXaqyJtlPhxoe+PBYl8xt/3knHAZ13U2pTv/jNArp2VUZ\nHuSY7Cpo6XGJIulNls0ulQ6+wGzB5CqC/iMTxsqZMYPcW9Z0lWN3x0lMjihk1LszlSAI8mv/\n3Vrm1MvJZMGOl3cHE+WErp0f5y56TiYhEUFMp5VVsySbX5n3x5tLdr6x+N/3TA4PGmoDoBgx\n27P2qHZmN0lwEARB0MQAs5GvFavnxuYmBf15pq2zX8tj03KTgudkhvpIPYXdC7dlvrvlQmFN\nr30lXcJ/ZU02ggCWmxGkbBolLowr4ATIVM7dBxgB5MzYYden2CRHBn7+1IzmblW3XC/i0SUh\nbMeycKlcRxhrwHH8fH1/ZPCQri8Noleh37CryimA0q80fLq94t0Hpw7xRZbkRkhCOZsP1jd2\nqcgkNDWad+fCeDHfk/ailS3ETR/Km33rOlhjl2rDrqryZjmG4Sw6ZcWM6FvnxNKG0LXOv+Qm\nBT99c/on2ytwoq4wJgtxKNCK4bsKWn8/2dwtJ854mp4mnnj/VhAEeaZnQF/TRtBuQ6u33LUw\n8Wx1z/n6S2NiObRHlqbkpYisGB4jZjdL1U5PWTgp3DakSaUz7Stsb+3RMAPI6RLBjDQxYabh\nVb26JufNzecrrvYFhKIIYTle9PDTGCEIGhcwHAl5BgY4riGSEM4TK9LGexeDYdEpb943paJF\nXt2qwHA8IZybFSu0RWESxcRVKkliNglFnr45/fX/nXPMj0VRZN1NqSPppkZCkbgwblwY1/Uu\nncsMi8t3DaeQxJ0zVb2ETdRKGmQavXno4zwTwrmv3zN55Psh3AwgagZR2ig7XNLZJdMFB9Jn\nZYRMTQ4e+bsPUXWb4oUNBfaGrxq9edOB+sqWgXcemOprgbyRu35q5PGybvsJhiN3BSxv/Xj+\nZIXU3QtGidiP3pTitf1BEOTnXK8Z2GEY9u6DUzv6tG29Gh6bJglh22KjJBR5/Z7J72654NjE\nal526LplqQCAotq+d7eUqHUX6+B+P9WSIRG8fs9kD76m+Rzah49MO1fXZ5sufKGhv6SBoP3H\n9BTRCZcPvWgxOyfB7ZQ3CIIgaAKAAQ7ILZMF213QWtU6YDRbJSGcZdOj3SW3e1daND8t2jkB\nNSGY3RamK++8oplofDA7PYwLAJiaHLz+6Zk/7K+raVdYrFhCeOCdC+M9mxE7FGIeg0xCCOuN\nI4KHN2gTw/A/z7adqpD2KQ0hfMbCSeGzM0PkauIjSxzH5WqjPcBR2ig7Ud7dpzCECBjzssMS\nwgliMV4RE8IhPJd2rNnBcfB/v1fsKmi1rxwo7pidGfry7Vmom5pt7/pmT7XrOJvz9f1nq3vy\niEqf/N2tc2Jd/1PIJGRZfrTrg8/X9xNGN9h0amYsPyNWsDQvEo6ugCDIbpCvez4nAAAQHsR0\nrSsR8xkfrcsvaehv6FTSqKTUKJ7tIoFSa3p783ntlRcAyppk6/+ofP7WTA+2hyBgSmLQlMQg\nAMCiyeHPfFHgNElt1SzJPUsSGdsr9p+7PNglOTLwpTXZ7tqIQBAEQRMDDHBAxKRy3UtfF9oL\nMc5W9+483fLKmuzcpLG7Ju9kQZIoIZhdLVUp9WZ2ADkhmB0bdLkYJErE/sddk8ZmJ4wA8pzM\n0IPnO53WhdyAYf37GM3Wl74+ay8Aae/VFNb0nqwISY8hLjBGEMR20Ilh+Efbyh0H8v1+quW2\nObH3Lkkc3t9kaG6cFrm7oNVodq59uHnm5Y76Jyu6HaMbNsdKuzIk/BunRY3GrhxZrJi74akX\nGmUTMsCREy98ZlXGV7ur7OcMXCb1yZXpsaEEjfrO1fYRvojOaH5lbc5QRjNCEHRNEfMZcWHc\nhk7nCWWMAPLkBOI5rzYIAnLihTnxV2RJnKqQaonSG49e6HpiRdoIi+MEnIAvn53569Gmc7V9\nSq0pIpi5fHqMLfbx3K0ZK2ZGV7cqdEZLXBjHnhMKQRAETWAwwDGhdMl0LVJVAJUcF8bheNRf\n0+6jreVObSZ0Bsv7P5f+8NLckdR9jFAknxHpURcJr3t8eZpCYzpXd/m8UcSjv3bnpGEdqG07\n3uza3uJYaXd6jIBKRl3zESYnCm3pG/uK2h2jGwAADMO3HG5Ii+ZPSRrs0NMzoQLm6/dM/u9v\nZb2Ki+0bWHTKgzckO77XkZIuwuceKekagwCHbZYN4V1DmSnop67LjZiWIjpf39czoA8VMCYl\nBLkrX3JXVGXFcIPJMvSiJwiCrh3P3ZLx4ldnHWeRkEnIkyvSBx9ETcgpvcLOZMFkKkOoYHiZ\nj67oVPLdixLuXpTgepckhAOHwkIQBF1TYIBjglBqTf/3e8Wx0ouDPAOopNvnxa2eG+fZxQq5\nynihkaAkQaUzFdX0znEzdPaawgggv/1A7rm6vvImudFsjRGz52SFDvcylOPgVUcXGvofX572\n6Y5yxyqY4ED6E8svtlA5WOycPGJz4HzHaAQ4AAA58cJvX5hdUt/fJdMJuQGZsQIu84oIWp+S\nuKzGHhMZVXQaWcgN6Cfaw8h7vvqyQBZ1XvbVhz2HCojDgoEsKjMARjcgCCIQG8r59oXZvxxp\nrGyRmyyYJISzem5shEefqEz3UVQYYIUgCIK8CwY4JgIcx/+58ZxjWy+Dyfr9vloAwO3zPBkE\n26fUu7kcPkbnq/5ickLQ4Mm6gxvQEE8zlauNS3IjkqN4W483tUjVDBo5Q8K/eZbE3nC+x83/\nQo+bq2ReQaOQBqn1cDdh1IPJozgODpd07ipo7ZJpA1m0KYlBa+bHXzVv6MZpUbafeUcsOmV+\nztXP/ye8edlhmw7UuxYZXT81EiZsQxDkDpdJfWhp8nCfhWE4giCOny1TEoO+3VPj+sjkyMAR\nZptCEARBkBMY4JgIShpkjtENu1+PNt4yW+LYOxDHwdHSrtNVPf1Kg4hHn58dZqtTdeJ0cf6K\nu4Z/vgq5I+AEyFUEMQ4hNwAAECViPXdLBuET2XRKH1GMg+P+P2605aeKz1b3uq7PSBcP96Xe\n+7nk8KWCF4XG1CJVHy/r/uSx6XzOYD1ub5sTK1MZdp9ps88FDAqkv7g6c5Af5muHkBvw6trs\nD34ttY8wAADMzgy9Y0H8OO4KgqAJpqi2b9OBuqYuFQlFEyK491+XlBQZCACQhHBWzIjZcbLZ\n8cE0Csk2YAWCIAiCvAgGOPyMxYqXNcna+zQ8Fi0thm9rOenaBsxGa7B09mujLo18t1ixf286\nb28bUduuOF7WvWhS+FMr052u4or5DEkIp6lb5fSCVDL6/+zdd3xb9fU//vfVXpZky3vvHdvZ\neydkELJIIYywCmUUCm2hm7bfjh8tpYUPtEDZFGiBAAkJJGSRvbfjeO8tT9nWHvf+/nBQFOnK\nlmXZku3X8w8e0blXV2+PBN2j8z5nerrfmoyOG2YrLeBxCCGL8qMrGll+doNuApqVHe760yGE\nzMryWzfNm6bFHitqOVt6QzPL7ITgDfOShnSdM6Vt37q081B3G979puynt7Gne/pxONTj63NX\nz4y/VNnZozPHh8vmTYocZu+68WRWdsS7P1u0/3xTfZtWIRVMTQ/NS1b5e1EAMH58dqT6ja9K\nvntEX67qfOrVE7++a/L8SVGEkEfXZmcnKD85VF2n7pMIefkpqgdWZbrbPQcAAOA1JDjGkrIG\nzd8+uVzfpu1/KBJw770p/dYFyQM+6XrqYsfJOsemmP32nm+cmh423+Vj9qdunfTzN04bzDf0\nJvzBmuyBP0WHAeiN1o8OVBy42NTVa1JIBQvzo+5cknapsuPsjRMuVs+Mn5c7SNXDbQtTTl5V\n17T2OQbzU1Qrpsf5ft2e4XKoP90//ZuzjQcuNDV36sKV4gV5UWvnJA51QsepYpYyEELIyWK1\nJ09HP7kByCWCjfOHlm8CAPBEj878nssmQZpm/rnt6uzsyP7/ESzMj16Yjx5eAAAwspDgGDM0\nWvMv3zqjNVyvMDeabf/+qkQuFWTEKVmfIhPzY0KvNyc/WsjW0pIhR660uCY4MuOVbz+z8P29\n5Vdru0wWOjkq6I4lqdkJwT74SiYkg9n65L9O1KmvpSR6dOYdJ+rOlra/8sS8s2VtJ662tmmM\nUSrJTdNiPWnqIRHxXn5i7qeHqo5eaVV3G2JCJcumxHqRTfAtiqJWzYhbNWNYSRbHjv2O+gxm\nmmY4HHSMAAAIOJerOl0nfxFCurWmquYed+9SAAAAfA4JjjFjz7kGx+yG3edHal57an5esqqw\nutPp0J1LUx3veDv72FpaUqTTefYE01/3EaoQuesBAUO140