---
title: "_systemPipeR_: NGS workflow and report generation environment"
author: "Author: Thomas Girke (thomas.girke@ucr.edu)"
date: "Last update: `r format(Sys.time(), '%d %B, %Y')`"
output:
BiocStyle::html_document:
toc: true
toc_depth: 3
fig_caption: yes
fontsize: 14pt
bibliography: bibtex.bib
---
```{r style, echo = FALSE, results = 'asis'}
BiocStyle::markdown()
options(width=100, max.print=1000)
knitr::opts_chunk$set(
eval=as.logical(Sys.getenv("KNITR_EVAL", "TRUE")),
cache=as.logical(Sys.getenv("KNITR_CACHE", "TRUE")),
warning=FALSE, message=FALSE)
```
Note: the most recent version of this tutorial can be found here and a short overview slide show [here](https://docs.google.com/presentation/d/175aup31LvnbIJUAvEEoSkpGsKgtBJ2RpQYd0Gs23dLo/embed?start=false&loop=false&delayms=60000).
# Introduction Slides
See [here](https://docs.google.com/presentation/d/175aup31LvnbIJUAvEEoSkpGsKgtBJ2RpQYd0Gs23dLo/embed?start=false&loop=false&delayms=60000).
# Background
[_`systemPipeR`_](http://www.bioconductor.org/packages/devel/bioc/html/systemPipeR.html) provides utilities for building and running automated end-to-end analysis workflows for a wide range of next generation sequence (NGS) applications such as RNA-Seq, ChIP-Seq, VAR-Seq and Ribo-Seq [@Girke2014-oy]. Important features include a uniform workflow interface across different NGS applications, automated report generation, and support for running both R and command-line software, such as NGS aligners or peak/variant callers, on local computers or compute clusters. The latter supports interactive job submissions and batch submissions to queuing systems of clusters. For instance, _`systemPipeR`_ can be used with most command-line aligners such as `BWA` [@Li2013-oy; @Li2009-oc], `TopHat2` [@Kim2013-vg] and `Bowtie2` [@Langmead2012-bs], as well as the R-based NGS aligners [_`Rsubread`_](http://www.bioconductor.org/packages/devel/bioc/html/Rsubread.html) [@Liao2013-bn] and [_`gsnap (gmapR)`_](http://www.bioconductor.org/packages/devel/bioc/html/gmapR.html) [@Wu2010-iq]. Efficient handling of complex sample sets (_e.g._ FASTQ/BAM files) and experimental designs is facilitated by a well-defined sample annotation infrastructure which improves reproducibility and user-friendliness of many typical analysis workflows in the NGS area [@Lawrence2013-kt].
Motivation and advantages of _`sytemPipeR`_ environment:
1. Facilitates design of complex NGS workflows involving multiple R/Bioconductor packages
2. Common workflow interface for different NGS applications
3. Makes NGS analysis with Bioconductor utilities more accessible to new users
4. Simplifies usage of command-line software from within R
5. Reduces complexity of using compute clusters for R and command-line software
6. Accelerates runtime of workflows via parallelzation on computer systems with mutiple CPU cores and/or multiple compute nodes
6. Automates generation of analysis reports to improve reproducibility
A central concept for designing workflows within the _`sytemPipeR`_ environment is the use of workflow management containers called _`SYSargs`_ (see Figure 1). Instances of this S4 object class are constructed by the _`systemArgs`_ function from two simple tabular files: a _`targets`_ file and a _`param`_ file. The latter is optional for workflow steps lacking command-line software. Typically, a _`SYSargs`_ instance stores all sample-level inputs as well as the paths to the corresponding outputs generated by command-line- or R-based software generating sample-level output files, such as read preprocessors (trimmed/filtered FASTQ files), aligners (SAM/BAM files), variant callers (VCF/BCF files) or peak callers (BED/WIG files). Each sample level input/outfile operation uses its own _`SYSargs`_ instance. The outpaths of _`SYSargs`_ usually define the sample inputs for the next _`SYSargs`_ instance. This connectivity is established by writing the outpaths with the _`writeTargetsout`_ function to a new _`targets`_ file that serves as input to the next _`systemArgs`_ call. Typically, the user has to provide only the initial _`targets`_ file. All downstream _`targets`_ files are generated automatically. By chaining several _`SYSargs`_ steps together one can construct complex workflows involving many sample-level input/output file operations with any combinaton of command-line or R-based software.
