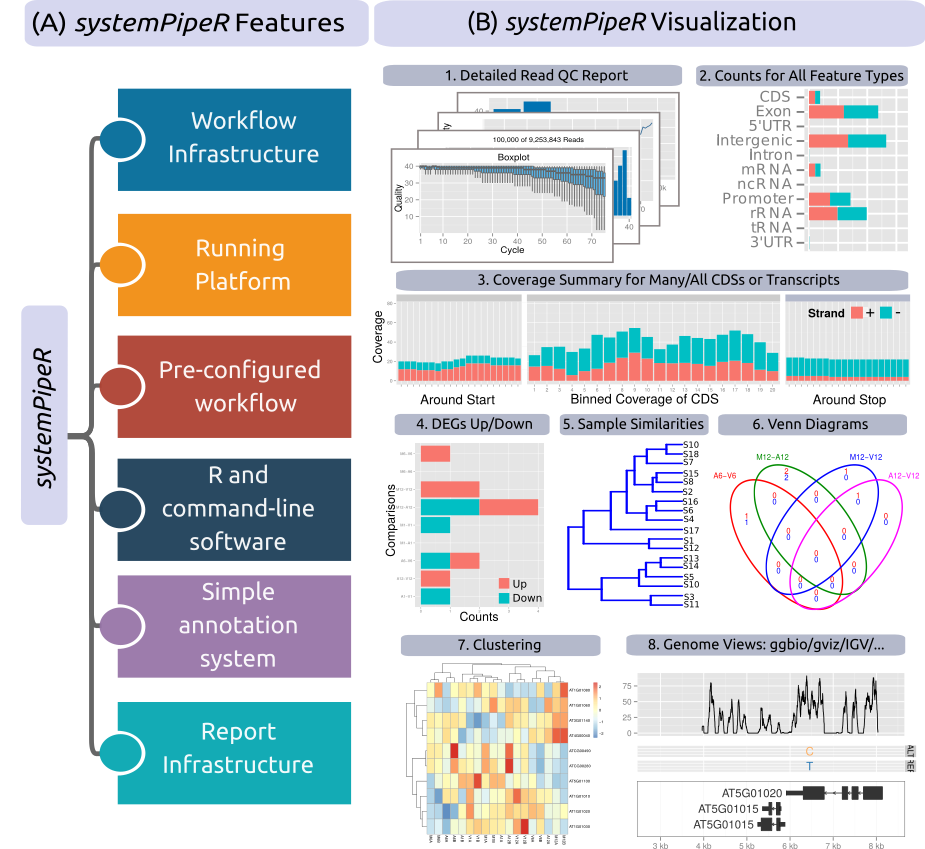
Figure 1: Relevant features in `systemPipeR`. Workflow design concepts are illustrated under (A). Examples of `systemPipeR's` visualization functionalities are given under (B).
Figure 1: Relevant features in `systemPipeR`. Workflow design concepts are illustrated under (A). Examples of `systemPipeR's` visualization functionalities are given under (B).
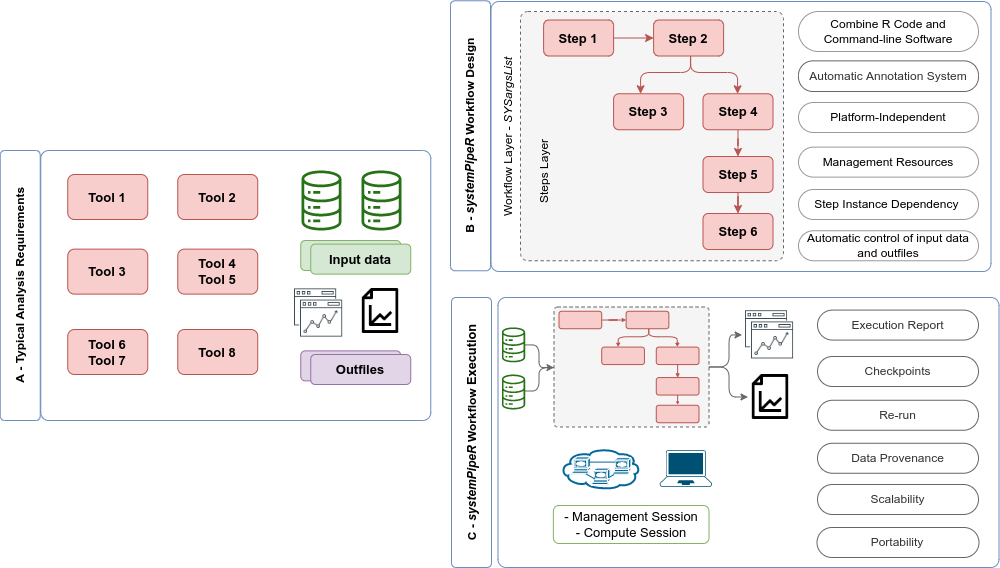
Figure 2: Overview of `systemPipeR` workflows management instances. A) A typical analysis workflow requires multiple software tools (red), as well the description of the input (green) and output files, including analysis reports (purple). B) `systemPipeR` provides multiple utilities to design and build a workflow, allowing multi-instance, integration of R code and command-line software, a simple and efficient annotation system, that allows automatic control of the input and output data, and multiple resources to manage the entire workflow. c) Options are provided to execute single or multiple workflow steps, while enabling scalability, checkpoints, and generation of technical and scientific reports.