SdPbth19Kl/+xI1QOrMr1ohCnk\nc7csT9+ynGUq3pgWrhSzxsMUYmQ3AAACk87IPouaEML61gUAYFAWtrQpwKA4g58CgaGmxfn2\nuF+tuo9hmN/fO3X51Fh7Kw2JiPfILdm3zr9h94pSxr675LtdJzaGqWaYEwxzkGGOM0w5IXhT\n4jPnypw3B12Lu2wamuCWTI6h2AZ7LJuKYSgAAAFqgG4ar39V/OePLlysZJk9DwAA4HOo4Aho\nDEPquvVtWpPZRveZ2NMNXA5FUZRMzH/m9vz7V2bUtPaJhdzkSLnEZazmvJwI1nak83IjCaEZ\n5gIh9hyKiZBGhumkqGmEYEa9DxhM7J9u6QxuP/WamFJj5A+vyXprV4nVdn1S8fSMMO8GHgMA\nwJCYLDYv2jPnJIbEhkkb23Wuh+patXWt2sOXWzbMS3p0bbYv1ggAAOAWEhyBy2yjj1Z3dn/X\nkkASxF5/kZMYbP/AO1Qh6p8wymr93MRzFR1FNV2OwfmTohbmRRHS5JDdsDMwTB1FTdwbyz69\nZdeZ+trWPpGAOykpZHFBNGtxgSeiQ6XlbANTYsOkrsEJbuP8pKnpYbvP1De260LkwukZYf1N\n+AEAYIR09Zne3V12ori1T28JVYhWTI/bvDjF80wHj0s9u2Xq798719Kld3fOtmM1M7PCp6SF\n+mjJAAAALJDgCFyFzT3dDg0XU5NCrpS1d2lu6JfB43LuW5Hh4QUFfO5fvj/jm3ONx4taO3oM\nEcGSZVNiFuRFEUIYxrl/x3c6CZmgCY7z5e3P/feSvefl16fqtx2r/dMD0xVSgRdXWzUj7tAl\n5+mnhJBVM+OHtcpxKiFC9sgt+KAPAGA0dPQYH3/lWFevyf7wo/0V58vb//HobB7X073MSZFB\nbz69cP/5xoqmnoMXm/VsdYtHCluQ4AAAgBGFBEeAYhjSqDE4Rrhc6ualqacvNlfUdDEMIYQk\nRQY9vj53SJNNOBxq9Yy41SxzLtxtlJigGyi0BsufP7ro1BqtrEHzz+1Fv75ritPJJott6+Hq\n40WtHT3GyBDJsqkxa2YlcG/siDk5NfQHa7Le/abM3jCJy6HuXJo66ETYAGc02y5VdjR36sOU\novwUlVziTfZn5Ki7Dd9ebGps16nkolnZ4eNyDFCf3nK4sLmhTRccJJiaFpYWq/D3igBgjPlw\nf4U9u2FXWq/Ze65x9VCy8AIep//8fecaWU/o7DWyxgEAAHwFCY4AZbLRVppxCopFvEWz4+++\nKV0l4AUHCQfYjTJ0YkJYNlAQwj7SYuxq7tR/e7GpuUMXqhDNyo5wd8d7sljN2vj92JVWndEq\ndehvojNaf/LqiZrW6/Nfyxo0J6+q//z9GU45jk0LkufkRBwpbG3p1EUES+bmRiREBPnuK/OD\nc+XtL352pf27TJxMzH/4lqwV04Y1JtZzJovtxFV1Q5s2OEhYkKKKC5c5nfD1qfrXdxabLLb+\nhx8frFw9M/7Jjble7zMKQCeL1f/YWtiju1Zn9N6e8tUz4h9fn4OJMwDgufNu2l2fK2sfUoLD\nLjhIqO42uMZVch++bwEAAGCBBEeA4nMpDkXRjHOOgxAilwrSfH1jTFHRDNPKGvftC/nX9mO1\nb+4qsddQfHywas2shCc25Lje8bZ2sbwzI4TYaKZdY5BGXv/+f3qoyp7dsLtQ0bH3XOMql0qZ\naJV08+KUYX0NAaOhTfu79845TvDSGix//7QwVC6amh420q9+uarz+U8u23MrXA61cX7Sg6uz\n7D/Jyqael7cVMTf+Ddp1uj4xMmj93MSRXt7oUHcb/vzhBbPDj4Cmma9O1UWGiG9bNE5+zQBg\nFLBuJ+mPd/YaP9hXcaWmy2CyJkUG3b44JS9ZNegFF+RFbT1c7RpfmI+GSgAAMLIwJjZAcSkq\ngq2rKEVI9Ih8AKKkqLQbfx8oQuIJ8fEGiqLarj9/dPGRF4/+4s3TWw9Xj+aA65J6zas7rjq9\n4len6r46Ve96cpDY7eyYIMkNh04Vq1lPO+kmPm7sPFnH+uP7/EjNSLxcU4fuxNXWi5UdfXpL\nV6/pt++da3fYw2Wjma2Hq7cdu/7Se881Mmz5wW/ONIzE8vxi77lGM9uP4Gu2X2kAAHeiVezt\nrmVi/oMvHN51ur6hTdvRYzxb1v7066c+OVQ16AXvWpqWEad0Cq6flzg5FQ04AABgZKGCI3Dl\nRyu69WbjjTcw6eEypft77+GJoygVIW0MYyBESFFhhLDXiRjMtn0XGmta+3hcTlqMYkl+lIdN\nyN7bU/bfA5X2hxcqOvaea3jhkdnete0cqj1n2e9svznbcMvsBKfgtIwwDoeiXXYJpcYonCps\ne/Xs43t7dWbW+LjhWrcycNxrXX2mf24rOlZ0rcJILODlJoewjt3dcaJ24/yk/j83d7J38m/u\nZJliOEY1dbB/La3dequN9rw1IABMcKtnxpc1aJyCHA5V36bVGZ3/sX1/T9nCvKjIEMkAF5SI\neC/9cM7Xp+pPFas7e42xYbKbZ8WjvSgAAIwCJDgCl0zIuykjoljd16Y1mW20XMRLD5VFjuz+\nVQkhiQM3KKho6vnrp4Xd2mvdyL691LzzVN2zd04OVw7SraOsQeOY3ehXp9a+s7v0x5vyhrFm\nT7W6mV3XwnYnHBsm3bw4xWnBQj73iQ25TmeGK0WsXdMigsdb+xInfDf3zzyuL7s/0DTz7Ltn\nKxwm7BrM1rOlbawnN3fqzVZawOMQQhz7pDiSjVR+0A/EQvYJjgIel8tBdgMAPLVyelx9m3bb\nsRp7Wl8k4N63IuP1ncWuJ1ttzJnS9rVznD8YcMLlUGvnJAx6GgAAgG8hwRHQBDxOQUwAzUSw\n2Oi/f37Fnt0ghDCENHfqX/7y6p/unTbwc49dYenxQQg5UtgyOgkOqYj9ztbdnfB9KzIy4pSf\nHKyqaekTi7iTklT3r8yIVjl/ZrVsamxJvfMHX/3xYS44wOUmhZxj60s3KSnEh69ytqzdMbsx\nMB6X4n+XXpmWEXaQbS7v9IwR7w8yaqakhbLuRpmcFjqO+qgCwIijKPLwmqxlU2JOl7S19xii\nVdLFBdE6I3t9IiGkR+c8csVf+vSWL47VlDdoCCEZccoN85KctpECwNhlHcWd7DCeIMEBQ1Bc\n193eY2QIsd899f+hpF7T2m2IHLBmoauP/f2Qzmg1mm0iAftn0a5OFqtPFas7eo3RIdIV02NT\nPU4ATc8IO3qlxTU+IzPc3VNmZ0fMzo4Y+LI3z4wvqevef6HJHuFwqNsXpYynG2lWa+ck7Dpd\n33bjMGOJiHfn0jQfvkpFk6fZDUJIXrLK3i926eSYvecaL1d1Op6gkou2LE/34fL8a15u5OTU\n0IuVHY5BiYj3/VUZ/loSAIxdKdHylGi5/aFIwGXdqkkICfdpiWJju27bsZpadV+QmJ+XrLpl\ndgKf51ENWnFd92/fPderv7Yh9GxZ+86TdX96YLpr+w8AAJg4kOCYKBjC2Giay+FQxPvPdts0\nRkLYn9+mGSTBEcLWM5UQIhPzPcxuWG30nz+6eLzIXgnSvvNU3V1L07Ys9+iOetnU2L3nG4tq\nuhyD4Urx3Z493R0Oh/rZ5oJlU2OPXmlp1xijVZKlU2Kc3l1ZbfSXx2vPlXf06EyxYbJ1cxJz\nEtnH044hMjH/H4/Nfn1Hsb07Rm5iyOMbcmLD2JvVjTQhn/vAqkz7Qw6Heu7BGZ8frdl9ur6l\nyxAsE8zKjrhvRYZSNhoNX0YHRVF/fGD61kNVO07UdWtNQj53anroQzdnxYT650cAAOOJTMyf\nmRnu2jBbKuINmvr33DdnGl7eVmS1Xfuc9sRV9den65//wcxBB8raaOav/7tkz27069GZn/vv\npXeeWYhR2QAAExYSHGNVab2mprVPIuRlJSgH7n9hZWx9Zq3Jdu1NgIgrDBLIuJQ3W/Td7eYg\nhMjcbACxW5AXxdp6fUGep0Pjvjha45DdIIQQmmY+2FeenxLiydQ6Hpf660MzPz9a/c2ZhpYu\ng0ounJ0dce+KdLnEB3e8U9JC3bVP69GZn/n3qdrvWm9WNvUeutR897K0e24a86UE4Urxb++Z\nqjNaWzr1YUrRSDSLdfdBXHCQYNmU2B0n6kwWG0VRuUnBj96SkxojdzyHx+Xcvijl9kUpNprh\njtM3uwIe565laXctS9MaLBIhD+/pAcCHfrQxt7lTV6fW2iNiAe+Z2wt88v9NQkibxvDK9uvZ\njX4NbdpXvyx+dsuUgZ9bWq9pYWut1dypK2vsyYpHEQcAwASFBMfYo+42vPDpZXvtPY/LuXV+\n0v0rM1jvbWyMrcvYTTvMyzTaTBajVSUK5gx9m35uYoiQzzVZbE7xULkoMVI28HPTYhVblqd9\nsK/CMZgYGfSAx+X0315kaalACNl/ocmTBAchhM/jbF6cunlxKk0zo3Yr+O43ZbUug0U+OlA5\nMyt8jJbRarRmmZhvbyYqFfGcMgs+NC09NCte6drl5O5l6bfMTnhwdWZ7j1EuEQxcBDResxuO\nxlPzVAAIECq56LWnFuw+U19Y3WU0W5Mig9bOSQxV+KzZ+fGiVtZx4yeLW00Wm5A/0D/sHT0G\nd4faNQYkOAAAJiwkOMYYq4359dtn6tu0DhH6k0NVAj6XdaeGzqJ3zG70szE2vdUg4w80442V\nXMK/Z1nam7tLHYNcDvXwzVmepEu2LE+fmh725fHahnatQiqcnhG2dk6C58Msndo9XI93u32X\n4w5FkRNXW8sbezgUlRGnnJEZPnJNGY8UsjT+YBjm6JXWsZXgMFlsnxys+vJEbZ/ewuNS+Smh\nD6/JSoxknyXMymyld52qL67vtlrp5Gj5ujmJg3aDoyjqD/dPf21H8bcXr3U5kYn5963I6J/s\nS1HUoON7JojmTt2p4rY2jSEyRDIvN9KHdyAAMJHxuNQtsxNch6n7REcPywwyQojVxnT1maIG\nnESrlLHveyWEjKetiAAAMFRIcIwxZ0rVjtkNu23Hau5Ykuo6odNMs3dBN9NmQoac4CCErJwW\nmxAu++RIdU1LH5dLZcQq7liUEh9+Q/lGcV3316fqG9u1IXLR9IywldPj7OUS2QnB2Qletp+Q\nS/haA8uXM9SdEepuwx8+OO84myMnMfjZLVPddQkZDrOVZl0zIYR1uGzAYhjm2XfPXqq8Vjdk\ntTHny9ufeKXrxcfmeFi+0dyp+9VbZ5q/G8p7rKh1+7Ha394zZdDqG4VU8Is7Cr6/KqO2VSsR\n8ZKjgsTCifIPV2evsbKpl2aYlGj5AKmcjw9W/mdvhb3M+53dpQ+vyb55VvxoLRMAwBvukhQc\nDqUc7P/s2QnBwTKh41i3fiFyoddvMwAAYByYKPcJ40ZVs/Nmh35ag0XdrXdtLuhSvTFI3BNZ\n8crf3+12c+y735T979tK+8PjRa17zzU+9+AMz29KGYacLlGXN/ZQFEmPvV5eMTsn8vMj1a7n\nz8kZQrczhiF/vDG7QQi5Wtv9l/9dfP4Hszy/jocEPI5MzJ6XGbSDWkA5flVtz27YmSy2t3eX\nPvfgDE+u8MInhfbsRr9evfm5/1567+eLBq5D7hemFIdNpGINi5V+a1fpjhO1NpohhFAUtXpm\n3MNrsl034xwran1nd5ljxGi2/d8XVxIjg8ZBL1sAGMdmZ0e8vbvUdVDL1LTQQd8z8Hmcn3wv\n7/f/OWezXX86RZE7Fqd5XhkKAADjDxIcY8wA3QRYD/E4XJvNuWUGIYTP8XQs6wA6eowf7q8o\nrdeYrbbUGMUdS1J1RotjdqNfcV33RwcqH1ydyXoR12v+8YPzjj0XshOCHl/odAAAIABJREFU\nn90yRSUX3bkk9Wxpm1MBy5yciAV50Z6vubxRU97IMnn0UmVnY7tuJCaALMiL2nW6njXu89ca\nOZdunEVqd7mqg2EYarAdPq1d+qLaLtd4Z6/xYkXHLN815B83/rm9aPeZBvtDhmG+PlWvNVh/\nfddkpzO/PsXy20UI+fpUHRIcABDIYsOk963IeOfGfa8hcuEP1+d48nSZmE9uzI0wDHl/b9m8\nSRFj6yMEAADwISQ4xhh3dyyhClFEMMuWEwlPbJ+f4hQf5kqKart+/dZZg9na/7CxXXe0sCU/\nhX2SyKFLzR4mOP704QWnjpLFdd3P/ffSC4/MCpLw//XkvE8OVp0qUbdrjDGh0pXT41ZMjx1S\n+4ymDp27Qw3t2pFIcNy/MqO4rtupz+hdy9LSYxX9fzaabR8frDx6pbWt2xCtkiybGrthXmKg\nfQBlNLOkyQghVhtjttKDlmC4659CCFG7PzRhdfWZvjnb6Bo/fLn5vhXpToVaje0se9YIIQ3t\nbn/VAQACxObFKZOSQrYerq5T90lFvPwU1R1LUj3smvzxwUqbS/WH1mDZcaLu/pWe9i8HAIBx\nBgmOMSYvWTUlLfRChfPH6fevzGC9zxdyBXJBUJ9Fy3y3KYVDceQCGY8zrB89w5B/bC20Zzf6\n2WjmchX75/wdvUaGIYNmIqpbeovrul3jhdWddWptQoRMyOfec1P6cAasDjBuQzIynR0UUsGr\nT87bfrz2XFm7RmuOC5etm5OQmxTSf1RrsPz41RP2IXw1rX1vfl1y8qr6+YdnBlSOIzaMfUpO\nRLDYkw0mrjMFk2MVi6fHRqikITJBu96sEvO9GOszXlU19TJudpFVNPY4JTjcFXKLhT6o0gIA\nGGk5icE5iVO9eGJFE0s9JiGEtU4TAAAmCCQ4xhiKIr+7Z+q7e8p2nqjr/+BCJRf9YE3W4gK3\n2zQkPJGIKzDZzDaG5nG4Ao5g+HeS9W19jWyfD7t+ltIvWCb05DVZr9mvoV2bEDHIJFpP5CSG\nCHgcs8tcOomIlzliU+V4XM6mBcmbFiS7Htp6uNqe3bArqu3afaZhhLrWe2fZlJj/Hah0SmkR\nQm6ZnXjoUvPRK63tPYaIYPHSyTGs+00SIoKiVRJ7D44VcxNXz0u0H+0wWHpM1gSFmD8B5rl6\nxP23wfXv0ZS0UNc5xISQKWlhPl0TAEBgodz8W4n/kQCMDxYLyxhpgEEhwTH2iIW8x9bm3L8i\no06tlYp40aHSARpz9ONQHDHPl/tRNVqWbS8DmJ8X6clpA5RXDHBoSBRSwd3L0512/BJCHlyd\n6Uklgs+dLmlzFw+oBEeoQvTbe6b87dPLXb3XWtZzONTqmfFFtV2nitX9kdJ6zeHLLcumxDxz\ne4HTfThFkR9vyvv122fMVjoyVLpyjvOXZqEZtc4UG4Rd04QQkh6r4HIo13QhRZHMeOdNarcv\nTjlS2OI0bTE+XLZubgD9/gAA+FxGnOLEVZZ5ZBkj9nEFAAAEPiQ4xiqxcAQrDgYVpnR7Izo9\nM+xsabtjJCky6J7lHm0qyU4IFvK5JotzuweRgJvtcl/ntc2LU6JVkvf3lje2awmhEiNkD6zK\nnJkV7qvrD0mfmyGyffqhpZBGwdT0sHefWXTkSku9WquUCaemhxZWdX11ss7ptP0XmqZlhC+Z\n7FxSlJ+ieuOnC97fUx6sknDYUnJas82TfUwTgUIquGVOwvZjtU7xm6bGRQQ7d88Jlgn/+cS8\nt3eXHrvSajBbpSLesimx965IFwvwzzsAjGebF6eeKW2z2m7IBSukgrUuOXQAAJg48A54vClr\n0Gw7VlvfppVL+JPTQjfMSxLwfN/KIVolzYhTljVonOLBMuGzd0+9XNX51am6pg5dSJBoRmbY\nhnlJfM/WIBPz77kp/c2vS5zi96/MkIh8+bu6IC9qQV6UwWSlKMpXtSHeiQgWt7N12YwMkWi0\n5g/3lxfVdGsNlsTIoNsWJeclq0Z/hY7EQt6KaXH2h69sK2I97fDlZtcEByEkWiX95Z2T1Tpz\nl5ElrcMQYmMY3gTOcKi7DRcrOrq1pv7JAhIh77PD1f3bqXhcasO8pHtXsLfNC5ELn7k9/+nb\n8nv1ZoXUud0JAEBgstpoi432OhubGa/84/3TX9l2tbnz2v7W7ITgJ2+dFCwT+m6NAAAwxiDB\nMa58fLDqvT1l9pHyFyo69pxteOGR2SFBvv+f/TO35//izdOOhfESEe/ndxSIBNyZWeFeF0R8\nb2FyZIj4vT3lDW1aQkh8uOy+lRnzcm/Y4VLWoDlb1t7dZ4pWSRYXxITIvfzq3HVnHE03TY0t\nqmGZn5qfovr+C4f69NcSAW0aw5nStgdWZWxenDq6CxxI53fbVZx09LLUDNvx3OyooqiBpiCP\nA1Ybc+Jqa1Vzr1jIzYoPzk+5IV31wb6Kjw9WWr7rDhMRLH76tvxNC5KrmntphkmOkg+auaAo\nguwGAIwJV6q73tpVUt7YY6OZaJX0zqUpSZHy1m59uFKcHCX38EMRQsjU9LC3nl5Y29rb3mOM\nVknjw2UTOEkOAACEIMExntSp+xyzG/0a23VvfFXyizsKfP5y8eGyt59ZuP1YbXFdt8VKp8Uo\nNsxP8kkmZf6kqPmTovRGKyHEqXCDYZh/br/69el6+5f5n30VP9qQu3RKzPBfd2Ami628oadN\nY4hSSdJjlTyub95DrZgeW9ao+fpUvT3C4VB3LU399mKzPbth9/6e8vmTopyGaPhRsEzQ2qV3\njQ/8axAk4