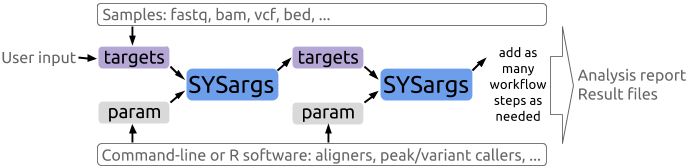
**Figure 1:** Workflow design structure of *systemPipeR*
The intended way of running _`sytemPipeR`_ workflows is via _`*.Rnw`_ or _`*.Rmd`_ files, which can be executed either line-wise in interactive mode or with a single command from R or the command-line using a [_`Makefile`_](https://github.com/tgirke/systemPipeR/blob/master/inst/extdata/Makefile). This way comprehensive and reproducible analysis reports can be generated in PDF or HTML format in a fully automated manner by making use of the highly functional reporting utilities available for R. Templates for setting up custom project reports are provided as _`*.Rnw`_ files by the helper package _`systemPipeRdata`_ and in the vignettes subdirectory of _`systemPipeR`_. The corresponding PDFs of these report templates are available here: [_`systemPipeRNAseq`_](https://github.com/tgirke/systemPipeR/blob/master/vignettes/systemPipeRNAseq.pdf?raw=true), [_`systemPipeRIBOseq`_](https://github.com/tgirke/systemPipeR/blob/master/vignettes/systemPipeRIBOseq.pdf?raw=true), [_`systemPipeChIPseq`_](https://github.com/tgirke/systemPipeR/blob/master/vignettes/systemPipeChIPseq.pdf?raw=true) and [_`systemPipeVARseq`_](https://github.com/tgirke/systemPipeR/blob/master/vignettes/systemPipeVARseq.pdf?raw=true). To work with _`*.Rnw`_ or _`*.Rmd`_ files efficiently, basic knowledge of [_`Sweave`_](https://www.stat.uni-muenchen.de/~leisch/Sweave/) or [_`knitr`_](http://yihui.name/knitr/) and [_`Latex`_](http://www.latex-project.org/) or [_`R Markdown v2`_](http://rmarkdown.rstudio.com/) is required.
[Back to Table of Contents]()
# Getting Started
## Download latest version of this tutorial
In case there is a newer version of this tutorial, download its `systemPipeR_Intro.Rmd` source and open it in your R IDE (e.g. vim-r or RStudio).
```{r download_latest, eval=FALSE}
download.file("https://raw.githubusercontent.com/tgirke/systemPipeRdata/master/vignettes/systemPipeR_Intro.Rmd", "systemPipeR_Intro.Rmd")
```
## Installation
The R software for running [_`systemPipeR`_](http://www.bioconductor.org/packages/devel/bioc/html/systemPipeR.html) can be downloaded from [_CRAN_](http://cran.at.r-project.org/). The _`systemPipeR`_ environment can be installed from the R console using the _`biocLite`_ install command. The associated data package [_`systemPipeRdata`_](https://github.com/tgirke/systemPipeRdata) can be installed the same way. The latter is a helper package for generating _`systemPipeR`_ workflow environments with a single command containing all parameter files and sample data required to quickly test and run workflows.