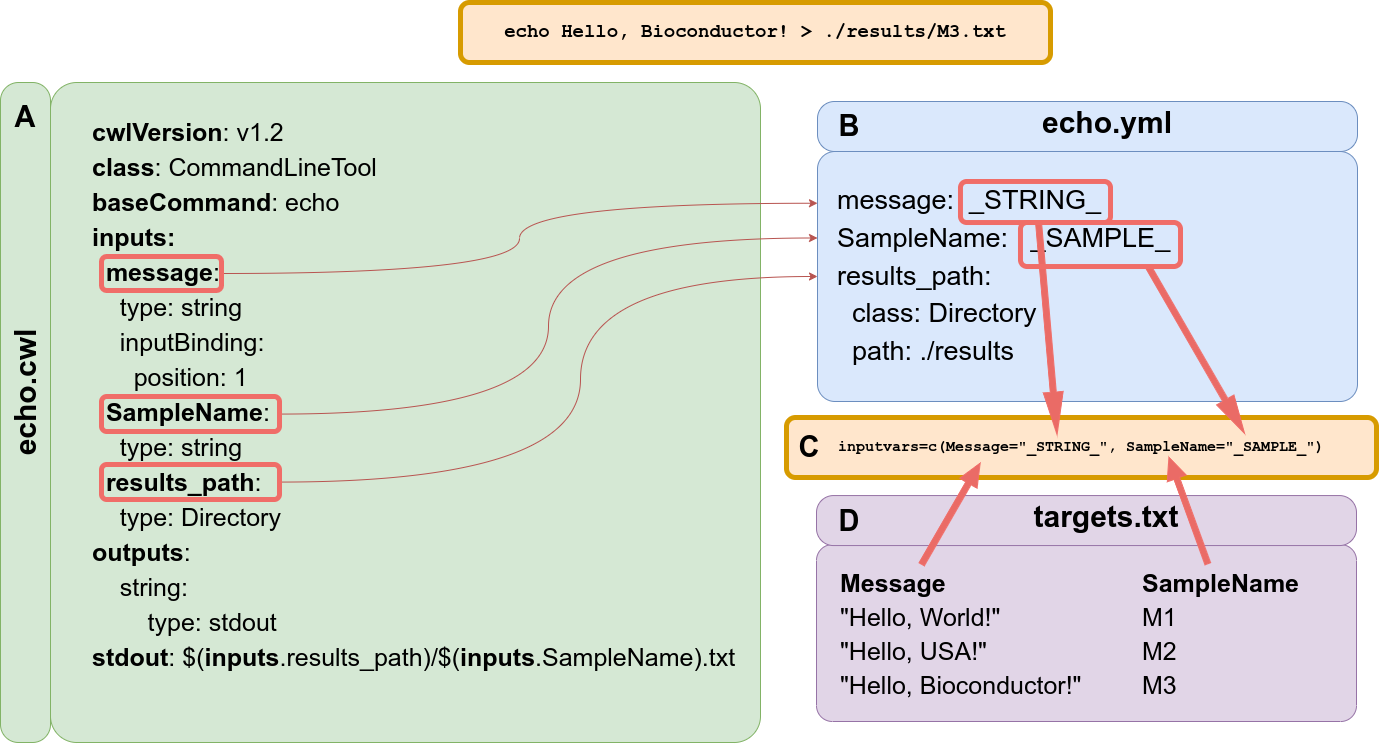
Figure 3: Example of ‘Hello World’ message using CWL syntax and demonstrating the connectivity with `systemPipeR`. (A) This file describes the command-line tool, here using ‘echo’ command. (B) This file describes all the parameters variables connected with the tool specification file. Here the reference value of the input parameter can be specific or can be filled dynamically, adding a variable that connects with the targets files from `systemPipeR`. (C) `SYSargsList` function provides the ‘inputvars’ arguments to build the connectivity between the CWL description of parameters and the targets files. The argument requires a named vector where each vector element is required to be defined in the CWL description of parameters file (B), and the names of the elements are needed to match the column names defined in the targets file (D).