LbriWs3WrmAN47fl9a29v35owuODWVnZUf88o6C/izbrtP1H+wrdzxf3W347Xvn\n3n56oVMeBABgrDt6peWPH1ywP2zu1L3waaH9YVSI5Ecbc6eme9ommcelUmMUqTEKH68SAADG\nJiQ4xo+jV1pptiEmx4tabTQzEp+NiwW8O5aMVEEB656U7cdrd97Y9MFgsr7w6eXUGIVPZqy4\nc7qk7ZVtRW3f7SWJD5f9eFNeTqIP2oJQFPXkxklLJ8ccK2pVdxuiVZIlk2PCleIP91e4nmyj\nmdMlbRvnJw3/dX1i3qSoknrnbUqEkAV5UQM8S8DlREgFap3Z8ZdVyOWEu4ySHTcsVvp3751r\nuTEZdKpY/c/tV5+5PZ8Qss2l3QYhxGCy7j3XeOfSAKrZAQAYJoZhXttRPMAJLV36Z989938/\nnJMWi5wFAAAMme+7M4C/dLnZL2Cy2LRuOlmOObvPNLgGbTSz9xxL3FdK6zX/7z/n2hw6ZdS3\naX/51ukWtuIF7+QmhTxyS/bv7pn60M1ZKdHyXr2ZYZ+3O+T5NSNq/dzEglTn+oK5uZHLBiuo\nCRbxk5RipYgn5nFlAm6EVJCkFLvbujIOnC9vZ/1tOXipSWuwMAzT0O48KrhfrZpl/isAwNhV\n36Z1mvrkymqjPzlUNTrrAQCAcQYVHOOHu1YUQj5XJuaP8mI81KMzyyUCz3fMNnfoWOON7exx\nn9h6uMqpSTshxGi2bTtW89janJF4xZAgIY9Lub4oISQ8OIAGqfJ5nL8+NHP3mYYjhS3tPcbI\nYPHSKbGLC6I9+YEKuZwo6UTpA9fg5vfTamOaO/XpsQo+l2OinYcHEUJGokMwAIAfGUws/9a5\nKm/sGemVAADAuIQEx/gxLzfyw/0VrrtU5uZGBlrvRq3B8v6e8v0XGnVGq0jAnZcb+f3VmSr5\n4LfuYiHPbGUpYZD6dMaKk8qmXtZ4xYi9/RILebOzI45eaXWOC3hzciJZn+IvFEWtnhm/ema8\nvxcS0AaY1CMWcAkheckhZ8vaXY+iAQcAjDNRKgmHQ7HuqHUUWO9aAABg7MDHg+NHYmTQfS5T\nJGNCpT9Yk+WX9bhjMFuf+teJL0/U6oxWQojRbNt/oenxl4939bFvsXE0LYO965i7uNd0Rmtp\nvaasQWMwW93VI1AuB3r15uK6bnW3wd0GE8/9cF1uQkSQY0TI5/7ke3kjMRAHRlpBisr1t4UQ\nEqYUx4ZJCSH3rcgQ8p2TIOmxisUFI949FwBgNCmkgvmTBs/UZ8QrR2ExAAAw/qCCY1zZvDhl\ncqrqi6M1dWqtQiqYnBa6YV6i642Tf+08UVff5txxoLPX+PG3lY+tG2THx703pZ8ra+/R3VDE\nkZesWpQfTQgxmm3bj9eWN2isNJMaLV8/L1E+9L6VVhv94f6Kzw5Xm600IUQs4EWHSVjPzHJ4\n+9XRY3x9Z/GRwpb+hwkRQU9syMlL9v7j9xC58LWn5u8+U19Y3akzWJOigtbNTQxXir2+IPhR\nXLhs7eyEL0/UOsUfXZvdn/hIi1W8+NjsV78sLqrtIoTwuJzVM+PuW5Hhq2E9AACB48mNk/r0\nlgsVHe5OEPA4mxenjOaSACAAWawe7WgDcIIEx3iTEaf85Z2T/b2KgVys7GSNO73XoWnmm7MN\nFys7+/TmuDDZ2jkJceGyyBDJa0/Nf2tX6ZnSNq3BEqoQ3Twz/nuLUjgcqk7d98u3zthbl50q\nVn95ovb390yblBwypOX93+dFexxalhrM1qqmXh6Hst5YTyuXCOzTTIxm29Ovn2ruvN5noU7d\n94s3z7zwyKzsBO8nrfC41C2zE26ZneD1FUYZw5CS+u46tVYm5uckBLtrCjMxPbYuOykq6KMD\nle0aA0VRaTHyh27OctyBkhqj+Mdjs/VGa1efKTJEgtQGAIxXMjH/Lw/NPFfefrW222yxSUX8\n40WtFU3Xdn3Ghkmf3DgpOUru30UCAMAYhQQHjDaDycoeN19P0/bpLb9467S9ycWFio6vT9c9\nti5nzayEUIXoF3cUEEJMFptjccpfP77k1Ji9T2957n8X3/v54v5OjWUNmq2Hq+vUfSIBLy85\n5I4lqa69V1u69HvPswxkYQiVECGtU18rPMlOCH7y1kn2piF7zjU4Zjf6WW30B/sqnntwxiDf\njvGisV33wqeXi+u6+x8K+dzNi1PuXJrmeQfZ8c3erKRPbxHwOe7qqiQiHuuAZACAcWZaeti0\n9GvbS+9YklKn1rZ06cMU4qSooEBrHAYAAGMI3knDaIsNk9pvgx3FhUntf35zV4lTC0+rjXn1\ny6sFKaGx353meItYp9aytgLt6DEWVndOSw/bdqzm31+V2LualTVo9p1v/Psjs+PCZY7nl9Zr\nWNtn2Gj6kVuyw5RidbchJlQSFSJ1vG8vYftyCCFXa7tY4+OPyWL75Vun1d0Gx8j7e8vFQp69\nzgX6BUkCdKQRAIC/UBSVGBmUGBk0+KkAAAADQpNRGG03z2Lfc7Hmu70YNM0cvtTieoLVxtib\nXDhp7zGwxgkhbd0Gdbfhza9LnXq2a7TmV7YXOZ3MuO8OyhASHy6bnhEWrZI6VSWwznMlhNjo\nAa43rhwpbHHMbth9dqR6gnwHAAAAAADA75DggNGWFa/88aY8seB69RCPy7l/Zca83Gtt1bVG\ni8HMvo2lTcOeyFBK3TYTVcoEp0rUVhvteuhyVWef3uIYSYlm3/RLUZS7Q4QQd1uFk6PkE2SD\nRlUz+yTdjh6jU0dYAAAAAACAEYItKuAHq2bETc8IO361tblDH64Uzc6JiFZd358iEfL5PI7F\nypKSUMrYExnJUfKIYLFrEYFUxMtPCd16uIr1WQxDurUmxy0DCRFBs7IjThWrnc5cOT02WOa2\nZeaqmXGfHanWGixO8dsWTZQm8APsl8ZWagAAAAAAGB2o4AD/CFWI1s1JfHRt9q0Lkh2zG4QQ\nHpeamRnO+qy5OZGscQ6H+vGmvP5moo7BJzbkSkW8UIXI3bNCgpzTFj/fXLB0SozjOWtmJfxw\nfe4AX0uwTPjcgzMSIq6385CKeD/amDt/Evtqx5+cRPZhMfHhMrScAAAAAACA0YEKDghED9+S\nXdqgcZqKcvuilLRYhbunTEkLfeOnCz/cV15cp6EZJi1GfteytP7NI7OzI/69s8RkcR6mPT0j\nzHWQilTE+/nmgruWplU29XA4VHqsIjJEMuiCM+KUrz21oKimq6Fdq5ILcxJDFO53zYw/s7Ii\nsuKVJfUap/j9KzP8sh4AAAAAAJiAkOCAQBQRLH7jJws+Plh1oaKjV2dOiJBtmJ9knyfnTrRK\n8rPNBa5xlVz05K2TXvys0HHbS7RK8sQGt3UZsWHS2DCpu6OseFyqIFVVkKoa0rPGBw6H+tMD\nM97aVfLN2cb+xqoRweJH12bPcVNxAwAAAAAA4HNIcECAkon5D67O9NXVlk2JyYpXfnG0pqa1\nTyLk5SWHrJub6DhoFoYpSML/8aa8h27Oqm/TyiWCaJWEg+4bAAAAAOAVq4WlHx/AoJDggIki\nJlQ6QMkG+IRMzM9OYO/HAQAAAAAAMKLQZBQAAAAAAAAAxjxUcACpaOwpb+xhCJMWo8iIU/p7\nOQAAAAAAAABDhgTHhKY1WP6xtfBYUas9Micn4qffy8doTwAAAAAAABhbsEVlQnv+k8uO2Q1C\nyImr6r98fNFf6wFXDKO3MS1Wut7GqBli9vdy/I8hjI1xnvgLAAAAAACACo6Jq7lTd6pY7Ro/\nW9pe36aND5eN/pLAiZVutDFqQhhC+v/TxOPEc6lQPy/LTyy0WWvptdAWQghFKAlPKuHLKIJZ\nLQAAAAAAQAgqOCay2tY+d4dqWtweglFjYzpsTOu17MY1tJWuYxi939bkP2ba1G3q7M9uEEIY\nwuis2h5zt39XBQAAAAAAgQMVHBMXj+s2vcXnBW7mq6VLf/hyS2uXPlwpnp0TkRQZ5O8VjRSa\naWcLMzamnUcljPZq/E1rYUm6mW0mM20ScISjvx4AAAAAAAg0SHBMXJnxSh6XY7XRTnEel8pO\nCPbLkgb1xdGad3aXmq3X1vzBvvLvLUx5YFWGf1c1QhjGxB4n7PFhvBAxW21CPte3l/UhhmGs\n39VuODHbzEhwAAAAAAAAQYJjIpNLBLctSv7vgUqn+KYFyUqZwC9LctLRY6xq7uVyqeQoeUiQ\nsLC68/WdxY4n2Gjm44OVSVFBiwui/bXIkcQlxOom7hsNbdo3vi65VNlpstgigsUb5yetnZPI\n5QRcVwvmhn06NzDZjAzhiLkiHidwEzQAAAAAADAKkOCY0O69KSMkSPT+3rI+vYUQIhPz71me\nvm6u/7c/GM22f39VvPtMA00zhBAel7NxflJ7j5H15F2n64ea4KhT9311sr6hXSuXCqalhy2f\nGkNRAXdXz6HkNrZdKhxK7pPrlzVonn79lMlybSKJutvw2o7iq7Xdv7l7ik+u7ysmi61OrZUH\nUxwOS5rDStMWWq+z6IMEMilP7PlltQbLN2cbalv7JCLepCTVvNzI4fwK0DRT09rX2qUPVYiS\no+SBvMkLAAAAIPBZrM5l5gCeQIJjQqMosnZOwppZ8S1deoYh0SoJJzA+vf/bJ5eOXrk+v9Zq\noz89VBUSxL4ToalDN6SLbztW88ZXJTb62t3yoUvNu07X/3/fnyERBdZfBy4nirZpGHLD1gyK\nkvpqisobX5fYsxt2RwpbLld15qeofPISw/fVqbp3vynr01tuXRy1YVGU6wnMd52S+8xaAYfP\n53j0Q7xQ0fHcfy/26K6N3d1+rHZScsj/u3eaTMz3YpHljT0vfV5Y2dTb/zBaJf3RxtwpaRN0\n2A0AAAAAgL/gY0YgHA4VEyqNDZMGSHajoU3rmN2w6+5j7z0hFQ3hprS2te/fDtmNfsV13e/t\nKRvSIkcBRQR8bjaXUvXvSaEIn0tFCTgZxBeDUU0WW1EN+wiS8+WszU39YPvx2pe/KOovL9p+\nuHX/2Xba4QfHEIphbtiWYrCy1/g40Rosf/rwgj270e9KdderX171YpFtGsPP3jhlz24QQpo7\ndb9552xFY48XVwMAAAAAAK8F1kfWAISQiib2O0N3bRimZ4R5fvFDl5tpmuVKBy42PbYux/Pr\nDF+bxvDR/oqyhh6aYVKj5XcuTYsNkzqdQxE+j5PEI4QQ2rfpSKPZxjDs31GdkbXxx2ijaeaj\n/RX2hzaaee+rhgNnOrISZavmRoYqRAwhTrkeG+NckMLq+NVWrYGlZenhwpYfbZwkEgytl8f2\nY7V6l++Y1UZ/cqgq0Db7AAAAAACMb0hwwFgSpZK0dOodIxHB4tsXp3h+hXYN+4f8fXqLwWwV\nC0bpb8Slys5n3z1r3yFS29p36HLLb+6ePCcn0s0zfFxsJZfw5RJBr97sesg1z+IXLV16pyIL\nQkhDm6GhzaCUi9fMY2m3waE8+i45/QrZWax0u8YQFy4b0jrL3VRqlDVohnQdAAAAAAAYJmxR\ngYCTHqtkjfO41AsPz7p9UYpKLiKEKKSCW2Yn/OvJeRYrvet0/X/2lu851+B6S+zE3YAYkYAr\n4o9SdoOmmb9vvezU/8Jqo1/87IprU4wRQlHUyhlxrnGJiLe4IGZ01jAwN/UlhBDCc5PIEHI9\nmv4zQKMNmcSbHhysArBtLQAAAADA+IYKDgg4sWHSxQXRBy81O8XXzU0KU4q/vzrz+6szTRab\nkM8lhGw/VvvON6VG87W8gEzMf3x9zpLJbm/R5+REbj1czRoftRvSiqYedbfBNd6jMxdWdw1p\nx81w3HtTepvGcMjh+xwsE/5sc36ADAmODBHLxHzWvSQ58WFciuu0IUXEFYq47G1onUxLD3uT\nKnXdoZMWqwiWeXQFRxlxisLqTtb4UC8FAAAAAADDgQQHBKKffC9PIRPsPFHX3w1UwONsWpi8\nZXm6/YT+7MbpkrZXd9zQGFJrsDz/yeXYMFl6LPvtZU5i8Pq5iduP1zoGw5XiB1dn+vyrcEej\ndVtmotGyN1JlZaMZk9nm9fAXPo/zqzsnr52dcLGyo09viY+QLS6IkQbMKBkel7NpQbJr89eM\nOOW09DBCiNaqN9lMNobmUVwJTyzmiTy8cmJk0IZ5iV8crXEMCvncH3rVhGX93MTdZxqcEjF8\nHuf2RaleXA0AAAAAALwWKDczAI6EfO5ja3PuWJJa09LHoaiUaHkQ296BHSdrXYM0zew8UffT\n2/LcXfyxdTlT0kK3H6+tb9MqZcJp6aGbF6eO5ozY/i02rEIVHt2lV7f0vvFVSWF1l9VGhypE\nmxYkr5ubyPVqCE5uUkhuUogXTxwFdyxJsdHMp4eq7Dt3ZmdHPLlxUv/ujyC+NIjvZbuQR27J\nzopXfnKoqrZVKxZy85JV31+V6V3zkTCl+PkfzHzx8yv2sSkxodIfbcxNjZF7tzYAAAAAAPAO\nEhwQuIJlwuC0gbYM1Ku1rPG6tr6BrzwrO2JWdoT3KxuelGh5fLisvs158WFK8aSkEEJIm8ZQ\n1qAxWeikyKCUaOf75KLarp+/cdpipfsfdvQYX99ZXFKv+fVdk4e5sPLGnjp1n1TEz4xXhgQN\nebOGz1EUtWV52prZ8eUNPXqTNTkqKCEiyFcXX5gfvTA/mmGY4TfLSI1R/POJuTWtfa1d+jCF\nOClKzuOiAQcAAAAAwGhDggNGXGuXvqxBYzTbkqPkaW52jninf6OK5/EAQVHk55sLfvX2GceW\nqFIR7+eb8ymKevPrkm3Haqy2ax0iZmSG/2RTXoj8errh3ztL7NkNu8OXm9fNSfC6FqO1S//3\nrYWXq671khDyuXcsSb1jSWogNMoMlglnZoWP0MV91QqUoqjkKHlyFKo2AAAAAAD8BgkOGEFW\nG/PWrpIvj9f2t9IghKRGy8OUYo3WFB4sWVwQPSdnWGUU+Skq1zoIQsjk1NDhXHYUpMUq3v3Z\nos+P1pTWa2iaSYtV3LogKVgmfGtXqVMP1DOlbb97/9zLj8/pvxXXG63ljezzR89XdHiX4LBY\n6V+9faaxXWePmCy29/aUiQTcjfOTvLjgGMKQXprpYIiRIkKKhHCoYH+vCAAAAACIdbRmC8I4\ngwQHjKB3dpc6tXKsbO6tbO4lhJTUaw5fbl5cEP2LOwq8/hT9jiWpR6+0OPXsjFZJ1s1N9HbJ\no0cm5t97U7pjxGSxfXlj99N+ZQ2aS1Wd/Vkbg9nqbn6qwWT1biXHi1odsxt2nx6q2jAvcRyP\nO7UxtTTT2v9nhhBC2hkSwqXSCBm3XzIAAAAAwDjG8fcCYNwymm07T9YNfM7BS837zjd5/RKh\nCtHLj8+dkxPZ31+Tx+Usnxr74g/njNooEIuV3nOu4fWdxW/vKj1xtdVd6sFDTR06k5tcdWVT\nb/8flDKhTMzSb5UQEhcm8+51q1t6WeNdfabO3iFMdRlbaKbbnt1wCHbRjNov6wEAAAAAgGFC\nBQeMlAFu1x0dutR807RYr18lMkTy+3unWm10Z69JJReNZnPH6pbeP/znQnPn9dqHnMTg3987\nTSEVeHfBAcag2A9xOdTK6XGfHal2OkEuESzIi/LudTnuX3eAQ2MdQzpY4zTTwaEiR3kxAAAA\nAAAwfKjggJHi4dTS9h7jAEd7dObtx2r/ub3ovwcqyxrYe08QQnhcTkSweDSzG1Yb/Yf/nHfM\nbhBCrtZ2/2NrodfXjAmVsU7DJYTkJF7vDXH/yoxFBdGOR0MVot/dM9XdcweVFc/eeCIqRBII\ns1RGCMOY3RxxFwcAAAAAgICGCo6JhiG6MsbURGgT4YdQskmEKx2hV4oNkwVJ+H16y8CnDXAL\nfaSw5aXPr2gN167wn33lq2fEP74+JxDKCi5WdjZ36l3jp0rUXb0mx6EnnuNxqS3L01/98qpT\nfE5OREac0v6Qz+P86s7Ja+ckFFZ19erNiRFBiwqiRQLvB8fMyAzLTggurut2it+3MsPrawY+\niuK52VLkZZ4IAAAAAAD8CwmOiYQ2Mm07iNneYqCK6b1EqZYTScpIvBqPS91zU/q/tjvfrjuZ\nn8e+HaCpQ/eX/12y2q7PQ6Vp5qtTdTGhklsXJPtyoV5p6mDpykkIYRjS3KnzLsFBCFk/N1HA\n47z7TVn/BFkel1o7J/G+FSyJhtzEkNxEL4fCOqEo6o/3T39zV8mes40MwxBCVHLRD9ZkLb6x\nTmScoUgIIc45HUIIBqkAAAAAAIxRSHBMIEz3UYfsRn/IwnTuo4QjpjOXAAAgAElEQVSRI1TH\nsW5OoojPfWtXaf/tOkVRzI0fmk/PDFs1I571uXvPNTpmN+y+Pl0fCAmOAfqYSobX4nT1zPib\npsXWt2lNFjo+XDY6DVODJPyfbMp7cHVmvVorFfPjwqQ87jjfv8ahQmnSyTA37HuiiIxDjee0\nDgAAAADAOIYEx4TBWIm+gi1uIfoqEpQ3Qi+7Ynrc0imxTR1ao9kWFy49crn10OXmNo0hXCle\nMjlm+dRYd0NIndpbOMT1DMP4fXZpfoqKy6FstPMmh1CFKDEiaJgX53E5yVHyYV7EC3KJIDfJ\nN1UhYwHFozJp0kYzHYQYCRFwKBWHisSMWAAAAACAMQoJjgnDpicM+0wTxtY7ord0PC6V8N09\n/8oZcStnxHnyLJGA/ZdTJOD6PbtBCAlXijcvSf1o/w05Iw6HemxdQLQIGSq90drarQ9Xit3N\noB2vOFQ4hwr39yoAAAAAAMAHkOCYMDgiQihCWNoqUhzx6C9nUNPSQ/ecbWCLh43+Yljde1N6\nYkTQh/srGtq1HIrKjFc+uDozO2GMdXBQdxte23H1xNVre5cKUlU/XJebECHz8OlWG11Sp2nt\n1ocpxFkJSiHf+16nAAAAAAAAw4EExwRBE3M1kagIYyKMjZhNhLZXc1BEnOjHlbkzf1JUfkr9\n5apOx6BMzL8/kEZ7LMyPWpgfZbHSHA7l4VjcgNKjMz/1rxOdvdcn9V6q7PzxqydefXJeZIhk\n0Kdfrup86fMr9n6roQrRjzbkzsqOGKnlAgAAAMDEYLGyNOMDGNQ47yMIhBDCmJie3Yz+HOHY\nCJdHeEIikROB6NpR+RTCV/l1few4HOrP359x34qMUIWIECIW8hbmR7321PyY0JGaa+s1Po8z\nzOwG01lPn/2M/vZV+vTHTGu5rxY2qG3HahyzG/20BsvHB6sGfW5ju+4375x1nCbT0WP8wwfn\nyxo0AzwLAAAAAABghKCCY/xj9BeJzWUcpkBMOEoqaBoRJ/ljUR4R8Dh3Lk29c2mqwWwVC3iE\nEKuNqW/TcjlUVIhkLLa6YEVf2M4U7ibMtSw1U3yAyljAmX03GflWI0U1LHNSCSFXaroGfe72\n4zUmi3NXF6uN2Xq4+jd3T/HB4gAAAAAAAIYCCY4JwFzHHg9KC+TshiOxgEfTzGdHqj86UGkw\nWQkhcong3hXpa2YlBEC/0WFhGouYy187B8uOMKFJVPq8kX511kG8hBCrBzWBlU29buI9w1oT\nAAAAAACAV5DgGO8YK2Es7Idow+guZVhe21H85Yla+8NevfmVbUW9OvNdy9L8tygfYCpPsMer\nTo5CgiMxMqi4jqWIIylq8Em37ipodCbrK9uKwoPF83IjR20/kdXG7DhRu+dsQ1OHLlQhnpMT\ncdeyNKkI/74BAAAAAEwg6MEx3lE8Qrm5zaNE7PHA06Yx7DjJUofyv28r9Ubr6K/Hl7SdrGGm\nr2MUXnztnAQelyVPsXH+4KU9WfFK1niP1rzzZN3bu0of+vuRTw8N3stj+Gw086u3T7++s7im\ntc9spZs7dZ8dqX7spaM9OvMovDoAAAAAAAQIJDgmAEE8a5gSJIzyQrxWUtfNMCwDbs1Wuqxx\njLe0FLLXOFBu4r6VHCV/dsvUELnQHgmS8H+2uSAvefC+sxvmJcklggFOsNrot3aVni1t98FC\nB3TgQtOlSuc8UUuX/qMDFSP90gAAAAAAEDhQwj3+UeLJjLWD2G7smCCeRHghflrRkFltLNmN\na4esbg+NCVRcHtN4heVAXN7oLGB2dkRBiupCRUdrlz48WFyQEhok4XvyxFCF6IVHZr38RVFR\n7UAdSXedqZ+eGeajxbI7U9rGHi9pe2xtDiFEZ7Reqe5UawzRIdK8lBAhnzui6wEAAAAAAL9A\ngmMC4Igp+c3EWMJYWghjIFwlJcwg/Ah/L2sI3LWEoCiPukUEMip9HlV9hlHfUGtABcdwJq0Y\ntTWIhby5uZFePDExMujvj85Wd+ubO/WvbLvS1KF3PaexXeca9C2tgb3LTK/eQgg5eKn51S+v\n2rerhCvFT22aNC19ZHMuAAAAAAAw+rBFZWKguEScS8mXU/JVlDiHcLhuO48GpOQo+eTUUNf4\novzoUMWY6STCjsPjrPwpZ/omKjiW8AREGUUVrOGs+SXhi/29Mo9QFIkMkUxJCw1ys13FtdOn\nzmitbOrVaH3WICMyRMIaj1JJCqs7n//4kmMzjjaN4Xfvnatv0/rq1QEAAAAAIECggmPioBlz\nGbFUEmIjhBDCIfwUSpA1VpJcv7pr8t+3Fp4qVtsjSyZHP3nrJL8shqYZhhCumzEiQ8bhUrkr\nqNzRK9kYCVPTw0rrWfqhTMu4XivR0WN8fWfxkcKW/oeZ8con1uemxSqG+dLLp8buOl3vGl8x\nLe6LozU22nkTk8VKf3m89okNucN8XQAAAAAACChIcEwUjLmIWKodAjSxVDCMmRJO9tuahkIh\nFfzhvmllDZrKpl4Oh8qMVyZF+mFzyuWqzvf2lFU09tAMSYmW37siHZsd+t06P+nQpeamjhs2\npMSFyzbOuzaQRW+0/vjVE+ru68OJS+s1P3nt5CtPzE0c3o8yJzH4oZuz3v2m1LFXy4ppcWtm\nxX92pJr1KVXNvaxxAAAAAAgEFovN30uAMQkJjomBMRFLDUvcWkcEmYQaG7shCCEZccqMOPbp\npKNgz7mGv39aaH9Y1qD51Vtnfrg+Z92cxOFclmHIsaKW8+UdPTpzXJjs5lnxEcFj5idiJxPz\nX3li7n/2lR+82NyjMyukgiWTY7YsT5N8t0Xlq1N1jtmNfiaL7YN9Fc9umTLMV//ewuRZWeH7\nzjc1d+pCFaLZ2RH5KSpCCOsQXEIIjzs2CpcAAAAAAMBzSHBMDLSGEDfTRmzdhDf2bqdHn8li\n+/fOEtf427tKl06OkYk9mjziymC2/v698xcrO+yRbcdqHt+Qs2JanJcL9R+ZmP/Y2pzH1uaY\nLDbXSSVFtd2sz7o64BAWz8WFyx5YleEUzEkIYe1ympsU7JMXBQAAAACAwIGPMSeIAWapju0x\nq6OmtF7DOq3DaLZddXPr7on395Q7ZjcIISaL7f8+L2ruHPHhIyOHdQ6rzUaznmxxE/eJzUtS\nJC5dTkPkwg3fbZwBAAAAAIBxAwmOiYGjJMRNR0yO33Z8jC16k9XdIZ3R+5E0+883uQatNvrg\nxWavrxmY3DXaSI6SD/VSNsaqt/b2Wbp01h4rPdA0lphQ6T8enZ2TeL1eY3pm2D8ena2Qss98\nAQAAAACAsQtbVCYGSkR4ccTqMmmCF0s4Un8saOyJVrn9RsWEevk9NJisvXr2+/PWLud2FWPd\nmtkJO07UmVz6RW1akDyk6xisWr21h/mu8shA+kRcmYzvNk+XHCV/8bE5XX0mdbchJlQidzPO\nFgAAAAAAxjpUcEwUlDCf8JMd6jgowkscKyNUAkFChCw7gaVxQ0q0PN3bQaciAVfAY/87KJd6\n2dQjYEWFSP54/3TH/qlSEe+pWyfNzAr3/CIW2qSzapgb91UZbVqjbZAdPSFBwqx4JbIbAAAA\nAADjGCo4Jg4uJcgj/DRCawjDEK6SUBJ/L2mM+eWdk3/33rnqlusTRuPCZb+5ewpFudn+MxiK\nomZkhh8ranU9NDs7wstVBrCCVNVbTy+8VNnZ1KELVYjyU1RD3SpisulZ40abTsRFLRIAAAAA\nwISGBMcEQ4kJFzNTvBQRLH71yXmHLjeXNmgYmqTHKRYXxLgbROqhB2/Oulrb3a01OQZXzYjL\nTQoZ3mJHnNVGHy9S16r7xALupGRVVrxHzVyEfO6QSjac2Bj2Tii0mzgAAAAAAEwcSHAADAGH\nQy2ZHLNkcoyvLhitkrzx0wX/2Vt+vryjR2eKD5etn5e0KD/aV9cfIVXNvX/+6ILjBNZFBdE/\n/V4e6/wUH6Io9h09FHbbAQAAAABMeEhwAPiZQip4YkOuv1cxBCaL7bfvnWvX3NAG9dClZrmE\n//j6kf1CBByR2cbSflXAEXl4hctVnR8frKpt7RMJuLmJIfeuSA9VePpcAAAAAAAIZEhwAHip\nR2f+9FBVSb3GYqVTouW3L06JCpkQbU1OFaudshv99pxtfOjmrBEt4hBxJSabznLjaFguxRPz\nPJo1+8mhqrd3ldofNnXojlxp+dvDs7xuEwsAAAAAI8Fqpf29BBiTUNcN4I2Kxp4H/nZo6+Hq\nopqusgbNrtP1D71wmLVd6PhT38Y+ssRksbV1j/R0W0ouCJPw5FyKRwjhUFwRV6YUhHPcbF1x\npO42vL+nzCloMFlf/uLKiKwUAAAAAABGFyo4ALzxwqeX+/QWx4jZSv9ja+GU1FCJaJz/tRIJ\n3NZoCN0f8hWKUBKeXOJZyYajM6VtVhvjGi9v7OnoMWKjCgAAAADAWIcKDoAha2jT1rT2uca1\nBsuFyo7RX88oK0hRscZjQqXhysCd0dOrM7s9pHd7CAAAAAAAxgokOACGrKvP5PZQr9tD40Za\nrGL51FinIJdDPXJLtg9fpaq598CFpqNXWln7fXghPJg9+UJRVJgicPMyAAAAAADgoXFeSw8w\nElRyt9sZVHLhaK7EX37yvbyUaPnHBys1WjNFkbQYxcNrsiclh/jk4l29phc/Lzxd