```{r install, eval=FALSE}
source("http://bioconductor.org/biocLite.R") # Sources the biocLite.R installation script
biocLite("systemPipeR") # Installs systemPipeR
biocLite("systemPipeRdata") # Installs systemPipeRdata
```
[Back to Table of Contents]()
## Loading package and documentation
```{r documentation, eval=FALSE}
library("systemPipeR") # Loads the package
library(help="systemPipeR") # Lists package info
vignette("systemPipeR") # Opens vignette
```
[Back to Table of Contents]()
## Load sample data and workflow templates
The mini sample FASTQ files used by this overview vignette as well as the associated workflow reporting vignettes can be loaded via the _`systemPipeRdata`_ package as shown below. The chosen data set [`SRP010938`](http://www.ncbi.nlm.nih.gov/sra/?term=SRP010938) contains 18 paired-end (PE) read sets from _Arabidposis thaliana_ [@Howard2013-fq]. To minimize processing time during testing, each FASTQ file has been subsetted to 90,000-100,000 randomly sampled PE reads that map to the first 100,000 nucleotides of each chromosome of the _A. thalina_ genome. The corresponding reference genome sequence (FASTA) and its GFF annotion files (provided in the same download) have been truncated accordingly. This way the entire test sample data set requires less than 200MB disk storage space. A PE read set has been chosen for this test data set for flexibility, because it can be used for testing both types of analysis routines requiring either SE (single end) reads or PE reads.
The following generates a fully populated _`systemPipeR`_ workflow environment (here for RNA-Seq) in the current working directory of an R session. At this time the package includes workflow templates for RNA-Seq, ChIP-Seq, VAR-Seq and Ribo-Seq. Templates for additional NGS applications will be provided in the future.
```{r genRna_workflow, eval=FALSE}
library(systemPipeRdata)
genWorkenvir(workflow="riboseq", bam=TRUE)
setwd("riboseq")
```
The working environment of the sample data loaded in the previous step contains the following preconfigured directory structure. Directory names are indicated in _**grey**_. Users can change this structure as needed, but need to adjust the code in their workflows accordingly.
* _**workflow/**_ (_e.g._ _rnaseq/_)
+ This is the directory of the R session running the workflow.
+ Run script ( _\*.Rnw_ or _\*.Rmd_) and sample annotation (_targets.txt_) files are located here.
+ Note, this directory can have any name (_e.g._ _**rnaseq**_, _**varseq**_). Changing its name does not require any modifications in the run script(s).
+ Important subdirectories:
+ _**param/**_
+ Stores parameter files such as: _\*.param_, _\*.tmpl_ and _\*\_run.sh_.
+ _**data/**_
+ FASTQ samples
+ Reference FASTA file
+ Annotations
+ etc.
+ _**results/**_
+ Alignment, variant and peak files (BAM, VCF, BED)
+ Tabular result files
+ Images and plots
+ etc.
The following parameter files are included in each workflow template:
1. _`targets.txt`_: initial one provided by user; downstream _`targets_*.txt`_ files are generated automatically
2. _`*.param`_: defines parameter for input/output file operations, _e.g._ _`trim.param`_, _`bwa.param`_, _`vartools.parm`_, ...
3. _`*_run.sh`_: optional bash script, _e.g._: _`gatk_run.sh`_
4. Compute cluster environment (skip on single machine):
+ _`.BatchJobs`_: defines type of scheduler for _`BatchJobs`_
+ _`*.tmpl`_: specifies parameters of scheduler used by a system, _e.g._ Torque, SGE, StarCluster, Slurm, etc.
[Back to Table of Contents]()
## Structure of _`targets`_ file
The _`targets`_ file defines all input files (_e.g._ FASTQ, BAM, BCF) and sample comparisons of an analysis workflow. The following shows the format of a sample _`targets`_ file included in the package. It also can be viewed and downloaded from _`systemPipeR`_'s GitHub repository [here](https://github.com/tgirke/systemPipeR/blob/master/inst/extdata/targets.txt). In a target file with a single type of input files, here FASTQ files of single end (SE) reads, the first three columns are mandatory including their column names, while it is four mandatory columns for FASTQ files of PE reads. All subsequent columns are optional and any number of additional columns can be added as needed.
### Structure of _`targets`_ file for single end (SE) samples
```{r targetsSE, eval=TRUE}
library(systemPipeR)
targetspath <- system.file("extdata", "targets.txt", package="systemPipeR")
read.delim(targetspath, comment.char = "#")
```
To work with custom data, users need to generate a _`targets`_ file containing the paths to their own FASTQ files and then provide under _`targetspath`_ the path to the corresponding _`targets`_ file.
[Back to Table of Contents]()
### Structure of _`targets`_ file for paired end (PE) samples
```{r targetsPE, eval=TRUE}
targetspath <- system.file("extdata", "targetsPE.txt", package="systemPipeR")
read.delim(targetspath, comment.char = "#")[1:2,1:6]
```
[Back to Table of Contents]()
### Sample comparisons
Sample comparisons are defined in the header lines of the _`targets`_ file starting with '``# ``'.
```{r comment_lines, eval=TRUE}
readLines(targetspath)[1:4]
```
The function _`readComp`_ imports the comparison information and stores it in a _`list`_. Alternatively, _`readComp`_ can obtain the comparison information from the corresponding _`SYSargs`_ object (see below).
Note, these header lines are optional. They are mainly useful for controlling comparative analyses according to certain biological expectations, such as identifying differentially expressed genes in RNA-Seq experiments based on simple pair-wise comparisons.
```{r targetscomp, eval=TRUE}
readComp(file=targetspath, format="vector", delim="-")
```
[Back to Table of Contents]()
## Structure of _`param`_ file and _`SYSargs`_ container
The _`param`_ file defines the parameters of a chosen command-line software. The following shows the format of a sample _`param`_ file provided by this package.
```{r param_structure, eval=TRUE}
parampath <- system.file("extdata", "tophat.param", package="systemPipeR")
```
The _`systemArgs`_ function imports the definitions of both the _`param`_ file
and the _`targets`_ file, and stores all relevant information in a _`SYSargs`_
object (S4 class). To run the pipeline without command-line software, one can
assign _`NULL`_ to _`sysma`_ instead of a _`param`_ file. In addition, one can
start _`systemPipeR`_ workflows with pre-generated BAM files by providing a
targets file where the _`FileName`_ column provides the paths to the BAM files.
Note, in the following example the usage of _`suppressWarnings()`_ is only relevant for
building this vignette. In typical workflows it should be removed.
```{r param_import, eval=TRUE}
args <- suppressWarnings(systemArgs(sysma=parampath, mytargets=targetspath))
args
```
Several accessor methods are available that are named after the slot names of the _`SYSargs`_ object.
```{r sysarg_access, eval=TRUE}
names(args)
```
Of particular interest is the _`sysargs()`_ method. It constructs the system
commands for running command-lined software as specified by a given _`param`_
file combined with the paths to the input samples (_e.g._ FASTQ files) provided
by a _`targets`_ file. The example below shows the _`sysargs()`_ output for
running TopHat2 on the first PE read sample. Evaluating the output of
_`sysargs()`_ can be very helpful for designing and debugging _`param`_ files
of new command-line software or changing the parameter settings of existing
ones.
```{r sysarg_access2, eval=TRUE}
sysargs(args)[1]
modules(args)
cores(args)
outpaths(args)[1]
```
The content of the _`param`_ file can also be returned as JSON object as follows (requires _`rjson`_ package).
```{r sysarg_json, eval=TRUE}
systemArgs(sysma=parampath, mytargets=targetspath, type="json")
```
[Back to Table of Contents]()
# More detail
See `systemPipeR` vignette [here](https://bioconductor.org/packages/release/bioc/vignettes/systemPipeR/inst/doc/systemPipeR.html#workflow-overview).
# Workflow demo
RIBO-Seq example [here](https://htmlpreview.github.io/?https://raw.githubusercontent.com/tgirke/systemPipeRdata/master/inst/extdata/workflows/riboseq/systemPipeRIBOseq.html)
[Back to Table of Contents]()
# Version information
```{r sessionInfo}
sessionInfo()
```
[Back to Table of Contents]()
# References