0tb/kMel\nNsxLemBVJpdDDeeys7IiZGK+1mBxis/MCg+S8IdzZQAAAAAACARIcAAMWWyYNDlKXt3S6xSX\nSwRT0kL9sqRRxuVQG+cnbZyf1K01iQW8AbpyDBXDML9972x5Y489YrUxWw9XcznUA6syh3Pl\nIAn/Z7fn/+XjS3qj1R5MiAh6cuNYmtELAAAAAADuIMEB4I2nb8v/xZunHXs3CHicn3xvkljo\nn79TJoutXq0V8LkxoVIed1iVDkMSLBtyxQrDkHPl7VXNvQIeJyshOCte6Xj0YmWnY3bDbvvx\n2ruWpQ1zBu2s7Ih3nl6042RtTUufUMCdlBSyakb8aH67AAAAAABg5CDBAeCN1Bj5Oz9b+Omh\n6pL6brOFTotR3LYoOTJEMvorMVvpD/dVfH602mKlCSEKqeD+lRmrZ8aP/kocVTT2HC5sUXcb\nIoLFC/Ki0mMV/fHWLv3/99+LpfUa+5nzciOfuT3fnhiqanaui+lnNNuaOnTJUUMenuIkRC68\nb0XGMC8CAAAAAAABCAkOAC/JJYIHVw9r04RP/O2Ty4cvN9sf9ujML31+xWi2bZyf5K8lvb2r\ndOuRapq+NpP1syPVt85PeujmLJpmfv/+eaetPceKWgV87i/uKOh/OECjjWH24AAAAAAAgPEN\nCQ6AMay6pdcxu2H3wb7yW2Yn8Hnej0kymK1dvaaIYEn/Do6jV1pOFbd19ZmiVZKV0+PSvqvI\ncHX0Susnh6ocIzTNbD1cnRmvlEsEro1LCCGHLjc/ujZbIRUQQrITglkvK5cIYsNkXn85AAAA\nAAAw7iHBATCGFdd1s8Z1Rmtta98AaYgB1Kn7XttRfLGyg2EIj0stmRLToTFeqOjoP3qekK9P\n19+1NG3L8jTWp+8738ga33uucUZmOOshmmYa2rSKpBBCSGa8clZ2xKlitdM599yUjgoOAAAA\ngAnCYqH9vQQYk7z/gBcA/M723TaQIR0aQH2b9kf/PHGhooNhCCHEamP2nm20Zzf60TTzwb7y\nopou1iu0dulZ4y2degHf7T84Aofuob+6c/LG+Un23p8KqeDHm/LWzkkY4pcCAAAAAAATCyo4\nAMawFDdNN/k8Tny4Nxs6PthXYTBZBz+PkAMXm3KTQlzjMjGf9XyZmD8pSUVRFMM4Z17kEoFj\n91C90XrbopQty9Pr27RCPic+XMbjIhULAAAAAACDQIIDYAzLSQzJTgh23ahy86x4icibv91X\nqjs9PLNNY2CNz8gMZy3umJUdHq2SrJuTsP14rdOhB1dn8rgUw5C95xre21Pe2WskhIQqRPev\nzFg+NXYIqwcAAAAAgAkMn4sCjGEURX57z1TH3hYcDrV2TsIPbs7y7oJmq6fbHZVSIWt8/dzE\n1BjnupKUaPn6uUmEkEduyX5sXU5/P1FCSLRK+rt7pq6cEUcI+WBf+d+3FvZnNwghHT3Gv31y\n+eODVQQAAAAAAMADqOAAGNtCgoR/emB6ZVNPRVOvkM/NjFdGqyReXy0hQna1lr1xqZO5uRGs\ncZGA++Jjc7Yerv72YpO62xARLF5cEH3bohQhn0sI4XCo9XMT189NbNcYBHyuPdPRozOz5jI+\n2l9xy+wEqVfVKAAAAAAAMKHgtgFgPEiNUaTGeDMzxcn6uUmuCQ6KEKe2GQvzo2dnR7q7iJDP\nvXtZ2t3L2Mes9AtTih0fFtd1W20sxSMmi62kvntaethgCwcAAAAAgIkOCQ4AuG5hflR7T9b7\ne8pNFlt/RCUXPb4hp7q571SJurPHGBMmXT0jfsnkGMqnM1vN373ckA4BAAAAAADYIcEBADfY\ntCB5UX70+Yr2Do0xNkw6IzNcLOTNzYncsnygioxhio8Icncowf0hAAAAAAAAOyQ4AAKUutsg\n4HOCZey9PEdUqEK0YlrcaL5iUmRQforqcpXzDJfpmWExodLRXAkAAAAAAIxRSHAABBaaZr48\nUfvR/spevZkQEhkieejmzPmTovy9rhH3q7smP/ffi5cqr+c4pqaH/ez2Aj8uCQAAAAAAxhAk\nOAACy6s7ru44UWd/2Nql/+MHF57cOOnmWfF+XNUoCJYJn//BrMtVnWUNGoqiMuOUk5JD/L0o\nAAAAAPADqxVd2MAbSHAA+F5Th+50SVubxhCtks7NjVDJRR4+sblTv/NkvWv87d2lK6bH8rgc\nny4zEOWnqPJTVP5eBQAAAAAAjD1IcAD42H8PVH64v8I+9PTtXaWPrs1eOcOjlhZFNV0Mw7jG\ntQZLVXNvRpzSlwsFAAAAAAAYR8b/B8IAo+lIYct7e8rs2Q1CiMFsfemLK6X1Gk+ebnZfjGe2\n0O4OAQAAAAAAACo4AHzpq1N1rkGaZr4+VZ8ZP3j9RUI4+0hUDoeKj5ANd3Gj7nhR695zjc2d\nulCFeG5uxOoZ8RwO5e9FAQAAAADA+IQEB4AvNbbrWOMN7VpPnp6bFJwao6hs6nGKLymIVkgF\nw13cKGIY8vwnlw5caOp/WKfWni9vP3Ch6S8PzRTyuf5dGwAAAAAAjEvYogLgS2Ihe9LQXdwJ\nRVG/v3dqdkKwY3D+pKgnNub6YHGj6FhRiz27YXe1tvvTQ9XeXVBntF6s7Dh8ubm6pXfYqwMA\nAAAAHzh69OjDDz+clZUllUpjY2M3bNhw6dIlfy8KJjRUcAD40uRUVUMbS7GGutvQ0qWPCpEM\neoVwpfjFx+ZcrOyobOrh8zjZCcFjsbfokcJW1vjRKy1blqcN9Wq7Tte/tatUa7D0P8xLVv14\n06SYUOmwlggAAAAAw/PXv/61vr7+tttuS09Pb2pqevnll2fOnHngwIF58+b5e2kwQSHBAeBL\ndyxJPV6k7uw1OsUb27U/+/epN366QCwY/C8dRZEpaaFT0kJHZo2jobvPxBrv6mWPD+DAhaaX\nPr/iGCms7vzFm6ff/OlCkQC7XQAAAAD85qWXXkpNTbU/3LRpU2Zm5vPPP48EB/gLtqgA+JJK\nLnrlibkKCd/1kLrbsO+c866N8UolF7LGQxUix4c0zTS26y5UdDR36tnG4xJCyMcHK12D6m6D\n6xYYAAAAABhNjtkNQkhycnJiYmJzc7O/1gOACg4AH1PJhTqTlfVQWYOGkIRRXo9fLMyPOniJ\n5f9tC/Oj7H8urut++Ysie0+NnMTgH22clBR5wxwZs5WuU7P3Z61wacUKAAAAAH7U1NRUW1u7\nZcsWfy8EJi4kOAB8z10xAuPuwLgzJydy9cz4XafrHYOTU0NvXZDc/+fa1r6fv3HaZLHZj16t\n7X769ZNv/GSBSn69yoMihKLYv58UBs4CAAAABAyaph966CGB4P9v7/5ju67vBI5/+gNaihSk\nA/FAT4pVoS0DwajVwG6RG4PkUn5sYVNuW8a5aYzhZJsSMWcCxGyJLsuZucUNK5B5TuwxUC9h\nfxwbIWXT7Txa3M2ptwwPnBvQWk4KtN/v/fH1vnRQoBu2n774Ph5/9fP5vFtf6NuvX5/9fj/f\n4atXrz7v4s9//vPbtm0729Xjx4+fPJn5UKejUAgc8CErKiq6euLoX+9vP/PSNZPi3S70L7Zy\nSf0tdRN2vLL/f/74/vgx5Q21E+bNmlj0/1niX/79zd51I6fz/ZPNu/77HxZOzZ8ZVlpcfXnl\nmwf6+OSU664soL+ZAADp6unp6ezszB9WVlYWF5+63UE2m7377rt37Njx7LPPnva+lT7df//9\nCxcuPNvVlStXXuC0FCyBAz58n/n41Q8//cppJz8yuvxvZ09KZZ603HDtuBuuHdfnpT4DUJIk\nv/rd6eeXz6t5+OlfnHbyivGX/M2MiRc+IQAA/dHa2jpz5szeh3V1dbmvs9nsXXfd9b3vfW/j\nxo1Llizpz0+bOnXq1KlTz3Z17dq1FzgtBUvggA9fQ+1lD3xmxne2v9Z+9ETuTN3ksfctnV5R\n7t+4D2Qyfb9b58x38TTUTnjw9uu/s/21/GfTNNRedk9j3fBS90gGABgkNTU1u3btyh9WV3/w\nvuNsNnvnnXdu2LDh6aef/uxnP5vSdPCBqP+7tXv37vXr17e0tBw/fvyqq6764he/uGrVqvzV\njo6O1atXNzc3t7e3T5s2bc2aNYsXL05xWgrQx2dOvLn2sjfefu/I0eOTxo2cPKHSPSN6u3ri\n6IOH3z/zfM2k0WeenPvRyxtqL/vtO53t/3viyvGXXHbpiIEfEACAU0aOHHnmh79ms9kVK1Y0\nNTU99dRTd9xxRyqDQW8hA8ezzz57++2333zzzevXrx81atRbb7317rvv5q9mMpmFCxfu3bt3\n/fr1U6ZM+f73v7906dLm5ubGxsYUZ6YAjRheWl89Nu0phqhPza1uee2d7p4/eb1G+fCSRbdO\n7nP9sNLiPtsHAABpue+++zZs2LB48eKKiootW7bkTlZUVCxYsCDdwShY8QLHO++8s2LFisbG\nxh/+8Ie9b2yT19zcvHv37qamps997nNJknziE5+4/vrrv/KVrwgcMHRcd+WYf/r72f/8r23v\nth/Lnbli/CX/uLT+8rEV6Q4GAEA/tbS0JEnS3Nzc3NycPzlx4sS33347vaEoaPECR1NT09Gj\nRx955JHi4uJMJnNm49i6dWt5efmyZctyhyUlJcuXL//qV7+6d+/e6dOnD/q8QN9unDp+xtVz\n/+t37e+2H/urqpHXXjGmtMTbeAAAwtizZ0/aI8CfiHeXvp/+9KeTJk169dVXr7vuutLS0qqq\nqjvvvLO9/dQnL+zbt6+mpqasrCx/pr6+PkmStra2FMYFzq5sWMlHp1TNmzWp9qpL1Q0AAOBC\nxHsFx4EDB9rb27/whS889NBDs2bN2rNnz7p161pbW3fv3p17NcehQ4fyN/XNGTt2bO78uX/y\nSy+9dLYXU3V1dSVJ0tPT8+H8GYAkef3tjl/vb+/uydRMHF032c1KAACACzKkA0dPT09nZ2f+\nsLKyMve2lKNHj37zm99cuXJlkiS33XZbUVHRmjVrduzYMX/+/HP8tKLzfYjFN77xjf379/d5\nKffRlSUlJX/2nwE4w/td3Y8+95+7Wt/Jn5l59UfuXzZjbGXZOb4LAADgHIZ04GhtbZ05c2bv\nw7q6uqqqqiRJereMBQsWrFmz5pe//GXuZFVV1eHDh3v/nNxh7nUc57Bz586zXTpx4kRZWVnu\nrS7ABXpsy97edSNJkv94449rN//isbsafJguAAAnuzNpj0BIQzpw1NTU7Nq1K3+Ye+PJ9OnT\nd+7cmcmc2vG5r/N3G62trd2yZUtXV1d5eXnuzN69e5MkqaurG7TJgbP5Y0fXrtaDZ57f99sj\nr7/dfu0VYwZ/JAAA4CIwpG8yOnLkyFt7qaioSJJkyZIlSZK88MIL+WXbtm1LkuSmm27KHS5a\ntOj48ePPPPNM7rCnp2fTpk1TpkzxESowFPzu3aPZbN+Xfvv7zr4vAAAAnM+QfgVHn+bMmbN0\n6dKHHnqos7Nz9uzZLS0tjz766Pz58z/2sY/lFixatKihoeHee+/t6Oiorq7esGFDW1tb709m\nBlI0rOSsXXWY29wAAAB/qXiBI0mSzZs3r127duPGjV//+tcvv/zyVatWPfzww/mrxcXFL774\n4urVqx955JGOjo6pU6du2bKlsbExvXmBU2omjS4fXtJ14vTPJCouLqqffGkqIwEAABeBkIGj\nrKxs3bp169atO9uCMWPGPPHEE0888cRgTgX0R/nwkuXzrnnyxV+ddn7RrZPHjRmRykgAAMBF\nIGTgAEL71NzqMZcM3/Bvvz70XleSJKMqhi2/7Zq/a/jrtOcCAAACEziAFMybNWnerEm/P3Is\nk81OuHREkY+HBQAALozAAaTmsku9JwUAAPhwDOmPiQUAAADoD4EDAAAACM9bVAAAABhCuk/2\npD0CIXkFBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4Qkc\nAAAAQHgCBwAAABBeadoDAAAAwCknuzNpj0BIXsEBAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQ\nnsABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQnsABAAAAhCdwAAAAAOGVpj0AAAAAnNLdnUl7\nBELyCg4AAAAgPIEDAAAACE/gAAAAAMITOAAAAIDwBA4AAAAgPIEDAAAACE/gAAAAAMITOAAA\nAIDwBA4AAAAgvNK0B4jkRz/6UVtbW9pTDJ7t27ePGjWqpKQk7UEoIO+99142mx09enTag1BA\nMpnMwYMHJ06cmPYgFJY//OEPlZWVZWVlaQ9CAenq6iorK5szZ07ag9CHn//852mPABcDgaNf\nSktLb7rppi1btqQ9yKB66623SkpKiou9zIfB09PTkySJrDp4UEkAAAv1SURBVMZgymaz3d3d\npaWlRUVFac9CAenu7i4uLvYfWQZTJpNJkqSlpSXtQejbJz/5ybRHGCpuuOGGnc/fm/YUXJC0\n9nNRNptN5S/MEJfJZEpKSn7yk5/I/AymFStWnDhxYuPGjWkPQgFpaWlpaGjI/WIz7VkoIPX1\n9V/60pfuueeetAehgHzrW9966qmnXn311bQHARgofm8AAAAAhCdwAAAAAOEJHAAAAEB4AgcA\nAAAQnsABAAAAhCdwAAAAAOEJHAAAAEB4AgcAAAAQXmnaAzBEFRcXL1iw4Morr0x7EArL7Nmz\nT548mfYUFJYrrrhi/vz5w4YNS3sQCsvcuXNra2vTnoLCUldXN2fOnLSnABhARdlsNu0ZAAAA\nAC6It6gAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEA\nAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AUqEOHDq1atWru3LmVlZVFRUWbN28+\nc01HR8fdd989YcKE8vLy66+/vrm5+c9dAOf2yiuvFJ1hz549vdfYZgwcu4uB5lGOQeBJHUCe\nwFGgDh482NTUNHz48Hnz5vW5IJPJLFy4cPPmzQ8++GBzc/PkyZOXLl26devW/i+AfnrggQee\n6+Waa67JX7LNGDh2F4PGoxwDypM6gFOyFKSenp7cFz/+8Y+TJNm0adNpC5577rkkSZqamnKH\n3d3d06dPnzJlSv8XwHm9/PLLSZJs3779bAtsMwaO3cUg8CjHIPCkDiDPKzgKVHHxef7Rb926\ntby8fNmyZbnDkpKS5cuXv/nmm3v37u3nAui/Y8eOZTKZM8/bZgwcu4vB5FGOgeNJHUCewEHf\n9u3bV1NTU1ZWlj9TX1+fJElbW1s/F0A/3XHHHRUVFWVlZbfccsuOHTt6X7LNGDh2F4PGoxzp\n8qQOKBwCB307dOjQ2LFje5/JHR46dKifC+C8KioqVqxY8e1vf/ull1567LHHDhw4MH/+/N5v\n+rXNGDh2F4PAoxxDgSd1QOEoTXsABlxPT09nZ2f+sLKy8rwvZTyHoqKiC1xAYepzH06bNu3J\nJ5/Mn1y2bFl9ff3Xvva1xsbGc/8024yBY3fxIfIox1DmSR1w8fEKjotfa2vrpb289tpr/fmu\nqqqqw4cP9z6TO8wH/vMugN76sw/HjRu3YMGC3/zmN0eOHMmdsc0YOHYXg8+jHKnwpA4oHALH\nxa+mpmZXL9XV1f35rtra2tdff72rqyt/Jnejqbq6un4ugN76uQ+7u7uTXvdLs80YOHYXqfAo\nx+DzpA4oHALHxW/kyJG39lJRUdGf71q0aNHx48efeeaZ3GFPT8+mTZumTJkyffr0fi6A3vrc\nhydPnuy9Zv/+/S+88MK0adNGjx6dO2ObMXDsLgaBRzmGAk/qgMLhHhyFa9u2bSdOnGhtbU2S\n5OWXXy4vL0+SZPHixblfKy1atKihoeHee+/t6Oiorq7esGFDW1tbc3Nz/tvPuwDO69Of/vSI\nESNmz549duzYN95447vf/W5nZ+cPfvCD/ALbjIFjdzEIPMoxODypA/hAlkKV//VRb8eOHcsv\nOHLkyJe//OXx48eXlZXNmDHj+eefP+0nnHcBnNvjjz9+4403VlVVlZaWjhs3rrGx8Wc/+9lp\na2wzBo7dxUDzKMfg8KQOIKcom80OfEUBAAAAGEDuwQEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAAABCewAEAAACEJ3AAAAAA4QkcAAAAQHgCBwAA\nABCewAEAAACEJ3AAAAAA4QkcAAAAQHj/B3ux/EkTkj2NAAAAAElFTkSuQmCC"},"metadata":{"image/png":{"width":720,"height":720}}}]}],"metadata":{"kernelspec":{"name":"ir","display_name":"R","language":"R"},"language_info":{"name":"R","codemirror_mode":"r","pygments_lexer":"r","mimetype":"text/x-r-source","file_extension":".r","version":"4.0.3"}},"nbformat":4,"nbformat_minor":2}